Embed Size (px)
Citation preview

A n n a l s o p C l i n i c a l a n d L a b o r a t o r y S c i e n c e , Vol. 4 , No. 6Copyright © 1974, Institute for Clinical Science
Biochemical Abnormalities in Reye’s Syndrome
MARIANO M. ALVIRA, M.D.* AND DONALD T. FORMAN, Ph.D.
Department of Pathology and Laboratory Medicine,Evanston Hospital,
Evanston, IL 60201 and
Departments of Biochemistry and Pathology, Northwestern University Medical School,
Chicago, IL 60611
ABSTRACT
Reye’s syndrome has been characterized by cerebral edema without inflammatory cell infiltration or demyelination, and fatty degeneration of the viscera, especially the liver.19 Serum from nine children (ages 7 months to 8 years) with Reye’s syndrome confirmed at autopsy were examined prior to death. From our chemical studies this entity is defined biochemically as a severe metabolic acidosis presenting with decreased glucose concentrations in blood and cerebrospinal fluid, high concentrations of serum uric acid, urea nitrogen, ammonia and associated with marked increase of aspartate and alanine aminotransferase, lactic dehydrogenase and creatine phosphokinase (C P K ).
Biochemically, these changes may be explained by a sudden insult to intracellular respiration causing an increased tissue catabolism resulting in an elevated endogenous nitrogen load. Elevations of CPK in all subjects studied implies a severe impairment of oxidative metabolism. Since CPK isoenzymes in two patients showed marked elevations in skeletal muscle CPK ( M M ) and minor increases in heart ( M B) CPK, it may be postulated that this is a result of the primary hypoxemia and hypocapnia which these children exhibit. Serum gamma-glutamyl transpeptidase activity and bilirubin concentrations in the same patients were not increased and it is possible that liver damage in Reye’s syndrome is not so important a factor as has been reported.22 A point under study at this time is the relationship of the increased CPK concentration and the time of demise. Preliminary findings indicate a progressive rise in CPK suggests a grave prognosis.
In tro d u ctio n
D ata are presented on the clinical and pathological features of Reye’s syndrome as seen in a series of nine patients. Reye’s
* Present address: Department of Pathology, University of Cincinnati Medical Center, Cincinnati, OH 45221.
syndrome has been characterized by cerebral edema without inflammatory cell infiltration or demyelination, and fatty degeneration of the viscera, especially the liver.19
Included are additional biochemical data found in nine fatal cases of Reye’s syndrome at Evanston Hospital between July 1966 and October 1973.
477

4 7 8 A L VIRA AND FO RM A N
A presumptive diagnosis of Reye’s syndrome is made in a pediatric patient presenting with acute encephalopathy of no apparent cause. Certain clinical laboratory tests tend to support this diagnosis.9 The cerebrospinal fluid is free of inflammatory cells, and hypoglycemia are severe metabolic acidosis with hyperkalemia are usually present. Most striking are biochemical abnormalities suggestive of severe hepatic disease.5
M ateria l an d M ethods
Sera from nine children (ages 7 months to 8 years) with Reye’s syndrome confirmed at autopsy were examined prior to death.
Serum glucose was measured by a modification of the method of Hoffman7 and serum potassium values were determined by flame photometry.22 Serum osmolality was measured by a freezing-point depression apparatus3 and bilirubin was determined by a modification of the method of Jendrassik and Grof.11 Serum urea nitrogen and creatinine were determined by a reference autoanalyzer* method.14’17 Uric acid was measured by the method of Liddle12 and plasma ammonia was determined by an ion-exchange technic as described by Forman.4 Serum aminotransferases were assayed according to Henry et al6 and lactic dehydrogenase following the method of Amador.1 Alkaline phosphatase was determined by the optimized technique described by McComb and Bowers15 and creatine phosphokinase (C PK ) by the method of Rosalki.21 CPK isoenzymes were separated by agarose electrophoresis at pH 8.6 and their migration patterns determined by fluorescence assay. Serum gamma-glutamyl transpeptidase activity was determined according to Lum and Gambino.13
* Technicon Instruments Corp., Tarrytown, NY 10591.
