Embed Size (px)
Citation preview

J. Mol. Biol. (1969) 43, 385406
Base-pairing Configurations between Purines and Pyrimidines in the Solid State
III. Crystal and Molecular Structure of two 2: 1 Hydrogen-bonded Complexes, 1-Methylthymine : 9-Ethyl-2,6-diaminopurine
and 1-Methyl+iodouracil : 9-Ethyl-2,64iaminopurine
T. D. SAKORE, HEXRY M. SOBELL, FERN~I)O UZA?
Department of Chemistry, The University of Rochester Rochester, N. Y. 14627, U.S.A.
and Department of Radiation Biology and Biophysics
The University of Rochester School of Meokine and Dentistry Rochester, N.Y. 14620, U.S.A.
AXD
GOPINATH MARTHA
Center for X-ray Crystdbgraphic Research Roswell Park Memorial Institute
Buffalo, N.Y., U.S.A.
(Received 25 March 1969)
9-Ethyl-2,6-diaminopurine forms 1: 2 hydrogen-bonded complexes with I-methyI- thymine and with I-methyl-5-iodouracil, Both structures have been determined by three-dimensional X-ray diffraction methods and refined by block diagonal least squares. The first complex, 9-ethyl-2,6-diaminopurine : 1-methylthymine, exhibits both Watson-Crick and “reversed” Hoogsteen type base pairing con- figurations, while the second complex, 9-ethyl-2,Bdiaminopurine : l-methyl-5 iodouracil, involves both “reversed” Watson-Crick and Hoogsteen type base pairing configurations. These base pairing configurations may reflect differences in crystal lattice forces stabilizing these structures; however, an alternative possibility is that the observed configurations are particularly stable ones which reflect the strong associations between these compounds in solution before co- crystallization. If the latter is true, then the diaminopurine : iodouracil inter- action involves a perturbation not present in the diaminopurine : thymine interaction, and this possibility is discussed in relation to previous structures described in thk series.
1. Introduction AB part of a general program aimed at deepening our understanding of the inter- molecular forces between purinee and pyrimidines which are responsible for their base pairing specificity in nucleic acid structures, we have extended the technique of purine-pyrimidine co-crystallization to study the base pairing properties of several synthetic baee analogue compounds. The previous papers in this series have described complexes between 9-ethyladenine : l-methyl-5-iodouracil and g-ethyl-S-
t Present address: Centro Di Studio Per La Strutturistica Cbimica, Del Consiglio Nazionale Delle Ricerehe, Istituto di C&mica F armaceutioa, Citta Universitaria, Rome, Italy.
336

386 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND Cr. KARTHA
bromoadenine : l-methyl-5-bromouracil (Sakore, Tavale & Sobell, 1969; Taval~, Sakore & Sobell, 1969). These structures both display the “reversed” Watson-Crick type base pairing configuration, and it is of interest to understand whether this
interaction reflects some perturbation in the adenine-iodouracil and adenine-bromo- uracil pairing not present in the adenine: thymine pair. This paper will demonstrate that 9-ethyl-2,6-diaminopurine (9-ethyl-2-aminoadenine) forms crystalline complexes with l-methyl-5-iodouracil and with I-methylthymine, the first involving “reversed” Watson-Crick and Hoogsteen type base pairing configurations, the second involving Watson-Crick and “reversed” Hoogsteen configurations. These different base pairing configurations have suggested that halogen substitution on the uracil ring may affect the stabilities of different types of purine-pyrimidine hydrogen bonded configurations, a possibility explored further in this and in the following paper. A preliminary com- munication describing a portion of this work has appeared (Labana & Sobell, 1967).
2. Materials and Methods 9-Ethyl-2,6-diaminopurine, I-methyl-5-iodouracil, and I-methylthymine were synthe-
sized by the Cycle Chemical Company, Los Angeles, California. Initial samples of O-ethyl- 2,6-diaminopurine were kindly supplied by Dr John Montgomery, Southern Research Institute, Birmingham, Ala. 1: 2 mixtures of these compounds were dissolved in 9576 ethanol and the solutions were allowed to evaporate slowly at room temperature. Both 9-ethyl-2,6-diaminopurine : I-methylthymine and 9-ethyl-2,6-diaminopurine : I-methyl-5- iodouracil complexes form plate-like crystals, and ultraviolet absorption spectra from solutions obtained from washed single crystals suggested 1:2 complexes. The same crystals could be obtained from equimolar mixtures of these compounds.
The unit cell constants and space groups for both structures were obtained initially from 30” precession photographs using CuK, radiation, and were then refined by several high-angle reflections measured on a Picker 4-circle computer controlled (PDP-81) S-ray diffractometer. The final crystal data for these structures are as follows:
9-ethyL2,6-diarninopurine : I-methylthymine. Hz0 a = 10.216 & 0.012 a b = 26.467 + 0*031 ii c = 16.235 + 0.013 A 0, = 50@ * O.1° /3 = 35.6” f O.1° y = 27-9O + O.1° Volume = 1139.8 A3 Spaoe group: PI Density (observed) = 1.39 g/cc. Density (calculated) = 1.387 g/cc
9-ethyl-2,6-dimninopurine : I-mthyl-5-iodouracil a = 9.167 & 0.011 A b = 12.637 rf: 0.013 d c = 20.463 + 0,020 A 6 = 96.6 + 0.6” Volume = 2355.1 A3 Space group : P2& Density (observed) = I.90 g/cc. Density (calculated) = 1.92 g/cc
The first structure contains two asymmetric units per unit cell, each asymmetric unit consisting of a 1: 2 9-ethyl-2,6-diaminopurine : I-methylthymine trimer complex and a water molecule. Adjacent trimers are related by inversion symmetry. The second structure contains four asymmetric units per unit cell, each asymmetric unit containing a 1:2
DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 387
9-ethyl-2,6-diaminopurine : I-methyl-Godouracil trimer complex. Adjacent trimers are related by either glide plane or 2-fold screw symmetry, as well as by inversion symmetry.
Intensity data for both structures were collected on a Picker 4.circle computer con- trolled X-ray diffractometer with nickel filtered CuK, radiation using the 20 scan teclr- nique. 3458 reflections were recorded to a maximum 28 angle of 130’ in the diaminopurine--- thymine complex, which represents 75% of the data theoretically accessible in the copper sphere. Of these, 2963 were recorded as non-zero. 2982 reflections were recorded to a maximum 20 angle of 110” in the diaminopurine-iodouracil complex, representing 65’1, of the unique data theoretically accessible. Of these, 2855 were non-zero. The intensitios were corrected with the Lorentz-polarization factor. No absorption corrections wcro applied. Computations were done on an IBM 360-65 high-speed digital computer.
