Embed Size (px)
Citation preview

— —< <
Basis Set Approach to Solution ofPoisson Equation for Small MoleculesImmersed in Solvent
LAURENT DAVID and MARTIN J. FIELD*Laboratoire de Dynamique Moleculaire, Institut de Biologie Structurale—Jean-Pierre Ebel, 41 Avenue´des Martyres, 38027 Grenoble Cedex 1, France
Received 9 November 1995; accepted 14 May 1996
ABSTRACT
The problem of how to calculate the electrostatic interactions between moleculesand a solvent is a very important one in theoretical chemistry and biophysics.One of the more commonly used methods has been to represent the solvent by
Ž .a dielectric continuum and then to solve the Poisson or the Poisson]Boltzmannequation for the potential due to the charge distribution of the solute. Thesolution of the equation has, up to now, been largely carried out using finite-difference grid-based methods. In this article, we investigate the use of analternative method, based on a basis set expansion of the potential. The choice ofbasis functions, the representation of the dielectric function and the accuracythat can be obtained are discussed and illustrated by example calculations onsmall molecules. Q 1997 by John Wiley & Sons, Inc.
Introduction
he behavior and properties of molecules areT profoundly changed when they are immersedin solution. These changes are due, in large part, tothe electrostatic interactions between the soluteand the solvent, and so an ability to calculate theseterms is essential if the effects of solvent are to beunderstood. One of the more successful models
*Author to whom all correspondence should be addressed.E-mail: [email protected]
which has been used to study solutersolvent inter-Žactions particularly for large macromolecular sys-
.tems, such as proteins has been to describe thesolvent as a dielectric continuum and to solve thePoisson]Boltzmann equation for the electrostaticpotential, and hence the energy of solvation, dueto the charge distribution in the solute.1 ] 4
Up to now, the methods of choice for the solu-tion of the Poisson]Boltzmann equation have beenthe numerical finite difference4 and, to a lesserextent, finite element5 and boundary element ap-proaches.6,7 These have been developed to thepoint where they can be used to rapidly obtain the
( )Journal of Computational Chemistry, Vol. 18, No. 3, 343]350 1997Q 1997 by John Wiley & Sons CCC 0192-8651 / 97 / 030343-08

DAVID AND FIELD
electrostatic potential for systems containing sev-eral thousands of atoms. The state-of-the-art issuch that the Poisson]Boltzmann equation is start-ing to be used to calculate the electrostatic energyof a system and the forces on its constituent parti-
Žcles during molecular dynamics simulations see,.e.g., ref. 8 . However, these algorithms have some
limitations. The precision of the finite differencemethods, for example, is limited by the size of thegrid that can be employed. For large systems theuse of a fine grid is expensive and so a coarser gridmust be used with a consequent reduction in theaccuracy of the potential that can be obtained.
In this article we explore an alternative methodto the solution of the Poisson equation for a so-lutersolvent system. It is based on the expansionof the electrostatic potential in terms of a linearcombination of basis functions. While basis setmethods have been used widely for the solution ofother partial differential equations of interest inchemical physics, such as the Schrodinger equa-¨tion,9 they have not been used, to our knowledge,for this application. There are several possible ad-vantages to using a basis set. These include thefact that the potential, when expressed in terms ofa basis set, has an analytic form and its derivativesand those of the solvation energy are straightfor-ward to evaluate—a fact which is important ifcontinuum models are to be useful for moleculardynamics simulations. On the other hand, one ofthe disadvantages of basis set methods is thatmany basis functions may be needed to provide areasonable approximation of the potential in thesystem.
The outline of this article is as follows: the nextsection describes the basis set expansion approachto the solution of the Poisson equation; the thirdsection presents the results of calculations on somemodel systems; and the last section concludes.
Methods
INTRODUCTION OF A BASIS SET
In this article we consider only the Poissonequation. The addition of the term dependent on
Žthe ionic strength is relatively straightforward at.least in its linear form and will be considered in
future work.The Poisson equation determines the electro-
Ž .static potent, f r , as a function of the positioncoordinate, r, in a system:
w Ž . Ž .x Ž . Ž .= ? « r =f r q 4pr r s 0 1
where r is the charge distribution and « is thedielectric constant. Both of these quantities, likethe potential, also depend upon the position coor-dinate.