Post-mortem examinations were performed in all nine cases. Post-mortem delay before fixation of tissues ranged from two hours to 18 hours. All tissues were fixed in 10 percent phosphate buffered formalin. In three more recent cases, tissue was fixed in 2 percent gluteraldehyde in sodium cacodylate buffer followed by post fixation in osmium tetroxide and 812 Epon embedding. The fixation time delays ranged two to four hours. Sections were studied with a Siemens Elmiskop 101 electron microscope. Hematoxylin and eosin, PAS, PTAH, Sudan IV, silver carbonate and gold sublimate stains were used to study tissue sections by light microscopy.
R esu lts
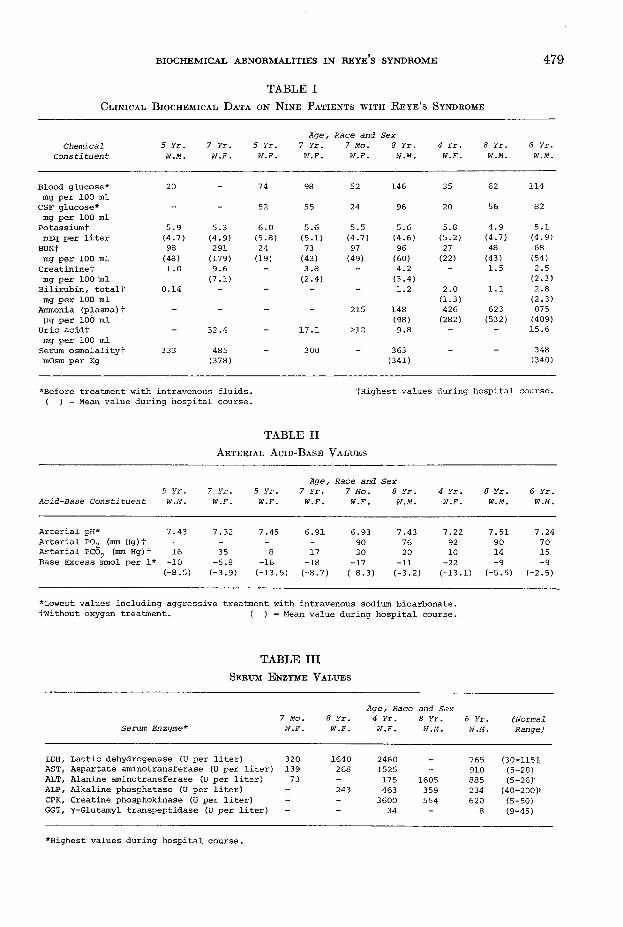
In table I are summarized the biochemical data obtained from nine patients with autopsy confirmed Reye’s syndrome. Four out of eight patients presented with serum hypoglycemia on admission and their cerebrospinal fluid (C S F ) glucose was lowered proportionally. Serum potassium was elevated in all these cases and mean values were also increased above normal. Serum urea nitrogen and creatinine analyses showed elevations in seven and five cases, respectively, and serum uric acid and plasma ammonia levels were elevated in five patients. Serum osmolality was measured in five cases and found to be very elevated in four subjects. Arterial blood gas analyses are summarized in table II. All subjects demonstrated a negative base excess, and decreased pH was found in five cases. Forty percent of subjects studied presented with mild hypoxemia prior to oxygen treatment. In eight cases, hypo- capnia was the rule.
In table III are illustrated the serum enzyme changes in the last five subjects studied. All patients showed marked elevations in serum lactic dehydrogenase (L D H ), aspartate aminotransferase (A ST) and alanine aminotransferase (A L T ). CPK
B IO C H EM IC A L A B N O R M A LIT IES IN R E Y E S SYNDROM E 4 7 9
T A B L E IC l i n i c a l B i o c h e m i c a l D a t a o n N i n e P a t i e n t s w i t h R e y e ’s S y n d r o m e
ChemicalConstituent
5 Yr. W.M.
7 Yr. 5 Yr. W.F. W.F.
A g e , 7 Yr. W.F.
Race and 7 Mo. W.F.
Sex 8 Yr. W.M.