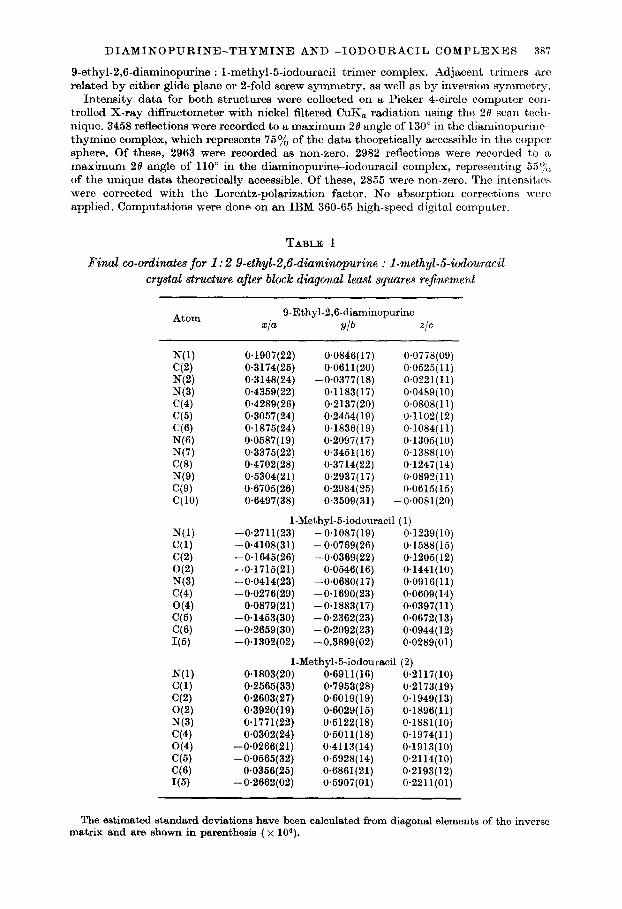
TABLE 1
Final co-ordinates for 1: 2 9.ethyl-2,6-diaminopurine : I-methyL5iodouracil crystal structure after block diagonal least squares refinement
Atom 9.Ethyl-2,6-diaminopurine
x/a ylb Z/C
N(l) cc4 N(2) N(3) C(4) C(5) ‘36) N(6) N(7) C(8) N(9) C(9) C(lO)
N(l) C(1) C(2) O(2) N(3) C(4) O(4) C(5) (36) I(5)
N(l) (31) C(2) O(2) N(3) C(4) O(4) (35) ‘3% I(5)
0.1907(22) 0.0846( 17) 0*0778(09) 0.3174(25) 0.0611(20) 0.0525(11) 0.3148(24) -0~0377(18) 0.0221(11) 0.4359(22) 0.1183(17) 0.0489(10) O-4289(26) 0.2137(20) O.OSOS( 11) 0.3057(24) 0.2454( 19) 0.1102(12) 0.1875(24) 0.1836(19) 0.1084(11) 0.0587(19) 0.2097(17) 0.1305(10) 0.3375(22) 0.3451(16) 0.1388(10) 0.4702(28) 0.3714(22) 0.1247(14) 0.5304(21) 0.2937( 17) 0.0892(11) 0.6705(26) 0.2984(25) 0.0615(15) 0.6497(38) 0.3509(31) -0.0081(20)
1-Methyl-5-iodouracil (1) -0*2711(23) -0.1087(19) 0.1239(10) -0.4108(31) -0.0769(26) 0.1588(15) -0*1645(26) -0.0369(22) 0.1205(12) -0.1715(21) 0.0546( 16) 0.1441(10) -0*0414(23) -0.0680( 17) 0.0916(11) -0*0276(29) -0.1690(23) 0.0609( 14)
0*0879(21) -0.1883(17) 0.0397( 11) -0.1453(30) -0.2362(23) 0.0672(13) -0.2659(30) -0.2092(23) 0*0944(12) -0.1302(02) -0.3899(02) 0*0289(01)
1-Methyl-5-iodouracil (2) 0*1803(20) O-6911(16) 0.2117(10) O-2565(33) 0.7953(28) 0.2173(19) 0*2603(27) 0.6019(19) 0.1949(13) 0*3920( 19) 0.6029(15) 0.1896(11) 0*1771(22) 0*6122(18) 0.1881(10) 0.0302(24) 0*5011(18) 0.1974(11)
-0*0266(21) 0.4113(14) 0.1913(10) -0.0565(32) 0.5928(14) 0.2114(10)
0.0356(25) O-6861(21) 0.2193(12) -0*2662(02) 0.5907(01) 0.2211(01)
The estimated standard deviations have been calculated from diagonal elements of the inverse matrix and are shown in parenthesis ( x 104).

388 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
3. Results
(a) 9-Ethyl-2,6-diaminopurine:l-methyl-Siodouracil
This structure was determined by the heavy-atom technique. An unsharpened Patterson function was calculated and this revealed interatomic vectors between iodine atoms both within the asymmetric unit, and between symmetry related mole- cules. A Fourier synthesis phased on the iodine atomic positions revealed the com- plete structure, and this was then refined isotropically by block diagonal least squares to a residual of 14.5%. The iodine atomic positions were next refined anisotropically,
TABLE 2
Anisotropic temperature prameters for the 9-ethyL2,6diaminopurine : I-methyl-5-iodouracil cystul structure after block diagonal least-squares re$nement
Atom B 11 B aa
9-Ethyl-2,6-diaminopurinc B 33 B 12 B 13 B23
N(l) 0.0083(28) C(2) O-0033(29) N(2) 0*0095(32) N(3) 0.0075(28) G(4) O-0068(33) C(5) 0.0014(27) (3’5) 0.0037(29) NW 0.0020(23) N(7) 0*0075(28) W3) 0*0063(36) NW 0.0041(26) C(Q) 0~0016(31) cw 0.0120(61)
NW C(l) C(2) O(2) N(3) C(4) O(4) (76) (76) I(5)
0*0068(28) O-0072( 19) 0.0078(40) 0*0068(26) 0.0041(32) 0*0066(21) 0.0103(27) 0.0063( 16) 0*0074(29) 0*0044(16) 0*0071(37) 0*0060(22) 0.0097(27) 0~0068(16) O-0089(39) 0*0063(22) 0*0091(29) 0*0029(11) 0*0206(04) 0.0047(01)
NW C(l) C(2) O(2) N(3) C(4) O(4) (35) C(6) I(6)
O-0044(26) 0.0073(43) O-0066(33) 0.0061(24) 0.0079(28) 0.0044(30) 0.0111(27) 0*0276(64) 0~0031(30) 0*0061(02)
0.0047( 16) 0*0049(19) O-0048(16) 0*00.56(17) 0*0037( 18) 0.0036(18) 0*0036( 18) 0*0066( 17) 0.0034(14) 0*0043(21) 0~0061(16) 0*0078(26) 0*0090(33)
O-0047(16) 0*0068(28) 0~0018(16) O-0047( 14) 0.0061(17) O-0028(17) 0~0041(13)
-0~0040(11) 0*0053(21) 0~0047(01)
O~OOll(O6) 0*0017(34) 0*0007(06) -0*0025(39) 0.0024(07) 0~0000(37) 0~0016(06) -0.0018(36) 0~0008(06) 0*0038(40) 0*0014(07) -0,0009(36) 0~0008(06) O-0039(37) 0~0018(06) 0.0022(32) 0.0017(06) -0.0011(32) 0~0024(09) 0~0010(44) 0.0023(06) -0*0016(33) 0*0030( 10) O.OOOl(46) 0.0044( 14) -0*0024(68)
I-Methyl-5-iodouracil (1) 0.0017(06) -0~0006(38) 0*0026(09) -0*0046(62) 0~0014(07) 0.0022(42) 0.0031(06) -0.0076(34) 0.0021(06) 0.0002(35) O.OOZO(OS) 0*0049(47) 0*0036(07) 0.0008(36) 0~0013(07) -0*0012(48) 0*0013(07) -0*0007(11) 0~0022(01) -0*0030(04)
I-Methyl-5-iodouracil (2) 0*0020(06) -O-0067(32) 0.0061(14) -0*0081(67) 0~0017(07) -0.0019(39) 0.0047(08) -0*0024(30)
-0~0012(05) -0*0010(36) O*OOlO(OS) 0.0009(37) 0*0030(06) 0*0010(31) 0~0001(05) 0.0009(37) 0.0012(07) -0*0073(41) 0~0022(01) -0*0002(02)
0.0017(19) -0.0006(21)
0.0018(23) 0.0023(20)
-0.0008(23) -0.0004(22) -0~0009(21)
0*0014(18) -O*OOOS(ZO)
0*0023(28) 0*0011(20) 0.0008(27) 0*0063(43)
0*0016(21) -0~0001(31)
0*0006(24) 0*0046(21) O-0022(22) 0*0002(28) 0*0033(22)
-0*0030(27) -0.0015(17)
O.OOOS(OZ)
0*0018(19) 0*0014(39)
-O-0002(25) O-0058(22) 0~0020(20)
-0~0001(22) 0.0053(21) 0.0020(27) 0*0004(23) 0*0024(02)
-0~0011(15) 0*0005(18)
-0.0044(17) -0.0017(16) -O.OOOO(lS) -0~0003(18)
0*0006(17) -0*0017(16) -0~0022(15) -0.0032(22) -0.0016(17) -0*0047(26)
0.0016(36)
0*0012(17) 0+0018(26) 0.0004(20)
-0~0016(16) -0*0002(16)
0*0003(22) -0.0029(18)
O-0017(21) 0.0023(16)
-0*0004(01)
-0.0022(16) -0*0029(33) -0~0012(18) -O-0039(17) -0*0023(16) -0~0001(17) -0~0005(15)
0~0002(12) -0~0012(19) -0~0002(01)
Estimated standard deviations arc shown in parenthesis ( x IO’). The temperature parameters shown arc coefficients in the expression,
T = exp ( - ULha + /La@ + t%P + BI& + B&J + Aa&) 1.
DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 389
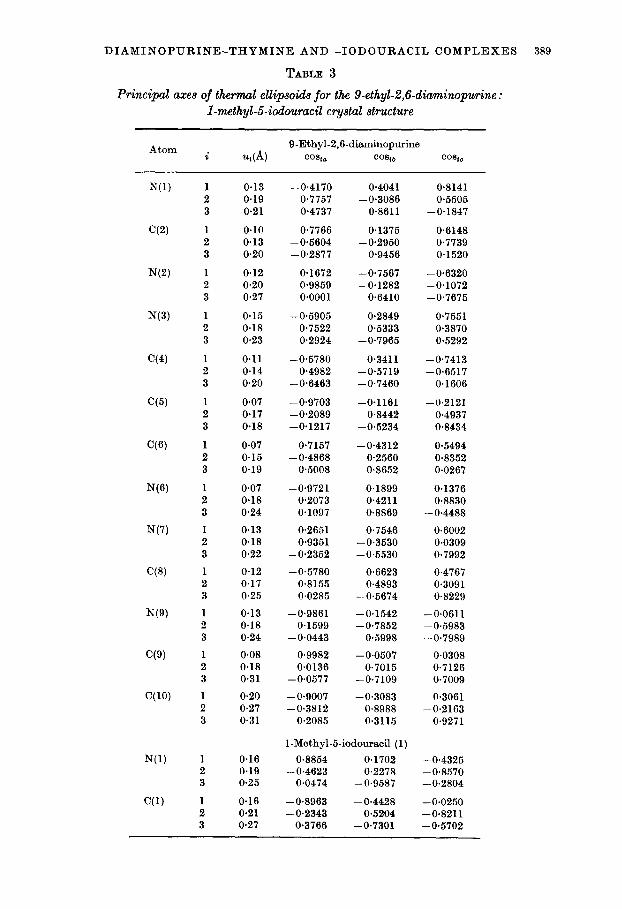
TABLE 3
Principal axes of thermal ellipsoids for the 9-ethyl-2,6-diuminopurine : I-methyl-5-iodouracil cystal structure
Atom Q-Ethyl-2,6diaminopurine i ~(4 two co*iiJ co*ic
N(1)
C(2)
N(2)
N(3)
C(4)
C(5)
N(7)
N(Q)
C(Q)
C(10)
N(1)
C(1)
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
0.13 0.19 0.21
0.10 0.13 0.20
0.12 0.20 0.27
0.15 0.18 0.23
0.11 0.14 0.20
0.07 0.17 0.18
0.07 0.15 0.19
0.07 0.18 0.24
0.13 0.18 0.22
0.12 0.17 0.25
0.13 0.18 0.24
0.08 0.18 0.31
0.20 0.27 0.31
0.16 0.19 0.25
0.16 o-21 0.27
-0.4170 0.7757 0.4737
0.7766 -0.5604 -0.2877
0.1672 -0.9859
0~0001
-0*5905 0.7522 0.2924
-0.5780 0.4982
-0.6463
-0.9703 -0.2089 -0.1217
0.7157 - 0.4868
0*5008
-0.9721 0.2073 0.1097
0.2651 0.9351
-0.2352
-0.5780 0.8155 0.0285
-0*9861 0.1599
- 0.0443
O-9982 0.0136
-0.0577
-0.9007 -0.3812
0.2085
0.4041 -0.3086
0.8611
0.1375 -0.2950
0.9456
-0.7567 -0.1282
0.6410
0.2849 0.5333
-0.7965
0.3411 -0.5719 -0.7460
-0*1161 0.8442
-0.5234
-0.4312 0.2560 0.8652
0.1899 0.4211 0.8869
0.7546 -0.3530 -0.5530
0.6623 0.4893
-0.5674
-0.1542 -0.7852
0.5998
- 0.0507 0.7015
-0.7109
-0.3083 0.8988 0.3115
0.6148 0.7739 0.1520
-0.6320 -0.1072 -0.7675
0.7551 0.3870 0.5292
-0.7413 -0.6517
0.1606
-0.2121 0.4937 O-8434
0.5494 0.8352 0.0267
0.1376 0.8830
-0.4488
0.6002 0.0309 0.7992
0.4767 0.3091 0.8229
-0.0611 -0.5983 -0.7989
0.0308 0.7126 o-7009
0.3061 -0.2163
0.9271
1-Methyl-5-iodouracil (1)
0.8854 0.1702 -0.4325 -0.4623 0.2278 -0.8570
0.0474 -0.9587 -0.2804
-0.8963 -0.4428 -0.0250 -0.2343 0.5204 -0.8211
0.3766 -0.7301 -0.5702
390 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
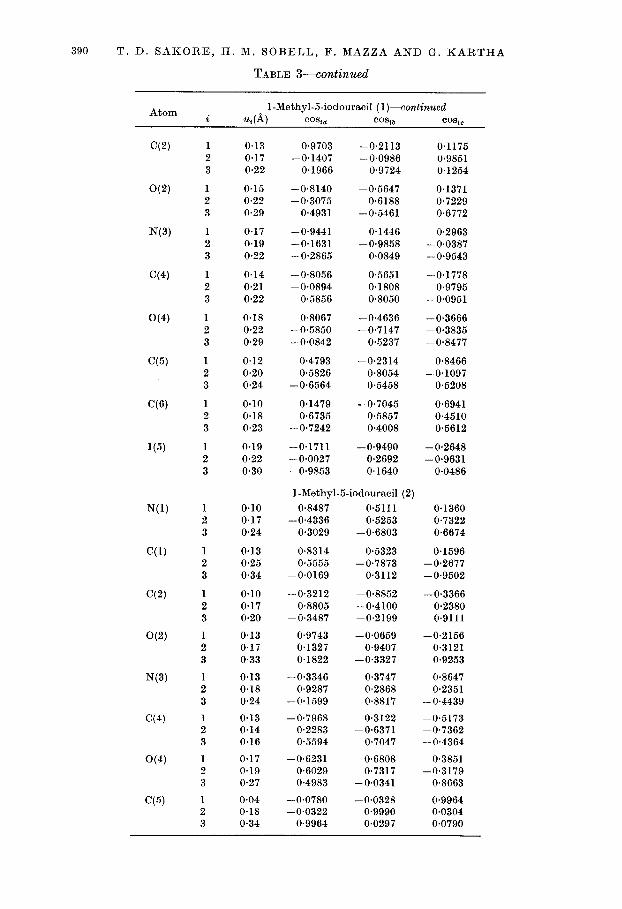
TABLE S-continued
Atom 1-Methyl-5-iodouracil (l)-continued i 4) CO% CO% b CO%0
C(2)
O(2)
N(3)
C(4)
O(4)
C(5)
C(6)
I(5)
N(l)
C(l)
w
O(2)
N(3)
C(4)
O(4)
C(5)
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
0.13 0.9703 -0.2113 0.1175 0.17 -0.1407 -0.0986 0.9851 0.22 0.1966 0.9724 0.1254
0.15 -0.8140 -0.5647 0.1371 0.22 -0.3075 0*6188 0.7229 0.29 0.4931 -0.5461 0.6772
0.17 -0.9441 0.1446 0.2963 0.19 -0.1631 -0.9858 -0.0387 0.22 -0.2865 0.0849 -0.9543
0.14 -0.8056 0.5651 -0.1778 0.21 -0.0894 0~1808 0.9795 0.22 0.5856 0.8050 -0.0951
0.18 0.8067 -0.4636 -0.3666 0.22 -0.5850 -0.7147 -0.3835 0.29 - 0.0842 0.5237 -0.8477
0.12 0.4793 -0.2314 0.8466 0.20 0.5826 0.8054 -0.1097 0.24 -0.6564 0.5458 0.5208
o-10 0.1479 -0.7045 0.6941 0.18 0.6735 0.5857 0.4510 0.23 -0.7242 0.4008 0.5612
0.19 -0.1711 -0.9490 -0.2648 0.22 -0.0027 0.2692 -0.9631 0.30 -0.9853 0.1640 0.0486
0.10 0.17 0.24
I-Methyl-5-iodouracil (2) 0.8487 0.5111
-0.4336 0.5253 0.3029 -0.6803
0.8314 0.5323 0.5555 -0.7873
-0.0169 0.3112
-0.3212 -0.8852 0.8805 -0.4100
-0.3487 -0.2199
0.9743 -0.0659 0.1327 0.9407 0.1822 -0.3327
-0.3346 0.3747 0.9287 0.2868
-0.1599 0.8817
-0.7968 0.3122 0.2283 -0.6371 0.5594 0.7047
-0.6231 0.6808 0.6029 0.7317 0.4983 -0.0341
-0.0780 -0.0328 -0.0322 0.9990
0.9964 0.0297
0.1360 0.7322 0.6674
0.13 0.25 0.34
0.1596 -0.2677 -0.9502
0.10 0.17 0.20
-0.3366 0.2380 0.9111
0.13 0.17 0.33
-0.2156 0.3121 0.9253
0.13 0.18 0.24
0.8647 0.2351
-0.4439
0.13 0.14 0.16
-0.5173 -0.7362 -0.4364
0.17 0.19 0.27
0.3851 -0.3179
0.8663
0.04 0.18 0.34
0.9964 0.0304 0.0790

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 391
TABLE $-continued
Atom
(76)
I(5)
I-Methyl-5-iodouracil (2)--con&nued %(K) CO% CO% CO%
0.041 0.8580 0.4702 0.2068 0.16 -0,284O 0.1008 0.9532 0.23 -0.4272 0.8769 -0.2204
0.13 0.9765 0.0127 -0.2153 0.19 0.0221 0.9871 0.1585 0.22 0.2146 -0.1695 0.9636
u, corresponds to the root-mean-squared displacement along the ith principal axis of the ellipsoid, and cost., cosu, and COB,,, the direction cosines which the ith axis makes with respect to the crystallographic axes, a, b and c*.
