Embed Size (px)
Citation preview

Vol. 169, No. 3
Cloning and Characterization of the Escherichia coli lit Gene, WhichBlocks Bacteriophage T4 Late Gene Expressiont
CHENG KAO, EFFIE GUMBS, AND LARRY SNYDER*
Department of Microbiology and Public Health, Michigan State University, East Lansing, Michigan 48824-1101
Received 28 July 1986/Accepted 16 December 1986
Escherichia coli lit mutations inhibit gene expression late in infection by bacteriophage T4. We cloned the litgene from wild-type E. coli and three independent lit mutants. We present evidence that lit mutations [renamedlit(Con) mutations] cause overproduction of the lit gene product and that overproduction of this product causes
the inhibition of gene expression. We also present evidence that the lit gene product is nonessential for E. coligrowth, although the gene is common to most E. coli K-12 strains.
The regulation of gene expression in bacteriophage T4 is a
well-orchestrated process involving a plethora of T4-encoded proteins as well as Escherichia coli gene products(2, 7). In our search for E. coli genes that are involved in T4late gene expression, we have isolated five independent E.coli mutants with mutations that block the expression ofwild-type T4 genes in infection and thus prevent propagationof the phage (5). These E. coli mutations, called lit mutationsfor late inhibitors of T4, all map at 25 min on the E. coli K-12genetic map and produce a seemingly specific inhibition ofgene expression, since neither DNA replication nor cell lysisis affected.Rare T4 point mutations overcome the inhibition of T4
gene expression imposed by lit mutations. These T4 muta-tions are called gol, for they permit growth on lit (5). Wesequenced five independent gol mutations, and they were allsingle base pair transitions within a 40-base-pair region aboutone-quarter of the way into the coding sequence for gene 23,the major capsid protein gene of T4 (4). However, althoughthe mutations are in gene 23, the Gol phenotype seems to beunrelated to the function of gene 23 protein itself, sinceamber mutations in gene 23 which male only the N-terminalone-fourth of the polypeptide do not affect this other pheno-type (3). The gol mutations seem to be cis- acting, at least forthe expression of gene 23, and maybe even other geneswhich are widely separated on the T4 genome (3). In additionto its cis effect on gene 23, the wild-type gol site may alsohave a trans-acting effect, possibly by inhibiting the expres-sion of other genes.
In this paper we present further characterization of E. colilit mutations. We present evidence that the lit mutations are
dominant and cause the overproduction of at least twopolypeptides of approximately 35 and 18 kilodaltons, one ofwhich may be the product of the lit gene. These polypeptidesare nonessential for E. coli growth, because wild-type cellsare phenotypically indistinguishable from cells in which theentire lit gene region has been deleted. To avoid confusion,we are changing our previous designation for lit mutations tolit(Con) mutations because they cause overproduction of thegene product rather than inactivating it, although we do notknow if they are analogous to operator-constitutive muta-tions in other systems. The wild-type gene will be designated
* Corresponding author.t Journal article no. 12065 from the Michigan Agricultural Exper-
iment Station.
lit', and the absence of a functional lit gene will be desig-nated lit'.
MATERIALS AND METHODSBacterial and phage strains. The strains used, their rele-
vant characteristics, and their source or reference are listedin Table 1.Media and antibiotics. E. coli was grown in either LB broth
(1% tryptone, 1% NaCl, 0.5% yeast extract) or tryptonebroth (0.5% NaCl, 1% tryptone, 1% Casamino Acids, 20 ,ugof thiamine per ml). Tryptone broth was used in geneticcrosses. Where appropriate, antibiotics were added in thefollowing concentrations: ampicillin, 25 jig/ml; chloram-phenicol, 20 ptg/ml; kanamycin, 50 p.g/ml; streptomycin, 100,ug/ml; and tetracycline, 10 V.g/ml.Conjugation and transduction. Hfr and F' mating proce-
dures were done as described previously (11). The recombi-nants were selected and purified once by patching on selec-tive plates before testing for the unselected markers. Gener-alized P1 phage transduction was done by the method ofMiller (15), but the bacteria were washed three times withsaline containing 10 mM sodium citrate prior to plating. T4transduction was done by the method of Takahashi and Saito(19) with a multiple T4 mutant, Nam55 (gene 42), Eam5l(gene 56), nd28 (denA), rH23 (denB), P-alc, obtained from E.Kutter.
Determination of the Lit phenotype. The qualitative pres-ence or absence of the lit(Con) mutation either in subclonesor in the E. coli genome was determined by streaking abacterial colony across a streak of wild-type T4 at a titer ofabout 107 per ml. A more sensitive test, spotting approxi-mately 105 T4+ phage onto a lawn of E. coli, was performedfor all promising cross-streaked clones. The same amount ofgol mutant phage was also spotted as a positive control. Theplates were usually incubated at 30°C. In determining theeffects of temperature, the cells were grown at 30°C andduplicate spot-test plates were incubated at 37 and 42°C aswell as 30°C.