4 Yr. W.F.
8 Yr. W.M.
6 Yr. W.M.
Blood glucose* 20 74 98 52 146 35 62 114mg per 100 mlCSF glucose* - 52 55 24 96 20 56 82mg per 100 ml
Potassiumf 5.9 5.3 6.0 5.6 5.5 5.6 5.8 4.9 5.1mEq per liter (4.7) (4.9) (5.8) (5.1) (4.7) (4.6) (5.2) (4.7) (4.9)
BUN+ 98 291 24 73 97 96 27 48 68mg per 100 ml (48) (179) (19) (43) (49) (60) (22) (43) (54)
Creatininet mg per 100 m l
1.0 9.6(7.1)
3.8(2.4)
4.2(3.4)
— 1.5 2.5(2.3)
Bilirubin, totalt mg per 100 ml
0.14 _ — “ “ 1.2 2.0(1-3)
1.1 2.8(2.3)
Ammonia (plasma)t Ug per 100 ml
“ _ _ “ 215 148(98)
426(282)
623(532)
875(409)
Uric acidt - 32.4 17.1 >12 9.8 - - 15.6mg per 100 mlSerum osmolalityt mOsm per Kg
333 485(378)
300 363(341)
348(340)
*Before treatment with intravenous fluids. ( ) = Mean value during hospital course.
tHighest values during hospital course.
T A B L E II Arterial Acid-Base Values
5 Yr.Age
7 Y r . 5 Yr. 7 Yr., Race and Sex
7 Mo. 8 Yr. 4 Yr. 8 Yr. 6 Yr.Acid-Base Constituent W.M. W.F. W.F. W.F. W.F. W.M. W.F. W.M. W.M.
Arterial pH* 7.43 7.32 7.45 6.91 6.93 7.43 7.22 7.51 7.24Arterial P02 (mm Hg)t - - - 90 76 92 90 70Arterial PC02 (mm Hg)f 16 35 8 17 30 20 10 14 15Base Excess mmol per 1* -10 -5.8 -16 -18 -17 -11 -22 -9 -9
C-8.5) (-3.9) (-13.5) (-8.7) (-8.3) (-3.2) (-13.1) (-5.5) (-2.5)
*Lowest values including aggressive treatment with intravenous sodium bicarbonate.fwithout oxygen treatment. ( ) = Mean, value during hospital course.
T A B L E III Serum E nzyme Values
Age, Race and Sex7 Mo. 8 Yr. 4 Yr. 8 Yr. 6 Yr. (Normal
Serum Enzyme* W.F. W.F. W.F. W.M. W.M. Range)
LDH/ Lactic dehydrogenase (U per liter) 320 1640 2460 765 (30-115);.AST, Aspartate aminotransferase (U per liter) 139 268 1525 - 910 (5-28).ALT, Alanine aminotransferase (U per liter) 73 - 175 1605 885 (5-26)'ALP, Alkaline phosphatase (U per liter) - 243 463 359 234 (40-200)'CPK, Creatine phosphokinase (U per liter) - - 3600 554 620 (5-50)GGT, ^-Glutamyl transpeptidase (U per liter) “ 34 “ 8 (9-45)
♦Highest values during hospital course.

480 A L VIRA AND FO RM AN
was assayed in the last three subjects and was remarkably elevated. In four patients studied, the serum alkaline phosphatase (ALP) was only slightly elevated. Serum y-glutamyl transpeptidase (G G T) was within normal limits in the two cases studied; in five cases examined, total bilirubin (table I) was within the normal range.
