Embed Size (px)
Citation preview

Bioinformatics Research OverviewLi Liao
Develop new algorithms and (statistical) learning methods that help solve biological problems
> Capable of incorporating domain knowledge
> Effective, Expressive, Interpretable
1Li Liao, SIG NewGrad, 09/29/2008

Motivations• Understanding correlations between genotype
and phenotype• Predicting genotype <=> phenotype• Some Phenotype examples:
– Protein function– Drug/therapy response– Drug-drug interactions for expression– Drug mechanism– Interacting pathways of metabolism
2Li Liao, SIG NewGrad, 09/29/2008

Bioinformatics in a … cell
3Li Liao, SIG NewGrad, 09/29/2008

Credit:Kellis & Indyk4Li Liao, SIG NewGrad, 09/29/2008

Projects– Genome sequencing and assembly (funded by NSF)– Homology detection, protein family classification (funded by a DuPont S&E award)
Support Vector Machines Hidden Markov models Graph theoretic methods
– Probabilistic modeling for BioSequence (funded by NIH) HMMs, and beyond Motifs finding Secondary structure
– Systems BioinformaticsPrediction of Protein-Protein InteractionsInference of Gene Regulatory NetworksPrediction of other regulatory elements
Pattern analysis for RNAi (funded by UDRF)
– Comparative Genomics Identify genome features for diagnostic and therapeutic purposes (funded by an Army grant)
5Li Liao, SIG NewGrad, 09/29/2008

PeopleCurrent members:
- Dr. Wen-Zhong Wang (Postdoc Fellow)
- Roger Craig (PhD student)
- Alvaro Gonzalez (PhD student)
- Kevin McCormick (PhD student)
- Colin Kern (PhD student)
Past members:
- Robel Kahsay (Ph.D. currently at DuPont Central Research & Development)
- Kishore Narra (M.S. currently at VistaPrint, Inc.)
- Arpita Gandhi (M.S. currently at Colgate-Palmolive Company)
- Gaurav Jain (M.S. currently at Institute of Genomics, Univ. of Maryland)
- Shivakundan Singh Tej (M.S.)
- Tapan Patel (B.S. currently in MD/PhD program at U Penn)
- Laura Shankman (B.S., currently in PhD program at U Virginia)
6Li Liao, SIG NewGrad, 09/29/2008

7Li Liao, SIG NewGrad, 09/29/2008

8Li Liao, SIG NewGrad, 09/29/2008

Hybrid Hierarchical Assembly
• Three types of reads: Sanger (~1000bp), 454 (~100bp), and SBS (~30bp).
• Assembly of individual types using the best suited assemblers.– Phrap, TIGR, etc. for Sanger reads– Euler assembler and Newbler for 454 reads– Euler short, Shorty for SBS reads
• Hybrid and hierarchical – Use longer reads as scaffolding to resolve repeat regions
that are difficult for shorter reads– Use contigs from shorter reads (pyrosequencing) as
pseudoreads to bridge gaps (nonclonable and hard stops) with Sanger reads.
9Li Liao, SIG NewGrad, 09/29/2008

Major Findings
• Hybrid hierarchical assembly is proved to be an effective way for assembling short reads
• Incremental approach to selecting ABI reads is more effective than random approach in generating high coverage contigs
• Staged assembly using Phrap is an effective alternative to the proprietary Newbler assembler.
Publications:Gonzalez & Liao, BMC Bioinformatics 2008, 9:102.
10Li Liao, SIG NewGrad, 09/29/2008

Blue lines are contigs generated from hybrid assembly11Li Liao, SIG NewGrad, 09/29/2008

Detect remote homologues
Attributes:- Sequence similarity, Aggregate statistics (e.g., protein families),
Pattern/motif, and more attributes (presence at phylogenetic tree).
How to incorporate domain specific knowledge into the model so a classifier can be more accurate?
Results:
- Quasi-consensus based comparison of profile HMM for protein sequences (Kahsay et al, Bioinformatics 2005)
- Using extended phylogenetic profiles and support vector machines for protein family classification (Narra & Liao, SNPD04, Craig & Liao, ICMLA’05, Craig & Liao SAC’06, Craig & Liao, Int’l J. Bioinfo & DM 2007)
- Combining Pairwise Sequence Similarity and Support Vector Machines for Detecting Remote Protein Evolutionary and Structural Relationships (JCB 2003)
12Li Liao, SIG NewGrad, 09/29/2008

Non-linear mapping to a feature spaceΦ( )
xi Φ(xi)Φ(xj)xj
L() = i ½ i j yi yj Φ (xi )·Φ (xj )
13Li Liao, SIG NewGrad, 09/29/2008

1 1 1 1 0
1 1 1 1 1= 3 0.5
0 1 1 1 1 = 3 0.1x =
y =
z =
Ham
min
g di
stan
ce
Tre
e-ba
sed
dist
ance
Data: phylogenetic profiles - How to account for correlations among profile components?
profile extension (Narra & Liao, SNPD 04)Transductive learning (Craig & Liao, ICMLA’05, SAC’06, IJBDM, 2007)
14Li Liao, SIG NewGrad, 09/29/2008

1 1 0 1 0 0 0 1 1
10.33
0.67
0.34
0.5
0.75
0.55
1 0.33 0.67 0.34 0.5 0.75 0.55
Post-order traversal
15Li Liao, SIG NewGrad, 09/29/2008

