Embed Size (px)
Citation preview

Calcium and Sulfate Retention by Two Oxisols of the Brazilian CerradoE. Marcano-Martinez and M. B. McBride
ABSTRACTThe ability of two representative oxisols from the Cerrado region
of Brazil to adsorb Ca2 and SO:, , when in their natural acidicstate, was determined by using dissolved CaCI,, K,SOj and CaSO4salts as adsorbates. Calcium adsorption from CaCI, and CaSCX, wasnearly linearly dependent on the equilibrium Ca! concentration,and was most successfully fitted to a Freundlich equation. Con-versely, sulfate adsorption from K2SO4 and CaSO4 was curvilinearlydependent on equilibrium SO2, concentration, conforming moreclosely to Langmuir adsorption behavior. Calcium adsorption wasexplained by nonspecific electrostatic attraction to soil clays, anddid not appear to be adsorbed to any significant degree by the nativeorganic matter. Sulfate adsorption was attributed at least in part toa ligand exchange mechanism, resulting in a measured increase inthe negative charge of the soil surfaces. Simultaneous adsorption ofCa:+ and SOj~ from CaSO4 solutions caused an enhancement inretention of both ions at higher concentrations, despite undersatur-ation with respect to gypsum precipitation. A mechanism ofCaSOJ ion pair adsorption on mineral surfaces is proposed in whichthe presence of one ion facilitates adsorption of the other.
SURFACE CHARGE in variable-charge soils is alteredby adsorption of anions with high affinities for the
surface, such as phosphate, sulfate, and silicate (Mott,1981), as well as by selectively adsorbed cations suchas Ca, Ba, Zn, and Cu (Breeuwsma and Lyklema, 1973;Huang and Stumm, 1973; Parfitt, 1980). Phosphateadsorption has received the most attention in the studyof anion adsorption by soils and effects on surfacecharge. Sulfate adsorption by acid soils has also beenstudied extensively (Chao et al. 1962; Couto et al. 1979;Gebhardt and Coleman, 1974; Harward and Reisen-auer, 1966; Johnson et al., 1979; Singh, 1984a,b,c),although the effect of sulfate adsorption on surfacecharge properties has not received much attention.Some workers considered sulfate to adsorb electro-statically (Marsh et al., 1987; Mott, 1981); others pos-tulated that sulfate coordinates to surfaces of oxidesby a ligand exchange mechanism (Parfitt and Smart,1978; Rajan, 1978), altering the point of zero charge(PZC) of the oxide. Rajan (1978, 1979) reported a de-crease in the PZC of both hydrous alumina and al-lophane after adsorption of sulfate. On the other hand,Breeuwsma and Lyklema (1973) reported the PZC ofhematite to increase after sulfate adsorption.
Couto et al. (1979) showed evidence that SO2." ad-sorption increased the retention of cations in two Ox-isols and an Alfisol. Similar results have been ob-served by Hue et al. (1985) and Zhang et al. (1987).As a result of the ligand exchange mechanism of ad-sorption, SO4" retention is usually accompanied by arise in pH of the soil solution due to the release ofOH~ groups from the surface (Couto et al., 1979; Mott,Dep. of Agronomy, Bradfield Hall, Cornell Univ., Ithaca, NY. Con-tribution from the Dep. of Agronomy, Cornell Univ. Research sup-ported in part by the TropSoil project. Received 15 June 1988.'Corresponding author.
Published in Soil Sci. Soc. Am. J. 53:63-69 (1989).
1981). This higher pH in itself increases the ability ofthe soil to retain cations because of the pH-dependentcharge of Oxisols (Bowden et al., 1980).
The adsorption of Ca2+ is believed to create positivecharge on oxides (Kinniburgh, 1983), but the magni-tude of this charge, detectable as an increase in anionretention, has not been reported. In addition, there isvery little information on the adsorption behavior ofthis ion in variable-charge soils. Most of the studiesinvolving Ca2+ adsorption have been limited to Ca-K (or another monovalent cation) or Ca-Mg exchangereactions, on layer silicates (Galindo and Bingham,1977; Goulding and Talibudeen, 1980; Hunsaker andPratt, 1971; Hutcheon, 1966).
The study of the behavior of Ca2+ and SO2" in soilsis of agricultural importance because of their majorrole as plant nutrients. In Oxisols, the amount of Ca2+
available to plants is usually low and tends to decreasewith depth; values of less than 0.1 cmolc/kg are notuncommon below a depth of 15 to 30 cm (Reeve andSumner, 1972; Ritchey et al., 1982). Sulfate is deficientin some tropical soils; the SO2," already present mightnot be readily available to plants because of strongadsorption (Fox, 1980). The application of gypsum(CaSO4-2H2O) has been recommended to overcomethe problems of Ca2+ deficiency in the lower horizonsof acid soils (Gillman and Sumner, 1987; Ritchey etal., 1980). The greater mobility in soils of gypsumcompared with lime makes gypsum a better source ofCa2+ for the subsoil.
The objective of this study was to compare the be-havior of Ca2+ and SO2." in two representative soilsof the Brazilian Cerrado when applied individually asCaCl2 and K2SO4, to behavior when applied togetheras gypsum, CaSO4-2H2O. For this reason, adsorptionisotherms were measured using solutions of CaCl2,K2SO4, and CaSO4 as adsorbates. It was initially as-sumed that both K+ and Cl~ were indifferent ions anddid not significantly affect the specific adsorption ofeither Ca2+ or SO2.". The effect of the adsorption ofthese ions on the development of surface positive ornegative charge was determined by measuring differ-ences in retention of anions and cations after Ca andsulfate adsorption.
