Embed Size (px)
DESCRIPTION
Problems With Clotting
Citation preview

BLEEDING DISORDERS

HemostasisRefers to the stoppage of bloodNormal when it seals a blood vessel to prevent blood loss/hemorrhageAbnormal when it causes inappropriate blood clotting, or is insufficient to stop blood flow from the vasculature Balance of pro-coagulants and anti-coagulants that maintains:
Blood flow Integrity of the vasculature
Hemostasis MechanismVessel spasm
Transient -1minTXA2 -prostaglandin f. Plts-vasoconstriction
Formation of the platelet plugPlts in contact with vessel wallAdhesion(vWF) and aggregation(granules) Plts
Blood coagulation/insoluble fibrin clotActivation of coagulation pathwayFibrinogen to Fibrin mesh

Clot retractionOccurs 20-60min after a clot has formed Squeezes serum from the clotJoining the edges of the broken vessel
Clot dissolutionFibrinolysis - begins shortly after its formationAllows blood flow to be reestablishedBalance between activators and inhibitorsPlasminogen to Plasmin - digest fibrin strandsAllows permanent tissue repair
Hemostasis Mechanism

Coagulation CascadeLiver is the major site of synthesisIn severe liver disease all coagulation factors are diminished except factor VIIIVitamin k dependent factors: II, VII, IX, and XProtein C and S are naturally occurring anticoagulants which synthesis is also vitamin k dependent Warfarin blocks liver uptake of vitamin K, decreasing the synthesis of vitamin k dependent factors

Coagulation cascadeArtificially divided into extrinsic and intrinsic pathways that converge into the common pathway leading to thrombin and fibrin generationExtrinsic pathway assessed in the laboratory by the Prothrombin time (PT) PT- sensitive to deficiencies of factors, II,VII, V, and X, all these associated with bleeding complications PT- to monitor warfarin (coumadin) therapy

Coagulation cascadePT-INR-international normalized ratio, the degree of prolongation of PT by warfarin depends on the strength of the reagents used in the lab ,which could vary among labs. INR created To standardize the variations and allow for global application of anticoagulant recommendationsINR = patient PT/ mean control PT
Coagulation cascadePTT- partial thromboplastin time, assess the intrinsic pathwaySensitive to deficiencies of factor, II, V,VIII,IX, X,XITo monitor Heparin therapyHeparin binds to antithrombin III, and increases its ability to inactivate thrombin, factor Xa and others


THROMBOCYTOPENIA

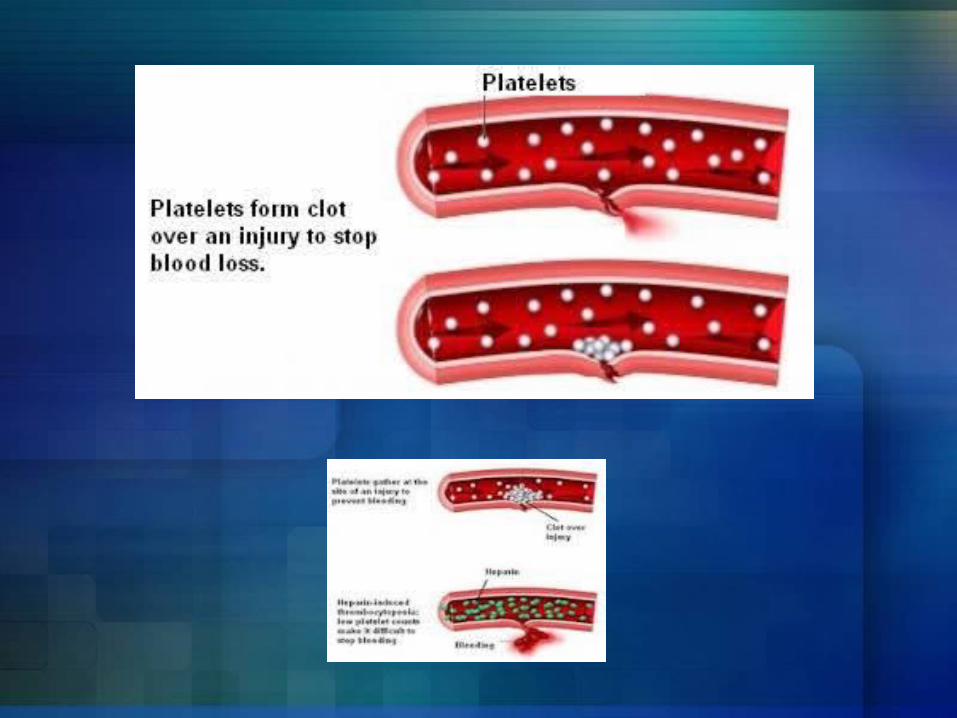
OverviewThrombocytopenia is the medical term for a low blood platelet count. It is the most common bleeding disorder.

Platelets Platelets are produced in bone marrow from MegakaryocytesGeneral platelet count ranges
150,000 – 450,000 microliterEach platelet lives only about 10 days.The risk of bleeding increase as number of platelet decreases.

Classification of thrombocytopeniaMild: 100,000-150,000 microliter
Moderate: 50,000-100,000 microliterSevere: less than 50,000 microliterThe greatest risk is when platelet count below 10,000 microliter

CausesCommon causes
PregnancyGestational thrombocytopenia
Drug-inducedHeparin, Quinidine, Quinine, Sulfonamides
Viral infectionHIV, Infectious mononucleosis, Hepatitis
Hypersplenism due to chronic liver disease
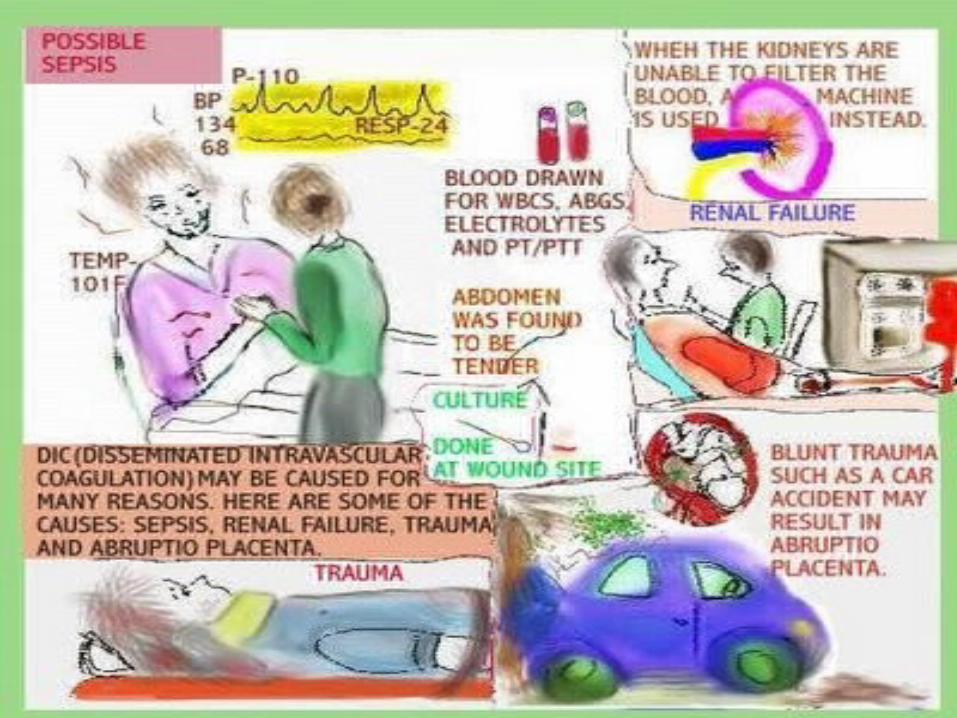
Other causes MyelodysplasiaCongenital thrombocytopeniaThrombotic thrombocytopenic purpura-hemolytic uremic syndromeChronic disseinated intravascular coagulation

Causes of ThrombocytopeniaDecreased production
Bone marrow suppression or damageLeukemia , Aplastic anemiaViral infection [Include HIV infection]Dengue feverChemo or radiation therapyCongenital or acquired bone marrow aplasia or hypoplasia

Causes of ThrombocytopeniaDecreased production
Vitamin B12 or folic deficiency Liver failure
Heavy alcohol consumption
Sepsis, systemic viral or bacterial infection

Causes of Thrombocytopenia
Increased destructionIdiopathic (Immune) Thrombocytopenic Purpura [ITP]Alloimmune destruction—Posttransfusion, Post-transplantationDisseminated Intravascular Coagulation [DIC]Thrombotic Thrombocytopenic Purpura [TTP]Hemolysis, elevated liver enzymes, low platelets [ HELLP]
Increased destructionSystemic lupus erythematosus [SLE]Dengue fever [cause shortened platelet survival and immunological platelet destruction]Pregnancy relate – Gestational thrombocytopenia [Mild]

Medication inducedHeparinValproic acid [Depakin]QuinidineSulfonamide antibioticMMR VaccineClopidogrel [Plavix]MethotrexateCarboplastin
VancomycinFamotidine [Pepcine , Pepcidine ]Mebeverine [Colofac]MetronidazoleInterferon