It has been shown that there exists an expres-sion which represents a free energy functional, G,for the Poisson equation. It is10 ] 13
Ž .« r 2Ž . Ž . Ž Ž .. Ž .G s dr r r f r y =f r 2H ž /8p
The Poisson equation results from the functionalvariation of this expression for the electrostatic freeenergy with respect to the potential. In other words,the electrostatic free energy is at an extremum forthe potential which obeys the Poisson equation.
Ž .Eq. 2 defines the variational principle that wecan use to obtain the electrostatic potential using abasis set approach. To do this, we assume that thepotential can be expanded as a linear combination
� 4of M potential basis functions, f :i
M
Ž . Ž . Ž .f r s c f r 3Ý i iis1
� 4where the c are the expansion coefficients for theipotential. Substituting the expression for the po-tential into the expression for the electrostatic en-
w Ž .xergy eq. 2 gives the matrix equation:
Ž .G s c P y c c F 4Ý ÝÝi i i j i ji i j
where P and F are integrals over the basis func-i i jtions which are defined as follows:
Ž . Ž . Ž .P s drr r f r 5Hi i
Ž .« rŽ Ž .. Ž . Ž .F s dr =f r ? =f r 6Ž .Hi j i j8p
Before we can minimize this expression with re-spect to the expansion coefficients we need to takeaccount of the fact that the total charge on thesystem, q, is fixed. That is:
Ž . Ž .drr r s q 7HThis equation constrains the value of the expan-sion coefficients because the charge density deter-mines the value of the potential through the Pois-son equation. Substituting the expression for the
Ž .charge density from eq. 1 and for the potential
VOL. 18, NO. 3344

SOLUTION OF POISSON EQUATION
Ž . Ž .from eq. 3 into eq. 7 gives a constraint conditionfor the expansion coefficients:
Ž .c K q q s 0 8Ý i ii
where the K are integrals defined as:i
1w Ž . Ž .x Ž .K s dr= ? « r =f r 9Hi i4p
We now minimize the expression for the freew Ž .xenergy eq. 4 with respect to the expansion coef-
w Ž .xficients, subject to the constraint condition eq. 8 .Solving for the vector of expansion coefficients, c,gives:
1 y1 Ž . Ž .c s F P y lK 102
where F is a matrix of the F integrals and P andi jK are vectors of the P and K integrals, respec-i itively. l is a Lagrange multiplier that has beenintroduced to satisfy the constraint condition. Ithas the form:
2 q q KT Fy1 PŽ .l s 11T y1K F K
Once the coefficients have been determined bysolving these equations the energy can be calcu-
Ž .lated by substituting for the vector c in eq. 4 .
CALCULATION OF DERIVATIVES
In molecular simulations it is often important toŽbe able to calculate the forces on the atoms and
hence the first derivatives with respect to the.atomic positions . In the basis set approach the
derivatives of the energy with respect to a parame-ter, x, such as the coordinate of an atom, arestraightforward to obtain. Differentiating the ex-pression for the energy gives:
G P F cTT Ž . Ž .s c y c q P y 2Fc 12ž / x x x x
The last term can be simplified by rearranging thew Ž .xexpression for c eq. 10 :
c cT TŽ . Ž .P y 2Fc s lK 13 x x
It is not necessary to calculate the derivatives ofthe expansion coefficients with respect to x. To see
wthis we differentiate the constraint condition eq.Ž .x8 :
c KT T Ž .K q c s 0 14
x x
which equates the term involving the derivativesof the expansion coefficients to one involving thederivatives of the K integrals. The final expres-ision for the derivatives is:
G P F KT Ž .s c y c y l 15ž / x x x x
Given suitable differentiable forms for the po-tential basis functions, such as the ones discussedin the following subsection, the derivatives should
Ž .be readily evaluated using eq. 15 , as all the otherquantities in the equation, such as the vector, c,and the Lagrange multiplier, l, will have beendetermined in a preceding variational calculation.