MATERIALS AND METHODSSoil Characteristics
The soils used in this study were the Dark Red AcrustoxA and B horizons (DRA and DRB) and the Red YellowAcrustox A and B Horizons (RYA and RYB) from Brazil.These soils have been described in detail elsewhere (Macedoand Bryant, 1987) and relevant soil chemical data are avail-able in a separate paper (Marcano-Martinez and McBride,in review). Because Soil Taxonomy does not distinguish thesesoils, the nomenclature of the Brazilian classification, basedupon soil color, is used in this paper.Calcium Adsorption Isotherms
Two grams of soil were suspended in centrifuge tubes in20 mL of 0.01 M KC1 with a Ca2+ concentration of 0, 0.05,
63
64 SOIL SCI. SOC. AM. J., VOL. 53, JANUARY-FEBRUARY 1989
0.1, 0.15, 0.2, 0.25, 1.0, 1.5, 2.0, 2.5, and 3.0 mMas CaCl2.The treatments were replicated four times. These were shakencontinuously for a predetermined equilibrium time of 1 h.At the end of the hour, samples were centrifuged and thesupernatants collected for measurement of pH, Ca2+, K+,C1-, and AP+. Next, the adsorbed Ca2+ and Cl~ were dis-placed by washing the soil four times with 20 mL of 0.25M Mg(NO3)2 and the supernatants collected in 100-mL vol-umetric flasks; after making up to volume, the displacedCa2+ and Cl~ were measured. The adsorbed ions were cal-culated after correcting for the KC1 solution entrained in thesoil volume. Calcium was determined by atomic absorptionand Cl~ with a chloride electrode.Sulfate Adsorption Isotherms
Two grams of soil were equilibrated with 20 mL of 0.01M KC1 with a SO2," concentration of 0, 0.1, 0.2, 0.3, 0.5,1.0,2.0, and 3.0 mMas K2SO4 in triplicated treatments. Twodrops of toluene were added to each sample to suppressmicrobial growth. The samples were shaken intermittentlyfor a predetermined equilibrium time of 24 h. After equi-libration, the samples were centrifuged and the supernatantscollected for determination of pH, SO2.-, K+, and Cl~. Theadsorbed K+ was extracted by washing the soil four timeswith 0.1 M NH4NO3 and collecting the supernatants aftereach wash in 100-mL flasks. Adsorbed sulfate was estimatedfrom the amount initially present in the equilibrium solu-tion minus the concentration found after equilibration. Sul-fate was determined with a Dionex 2000i ion chromato-graph (Dionex Corp., Sunnyvale, CA) with the AS3 columnusing an eluting solution of 0.003 M NaHCO3 + 0.003 MNa2CO3.
Simultaneous Ca2+ and S(y4~ AdsorptionThe concurrent adsorption of Ca2+ and SO2," was studied
by a similar procedure to Ca2+ and SOi~ adsorption. Thelevels of CaSO4 dissolved in solution were 0, 0.1, 0.2, 0.3,0.5, 1.0, 2.0, and 3.0 mM in triplicate. Equilibration timewas 24 h, after which time samples were centrifuged, andSO^r and Ca2+ analyzed in the supernatants. The sampleswere then extracted with 0.25 M Mg(NO3)2 to displace andmeasure the adsorbed Ca2+ and K+.
RESULTS AND DISCUSSIONCalcium Adsorption
A kinetic study assessed the minimum requiredequilibration time for Ca2+ adsorption. A concentra-tion of 0.5 mM CaCl2 was added to the four soils ina 0.01 M KC1 background. The change in Ca2+ con-centration in solution, as well as pH, was measuredafter 5 min, 35 min, 1 h, 2 h and 3.5 h.
Adsorption of Ca2+ occurred quickly; Ca2+ concen-tration was constant 5 min after adding Ca2+ to thesoil. Adsorption did not change the pH, suggestingthat Ca2+ was adsorbing on sites previously occupiedby K+. Negligible quantities of A13+ were exchangedinto solution by Ca2+ adsorption. Thereafter, an equi-libration time of 1 h was selected for the CaCl2 sorp-tion studies as a matter of convenience.