Test of dominance of lit(Con) mutations. A merodiploid wasmade by crossing the F'-containing strain KLF25/KL181(14) with a pBR322-containing recA derivative of the lit(Con)mutant MPH6 (5), selecting for Trp+ recombinants andcounterselecting the donor with ampicillin. The apparentrecombinants that grew on the selective plates were patchpurified and then tested for the Lit(Con) phenotype. Toascertain that the F' factor included the region of the lit(Con)mutations, we crossed an apparent recombinant which
1232
JOURNAL OF BACTERIOLOGY, Mar. 1987, p. 1232-12380021-9193/87/031232-07$02.00/0Copyright © 1987, American Society for Microbiology
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

E. COLI lit GENE AND T4 GENE EXPRESSION 1233
TABLE 1. Bacterial and phage strains and plasmids
Strain, phage, Relevant characteristics Source or referenceor plasmid
E. coliAB2495 F- multiple auxotroph, parent to MPH Lit(Con) mutants 5MPH5 NTG-induced Lit(Con) mutant 5MPH6 NTG-induced Lit(Con) mutant 5MPH21 NTG-induced Lit(Con) mutant 5X1081T1 minB; no sequence homologous to lit gene region 61046 recA rk- mk+ B. ChelmAB484 F- multiple auxotroph B. BachmannRAMP1 AB484 transduced to polA(Ts) This workHfr JC158 Clockwise transfer starting at 0 min B. BachmannHfr 4206 Counterclockwise transfer between serA and lysA B. BachmannKLF25/KL181 F' covering purB region 14MM383 polA(Ts) (polA12) B. BachmannRS3087 TnJO infadABfad-751: :TnlO B. BachmannRS3032 TnlO in fadR R. SimonsJK258 purB58 R. Simons
BacteriophageT4 gol 6B T4 with gol mutation, spontaneous 5T4 a Alc T4 endoIl-, endoIV-, 42-, 56-, alc- E. Kutter
PlasmidspBR328 Camr Tetr Ampr; unique PstI site in Ampr gene; unique Boehringer Mannheim
EcoRV site in Tetr genepM6P1 6.1-kb PstI subclone from MPH6 [Lit(Con)] This workpM6E1 2.3-kb EcoRV subclone from MPH6 This workpM6E2 2.3-kb EcoRV subclone in reverse orientation to pM6E1 This workpAP1 6.1-kb PstI subclone from AB2495 (Lit+) This workpAE1 2.3-kb EcoRV subclone from AB2495, same orientation This work
as pM6E1pWH4 Cosmid vector; Kanr; single BamHI site 10pA83 gol+ HindIlI fragment in Tetr gene of pACYC184 4
showed the Lit(Con) phenotype with a strain containing apurB mutation and obtained Pur+ recombinants.
Preparation of DNA and Southern hybridization. Plasmidswere isolated with an alkaline lysis protocol (1). Restrictionenzymes (purchased from Boehringer Mannheim) were usedwith the recommended buffers. The protocol of Rodriguezand Tait (16) was followed in the purification of chromo-somal DNA. Three micrograms of the purified chromosomalDNAs was digested with the appropriate restriction enzymeand electrophoresed in 0.8% agarose buffered with 0.089 MTris, 0.089 M boric acid, and 2.8 mM EDTA, pH 8.3. Thegel-fractionated DNAs were then transferred onto nitrocel-lulose paper (Millipore) by Southern transfer (18) and probedwith [32P]dATP-labeled nick-translated DNA (Amershamkit) of at least 5 x 107 cpm/,g. Hybridization was done at42°C for 15 to 18 h in a 10-ml volume containing 50%deionized formamide, 5x Denhardt solution (1% Ficoll, 1%polyvinylpyrrolidone, 1% bovine serum albumin), 6x SSC,and 100 p,g of sheared, heat-denatured salmon sperm DNAper ml. Thereafter, the filter was washed for 30 min with a
solution of 0.1% sodium dodecyl sulfate (SDS)-2x SSC atroom temperature, 42°C, and 65°C in succession, with one
change of wash buffer at each temperature. Autoradiographswere made on Kodak XAR-5 film.Cosmid cloning. Genomic DNAs were purified as above,
partially digested with restriction endonuclease Sau3A togive fragment sizes of ca. 40 kilobases (kb), dephosphoryl-ated with calf intestinal phosphatase (Boehringer Mann-heim), and then ligated to the compatible unique BamHI sitein the cosmid vector pWH4, carrying kanamycin resistance(10). The ligated DNA was then packaged into lambda phage
(Vector Cloning Systems, Gigapack), and the phage wereused to infect E. coli, selecting for kanamycin resistance.The kanamycin-resistant clones were then screened for theLit(Con) phenotype or by plate hybridization as below.Colony hybridizations. To clone the lit' gene, we prepared
DNA from AB2495, parent to the Lit(Con) mutants, andligated it into pWH4 as described above. The cosmids werescreened by colony hybridization in X1O81T1, a Lit0 deletionstrain, by a modification of a published procedure (8). First,the kanamycin-resistant colonies were replica plated bytoothpicks onto nitrocellulose filters on top of LB platesalong with positive and negative controls. The plates wereincubated for 5 h at 37°C and then lysed in situ by blotting on3mm Whatman filters soaked with 0.2 N NaOH and 0.5 MNaCl. The nitrocellulose filters were neutralized by repeatedblotting on 0.5 M Tris (pH 8.0)-1.0 M NaCl-soakedWhatman filters and dried, and then the DNA was fixed bybaking at 80°C under vacuum. The filters were hybridized tothe 2.3-kb EcoRV fragment containing the lit gene which hadbeen isolated from pM6E1 and nick-translated to a specificactivity of >7 x 107 cpm/,ug of DNA.