Clinical manifestationsUsually asymptomaticWhen platelet count is below 20,000m3- Petechiae occur
spontaneously- Ecchymoses occur
at sites of minor trauma
- Bleeding from mucosal surfaces, nose, GIT, genitourinary tract, respiratory system, and within CNS
When platelet count is below 20,000m3- Menorrhagia- Excessive bleeding
after procedures (dental extractions, minor surgery, biopsies
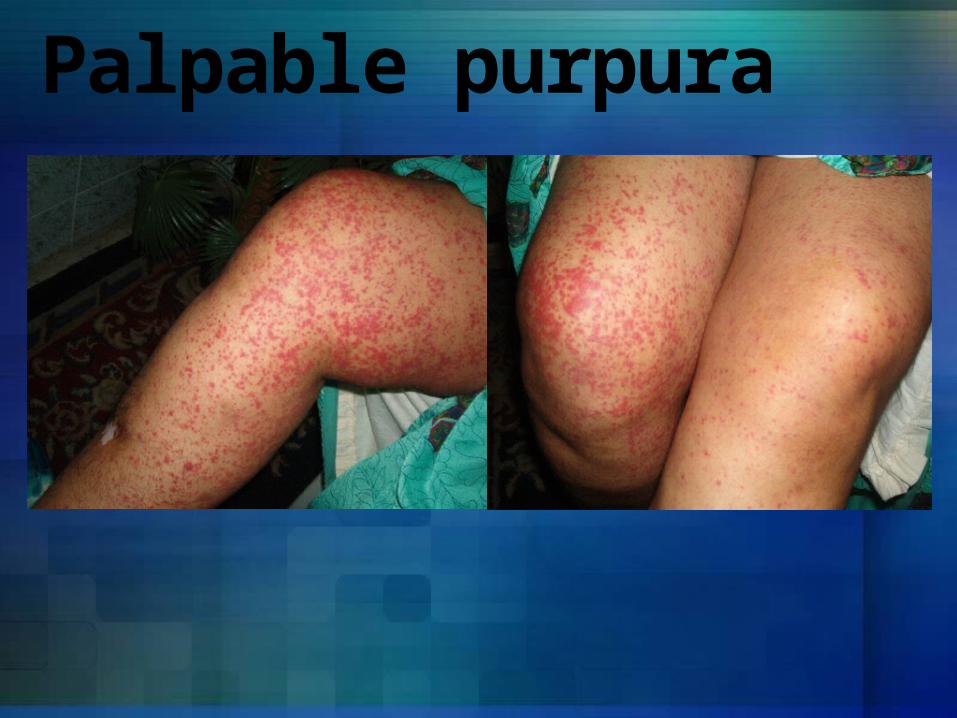
Palpable purpura
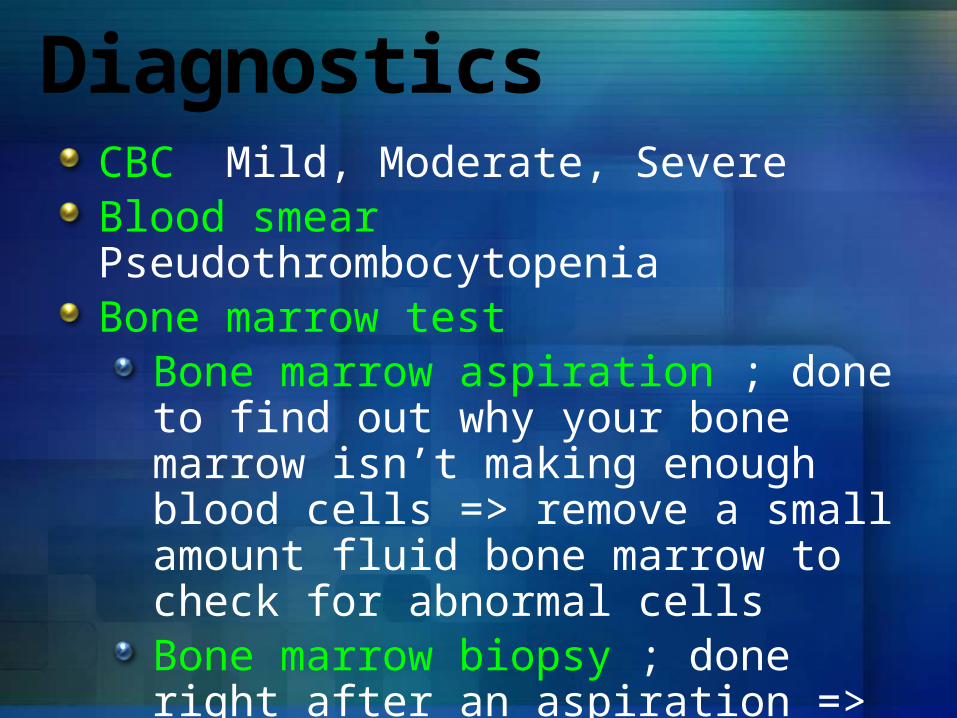
DiagnosticsCBC Mild, Moderate, Severe Blood smear Pseudothrombocytopenia Bone marrow test
Bone marrow aspiration ; done to find out why your bone marrow isn’t making enough blood cells => remove a small amount fluid bone marrow to check for abnormal cellsBone marrow biopsy ; done right after an aspiration => remove bone marrow tissue to check the number and types of cells in the bone marrow
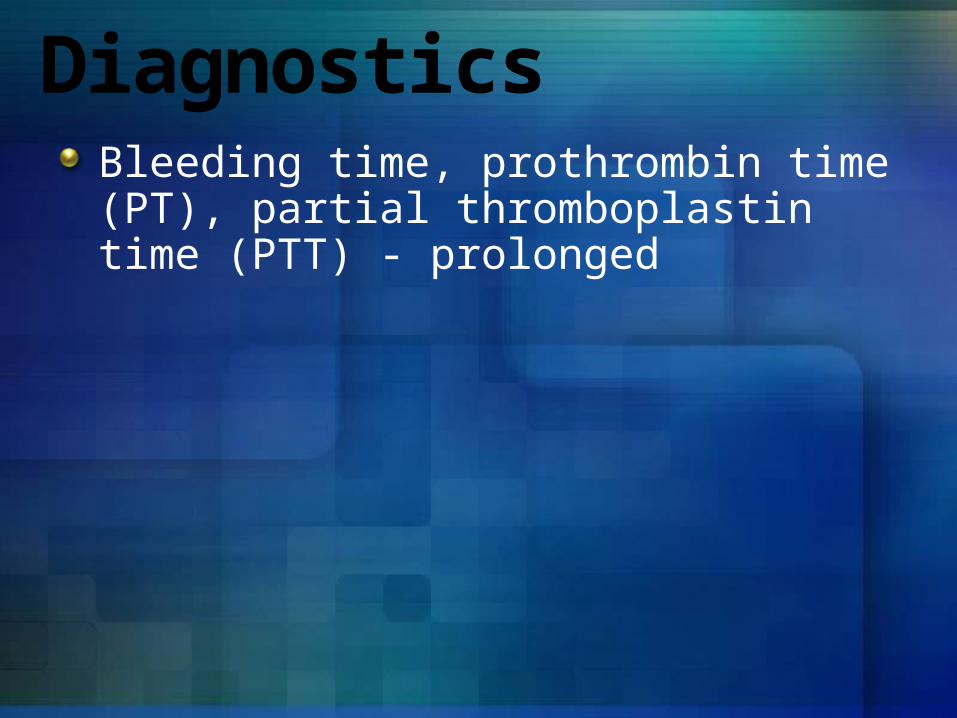
DiagnosticsBleeding time, prothrombin time (PT), partial thromboplastin time (PTT) - prolonged

Management: MedicalTreat underlying causePlatelet transfusionSteroids or IV immunoglobulins

Management: NursingInstitute bleeding precautions- Avoid use of plain razor, hard toothbrush or floss,
intramuscular injections, tourniquets, rectal procedures or suppositories
- Administer stool softeners- Restrict activity and exercise when platelet
count is below 20,000m3

Management: NursingMonitor pad count/amount of saturation during mensesAdminister blood products as orderedEvaluate urine, stool and emesis for gross and occult bloodHealth teachings on:- Avoid blowing nose- Avoid use of aspirin and NSAIDs- Demonstrate use of direct, steady pressure at
bleeding site- Encourage routine follow-up for platelet counts
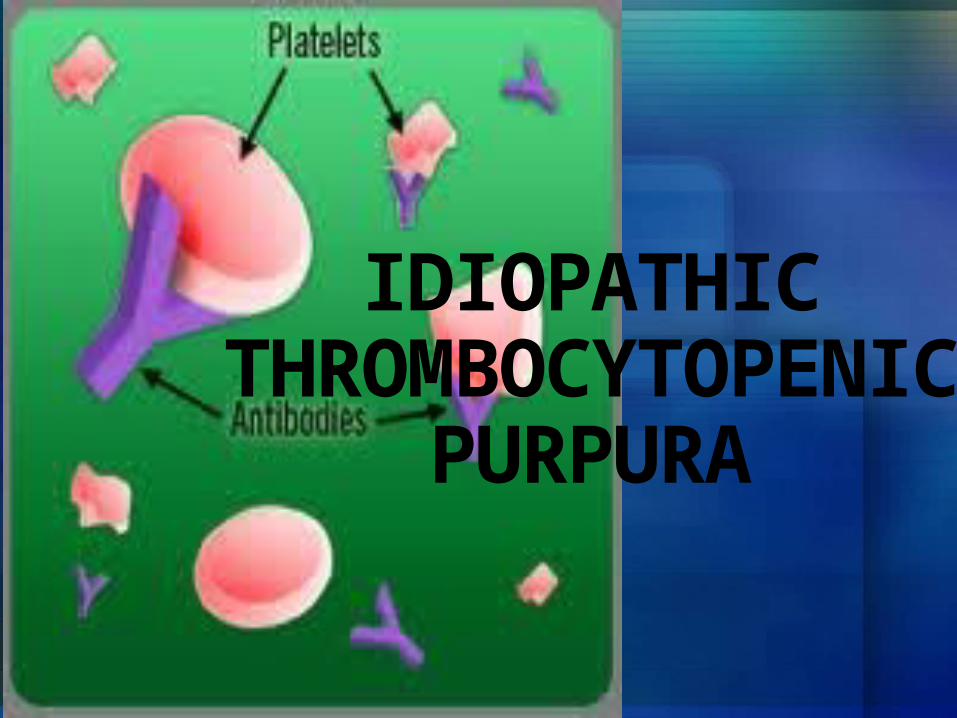
IDIOPATHIC THROMBOCYTOPENIC
PURPURA

OverviewIdiopathic thrombocytopenic purpura is a bleeding disorder in which the immune system destroys platelets, which are necessary for normal blood clotting. Persons with the disease have too few platelets in the blood.ITP is sometimes called immune thrombocytopenic purpura.

