Embed Size (px)
Citation preview

CH. 19
THERMO--ENTROPY
FREE ENERGY
Spont. equilibrium Qc:Kc
1st LawH; S2nd 3rd Law
SSpont/non exo/endo
G = H - TSSpont/non exo/endoWork; temp
Rxn DirEquilibriumQ vs K
Why does a change occur????Spont/Non revist 1st law, H, exo/endo, Q/K equilibrium, rxn dir, Hess’ Law, state fcts

1st LAW: E conserved E = PE + KEE when add/remove q/w E = q + w
+ heat gain sys - heat lost surr
1st Law Does: acct’s for E Limitation: not predict dir
ENTHALPY H < 0 Spont (-H, exo)combustion, metal oxidation, ionic solid formation
H > 0 Spont (+H, endoo)????? Under right conditionsmelting, vaporization, H2O soluble salts

2nd Law: spont. change
Spont : process that causes a chem/phy , occurs by itself w/o external input of E outside the sys
----------><---------
Sys releases enough E to keep going
Nonspont : sys must have constant input of E from surroundings
----------><---------
NON +H
SPONT
SPONT - H
NON

Spont Endo Rxnfreedom of motion after occur;E more dispersed
Predict dir: compare Q to K
Incr freedom: g ---> l ---> s
Direction Key Factor: freedom of motion, dispersion of E
Microstates (W): # of states (ways) a sys has to disperse E
S = k LnW k: Boltzman’s Constant R/N = 1.38*10-23 J/K
S: Entropy; measure disorder of sys

S = k LnW
W?: fewer microstates to spread E, lower S value
Incr S; incr freedom s ---> l ---> g incr # mols (particles)
Then, which is lower? N2O4 or 2 NO2 ??
S: state fct S = Sf - Si
LOWER ENTROPYa) 1 mol CaF2(s) or 1 mol BaCl2(s) lower molar massb) Br2(g) or Br2(l) (l):less freedomc) 2 mol NaCl(s) or 2 mol NaCl(aq) fewer particles less freedomd) NO2(g) or NO3(g) fewer W

W = 1 S = k LnW Ln W = 0 S = 0
3rd Law: perfect crystal; Ssys= 0 @ 0 K
2 Approaches:1. # microstates 2. heat
Restate 2nd Law:spont rxn moves in dir to incr ENTROPYSuniv = Ssys + Ssurr > 0

Incr T (absorb q) *incr So KE more * incr KE KE more disperse, more W
Other trends dissolve: s - l - g atomic size/molecular complexity
So VALUES effects of heat; affeted by Temp, state, …. J/molK @ 298 K
State (absord q) *more W E more dispersed *incr So

N2 (g) + 3 H2 (g) ----> 2 NH3 (g)
Sorxn = So
pdts - Soreact < 0
S OF RXN
CH2
------> H3C---CH2=CH2 (g) So > 0H2C---CH2 (g)
Use Hess’ Law, combine Hof find Ho
rxn
oreact
opdts
orxn S - S S nm

N2 (g) + 3 H2 (g) ----> 2 NH3 (g)
Sorxn = (2*So
NH3) - [(1*SoN2) + (3*So
H2)
So Values from Table
= (2*193) - [(1*191.5) + (3*130.6)
= -197 J/Kas predicted So < 0
Predict sign ∆S +/-; increase/decreasea) H2O (l) <----> H2O (g)
b) CO2 (s) <----> CO2 (g)
c) CaO (s) + CO2 (g) ----> CaCO3 (s)
d) Ag+ (aq) + Cl- (aq) ----> AgCl (s)
e) N2 (g) + O2 (g) ----> 2 NO (g)
a) +; increaseb) +; increasec) -; decreased) -; decreasee) +; increase

GIBBS FREE ENERGY Go
Determine w/ 1 criteria
Suniv = Ssys + Ssurr
tells dir
G: measure of spontaneity of process & E available
G = H - TSG = H - TS
Predict Spontaneity 1 variable G 2 variables Ssys + Ssurr