ash/3
OL1 2 3 45ii
FIG. 1. The O-ethyl-2,6-diaminopurine : I-methyl-S-iodouracil crystal structure as viewed down the c-axis. For clarity, only half the unit cell is shown. Hydrogen-bonding contacts are indicated with dashed lines. Dots indicate centers of symmetry.
reducing the residual to 12*4o/o. At this stage, a difference Fourier synthesis was calculated and an attempt was made to locate hydrogen atoms in the structure. Although several peaks were found in expected hydrogen positions, numerous other background peaks were found and therefore no attempt was made in assigning hydrogen atomic positions. The complete structure, with the exception of hydrogen atoms, was then refined anisotropically, varying individual light atom and iodine temperature parameters. The final unweighted residual is ll*S%, while the weighted residual is 13*2%, reflections being weighted according to a weighing scheme based
26

392 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
on counting statistics, as described by Stout & Jensen, 1968. Tables 1, 2 and 3 show the final co-ordinates and temperature parameters obtained from the structure analysist.
Figure 1 shows a portion of the crystal structure as viewed down the c-axis. One 2 : 1 l-methyl-5-iodouracil : 9-ethyl-2,6-diaminopurine trimer is shown in dark outline, while symmetry related molecules (related by inversion symmetry) are further behind and are shown with lighter lines. It is seen that the diaminopurine and iodouracil bases form a planar complex, joined by hydrogen bonds in which two types of base pairing configurations are seen. One configuration involves an N-H . . , N bond be- tween the imidazole N(7) of diaminopurine and N(3) of iodouracil (2.83 A), and an N-H. . . 0 bond involving the N(6) amino group on diaminopurine interacting with the O(4) carbonyl oxygen of iodouracil (2.98 A). This base pairing configuration is very similar to that described previously between 9-ethyladenine : 1-methyl-5-iodo- uracil (Sakore et al., 1969) and has been seen in other adenine-thymine (or uracil) crystalline complexes (Hoogsteen, 1959,1963; Mathews & Rich, 1964; Tomita, Katz & Rich, 1967; Baklagine, Volkenshtein & Kondrashev, 1966). The other configuration involves three hydrogen bonds on the N(1) diaminopurine side, an N-H . . . N bond between N(3) of uracil and N(1) of diaminopurine (2.91 A), and two N-H . . . 0 bonds connecting the two amino groups of diaminopurine with the carbonyl oxygen atoms on the second iodouracil residue (2.92 A, 2.87 A). This base pairing configuration is analogous to that occurring in the 9-ethyladenine : 1-methyl-5-iodouracil and g-ethyl- B-bromoadenine: 1-methyl-5-bromouracil structures, except that an additional hydro- gen bond connects the bases (Sakore et al., 1969; Tavale et aE., 1969). The over-all crystal structure consists of trimer base pairs hydrogen bonded together between diaminopurine residues across centers of symmetry. This planar hexamer structure then packs together to form the angulated structure shown in Figure 2, which depicts
FICA 2. A view of the 9-ethyl-2,6-diaminopurine : 1-methyl-5-iodouracil crystal structure down the b-axis. The molecules drawn with heavier lines are related to those drawn with lighter lines by 2-fold screw symmetry.
t The observed and calculated structure factors for this and for the following structure have been microfilmed and stored at ASIS of Auxiliary Publication Service, 22 West 34th Street, New York, N.Y., U.S.A. under document number 00437.

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 393
the structure viewed down the b-axis. In this projection, it is noticed that the trimer itself is not completely planar. The diaminopurine-ioclouracil interaction on the N(7) imiclazole side involves a dihedral angle of l&2”, the largest deviation from planarity yet observed in complexes of this type, and probably reflects the lattice forces and packing requirements in the crystal structure. There is little direct stacking between adjacent trimer base pairs, except along the b-axis, where the bases stack acutely upon one another.
Figure 3 shows a schematic diagram of the trimer complex with the bond distances and angles as obtained from this analysis. The estimated standard deviations are
Fm. 3. A schematic diagram of the 9-ethyl-2,6-diaminopurine : 1-methyl-5-iodouracil trimer complex showing bond distances and angles, as well as other contacts of interest. Dashed lines indicate hydrogen bonds. A portion of a symmetry related diaminopurine residue is also shown.
0.05 A for bond distances and 2.3” for bond angles involving light atomst. There is reasonable agreement between the bond distances calculated for this structure and the more accurate values described for the 9-ethyl-2,6-cliaminopurine : l-methylthy- mine structure, described below. The rather large standard deviations of bond lengths and angles in this structure determination is not entirely unexpected, considering
t The standard deviations reported for both structures in this paper have been calculated from co-ordinate standard deviations obtained from diagonal elements of the inverse matrix after block diagonal least-squares refinement.
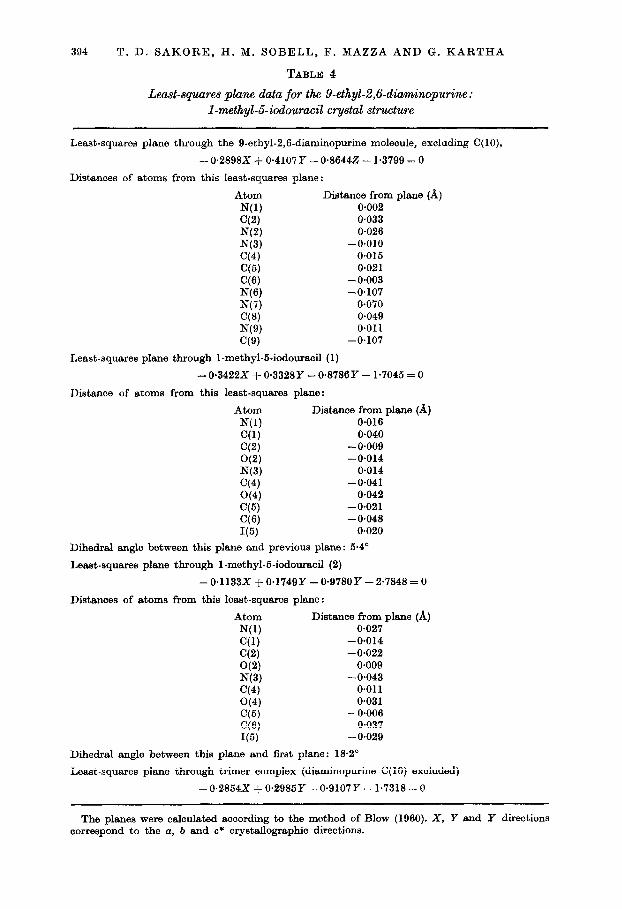
394 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
TABLE 4
Least-squares plane data for the 9-ethyl-2,6-diami~urine : I-methyl-5-iodouraeil crystal structure
Least-squares plane through the 9-ethyl-2,6-diaminopurine molecule, excluding C(lO),
-0.2898X + 0.4107Y - 0.86442 - 1.3799 = 0
Distances of atoms from this least-squares plane:
Atom Distance from plane (A) N(1) 0.002 C(2) 0.033 NC4 0.026 N(3) -0.010 C(4) 0.015 C(5) 0.021 C(6) - 0.003 NW -0.107 N(7) 0.070 ‘78) 0.049 N(9) 0.011 C(9) -0.107
Least-squares plane through I-methyl-5-iodouracil (1)
-0.3422X + 0*3328Y - 0.8786Y - 1.7045 = 0
Distance of atoms from this least-squares plane:
Atom Distance from plane (A) N(1) 0.016 C(1) 0.040 C(2) -0.009 WV -0.014 N(3) 0.014 C(4) -0.041 C(4) 0.042 C(h) -0.021 (3’3) -0.048 I(5) 0.020
Dihedral angle between this plane and previous plane: 6.4”
Least-squares plane through I-methyl-6.iodouraoi (2)
- 0.1133X + 0*1749Y - 0.978OY -2.7848 = 0
Distances of atoms from this least-squares plane:
Atom Distance from plane (A) N(1) 0.027 C(1) -0.014 cc3 -0.022 O(2) O-009 N(3) -0.043 C(4) 0.011 G(4) 0.031 C(5) - 0.006 CC’31 0.037 I(5) - 0.029
Dihedral angle between this plane and first plane: 18.2’
Least-squares plane through trimer complex (diaminopurine C( 10) excluded)
-0.2854X + 0.2985Y - 0.9107Y - 1.7318 = 0
The planes were calculated according to the method of Blow (1960). X, Y and Y directions correspond to the a, b and c* orystallographic directions.
