Embed Size (px)
Citation preview

www.pei.de
CMC Strategy Forum Europe 2015
22-24 May Killarney, Ireland.
Technical innovations – impact on regulatory expectations for product
characterization
Steffen GrossHead, Section Monoclonal and Polyclonal Antibodies,Paul-Ehrlich-Institut

Disclaimer
The view expressed in the following is the ones of the presenter and does not necessary express the view of either the CHMP, BWP, EDQM or the Paul-Ehrlich-Institut

3
Outline- Information in the documentation
- CTA- MAA
- Characterisation- Assay sensitivity- Setting specifications, define acceptance criteria- Clinical qualification
- Control strategy- RTRM and RTRT
- Legal requirements for testing- Monographs

Understanding of the molecule is a prerequiste forclinical trials
Characterisation of a biotechnological or biological substance (which includes the determination ofphysico-chemical properties, biological activity, immuno-chemical properties, purity and impurities) byappropriate techniques is necessary to allow relevant specification to be established. Reference to theliterature data only is not acceptable. Adequate characterisation is performed in the developmentphase prior to phase I and, where necessary, following significant process changes.

FIM/Phase I studies
5.1. Determination of strength and potencyTo determine a safe starting dose, the methods used for determination of the strength and/or the potency of the product need to be relevant, reliable and qualified.
