Embed Size (px)
Citation preview

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Feb. 2007, p. 1089–1100 Vol. 73, No. 40099-2240/07/$08.00�0 doi:10.1128/AEM.01577-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Characterization of Strong Promoters from an EnvironmentalFlavobacterium hibernum Strain by Using a Green
Fluorescent Protein-Based Reporter System�
S. Chen,1* M. Bagdasarian,1 M. G. Kaufman,2 and E. D. Walker1
Department of Microbiology and Molecular Genetics1 and Department of Entomology,2
Michigan State University, East Lansing, Michigan 48824
Received 7 July 2006/Accepted 9 December 2006
We developed techniques for the genetic manipulation of Flavobacterium species and used it to charac-terize several promoters found in these bacteria. Our studies utilized Flavobacterium hibernum strain W22,an environmental strain we isolated from tree hole habitats of mosquito larvae. Plasmids from F. hibernumstrain W22 were more efficiently (�1,250-fold) transferred by electroporation into F. hibernum strain W22than those isolated from Escherichia coli, thus indicating that an efficient restriction barrier exists betweenthese species. The strong promoter, tac, functional in proteobacteria, did not function in Flavobacteriumstrains. Therefore, a promoter-trap plasmid, pSCH03, containing a promoterless gfpmut3 gene wasconstructed. A library of 9,000 clones containing chromosomal fragments of F. hibernum strain W22 inpSCH03 was screened for their ability to drive expression of the promoterless gfpmut3 gene. Twenty strongpromoters were used for further study. The transcription start points were determined from sevenpromoter clones by the 5� rapid amplification of cDNA ends technique. Promoter consensus sequencesfrom Flavobacterium were identified as TAnnTTTG and TTG, where n is any nucleotide, centered approx-imately 7 and 33 bp upstream of the transcription start site, respectively. A putative novel ribosomebinding site consensus sequence is proposed as TAAAA by aligning the 20-bp regions upstream of thetranslational start site in 25 genes. Our primary results demonstrate that at least some promoter andribosome binding site motifs of Flavobacterium strains are unusual within the bacterial domain andsuggest an early evolutionary divergence of this bacterial group. The techniques presented here allow formore detailed genetics-based studies and analyses of Flavobacterium species in the environment.
Flavobacteria belong to a systematically diverse yet phylo-genetically coherent group within the phylum Bacteroidetes (5,12, 51). In the past referred to as the Cytophaga-Flavobacteri-um-Flexibacter-Bacteroides, Cytophaga-Flavobacterium-Bacte-roides, or CFB group, this cluster represents an abundant andubiquitous assemblage of heterotrophic bacteria, with Fla-vobacterium being one of many genera (5, 22). Bacteria in theBacteroidetes are widespread in freshwater and marine ecosys-tems and inhabit the water column, biofilms, sediments, soils,and feces (7, 12, 22–25). An important functional role of theseorganisms is the transformation of high-molecular-weight, dis-solved organic matter in aquatic environments (12, 24, 25).Some flavobacteria, in particular Flavobacterium psychrophi-lum and Flavobacterium columnare, are pathogens of fish (36,48), while other bacteria in Bacteroidetes are facultative patho-gens of immunocompromised adult humans (28).
Molecular genetic manipulation studies of bacteria in Bac-teroidetes have proven to be difficult and are limited in scope.Plasmids, selectable markers, and transposons functional inproteobacteria fail to function in Bacteroidetes (2, 31, 43). Ge-netic techniques, including cosmid complementation andtransposon-mediated mutagenesis, were used to identify sev-
eral gld genes associated with gliding motility in Flavobacteriumjohnsoniae (1, 8, 9, 15–17, 21, 29–31). These techniques wererecently extended to F. psychrophilum, leading to characteriza-tion of the tlp gene possibly involved in gliding motility andbiofilm formation (3). An expression system was developed toexpress homologous and heterologous genes under the controlof the hepA regulatory region using the Pedobacter heparinus(previously Flavobacterium heparinum) conjugation-integra-tion plasmid pIBXF1 (6, 43). Modifications of this plasmidpermitted in vivo expression of several heparinase genes aswell as the gene cslA encoding the chondroitinase A enzyme asa reporter (6). Expression of Escherichia coli lacZY in F. psy-chrophilum was recently investigated by Alvarez et al. by in-serting this reporter gene under a putative open reading frame1 (ORF1) promoter on the pCP23 plasmid (2). A xylosidase/arabinosidase bifunctional reporter system has also been de-veloped for Bacteroides and Porphyromonas (49). The afore-mentioned studies notwithstanding, genetic manipulation ofthis important group of bacteria is underdeveloped. Remark-ably, there are only few selectable antibiotic resistance markersavailable (41), and very few studies of native promoters andribosome binding sites (RBSs) of this group have been under-taken. Given the ubiquity of Bacteroidetes and their importancein ecological processes, as well as the association of the groupwith pathogenesis in many fish species, further development ofgenetic tools is warranted. In particular, there is a need toidentify and characterize native promoters that will allow forthe expression of gene inserts in these bacteria. Because larval
* Corresponding author. Mailing address: Department of Microbi-ology and Molecular Genetics, 2215 Biomedical and Physical SciencesBuilding, Michigan State University, East Lansing, MI 48824. Phone:(517) 355-6463, ext. 1548. Fax: (517) 353-8957. E-mail: [email protected].
� Published ahead of print on 22 December 2006.
1089
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

mosquitoes such as Ochlerotatus triseriatus, dwelling in water-filled tree holes and similar habitats, feed upon Flavobacteriumand other microorganisms (19, 20, 47; M. Kaufman, E. Walker,and S. Chen, unpublished observations), our interest is in thepotential use of these highly ingestible bacteria to expressgenes encoding proteins toxic to mosquito larvae. Here, wereport progress towards this end, with emphasis on devel-opment of new plasmid systems for genetic transformationin an environmental isolate of Flavobacterium, analysis ofnative promoter structure and function using the green flu-orescent protein (GFP) reporter, and an investigation ofproperties of the RBS.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions. Bacterial strains and plas-mids used in this study are listed in Table 1. Flavobacterium strain W22, hereclassified as F. hibernum, was initially isolated from a water-filled tree hole in anAmerican beech tree located near the Michigan State University campus. Fla-vobacterium johnsoniae UW101 (ATCC 17061) was obtained from MarkMcBride of the University of Wisconsin—Milwaukee. Strains of E. coli wereroutinely grown in Luria-Bertani broth at 37°C. Flavobacterium strains weregrown at 26°C in Casitone yeast extract (CYE) medium as previously described(31). Liquid cultures were incubated with shaking at 200 rpm. Solid CYE me-dium contained 20 g of agar per liter. Antibiotics were used as indicated at thefollowing concentrations: ampicillin, 100 �g/ml; erythromycin, 100 �g/ml; tetra-cycline, 10 �g/ml; rifampin, 50 �g/ml; chloramphenicol, 25 �g/ml; streptomycin,100 �g/ml; and kanamycin, 30 �g/ml.