CHOICE OF BASIS SET
� 4The choice of forms for the basis functions, f ,i
and for the charge density, r, and the dielectricconstant, « , will determine the efficiency and theprecision of the basis set approach to the solutionof the Poisson equation.
To start, a few general remarks on the choice ofbasis functions can be made:
B For the variational principle to be valid, thepotential, and hence the basis functions, must
Ž .tend to zero to infinity; that is f r ª 0 asir ª `.
B At large distances, the potential tends to-ward the same potential produced by thesame charge distribution but in a dielectricconstant equal to that outside the molecule.Therefore, the basis set should include func-tions that give this limiting behavior.
B w Ž .xThe K integrals eq. 9 can be evaluated,i
regardless of the form of the basis functions,by transformation of the volume integral tothat over a surface at infinity. For basis func-tions that have the limiting behavior of ry1
the integral will equal y« where « is the0 0dielectric constant outside the molecule. Forbasis functions that decay more rapidly thanthis, the integrals are zero. Due to theseresults, some limitations are imposed on thechoice of functions in a basis set. If the totalcharge on the system is nonzero then it isessential to use at least some basis functionswith the ry1 limiting behavior; that is, withnonzero K integrals, so that the constrainti
JOURNAL OF COMPUTATIONAL CHEMISTRY 345

DAVID AND FIELD
Ž .condition of eq. 8 can be satisfied. If thetotal system charge is zero then there is nosuch limitation.
In this initial work we have chosen to useŽGaussian functions or functions derived from
.them, such as error functions as much as possible.This is because they are very amenable to variousmanipulations, including integrations, and becausethere exists a large body of experience from thequantum chemistry field on their use.9 Thus, in-stead of the point charge model that is usual forthe charge density, we express the charge densityof the molecule as a sum of Gaussian densities foreach atom, r :a
3 22N Ž .1 r y R aŽ . Ž .r r s q exp y 16Ý a 2 2ž / ž /2ps 2sa aas1
where q is the atomic charge; s is the width ofa a
the distribution for the atom, a; and the number ofatoms in the system is N.
For the basis functions we tried a number offorms which are all based upon the Gaussian andwhich are discussed more fully in the Resultssection. In particular, we used functions whichrepresent the potential due to a Gaussian chargedistribution; that is:
'Ž . < <r r9 erf r y R r 2 sŽ .a a a Ž .dr9 s q 17H a< < < <r y r9 r y R a
2Ž .2 r y R a Ž .s F 1802 2(ps 2sa a
w xwhere the function, F x , is given by:0
1 2w x Ž . Ž .F x s exp yxs ds 19H00
With these forms for the basis functions and thew Ž .xcharge distribution the P integrals eq. 5 arei
readily evaluated.The remaining problem is how to represent the
Ž .dielectric function, « r . It is well known that thevolume of a molecule comprised of hard-sphereatoms can be obtained by integrating a ‘‘matter’’
Ž . 14,15density, m r , of the form :
N
Ž . Ž Ž .. Ž .m r s 1 y 1 y n r 20Ł aa
where n is a step function centered at atom aa
which has the value of 1 inside the hard sphere
and a value of 0 outside. Thus, the matter densityhas a value of 1 inside the molecule and a value of0 outside. We can use this function to define thedielectric function as:
Ž . Ž . Ž Ž .. Ž .« r s « m r q « 1 y m r 21i 0
where « and « are the interior and exteriori 0dielectric constants, respectively. To make the ma-nipulation of this function easier we replace thestep function form for n by products of polynomi-a
als and Gaussians, the exact forms for which arediscussed next. We note that Grant and Pickuphave recently used single Gaussian functions intheir calculations to determine the volumes andsurface areas of molecules.15 With this form for the
w Ž .xdielectric function, the F integrals eq. 6 , al-i jthough not trivial, can, in principle, be evaluatedrelatively efficiently.