5.2. Qualification of the material used The material used in non-clinical studies should be representative of the material to be used for FIH/early CT administration. It is important to have an adequate level of quality characterisation even at this early point of development. A characterisation of the product including its heterogeneity, degradation profile and process-related impurities should be performed.
Investigator Brochure (CTA)
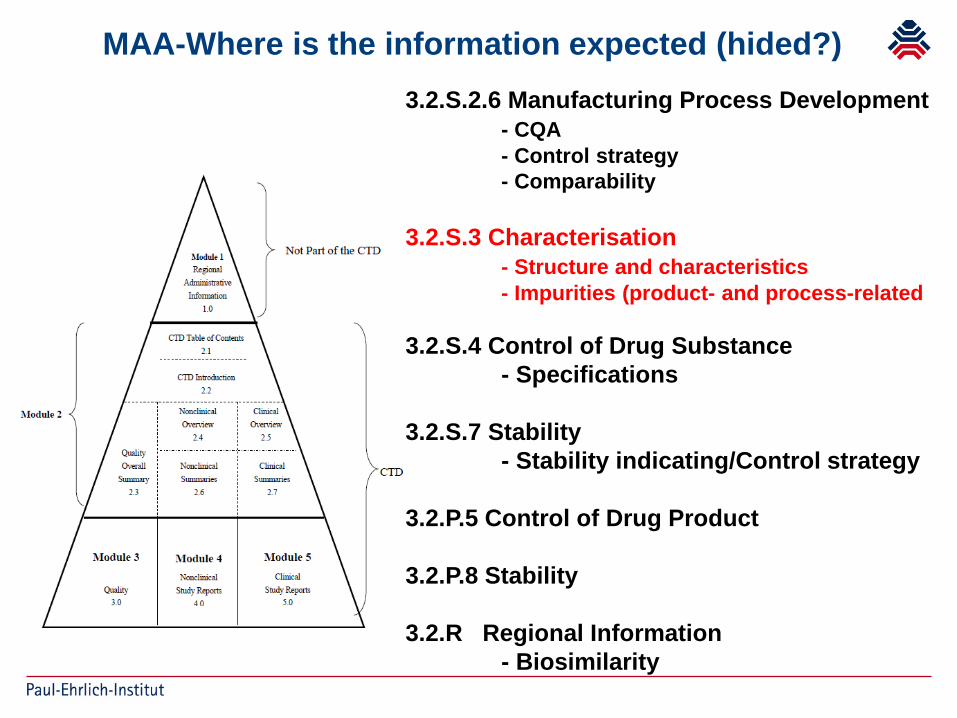
MAA-Where is the information expected (hided?)3.2.S.2.6 Manufacturing Process Development
- CQA- Control strategy- Comparability
3.2.S.3 Characterisation- Structure and characteristics- Impurities (product- and process-related
3.2.S.4 Control of Drug Substance- Specifications
3.2.S.7 Stability- Stability indicating/Control strategy
3.2.P.5 Control of Drug Product
3.2.P.8 Stability
3.2.R Regional Information- Biosimilarity
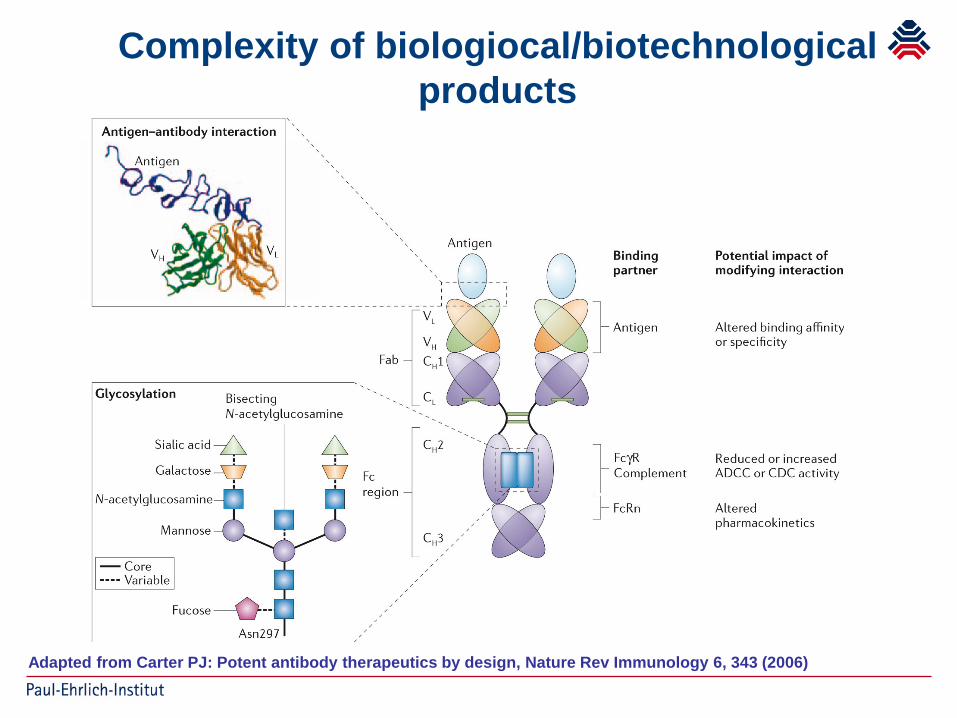
Complexity of biologiocal/biotechnologicalproducts
Adapted from Carter PJ: Potent antibody therapeutics by design, Nature Rev Immunology 6, 343 (2006)
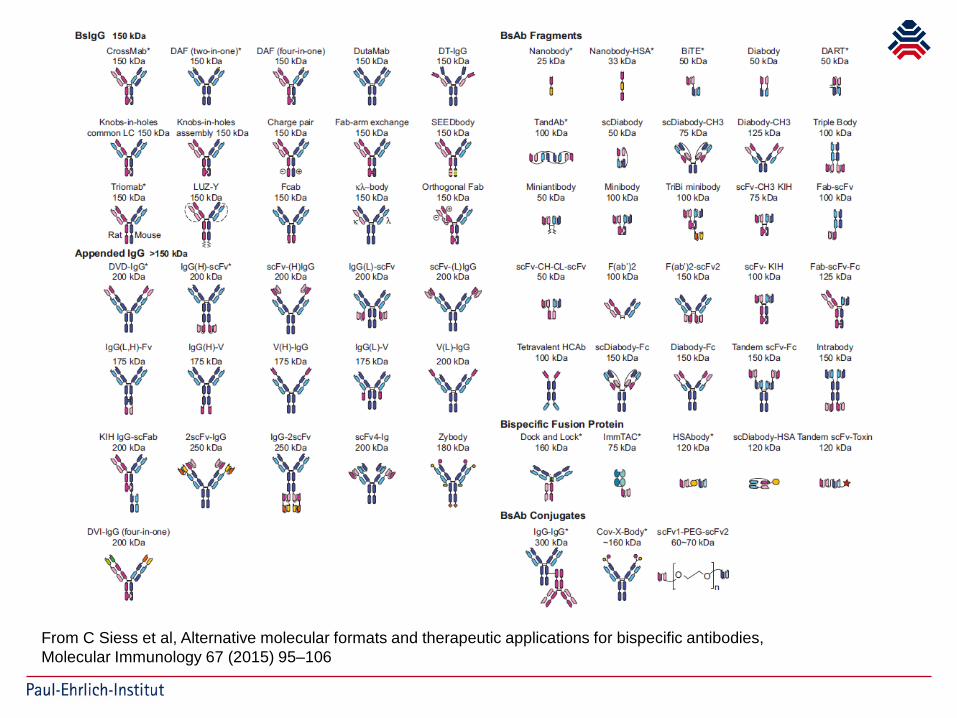
From C Siess et al, Alternative molecular formats and therapeutic applications for bispecific antibodies,Molecular Immunology 67 (2015) 95–106
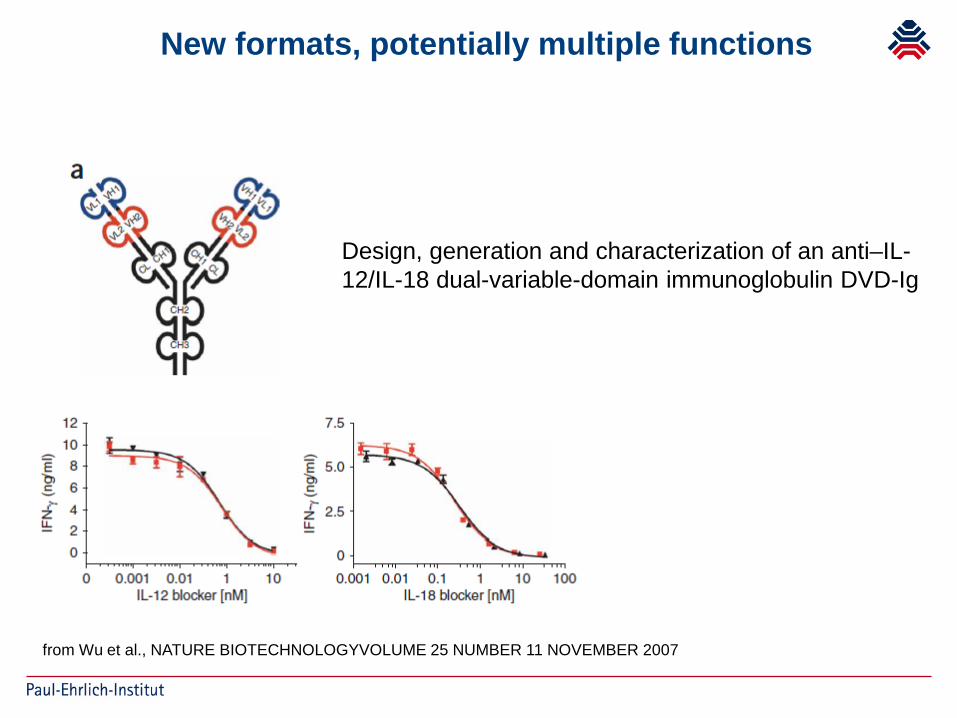
New formats, potentially multiple functions
Design, generation and characterization of an anti–IL-12/IL-18 dual-variable-domain immunoglobulin DVD-Ig
from Wu et al., NATURE BIOTECHNOLOGYVOLUME 25 NUMBER 11 NOVEMBER 2007
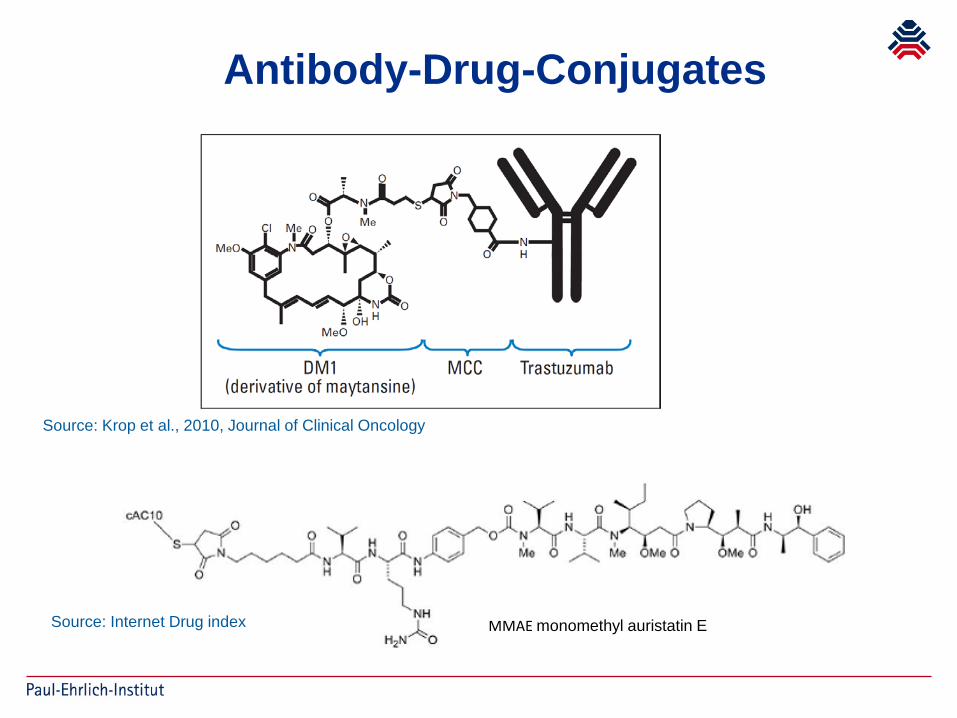
Antibody-Drug-Conjugates
Source: Krop et al., 2010, Journal of Clinical Oncology
Source: Internet Drug index MMAE monomethyl auristatin E
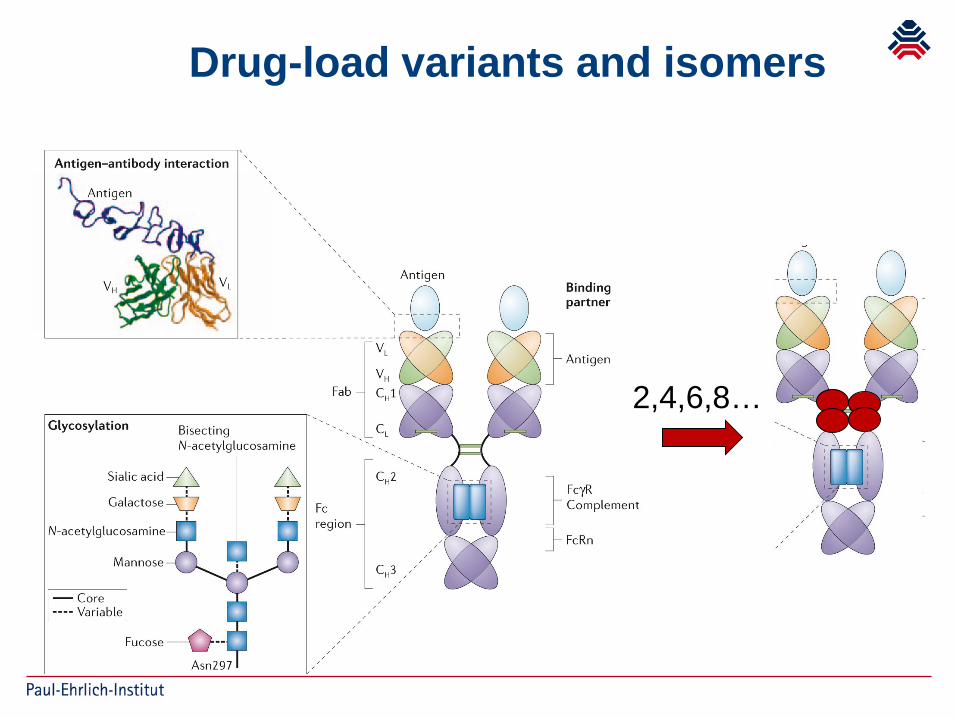
Drug-load variants and isomers
2,4,6,8…