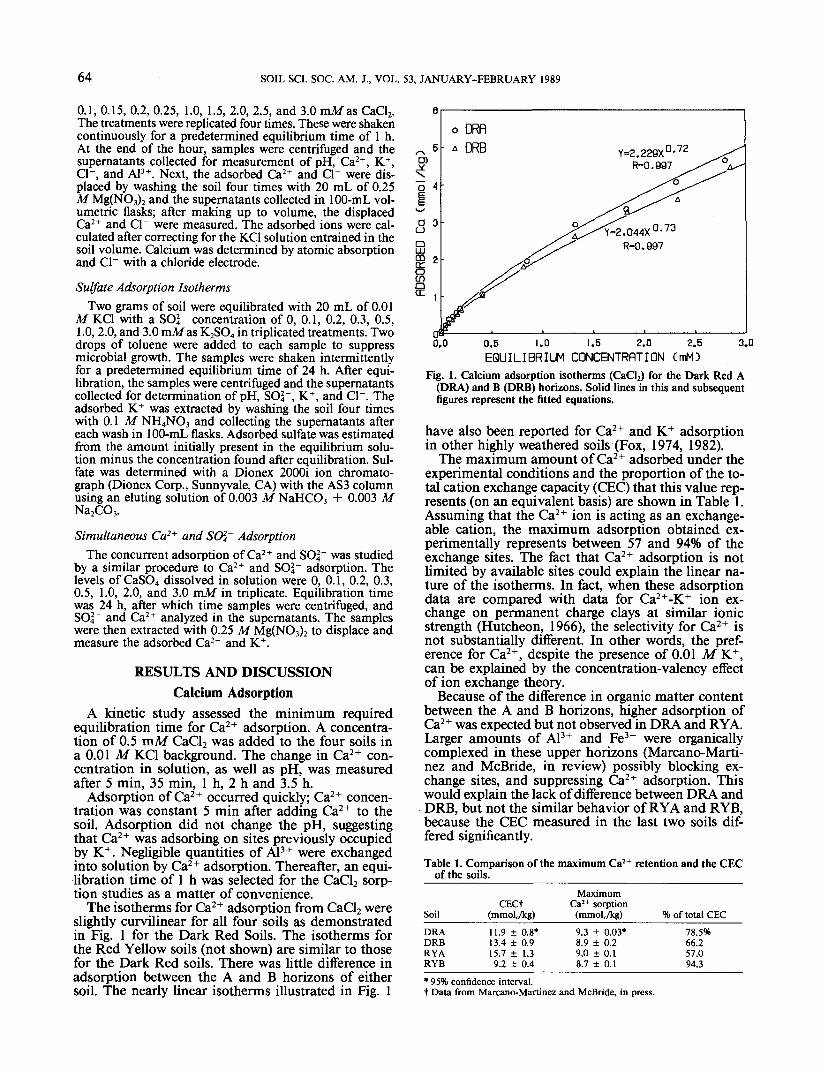
The isotherms for Ca2+ adsorption from CaCl2 wereslightly curvilinear for all four soils as demonstratedin Fig. 1 for the Dark Red Soils. The isotherms forthe Red Yellow soils (not shown) are similar to thosefor the Dark Red soils. There was little difference inadsorption between the A and B horizons of eithersoil. The nearly linear isotherms illustrated in Fig. 1
o.o 0.5 1.0 1.5 2.0 2.5EQUILIBRIUM CONCENTRflTION CmM)
Fig. 1. Calcium adsorption isotherms (CaCl2) for the Dark Red A(DRA) and B (DRB) horizons. Solid lines in this and subsequentfigures represent the fitted equations.
have also been reported for Ca2+ and K+ adsorptionin other highly weathered soils (Fox, 1974, 1982).
The maximum amount of Ca2+ adsorbed under theexperimental conditions and the proportion of the to-tal cation exchange capacity (CEC) that this value rep-resents (on an equivalent basis) are shown in Table 1.Assuming that the Ca2+ ion is acting as an exchange-able cation, the maximum adsorption obtained ex-perimentally represents between 57 and 94% of theexchange sites. The fact that Ca2+ adsorption is notlimited by available sites could explain the linear na-ture of the isotherms. In fact, when these adsorptiondata are compared with data for Ca2+-K+ ion ex-change on permanent charge clays at similar ionicstrength (Hutcheon, 1966), the selectivity for Ca2+ isnot substantially different. In other words, the pref-erence for Ca2+, despite the presence of 0.01 M K+,can be explained by the concentration-valency effectof ion exchange theory.
Because of the difference in organic matter contentbetween the A and B horizons, higher adsorption ofCa2+ was expected but not observed in DRA and RYA.Larger amounts of A13+ and Fe3+ were organicallycomplexed in these upper horizons (Marcano-Marti-nez and McBride, in review) possibly blocking ex-change sites, and suppressing Ca2+ adsorption. Thiswould explain the lack of difference between DRA andDRB, but not the similar behavior of RYA and RYB,because the CEC measured in the last two soils dif-fered significantly.
Table 1. Comparison of the maximum Ca2+ retention and the CECof the soils.
Soil
DRADRBRYARYB
CECf(mmol</kg)
11.9 ± 0.8*13.4 ± 0.915.7 ± 1.39.2 ± 0.4
MaximumCa2+ sorption
(mmolc/kg)
9.3 ± 0.03*8.9 ± 0.29.0 ± 0.18.7 ± 0.1
% of total CEC
78.5%66.257.094.3
* 95% confidence interval.t Data from Marcano-Martinez and McBride, in press.

MARCANO-MARTINEZ & MCBRIDE: CALCIUM AND SULFATE RETENTION BY OXISOLS OF THE BRAZILIAN CERRADO 65
Numerous equations (including linear, quadratic,Langmuir, Freundlich, Tempkin and Gunary) were fitto the adsorption data by nonlinear regression anal-ysis. The Freundlich and quadratic equations fit theCa adsorption data most satisfactorily, with compa-rable R2 and residual sum of squares (RSS) values.Consequently, the Freundlich was selected to describeadsorption because of its simplicity. The best-fitFreundlich equations are plotted in Fig. 1 for the DarkRed soils. The coefficients of these equations for allthe soils are reported in Table 2.
Adsorption of Ca2+ and other metals on pure Feand Al oxides results in the release of H+ to solutionwith the surface becoming more positive (Huang andStumm, 1973; Kinniburgli, 1983; Kinniburgh et al.,1975). In the present experiments Ca2+ adsorption didnot decrease pH, i.e., displacement of protons by Ca2+.This implies that Ca2+ was displacing exchangeableK+ formed in the 0.01 M KC1 medium. It is likelythat these sites were on kaolinite because adsorptionof Ca2+ on oxides is unlikely to be significant at thelow native pH values of these soils (Kinniburgh andJackson, 1982). Adsorption of Ca2+ occurred at a pHof 4.2 to 4.3 in the A horizons, and 4.7 to 4.8 in theB horizons.