TABLE 1. Bacterial strains and plasmids used in this study
Strain or plasmid Relevant characteristic(s) and/or plasmid constructiona Source orreference
Strains
E. coliDH5� endA1 hsdR17 supE44 thi-1 recA1 gyrA96 (Nalr) relA1�(lacIZYA-argF)U169
deoR(�80dlacZ�M15)Clontech
S17-1 hsdR17(rK� mK
�) recA RP4-2 (Tcr::Mu-Kmr::Tn7 Strr) 42JM109 F� traD36 proAB� lacIq lacZ�M15/recA1 supE44 endA1 hsdR17 gyrA96 relA1
thi-1 mcrA �(lac-proAB)Promega
F. hibernumW22 Wild type This studyW22 R1 Rifampin-resistant mutant This study
F. johnsoniae UW101 ATCC 17061 1
PlasmidspGEM-T easy vector Cloning vector, Apr PromegapKEN2 gfpmut3, Apr 11pMMB66EH IncQ RSF1010�(PstI-PvuII, 2.87 kb) � lacIq tacP rrnB Apr 13pMMB207 IncQ RSF1010 derived, Cmr 33pMMB503 IncQ RSF1010 derived, Smr 32pCP23 ColE1 ori (pCP1 ori), E. coli-Flavobacterium shuttle plasmid, Apr (Tcr) 1pCP29 ColE1 ori, (pCP1 ori), E. coli-Flavobacterium shuttle plasmid, Apr (Cfxr, Emr) 21pNJR5 IncQ, E. coli-Bacteroides shuttle vector, Kmr (Emr) 41pSCH01 gfpmut3 gene inserted in BamHI and PstI sites in pMMB603EH, Apr This studypSCH02F Expression cassettes including lacIq, ptac, and gfpmut3 fragment in pCP23
(ORF1, forward), Apr (Tcr)This study
pSCH02R Expression cassettes in pCP23 (ORF1, reverse), Apr (Tcr) This studypSCH03 Promoterless gfpmut3 including E. coli RBS in pCP23, Apr (Tcr) This studypSCH41 HU fragment trapped in pSCH03, Apr (Tcr) This studypSCH41.�210 D1, 375-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.�100 D2, 300-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.�52 D3, 275-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.�23 D4, 217-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.34 D5, 98-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.67 D6, 68-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studypSCH41.97 D7, 36-bp fragment of HU fused to gfpmut3, Apr (Tcr) This studyWT RBS Wild-type RBS, Apr (Tcr) This studyM1 RBS mutant, TAATA3TCGTT, Apr (Tcr) This studyM2 RBS mutant, TAATA3TTGTT, Apr (Tcr) This studyM3 RBS mutant, TAATA3TATTA, Apr (Tcr) This studyM4 RBS mutant, TAATA3TTTTA, Apr (Tcr) This studyM5 RBS mutant, TAATA3TTTTT, Apr (Tcr) This studyM6 RBS mutant, TAATA3TGATA, Apr (Tcr) This studyM7 RBS mutant, TAATA3TAATG, Apr (Tcr) This studyM8 RBS mutant, TAATA3TAAAA, Apr (Tcr) This studyM9 RBS mutant, TAATA3TAGTA, Apr (Tcr) This study
a Antibiotic resistance phenotype: Apr, ampicillin; Tcr, tetracycline; Emr, erythromycin; Cfxr, cefoxitin; Cmr, chloramphenicol; Kmr, kanamycin; and Smr, strepto-mycin. Unless indicated otherwise, antibiotic resistance phenotypes are those expressed in E. coli. Antibiotic resistance phenotypes listed in parentheses are thoseexpressed in Flavobacterium strains but not in E. coli.
1090 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

Conjugal transfer of plasmid DNA from E. coli to F. hibernum strain W22.Rifampin resistance was used as a counterselectable marker for conjugation.Rifampin-resistant F. hibernum strain W22 mutants were obtained by platingmore than 109 cells on CYE medium supplemented with 50 �g of rifampin perml. Equal amounts of F. hibernum strain W22 cells and E. coli cells (approxi-mately 108 cells of each strain) at mid-log phase were harvested by centrifuga-tion, washed with CYE medium, mixed, spotted onto CYE agar containing 5 mMCaCl2, and incubated at 26°C for 12 h. After conjugation, cells were scraped offthe plates, diluted, and plated on CYE agar containing the appropriate antibi-otic. For conjugal transfer of plasmids pCP23 and pCP29, counterselection wasunnecessary because the tetracycline resistance and erythromycin resistancewere not expressed in E. coli.
Electroporation. Flavobacterium cells were harvested during exponentialgrowth, washed three times in 10% (vol/vol) glycerol at 4°C, and resuspended toa cell density of approximately 1011/ml in GYT medium (10% [vol/vol] glycerol,0.125% [wt/vol] yeast extract, and 0.25% [wt/vol] tryptone, pH 7.0). Approxi-mately 80 ng of plasmid DNA was added to 100 �l of cells. Electroporation wasconducted by using a Bio-Rad pulser in a 2-mm cuvette according to the man-ufacturer’s instructions. Following electroporation, 1 ml of CYE medium wasimmediately added to each cuvette. Cells were incubated at 26°C for 1.5 h toallow expression of antibiotic resistance and plated on CYE agar with theappropriate antibiotic. Colonies were counted after 2 to 3 days of incubationat 26°C.
Plasmid constructions. An expression vector using tac promoter was con-structed as follows. A 750-bp, promoterless version of gfpmut3 was amplified byPCR from plasmid pKEN2 containing the E. coli ribosome binding site by usingthe primers gfpF1 and gfpR1 (Table 2). The gfpmut3 fragment was digested withBamHI and PstI and inserted into pMMB66EH at the same restriction sites. Theresulting plasmid was named pSCH01. The expression cassette (including thelacIq, tac promoter, gfpmut3, and transcriptional terminator regions) from plas-mid pSCH01 was amplified by PCR with primers 66EHF and 66EHR. Theresultant fragment was ligated into pCP23, which had been digested with KpnIand SphI, blunt ended with T4 DNA polymerase, dephosphorylated, and gelpurified. The constructs were transformed into E. coli DH5�, and the inserts inboth orientations were screened, yielding plasmids pSCH02F and pSCH02R.
These plasmids were extracted from E. coli and transformed into F. hibernumstrain W22 and F. johnsoniae by electroporation.
A promoter-probe vector was constructed as follows. The promoterless gfpmut3construct was released from pSCH01 by BamHI and PstI and ligated into thesame sites of pCP23, resulting in the promoter-probe vector herein designatedpSCH03.
In order to analyze promoter structure in detail, promoter deletion derivativesfor the strongest promoter found in the analysis shown below (on plasmidpSCH41) were constructed. The positions of N-terminal primers on the putativepSCH41 promoter region were as follows, with the TSP assigned as �1: D1,�210 bp; D2, �100 bp; D3, �52 bp; D4, �23 bp; D5, �34 bp; D6, �67 bp; andD7, �97 bp. Amplification of the deletion derivatives and gfpmut3 was per-formed with the above N-terminal primers (containing an N-terminal KpnI site)and C-terminal primer gfpSphIR (with an SphI site) complementary to the 3�end of gfpmut3. The PCR products were inserted into the T-easy vector andsequenced. The inserts were released from this vector by KpnI and SphI andcloned in the same sites of pCP23 to create the following deletion vector series:pSCH41.�210, pSCH41.�100, pSCH41.�52, pSCH41.�23, pSCH41.34, pSCH41.67,and pSCH41.97.
In order to elucidate properties of the ribosome binding site, site-specificmutagenesis was performed by using the oligonucleotide-directed PCR method.The sequences of the nine mutagenic oligonucleotides used for mutagenesis ofthe RBS, labeled RBSM1 through RBSM9, are shown in Table 2. These N-terminal oligonucleotides were designed to mutagenize the 4-bp putative RBSregion upstream of the translation start codon bearing a BamHI site, whereas theC-terminal primer gfpSphIR was designed to have a SphI site. The product of thePCR with the mutagenic primer and C-terminal primer was purified, insertedinto T-easy vector, and sequenced. The inserts were released by BamHI and SphIand cloned into the same sites on pSCH41, resulting in the desired RBS mu-tagenesis series (RBSM1 to RBSM9).