Results
SYSTEMS WITH SPHERICAL SYMMETRY
To obtain insights into the basis set approach itis useful to consider spherically symmetric sys-tems with a single positive unit charge because thepotential and the energy can be solved for numeri-cally using standard techniques for arbitrary formsof the dielectric and charge distribution functions.The Poisson equation in this case reduces to asingle variable equation:
1 d df2 Ž . Ž . Ž .r « r q 4pr r s 0 222 ž /dr drr
For the calculations in this section we repre-sented the charge distribution as a single Gaussian
˚w Ž .xfunction eq. 16 with s s 0.5 A. For the dielec-tric function we used the expression given in eq.Ž .21 with values for the dielectric constants, « andi« , of 1 and 78.36, respectively. A number of forms0for the function, n , were tried:
Ž . Ž . Ž .n s s Q s y 1 23h s
Ž . Ž 2 . Ž .n s s exp ys 240
Ž . Ž 2 . Ž 2 . Ž .n s s 1 q s exp ys 252
12 4 2Ž . Ž . Ž .n s s 1 q s q s exp ys 26Ž .4 2
1 12 4 6 2Ž . Ž . Ž .n s s 1 q s q s q s exp ys 27Ž .6 2 5
Žwhere Q is the step function with a value of 0 if.the argument is greater than 0 and 1 otherwise .
VOL. 18, NO. 3346
SOLUTION OF POISSON EQUATION
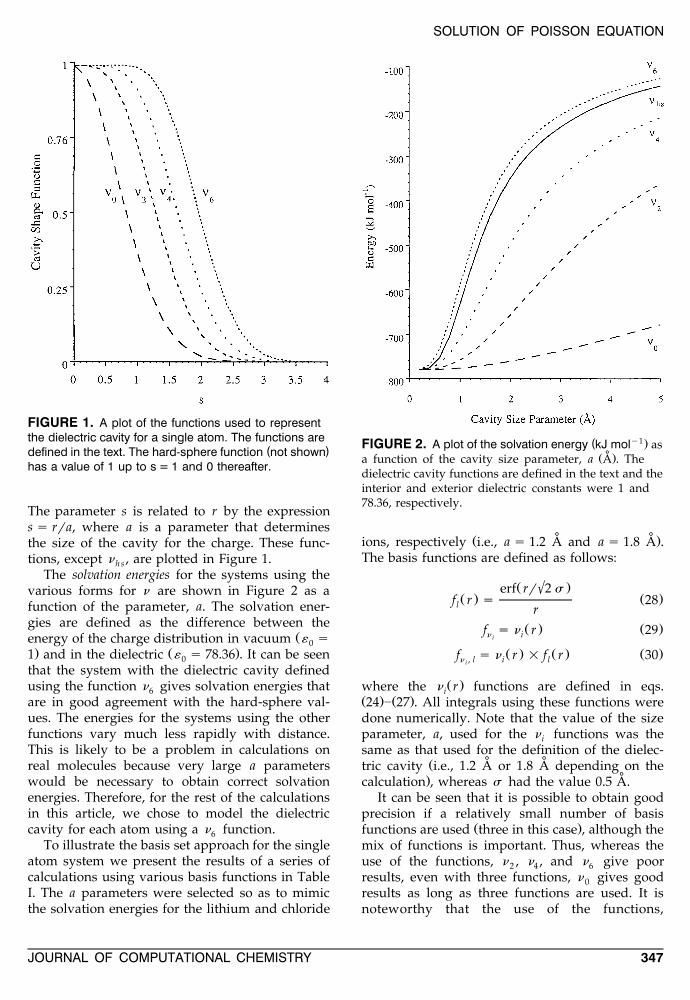
FIGURE 1. A plot of the functions used to representthe dielectric cavity for a single atom. The functions are
( )defined in the text. The hard-sphere function not shownhas a value of 1 up to s = 1 and 0 thereafter.
The parameter s is related to r by the expressions s rra, where a is a parameter that determinesthe size of the cavity for the charge. These func-tions, except n , are plotted in Figure 1.h s
The solvation energies for the systems using thevarious forms for n are shown in Figure 2 as afunction of the parameter, a. The solvation ener-gies are defined as the difference between the
Ženergy of the charge distribution in vacuum « s0. Ž .1 and in the dielectric « s 78.36 . It can be seen0
that the system with the dielectric cavity definedusing the function n gives solvation energies that6are in good agreement with the hard-sphere val-ues. The energies for the systems using the otherfunctions vary much less rapidly with distance.This is likely to be a problem in calculations onreal molecules because very large a parameterswould be necessary to obtain correct solvationenergies. Therefore, for the rest of the calculationsin this article, we chose to model the dielectriccavity for each atom using a n function.6
To illustrate the basis set approach for the singleatom system we present the results of a series ofcalculations using various basis functions in TableI. The a parameters were selected so as to mimicthe solvation energies for the lithium and chloride
( y1.FIGURE 2. A plot of the solvation energy kJ mol as˚Ž .a function of the cavity size parameter, a A . The
dielectric cavity functions are defined in the text and theinterior and exterior dielectric constants were 1 and78.36, respectively.