--- + always --- Spont @ all temps; 2 O3 (g) ----> 3 O2 (g) reverse always nonspont.
+ --- always + Nonspont @ all temps; 3 O2 (g) ----> 2 O3 (g) reverse rxn occurs
--- --- -- low temp Spont @ low temps; CaO(s) + CO2 (g) ----> + hi temp reverse nonspont CaCO3 (s)
+ + + low temp Nonspont @ low temps; CaCO3 (s) ----> CaO(s) + -- hi temp becomes spont as temp CO2 (g) increases
H S G RXN characteristics Example
Kelvinin Temp.
ST - H G
809
pg

Calculate G -- using 2 methods
Hsys from Hof values
Ssys from Sof values
Method #1 calcu Hsys & Ssys from table values
3 NO (g) ---> N2O (g) + NO2 (g)
Hsys = [(1 mol NO2*33.2) + (1 mol N2O*82.05)] - (3 mol NO*90.29) kJ/mol = -155.62 kJ/mol
Ssys = [(1 mol NO2*239.9) + (1 mol N2O*219.7)] - (3 mol NO*210.65) J/mol = -172.35 J/mol = -0.172 J/mol
Gsys = (-155.62 kJ/mol) - [(298)*(-0.172 kJ/mol)] = -104.4 kJ/mol

Gorxn= mGo
f PDTS - nGof REACTS
N2O (g) + NO2 (g) ---> 3 NO (g)
Gorxn = (3 mol NO*86.60) - [(1 mol NO2*104.2) + (1 mol N2O*82.05)]
kJ/mol = 104.6 kJ/mol
Method #2 calcu using Gof table values
#1: -G #2: +Gas written: fwd rxn spont rev rxn nonspont

Problem 1: Using Appendix C, calculate Go @ 298 K for combustion of methane: CH4(g) + 2 O2(g) ---- 2 H2O(g) + CO2(g)
Solution: Go = [2∆Go[H2O (g)] + 1 ∆Go[CO2(g)]] - [1∆Go[CH4(g)] + 2∆Go[O2(g)]] = [2[-228.57] + 1 [-394.4]] - [1[-50.8] + 2[0]] = [[-483.64] + [-393.5]] - [[-50.8] + [0]] = [-851.54] – [-50.8] = -800.7 kJ

G = -wmax
-Gsys
spont, work “on sys” Erxn < wmax, some G lost to q
G: amt available E for sys to do work w/o need outside source E
+Gsys
NONspont, unless surr does work “on sys” Erxn need > wmax, some G added as q

TEMPERATURE
1 & 2 Temp Independent#1 H < 0 & S > 0 -H - TS ------> “-” concludes -G combustion, decomposition
#2 H > 0 & S < 0 +H - [-(TS)] ------> “+” concludes +G
3 & 4 Temp Dependent#3 @ low temp H < 0 & S < 0 -H - [-(TS)] ------> “-” concludes -G -H overcome +TS
#4 @ high temp H > 0 & S > 0 +H - TS ------> “-” concludes -Gif T large so -TS = “-”
--- + always ---
+ --- always +
--- --- -- / +
+ + + / --
H S G
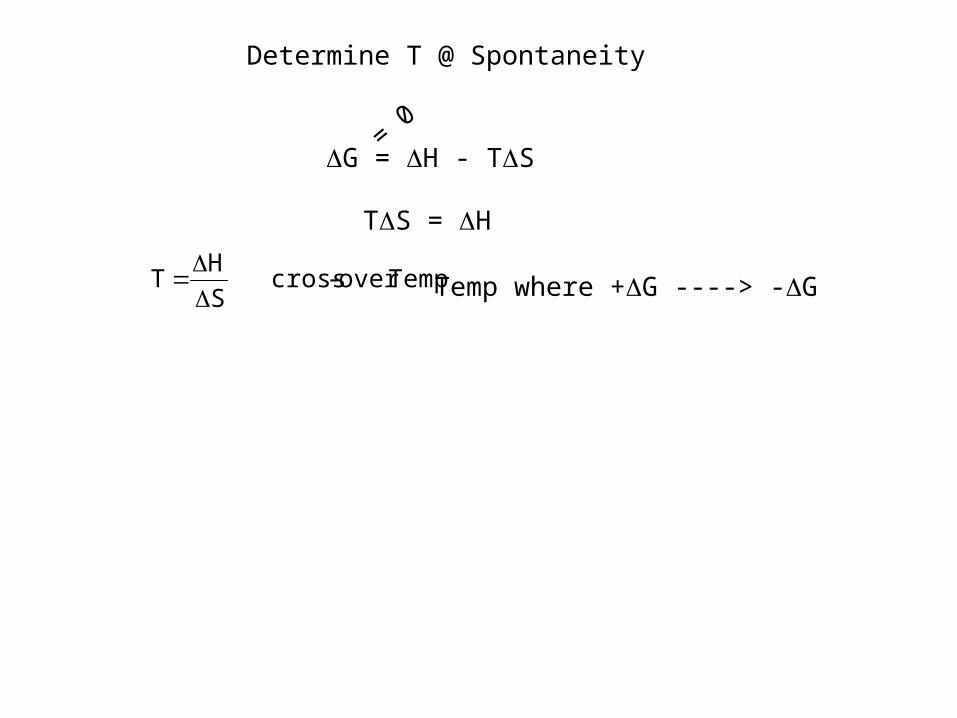
G = H - TS
Determine T @ Spontaneity
= 0
TS = H
Tempover -cross S
H T
Temp where +G ----> -G