The positive charge of the surface, measured by Cl~retention, was not greatly affected by the adsorptionof Ca2+. In the four soil materials, Cl~ retention wasnearly constant up to a Ca2+concentration of 1 mM;at higher levels, Cl~ adsorption was higher but did notincrease in proportion to increases in Ca2+. The meanvalues of Cl~ retention at Ca2+ concentrations < 1mMwere 1.12, 3.81, 1.06, and 3.89 mmol/kg for DRA,DRB, RYA, and RYB respectively. For Ca2+ concen-trations > 1 mM, Cl~ adsorption averaged 1.76, 3.85,3.25, and 5.96 for these same soil materials. HigherCl~ retention in the B horizons may reflect the lowerorganic matter, and greater number of positivelycharged oxide surface sites. These sites are involvedin ligand exchange with functional groups of organicmatter, reducing surface positive charge (Parfitt, 1978).
The increased Cl~ adsorption at high Ca2+ levelscould be due to an increase in surface charge uponCa2+ adsorption on oxides (Huang and Stumm, 1973).Table 2. Freundlich function parameters (Y=AX S ) used to describe
Ca2+ and SOj" adsorption isotherms, and 95% confidence limits.
Parameter
Horizon Adsorbed lonfDRA
DRB
RYA
RYB
Ca(Cl)Ca(S04)S04 (K)S04 (Ca)Ca (Cl)Ca(S04)S04 (K)SO4 (Ca)Ca (Cl)Ca(SO4)S04 (K)SO4 (Ca)Ca(Cl)Ca(SO4)S04 (K)S04 (Ca)
2.23 ± 0.052.99 ±0.153.50 ±0.175.65 ± 0.232.04 ± 0.05
ND4.86 ± 0.24
ND1.96 ± 0.052.69 ±0.183.14 ± 0.164.57 ± 0.251.92 ± 0.05
ND3.93 ± 0.22
ND
0.72 ± 0.031.03 ± 0.060.25 ± 0.040.35 ± 0.040.73 ± 0.03
ND0.26 ± 0.04
ND0.87 ± 0.031.08 ± 0.080.23 ± 0.040.45 ± 0.060.81 ± 0.03
ND0.17 ± 0.03
ND
This mechanism should have been apparent at lowerCa2+ levels, however, requiring the displacement ofprotons from the oxide surface. Alternatively, adsorp-tion of the ion pair CaCl+ would have been significantat the highest CaCl2 levels. Although the concentra-tion of CaCl+ in the aqueous phase would be very loweven at the highest CaCl2 additions (~2 X 10~6 M),this ion pair may be preferentially adsorbed at claysurfaces. Selective adsorption of CaCl+ or MgCl+
complexes by smectites has been documented (Spositoet al., 1983). However, the simplest explanation of atleast part of this Cl~ adsorption is the substantiallyincreased Cl~ concentrations at the highest CaQ2 lev-els, which could effect greater retention on positivecharge sites.
Sulfate AdsorptionThe reaction rate for sulfate adsorption was slower
than for Ca. This is shown in Fig. 2 as a reduction inSO2." concentration with time after mixing the soilswith 0.5 mM K2SO4. An equilibration time of 24 hwas chosen for both K2SO4 and CaSO4 adsorptionstudies based on these data.
Unlike Ca2+, sulfate adsorption followed a high af-finity isotherm, as seen in Fig. 3 for the Dark Red soilmaterial. The Red Yellow soil materials producedsimilar isotherms (not shown). The B horizons hadsignificantly higher adsorption than the A horizons.Both horizons of the Dark Red Acrustox adsorbed sig-nificantly higher amounts of SO4~ than the corre-sponding Red Yellow horizons; this effect was morenoticeable in the B horizons.
The differences in SO4~ adsorption between the Aand B horizons are probably an effect of organic mat-ter content, since neither pH nor free Fe oxides canbe related to these differences. Similar results havebeen reported previously (Bornemisza and Llanos,1967; Couto et al., 1979; Johnson and Todd, 1983;Singh, 1984b). Organic anions in the A-horizons maycompete with SO4~ for positively charged sites (Par-fitt, 1978). The higher adsorption of sulfate in the DarkRed soil materials is probably attributable to the greateramount of Fe oxides in these soils compared to the
0.225
0.12520 30
TIME (h)t Brackets denote the counterion associated with the adsorbing ion.ND = these adsorption data were not collected.
Fig. 2. Reduction of sulfate concentration with time in B horizonmaterials.