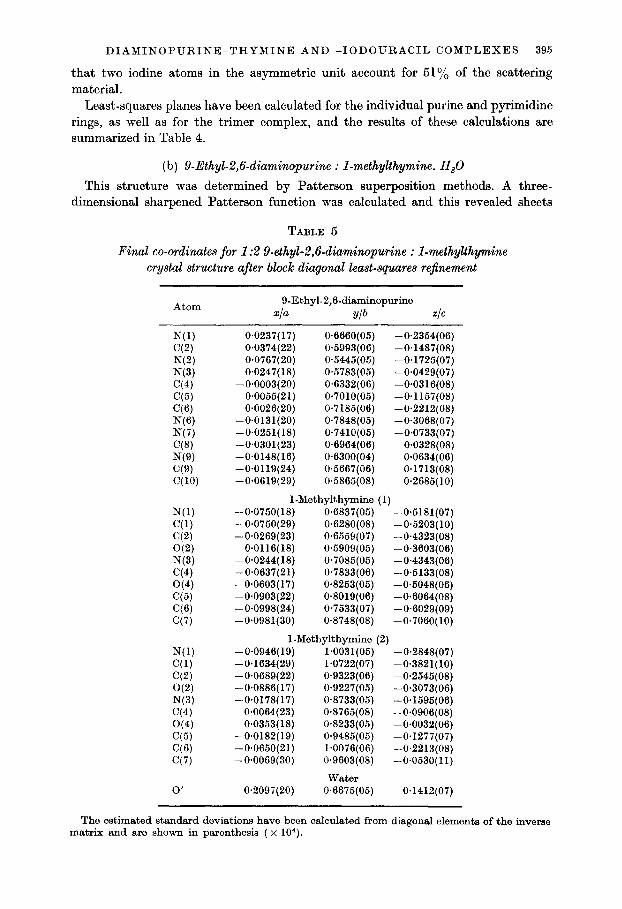
DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 395
that two iodine atoms in the asymmetric unit account for 510/ of the scattering material.
Least-squares planes have been calculated for the individual purine and pyrimidine rings, as well as for the trimer complex, and the results of these calculations are summarized in Table 4.
(b) 9-Ethyl-2,6-diaminopurine : I-methylthymine. H,O
This structure was determined by Patterson superposition methods. A three- dimensional sharpened Patterson function was calculated and this revealed sheets
TABLE 5
Final co-ordinates for 1:2 9-ethyl-2,6-diuminopurine : I-methylthymine crystal structure after block diagonal least-squares re$nement
Atom 9-Ethyl-2,6-diaminopurine
xIa Ylb ZIG
N(1) C(2) N(2) N(3) C(4) C(5) (26) N(6) N(7) (78) NW WV C(10)
N(l) (31) C(2) O(2) N(3) C(4) O(4) C(5) (26) C(7)
N(l) C(l) C(2) O(2) N(3) C(4) O(4) C(5) C(6) C(7)
0’
0.0237(17) 0.6660(05) -0.2354(06) 0.0374(22) 04993(06) -0.1487(08) 0.0767(20) 0.5445(05) -0*1725(07) 0.0247( 18) 0x5783(06) -0*0429(07)
-0.0003(20) 0.6332(06) -0~0316(08) -0.0055(21) 0~7010(05) -0.1157(08)
0.0026(20) 0.7185(06) -0.2212(08) -0.0131(20) 0*7848(05) -0.3068(07) -0.0251(18) 0*7410(05) -0.0733(07) -0.0301(23) 0.6964(06) 0*0328(08) -0.0148(16) 0.6300(04) 0.0634(06) -0.0119(24) 0.5667(06) 0.1713(08) -0.0619(29) 0.5865(08) 0.2685( 10)
1 -Methylthymine (1) -0.0750(18) 0.6837(05) -0.5181(07) -0.0750(29) 0.6280(08) -0.5203(10) -0.0269(23) 0.6559(07) -0.4323(08)
0~0116(18) W5909(05) -0.3603(06) -0.0244( 18) 0*7085(05) -0.4343iOBj -0.0637(21) -0.0603~17j
0.7833fO6) 0.8253(‘05)
-0.5133(08) -04048iO6j
-0.0903(22) 0.8019(06) -0*6064(08) -0.0998(24) O-7533(07) -0.6029(09) -0.0981(30) 0.8748(08) -0*7060(10)
1-Methylthymine (2) -0.0946( 19) 1~0031(06) -0*2848(07) -0*1634(29) 1.0722(07) -0*3821(10) -0~0689(22) 0*9323(06) -0.2545(08) -0.0886(17) 0.9227(05) -0.3073(06) -0.0178(17) 0.8733(05) -0.1595(06)
0.0064(23) 0.8765(08) -0.0906(08) 0*0353(18) 0.8233(05) -0.0032(06)
-0.0182(19) 0.9485(05) -0+1277(07) -0.0650(21) 1.0076(06) -0.2213(08) -0*0069(30) 0.9603(08) -0~0530(11)
water 0.2097(20) 0.6675(05) 0.1412(07)
The estimated standard deviations have been calculated from diagonal elements of the inverse matrix and are shown in parenthesis ( x 104).
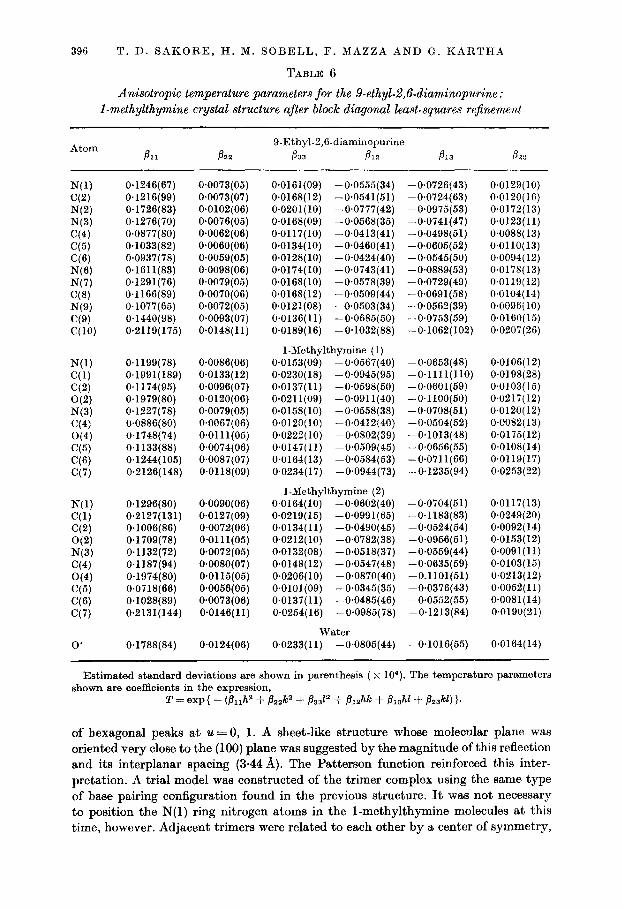
396 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
TABLE 6
Anisotropic temperature parameters for the 9-ethyL2,fMiaminopurine : I-methylthymine crystal structure after block diagonal least-squares rejkement
Atom B 21
9.Ethyl-2,6-diaminopurine 833 B 12
N(1) C(2) N(2) N(3) C(4) C(5) C(6) N(6) N(7) C(8) N(9) C(9) C(W
0.1246(67) 0.1216(99) 0.1726(83) 0.1276(70) 0.0877(80) 0.1033(82) 0.0937(78) 0.1611(83) 0.1291(76) 0.1166(89) 0.1077(65) 0.1440(98) 0.2119(175)
0~0073(05) 0.0073(07) 0.0102(06) 0.0076(05) 0*0062(06) O*OOSO(OS) 0*0069(05) 0.0098(06) 0.0079(05) 0~0070(06) 0*0072(05) 0.0093(07) 0.0148(11)
N(l) 0.1199(78) O~OOSS(O6) C(l) 0.1991(189) 0.0133(12) C(2) 0.1174(95) 0.0096(07) O(2) 0.1979(80) 0~0120(06) N(3) 0.1227(78) 0.0079(05) C(4) 0~0886(80) 0,0067(06) O(4) 0.1748(74) 0~0111(05) C(5) 0.1133(88) 0.0074(06) CC’31 0.1244(105) 0~0087(07) C(7) 0.2126(148) 0.0118(09)
N(1) C(1) C(2) O(2) N(3) C(4) O(4) C(6) ‘36) C(7)
0.1296(80) 0.2127(131) 0.1006(86) 0.1709(78) 0.1132(72) 0.1187(94) 0.1974(80) 0.0718(66) 0.1028(89) 0.2131(144)
0.0090(06) 0.0127(09) 0.0072(06) 0~0111(05) 0.0072(05) 0.0080(07) 0~0115(05) 0~0056(05) 0.0073(06) 0.0146(11)
0’ 0*1788(84) 0.0124(06)
0.0161(09) -0*0555(34) 0.0168(12) -0*0541(51) 0~0201(10) -0*0777(42) 0*0168(09) -0.0568(35) 0~0117(10) -0.0413(41) 0.0134(10) -0*0460(41) 0*0128(10) -0*0424(40) 0.0174(10) -0*0743(41) 0.0168( 10) -0.0578(39) 0.0168(12) -0.0509(44) 0~0121(08) -0.0503(34) 0.0136(11) -0~0685(50) 0.0189( 16) -0.1032(88)
1-Methylthymine (1) 0.0153(09) -0*0667(40) 0.0230( 18) -0*0945(95) 0*0137(11) -0~0598(50) 0*0211(09) -0~0911(40) 0.0158(10) -0.0558(38) 0~0120(10) -0~0412(40) 0.0222( 10) -0+0802(39) 0.0147(11) -0*0509(45) 0.0164( 13) -0*0584(53) 0.0234( 17) -0*0944( 73)
I-Methylthymine (2) 0.0164(10) -0.0602(40) 0.0219(16) -0*0991(65) 0.0134(11) -0*0490(45) 0.0212(10) -0,0782(38) 0.0132(08) -0*0618(37) 0.0148(12) -0.0547(48) 0.0206(10) -0*0870(40) 0~0101(09) -0.0345(35) 0.0137(11) -0.0485(46) 0.0254( 16) -0.0985(78)
water 0.0233( 11) -0~0805(44)
-0.0726143) -0.0724(63) -0.0975(53) -0.0741(47) -0.0498(51) -0.0605(52) - 0.0545( 50) -0.0889(53) -O-0729(49) -0.0691(58) -0*0662(39) -0.0763(59) -0.1062(102)
-0.0653(48) -0~1111(110) -0.0601(59) -0~1100(50) -0~0708(51) -0~0504(52) -0.1013(48) -0.0656(55) -0*0711(66) -0*1235(94)