Characterisation/Specifications
Maytansinol• MayOH is a well-characterized, stable compound• MayOH is a well-defined single compound that is produced and tested for conformance against an appropriate specification using qualified reference standards.• MayOH has a well-defined and consistent impurity profile
MMAE monomethyl auristatin ESpecifications have been established based on principles outlined in ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances Specifications.
Antibody SpecificationsThese specifications have to be established based on principles outlined in ICH Q6B.
The specifications set for linker and cytotoxic drug should include the recommend acceptable amounts for residual solvents guidance given in the ICH Q3C “Impurities: Guideline for residual solvents” should be followed.
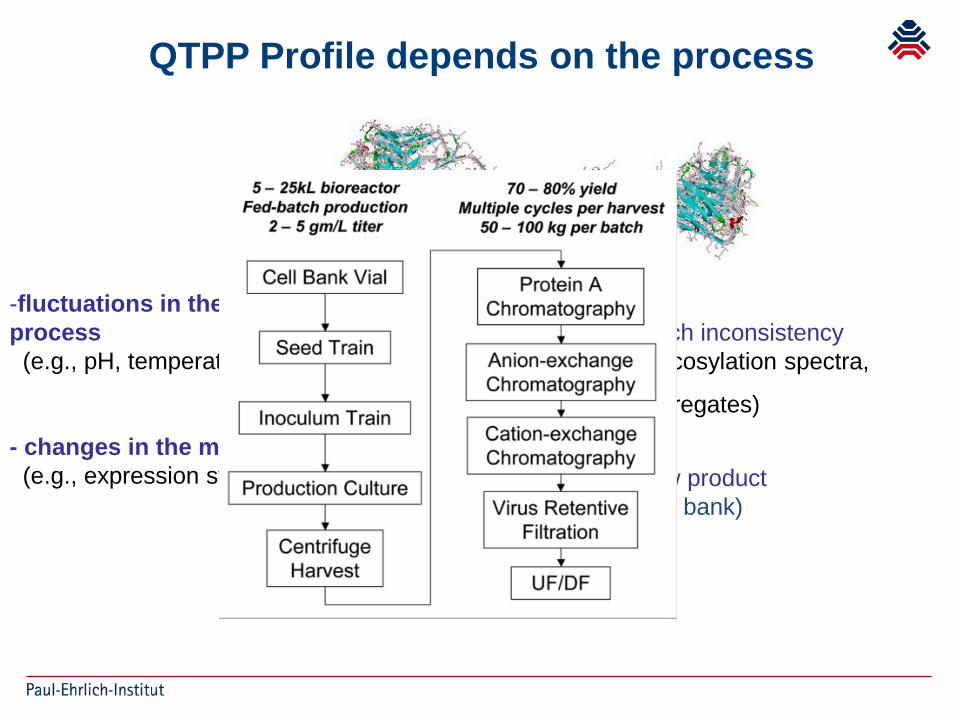
-fluctuations in the manufacturing process(e.g., pH, temperature, culture media):
- changes in the manufacturing process(e.g., expression system): New product
(cell bank)
Batch inconsistency(glycosylation spectra,
aggregates)
QTPP Profile depends on the process
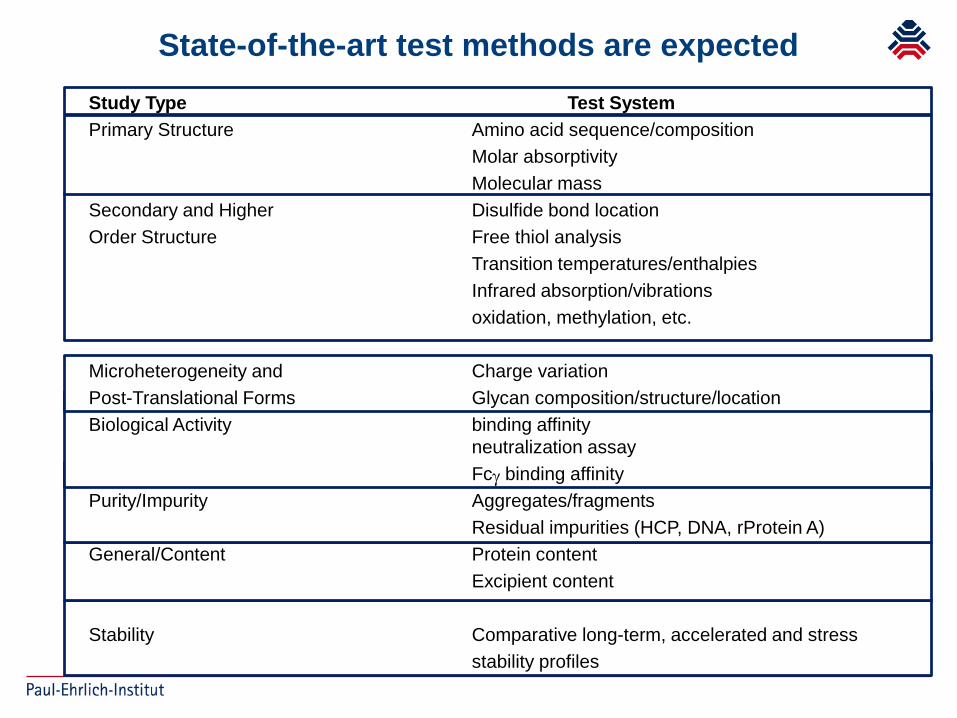
State-of-the-art test methods are expectedStudy Type Test System Primary Structure Amino acid sequence/composition
Molar absorptivityMolecular mass
Secondary and Higher Disulfide bond locationOrder Structure Free thiol analysis
Transition temperatures/enthalpiesInfrared absorption/vibrationsoxidation, methylation, etc.
Microheterogeneity and Charge variationPost-Translational Forms Glycan composition/structure/locationBiological Activity binding affinity
neutralization assayFcγ binding affinity
Purity/Impurity Aggregates/fragmentsResidual impurities (HCP, DNA, rProtein A)
General/Content Protein contentExcipient content
Stability Comparative long-term, accelerated and stressstability profiles