66 SOIL SCI. SOC. AM. J., VOL. 53, JANUARY-FEBRUARY 1989
7.0
6.5 •
0.5 1.0 1.6 2.0 2.5EQUILIBRIUM CONCENTRflTION CmM)
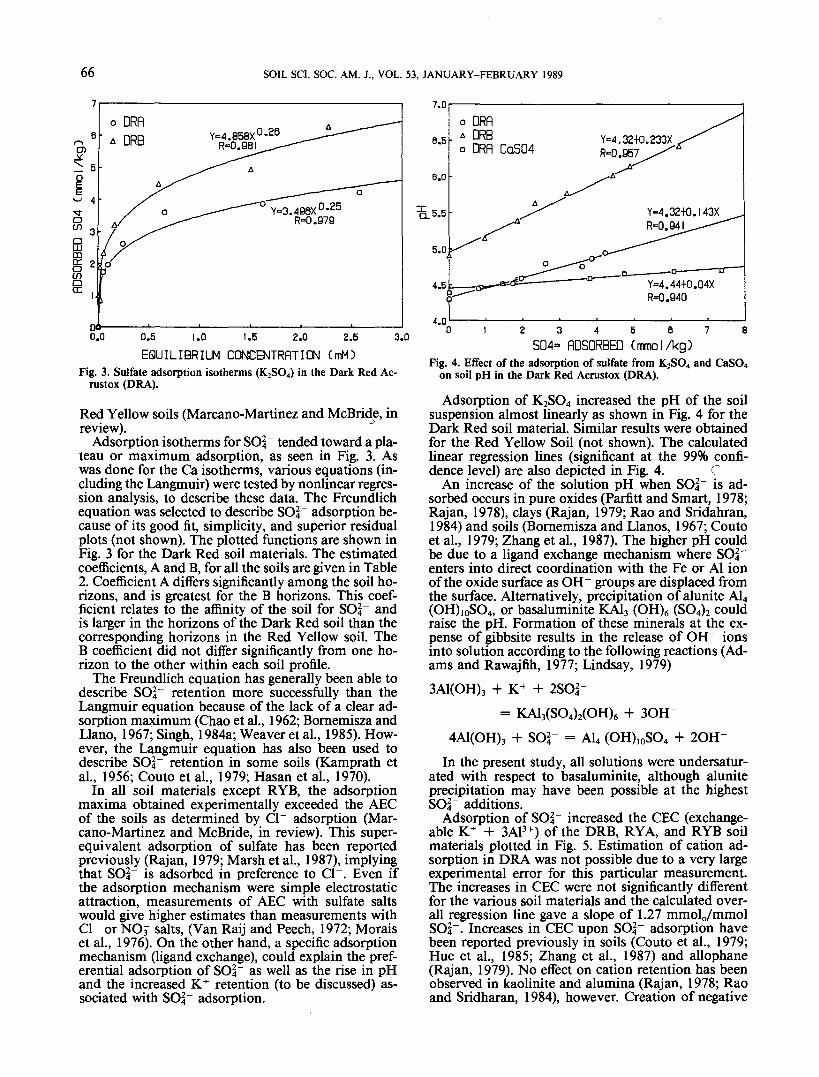
Fig. 3. Sulfate adsorption isotherms (K2SO4) in the Dark Red Ac-rustox (DRA).
Red Yellow soils (Marcano-Martinez and McBride, inreview).
Adsorption isotherms for SO^" tended toward a pla-teau or maximum adsorption, as seen in Fig. 3. Aswas done for the Ca isotherms, various equations (in-cluding the Langmuir) were tested by nonlinear regres-sion analysis, to describe these data. The Freundlichequation was selected to describe 804" adsorption be-cause of its good fit, simplicity, and superior residualplots (not shown). The plotted functions are shown inFig. 3 for the Dark Red soil materials. The estimatedcoefficients, A and B, for all the soils are given in Table2. Coefficient A differs significantly among the soil ho-rizons, and is greatest for the B horizons. This coef-ficient relates to the affinity of the soil for SOI" andis larger in the horizons of the Dark Red soil than thecorresponding horizons in the Red Yellow soil. TheB coefficient did not differ significantly from one ho-rizon to the other within each soil profile.
The Freundlich equation has generally been able todescribe 864" retention more successfully than theLangmuir equation because of the lack of a clear ad-sorption maximum (Chao et al., 1962; Bornemisza andLlano, 1967; Singh, 1984a; Weaver et al., 1985). How-ever, the Langmuir equation has also been used todescribe SO^ retention in some soils (Kamprath etal., 1956; Couto et al., 1979; Hasan et al., 1970).
In all soil materials except RYB, the adsorptionmaxima obtained experimentally exceeded the AECof the soils as determined by Cl~ adsorption (Mar-cano-Martinez and McBride, in review). This super-equivalent adsorption of sulfate has been reportedpreviously (Rajan, 1979; Marsh et al., 1987), implyingthat SOI" is adsorbed in preference to Cl~. Even ifthe adsorption mechanism were simple electrostaticattraction, measurements of AEC with sulfate saltswould give higher estimates than measurements withCl~ or NOf salts, (Van Raij and Peech, 1972; Moraiset al., 1976). On the other hand, a specific adsorptionmechanism (ligand exchange), could explain the pref-erential adsorption of SO|" as well as the rise in pHand the increased K+ retention (to be discussed) as-sociated with SO4~ adsorption.
o DRflA DRBD DRR CaS04
3.04.0
8I 2 3 4 5 6 7S04= RDSORBED (mmol/kg)
Fig. 4. Effect of the adsorption of sulfate from K,SO4 and CaSO4on soil pH in the Dark Red Acrustox (DRA).