Promoter-trapping and native promoter analysis. Chromosomal DNA of F.hibernum W22, extracted with the genomic DNA extraction kit (Promega), waspartially digested with Sau3AI and size fractionated by agarose gel electrophore-sis. DNA fragments ranging from 0.3 to 2.0 kb were purified and ligated into theBamHI site of pSCH03 (extracted from F. hibernum strain W22). The ligation
TABLE 2. Primers used in this study
Primer Sequencea
63f ..................................................................................................5�-CAGGCCTAACACATGCAAGTC-3�1387r..............................................................................................5�-GGGCGGWGTGTACAAGGC-3�gfpF1 .............................................................................................5�-CGCGGATCCTTTAAGAAGGAGATATACATATGAGTAAAGGAGAAG-3�gfpR1.............................................................................................5�-AAACTGCAGGAATTCTTATTTGTATAGTTC-3�66EHf ............................................................................................5�-CCTGCTAATTGGTAATACC-3�66EHr............................................................................................5�-CGGAAATGTTGAATACTCATAC-3�D1 ..................................................................................................5�-CGGGGTACCTTACCAGCAGATGCGG-3�D2 ..................................................................................................5�-CGGGGTACCCAGGATCGACAAGCGAC-3�D3 ..................................................................................................5�-CGGGGTACCCTAAATTTAAAGAAAACACTTGC-3�D4 ..................................................................................................5�-CGGGGTACCCGGATTTCCTATTAAATTTGTG-3�D5 ..................................................................................................5�-CGGGGTACCCTAATTATTATGAACAAATCAG-3�D6 ..................................................................................................5�-CGGGGTACCGCTATCGCTGCTGATGCAGG-3�D7 ..................................................................................................5�-CGGGGTACCGCTGCAGCTAAATTAGC-3�gfpSphIR.......................................................................................5�-ACATGCATGCGAATTCTTATTTGTATAGTTC-3�RBSM1..........................................................................................5�-CGCGGATCCAATTAATAATTCGTTTTTATGAGTAAAGGAGAAG-3�RBSM2..........................................................................................5�-CGCGGATCCAATTAATAATTTGTTTTTATGAGTAAAGGAGAAG-3�RBSM3..........................................................................................5�-CGCGGATCCAATTAATAATTATTATTTATGAGTAAAGGAGAAG-3�RBSM4..........................................................................................5�-CGCGGATCCAATTAATAATTTTTATTTATGAGTAAAGGAGAAG-3�RBSM5..........................................................................................5�-CGCGGATCCAATTAATAATTTTTTTTTATGAGTAAAGGAGAAG-3�RBSM6..........................................................................................5�-CGCGGATCCAATTAATAATTGATATTTATGAGTAAAGGAGAAG-3�RBSM7..........................................................................................5�-CGCGGATCCAATTAATAATTAATGTTTATGAGTAAAGGAGAAG-3�RBSM8..........................................................................................5�-CGCGGATCCAATTAATAATTAAAATTTATGAGTAAAGGAGAAG-3�RBSM9..........................................................................................5�-CGCGGATCCAATTAATAATTAGTATTTATGAGTAAAGGAGAAG-3�WT.................................................................................................5�-CGCGGATCCAATTAATAATTAATATTTATGAGTAAAGGAGAAG-3�SCH08race....................................................................................5�-CCGCTCTGCCTCTTGCTCCAGGTCTTG-3�SCH13race....................................................................................5�-CCACATTGAAGTGTAAGCTGCCTGAACAGCTGC-3�SCH17race....................................................................................5�-GTCAATAAGTTCGTCTTTGTTAGCAGGCC-3�SCH36race....................................................................................5�-TGGCATTTGGATTGGATTACCCGTTCC-3�SCH40race....................................................................................5�-GTCTACTTCTCCCCAAACCGGTTTCCATG-3�SCH41race....................................................................................5�-GTAAGTAGCATCACCTTCACCTTCACCGG-3�SCH52race....................................................................................5�-CAACGCTTCGTACCCGTCAACACCTTTGG-3�
a Restriction sites on the primers are underlined.
VOL. 73, 2007 PROMOTERS IN FLAVOBACTERIUM 1091
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

mixture was electroporated into electrocompetent Flavobacterium cells. Quali-tative screening of transformants for expression of the gfpmut3 gene was per-formed by fluorescence microscopy (excitation wavelength of 485 nm and emis-sion length of 510 nm). Quantitative analysis of GFP production was performedusing a microtiter fluorescence spectrometer (Dynex, Chantilly, VA). Strainswere grown overnight in CYE medium supplemented with 10 �g/ml tetracycline.Two hundred microliters of each culture was centrifuged, washed with phos-phate-buffered saline, and diluted to an optical density at 600 nm of 0.4. Thesamples were analyzed under the following conditions: excitation wavelength of485 nm, emission wavelength of 510 nm, and 0.2-s interval at 25°C. To ensurethat the values recorded were due to GFP, cultures of untransformed strainswere used as the appropriate blanks for calculation of the relative units offluorescence. Colonies showing high fluorescence were subcultured and stored inCYE-glycerol medium at �80°C.
RNA isolation. Flavobacterium cells (2 ml) from exponentially growing cul-tures (turbidity at 650 nm of 0.3 to 0.4) were stabilized using 2 volumes ofRNAprotect bacterial reagent (QIAGEN) for 5 to 10 min. Total RNA wasextracted by using the RNeasy kit (QIAGEN) according to the protocol of themanufacturer. Following extraction, total RNA was treated with DNase I. TheDNase I was later heat inactivated at 70°C for 15 min. To concentrate thesamples, total RNA was precipitated with ethanol and resuspended in 30 �l ofRNase-free water. Samples were stored at �80°C.
Transcriptional start site. The transcriptional start site was determined byusing the method 5� rapid amplification of cDNA ends based on the switchingmechanism at 5� end of RNA transcript (SMART-RACE system), as recom-mended by the supplier (Clontech, Mountain View, CA) with 3 �g of total RNA(DNA free). A gfpmut3 gene-specific primer (gfpSphIR) was used to initiate thefirst strand of cDNA synthesis for 1.5 h at 42°C. Small aliquots of the abovecDNA as a template were amplified using SMART PCR primer UP and gene-specific primers (Table 2). The PCR products were cloned into the T-easy vectoraccording to standard procedures and sequenced.
DNA sequencing. Plasmid DNA was prepared using QIAGEN Mini-Prep spincolumns. The insert fragment was sequenced by the dideoxy termination methodusing an automated sequencing system (Applied Biosystem). GenBank databasesearches were carried out using the National Center for Biotechnology Infor-mation BLAST web server (http://www.ncbi.nlm.nih.gov/BLAST). Multiple se-quence alignments were carried out with the ClustalW program and later ad-justed manually.
Nucleotide sequence accession number. The inserter sequences of the pro-moter clones reported in this paper have been deposited in the GenBank data-base under the following accession numbers: SCH08, DQ834946; SCH13,DQ834947; SCH14, DQ834948; SCH15, DQ834949; SCH16, DQ834950;SCH17, DQ834951; SCH19, DQ834952; SCH24, DQ834953; SCH25, DQ834954;
SCH28, DQ834955; SCH29, DQ834956; SCH35, DQ834957; SCH36, DQ834958;SCH40, DQ834959; SCH41, DQ834960; SCH42, DQ834961; SCH45, DQ834962;SCH47, DQ834963; SCH51, DQ834964; and SCH52, DQ834965.
RESULTS
Phylogenetic placement of F. hibernum strain W22. GenomicDNA was extracted from strain W22, and the 16S rRNA gene wasamplified using primers 63f and 1387r (Table 2). Both strands ofthe amplified fragment were sequenced. BLAST analysis of the16S rRNA gene indicated that the strain was a Flavobacteriumsp. Sequence analysis showed that the sequence was 99% iden-tical to the 16S rRNA gene of the Antarctic psychrotroph F.hibernum (GenBank accession no. L39067.1). Placement of thesequence into a phylogenetic tree using ARB (26) revealed aclose relationship to other F. hibernum sequences (Fig. 1).