Problem #2: What is the Free E value for the equilibrium the normal BP of CCl4 (l) & estimate the normal BP (oC).
@ equilibrium ∆G = 0, as at normal BP both liquid and vapor at standard states
G = H - TS ∆G = 0 CCl4(l) - CCl4(g) T = H/S ∆H = [1*(-106.7)]- [1*(-139.3)] = 32.6 kJ ∆S = [1*(309.4)]- [1*(214.4)] = 95.0 J/K = 0.0950 kJ/K T = (32.6)/(0.0950) = 343 K – 273 = 70 oC

Problem #3: Methanol boils at 64.7oC & ∆Hvap = 71.8kJ/molWill the boiling increase or decrease the entropy & what is the ∆S of 32.0 g when methanol is vaporizeg at its BP? CH3OH(l) --- CH3OH(g) ∆S increases
S = H/T ∆H = (32.0 g)*(1 mol/32.0 g)*(71.8 kJ/mol) = 71.8 kJ
S = H/T = 71.8 kJ/(64.7+273K)*(1000 J/1 kJ) = ∆S = 213 J/K
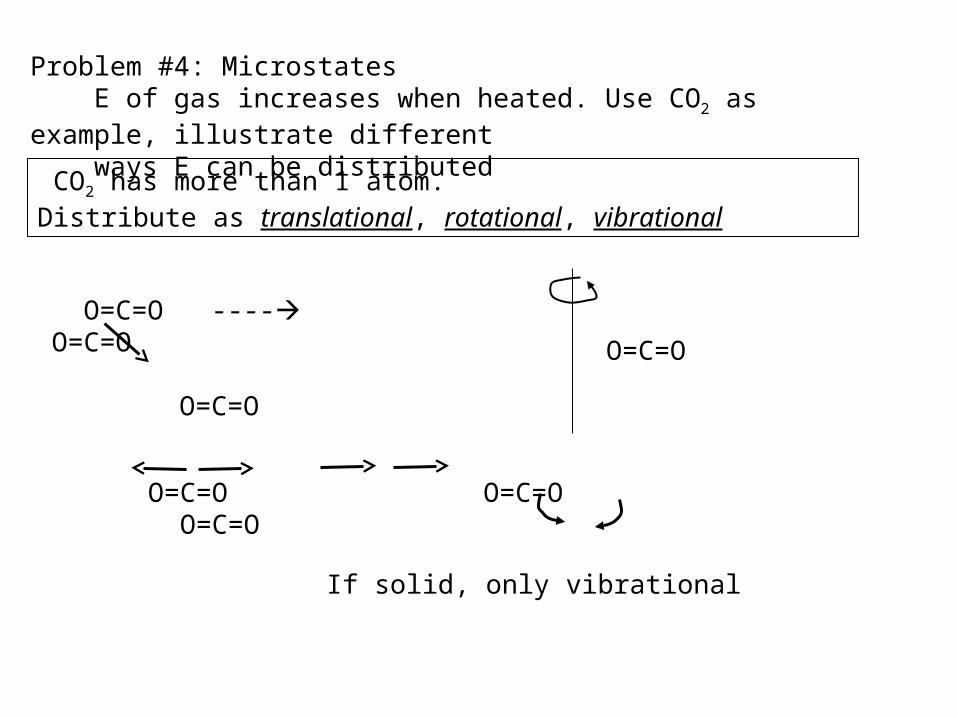
Problem #4: Microstates E of gas increases when heated. Use CO2 as example, illustrate different ways E can be distributed
CO2 has more than 1 atom.Distribute as translational, rotational, vibrational
O=C=O ---- O=C=O O=C=O
O=C=O
O=C=O O=C=O O=C=O
If solid, only vibrational