OverviewITP occurs when certain immune system cells produce antibodies against platelets. Platelets help your blood clot by clumping together to plug small holes in damaged blood vessels.The antibodies attach to the platelets. The spleen destroys the platelets that carry the antibodies.
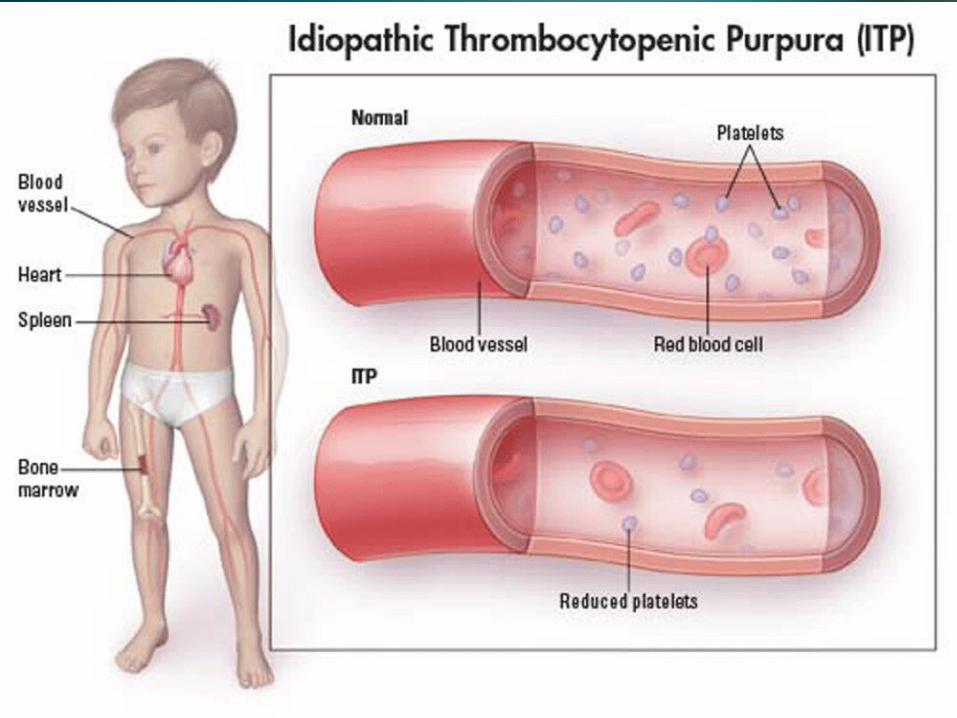

OverviewIn children, the disease sometimes follows a viral infection. In adults, it is more often a chronic (long-term) disease and can occur after a viral infection, with use of certain drugs, during pregnancy, or as part of an immune disorder.ITP affects women more frequently than men, and is more common in children than adults. The disease affects boys and girls equally.

Clinical manifestationsMenorrhagiaBleeding into the skin causes a characteristic skin rash that looks like pinpoint red spots (petechial rash)Easy bruisingNosebleed or bleeding in the mouth
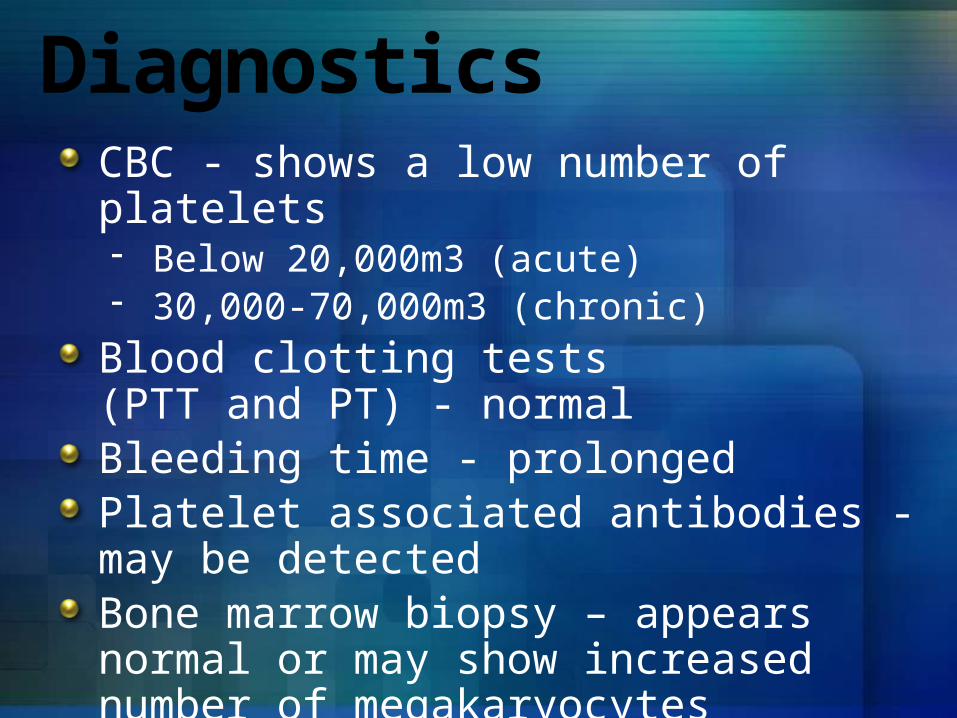
DiagnosticsCBC - shows a low number of platelets- Below 20,000m3 (acute)- 30,000-70,000m3 (chronic)
Blood clotting tests (PTT and PT) - normalBleeding time - prolongedPlatelet associated antibodies - may be detectedBone marrow biopsy – appears normal or may show increased number of megakaryocytes

Management: MedicalAnti-inflammatory steroid medicine – prednisoneSplenectomy - will increase the platelet count in about half of all patientsDanazol (Danocrine) - taken orallyHigh-dose gamma globulin Drugs that suppress the immune systemFiltering antibodies out of the blood streamAnti-RhD therapy for people with certain blood types

Management: NursingInstitute bleeding precautions- Avoid use of plain razor, hard toothbrush or floss,
intramuscular injections, tourniquets, rectal procedures or suppositories
- Administer stool softeners- Restrict activity and exercise when platelet
count is below 20,000m3

Management: NursingMonitor pad count/amount of saturation during mensesAdminister blood products as orderedEvaluate urine, stool and emesis for gross and occult bloodHealth teachings on:- Avoid blowing nose- Avoid use of aspirin and NSAIDs- Demonstrate use of direct, steady pressure at
bleeding site- Encourage routine follow-up for platelet counts

DISSEMINATED INTRAVASCULAR COAGULATION

OverviewDisseminated intravascular coagulation (DIC) is a serious disorder in which the proteins that control blood clotting become abnormally active. Normally when you are injured, certain proteins in the blood become activated and travel to the injury site to help stop bleeding. However, in persons with DIC, these proteins become abnormally active. This often occurs due to inflammation, infection, or cancer.
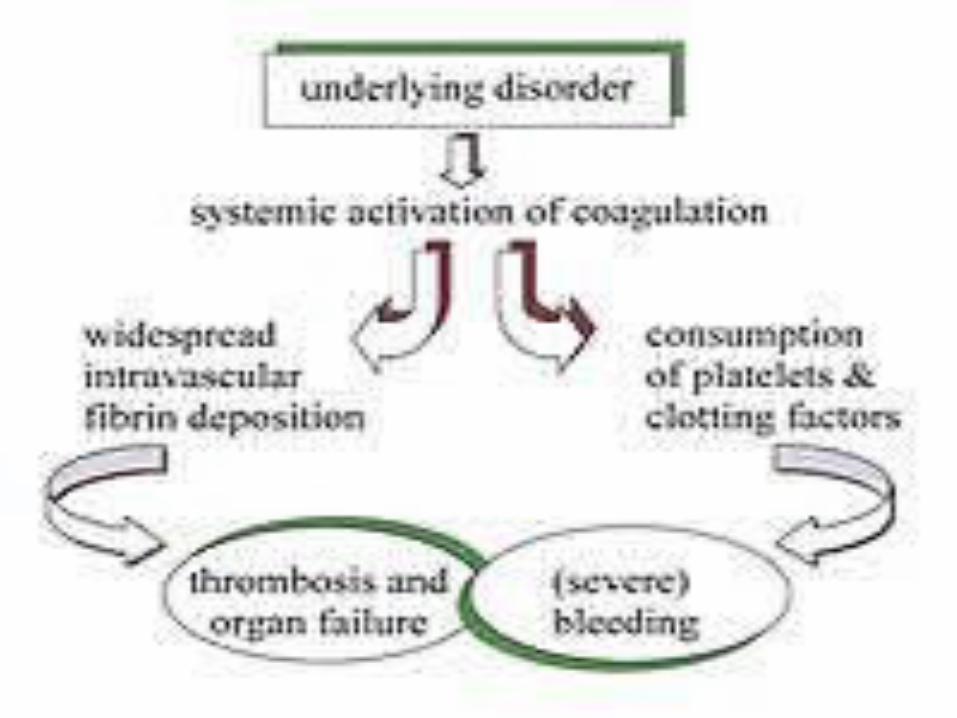