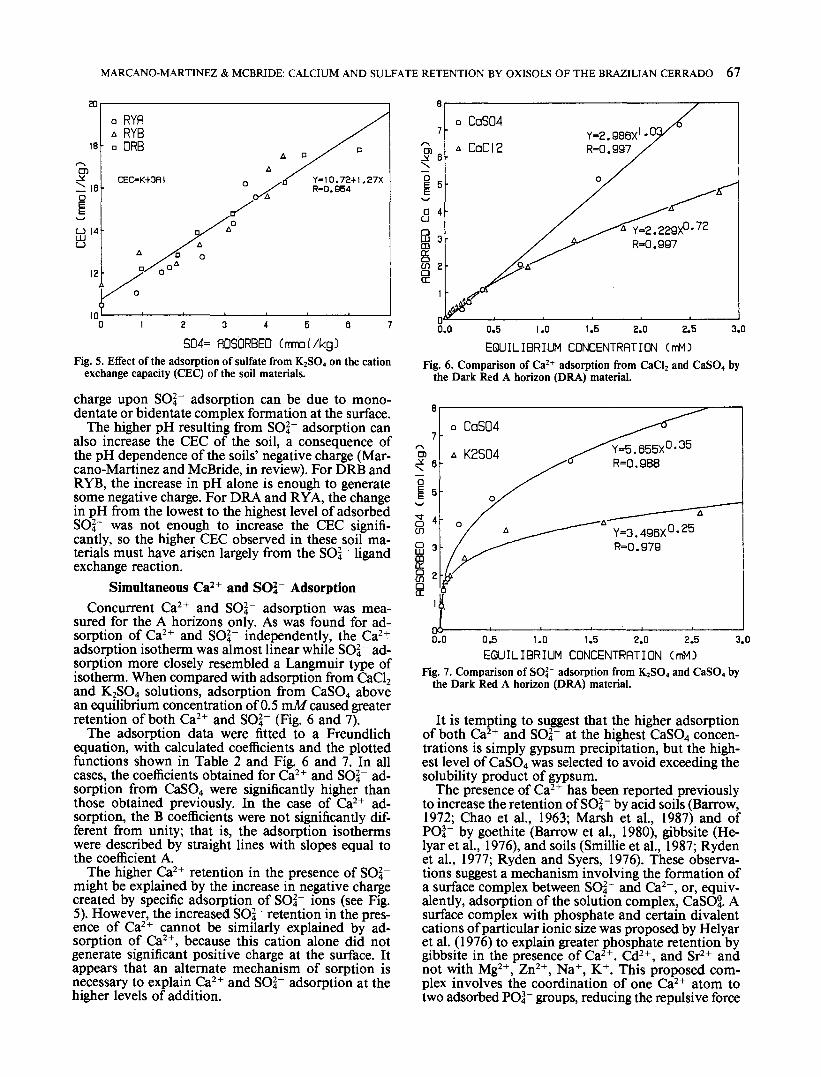
Adsorption of K2SO4 increased the pH of the soilsuspension almost linearly as shown in Fig. 4 for theDark Red soil material. Similar results were obtainedfor the Red Yellow Soil (not shown). The calculatedlinear regression lines (significant at the 99% confi-dence level) are also depicted in Fig. 4. C
An increase of the solution pH when SO|" is ad-sorbed occurs in pure oxides (Parfitt and Smart, 1978;Rajan, 1978), clays (Rajan, 1979; Rao and Sridahran,1984) and soils (Bornemisza and Llanos, 1967; Coutoet al., 1979; Zhang et al., 1987). The higher pH couldbe due to a ligand exchange mechanism where SO|~enters into direct coordination with the Fe or Al ionof the oxide surface as OH~ groups are displaced fromthe surface. Alternatively, precipitation of alunite A14(OH)10SO4, or basaluminite KA13 (OH)6 (SO4)2 couldraise the pH. Formation of these minerals at the ex-pense of gibbsite results in the release of OH~ ionsinto solution according to the following reactions (Ad-ams and Rawajfih, 1977; Lindsay, 1979)3A1(OH)3 + K+ + 2SO|"
= KA13(SO4)2(OH)6 + 3OH"4A1(OH)3 + S0|" = A14 (OH)10SO4 + 2OH"
In the present study, all solutions were undersatur-ated with respect to basaluminite, although aluniteprecipitation may have been possible at the highestSO|" additions.
Adsorption of 804" increased the CEC (exchange-able K+ + 3A13+) of the DRB, RYA, and RYB soilmaterials plotted in Fig. 5. Estimation of cation ad-sorption in DRA was not possible due to a very largeexperimental error for this particular measurement.The increases in CEC were not significantly differentfor the various soil materials and the calculated over-all regression line gave a slope of 1.27 mmol0/mmolSOI". Increases in CEC upon SO|~ adsorption havebeen reported previously in soils (Couto et al., 1979;Hue et al., 1985; Zhang et al., 1987) and allophane(Rajan, 1979). No effect on cation retention has beenobserved in kaolinite and alumina (Rajan, 1978; Raoand Sridharan, 1984), however. Creation of negative
MARCANO-MARTINEZ & MCBRIDE: CALCIUM AND SULFATE RETENTION BY OXISOLS OF THE BRAZILIAN CERRADO 67
0 1 2 3 4 5 6 7
504= flDS'ORBED (mmol/kg)Fig. 5. Effect of the adsorption of sulfate from K2SO4 on the cation
exchange capacity (CEC) of the soil materials.
charge upon SO^" adsorption can be due to mono-dentate or bidentate complex formation at the surface.
The higher pH resulting from SO%~ adsorption canalso increase the CEC of the soil, a consequence ofthe pH dependence of the soils' negative charge (Mar-cano-Martinez and McBride, in review). For DRB andRYB, the increase in pH alone is enough to generatesome negative charge. For DRA and RYA, the changein pH from the lowest to the highest level of adsorbedSO|~ was not enough to increase the CEC signifi-cantly, so the higher CEC observed in these soil ma-terials must have arisen largely from the SO^" ligandexchange reaction.