EQUILIBRIUM; RXN DIR
G = RT LnQ/K = RT Ln Q - RT Ln K
Recall, ch 17 Q < K Q/K < 1 rxn rgt Q > K Q/K > 1 rxn left Q = K Q/K = 1 @ equil
Relate to G Q/K < 1 Ln Q/K < 0 ====> G < 0 rxn rgt Q/K > 1 Ln Q/K < 0 ====> G > 0 rxn left Q/K = 1 Ln Q/K < 0 ====> G = 0 @ equil

@ STD STATES (1 M 1 atm) then G = Go, Q = 1
then, G = RT Ln 1 - RT Ln K = -RT Ln K
As G becomes more “+”, K more “-”; equil more reactAs G becomes more “-”, K more “+”; equil more pdt
@ any starting condition G = Go + Ln Q
Concentrations expressed asGases: partial pressures, atmSolutes: molarity, M

Problem #5: Nonstandard ConditionsCalculate ∆G at 298 K for the formation of 2.0 atm ammonia gas from 0.50 atm N2 & 0.75 atm H2
Find rxn quotient Q for specific partial pressures, evaluate ∆Go
N2 (g) + 3 H2 (g) <----> 2 NH3 (g)
18.96 (0.50)(0.75)
(2.0)
))(P(P
P Q
3
2
N3H
2NH
22
3
∆Go = 2*Gof = 2*(-16.66) = -33.3 kJ
Nonstd conditions: G = Go + RT Ln Q
G = (-33.3 kJ) + [(8.314 J/mol∙K)(298 K)(Ln 18.96)(1 kJ/1000J)] = (-33.3 kJ) + (2.4778 kJ)(2.94) = -33.3 + 7.28 = -26.0 kJ

Problem #6: Find “K” constant at 25oC for the formation of ammonia gas
N2 (g) + 3 H2 (g) <----> 2 NH3 (g)
∆Go = -33.3 kJ/mol = -33,000 J/mol
@ equilibrium, ∆G = 0 then ∆Go = -RT LnK then, K = e-∆Go/RT
13.4 K) K)(298J/mol (8.314
J/mol) (-33,000-
RT
G- o
K = e13.4 = 6.6*105

SUMMARYsys w/ fewer microstates (W), lower S “ ‘ higher “ “ , higher S
incr T, incr S
states s-->l-->--g, incr Sincr # particles (mols), incr S
solid dissolve in liquid nature of solute & solvent E can become localized around ions, can lead to “-S”
dissolve gas fewer W’s available, decr S
atomic size group; complexity, ion size, incr E-levels, incr W, incr S
Spontaneity no relation to “rate”
LeChatilier incr P, rxn dir fewest # mols gas N2(g) + 3 H2(g) ----> 2 NH3(g)
Sorxn states: # mols gas incr, +So (usually)
# mols gas decr, -So (usually)
Go = - RT Ln KG = Go + RT Ln Q -Go K > 1 +Go K < 1