-0~0704(51) -0.1183(83) -0.0524(54) -0.0956(51) -O-0559(44) -0.0635(59) -0.1101(51) -0.0376(43) -0.0552(55) -0.1213(84)
-0.1016(65)
0.0129(10) O.OlZO( 16) 0.0172(13) 0.0123(11) 0~0088( 13) 0~0110(13) 0.0094( 12) 0.0178(13) 0.0119(12) 0.0104(14) 0.0096( 10) O.OlSO( 15) 0.0207(26)
0.0106(12) 0.0198(28) 0.0103(15) 0.0217(12) 0.0120(12) 0.0082( 13) 0.0175(12) 0.0108(14) 0.0119(17) 0.0253(22)
0.0117(13) 0.0249(20) 0.0092( 14) 0.0153( 12) 0~0091(11) 0.0103(15) 0.0213(12) 0.0052(11) 0~0081(14) 0.0190(21)
0.0164(14)
Estimated standard deviations are shown in parenthesis ( x 104). The temperature parameters shown are coefficients in the expression,
of hexagonal peaks at u = 0, 1. A sheet-like structure whose molecular plane was oriented very close to the (100) plane was suggested by the magnitude of this reflection and its interplanar spacing (344 A). The Patterson function reinforced this inter- pretation. A trial model was constructed of the trimer complex using the same type of base pairing configuration found in the previous structure. It was not necessary to position the N(1) ring nitrogen atoms in the 1-methylthymine molecules at this time, however. Adjacent trimers were related to each other by a center of symmetry,
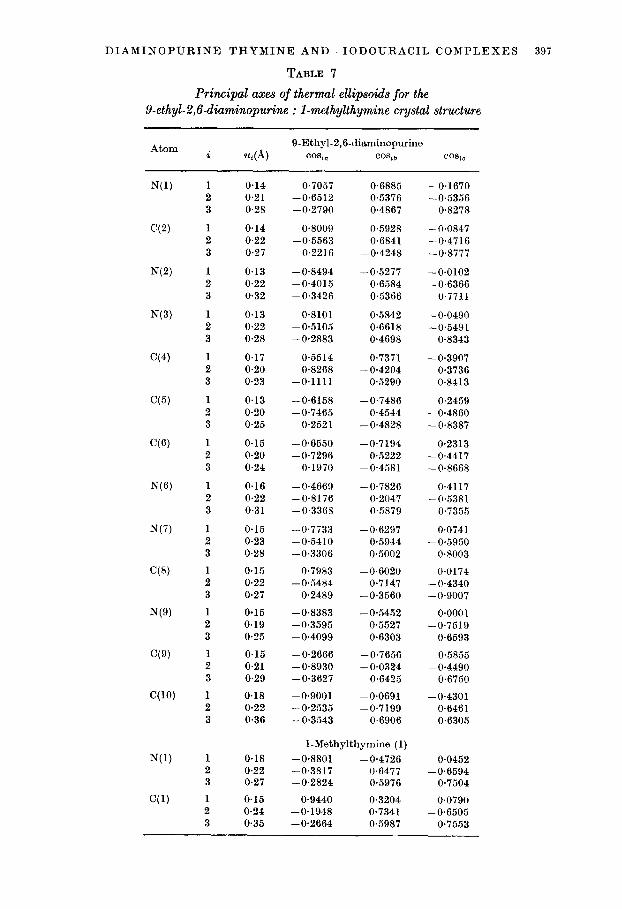
DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 397
TABLE 7
Principal axes of thermal ellipsoids for the 9-ethyl-2,6-diaminopurine : l-methylthymine crystal structure
Atom 9-Ethyl-2,&diaminopwine i u,(A) CO8j, co%3 CO&c
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
0.14 0.7057 0.6885 -0.1670 0.21 -0.6512 0.5376 -0.5356 0.28 -0.2790 0.4867 0.8278
0.14 0.8009 0.5928 -0.0847 0.22 -0.5563 0.6841 -0.4716 0.27 0.2216 -0.4248 -0.8777
0.13 -0.8494 -0.5277 -0.0102 0.22 -0.4015 0.6584 -0.6366 0.32 -0.3426 0.5366 0.7711
0.13 0.8101 0.5842 -0.0490 0.22 -0.5105 0.6618 -0.5491 0.28 -0.2883 0.4698 0.8343
0.17 0.5514 0.7371 -0.3907 0.20 0.8268 -0.4204 0.3736 0.23 -0.1111 0.5290 0.8413
0.13 -0.6158 -0.7486 0.2459 0.20 -0.7465 0.4544 -0.4860 0.25 0.2521 -0.4828 -0.8387
0.15 -0.6550 -0.7194 0.2313 0.20 -0.7296 0.5222 -0.4417 0.24 0.1970 -0.4581 -0.8668
0.16 -0.4669 -0.7826 0.4117 0.22 -0.8176 0.2047 -0.5381 0.31 -0.3368 0.5879 0.7355
0.15 0.23 0.28
0.15 0.22 0.27
0.15 0.19 0.25
0.15 0.21 0.29
0.18 0.22 0.36
-0.7733 -0.6297 -0.5410 0.5944 -0.3306 0.5002
-0.7983 -0.6020 -0.5484 0.7147
0.2489 -0.3560
-0.8383 -0.5452 -0.3595 0.5527 -0.4099 0.6303
-0.2666 -0.7656 -0.8930 -0.0824 -0.3627 0.6425
-0*9001 -0.0691 -0.2535 -0.7199 -0.3543 0.6906
0.0741 -0.5950
0.8003
0.0174 -0.4340 -0.9007
0~0001 -0.7519
0.6593
0.5855 -0.4490
0.6750
-0.4301 0.6461 0.6305
0.18 0.22 0.27
0.15 0.24 0.35
I-Methylthymine (1)
-0.8801 -0.4726 -0.3817 0.6477 -0.2824 0.59'76
0.9440 0.3204 -0.1948 0.7341 -0.2664 0.5987
0.0452 -0.6594
0.7504
0.0790 -0.6505
0.7553

398 T. D. SAKORE, H. MI. SOBELL, F. MAZZA AND G. KARTHA
TABLE 7-continued
Atom I-Msthylthymino ( l)--continued i 44 COf40 CO%7 CO%
WY
O(2)
N(3)
C(4)
O(4)
C(5)
C(6)
C(7)
N(l)
(71)
C(2)
WI
N(3)
C(4)
O(4)
C(5)
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
1 2 3
0.18 -0.9768 -0.2123 -0.0271 0.22 -0.1023 0.5745 -0.8121 0.27 0.1880 -0.7905 -0.5829
0.18 0.6183 0.7282 -0.2957 0.22 -0.7133 0.3620 -0.6001 0.34 -0.3299 0.5820 0.7432
0.16 -0.8310 -0.5525 0.0648 0.21 -0.4871 0.6665 - 0.5644 0.27 -0.2686 0.5005 0.8229
0.19 -0.1026 0.8347 -0.5410 0.21 -0.9885 -0.1465 - 0.0384 0.23 0.1113 -0.5308 -0.8401
0.17 -0.8725 -0.4872 0.0372 0.24 -0.4192 0.7072 -0.5694 0.33 0.2511 -0.5124 -0.8212
0.18 -0.8025 -0.5944 0.0509 0.20 -0.5167 0.6498 -0.5574 0.26 -0.2983 0.4736 0.8287
0.18 -0.8972 -0.4365 0.0672 0.22 -0.3962 0.7280 -0.5595 0.28 0.1953 -0.5286 -0.8261
0.17 0.3027 0.8410 -0.4485 0.25 -0.8943 0.0880 -0.4387 0.36 -0.3294 0.5339 0.7788
0.20 0.24 0.28
1-Mothylthymine (2)
-0.6910 -0.6883 - 0.6362 0.4340 -0.3432 0.5812
-0.5124 -0.7577 -0.8053 0.2604 -0.2983 0.5984
-0.6745 -0.6469 -0.6325 0.2574 -0.3810 0.7180
-0.9050 -0.4147 -0.2738 0.7387 -0.3257 0.5314
-0.6557 -0.7388 - 0.4002 0.1655 - 0.6402 0.6533
-0.7175 -0.6803 -0.5564 0.4301 -0.4192 0.5935
-0.0691 -0.8299 -0.8857 -0.2044 -0.4591 0.5191
-0.8574 -0.5047 -0.3116 0.3530 -0.4097 0.7878
0.2208 -0.6379
0.7378
0.16 0.23 0.36
0.4042 -0.5327
0.7436
0.16 0.24 0.25
0.3561 -0.7306
0.5825
0.17 0.25 0.32
-0.0951 -0.6160
0.7820
0.16 0.23 0.27
0.1555 -0.9013
0.4042
0.18 0.23 0.26
0.1499 -0.7110
0.6870
0.19 0.25 0.35
0.5536 -0.4169
0.7209
0.16 0.20 0.21
0.1008 -0.8822
0.4699

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 399
TABLE ‘l-continued
Atom
(36)
C(7)
0’
i
1 2 3
1 2 3
1 2 3
1-Methylthymine (2)-continued Ul(4 CWa CO% CO%
0.14 -0.8833 -0.4688 -0.0041 0.22 -0.3385 0.6439 -0.6861 0.25 -0.3243 0.6047 -0.7274