OverviewSmall blood clots form in the blood vessels. Some of these clots can clog up the vessels and cut off blood supply to various organs such as the liver, brain, or kidney. These organs will then be damaged and may stop functioning.Over time, the clotting proteins are consumed or "used up“. When this happens, the person is then at risk for serious bleeding, even from a minor injury or without injury. This process may also break up healthy red blood cells.
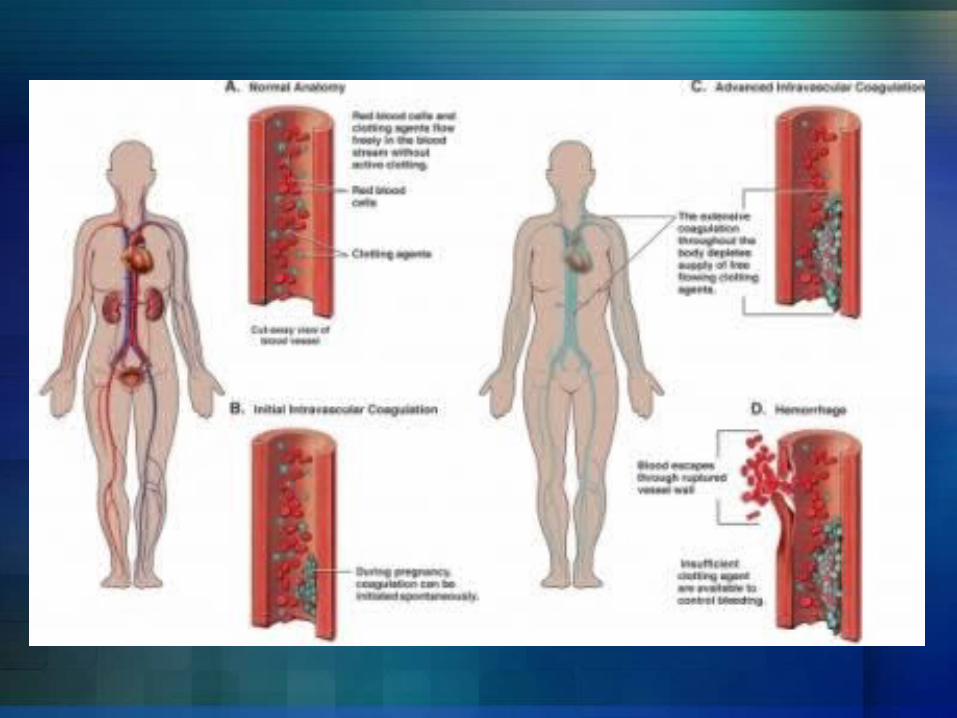

Risk factorsBlood transfusion reactionCancer - especially certain types of leukemiaInfection in the blood by bacteria or fungusLiver diseasePregnancy complications – abruptio placenta and retained placentaRecent surgery or anesthesiaSepsis Severe tissue injury – burns and head injury
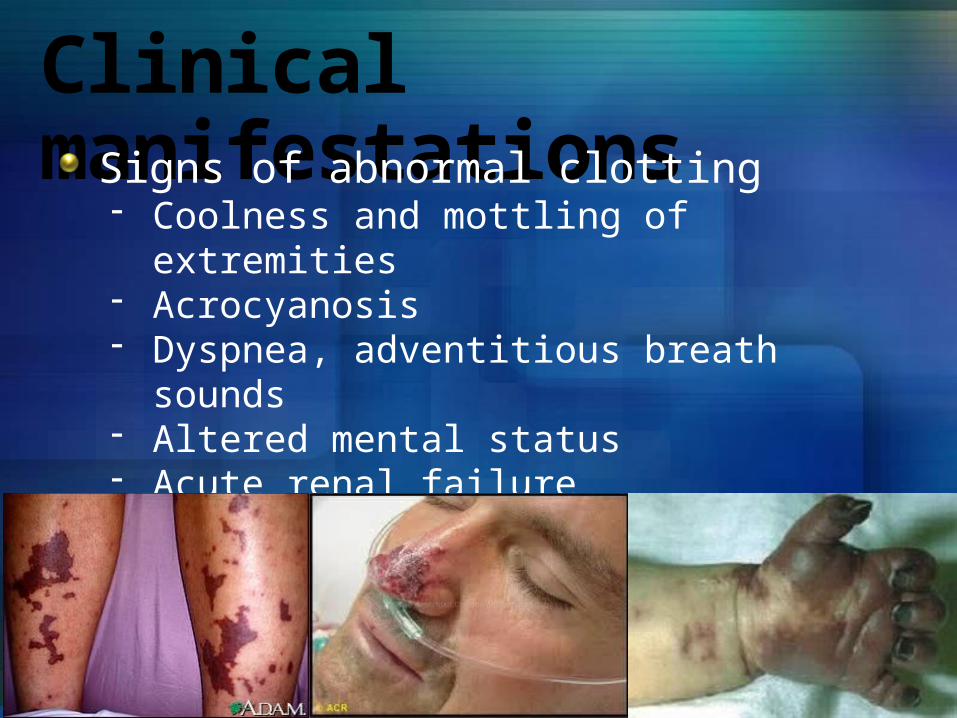
Clinical manifestationsSigns of abnormal clotting- Coolness and mottling of extremities- Acrocyanosis- Dyspnea, adventitious breath sounds- Altered mental status- Acute renal failure- Pain
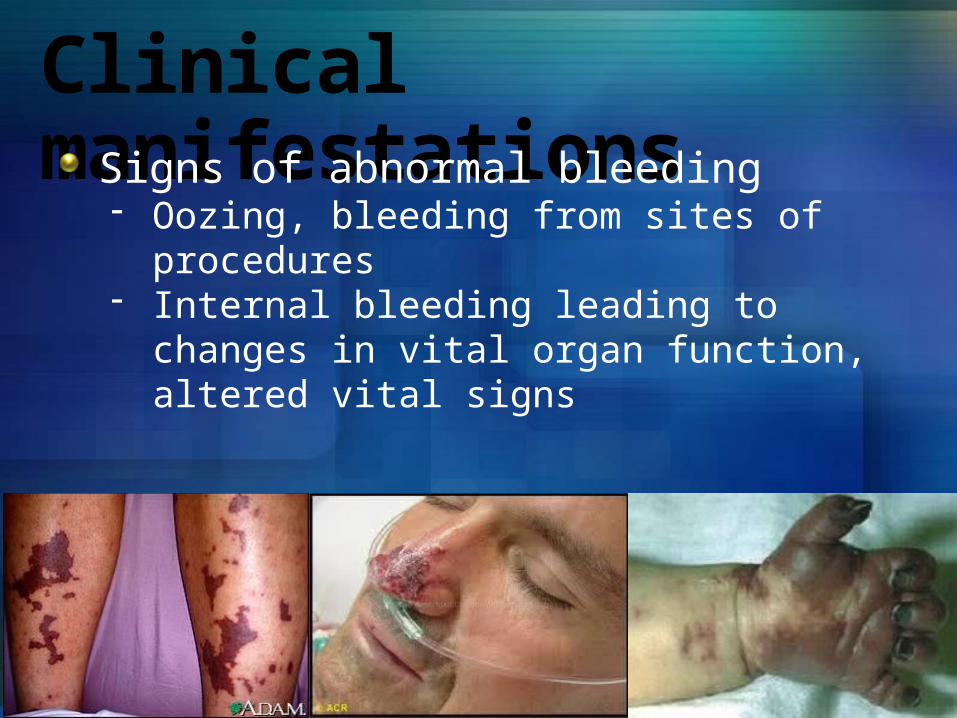
Clinical manifestationsSigns of abnormal bleeding- Oozing, bleeding from sites of procedures- Internal bleeding leading to changes in vital organ
function, altered vital signs

DiagnosticsComplete blood count with blood smear examinationFibrin degradation products – increased Partial thromboplastin time (PTT) – prolonged Platelet count – diminshed Prothrombin time (PT) – prolonged Serum fibrinogen – decreasedAntithrombin III – decreased

Management: MedicalThe goal is to determine and treat the cause of DIC.Replacement therapy- Fresh frozen plasma- Platelet transfusion- Cryoprecipitate
Supportive measures- Fluid replacement- Oxygenation replacement and monitoring- Maintenance of blood pressure and renal perfusion
Heparin therapy

Management: NursingInstitute bleeding precautions- Avoid use of plain razor, hard toothbrush or floss,
intramuscular injections, tourniquets, rectal procedures or suppositories
- Administer stool softeners- Restrict activity and exercise when platelet
count is below 20,000m3

Management: NursingMonitor pad count/amount of saturation during mensesAdminister blood products as orderedAvoid dislodging clots. Apply pressure to sites of bleeding for at least 20 minutesMaintain bed rest during bleeding episodesIf internal bleeding is suspected, assess bowel sounds and abdominal girthEvaluate fluid status and bleeding by frequent measurement of vital signs, CVP, intake and output

Management: NursingPromoting tissue perfusion- Keep warm- Avoid vasoconstrictive agents- Change position frequently and perform ROM- Monitor ECG and laboratory tests for dysfunction
of vital organs caused by ischemia- Monitor for signs of vascular occlusion
Eyes – visual deficits Bone – bone pain Extremities – cold, mottling, numbness