16Li Liao, SIG NewGrad, 09/29/2008

Sequence Models (HMMs and beyond)
Motivations: What is responsible for the function?
– Patterns/motifs
– Secondary structure
To capture long range correlations of bio sequences
– Transporter proteins
– RNA secondary structure
Methods: generative versus discriminative
– Linear dependent processes
– Stochastic grammars
– Model equivalence
17Li Liao, SIG NewGrad, 09/29/2008

TMMOD: An improved hidden Markov model for predicting transmembrane topology (Kahsay, Gao & Liao. Bioinformatics 2005)
18Li Liao, SIG NewGrad, 09/29/2008

Mod. Reg.Dataset
Correcttopology
Correctlocation
Sens-itivity
Speci-ficity
TMMOD 1(a)(b)(c)
S-8365 (78.3%)51 (61.4%)64 (77.1%)
67 (80.7%)52 (62.7%)65 (78.3%)
97.4%71.3%97.1%
97.4%71.3%97.1%
TMMOD 2(a)(b)(c)
S-8361 (73.5%)54 (65.1%)54 (65.1%)
65 (78.3%)61 (73.5%)66 (79.5%)
99.4%93.8%99.7%
97.4%71.3%97.1%
TMMOD 3(a)(b)(c)
S-8370 (84.3%)64 (77.1%)74 (89.2%)
71 (85.5%)65 (78.3%)74 (89.2%)
98.2%95.3%99.1%
97.4%71.3%97.1%
TMHMM S-8364 (77.1%) 69 (83.1%) 96.2% 96.2%
PHDtm S-83(85.5%) (88.0%) 98.8% 95.2%
TMMOD 1(a)(b)(c)
S-160117 (73.1%)92 (57.5%)117 (73.1%)
128 (80.0%)103 (64.4%)126 (78.8%)
97.4%77.4%96.1%
97.0%80.8%96.7%
TMMOD 2(a)(b)(c)
S-160120 (75.0%)97 (60.6%)118 (73.8%)
132 (82.5%)121 (75.6%)135 (84.4%)
98.4%97.7%98.4%
97.2%95.6%97.2%
TMMOD 3(a)(b)(c)
S-160120 (75.0%)110 (68.8%)135 (84.4%)
133 (83.1%)124 (77.5%)143 (89.4%)
97.8%94.5%98.3%
97.6%98.1%98.1%
TMHMM S-160 123 (76.9%) 134 (83.8%) 97.1% 97.7%
19Li Liao, SIG NewGrad, 09/29/2008
20Li Liao, SIG NewGrad, 09/29/2008

21Li Liao, SIG NewGrad, 09/29/2008
Li Liao, SIG NewGrad, 09/29/2008 22
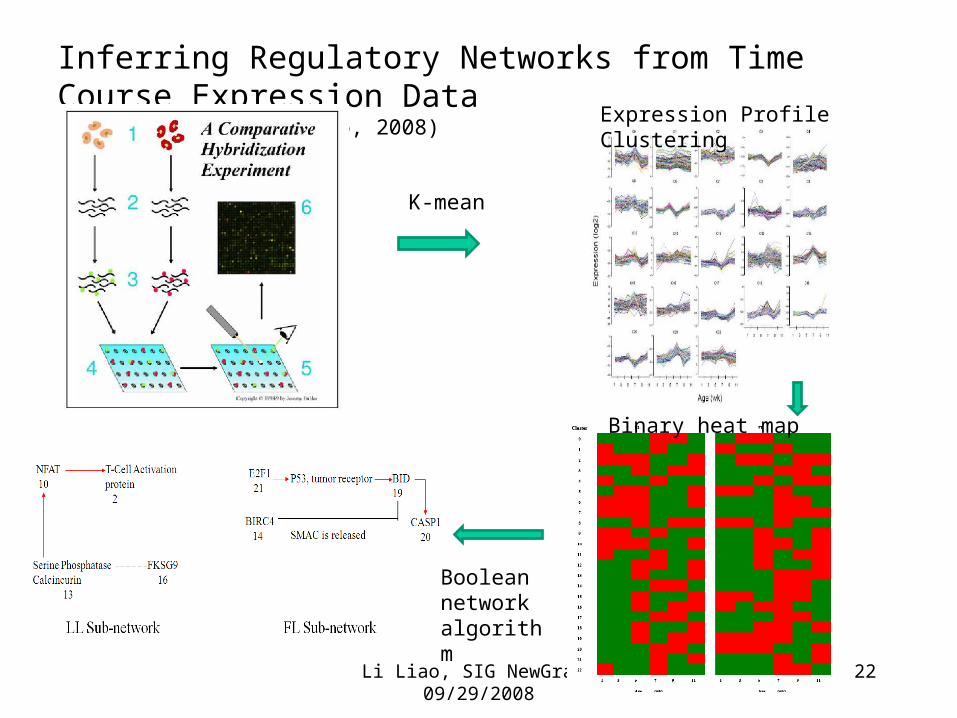
Inferring Regulatory Networks from Time Course Expression Data(Gandhi, Cogburn & Liao, 2008)
Expression Profile Clustering
Binary heat map
Boolean network algorithm
K-mean





