˚ ˚Ž .ions, respectively i.e., a s 1.2 A and a s 1.8 A .The basis functions are defined as follows:
'Ž .erf rr 2 sŽ . Ž .f r s 28l r
Ž . Ž .f s n r 29n ii
Ž . Ž . Ž .f s n r = f r 30n , l i li
Ž .where the n r functions are defined in eqs.iŽ . Ž .24 ] 27 . All integrals using these functions weredone numerically. Note that the value of the sizeparameter, a, used for the n functions was theisame as that used for the definition of the dielec-
˚ ˚Žtric cavity i.e., 1.2 A or 1.8 A depending on the˚.calculation , whereas s had the value 0.5 A.
It can be seen that it is possible to obtain goodprecision if a relatively small number of basis
Ž .functions are used three in this case , although themix of functions is important. Thus, whereas theuse of the functions, n , n , and n give poor2 4 6results, even with three functions, n gives good0results as long as three functions are used. It isnoteworthy that the use of the functions,
JOURNAL OF COMPUTATIONAL CHEMISTRY 347

DAVID AND FIELD
TABLE I.Energies Obtained with an External DielectricConstant of 78.36 by Solving the PoissonEquation Using Various Forms for Basis
aFunctions for Single Particle System.
y1( )Energies kJ mol
˚ ˚Basis set a = 1.2 A a = 1.8 A
Exact 277.0 442.7f 3.8 4.1l
f , f 98.7 107.9l n0
f , f 198.3 331.0l n0 , lf , f , f 263.0 427.6l n0 n0 , lf , f , f 202.9 355.6l n2 ns, lf , f , f 135.1 249.8l n4 n4 , lf , f , f 90.8 168.6l n6 n6 , l
aThe basis functions are defined in the text. The dielectriccavity function used was n with values for a of 1.2 and 1.86A, respectively. The value of the internal dielectric constant= 1.
Ž . Ž .f r r« r , as basis functions, give much worselresults that the result cited in Table I, despite theiradditional complexity.
Instead of discussing further the symmetricalcase we move on to the discussion of generalmolecular systems and see if an equivalent accu-racy can be obtained.
MOLECULAR SYSTEMS
For the calculations in this section a set of sixsmall molecules were selected. They were:
˚1. A diatomic consisting of two atoms 2 A apart,each with a single positive charge and cavity
˚size parameters of 1.5 A.2. The same diatomic but with one atom having
a positive charge and the second a negativecharge.
3. Methanol. The geometry and parameters forthe molecule were taken from the examplesin the test suite provided with the UHBDprogram.16 The OPLS charges and atomicradii were used17 and the atoms in the methylgroup were treated as a single ‘‘united atom.’’
4. Formate anion. The parameters and geome-try were obtained from the QUANTA molec-ular modeling program.18 The charges were0, y0.628; C, 0.373; H, y0.117; the cavity
˚ ˚ ˚sizes: O, 1.655 A; C, 1.875 A; H, 1.2 A; and˚ ˚the geometry: C O , 1.26 A; H C , 1.14 A;
H C O , 117.28.
5. Acetic acid. The geometry and parametersfor this molecule were taken from the UHBDprogram test suite, although, in this case, themethyl group was treated as four separateatoms.
6. Methazolamide. The geometry and parame-ters were taken from the UHBD example testsuite.
The charge distributions of the atoms in all themolecules were represented by Gaussians with
˚widths of 0.5 A. Note that the variable parameters—the charges on the atoms, the width of the chargedistribution, and the cavity sizes—were all usedwithout optimization for these calculations. Noattempt was made to vary the parameters to ob-tain realistic values of the solvation energy, forexample. This will be the aim of future work if thebasis set method appears to be competitive withthe finite difference approaches.