Simultaneous Ca2+ and SO2." AdsorptionConcurrent Ca2+ and SO^" adsorption was mea-
sured for the A horizons only. As was found for ad-sorption of Ca2+ and SO2^ independently, the Ca2+
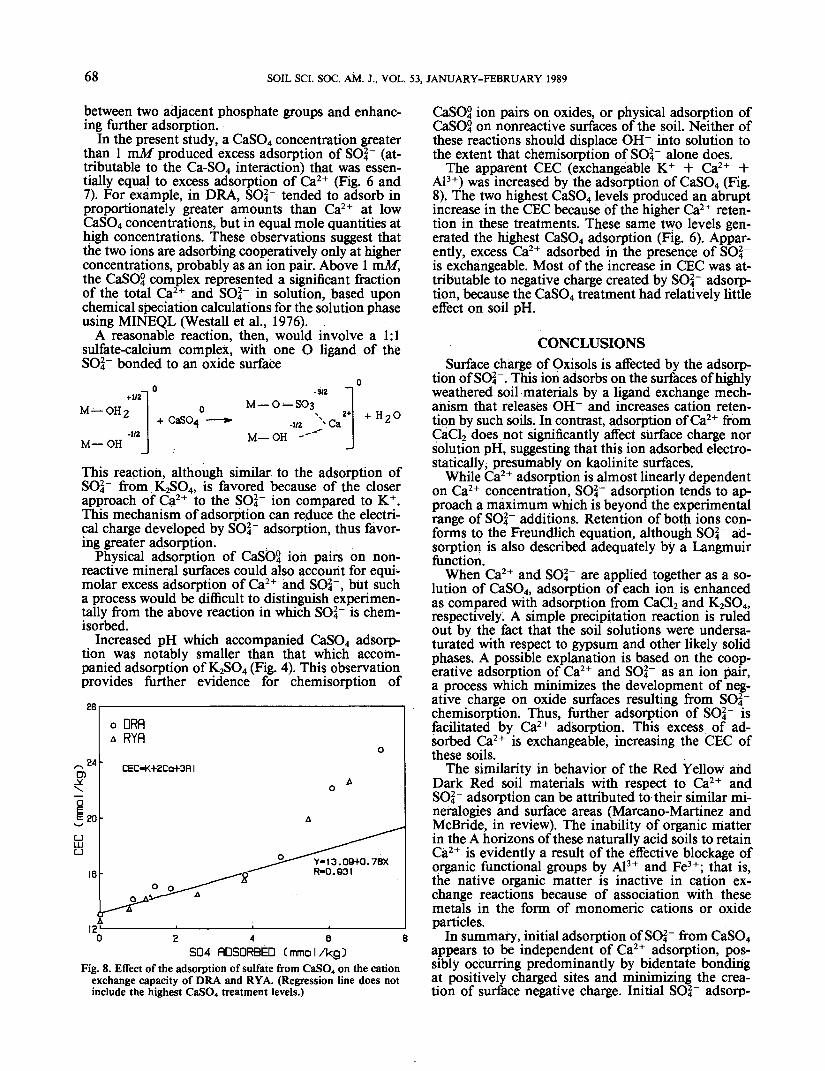
adsorption isotherm was almost linear while SC>4~ ad-sorption more closely resembled a Langmuir type ofisotherm. When compared with adsorption from CaCl2and K?SO4 solutions, adsorption from CaSO4 abovean equilibrium concentration of 0.5 mM caused greaterretention of both Ca2+ and SO2.- (Fig. 6 and 7).
The adsorption data were fitted to a Freundlichequation, with calculated coefficients and the plottedfunctions shown in Table 2 and Fig. 6 and 7. In allcases, the coefficients obtained for Ca2+ and SO2." ad-sorption from CaSO4 were significantly higher thanthose obtained previously. In the case of Ca2+ ad-sorption, the B coefficients were not significantly dif-ferent from unity; that is, the adsorption isothermswere described by straight lines with slopes equal tothe coefficient A.
The higher Ca2+ retention in the presence of SO2."might be explained by the increase in negative chargecreated by specific adsorption of SO2" ions (see Fig.5). However, the increased 804" retention in the pres-ence of Ca2+ cannot be similarly explained by ad-sorption of Ca2+, because this cation alone did notgenerate significant positive charge at the surface. Itappears that an alternate mechanism of sorption isnecessary to explain Ca2+ and 804" adsorption at thehigher levels of addition.
0.5 1.0 1.5 2.0 2.5
EQUILIBRIUM CONCENTRflTION CmM33.0
Fig. 6. Comparison of Ca2+ adsorption from CaCl2 and CaSO4 bythe Dark Red A horizon (DRA) material.
o CaS04
A K2S04 Y=5.655X°'35
R=0.988
Y=3.49BX°-25
R=0.979
0.5 1.0 1.5 2.0 2.5EQUILIBRIUM CONCENTRRTION CmM)
3.0
Fig. 7. Comparison of SOJ" adsorption from K2SO4 and CaSO4 bythe Dark Red A horizon (DRA) material.
It is tempting to suggest that the higher adsorptionof both Ca2+ and SO2" at the highest CaSO4 concen-trations is simply gypsum precipitation, but the high-est level of CaSO4 was selected to avoid exceeding thesolubility product of gypsum.
The presence of Ca2+ has been reported previouslyto increase the retention of SO2^ by acid soils (Barrow,1972; Chao et al., 1963; Marsh et al, 1987) and ofPO3
4- by goethite (Barrow et al., 1980), gibbsite (He-lyar et al., 1976), and soils (Smillie et al., 1987; Rydenet al., 1977; Ryden and Syers, 1976). These observa-tions suggest a mechanism involving the formation ofa surface complex between SOI" and Ca2+, or, equiv-alently, adsorption of the solution complex, CaSOSJ. Asurface complex with phosphate and certain divalentcations of particular ionic size was proposed by Helyaret al. (1976) to explain greater phosphate retention bygibbsite in the presence of Ca2+. Cd2+, and Sr2+ andnot with Mg2+, Zn2+, Na+, K+. This proposed com-plex involves the coordination of one Ca2+ atom totwo adsorbed POl~ groups, reducing the repulsive force
68 SOIL SCI. SOC. AM. J., VOL. 53, JANUARY-FEBRUARY 1989
between two adjacent phosphate groups and enhanc-ing further adsorption.
In the present study, a CaSO4 concentration greaterthan 1 mM produced excess adsorption of SO4~ (at-tributable to the Ca-SO4 interaction) that was essen-tially equal to excess adsorption of Ca2+ (Fig. 6 and7). For example, in DRA, SOl~ tended to adsorb inproportionately greater amounts than Ca2+ at lowCaSO4 concentrations, but in equal mole quantities athigh concentrations. These observations suggest thatthe two ions are adsorbing cooperatively only at higherconcentrations, probably as an ion pair. Above 1 mM,the CaSO4 complex represented a significant fractionof the total Ca2+ and SO2," in solution, based uponchemical speciation calculations for the solution phaseusing MINEQL (Westall et al., 1976).