All the nine cases showed fatty degeneration of liver cells and peculiar cerebral edema without inflammatory changes as described.19 Seven cases had varying degrees of fatty degeneration of kidneys and heart. All the cases showed extensive acute neuronal necrosis and early astrocytosis of the central nervous system. The latter was best shown with silver carbonate and gold sublimate stains. Ultrastructural features in three cases showed severe mitochondrial round swelling of liver, kidney, heart and brain (figures 1 and 2). The matrix space was increased and coarsely granular matrix substance was present. The mitochondrial cristae were remarkably well preserved and the intracristae spaces were not expanded. Intramitochondrial dense bodies were always present. The outer mitochondrial membrane was generally well pre
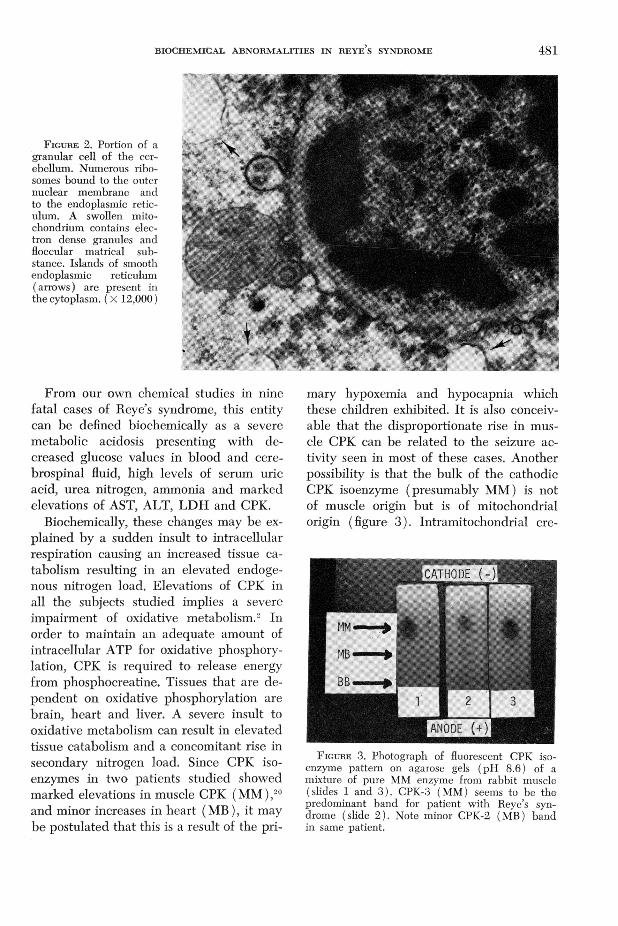
served. Ameboid mitochondrial shapes were generally absent.18 Organelle changes were also present as shown in figure 2. Proliferation of ribosomes and smooth endoplasmic membranes were very prominent, particularly in the central nervous system.
D isc u ss io n
Reye et al19 described the syndrome of acute encephalopathy and fatty degeneration of the viscera in children. In addition to the clinical and pathological findings, elevations of serum aspartate aminotransferase and alanine aminotransferase were reported in seven patients. Hypoglycemia, low glucose in cerebrospinal fluid and elevated serum urea nitrogen were also reported. Five years later, Huttenlocher et al8 reported plasma ammonia elevations in three children in whom blood ammonia levels were serially determined. Huttenlocher et al suggested that ammonia intoxication might be a causative factor in the onset of the acute encephalopathy. Since these reports, further elucidation of Reye’s syndrome has been lacking.
F ig u r e 1. Representative section from myocardium, left ventricle. Numerous swollen mito- chondriae disrupt myocardial cross striation. The Z bands are well preserved. Some mitochondria contain electron dense granules. (X 2,400)
BIO C H EM IC A L A B N O R M A LIT IES IN R EY E’S SYNDROME 481
F ig u r e 2. Portion of a granular cell of the cerebellum. Numerous ribosomes bound to the outer nuclear membrane and to the endoplasmic reticulum. A swollen mito- chondrium contains electron dense granules and fioccular matrical substance. Islands of smooth endoplasmic reticulum ( arrows) are present in the cytoplasm. ( X 12,000)
From our own chemical studies in nine fatal cases of Reye’s syndrome, this entity can be defined biochemically as a severe metabolic acidosis presenting with decreased glucose values in blood and cerebrospinal fluid, high levels of serum uric acid, urea nitrogen, ammonia and marked elevations of AST, ALT, LDH and CPK.