ComplicationsBleedingLack of blood flow to extrimeties and vital organsStroke

HEMOPHILIA

OverviewHemophilia is a bleeding disorder that slows down the blood clotting process. People who have Hemophilia often have longer bleeding after some sort of contact to injury. People who have severe Hemophilia start to have spontaneous bleeding in the joints and muscles all around your body. Hemophilia is more common in males than females.
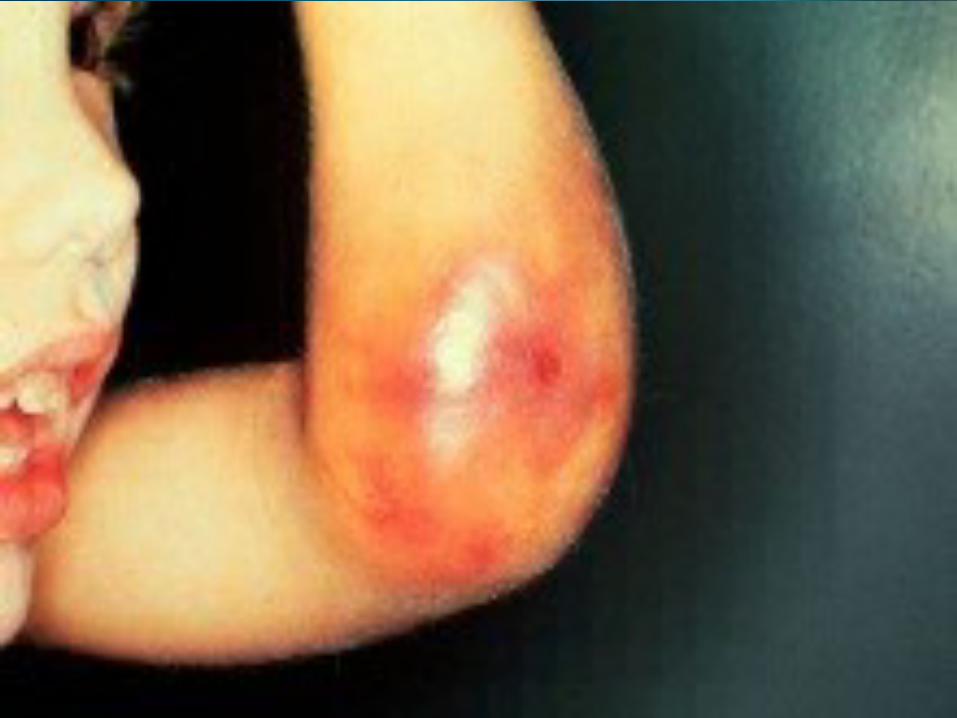
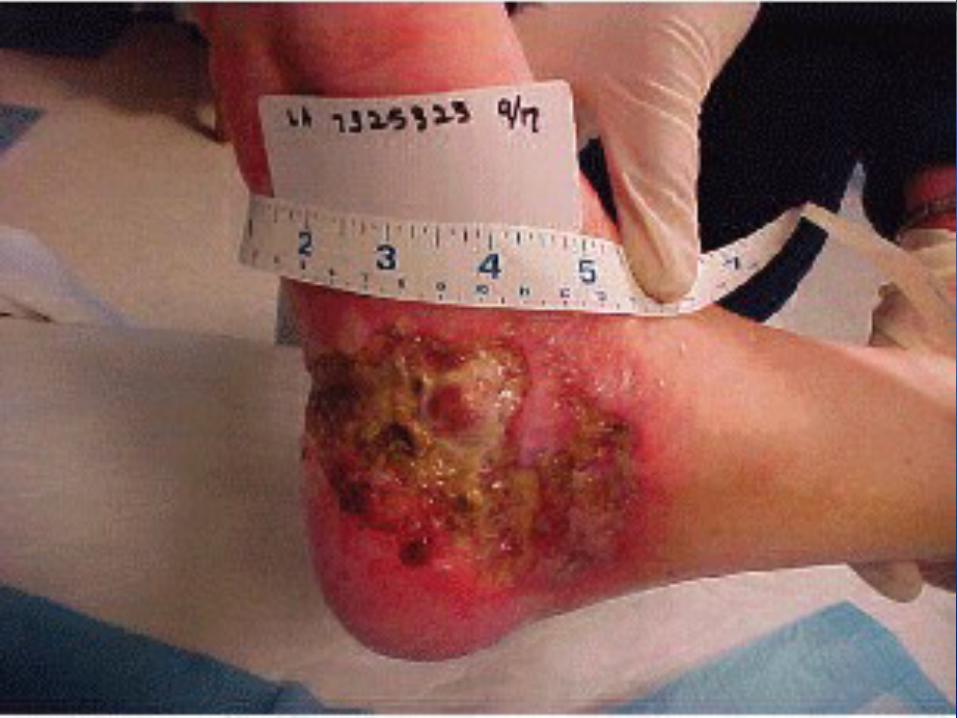
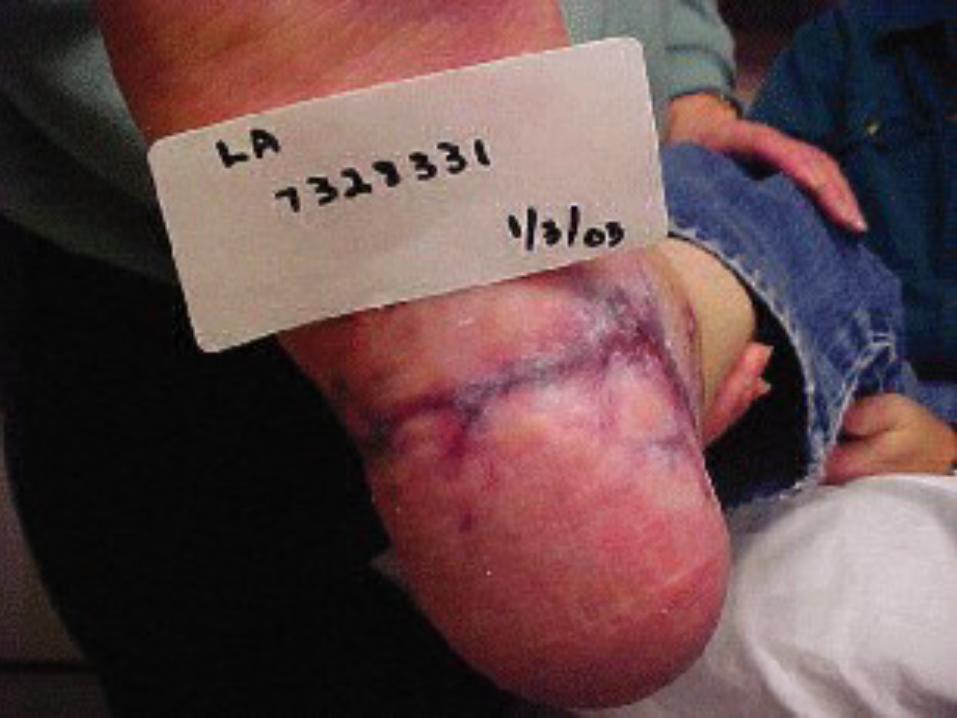
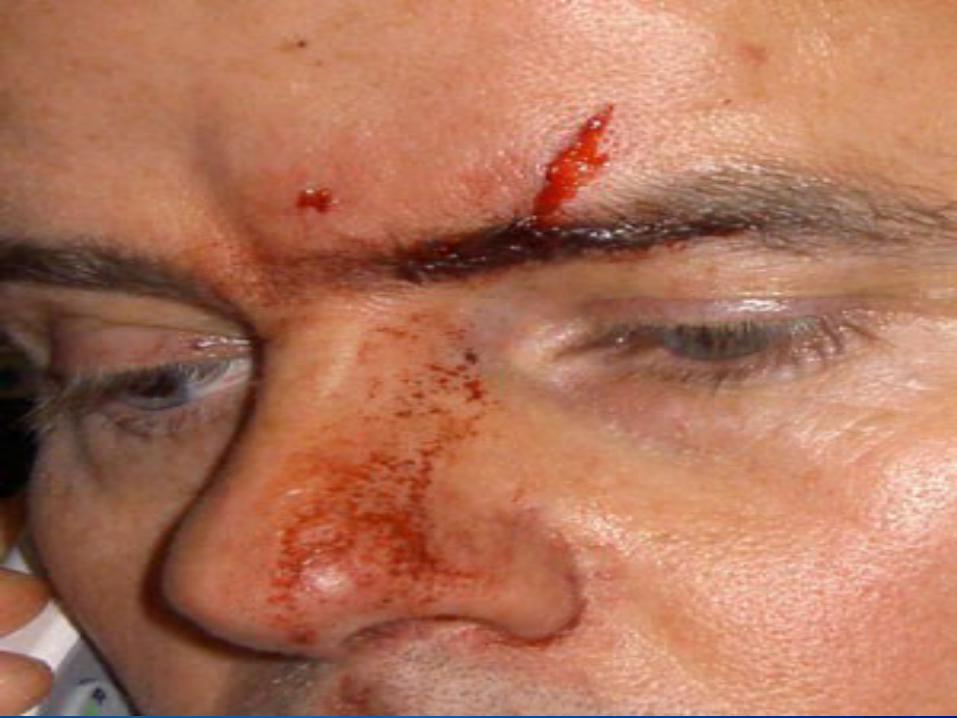
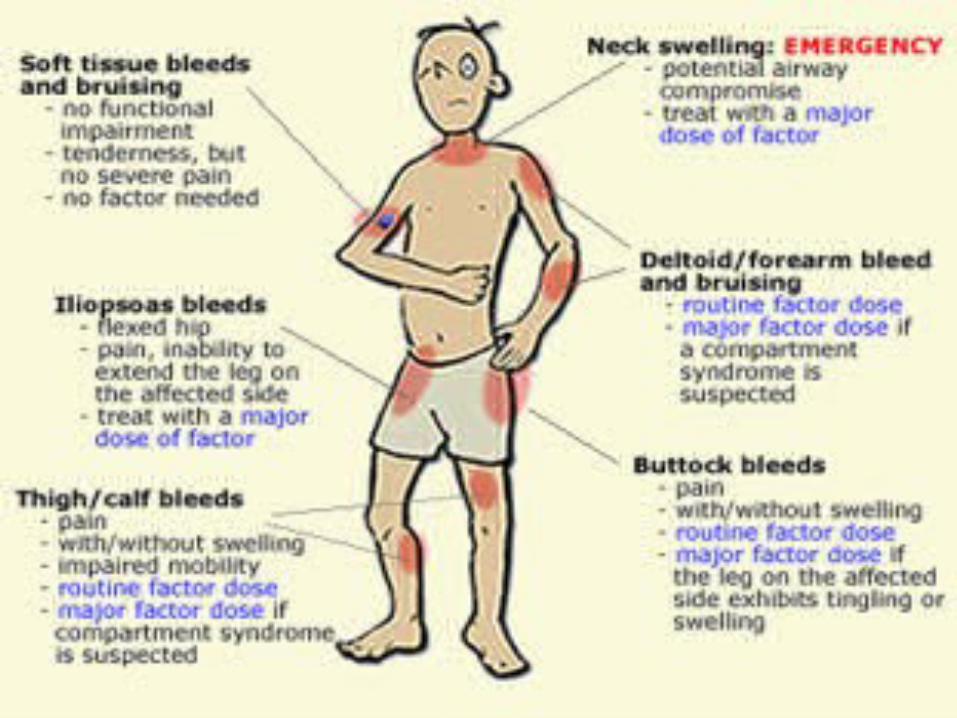

Types of HemophiliaHemophilia A:
Also known as classic hemophilia or Factor VIII DeficiencyPeople with this type of hemophilia have low levels of a blood clotting factor called figure 8 (FVIII)
Hemophilia B:Also known as Christmas disease or Factor IX DeficiencyPeople with this type of hemophilia have low levels of a blood clotting factor called figure 9 (FIX)
-The two different types of hemophilia are caused by permanent gene changes (mutations). Mutations in the FVIII gene cause Hemophilia A. Mutations in the FIX gene cause Hemophilia B.