The results of four sets of calculations, per-formed using different methods of solution, willbe presented for these molecules. The methods are:
Ž .1. Finite differences FD . To compare the re-sults calculated using various basis sets wemodified a finite difference program that wehad written previously, which solves thePoisson equation.19 To do this, the values ofthe dielectric on the grid are assigned using
w Ž .the form of the dielectric function eqs. 21Ž .xand 27 and the charges for each grid point
are calculated by integrating the charge den-w Ž .xsity eq. 16 over a cube centered on each
grid point. The method of iterative solutionis the same, although, in passing, we note
Ž .that convergence as a function of grid sizewas found to be much more rapid with aGaussian distribution of charge than whenusing the more traditional representation of aset of point charges. The values of the self-energies found when using small grid sizeswere also much reduced. For the smallmolecules we used a grid of size 1003 with a
˚spacing of 0.2 A. For the larger moleculesŽ .formate, acetic acid, and methazolamide ,we used a grid of size 1203 with spacings of
˚ ˚ ˚0.18 A, 0.186 A, and 0.227 A, respectively.Ž .2. Basis set 1 BS 1 . This basis set consisted of
Ž3N functions for each molecule where N is.the number of atoms in the molecule . Each
atom was assigned three functions of theŽ . Ž .forms given in eqs. 28 ] 30 with the n 0
w Ž .xfunction eq. 24 being chosen for n .i
VOL. 18, NO. 3348
SOLUTION OF POISSON EQUATION
Ž .3. Basis set 2 BS 2 . This basis set consisted ofonly three functions for each molecule. Thesewere:
N '< <q erf r y R r 2 sŽ .a aŽ . Ž .f r s 31ÝT < <r y R aas1
N
Ž . Ž < <. Ž .f r s 1 y 1 y n r y R 32Ž .Łn 0 aaas1
Ž . Ž . Ž . Ž .f r s f r = f r 33n , T n T
The choice of f ensures that the potential atTlong range has the correct form. This is be-
w Ž .xcause the constraint condition eq. 8 for acharged system can only be satisfied if thecoefficient for this basis function has the value
Ž1r« because all the K integrals are zero0 i.except for f .T
Ž .4. Basis set 3 BS 3 . As a result of our experi-ence with the previous two basis sets wecombined the three functions of BS 2 with the
w Ž .2 N short-range basis functions eqs. 29 andŽ .x30 of basis set 1 giving 2 N q 3 basis func-tions for each molecule.
The electrostatic energies for the molecule in vac-Ž .uum i.e., with a dielectric of 1 everywhere can, of
course, be calculated analytically and provide a
check on the accuracy of the finite difference calcu-lations. With the choice of the basis functions givenabove it is, in principle, possible to calculate the
20 Žintegrals P and F analytically because all thei i j.functions are Gaussians . However, for this work
we only evaluated the integrals analytically whichŽ . w Ž .did not involve the functions m r or f eqs. 20n
Ž . xand 32 , respectively . For integrals involvingthese functions we used an adaptive numericalintegration algorithm in three dimensions.21 Wechecked the values produced by the integrationand believe they are accurate, in all cases, to 10y3.