Transfer of plasmids into F. hibernum strain W22 by con-jugation and electroporation. Attempts to transfer broad-host-range plasmids pMMB66EH, pMMB207, and pMMB503, de-rived from IncQ plasmid RSF1010, or the E. coli-Bacteroidesshuttle plasmid pNJR5, by conjugation into F. hibernum strainW22 did not result in any transconjugants (frequency of�10�9). On the other hand, plasmids pCP23 and pCP29, de-rived from cryptic F. psychrophilum D12 plasmid pCP1, weretransferred by conjugation from E. coli to F. hibernum strainW22 at frequencies of 2.8 10�5 per recipient cell. A proce-dure was developed to allow for the direct introduction ofDNA into F. hibernum strain W22 cells by electroporation.Plasmid pCP23, isolated from F. hibernum strain W22, wasused to optimize this procedure. The optimal parameters fortransfer by electroporation were determined to be 10.0 kV/cmand 400 �. Under these conditions, 2.5 105 tetracycline-resistant transformants per �g of DNA were obtained. Only�200 colonies per �g of DNA were obtained when pCP23isolated from E. coli was used. This indicated that a restrictionbarrier existed between E. coli and Flavobacterium.
FIG. 1. Placement of the Flavobacterium 16S rRNA sequence into a phylogenetic tree using ARB, revealing affinity with F. hibernum. Anapproximately 1.3-kb region of the 16S rRNA gene was amplified using forward primer 63f and reverse primer 1387r and sequenced. Thephylogenetic tree was constructed by neighbor-joining analysis of partial 16S rRNA sequences according to the ARB manual (27). The barrepresents 10% sequence divergence.
1092 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from
Lack of E. coli lac promoter function in Flavobacteriumstrains. Plasmids pSCH02F and pSCH02R, both constructedin this study and containing the expression cassette includingthe lacIq, tac promoter, gfpmut3, and transcriptional termina-tor regions, resulted in intensive green fluorescence of thecolonies when introduced into E. coli DH5� grown on LB agarplates supplemented with isopropyl-�-D-thiogalactopyranoside(IPTG). However, no detectable fluorescence was recorded inF. hibernum strain W22 or F. johnsoniae, into which pSCH02Fand pSCH02R had been transferred after 48 h on the CYEagar plates with IPTG as an inducer (data not shown).
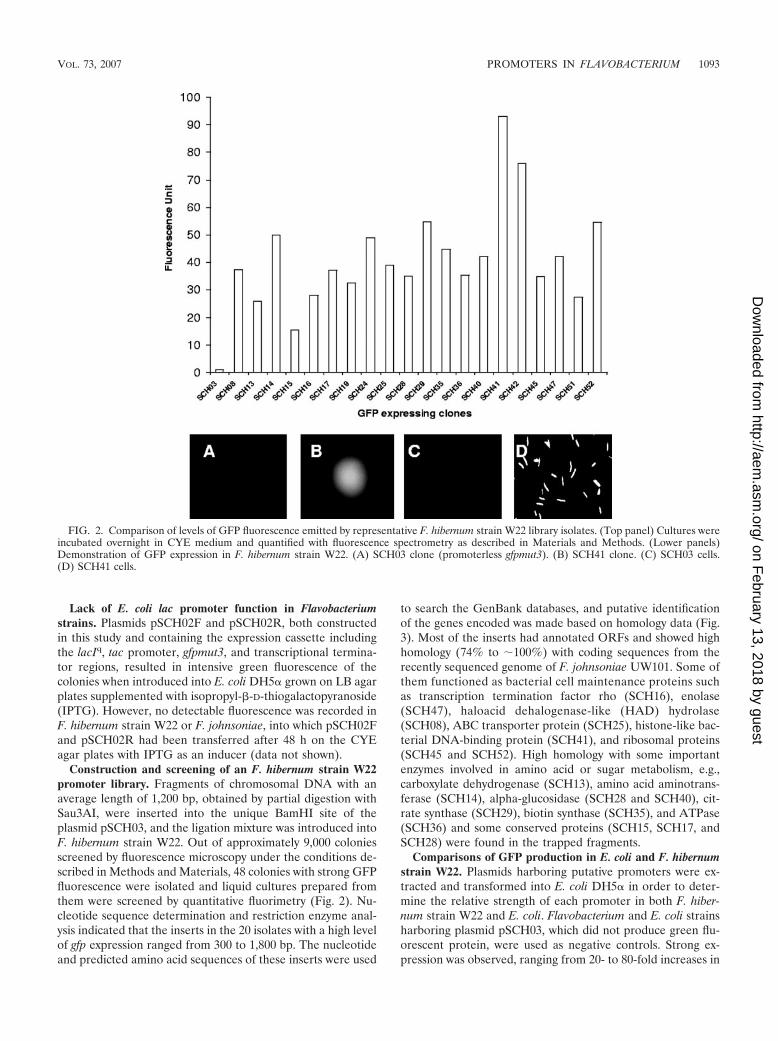
Construction and screening of an F. hibernum strain W22promoter library. Fragments of chromosomal DNA with anaverage length of 1,200 bp, obtained by partial digestion withSau3AI, were inserted into the unique BamHI site of theplasmid pSCH03, and the ligation mixture was introduced intoF. hibernum strain W22. Out of approximately 9,000 coloniesscreened by fluorescence microscopy under the conditions de-scribed in Methods and Materials, 48 colonies with strong GFPfluorescence were isolated and liquid cultures prepared fromthem were screened by quantitative fluorimetry (Fig. 2). Nu-cleotide sequence determination and restriction enzyme anal-ysis indicated that the inserts in the 20 isolates with a high levelof gfp expression ranged from 300 to 1,800 bp. The nucleotideand predicted amino acid sequences of these inserts were used
to search the GenBank databases, and putative identificationof the genes encoded was made based on homology data (Fig.3). Most of the inserts had annotated ORFs and showed highhomology (74% to �100%) with coding sequences from therecently sequenced genome of F. johnsoniae UW101. Some ofthem functioned as bacterial cell maintenance proteins suchas transcription termination factor rho (SCH16), enolase(SCH47), haloacid dehalogenase-like (HAD) hydrolase(SCH08), ABC transporter protein (SCH25), histone-like bac-terial DNA-binding protein (SCH41), and ribosomal proteins(SCH45 and SCH52). High homology with some importantenzymes involved in amino acid or sugar metabolism, e.g.,carboxylate dehydrogenase (SCH13), amino acid aminotrans-ferase (SCH14), alpha-glucosidase (SCH28 and SCH40), cit-rate synthase (SCH29), biotin synthase (SCH35), and ATPase(SCH36) and some conserved proteins (SCH15, SCH17, andSCH28) were found in the trapped fragments.
Comparisons of GFP production in E. coli and F. hibernumstrain W22. Plasmids harboring putative promoters were ex-tracted and transformed into E. coli DH5� in order to deter-mine the relative strength of each promoter in both F. hiber-num strain W22 and E. coli. Flavobacterium and E. coli strainsharboring plasmid pSCH03, which did not produce green flu-orescent protein, were used as negative controls. Strong ex-pression was observed, ranging from 20- to 80-fold increases in
FIG. 2. Comparison of levels of GFP fluorescence emitted by representative F. hibernum strain W22 library isolates. (Top panel) Cultures wereincubated overnight in CYE medium and quantified with fluorescence spectrometry as described in Materials and Methods. (Lower panels)Demonstration of GFP expression in F. hibernum strain W22. (A) SCH03 clone (promoterless gfpmut3). (B) SCH41 clone. (C) SCH03 cells.(D) SCH41 cells.
VOL. 73, 2007 PROMOTERS IN FLAVOBACTERIUM 1093
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

F. hibernum strain W22 compared with the control strainSCH03 (Fig. 4). Very little GFP was detected in E. coli trans-formants, although plasmid DNA isolated from E. coli indi-cated that their copy number was up to 10-fold higher than thatin F. hibernum strain W22 when the same concentration ofcells was used (data not shown). Reintroduction of these plas-mids into F. hibernum strain W22 resulted in intensive greenfluorescent colonies, showing that weak GFP production in E.coli was not due to mutation.