Hemophilia AAlso known as classic hemophiliaPeople with this type of hemophilia have low levels of a blood clotting factor called figure 8 (FVIII)
-Severe Hemophilia A: Spontaneous joint or deep muscle bleeding. Usually diagnosed within first two years of life.-Moderate Hemophilia A: spontaneous bleeding, delayed oozing after minor injury, and usually diagnosed before they are 5 to 6 years old. - Mild Hemophilia A: Do NOT have spontaneous bleeding but unusual bleeding occurs with surgery and tooth extractions. People are usually diagnosed with this in later life.
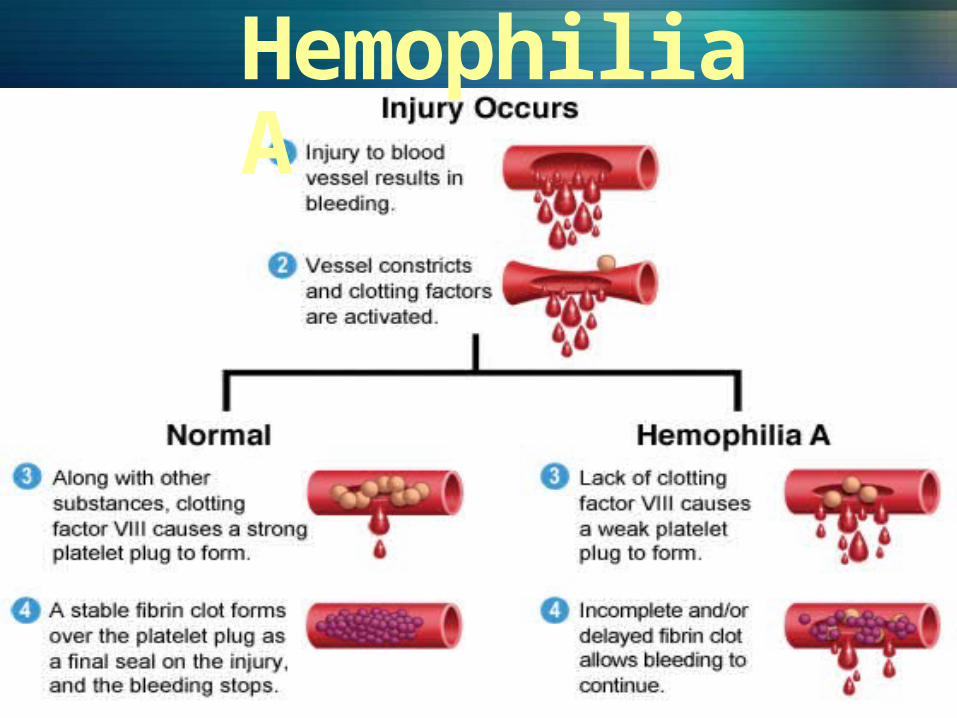
Hemophilia A

Hemophilia BAlso known as Christmas diseasePeople with this type of hemophilia have low levels of a blood clotting factor called figure 9 (FIX)
-Severe Hemophilia B: Spontaneous joint or deep muscle bleeding is the most frequent symptom. People are usually diagnosed in the first 2 years of life.-Moderate Hemophilia B: spontaneous bleeding, delayed oozing after minor injury, and usually diagnosed before they are 5 to 6 years old-Mild Hemophilia B: Do NOT have spontaneous bleeding but unusual bleeding occurs with surgery and tooth extractions. People are usually diagnosed with this in later life.

Which type is more common?Hemophilia A is more common than Hemophilia B. One in 5000-10000 males around the world have Hemophilia A. One in 20,000- 34,500 males around the world have Hemophilia B.
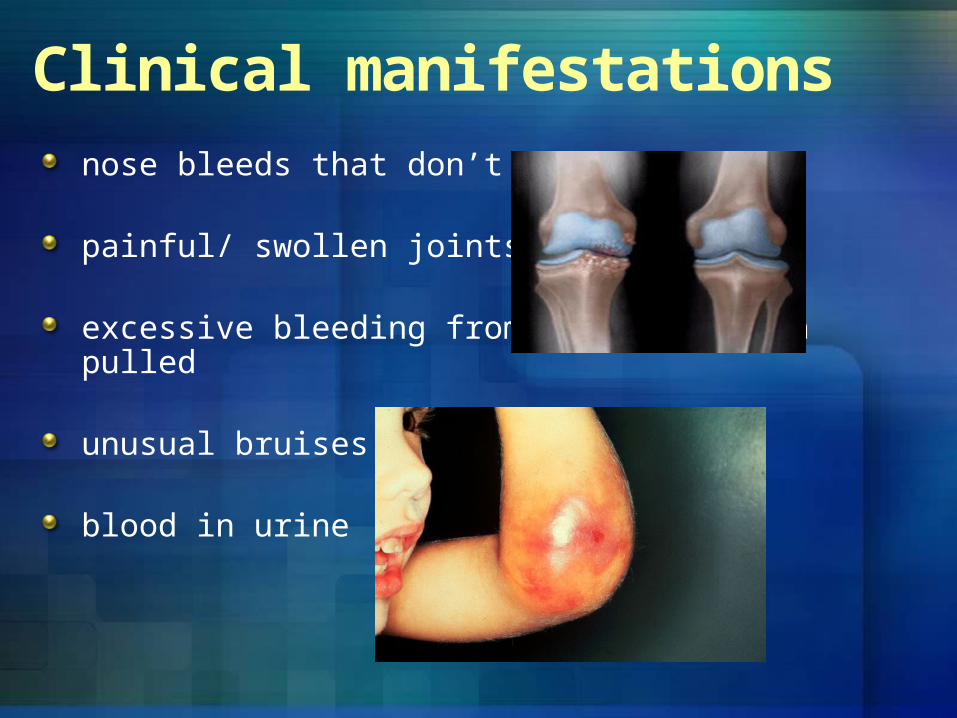
nose bleeds that don’t stop
painful/ swollen joints
excessive bleeding from having a tooth pulled
unusual bruises
blood in urine
Clinical manifestations

DiagnosticsHemophilia A&B are diagnosed by measuring factor clotting activity. Individuals who have Hemophilia A have low factor VIII clotting activity. Individuals who have hemophilia B have low factor IX clotting activity. Genetic testing is also available for the factor VIII gene and the factor IX gene. • Genetic testing of the FVIII gene finds a disease-causing
mutation in up to 98 percent of individuals who have hemophilia A.
• Genetic testing of the FIX gene finds disease-causing mutations in more than 99 percent of individuals who have hemophilia B.
• Used to identify women who are carriers of a type FVIII or FIX gene mutation, diagnose hemophilia in a fetus during a pregnancy, and sometimes used to diagnose individuals who have mild symptoms of hemophilia A or B.