Sequence variantsBy examining the crystal structure structural predictions for the variant were made
– position is on the surface of the molecule
– variant is not predicted to impact the structure of the molecule
– variant is not expected to affect the folding of the protein
– variant is not predicted to affect the hinge region
– Position is not in a CDR, so it is not expected to affect antigen binding.
– variant does not affect the potency (91%) of the molecule using the cell based potency assay.
– variant is not in the FcRn binding region, so it is not expected to affect clearance
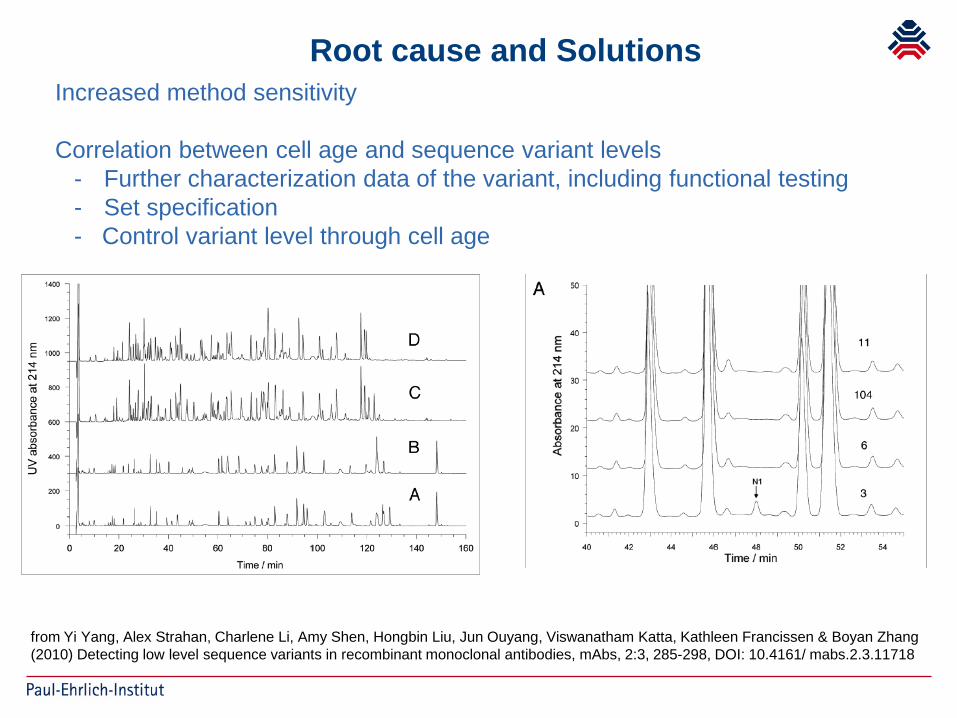
from Yi Yang, Alex Strahan, Charlene Li, Amy Shen, Hongbin Liu, Jun Ouyang, Viswanatham Katta, Kathleen Francissen & Boyan Zhang (2010) Detecting low level sequence variants in recombinant monoclonal antibodies, mAbs, 2:3, 285-298, DOI: 10.4161/ mabs.2.3.11718
Root cause and SolutionsIncreased method sensitivity
Correlation between cell age and sequence variant levels- Further characterization data of the variant, including functional testing- Set specification- Control variant level through cell age