0.14 0.9502 0.1074 0.2926 0.27 0.0711 0.8394 -0.5388 0.36 -0.3035 0.5323 0.7899
Water 0.26 o*oooo -0.8657 0~5006 0.28 -0.9287 -0.1856 -0.3211 0.33 -0.3709 0.4648 0.8040
u, corresponds to the root-mean-squared displacement along the ith principal axis of the ellipsoid, and COS,~, coslb and COSTS, the direction cosines which the ith axis makes with respect to the crystallographic directions, n, (c* X a) and c*.
forming hydrogen bonds between the aminopurine residues. This hexamer structure was then oriented in the V-U (100) Patterson plane with careful consideration given to the orientation of hexagonal peaks surrounding the origin of the Patterson function. Inter-ring vectors between the six-membered rings in diaminopurine and methyl- thymine could be predicted from the model structure, and heavy peaks corresponding to these were observed in the Patterson function. Further consideration of the Patterson function revealed that this basic trimer structure could explain the majority of the peaks and rings by repeated superposition, and it was decided to calculate structure factors based on this trial model. Preliminary calculations revealed a good fit between the observed and calculated structure factors, and the structure was therefore refined directly by block diagonal least squares using individual iso- tropic temperature factors for the light atoms. At this stage, however, no attempt was made to assign the N(1) ring nitrogen atom positions and these were simply given carbon form factors. The structure refined rapidly to a residual of 22.0%. At this point, a difference Fourier synthesis was calculated and this revealed the position of t,he C(l0) ethyl carbon atom and an additional peak, tentatively assigned as a water molecule. With these atoms included, the residual dropped to lS.lO/,, and to 1694, after several further cycles of isotropic refinement. At this point, the base pairing configurations could be determined unambiguously from the Fourier electron density map, which clearly fitted a “reversed” Hoogsteen structure and a Watson- Crick structure. Additional evidence which favored this assignment, were the iso- tropic temperature factors for the N(1) ring atoms, which had been assigned carbon form factors in the least squares refinement up until this time. These temperature factors were significantly lower than the corresponding C(5) temperature factors (in the Watson-Crick pairing, the temperature factors were 2.2 and 3.5 AZ, respectively; while in the “reversed” Hoogsteen pairing, they were 2.7 and 3-O AZ, respectively), suggesting that the magnitude of the form factor for these atoms was underestimated, i.e. the correct form factor corresponding to a nitrogen atom. It is important to realize, however, that these assignments do not conclusively exclude the possibility that pairing disorder exists, and, even after extensive refinement, we estimate that a pairing disorder of as much as 20% Hoogsteen and 80°& reversed Hoogsteen base

400 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
TABLE 8
Positional parameters for hydrogen atoms in I-methylthymine : g-ethyl- 2,6-diaminopurine crystal structure a.s obtained from d{fference Fourier synthesis
Atom 9-Ethyl-2,6-diaminopurine Xb Yib +
WW Wb) H(W Web) H(8) HW H( 10~) H( lob)
WW H(lb) H(W H(3) H(7a) Wb) H(7c) Jw)
WW Wlb) H(3) H(7a) H(6)
0.0500 0.5100 -0*1000 0.0750 0.5500 -0.2400
-0~0500 0*8300 -0~3000 -0.0370 0.8040 -0.3800 - 0.0330 0.7100 0~0800 -0.0830 0.5400 0.2000 -0.0410 0.5370 0.3200
0~0000 0.6050 0.2600
1 -Methylthymine (1)
-0.1900 0.6750 -0.5700 -0.2700 0.6460 -0.4200 - 0.0830 0.5750 -0.4400
0~0000 0.6930 -0.3600 -0.1670 0~8800 -0.7400 -0.1670 0.8580 -0.6300 -0.2670 0.9500 -0.6700 -0.1300 0.7740 -0.6700
l.Methylthymine (2)
-0.1400 1.1040 -0.3870 - 0.2500 1.0470 -0.3500 - 0.0250 0.8250 -0.1300
0.0170 1~0000 -0~0700 - 0*0500 I.0600 -0.2600
pairing configurations could go unnoticed in this analysis, The structure was then refined using individual anisotropic temperature parameters for all atoms and a Fourier and a difference Fourier synthesis were calculated. This revealed the positions of 21 hydrogen atoms, and the structure was again refined with several cycles of least squares in which anisotropic temperature parameters for carbon, nitrogen and oxygen atoms were allowed to vary while hydrogen atom parameters were kept constant. The final unweighted residual is 10.O”/O, while the weighted residual is 11.5%, reflections being weighted according to the weighting scheme based on counting statistics referred to previously. Tables 5, 6, 7 and 8 show the final co- ordinates and temperature factors which have been obtained from this analysis.