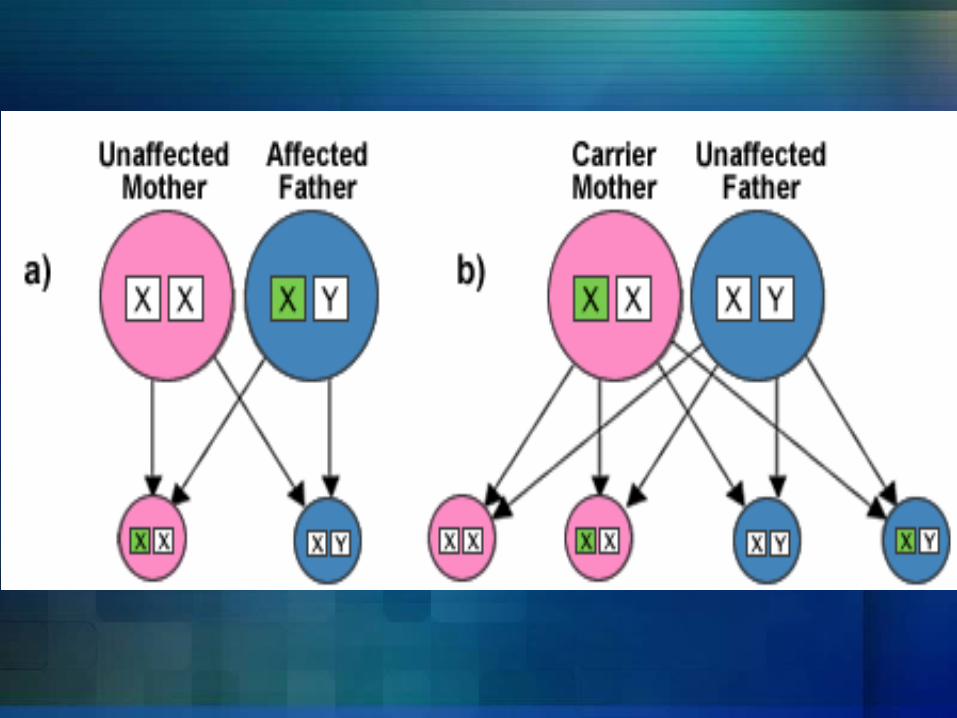
Hemophilia A and hemophilia B are inherited in an X-linked recessive pattern. The genes associated with these conditions are located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one changed copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two changed copies of this gene, it is very rare for females to have hemophilia. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons. In X-linked recessive inheritance, a female with one changed copy of the gene in each cell is called a carrier. Carrier females have about half the usual amount of clotting factor VIII or clotting factor IX, which is generally enough for normal blood clotting.
Is Hemophilia inherited?
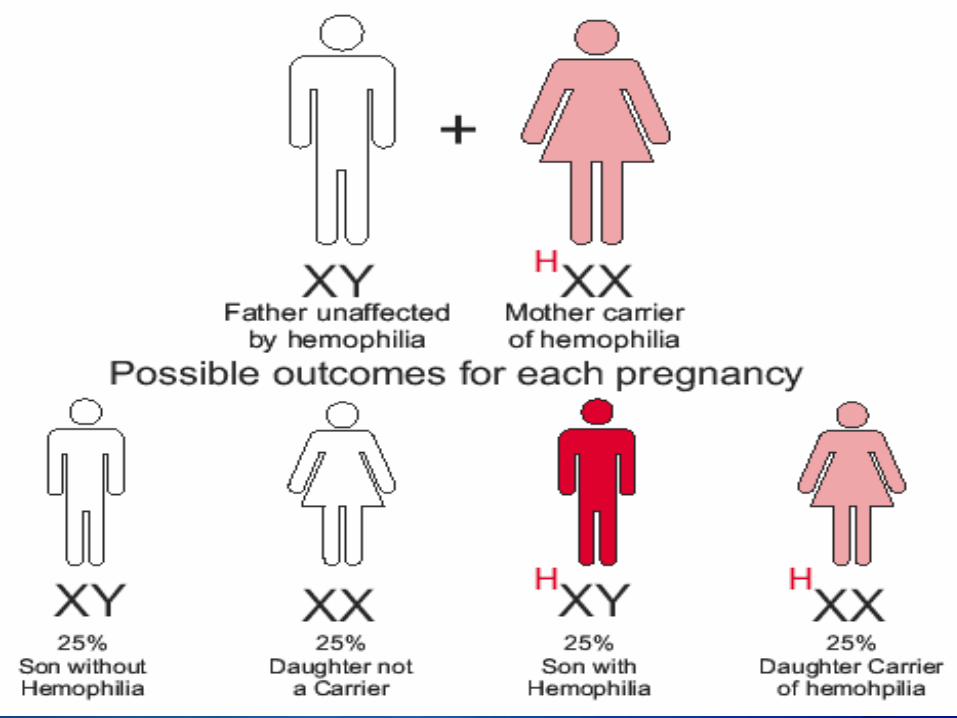
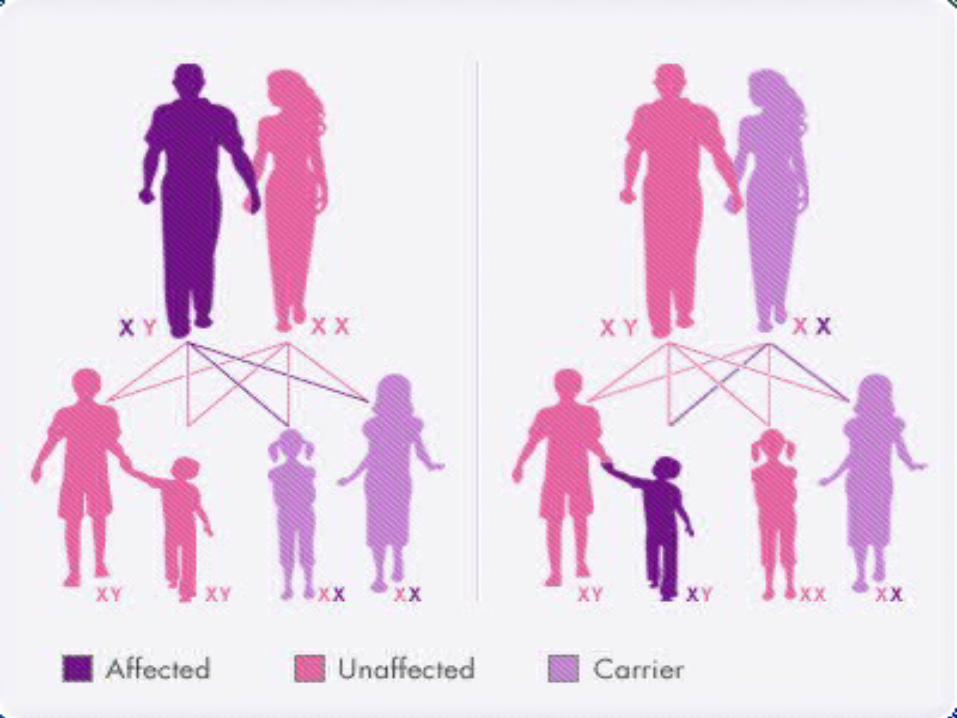
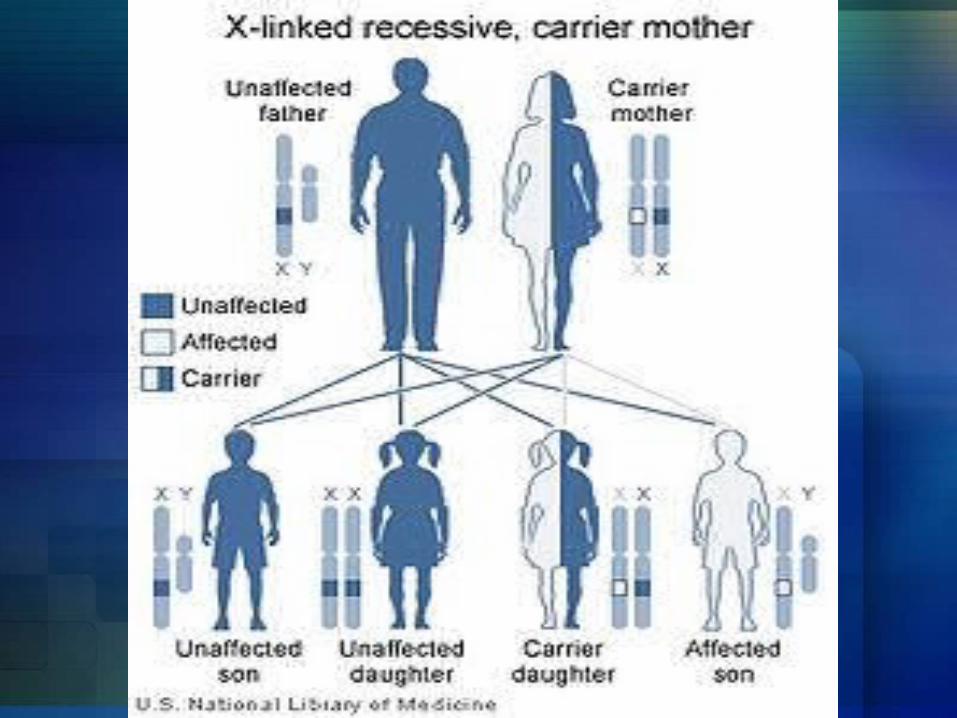
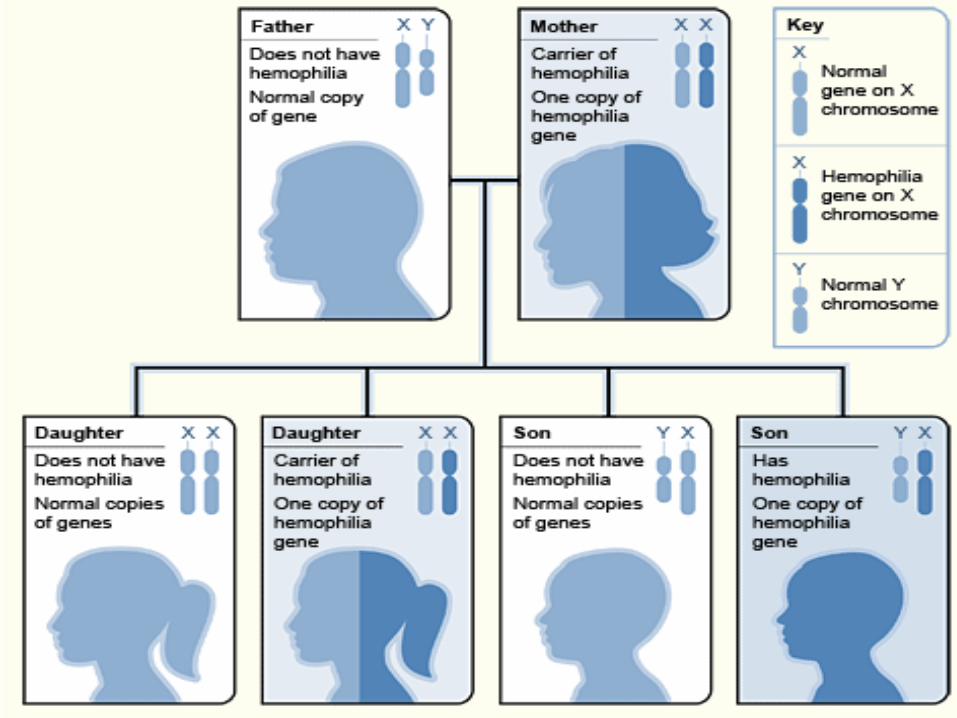
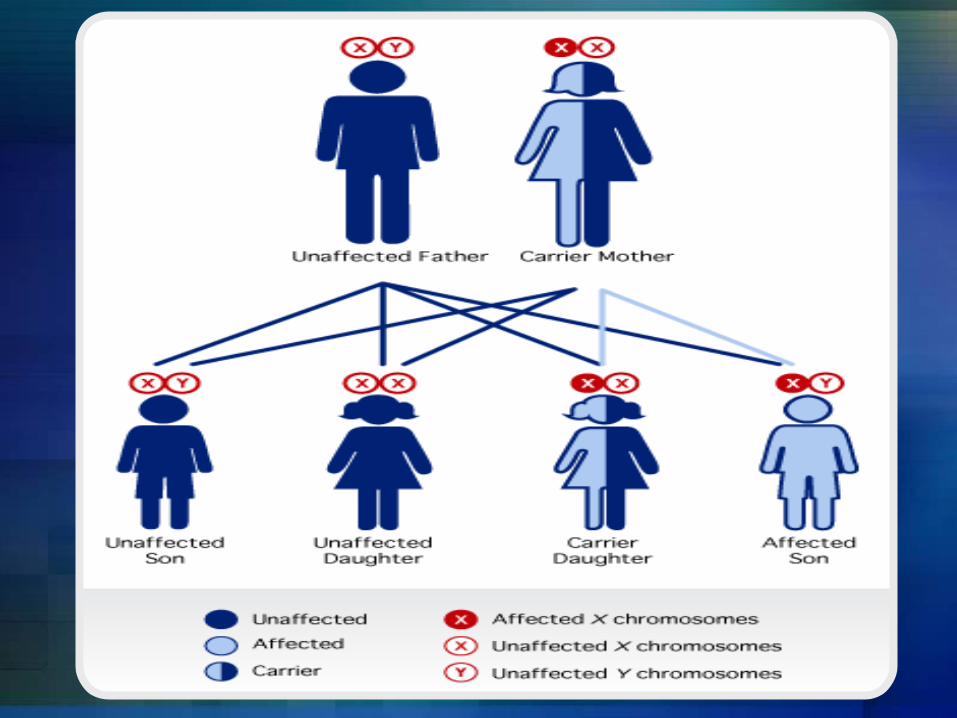
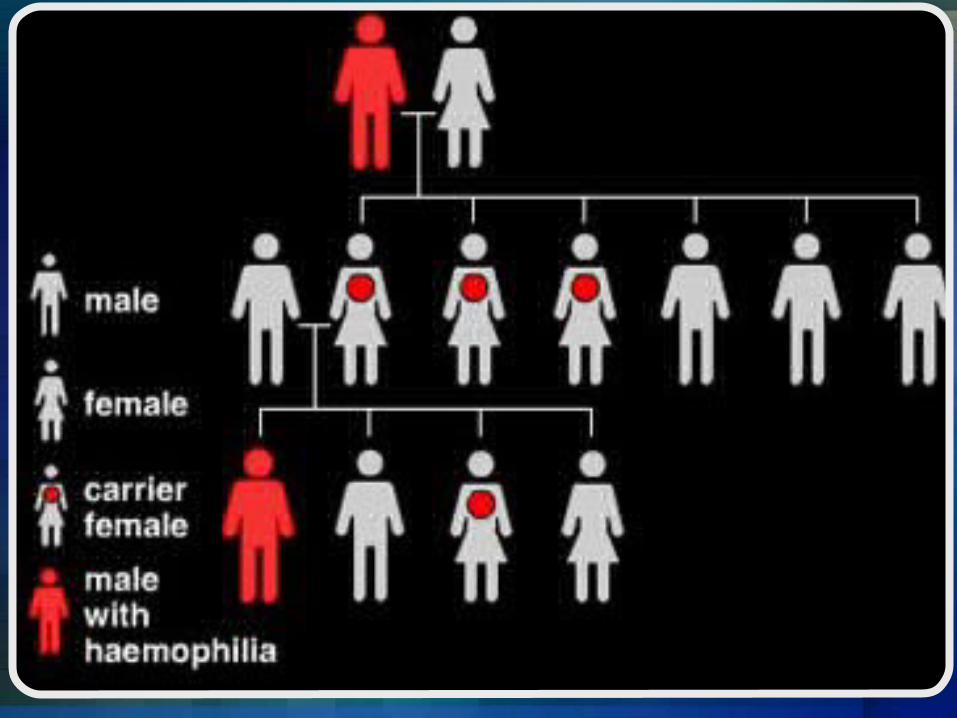
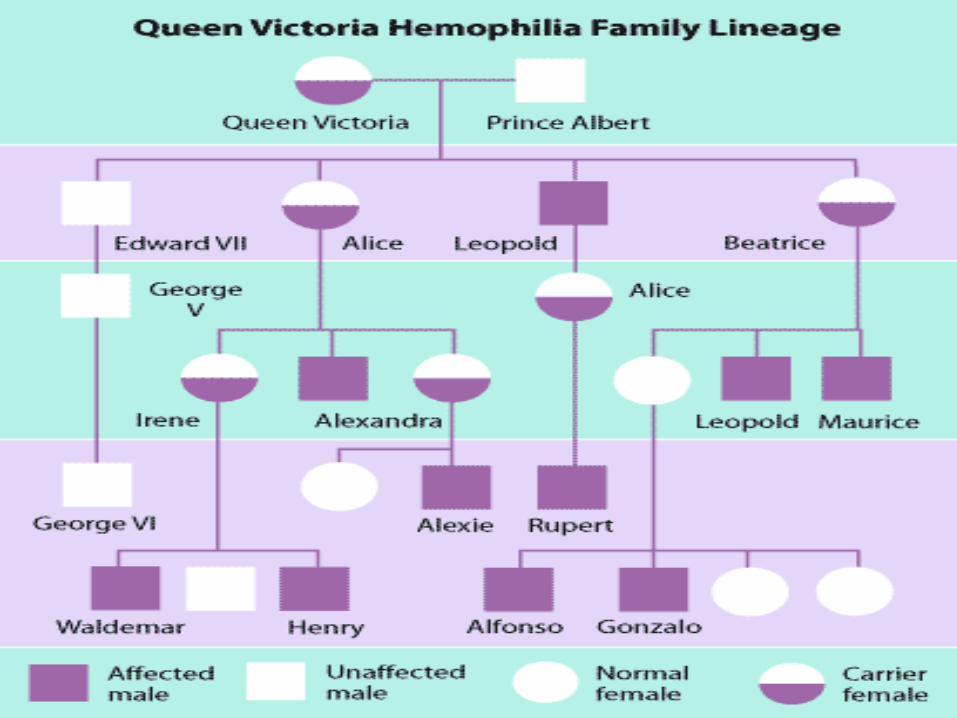