Identification of putative TSPs and consensus promoters.The following criteria were used for selection of “hot” pro-moter clones in this study: (i) clones that exhibited the highestfluorescence, (ii) sequences upstream of the reporter havinghomology to known bacterial genes, and (iii) fragments whereputative regulatory sequences were close enough to the re-porter gene (gfp) to facilitate the transcriptional start point(TSP) determination. In order to determine the TSP,
SMART-5� RACE PCR analysis was performed in seven pu-tative promoter clones. For each clone, we obtained a singledominant 5�RACE PCR product (data was not shown), indi-cating there was no alternative transcriptional start site. Toensure there was no contaminating DNA present in the RNApreparation, a negative control was included using the RNAthat had not been reverse transcribed. The amplified 5�-RACEproducts were cloned into T-easy vectors and sequenced asdescribed previously. Nucleotide sequences adjacent to theTSP were aligned with 10 promoters functional in Flavobacte-rium which were either experimentally identified or proposedin other studies, as shown in Fig. 4. The consensus sequencedetermined from this alignment consists of nucleotides thatoccur in more than 50% of the clones at any position. Themapped TSP was A, C, or G, with A present in 9 of 17 pro-moters examined. A putative �7 region matching the consen-sus Bacteroides sequence (TAnnTTTG) (4) was located from 2
FIG. 3. Description of clones containing putative promoter regions from F. hibernum strain W22. The diagrams show the F. hibernum strainW22 chromosomal DNA inserts containing the gene or protein sequence identity, as determined by BLASTN or BLASTX searches. ORFs wereidentified with Gene Finder. The solid arrows represent the direction of putative ORFs with the putative promoter(s), the single lines representcoding regions without promoter(s), and the dashed lines represent noncoding fragments. The solid thick lines represent promoterless gfpmut3.DNA inserts are not drawn to scale relative to each other. The insert size was determined by restriction digestion analysis with KpnI and BamHI.The database homology is only given to the ORFs with the putative promoter. The abbreviations of annotated genes from promoter clones wereassigned like those previously published. The GenBank accession numbers for the annotated genes are as follows: had, EAS57413; p5cd,EAS59076.1; bcat, EAS58378; cp1, EAS58170; rho, EAS57797; cp2, EAS57669; 2ogfo, EAS57503; abt, EAS59161; malt1, EAS61144; cp3,EAS57757; cs1, EAS59912; bts3r, EAS60446; iop, EAS60447; atp, EAS58692; HU, EAS59688; rpl2, EAS59936; esp, EAS58086; eno, EAS59913;alcd, EAS61717; rps10, EAS59940; and rpl3, EAS59939. N/A, data not available.
1094 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

to �16 nucleotides upstream from the TSP, and a putative �33region matching the Bacteroides conserved sequence (TTG)(4) was centered on �33 region from the TSP. The spacebetween �7 and �33 regions was 17 to �23 bases.
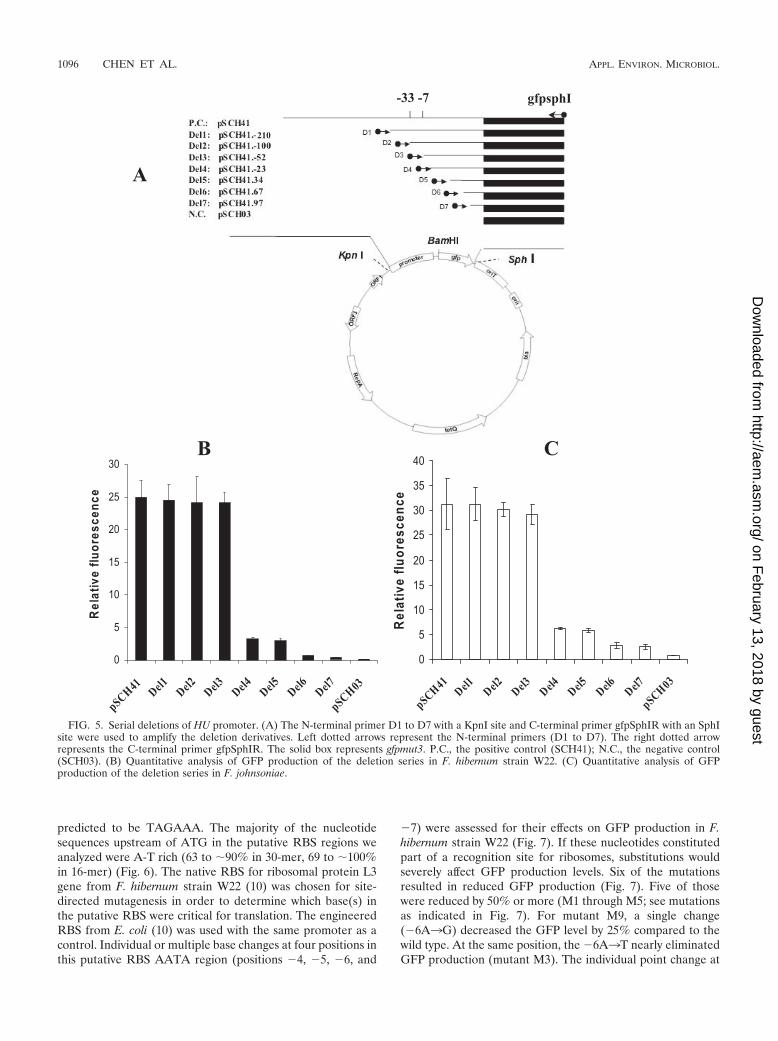
Deletion analysis of a strong promoter. In order to deter-mine which portion at the 5� end of the promoter was criticalfor maximum promoter activity, we chose the strongest pro-moter clone (SCH41) for the following experiment. Deletionderivatives of the trapped fragment encoding histone-like bac-terial DNA-binding protein (HU) were generated by PCR,inserted into the T-easy vector, and sequenced. The individualdeletion promoter fragments were released from T-easy vec-tors and fused to the same position on pCP23. These plasmidswere transformed into F. hibernum strain W22 and F.johnsoniae, and cells harboring these constructs were grown tolog phase and subjected to quantitative fluorescence analysis.Results from sequential deletion of the strongest promoter
clone (SCH41; Fig. 5) showed that there was no significantdifference in the level of gfpmut3 expression in plasmidspSCH41.�210 through pSCH41.�52 in both F. hibernumstrain W22 and F. johnsoniae. Deletion plasmid pSCH41.�23exhibited an eightfold lower level of fluorescence thanpSCH41.�210 in F. hibernum strain W22 and a fivefold lowerlevel in F. johnsoniae. These data indicate that the 52-bp frag-ment upstream of the TSP in the HU promoter is required formaximal expression of gfpmut3 in log-phase F. hibernum strainW22 and F. johnsoniae under our testing conditions.
Putative RBS in F. hibernum strain W22. Alignment of 12putative RBSs from this study with 13 RBSs from variouspublished Flavobacterium strains revealed that TAAAA wasthe consensus sequence (Fig. 6), with some variations. Theputative RBS was followed by a possible initiation codon ATGlocated after a spacer of 2 to 12 bp. The RBS consensussequence of the 16S rRNA of our F. hibernum strain W22 was
FIG. 4. Alignment of experimentally determined or proposed promoters functional in Flavobacterium and determination of GFP fluorescencefrom different promoter clones in E. coli and F. hibernum strain W22. The consensus sequence derived from this alignment is given at the bottom.It was defined as nucleotides that are present at any given position in more than 50% of the sequences. The promoters marked with an “a” wereidentified in this study; the other letters and associated reference numbers are as follows: b, Chen et al., unpublished data; c, reference 17; d andg, reference 8; e, reference 34; f, reference 35; h, reference 6; i, reference 37; j, reference 4. The �7 and �33 consensus regions are capitalizedand boldface. The relative GFP production level of each promoter clone is given as relative fluorescence values. Triplicate samples were used, andthe standard deviations are shown. The SCH03 clone is used as a negative control. N/A, data not available.