Setting specifications
All features of the molecule, relevant for safety/efficiacy
Possibility to define different specifications for biologicalactivity depending on the MoA (and indication)
Binding might not be sufficient
Define pre-specified acceptance criteria
for biosimilars within the originators range but tight(justified) for process related impurities, state of the artlimits
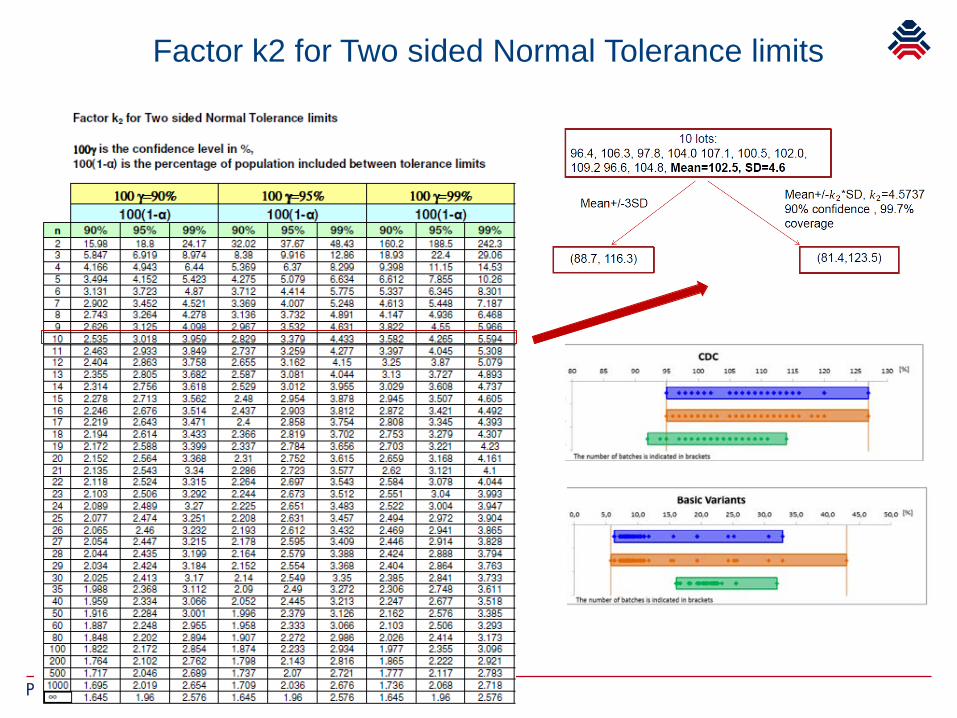
Factor k2 for Two sided Normal Tolerance limits
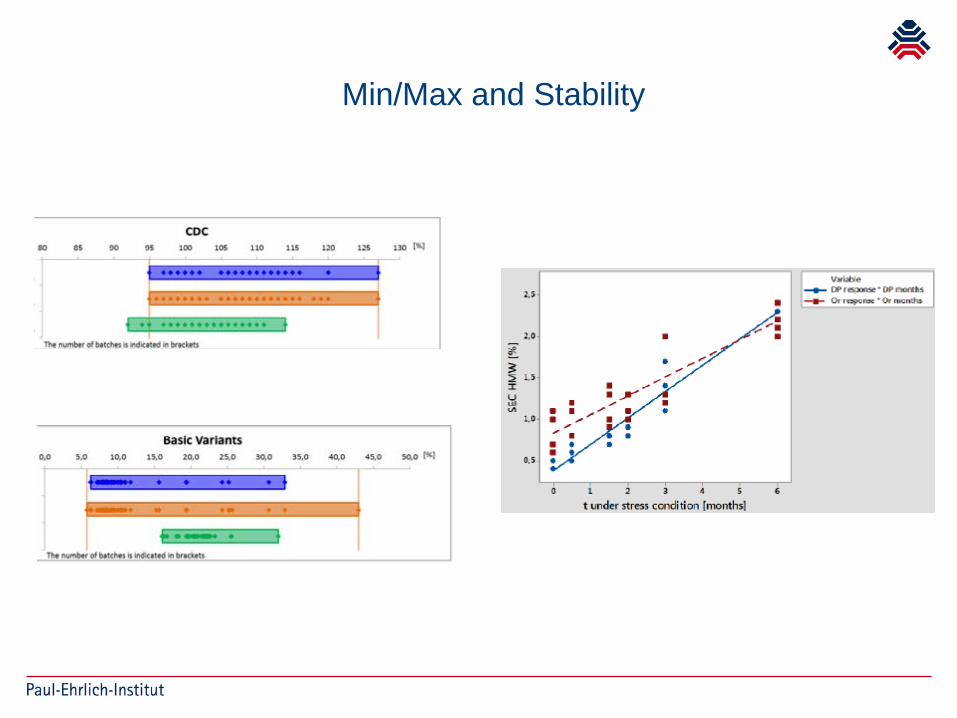
Min/Max and Stability
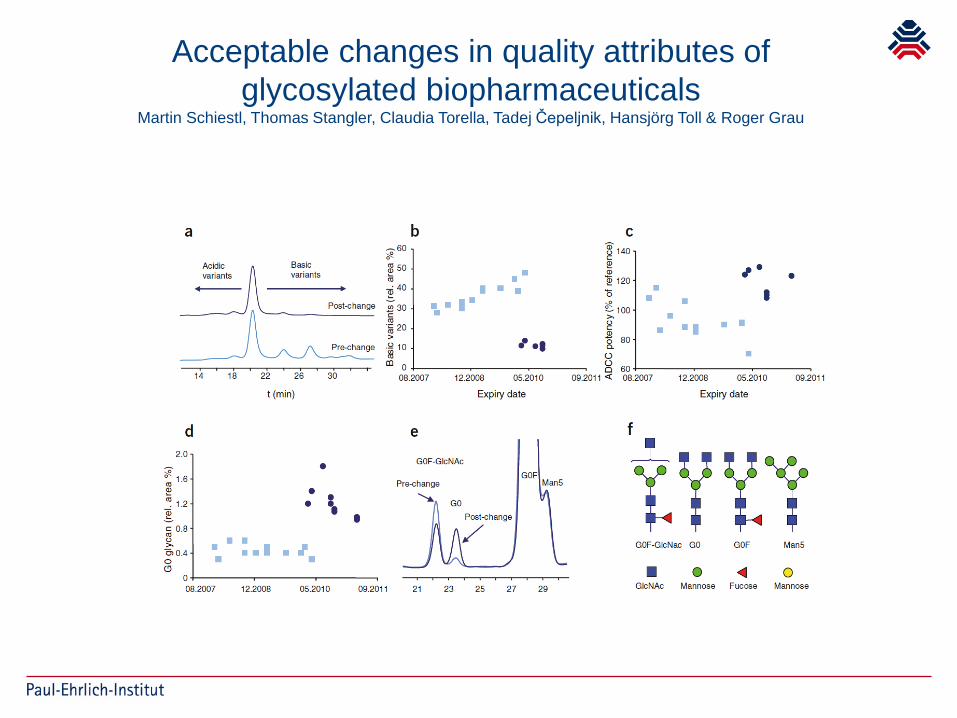
Acceptable changes in quality attributes ofglycosylated biopharmaceuticals
Martin Schiestl, Thomas Stangler, Claudia Torella, Tadej Čepeljnik, Hansjörg Toll & Roger Grau
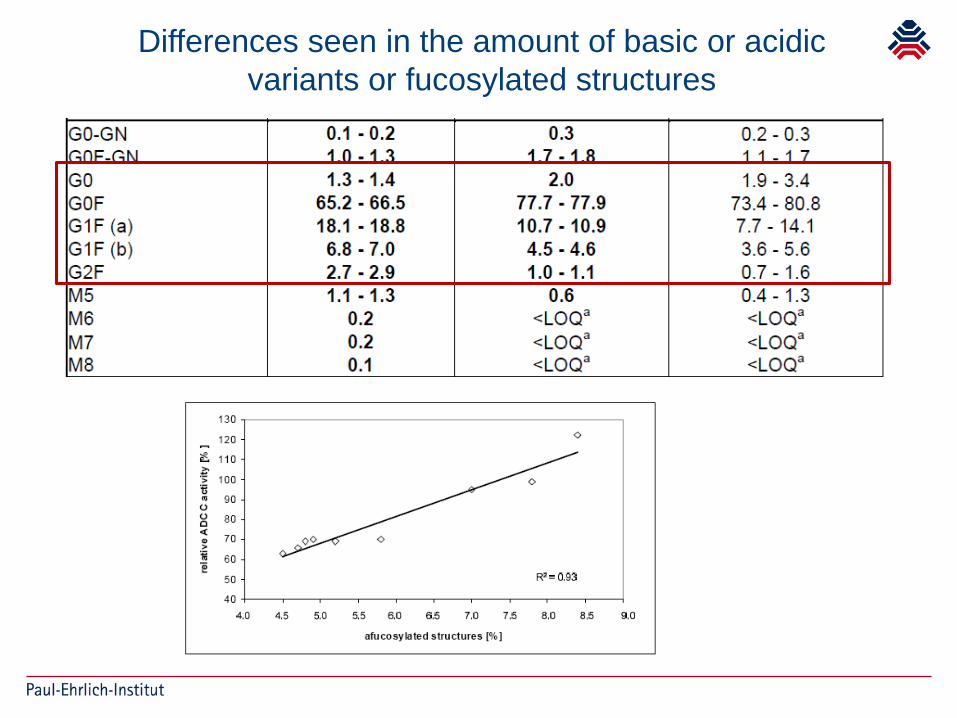
Differences seen in the amount of basic or acidic variants or fucosylated structures