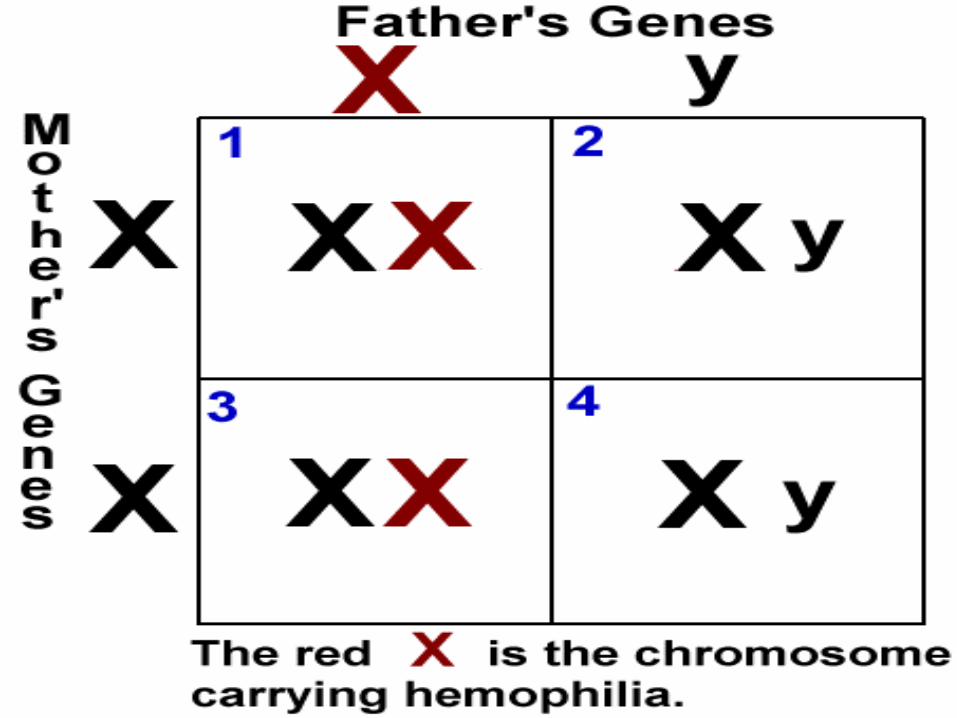

Right now, there is no cure for Hemophilia but, there are some treatments being used depending on the` severity of hemophilia

Management: NursingInstitute bleeding precautions- Avoid use of plain razor, hard toothbrush or floss,
intramuscular injections, tourniquets, rectal procedures or suppositories
- Administer stool softeners- Restrict activity and exercise when platelet
count is below 20,000m3

Management: NursingMonitor pad count/amount of saturation during mensesAdminister blood products as orderedEvaluate urine, stool and emesis for gross and occult bloodHealth teachings on:- Avoid blowing nose- Avoid use of aspirin and NSAIDs- Demonstrate use of direct, steady pressure at
bleeding site- Encourage routine follow-up for platelet counts

When people that have a serious case of hemophilia they have the option of getting a series of shots. They are known as clotting factor replacement therapy.
Preventions

END





