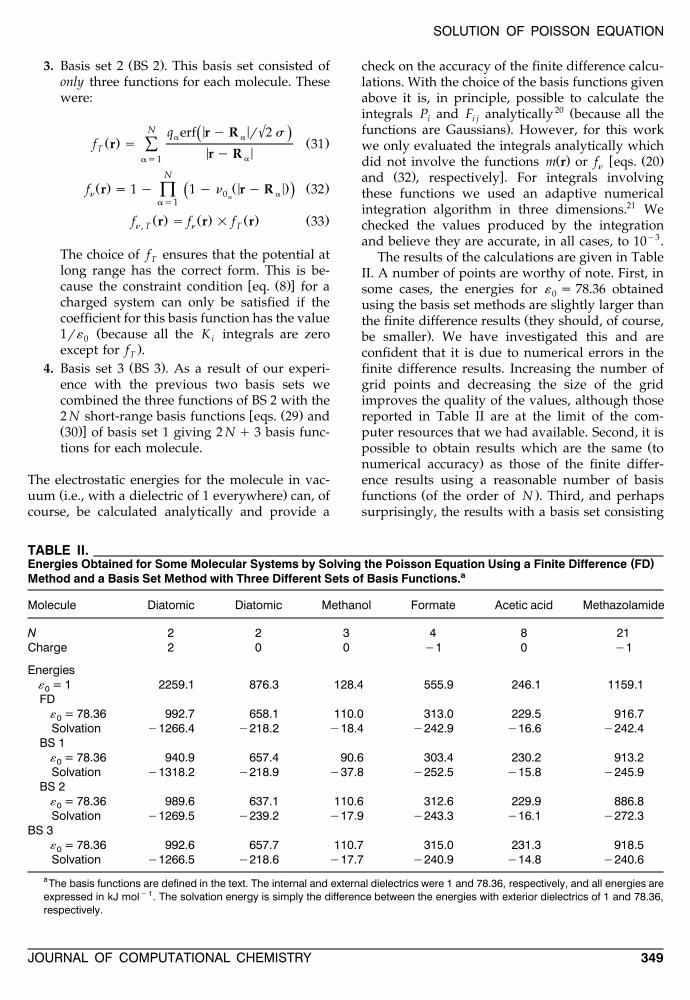
The results of the calculations are given in TableII. A number of points are worthy of note. First, insome cases, the energies for « s 78.36 obtained0using the basis set methods are slightly larger than
Žthe finite difference results they should, of course,.be smaller . We have investigated this and are
confident that it is due to numerical errors in thefinite difference results. Increasing the number ofgrid points and decreasing the size of the gridimproves the quality of the values, although thosereported in Table II are at the limit of the com-puter resources that we had available. Second, it is
Žpossible to obtain results which are the same to.numerical accuracy as those of the finite differ-
ence results using a reasonable number of basisŽ .functions of the order of N . Third, and perhaps
surprisingly, the results with a basis set consisting
TABLE II.( )Energies Obtained for Some Molecular Systems by Solving the Poisson Equation Using a Finite Difference FD
aMethod and a Basis Set Method with Three Different Sets of Basis Functions.
Molecule Diatomic Diatomic Methanol Formate Acetic acid Methazolamide
N 2 2 3 4 8 21Charge 2 0 0 y1 0 y1
Energies« = 1 2259.1 876.3 128.4 555.9 246.1 1159.10FD
« = 78.36 992.7 658.1 110.0 313.0 229.5 916.70Solvation y1266.4 y218.2 y18.4 y242.9 y16.6 y242.4
BS 1« = 78.36 940.9 657.4 90.6 303.4 230.2 913.20Solvation y1318.2 y218.9 y37.8 y252.5 y15.8 y245.9
BS 2« = 78.36 989.6 637.1 110.6 312.6 229.9 886.80Solvation y1269.5 y239.2 y17.9 y243.3 y16.1 y272.3
BS 3« = 78.36 992.6 657.7 110.7 315.0 231.3 918.50Solvation y1266.5 y218.6 y17.7 y240.9 y14.8 y240.6
aThe basis functions are defined in the text. The internal and external dielectrics were 1 and 78.36, respectively, and all energies areexpressed in kJ moly 1. The solvation energy is simply the difference between the energies with exterior dielectrics of 1 and 78.36,respectively.
JOURNAL OF COMPUTATIONAL CHEMISTRY 349

DAVID AND FIELD
Ž .of only three basis functions per molecule BS 2are only significantly worse than those evaluated
Ž .with the best basis set BS 3 in the case of theneutral diatomic and methazolamide. This raisesthe possibility of being able to find a basis setmethod consisting of a small number of functionsŽ .<N that can be used for calculations on largesystems when the solution of the matrix equations
w Ž .xfor the coefficients eq. 10 with a large numberof basis functions could become a limiting factor.