Isolation of apparent revertants. Plasmids containingcloned gol+ sequence from bacteriophage T4 do not trans-form Lit(Con) hosts. The rare transformants are found eitherto have deleted the gol sequence on the plasmid or to have amutation in the chromosome which makes them apparentlyLit' (4) but are most probably Lit0 deletion mutants whichhave lost the lit gene. The latter occur at a frequency ofabout 1 per 5,000. To isolate Lit0 mutants, we transformed aLit(Con) mutant, BKL403 (4), with plasmid pA83 (4) con-taining the gol+ sequence in pACYC184. The chloramphen-
VOL. 169, 1987
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

1234 KAO ET AL.
icol-resistant transformants were tested to distinguish muta-tions in the plasmid from apparent Lit' revertants. Fourindependent apparent revertants that propagated wild-typeT4 were isolated by this protocol. A spontaneous apparentrevertant of the Lit(Con) mutant, MPH24 (5), was found inour frozen stock.
Integrating plasmids with a poLA(Ts) mutant. E. coli with a
temperature-sensitive DNA polymerase I only allows repli-cation of ColEl-derived plasmids at lower temperatures. Wetransduced the polA(Ts) mutation of strain MM383 into a
multiply marked F- background to facilitate integration ofplasmids in a background convenient for genetic mapping.The advantage of this strain is that plasmids can be trans-formed into the cell and replicate stably at 30°C, but will notreplicate at 42°C. Strains that retain the plasmids' antibioticresistance at 42°C should have the plasmid integrated by asingle crossover between the plasmid insert sequence andthe homologous genomic sequence (9). The polA(Ts) mutanthas the additional advantage that it is also possible to allowthe excision and replication of the integrated plasmid bylowering the growth temperature to 30°C. The excised plas-mid should sometimes recombine out with the genomicsequence rather than the original insert, depending on wherethe excising crossover occurs.The polA(Ts) mutant was made in two steps. First, a
tetracycline resistance marker in transposon TnWO was intro-duced in the fadAB gene close to the polA(Ts) locus ofMM383 by P1 transduction from RS3087. A Tetrtransductant which still had the polA(Ts) mutation, as evi-denced by UV sensitivity, was used as a donor to transduceAB484 to Tetr. polA(Ts) cotransductants were detected bytheir UV sensitivity and one named RAMP1 was chosen forthe experiments below.
Plasmids containing the E. coli lit(Con) gene were trans-formed into E. coli RAMP1, and the cells were plated on LBchloramphenicol plates at 30°C. Transformants were cul-tured overnight in TB plus chloramphenicol at 30°C to allowintegration of some of the plasmids. The overnight culturewas diluted 100-fold and incubated overnight at 42°C. Theprocess was repeated at least twice more at 42°C to dilute outexisting plasmids. Finally, the culture was streaked out forsingle colonies at 42°C on chloramphenicol plates. Twosingle colonies were then grown up to log phase in TB andtested for the Lit(Con) phenotype.To excise the integrated plasmid, one of the colonies
above was cultured overnight at 30°C in TB plus chloram-phenicol. The overnight culture was diluted 1- to 100-fold,and the process was repeated three times. Plasmids were
prepared by the lysis procedure from 50 ml of the lastovernight culture and used to transform E. coli 1046, select-ing for chloramphenicol resistance. The transformants werescreened for the Lit(Con) or Lit' phenotype.
Preparation and labeling of minicells. Minicells were pre-pared essentially by the method of Russel and Model (17).Two additional sets of centrifugations (pelleting whole cellsat 2,000 x g, 10 min; pelleting minicells at 12,000 x g, 20min) were added before loading samples onto sucrose gradi-ents and before labeling cells. The labeling mix consisted ofM9 growth medium plus 0.5% glucose, adenine (50 ,ug/ml),thiamine (50 ,ug/ml), and 10 Vxg/ml each of all 20 amino acidsexcept methionine. [35S]methionine (Amersham) was addedat 50 ,uCi/ml.
Electrophoretic separation of labeled minicell proteins. Gelelectrophoresis was performed by the method of Laemmli(13). Minicell lysates were loaded on a 13% SDS-polyacrylamide gel and electrophoresed at 100 V with con-
TABLE 2. Mapping of the lit gene by a three-factor cross andsite of integration of the lit gene clone by transductiona
Recombinant types
Expt Selectedmarker Unselected No. of coloniesmarkers (no./no. tested)
I PurB+ Tetr Lit' 3/39PurB+ Tetr Lit(Con) 11/39PurB+ Tets Lit+ 9/39PurB+ Tet' Lit(Con) 16/39
II PurB + Camr 91/212Camr PurB + 62/367
aExperiment I shows the number of each recombinant type in a three-factor cross. The recipient was JK268 and the donor was MPH6 with a TnJOinsertion infadR. T4 ,Balc was used for the transduction. Experiment II showsthe frequency of cotransduction ofpurB58 and the integrated plasmid. P1 wasused for the transduction.
stant cooling. The gel was fixed by soaking in 12% trichlo-roacetic acid for 0.5 h and stained with Coomassie blue(0.1% Coomassie blue, 10% methanol, and 7% acetic acid)overnight. The gel was then destained in 12% aceticacid-12% methanol, dried, and autoradiographed. Theamount of labeling in proteins of interest was determined bycutting out the bands from the gel, using the autoradiographas a guide, and counting in a scintillation counter (BeckmanL57000).