Problem #7: Appendix C, compare So values for graphite & diamond. Why the difference?
∆S graphite = 5.69 J/K ∆S diamond = 2.43 J/K
diamond is network covalent solid; 4-C bondedgraphite is planar sheets of C
Problem #8: The Gfo for NO2 is 51 kJ/mol. Calculate the value of Kp @ 25oC.
2 O2(g) + N2(g) -- 2 NO2(g) form 2 mols, so = 2 Gf
o * 51 = 102 kJ
Gfo = -RT ln(Kp)
Ln K = -(102,000 J)/[(8.31)(298)] = -41.189 e-41.189 = 1.3*10-18 J
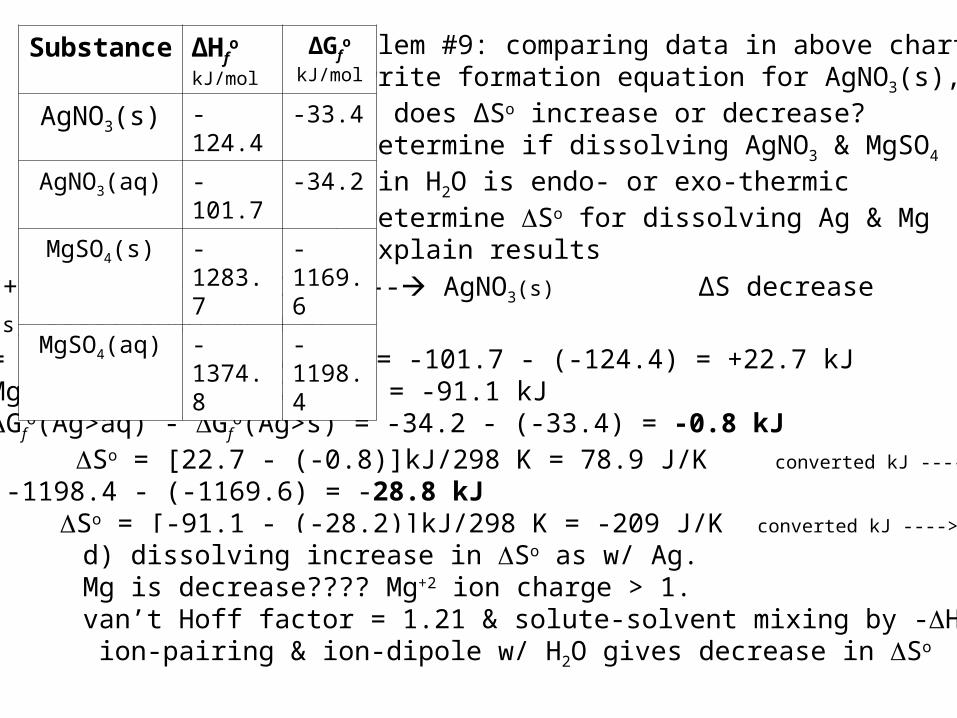
Problem #9: comparing data in above chart;a) write formation equation for AgNO3(s), does ∆So increase or decrease?b) Determine if dissolving AgNO3 & MgSO4
in H2O is endo- or exo-thermicc) Determine So for dissolving Ag & Mgd) Explain results
a) Ag(s) + 0.5 N2(g) + 3/2 O2(g) --- AgNO3(s) ∆S decrease b) AgNO3(s) ----> AgNO3(aq, 1 M) Ho = Ho(Ag>aq) - Ho(Ag>s) = -101.7 - (-124.4) = +22.7 kJ Ho Mg = -1374.8 - (-1283.7) = -91.1 kJc) Go = Gf
o(Ag>aq) - Gfo(Ag>s) = -34.2 - (-33.4) = -0.8 kJ
So = [22.7 - (-0.8)]kJ/298 K = 78.9 J/K converted kJ ----> J
Mg: Go = -1198.4 - (-1169.6) = -28.8 kJ So = [-91.1 - (-28.2)]kJ/298 K = -209 J/K converted kJ ----> J
d) dissolving increase in So as w/ Ag.Mg is decrease???? Mg+2 ion charge > 1.van’t Hoff factor = 1.21 & solute-solvent mixing by -Ho; ion-pairing & ion-dipole w/ H2O gives decrease in So
Substance ∆Hfo
kJ/mol
∆Gfo
kJ/mol
AgNO3(s) -124.4 -33.4
AgNO3(aq) -101.7 -34.2
MgSO4(s) -1283.7 -1169.6
MgSO4(aq) -1374.8 -1198.4

Problem #10: Appendix C, calculate the change in Gibbs E for each.State if spontaneous under std conditions.
a) H2(g) + Cl2(g) HCl(g) b) MgCl2(s) + H2O(l) MgO(s) + HCl(g)
#10. a) G = 2G(HCl(g)) – [G(H2(g)) + G(Cl2(g)] = 2(-95.27 kJ) – 0 – 0 = –190.5 kJ SPONTANEOUS
b) G = [G(MgO(s) + 2G(HCl(g)] – [G(MgCl2(s)) + G(H2O(l)] = [-569.6 + 2(-95.27)] – [(-592.1) + (-237.13)] = +69.1 kJ NONSPONT





