Clinical studies with Biosimilars may provideevidence that acceptance criteria/ranges are
clinically qualified (or not)
Further implications for future manufacturing process changes if Fc function plays a role
Ask for tightening acceptance ranges, what are acceptable criteria without clinical data?
If unclear, ask for contribution of activity to overall MoA, if unclear ask for clinical studies or reject changes?

ICH Q8 (R2)• A comprehensive pharmaceutical development approach will generate
process and product understanding and identify sources of variability. • Sources of variability that can impact product quality should be identified,
appropriately understood, and subsequently controlled. • Understanding sources of variability and their impact on downstream
processes or processing, in-process materials, and drug product quality can provide an opportunity to shift controls upstream and minimize the need for end product testing.
Control Strategy

Knowledge of process performance when operated under worst-case conditions for each CQA.
Small scale studies are considered essential in order to address multivariate parameters
Provide scientific rationale for worst case All DoEs to be provided? Moving outside of worst-case conditions
The Design Space is limited by the multivariate ranges for all critical process parameters (CPPs).
DoE and Small scale studies/linkage studies

Initial ConditionsSet Points
Raw MaterialAttributes
Process variables- Equipment preparation- Manufacturing process
Performance and Quality Variables
Real-time Multivariate Statistical Monitoring (RT-MSPM)
• Need for multivariate analysis arises since most process data are complex and multivariate in nature
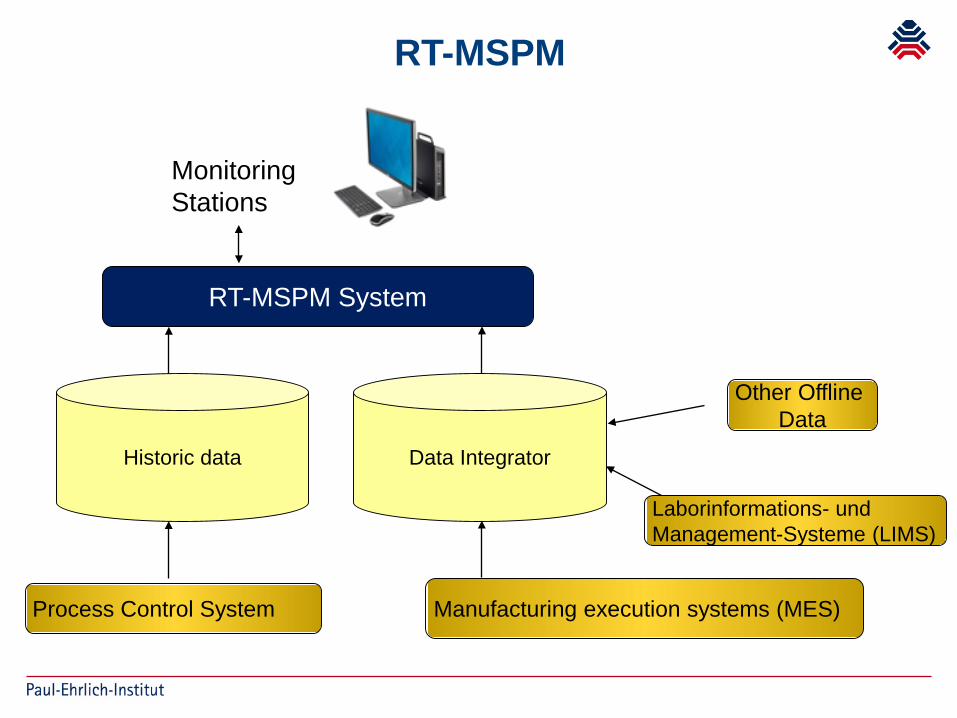
RT-MSPM
Historic data
RT-MSPM System
Process Control System
Monitoring Stations
Manufacturing execution systems (MES)
Laborinformations- und Management-Systeme (LIMS)
Other OfflineData
Data Integrator
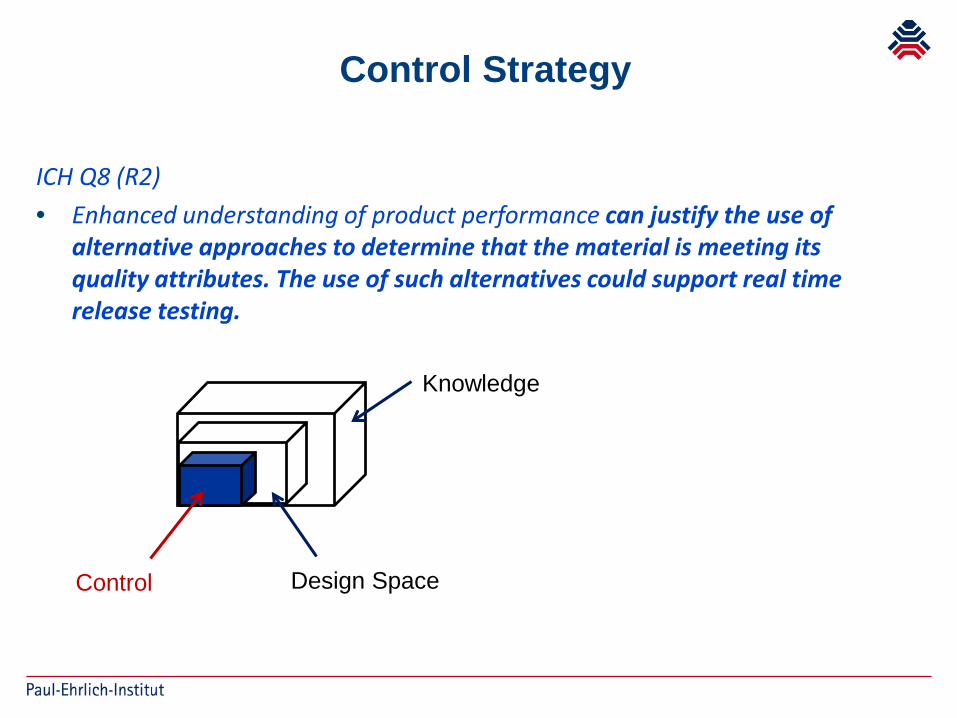
ICH Q8 (R2)• Enhanced understanding of product performance can justify the use of
alternative approaches to determine that the material is meeting its quality attributes. The use of such alternatives could support real time release testing.
Control Strategy
Knowledge
Design SpaceControl