RESULTS
Mapping lit(Con) mutations. The lit(Con) mutations havebeen mapped to about 25 min on the E. coli K-12 genetic map(5). To further refine the mapping, we performed three-factorcrosses such as the one shown in Table 2. The cross uses T4to transduce JK268 purB58 to Pur+ with the Lit(Con) mu-tant, MPH6, which contains a TnJO transposon infadR by aprevious transduction from R5302, as the donor. The Pur+transductants were tested for tetracycline resistance and thepresence of the Lit(Con) phenotype. purB cotransduced withfadR (TnJO) at a frequency of 36% and with lit(Con) at afrequency of 69%. Therefore, purB is closer to lit(Con) thanit is to fadR. This map order is also suggested by thefrequency of the individual recombinant types. From Table2, the rarest recombinant type is Pur+ Lit' TnJO Tetr. Withthe order above, this recombinant type would require fourcrossovers. We thus conclude that the lit gene is probablybetween the purB and fadR (TnJO) markers.Complementation test of dominance or recessivity. To de-
sign a strategy for cloning the lit gene, we had to knowwhether lit(Con) mutations are dominant or recessive. Todetermine this, we first crossed a recA derivative of theLit(Con) strain MPH6 (5) with a strain with an F' factorcovering this region as described in Materials and Methods.The merodiploids were then tested for the Lit(Con) pheno-type. Ten of 10 merodiploids were Lit(Con). This suggeststhat the lit(Con) mutations are dominant but does not proveit, since the lit' gene may be missing from the F' factor.However, we were sufficiently convinced to begin cloningthe lit gene with the assumption that lit(Con) mutations aredominant.
Cloning the lit(Con) gene. Since we thought that lit(Con)mutations are dominant, we tried to clone the DNA from aLit(Con) mutant by screening for cosmid clones that conferthe Lit(Con) phenotype in Lit' E. coli. We found thatapproximately 1 cosmid clone in 150 conferred the Lit(Con)
J. BACTERIOL.
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

E. COLI lit GENE AND T4 GENE EXPRESSION 1235
phenotype by the cross-streak test. We chose one of threesuch clones to subclone the lit gene into pBR328. We foundthat PstI endonuclease digestion of the 49-kb cosmid DNAgenerated eight visible fragments ranging from ca. 17 to 1 kb.We ligated the entire PstI digest into the unique PstI site inthe ampicillin resistance gene of pBR328. The ligation mix-ture was transformed into E. coli 1046, selecting for chlor-amphenicol resistance. The ampicillin-sensitive transform-ants, most of which had insert DNA, were screened for theLit(Con) phenotype by cross-streaking with T4. We foundone of nine subclones which conferred the Lit(Con) pheno-type. The plasmid had an insert of 6.1 kb and was used forfurther subcloning.
Restriction endonuclease EcoRV was found to cleave the6.1-kb PstI fragment and to leave four convenient-sizedpieces. These were ligated into the unique EcoRV site in thetetracycline resistance gene of pBR328, and clones withinserts were screened for the lit(Con) gene as above. Wefound that 7 of the 46 transformants conferred the Lit(Con)phenotype. These had an insert of 2.3 kb, and two of themwhich had the EcoRV fragment in opposite orientations werechosen for further experimentation.We also subcloned the lit gene from two other Lit(Con)
mutants, MPH5 and MPH21 (5), following the procedureabove. In both cases, the 6.1-kb PstI fragment and the 2.3-kbEcoRV fragment were found to confer the Lit(Con) pheno-type. We conclude that lit(Con) mutations are on thesefragments in all three mutants, so they must be very closelylinked. For the remaining experiments, we used the pBR328clones of the PstI and EcoRV fragments with the MPH6lit(Con) (lit-6) mutation. We refer to the plasmids as pM6P ifthe plasmid contains the PstI fragment and pM6E1 andpM6E2 if they contain the EcoRV fragment; 1 and 2 refer tothe two different orientations of the insert as determined byrestriction mapping. Although we only used clones of thelit-6 mutation, we assume the results would be identical withclones of the other mutations since they are indistinguishablein other respects.
Putative lit clone maps to the site of lit(Con) mutations. Toprove that we have cloned the lit gene, we integrated thepMP61 plasmid into the chromosome by a single crossoveras described in Materials and Methods and mapped itsposition of integration by Hfr crosses and P1 transduction,using the chloramphenicol resistance gene on the plasmid asa genetic marker. It seems quite certain that the plasmidintegrated by recombination with homologous sequences inthe chromosome, because we obtained no chloramphenicol-resistant colonies at 42°C with just the pBR328 cloningvector without an insert. Furthermore, from Southern hy-bridizations, there were no other regions of significant ho-mology to the lit gene (see below). The results of Hfr crossesput the site of integration of the plasmid close to the trp locusin the expected region and also showed that there was aunique site of integration (data not shown). The position ofthe plasmid integration was further localized by P1 transduc-tion (Table 2). We found the purB mutation to cotransducewith Camr at a frequency of 43 and 17%, selecting for purB+and Camr, respectively. This value is low compared with thepublished cotransduction data (5) and with the data in Table2, but the difference may be due to the presence of theplasmid vector in this experiment. Moreover, the trans-ductional data in Table 2 were obtained with T4 rather thanP1. These small discrepancies aside, the mapping datasupport the conclusion that our clones contain the lit gene.