Biochemically, these changes may be explained by a sudden insult to intracellular respiration causing an increased tissue catabolism resulting in an elevated endogenous nitrogen load. Elevations of CPK in all the subjects studied implies a severe impairment of oxidative metabolism.2 In order to maintain an adequate amount of intracellular ATP for oxidative phosphorylation, CPK is required to release energy from phosphocreatine. Tissues that are dependent on oxidative phosphorylation are brain, heart and liver. A severe insult to oxidative metabolism can result in elevated tissue catabolism and a concomitant rise in secondary nitrogen load. Since CPK isoenzymes in two patients studied showed marked elevations in muscle CPK (M M ),20 and minor increases in heart (MB), it may be postulated that this is a result of the pri
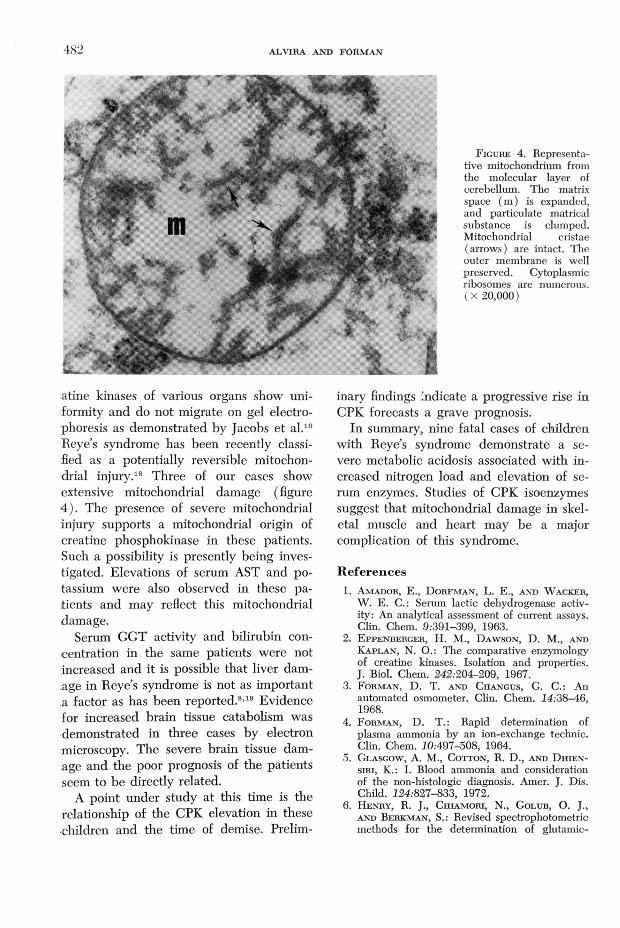
mary hypoxemia and hypocapnia which these children exhibited. It is also conceivable that the disproportionate rise in muscle CPK can be related to the seizure activity seen in most of these cases. Another possibility is that the bulk of the cathodic CPK isoenzyme (presumably MM) is not of muscle origin but is of mitochondrial origin (figure 3). Intramitochondrial cre-
F ig u r e 3. Photograph of fluorescent CPK isoenzyme pattern on agarose gels (pH 8.6) of a mixture of pure MM enzyme from rabbit muscle (slides 1 and 3 ). CPK-3 (MM) seems to be the predominant band for patient with Reye’s syndrome (slide 2 ). Note minor CPK-2 (MB) band in same patient.
482 A L V IR A AND FORM AN
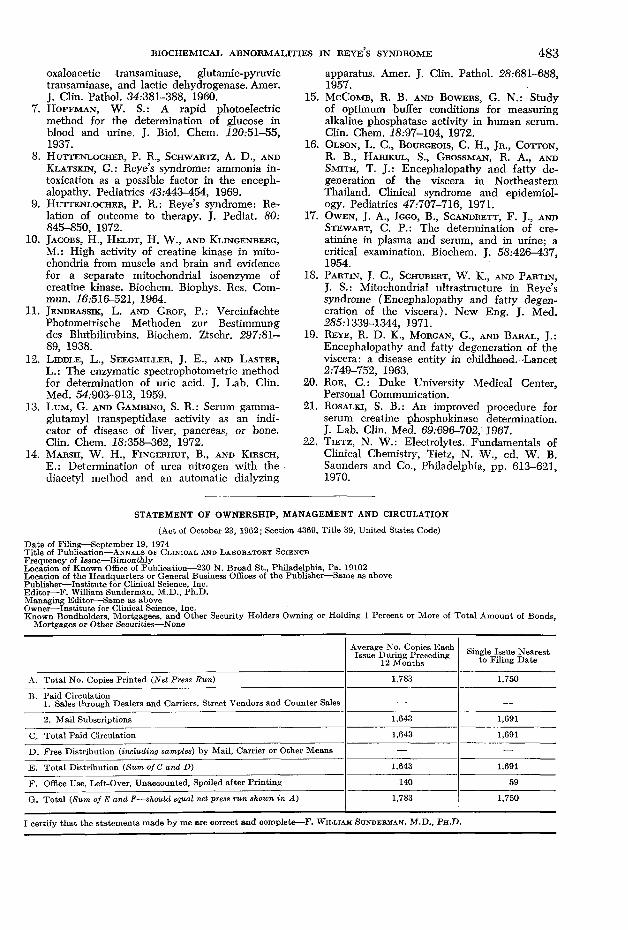
F ig u r e 4. Representative mitochondrium from the molecular layer of cerebellum. The matrix space ( m ) is expanded, and particulate matrical substance is clumped. Mitochondrial cristae (arrows) are intact. The outer membrane is well preserved. Cytoplasmic ribosomes are numerous. (X 20,000)
atine kinases of various organs show uniformity and do not migrate on gel electrophoresis as demonstrated by Jacobs et al.10 Reye’s syndrome has been recently classified as a potentially reversible mitochondrial injury.18 Three of our cases show extensive mitochondrial damage (figure4 ). The presence of severe mitochondrial injury supports a mitochondrial origin of creatine phosphokinase in these patients. Such a possibility is presently being investigated. Elevations of serum AST and potassium were also observed in these patients and may reflect this mitochondrial damage.