VOL. 73, 2007 PROMOTERS IN FLAVOBACTERIUM 1095
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from
predicted to be TAGAAA. The majority of the nucleotidesequences upstream of ATG in the putative RBS regions weanalyzed were A-T rich (63 to �90% in 30-mer, 69 to �100%in 16-mer) (Fig. 6). The native RBS for ribosomal protein L3gene from F. hibernum strain W22 (10) was chosen for site-directed mutagenesis in order to determine which base(s) inthe putative RBS were critical for translation. The engineeredRBS from E. coli (10) was used with the same promoter as acontrol. Individual or multiple base changes at four positions inthis putative RBS AATA region (positions �4, �5, �6, and
�7) were assessed for their effects on GFP production in F.hibernum strain W22 (Fig. 7). If these nucleotides constitutedpart of a recognition site for ribosomes, substitutions wouldseverely affect GFP production levels. Six of the mutationsresulted in reduced GFP production (Fig. 7). Five of thosewere reduced by 50% or more (M1 through M5; see mutationsas indicated in Fig. 7). For mutant M9, a single change(�6A3G) decreased the GFP level by 25% compared to thewild type. At the same position, the �6A3T nearly eliminatedGFP production (mutant M3). The individual point change at
FIG. 5. Serial deletions of HU promoter. (A) The N-terminal primer D1 to D7 with a KpnI site and C-terminal primer gfpSphIR with an SphIsite were used to amplify the deletion derivatives. Left dotted arrows represent the N-terminal primers (D1 to D7). The right dotted arrowrepresents the C-terminal primer gfpSphIR. The solid box represents gfpmut3. P.C., the positive control (SCH41); N.C., the negative control(SCH03). (B) Quantitative analysis of GFP production of the deletion series in F. hibernum strain W22. (C) Quantitative analysis of GFPproduction of the deletion series in F. johnsoniae.
1096 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

position �7 (�7A3G) in mutant M6 had no significant effecton the expression level compared to the wild type. The dualmutations at positions �6 and �7 (�6A3G and �7A3C) inmutant M4 resulted in 40% of the expression observed in thewild-type gene. The triple mutations (�4A-6A-7A3TTT) inmutant M5 decreased GFP production dramatically to about5% of the wild type. In contrast, in mutant M8, the T3Achange at position �5 of the predicted consensus region, re-sulting in the sequence TAAAA, increased the GFP produc-tion by 50% compared to the wild-type RBS. Similarly, a singlechange in mutant M7 (�6A3G) resulted in a 25% increase inGFP production. The same GFP production pattern was re-corded in F. johnsoniae when these constructs were trans-formed (data not shown).
DISCUSSION
Optimization of gene transfer techniques using electropora-tion and modified plasmid constructs allowed us to develop apromoter-trap system (herein named pSCH03), isolate andanalyze several strong promoters, and use GFPmut3 as a re-porter in F. hibernum strain W22. The gfp construct is wellestablished as a marker for gene expression in other bacteria(14, 27, 38), but had not been used previously with species of
Bacteroidetes. Promoter trapping has been used frequently toisolate and characterize native promoters (14, 27) and is de-pendent on functional reporter systems such as GFPmut3.GFPmut3 has several advantages as a reporter (11): it is non-toxic to its host, allows the in situ study of gene expression inenvironmental samples without the additional exogenous sub-strates, and is compatible with high-throughput screening tech-niques such as fluorescence-activated cell sorting. Sequenceanalysis shows that most of the trapped fragments containannotated ORFs that show high homology with other Fla-vobacterium strains. In this study, GFP production in F. hiber-num strain W22 was often associated with sequences that re-sembled those of authentic promoters. Notably, expression ofgfp in E. coli was nil for all of these promoter constructs, eventhough plasmid copy number was demonstrably higher in thathost compared to F. hibernum strain W22 (Fig. 4).
Promoters in F. hibernum strain W22 harbor RNA polymer-ase (sigma factor) binding sequences very similar to those of B.fragilis (4). This sigma factor, named �ABfr, with binding prop-erties to upstream regions of the promoter was found to bewidespread in the Bacteroidetes phylum as well as in the phy-lum Chlorobi (46). The TSPs observed in Flavobacteriumstrains indicated that the preferred �1 base is A. A conservedconsensus sequence, TAnnTTTG, was found 2 to 16 nucleo-
FIG. 6. Alignment of putative Flavobacterium RBS regions. The 30-bp nucleotide sequences upstream of the start codon were chosen forcomparison. The sources of the selected genes are as follows: a, F. hibernum strain W22; b, F. johnsoniae; c, F. columnare; d, P. heparinus; and e,F. psychrophilum. The putative consensus RBSs are indicated as boldface and capitalized. These were defined as nucleotides that are present atany given position in more than 50% of the sequences. The AT contents within 30 bp and 16 bp upstream of the start codon are given aspercentages.
VOL. 73, 2007 PROMOTERS IN FLAVOBACTERIUM 1097
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

tides upstream of the TSP, and a shorter, less conserved se-quence (TTG) was found centered at the �27 to �41 regionupstream of the TSP. Deletion of the �33 region (pSCH.�23)did not completely abolish expression of gfp, but dramaticallydecreased it, indicating that the �33 region is necessary for themaximum expression of the reporter gene (Fig. 5). This resultis unlike that found in Campylobacter jejuni, where only the�10 region is required (50). The space between the two con-served regions ranged from 17 to 23 bases. This space betweenthe �7 motif and the �33 region seems to be unusually large,particularly since eubacterial RNA polymerase works withinthe short interval of 17 � 1 bp (44). However, the two con-served regions and the distance between them in Flavobacte-rium promoters are similar to most known promoter structuresfound in B. fragilis (4). In addition, the TAnnTTTG motifwithin 20 nucleotides of TSP could also be found in manypromoters in Porphyromonas gingivalis, a member of the phy-lum Bacteroidetes (18). In B. fragilis, mutagenesis of the �7region completely disrupted expression of the reporter gene.Deletion of the �33 TTTG did not abolish expression as it didin the �7 element but resulted in a sharp reduction of expres-sion (4). We have further investigated the spacer length be-tween the �7 and �33 regions in Flavobacterium by site-di-rected mutagenesis and found the optimal length to be 19 bp(Chen et al., unpublished data). The differences in promoterconsensus sequences found upstream of several “housekeep-ing” genes in our Flavobacterum strain, compared to thosefound in E. coli and other gram-negative bacteria (44), suggestdifferences in the respective sigma factors. Recently, the puta-tive gene of a sigma factor in F. johnsoniae was cloned and thesequence showed high homology with the primary sigma factor�ABfr in B. fragilis. �ABfr in B. fragilis has been biochemicallyidentified and exhibits several unusual features compared withthose in other prokaryotes (46). The primary structure analysisshows that the protein completely lacks region 1.1, a highly
acidic N-terminal domain present exclusively in primary sigmafactors. In E. coli, region 1.1 has been proven to have severalmodulatory effects on RNA polymerase (RNAP) function andto constitute an autoinhibitory domain that prevents DNAbinding and promoter recognition. �ABfr did not support tran-scription from any promoter of B. fragilis or E. coli in associ-ation with the E. coli RNAP core. In contrast, it formed anactive holoenzyme only with its cognate core RNAP and rec-ognized only Bacteroidetes promoters.