Cloning the wild-type lit gene. To clone the wild-type litgene, we first tried the easy method described in Materials
A C D E F G H IFIG. 1. Southern hybridization of EcoRV-digested E. coli DNA
probed with pM6P1. Lanes: A, plasmid pM6E1 digested withEcoRV to show position of 2.3-kb EcoRV fragment containing the litgene (bottom band); B, C, and D, DNAs from Lit(Con) mutantsMPH6, 21, and 5, respectively; E, F, and G, DNAs from K-12 Lit'strains K99, AB484, and AB2495, respectively; H and I, twoindependent Lit' revertants.
and Methods of excising the integrated plasmid. Since theplasmid was inserted by a single crossover with the wild-type lit region, the integration event should yield the plasmidbracketed by two copies of the inserted sequence (9), one ofwhich has the lit(Con) mutation and the other the wild-typesequence. Excision can occur by recombination between therepeated sequences, and some percentage of the time willyield the plasmid with the wild-type sequence rather than themutant sequence, depending on the location of the crossoverrelative to the site of the lit(Con) mutation. All 50 excisedplasmids tested conferred the Lit(Con) phenotype afterretransformation into a Lit' recipient, and by this criterionstill had the lit(Con) allele in the clone. A likely reason forour failure to find clones of the lit' gene by this methodbecame apparent later (see below).We next used plate hybridization to try to clone the
wild-type lit gene. We used the 2.3-kb EcoRV fragment frompM6E1 as the probe and screened a cosmid library madefrom E. coli AB2495, the Lit' parent of Lit(Con) mutants.We found 1 positive colony out of 220 screened. The DNAfrom this cosmid clone was digested with PstI and EcoRV,electrophoresed, and subjected to Southern blot hybridiza-tion, again using the insert from pM6E1 as the probe. A PstIfragment of 6.1 kb and an EcoRV fragment of 2.3 kb werefound to be homologous to the probe. We ligated thedigested mixtures into pBR328 and identified clones withthese inserts by their size and restriction pattern on agarosegels. We refer to the clones of the wild-type lit gene as pAE1and pAE2 or pAP1 and pAP2, depending on whether theEcoRV or PstI fragment was cloned, respectively, and theorientation of the clone (see above).
Molecular characterization of the lit gene. We used thepM6P1 subclone as a probe in Southern hybridization exper-iments to determine the nature of lit(Con) mutations. Iflit(Con) mutations are large rearrangements, the size of therestriction fragments containing the lit gene should be dif-ferent for the Lit(Con) mutants and their parent. In the sameexperiment, we also determined whether the lit gene iscommon to most E. coli K-12 strains. The genomic DNAs ofthree independently isolated Lit(Con) mutants and threerandomly chosen K-12 strains were digested with EcoRV
VOL. 169, 1987
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
1236 KAO ET AL.
and probed with pM6P1 in a Southern hybridization exper-iment (Fig. 1). In all cases, the 2.3-kb EcoRV fragment lit upas well as the other fragments found in the PstI fragment.Therefore, there seems to be no gross rearrangement in thelit(Con) mutations. It also is apparent that most E. coli K-12strains have a lit gene, since all three of the wild-type K-12strains had the 2.3-kb EcoRV fragment. However, anotherK-12 strain, X1081T1, was found to have a deletion whichremoved all of the restriction fragments in the 6.1-kb PstIclone (data not shown). Because this strain has no chromo-somal sequences homologous to the lit gene region, it wasused for the plate hybridizations above to eliminate thebackground due to the endogenous lit gene (see Materialsand Methods). Because X1081T1 has lost the lit gene and itsgrowth is not affected, the lit gene product may not beessential for E. coli growth. This conclusion was substanti-ated by the characterization of Lit(Con) apparent revertants.
Characterization of apparent Lito revertants. The Lit'revertants are derived from Lit(Con) mutants for having lostthe ability to restrict the growth of T4 and so are phenotyp-ically Lit' or wild type. This reversion to wild type couldhave been due to suppression or further changes in the litgene. To determine the cause of reversion, the EcoRV-digested DNA from two apparent revertants, selected asdescribed in Materials and Methods, were probed withpM6P1 (Fig. 1, lanes H and I). Both strains were deleted forthe 2.3-kb EcoRV fragment as well as other fragments. Thelarger-sized hybridizing band was due to the linearizedplasmid, pA83, which was used to select the Lit0 derivatives,and had homology with the probe. We conclude that theentire 6.1-kb PstI fragment is lost in these Lit0 derivatives.Because the apparent revertants can have deletions whichremove the lit gene, they are not true revertants of thelit(Con) mutation, and either the wild-type gene, lit', or thenull allele, lit°, can confer the Lit' phenotype.To see the extent of the deletions in the apparent rever-
tants, we used the larger cosmid clone to probe EcoRV-digested DNAs from four independently isolated Lit0 strains(Fig. 2). In this case, only bands with homologous chromo-somal sequences will be detected because the cosmid has nohomology with pA83. Three of the Lit0 strains (Fig. 2, lanes
..."
*
B C D E FFIG. 2. Southern hybridization of EcoRV-digested E. coli DNA
probed with cosmid clone of lit(Con) gene from MPH6. Lanes A,cosmid clone digested with EcoRV; B, EcoRV-digested Lit' DNAfrom strain K99; C, D, E, and F, DNAs from four independentlyisolated Lit' strains digested with EcoRV. *, Position of the 2.3-kbEcoRV band.