Serum GGT activity and bilirubin concentration in the same patients were not increased and it is possible that liver damage in Reye’s syndrome is not as important a factor as has been reported.8’19 Evidence for increased brain tissue catabolism was demonstrated in three cases by electron microscopy. The severe brain tissue damage and the poor prognosis of the patients seem to be directly related.
A point under study at this time is the relationship of the CPK elevation in these children and the time of demise. Prelim
inary findings indicate a progressive rise in CPK forecasts a grave prognosis.
In summary, nine fatal cases of children with Reye’s syndrome demonstrate a severe metabolic acidosis associated with increased nitrogen load and elevation of serum enzymes. Studies of CPK isoenzymes suggest that mitochondrial damage in skeletal muscle and heart may be a major complication of this syndrome.
R eferen ces1. A m a d o r , E., D o r f m a n , L. E., a n d W a c k e r ,
W. E. C.: Serum lactic dehydrogenase activity: A n analytical assessment of current assays. Clin. Chem. 9:391-399, 1963.
2. E p p e n b e r g e r , H. M., D a w s o n , D . M., a n d K a p l a n , N. O.: The comparative enzymology of creatine kinases. Isolation and properties. J. Biol. Chem. 242:204-209, 1967.
3. F o r m a n , D. T. a n d C h a n g u s , G. C .: A n a u to m a te d o sm o m e te r . C lin . C h e m . 14:38-46,1968.
4. F o r m a n , D. T.: Rapid determination of plasma ammonia by an ion-exchange technic. Clin. Chem. 10:497-508, 1964.
5. G l a s g o w , A. M., C o t t o n , R. D ., a n d D h i e n - s i r i , K.: I. Blood ammonia and consideration of the non-histologic diagnosis. Amer. J. Dis. Child. 124:827-833, 1972.
6. H e n r y , R. J., C h ia m o r i , N., G o l u b , O. J., a n d B e r k m a n , S.: Revised spectrophotometric methods for the determination of glutamic-
BIO C H EM IC A L A B N O R M A LIT IES IN REY e ’s SYNDROM E 4 8 3
oxaloacetic transaminase, glutamic-pyruvic transaminase, and lactic dehydrogenase. Amer. J. Clin. Pathol. 34:381-388, 1960.
7. Hoffman , W. S.: A rapid photoelectric method for the determination of glucose in blood and urine. J. Biol. Chem. 120:51-55, 1937.
8. Huttenlocher, P. R., Schwartz, A. D., and Klatskin , G.: Reye’s syndrome: ammonia intoxication as a possible factor in the encephalopathy. Pediatrics 43:443-454, 1969.
9 . H u t t e n l o c h e r , P. R.: Reye’s syndrome: Relation of outcome to therapy, j . Pediat. 80: 8 4 5 - 8 5 0 , 1 9 7 2 .
10. J acobs, H., Heldt, H. W., and Klingenberg, M.: High activity of creatine kinase in mitochondria from muscle and brain and evidence for a separate mitochondrial isoenzyme of creatine kinase. Biochem. Biophys. Res. Com- mun. ¿6:516-521, 1964.
11. J endrassik, L . and Grof, P.: Vereinfachte Photometrische Methoden zur Bestimmung des Blutbilirubins. Biochem. Ztschr. 297:81- 89, 1938.