The signal for initiation of protein synthesis in bacteria con-sists primarily, but not exclusively, of an AUG codon and anrRNA-complementary sequence, the Shine-Dalgarno se-quence (39). This sequence is usually located 4 to 9 nucleotidesupstream of the initiator AUG in many mRNAs, where it iscomplementary to the 3� end of 16S rRNA. When constructingthe promoter-trap vector pSCH03, we originally assumed thatthe E. coli RBS sequence could be recognized by Flavobacte-rium strains. We examined the 30-bp regions upstream of thestart codon from 25 ORFs from different Flavobacteriumstrains (Fig. 6). One of the striking features observed in mostof the genes is that the majority of the nucleotide sequencesupstream of AUG were A�T rich (63 to �90% in 30-mer, 68to �100% in 16-mer). This finding contrasts with the well-characterized RBS consensus (AGGAGG) in other pro-karyotes, where the RBS is typically centered in a purine-richregion (45). A consensus sequence, “TAAAA,” is proposedherein as the novel putative RBS in Flavobacterium strains(Fig. 6). In contrast, no typical “AGGAGG” RBS-like se-quence existed within the 16 bp upstream of any of the putativeORFs isolated from F. hibernum strain W22. The 3� end of the16S rRNA of Flavobacterium strains is 5�-CUGGAUCACCUCCUUUCUA-3�. This does include a sequence complemen-tary to the typical RBS sequence “AGGAGG” of gram-nega-tive bacteria (40). It includes, however, an extra sequencepartially complementary to the sequence “TAAAA” found
FIG. 7. Effects of various mutations in the putative RBS region on GFP production level. Oligonucleotide-directed mutagenesis in the putativeRBS was created by PCR, and products were transcriptionally fused to HU promoter in SCH41. Replacement bases are underlined. The GFP levelwas expressed relative to that measured with the wild type.
1098 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

upstream of the 25 open reading frames from various Fla-vobacterium strains. In addition, our site-directed mutagenesisanalysis of the TAAAA sequence rendered further support forthe proposed consensus RBS motif in Flavobacterium strains.Why the putative RBS sequences found in this work do nothave a perfect complement in the 3� end of Flavobacterium 16SrRNA requires further study.
This study is the first to characterize the consensus promoterstructures and putative RBS motifs for general housekeepinggenes in a Flavobacterium species using the GFP-based re-porter system. This will lead to an understanding of specificgene regulation for both transcriptional and translational anal-ysis in Bacteroidetes. Results of this study can also be extendedto achieve stable protein production in Flavobacterium andrelated bacteria, especially when inducible systems are notapplicable. The evolutionary advantage of the novel promoterand RBS motifs for modulating gene expression in F. hibernumstrain W22 and related bacteria remains unclear, but it isreasonable to conclude that the departure from typical tran-scription and translation start signals reflects an early diver-gence of the Bacteroidetes group from the main bacterialbranch.
ACKNOWLEDGMENTS
We gratefully acknowledge Mark McBride for his generous adviceand supply of plasmids pCP23 and pCP29 and Flavobacterium strains.We also thank Angela Sosin and Blair Bullard for their assistance.
This project was funded by NIH grant AI21884.
REFERENCES
1. Agarwal, S., D. W. Hunnicutt, and M. J. McBride. 1997. Cloning and char-acterization of the Flavobacterium johnsoniae (Cytophaga johnsonae) glidingmotility gene, gldA. Proc. Natl. Acad. Sci. USA 94:12139–12144.
2. Alvarez, B., P. Secades, M. J. McBride, and J. A. Guijarro. 2004. Develop-ment of genetic techniques for the psychrotrophic fish pathogen Flavobac-terium psychrophilum. Appl. Environ. Microbiol. 70:581–587.
3. Alvarez, B., P. Secades, M. Prieto, M. J. McBride, and J. A. Guijarro. 2006.A mutation in Flavobacterium psychrophilum tlpB inhibits gliding motilityand induces biofilm formation. Appl. Environ. Microbiol. 72:4044–4053.
4. Bayley, D. P., E. R. Rocha, and C. J. Smith. 2000. Analysis of cepA and otherBacteroides fragilis genes reveals a unique promoter structure. FEMS Micro-biol. Lett. 193:149–154.
5. Bernardet, J. F., Y. Nakagawa, and B. Holmes. 2002. Proposed minimalstandards for describing new taxa of the family Flavobacteriaceae andemended description of the family. Int. J. Syst. Evol. Microbiol. 52:1049–1070.
6. Blain, F., A. L. Tkalec, Z. Shao, C. Poulin, M. Pedneault, K. Gu, B. Eggi-mann, J. Zimmermann, and H. Su. 2002. Expression system for high levelsof GAG lyase gene expression and study of the hepA upstream region inFlavobacterium heparinum. J. Bacteriol. 184:3242–3252.
7. Borriss, M., E. Helmke, R. Hanschke, and T. Schweder. 2003. Isolation andcharacterization of marine psychrophilic phage-host systems from Arctic seaice. Extremophiles 7:377–384.
8. Braun, T. F., M. K. Khubbar, D. A. Saffarini, and M. J. McBride. 2005.Flavobacterium johnsoniae gliding motility genes identified by mariner mu-tagenesis. J. Bacteriol. 187:6943–6952.
9. Braun, T. F., and M. J. McBride. 2005. Flavobacterium johnsoniae GldJ is alipoprotein that is required for gliding motility. J. Bacteriol. 187:2628–2637.
10. Chen, S., M. Bagdasarian, M. G. Kaufman, and E. D. Walker. 2007. Orga-nization of a partial S10 operon and its transcriptional analysis in Flavobac-terium hibernum strain W22. FEMS Microbiol. Lett. 267:38–45.
11. Cormack, B. P., R. H. Valdivia, and S. Falkow. 1996. FACS-optimizedmutants of the green fluorescent protein (GFP). Gene 173:33–38.
12. Cottrell, M. T., and D. L. Kirchman. 2000. Natural assemblages of marineproteobacteria and members of the Cytophaga-Flavobacter cluster consum-ing low- and high-molecular-weight dissolved organic matter. Appl. Environ.Microbiol. 66:1692–1697.
13. Furste, J. P., W. Pansegrau, R. Frank, H. Blocker, P. Scholz, M. Bagdasar-ian, and E. Lanka. 1986. Molecular cloning of the plasmid RP4 primaseregion in a multi-host-range tacP expression vector. Gene 48:119–131.
14. Handfield, M., H. P. Schweizer, M. J. Mahan, F. Sanschagrin, T. Hoang, andR. C. Levesque. 1998. ASD-GFP vectors for in vivo expression technology in
Pseudomonas aeruginosa and other gram-negative bacteria. Bio/Technology24:261–264.
15. Hunnicutt, D. W., M. J. Kempf, and M. J. McBride. 2002. Mutations inFlavobacterium johnsoniae gldF and gldG disrupt gliding motility and inter-fere with membrane localization of GldA. J. Bacteriol. 184:2370–2378.
16. Hunnicutt, D. W., and M. J. McBride. 2000. Cloning and characterization ofthe Flavobacterium johnsoniae gliding-motility genes gldB and gldC. J. Bac-teriol. 182:911–918.
17. Hunnicutt, D. W., and M. J. McBride. 2001. Cloning and characterization ofthe Flavobacterium johnsoniae gliding motility genes gldD and gldE. J. Bac-teriol. 183:4167–4175.
18. Jackson, C. A., B. Hoffmann, N. Slakeski, S. Cleal, A. J. Hendtlass, and E. C.Reynolds. 2000. A consensus Porphyromonas gingivalis promoter sequence.FEMS Microbiol. Lett. 186:133–138.
19. Kaufman, M. G., S. N. Bland, M. E. Worthen, E. D. Walker, and M. J. Klug.2001. Bacterial and fungal biomass responses to feeding by larval Aedestriseriatus (Diptera: Culicidae). J. Med. Entomol. 38:711–719.