.U--gillow
2"'.~~US ~=35am~~jji' am, -a..aui_igs
-18
9A B C D E F G H
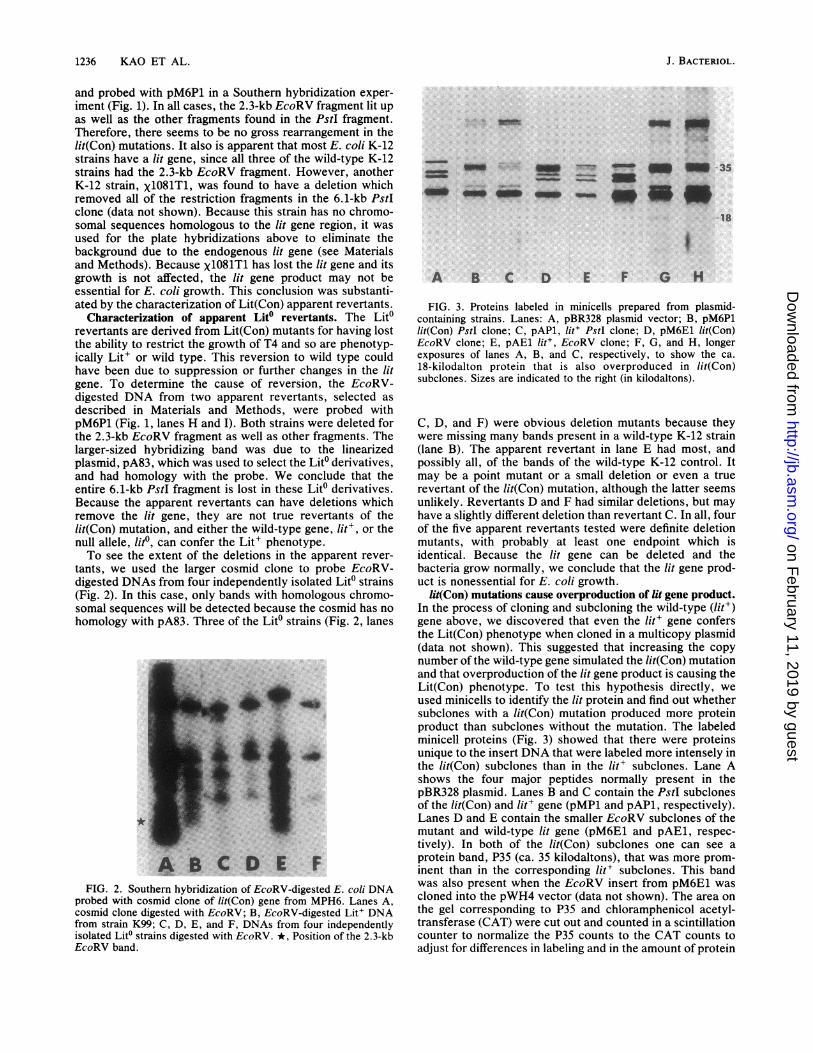
FIG. 3. Proteins labeled in minicells prepared from plasmid-containing strains. Lanes: A, pBR328 plasmid vector; B, pM6P1lit(Con) PstI clone; C, pAP1, lit' PstI clone; D, pM6E1 lit(Con)EcoRV clone; E, pAE1 lit', EcoRV clone; F, G, and H, longerexposures of lanes A, B, and C, respectively, to show the ca.18-kilodalton protein that is also overproduced in lit(Con)subclones. Sizes are indicated to the right (in kilodaltons).
C, D, and F) were obvious deletion mutants because theywere missing many bands present in a wild-type K-12 strain(lane B). The apparent revertant in lane E had most, andpossibly all, of the bands of the wild-type K-12 control. Itmay be a point mutant or a small deletion or even a truerevertant of the lit(Con) mutation, although the latter seemsunlikely. Revertants D and F had similar deletions, but mayhave a slightly different deletion than revertant C. In all, fourof the five apparent revertants tested were definite deletionmutants, with probably at least one endpoint which isidentical. Because the lit gene can be deleted and thebacteria grow normally, we conclude that the lit gene prod-uct is nonessential for E. coli growth.
lit(Con) mutations cause overproduction of lit gene product.In the process of cloning and subcloning the wild-type (lit')gene above, we discovered that even the lit' gene confersthe Lit(Con) phenotype when cloned in a multicopy plasmid(data not shown). This suggested that increasing the copynumber of the wild-type gene simulated the lit(Con) mutationand that overproduction of the lit gene product is causing theLit(Con) phenotype. To test this hypothesis directly, weused minicells to identify the lit protein and find out whethersubclones with a lit(Con) mutation produced more proteinproduct than subclones without the mutation. The labeledminicell proteins (Fig. 3) showed that there were proteinsunique to the insert DNA that were labeled more intensely inthe lit(Con) subclones than in the lit' subclones. Lane Ashows the four major peptides normally present in thepBR328 plasmid. Lanes B and C contain the PstI subclonesof the lit(Con) and lit' gene (pMP1 and pAP1, respectively).Lanes D and E contain the smaller EcoRV subclones of themutant and wild-type lit gene (pM6E1 and pAE1, respec-tively). In both of the lit(Con) subclones one can see aprotein band, P35 (ca. 35 kilodaltons), that was more prom-inent than in the corresponding lit' subclones. This bandwas also present when the EcoRV insert from pM6E1 wascloned into the pWH4 vector (data not shown). The area onthe gel corresponding to P35 and chloramphenicol acetyl-transferase (CAT) were cut out and counted in a scintillationcounter to normalize the P35 counts to the CAT counts toadjust for differences in labeling and in the amount of protein
J. BACTERIOL.
*.; .....
.......