Conclusions
In this article we have explored the use of abasis set expansion approach for the solution ofthe Poisson equation for small molecules im-mersed in a solvent that is represented by a dielec-tric continuum. We have found that a basis setmethod, with a relatively small number of basis
w Ž .xfunctions ; O N , can give results that are invery good agreement with those obtained by solv-ing the Poisson equation using a finite differencemethod. To make the basis function method practi-cal for application to larger systems we used func-tions of Gaussians to represent the charge distribu-tion of the molecule and the cavity formed by thesolute in the solvent.
The aim of the work presented here has been todemonstrate that the basis set expansion method isfeasible for the solution of the Poisson equation.As a result we have not focused on issues such asthe computer time taken by the algorithm or bythe optimization of the program, although the cal-culations reported here, even with the numericalintegration algorithm, took about the same amountof time as the finite difference methods. We expectthese times to be significantly reduced when moreadvanced methods are employed.
The main objective of our future work will be tosee if the method can be competitive with finitedifference methods for applications to proteins andother macromolecular systems. In particular wewill investigate alternative forms for the cavityfunction and the basis functions and look at waysin which the integrals involving these functionscan be efficiently evaluated. We will also investi-
gate the parameterization of the basis sets. Allthese aspects will be discussed in another publica-tion.
Acknowledgments
The authors thank the Institut de Biologie Struc-turale—Jean-Pierre Ebel, the Commissariat a l’En-`ergie Atomique, and the Centre National de laRecherche Scientifique for support of this work.
References
Ž .1. S. C. Harvey, Prot. Struct. Function Genet., 5, 78 1989 .Ž .2. K. A. Sharp, Curr. Opin. Struct. Biol., 4, 234 1994 .Ž .3. M. K. Gilson, Curr. Opin. Struct. Biol., 5, 216 1995 .Ž .4. B. Honig and A. Nicholls, Science, 268, 1144 1995 .
Ž .5. T. J. You and S. C. Harvey, J. Comput. Chem., 14, 484 1993 .Ž .6. H.-X. Zhou, Biophys. J., 65, 955 1993 .
7. R. Bharadwaj, A. Windemuth, S. Sridharan, B. Honig, andŽ .A. Nicholls, J. Comput. Chem., 16, 898 1995 .
8. M. K. Gilson, J. A. McCammon, and J. D. Madura, J.Ž .Comput. Chem., 16, 1081 1995 .
9. W. J. Hehre, L. Radom, P. v. R. Schleyer, and J. A. Pople,Ab Initio Molecular Orbital Theory, Wiley, New York, 1986.
Ž .10. K. A. Sharp and B. Honig, J. Phys. Chem., 94, 7684 1990 .11. E. S. Reiner and C. J. Radke, J. Chem. Soc. Faraday Trans., 86,
Ž .3901 1990 .12. M. K. Gilson, M. E. Davis, B. A. Luty, and J. A. McCammon,
Ž .J. Phys. Chem., 97, 3591 1993 .13. T.-S. Lee, D. M. York, and W. Yang, J. Chem. Phys., 102,
Ž .7549 1995 .14. K. D. Gibson and H. A. Scheraga, Mol. Phys., 62, 1247
Ž .1987 .Ž .15. J. A. Grant and B. T. Pickup, J. Phys. Chem., 99, 3503 1995 .
16. M. E. Davis, J. D. Madura, B. A. Luty, and J. A. McCam-Ž .mon, Comp. Phys. Commun., 62, 187 1991 .
17. W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 110,Ž .1657 1988 .
18. The QUANTA Molecular Modeling Program, MolecularSimulations Inc.
19. L. David, unpublished work.20. Note that by ‘‘analytic’’ we mean that the integrals can be
expressed in terms of well-known functions even if some ofthese, such as the error function or F , must be calculated0numerically.
21. The NAG Fortran Library, Mark 16, The Numerical Algo-rithms Group Ltd., Oxford, 1993.
VOL. 18, NO. 3350




![EXPLORING UNSTRUCTURED POISSON SOLVERS FOR FDS · Forcing Immersed Boundary Method [2] is used. In every time step it requires ... Both variants are able to scope with unstructured](https://img.pdfslide.us/doc/110x75/60d087a1286ead0c510db316/exploring-unstructured-poisson-solvers-for-fds-forcing-immersed-boundary-method.jpg)