12. L iddle, L ., Seegm iller , J. E., and L aster, L .: The enzymatic spectrophotometric method for determination of uric acid. J. Lab. Clin. Med. 54:903-913, 1959.
13. L um , G. and Gambino, S. R.: Serum gamma- glutamyl transpeptidase activity as an indicator of disease of liver, pancreas, or bone. Clin. Chem. 18:358-362, 1972.
14. Marsh, W. H., F ingerhut, B., and K irsch, E.: Determination of urea nitrogen with the diacetyl method and an automatic dialyzing
apparatus. Amer. J. Clin. Pathol. 28:681-688, 1957.
15. McComb, R. B. and B owers, G. N .: Study of optimum buffer conditions for measuring alkaline phosphatase activity in human serum. Clin. Chem. ¿8:97-104, 1972.
16. Olson, L. C., Bourgeois, C. H., J r ., Cotton, R. B., Harikul, S., Grossman, R. A., and Smith , T. J . : Encephalopathy and fatty degeneration of the viscera in Northeastern Thailand. Clinical syndrome and epidemiology. Pediatrics 47:707-716, 1971.
17. Owen, J. A., Iggo, B., Scandrett, F. J., and Stewart, C. P.: The determination of creatinine in plasma and serum, and in urine; a critical examination. Biochem. J. 58:426-437, 1954.
18. Partin, J. C., Schubert, W. K., and Partin, J. S.: Mitochondrial ultrastructure in Reye’s syndrome (Encephalopathy and fatty degeneration of the viscera). New Eng. J . Med. 285:1339-1344, 1971.
19. Reye, R. D. K., Morgan, G., and B aral, J.: Encephalopathy and fatty degeneration of the viscera: a disease entity in childhood. Lancet 2:749-752, 1963.
20. Roe, C.: Duke University Medical Center, Personal Communication.
21. Rosalki, S. B.: An improved procedure for serum creatine phosphokinase determination. J. Lab. Clin. Med. 69:696-702, 1967.
22. T ietz, N. W.: Electrolytes.. Fundamentals of Clinical Chemistry, Tietz, N. W., ed. W. B. Saunders and Co., Philadelphia, pp. 613-621,1970.
STATEM EN T OF O W NERSH IP, M ANAGEM ENT AND CIRCULATION
(Act of October 23, 1962 ; Section 4369, Title 39, United States Code)
Date of Filing— September 19, 1974Title of Publication—A n n a l s o f C l i n i c a l a n d L a b o r a t o r y S c i e n c e Frequency of Issue—BimonthlyLocation of Known Office of Publication— 230 N. Broad St., Philadelphia, Pa. 19102 Location of the Headquarters or General Business Offices of the Publisher—Same as above Publisher— Institute for Clinical Science, Inc.Editor—F . William Sunderman, M .D., Ph.D.Managing Editor—Same as above Owner—Institute for Clinical Science, Inc.Known Bondholders, Mortgagees, and Other Security Holders Owning or Holding 1 Percent or More of Total Amount of Bonds,
Mortgages or Other Securities—None
Average No. Copies Each Issue During Preceding
12 MonthsSingle Issue Nearest
to Filing Date
A. Total No. Copies Printed (Net Press Run) 1,783 1,750
B . Paid Circulation1. Sales through Dealers and Carriers, Street Vendors and Counter Sales — —
2. M ail Subscriptions 1,643 1,691
C. Total Paid Circulation 1,643 1,691
D. Free Distribution (including samples) by Mail, Carrier or Other Means — —
E . Total Distribution (Sum of C and D) 1,643 1,691
F. Office Use, Left-Over, Unaccounted, Spoiled after Printing 140 59
G. Total (Sum of E and F —should equal net press run shown in A) 1,783 1,750
I certify that the statements made by me are correct a n d complete— F. W iLLrA M S u n d e r m a n , M .D., Ph.D .












![Ursodeoxycholic acid versus placebo in the treatment of ...ursodeoxycholic acid (UDCA) [9, 10] to improve mater-nal pruritus and biochemical abnormalities. However, there are currently](https://img.pdfslide.us/doc/110x75/61033dc0eaa17e1cf4173857/ursodeoxycholic-acid-versus-placebo-in-the-treatment-of-ursodeoxycholic-acid.jpg)