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

E. COLI lit GENE AND T4 GENE EXPRESSION 1237
of each minicell sample. The ratio of P35 in lit(Con) to that inlit' clones was 6.9 in the PstI subclones. In the EcoRVsubclones the ratio was 1.6 after adjustment for the CATband.
After longer exposure of the same gel at least one smallerband (ca. 18 kilodaltons) appeared (lanes F, G, and H, Fig.3). This protein was also synthesized at a higher rate inlit(Con) subclones than in lit' subclones. It is not clearwhether this other polypeptide(s) is also overproduced as aresult of the lit(Con) mutations or is a cleavage product ofthe 35-kilodalton polypeptide. The 35-kilodalton polypeptideseems to migrate as a fuzzy band relative to the otherpolypeptides, for which we have no explanation at present.
DISCUSSION
E. coli lit(Con) mutations (originally called lit mutations)were detected because they cause a severe inhibition of T4bacteriophage gene expression late in infection (5). We haverefined the mapping of the lit gene to between the purB andfadR genes of E. coli, just clockwise of purB at 25 min. Wehave found that lit(Con) mutations are dominant over thewild-type (lit') allele in merodiploids. We have cloned the litgene from three different Lit(Con) mutants and from wild-type E. coli, first in approximately 40-kb cosmid clones andthen as subclones of a PstI fragment of 6.1 kb and an EcoRVfragment of 2.3 kb. The three lit(Con) mutations all cloned tothe same 2.3-kb EcoRV fragment, indicating that lit(Con)mutations are closely linked to each other.We have used the clones of the lit gene to identify some
possible lit gene products and to demonstrate that lit(Con)mutations probably cause the overproduction of the lit geneproduct and that this overproduction is responsible for thephenotypes. We have also used the clones to demonstratethat the lit gene product is nonessential for E. coli growth,even though the lit gene is common to most E. coli K-12strains. To summarize our conclusions thus far, the lit generesides at 25 min and can be deleted without adverse effectsand so is nonessential for E. coli growth. The gene isnormally expressed at low levels at most, but when itsexpression is increased it inhibits the development of T4 byinhibiting gene expression late in infection.Our evidence that lit(Con) mutations cause the overpro-
duction of the lit gene product is twofold. One line ofevidence is the behavior of clones of the wild-type (lit+) genein multicopy plasmids. In the original large cosmid clone,which is presumably low copy, the phenotype was Lit+.When the gene was subcloned into pBR328, a high-copyplasmid, the phenotype was Lit(Con). Furthermore, whenpAP1 was integrated into the chromosome by homologousrecombination, the phenotype was Lit'. Therefore, whenthe copy number of the lit' gene clone is again low, thephenotype is Lit'. Therefore, the Lit(Con) phenotype of thelit' gene cloned in pBR328 seems to be due to the copynumber and not, for example, to altered expression of thegene in the clone. In contrast to the larger PstI clone, the lit+gene in the smaller EcoRV clone showed the Lit(Con)phenotype even when crossed back into the chromosome.We think that in the smaller clone, more of the expression ofthe lit gene is occurring from promoters in the cloningvector. This conclusion is supported by the results of theminicell experiments (Fig. 3), which indicate greater expres-sion of the lit+ gene in the EcoRV clone than in the PstIclone.The minicell experiments also offer convincing evidence
that lit(Con) mutations cause overproduction of the lit pro-tein. We have identified polypeptides of 35 and 18kilodaltons as possible products of the lit gene. From boththe 6.1-kb PstI clone and the 2.3-kb EcoRV clone, thelit(Con) gene makes more of both the 35- and 18-kilod,altonpolypeptides than its lit' counterpart. Thus, the lit(Con)mutation is enhancing the expression of the genes even in themulticopy plasmid. It is not clear, however, how this en-hanced expression is achieved. For example, lit(Con) muta-tions could be "up promoter" or "up translational" mu-tants. A sequence determination of the base pair changes inlit(Con) mutations should distinguish the many possibilities.From the results discussed above, it is not obvious that the
lit(Con) mutations are in the EcoRV fragment, since thereare only slight differences between the lit' and lit(Con)clones in the rate of expression of genes in the clone.However, the EcoRV clones of the lit' gene and the lit(Con)gene do show differences when they are tested at highertemperatures. While the wild-type clone [or lit(Con) muta-tions in the chromosome] are only restrictive at tempera-tures at or below 37°C, the lit(Con) gene cloned in multicopyplasmids prevents the multiplication of T4 even at 42°C. Infact, the pM6E1 plasmid can even inhibit the multiplicationof some T4 gol mutants (data not shown), indicating that theseverity of the restriction is a function of the amount of litprotein. Apparently, less lit gene product is required toinhibit T4 late gene expression at lower than at highertemperatures, but the inhibition occurs at all temperatures.