RTRT as part of a Control Strategy System of release that gives assurance that the product is of
intended quality, based on the information collected during the manufacturing process, through product knowledge and on process understanding and control
Product knowledge and process understanding, the use of quality risk management principles and the application of an appropriate pharmaceutical quality system, as defined within ICH Q8,Q9 and Q10 provide the platform for establishing RTRT mechanisms
combination of a RTR approach for certain critical quality attributes (CQAs) and a more conventional evaluation for other CQAs (partial RTR).
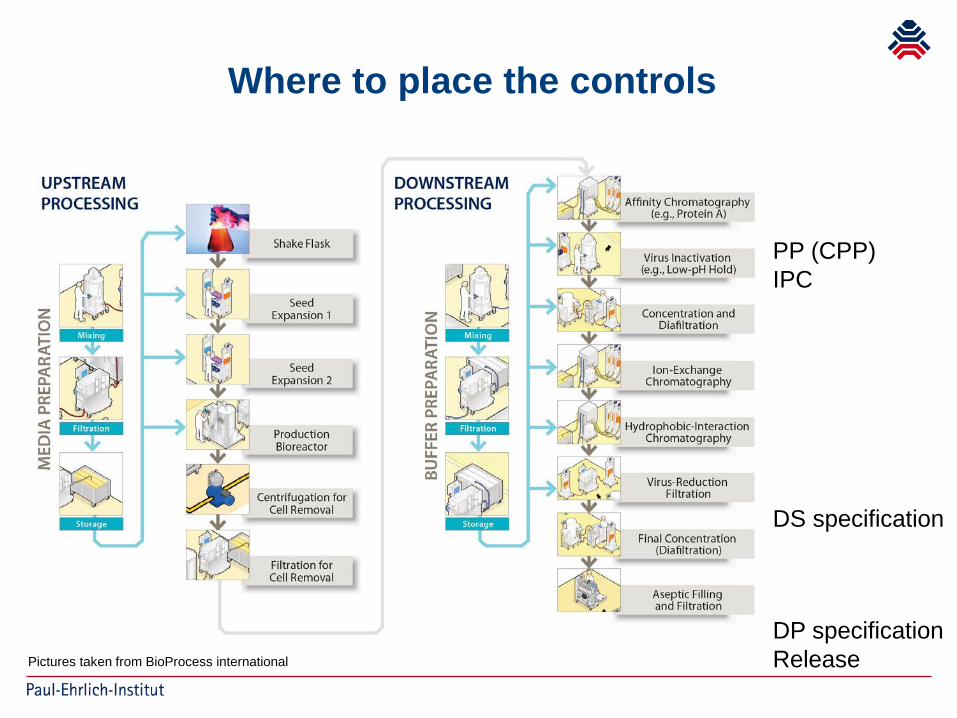
Where to place the controls
Pictures taken from BioProcess international
PP (CPP)IPC
DS specification
DP specificationRelease
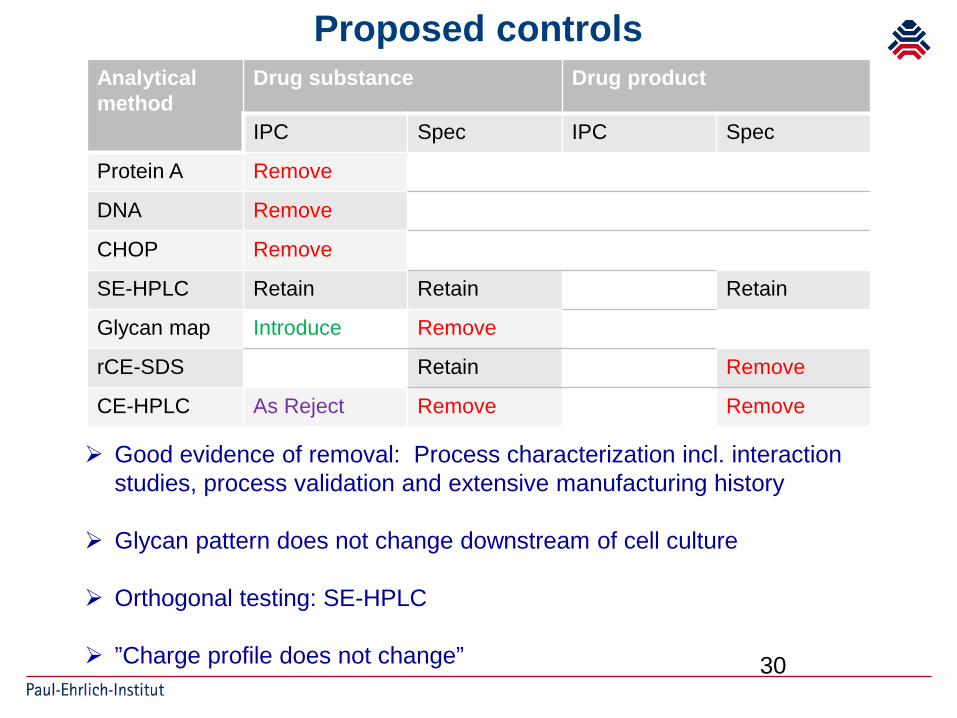
Proposed controls
30
Analyticalmethod
Drug substance Drug product
IPC Spec IPC Spec
Protein A Remove
DNA Remove
CHOP Remove
SE-HPLC Retain Retain Retain
Glycan map Introduce Remove
rCE-SDS Retain Remove
CE-HPLC As Reject Remove Remove
Good evidence of removal: Process characterization incl. interaction studies, process validation and extensive manufacturing history
Glycan pattern does not change downstream of cell culture
Orthogonal testing: SE-HPLC
”Charge profile does not change”
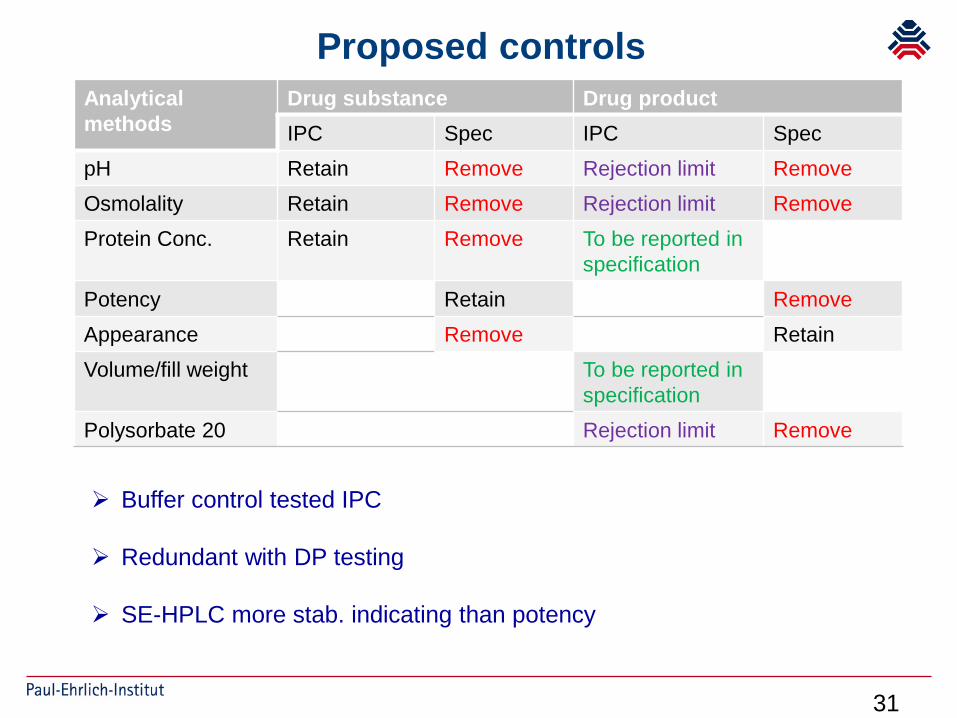
31
Proposed controlsAnalytical methods
Drug substance Drug productIPC Spec IPC Spec
pH Retain Remove Rejection limit RemoveOsmolality Retain Remove Rejection limit RemoveProtein Conc. Retain Remove To be reported in