A reasonable reaction, then, would involve a 1:1sulfate-calcium complex, with one O ligand of theSO2," bonded to an oxide surface
+1/2
M— OH 2
M—OH-112
-312M— O — S03
M— OH -""
2+Ca
HoO
This reaction, although similar to the adsorption ofSO4~ from K2SO4, is favored because of the closerapproach of Ca2+ to the SO4~ ion compared to K+.This mechanism of adsorption can reduce the electri-cal charge developed by SO4~ adsorption, thus favor-ing greater adsorption.
Physical adsorption of CaSO° ion pairs on non-reactive mineral surfaces could also account for equi-molar excess adsorption of Ca2+ and SO4~, but sucha process would be difficult to distinguish experimen-tally from the above reaction in which SO4~ is chem-isorbed.
Increased pH which accompanied CaSO4 adsorp-tion was notably smaller than that which accom-panied adsorption of K2SO4 (Fig. 4). This observationprovides further evidence for chemisorption of
2 4 6BOA flDSORBED Cmmol/kgD
Fig. 8. Effect of the adsorption of sulfate from CaSO4 on the cationexchange capacity of DRA and RYA. (Regression line does notinclude the highest CaSO4 treatment levels.)
8
CaSO4 ion pairs on oxides, or physical adsorption ofCaSO4 on nonreactive surfaces of the soil. Neither ofthese reactions should displace OH~ into solution tothe extent that chemisorption of SO4" alone does.
The apparent CEC (exchangeable K+ + Ca2+ +A13+) was increased by the adsorption of CaSO4 (Fig.8). The two highest CaSO4 levels produced an abruptincrease in the CEC because of the higher Ca2+ reten-tion in these treatments. These same two levels gen-erated the highest CaSO4 adsorption (Fig. 6). Appar-ently, excess Ca2+ adsorbed in the presence of SO4~is exchangeable. Most of the increase in CEC was at-tributable to negative charge created by SO4~ adsorp-tion, because the CaSO4 treatment had relatively littleeffect on soil pH.
CONCLUSIONSSurface charge of Oxisols is affected by the adsorp-
tion of SO4~. This ion adsorbs on the surfaces of highlyweathered soil materials by a ligand exchange mech-anism that releases OH~ and increases cation reten-tion by such soils. In contrast, adsorption of Ca2+ fromCaQ2 does not significantly affect surface charge norsolution pH, suggesting that this ion adsorbed electro-statically^ presumably on kaolinite surfaces.
While Ca2+ adsorption is almost linearly dependenton Ca2+ concentration, SO4~ adsorption tends to ap-proach a maximum which is beyond the experimentalrange of SO4~ additions. Retention of both ions con-forms to the Freundlich equation, although SO4~ ad-sorption is also described adequately by a Langmuirfunction.
When Ca2"1" and SO4~ are applied together as a so-lution of CaSO4, adsorption of each ion is enhancedas compared with adsorption from CaCl2 and K2SO4,respectively. A simple precipitation reaction is ruledout by the fact that the soil solutions were undersa-turated with respect to gypsum and other likely solidphases. A possible explanation is based on the coop-erative adsorption of Ca2+ and SO4~ as an ion pair,a process which minimizes the development of neg-ative charge on oxide surfaces resulting from SO4~chemisorption. Thus, further adsorption of SO4~ isfacilitated by Ca2+ adsorption. This excess of ad-sorbed Ca2+ is exchangeable, increasing the CEC ofthese soils.
The similarity in behavior of the Red Yellow andDark Red soil materials with respect to Ca2+ andSO4~ adsorption can be attributed to their similar mi-neralogies and surface areas (Marcano-Martinez andMcBride, in review). The inability of organic matterin the A horizons of these naturally acid soils to retainCa2+ is evidently a result of the effective blockage oforganic functional groups by A13+ and Fe3+; that is,the native organic matter is inactive in cation ex-change reactions because of association with thesemetals in the form of monomeric cations or oxideparticles.
In summary, initial adsorption of SO4" from CaSO4appears to be independent of Ca2+ adsorption, pos-sibly occurring predominantly by bidentate bondingat positively charged sites and minimizing the crea-tion of surface negative charge. Initial SO4" adsorp-

MARCANO-MARTINEZ & MCBRIDE: CALCIUM AND SULFATE RETENTION BY OXISOLS OF THE BRAZILIAN CERRADO 69
tion is accompanied by relatively little Ca2+ adsorp-tion. At higher SO2," adsorption levels, the weakermonodentate bonding mechanism may be operative,creating significant negative charge on oxides. Thepresence of Ca2+ assists this mechanism since co-ad-sorbed Ca2+ can efficiently compensate the surfacenegative charge by forming an ion pair with SO2,".Consequently, adsorption at high CaSO4 concentra-tions is characterised by eguimolar retention of Ca2+
and SO2," in the soil.



![THE ECOLOGICAL FOOTPRINT OF CAMPO GRANDE … · Instituto de Permacultura Cerrado/ Pantanal (IPCP) [Pantanal Cerrado Permaculture Institute] Secretaria de Desenvolvimento Socioeconômico](https://img.pdfslide.us/doc/110x75/5c10475309d3f20c238c4d7a/the-ecological-footprint-of-campo-grande-instituto-de-permacultura-cerrado.jpg)

