It is not clear from this work why E. coli has a lit gene. Theproduct of the gene is nonessential for E. coli growth underlaboratory conditions. Of the five lit° mutations we tested,four had deletions of the entire region and only one hadinactivated the gene by a point mutation or other smallchange undetectable by our method. This high frequency ofdeletions suggests that the lit gene may reside on a crypticprophage or transposon. Furthermore, at least one endpointseems to be common to all the deletions, arguing that the lit°mutations may be due to excision of a prophage or todeletions promoted by a nearby transposon. However, the litgene may merely reside in a region where deletions arefrequent, since one can also find point mutations at about thesame frequenlcy which presumably inactivate the gene.There is also no phenotype in uninfected cells of overpro-
ducing the lit protein, at least to the extent achieved here.We are attempting to further elevate expression of the litgene to see whether even higher levels have no effect on theuninfected bacterium.At present, the lit gene product achieves its special
interest through its interaction with T4 bacteriophage. Byinteracting with the T4 gol site, a small, approximately40-base-pair sequence in gene 23 about one-fourth of the wayin from the N terminus of the gene, the lit protein can causethe cessation of all gene expression late in T4 infection. Ourpresent evidence suggests that the lit protein interacts withRNA from this region and somehow blocks the translation ofthe RNA and, by an unknown mechanism, causes a transinhibition of other gene expression in the cell (K. Bergslandand L. Snyder, manuscript in preparation). An interactionwith the RNA was also suggested by the isolation of lit(Con)mutations. They were originally isolated (5) in a search forE.coli genes which interact with the T4 RNA-processing en-zymes polynucleotide kinase and RNA ligase (12), althoughthe relationship to these T4-induced enzymes remains un-clear. For the present, we think that the best clties to thefunction of the lit gene product lie in studies of what happensat the T4 gol site when the lit gene product is overproduced.
VOL. 169, 1987
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

1238 KAO ET AL.
ACKNOWLEDGMENTS
We thank Kristin Bergsland for careful reading of this manuscriptand communicating unpublished results and Barbara Bachmann ofthe Yale Stock Collection and Robert Simons for providing strainsand advice.
This work was supported by Public Health Service grantGM-28001 from the National Institutes of Health.
LITERATURE CITED1. Birnboim, H. C., and J. Doly. 1979. A rapid alkaline extraction
procedure for screening recombinant plasmid DNA. NucleicAcids Res. 7:1513-1523.
2. Brody, E., D. Rabussay, and D. H. Hall. 1983. Regulation oftranscription of prereplicative genes, p. 174-183. In C. K.Mathews, E. M. Kutter, G. Mosig, and P. B. Berget (ed.),Bacteriophage T4. American Society for Microbiology, Wash-ington, D.C.
3. Champness, W. C., and L. Snyder. 1982. The gol site: acis-acting bacteriophage T4 regulator region that can affectexpression of all the T4 late genes. J. Mol. Biol. 155:395-407.
4. Champness, W. C.; and L. Snyder. 1984. Bacteriophage T4 golsite: sequence analysis and effects of the site on plasmidtransformation. J. Virol. 50:555-562.
5. Cooley, W., K. Sirotkin, R. Green, and L. Snyder. 1979. A newgene of Escherichia coli K-12 whose product participates in T4bacteriophage late gene expression: interaction of lit with theT4-induced polynucleotide 5'-kinase 3'-phosphatase. J. Bacte-riol. 140:83-91.
6. Davie, E., K. Sydnor, and L. I. Rothfield. 1984. Genetic basis ofminicell formation in Estherichia coli K-12. J. Bacteriol.158:1202-1203.
7. Geiduschek, E. P., T. Elliott; and G. A. Kassavetis. 1983.Regulation of late gene expression, p. 189-192. In C. K.Mathews, E. M. Kutter, G. Mosig, and P. B. Berget (ed.),Bacteriophage T4. American Society for Microbiology, Wash-
ington, D.C.8. Grunstein, M., and D. S. Hogness. 1975. Colony hybridization: a
method for the isolation of cloned DNA that contains a specificgene. Proc. Natl. Acad. Sci. USA 72:3961-3965.
9. Gutterson, N. I., and D. E. Koshland, Jr. 1983. Replacement andamplification of bacterial genes with sequences altered in vitro.Proc. Natl. Acad. Sci. USA 80:4894-4898.
10. Herrero, A., J. Elhai, B. Hohn, and C. P. Wolk. 1984. Infrequentcleavage of cloned Anabaena variabilis DNA by restrictionendonuclease from A. variabilis. J. Bacteriol. 160:781-784.
11. Jabbar, M. A., and L. Snyder. 1984. Genetic and physiologicalstudies of an Escherichia coli locus that restricts polynucleotidekinase and RNA ligase-deficient mutants of bacteriophage T4. J.Virol. 51:522-529.
12. Kaufmann, G., M. David, G. D. Borasio, A. Teichmann, A. Paz,M. Amitsur, R. Green, and L. Snyder. 1986. Phage and hostgenetic determinants of the specific anticodon loop cleavages inbacteriophage T4-infected Escherichia coli CTr5X. J. Mol. Biol.188:15-22.
13. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
14. Low B. 1972. Escherichia coli K-12 F-prime factors old andnew. Bacteriol. Rev. 36:587-607.
15. Miller, J. H. 1972. Experiments in molecular genetics. ColdSpring Harbor Laboratory, Cold Spring Harbor, N.Y.
16. Rodriguez, R. L., and R. C. Tait. 1983. Recombinant DNAtechniques: an introduction, p. 45-46. Addison-Wesley Publish-ing Co., London.
17. Russel, M., and P. Model. 1984. Characterization of the clonedfip gene and its product. J. Bacteriol. 157:526-532.
18. Southern, E. M. 1975. Detection of specific sequences amongDNA fragments separated by gel electrophoresis. J. Mol. Biol.98:503-517.
19. Takahashi, H., and H. Saito. 1982. Mechanism of pBR322transduction mediated by cytosine-substituting T4 bacterio-phage. Mol. Gen. Genet. 186:497-500.
J. BACTERIOL.
on February 11, 2019 by guest
http://jb.asm.org/
Dow
nloaded from





