specificationPotency Retain RemoveAppearance Remove RetainVolume/fill weight To be reported in
specificationPolysorbate 20 Rejection limit Remove
Buffer control tested IPC
Redundant with DP testing
SE-HPLC more stab. indicating than potency
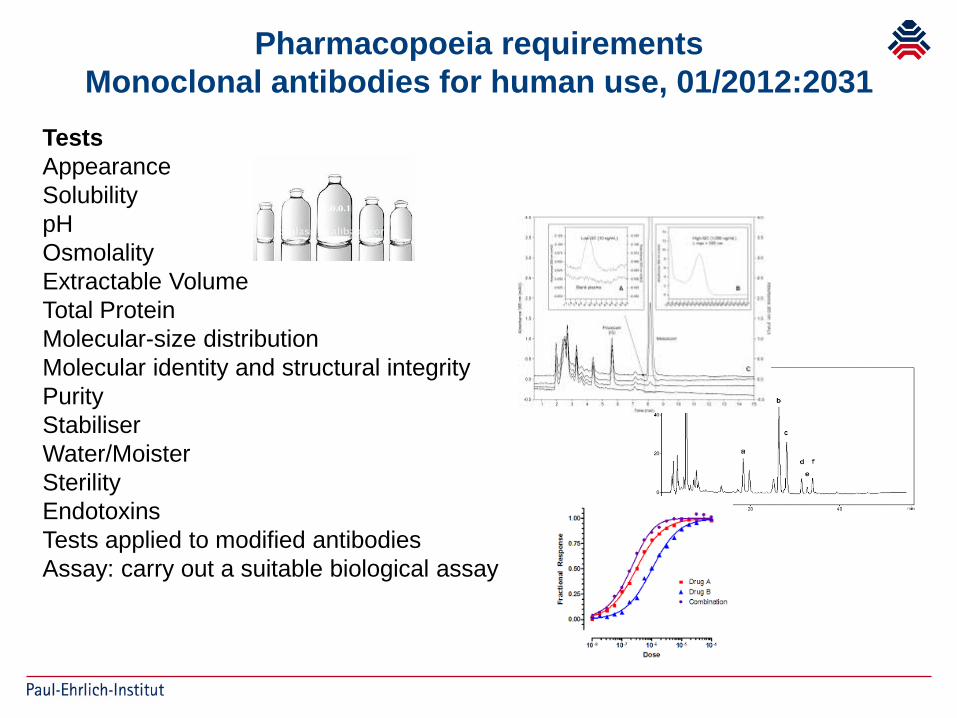
Pharmacopoeia requirementsMonoclonal antibodies for human use, 01/2012:2031
TestsAppearanceSolubilitypHOsmolalityExtractable VolumeTotal ProteinMolecular-size distributionMolecular identity and structural integrityPurityStabiliserWater/MoisterSterilityEndotoxinsTests applied to modified antibodiesAssay: carry out a suitable biological assay
INFLIXIMAB CONCENTRATED SOLUTION XXXX:2928
Product specific monographs (mAbs) Block innovation May inhibit product development May not reflect approved dossier Increase burden to industry and regulators
International Standard? Own in house standards (qualified)

Summary
Adequate characterisation is performed in the development phase prior to phase I
Advanced process analytics First principles modeling provides enhanced process understanding
and control opportunities Real-time multivariate statistical process monitoring enables proactive
monitoring of bioprocess operations, increases our understanding of raw material’s role in variation, and enables quality by design principles
Process analytical chemistry tools are important for controlling product quality and process performance
A “minimum” control system for a QbD product is considered necessary.
Legal requirements (monographs) and guidance documents for testing

Thank You!





