20. Kaufman, M. G., W. Goodfriend, A. Kohler-Garrigan, E. D. Walker, andM. J. Klug. 2002. Soluble nutrient effects on microbial communities andmosquito production in Ochlerotatus triseriatus habitats. Aquat. Microb.Ecol. 29:73–88.
21. Kempf, M. J., and M. J. McBride. 2000. Transposon insertions in the Fla-vobacterium johnsoniae ftsX gene disrupt gliding motility and cell division. J.Bacteriol. 182:1671–1679.
22. Kirchman, D. L. 2002. The ecology of Cytophaga-Flavobacteria in aquaticenvironments. FEMS Microbiol. Ecol. 39:91–100.
23. Kirchman, D. L., L. Y. Yu, and M. T. Cottrell. 2003. Diversity and abundanceof uncultured Cytophaga-like bacteria in the Delaware Estuary. Appl. Envi-ron. Microbiol. 69:6587–6596.
24. Kisand, V., R. Cuadros, and J. Wikner. 2002. Phylogeny of culturable estu-arine bacteria catabolizing riverine organic matter in the northern Baltic Sea.Appl. Environ. Microbiol. 68:379–388.
25. Kisand, V., and J. Wikner. 2003. Combining culture-dependent and -inde-pendent methodologies for estimation of richness of estuarine bacterio-plankton consuming riverine dissolved organic matter. Appl. Environ. Mi-crobiol. 69:3607–3616.
26. Ludwig, W., O. Strunk, R. Westram, L. Richter, H. Meier, Yadhukumar, A.Buchner, T. Lai, S. Steppi, G. Jobb, W. Forster, I. Brettske, S. Gerber, A. W.Ginhart, O. Gross, S. Grumann, S. Hermann, R. Jost, A. Konig, T. Liss, R.Lussmann, M. May, B. Nonhoff, B. Reichel, R. Strehlow, A. Stamatakis, N.Stuckmann, A. Vilbig, M. Lenke, T. Ludwig, A. Bode, and K. H. Schleifer.2004. ARB: a software environment for sequence data. Nucleic Acids Res.32:1363–1371.
27. Lun, S. C., and P. J. Willson. 2004. Expression of green fluorescent proteinand its application in pathogenesis studies of serotype 2 Streptococcus suis. J.Microbiol. Methods 56:401–412.
28. Manfredi, R., A. Nanetti, M. Ferri, A. Mastroianni, O. V. Coronado, and F.Chiodo. 1999. Flavobacterium spp. organisms as opportunistic bacterialpathogens during advanced HIV disease. J. Infect. 39:146–152.
29. McBride, M. J., and T. F. Braun. 2004. GldI is a lipoprotein that is requiredfor Flavobacterium johnsoniae gliding motility and chitin utilization. J. Bac-teriol. 186:2295–2302.
30. McBride, M. J., T. F. Braun, and J. L. Brust. 2003. Flavobacteriumjohnsoniae GldH is a lipoprotein that is required for gliding motility andchitin utilization. J. Bacteriol. 185:6648–6657.
31. McBride, M. J., and M. J. Kempf. 1996. Development of techniques for thegenetic manipulation of the gliding bacterium Cytophaga johnsoniae. J. Bac-teriol. 178:583–590.
32. Michel, L. O., M. Sandkvist, and M. Bagdasarian. 1995. Specificity of theprotein secretory apparatus—secretion of the heat-labile enterotoxin-B sub-unit pentamers by different species of Gram� bacteria. Gene 152:41–45.
33. Morales, V. M., A. Backman, and M. Bagdasarian. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recom-binants. Gene 97:39–47.
34. Naas, T., S. Bellais, and P. Nordmann. 2003. Molecular and biochemicalcharacterization of a carbapenem-hydrolysing beta-lactamase from Fla-vobacterium johnsoniae. J. Antimicrob. Chemother. 51:267–273.
35. Nelson, S. S., and M. J. McBride. 2006. Mutations in Flavobacteriumjohnsoniae secDF result in defects in gliding motility and chitin utilization. J.Bacteriol. 188:348–351.
36. Nematollahi, A., A. Decostere, F. Pasmans, and F. Haesebrouck. 2003. Fla-vobacterium psychrophilum infections in salmonid fish. J. Fish Dis. 26:563–574.
37. Rasmussen, J. L., D. A. Odelson, and F. L. Macrina. 1986. Complete nu-cleotide sequence and transcription of ermF, a macrolide-lincosamide-strep-togramin B resistance determinant from Bacteroides fragilis. J. Bacteriol.168:523–533.
38. Rediers, H., P. B. Rainey, J. Vanderleyden, and R. De Mot. 2005. Unravelingthe secret lives of bacteria: use of in vivo expression technology and differ-ential fluorescence induction promoter traps as tools for exploring niche-specific gene expression. Microbiol. Mol. Biol. Rev. 69:217–261.
39. Ringquist, S., S. Shinedling, D. Barrick, L. Green, J. Binkley, G. D. Stormo,
VOL. 73, 2007 PROMOTERS IN FLAVOBACTERIUM 1099
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from

and L. Gold. 1992. Translation initiation in Escherichia coli: sequences withinthe ribosome-binding site. Mol. Microbiol. 6:1219–1229.
40. Schneider, T. D., G. D. Stormo, L. Gold, and A. Ehrenfeucht. 1986. Infor-mation content of binding sites on nucleotide sequences. J. Mol. Biol. 188:415–431.
41. Shoemaker, N. B., R. D. Barber, and A. A. Salyers. 1989. Cloning andcharacterization of a Bacteroides conjugal tetracycline-erythromycin resis-tance element by using a shuttle cosmid vector. J. Bacteriol. 171:1294–1302.
42. Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilizationsystem for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Bio/Technology 1:784–791.
43. Su, H. S., Z. Q. Shao, L. Tkalec, F. Blain, and J. Zimmermann. 2001.Development of a genetic system for the transfer of DNA into Flavobacte-rium heparinum. Microbiology 147:581–589.
44. Typas, A., and R. Hengge. 2006. Role of the spacer between the �35 and�10 regions in �s promoter selectivity in Escherichia coli. Mol. Microbiol.59:1037–1051.
45. Vellanoweth, R. L., and J. C. Rabinowitz. 1992. The influence of ribosome-
binding-site elements on translational efficiency in Bacillus subtilis and Esch-erichia coli in vivo. Mol. Microbiol. 6:1105–1114.
46. Vingadassalom, D., A. Kolb, C. Mayer, T. Rybkine, E. Collatz, and I. Pod-glajen. 2005. An unusual primary sigma factor in the Bacteroidetes phylum.Mol. Microbiol. 56:888–902.
47. Walker, E. D., D. L. Lawson, R. W. Merritt, W. T. Morgan, and M. J. Klug.1991. Nutrient dynamics, bacterial populations, and mosquito productivity intree hole ecosystems and microcosms. Ecology 72:1529–1546.
48. Welker, T. L., C. A. Shoemaker, C. R. Arias, and P. H. Klesius. 2005.Transmission and detection of Flavobacterium columnare in channel catfishIctalurus punctatus. Dis. Aquat. Org. 63:129–138.
49. Whitehead, T. R. 1997. Development of a bifunctional xylosidase/arabinosi-dase gene as a reporter gene for the gram-negative anaerobes Bacteroidesand Porphyromonas, and Escherichia coli. Curr. Microbiol. 35:282–286.
50. Wosten, M., M. Boeve, M. G. A. Koot, A. C. van Nuenen, and B. A. M. vander Zeijst. 1998. Identification of Campylobacter jejuni promoter sequences.J. Bacteriol. 180:594–599.
51. Yoon, J. H., S. J. Kang, and T. K. Oh. 2006. Flavobacterium soli sp nov.,isolated from soil. Int. J. Syst. Evol. Microbiol. 56:997–1000.
1100 CHEN ET AL. APPL. ENVIRON. MICROBIOL.
on February 13, 2018 by guest
http://aem.asm
.org/D
ownloaded from