Figure 4 shows the 9-ethyl-2,6-diaminopurine : I-methylthymine crystal structure in the b-c (109) plane. The basic trimer structure is shown with the van der Waal’s radii inscribed, as well as the symmetry related trimer and other surrounding mole- cules. The diaminopurine and thymine bases form a planar complex, joined by hydrogen bonds involving two different base pairing configurations. One configuration involves an N-H . . . N bond between the imidazole N(7) of diaminopurine and N(3) of thymine (2.88 A), and an N-H . . . 0 bond involving the N(6) amino group on diaminopurine interacting with the O(2) carbonyl oxygen on thymine (2.99 A). This base pairing configuration is reversed in its orientation from the preceding structure, and has been seen in numerous other adenine-thymine (or uracil) crystalline com-

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 401
I\ /47* ..\ (O.l.0)
Fm. 4. A van der Waal’s packing diagram of the 9-ethyl-2,6-diaminopurine : 1-methylthymine crystal structure in the (100) plane. Dashed lines indicate hydrogen bonds. 0’ represents the oxygen atom of a water molecule and lies 0.6 A above and below the purine-pyrimidine molecular sheet. Dots show centers of symmetry.
plexes (Haschemeyer & Sobell, 1963,1965; Katz, Tomita & Rich, 1965,1967; Sakore $ Sobell, manuscript in preparation). The second configuration is also similar to the previous structure, except that the purine and pyrimidine rings are reversed in orientation, hydrogen bonding occurring between the O(4) and O(2) methylthymine carbonyl oxygens and the N(6) and N(2) diaminopurine amino groups, respectively (2.99 A, 2.89 A). A third hydrogen bond connects the thymine ring nitrogen, N(3),
FIQ. 5. A view of the 9-ethyl-2,6-diaminopurine : 1-methylthymine crystal structure perpendi- cular to the (100) plane. Small circles indicate water molecule position. Dashed lines indicate probable hydrogen bonding contacts.

402 T. D. SAKORE, H. M. SOBELL, .I?. MAZZA AND G. KARTHA
with the N(1) ring nitrogen on diaminopurine (2.99 A). This configuration is very similar to the Watson-Crick adenine-thymine base pairing configuration, differing only in that a third hydrogen bond is formed between the diaminopurine and thymine bases. As in the previous structure, adjacent trimers are related to each other by a center of symmetry, and are connected by hydrogen bonds between diaminopurine residues. The over-all crystal structure consists of sheets of base pairs which pack
0 c7
n I.52
0 I2 34
Pm. 6. A schemetio diagram of the 9-ethyl-2,6-diaminopurine : I-methylthymine trimer com- plex showing bond distances and angles, aa well aa other oontacts of interest. Dashed lines indicate hydrogen bonds. A portion of a symmetry related diaminopurine residue is also shown.
together in the (100) plane maximizing van der Waal’s contacts. In addition, there exists a water molecule which is intermediate in position between these molecular planes. This is shown in Figure 5, which is a view of the structure perpendicular to the (100) plane showing overlapping molecules from neighboring unit cells. The water molecule forms two hydrogen bonds between molecules in neighboring sheets, A strong hydrogen bond is formed involving the O(4) carbonyl oxygen on the l- methylthymine molecules involved in reversed Hoogsteen base pairing (2.68 A). Another possible hydrogen bond involves interaction with the N(3) ring nitrogen on diaminopurine in the sheet above (3.01 A). These distances correspond to shortened

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 403
TABLE 9
Least-squares plane data for the 9.ethyl-2,6-diaminopurine: I-methylthymine crystal structure
Least-squares plane through the 9-ethyl-2,6-diaminopurine molecule, excluding C( 10).
0.3939x - 0+3093Y - 0~68812 + 1.0364 = 0
Distances of atoms from this least-squares plane:
Atom Distance from plane (A) N(l) 0.017 C(2) 0.007 NW -0.043 N(3) 0.014 C(4) 0.015 C(5) -0.013 CC’3 0.006 N(6) 0.023 N(7) -0.032 C(8) -0.018 NW 0.007 C(9) 0.019
Least-squares plane through 1-methylthymine (1)
0.3232X - 0.5755Y - 0.76122 f 0.5696 = 0
Distances of atoms from this least-squares plane:
Atom Distance from plane (A) N(l) 0.092 C(1) -0.019 C(2) 0.018 O(2) -0.021 N(3) -0.014 C(4) 0.018 O(4) - 0.004 C(5) 0.019 ‘76) 0.073 C(7) -0.071
Dihedral angle between this plane and previous plane: 5.8”
Least-squares plane through 1-methylthymine (2)
0.3186X - 0.5436Y - 0.77662 + 0.7409 = 0
Distance of atoms from this least-squares plane:
Atom Distance from plane (A) N(1) -0.011 C(l) 0.045 C(2) - 0.007 O(2) -0.001 N(3) -0.015 C(4) -0.011 O(4) 0.040 C(5) -0.028 C(6) -0.032 C(7) 0.019
Dihedral angle between this plane and the first plane: 7.6”
Least-squares plane through complex (excluding C( 10)) :
0.3425X - 0.5982Y - 0.72442 -f 0.4250 = 0
The planes were calculated according to the method of Blow (1960). X, Y and 2 directions correspond to the a, b’ and C’ directions, where the 6’ direction is perpendicular to a and lies in the a-b plane, and where c’ is perpendicular to both a and b’.

404 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
van der Waal’s contacts and are reasonable O-H . . . 0 and O-H . . N hydrogen bonding contacts (Pauling, 1960). The angle subtended by O(4)-O’-N(3) is 131.2”, and this is a bit larger than the H-O-H angle found in water (104~5”) ; however, t,he distances and angles suggest that this is a reasonable hydrogen bonding assignment.
Figure 6 shows a schematic diagram of the 9-ethyl-2,6-diaminopurine: l-methyl- thymine trimer complex with the bond distances and angles as obtained from this analysis. The estimated standard deviations are O-013 A for bond distances and 0.8” for bond angles.
Least-squares planes have been calculated for the individual purine and pyrimidine rings, as well as for the trimer complex, and the results of these calculations are summarized in Table 9.
4. Discussion A major incentive for studies on base pairing configurations between purines and
pyrimidines in the solid state has been to understand whether halogen substitution on the uracil ring alters the relative stabilities of different types of purine-pyrimidine hydrogen bonded configurations existing in solution prior to co-crystallization (Miller & Sobell, 1967; Kyogoku, Lord & Rich, 1967). Although it is not possible to correlate configurations found in the solid state with those prevalent in solution directly, there is now a large body of information concerning the interactions of purines and pyri- midines which strongly suggests this (for recent reviews in this area, see Hoogsteen, 1968; Sobell, 1969; Voet & Rich, 1969). For these reasons, the structures described here are of particular interest since changes in base pairing are seen which may reflect perturbations in the interaction due to halogen substitution on the uracil ring. Similar observations were noted previously in complexes between 9-ethyladenine : I-methyl-5-iodouracil and 9-ethyl-8-bromoadenine : I-methyl-5bromouracil (Sakore et al., 1969; Tavale et al., 1969). These structures both demonstrated “reversed” Watson-Crick type base pairing configurations which are analogous to that found in the 9-ethyl-2,6-diaminopurine : l-methyl-5-iodouracil structure, and this suggests that the intermolecular forces stabilizing these complexes may be very similar. The substitution of a methyl group for the iodine atom at the 5 position on the uracil ring, however, is associated with a base pairing change on both the N(1) and the N(7) diaminopurine side. This base pairing alteration cannot be due to steric factors alone, since the I-methylthymine molecule has a pseudo-2-fold axis of symmetry connecting the N(3) and C(6) atoms which allows it to form other base pairing con- figurations with 9-ethyl-2,6-diaminopurine which are isomorphous to one another. Although we cannot rule out the possibility that a small number of base pairs use, for instance, Hoogsteen instead of reversed Hoogsteen base pairing configurations, our analysis has demonstrated that the majority of base pairs use the Watson-Crick and reversed Hoogsteen structures. This base pairing preference may therefore reflect some subtle difference in hydrogen bonding stability between these compounds for reasons which are still unclear.
It is of interest that Pullman & Caillet (1967) have calculated the energies associated with various hydrogen-bonded configurations of 2,6-diaminopurine and thymine and have found that the most stable base pairing configurations in vacuum are the opposite of those described here. The discrepancy between their theoretical predic- tions and our experimental observations could, perhaps, reflect the presence of alkyl groups on the glycosidic ring nitrogen in this crystal study, which may alter the

DIAMINOPURINE-THYMINE AND -1ODOURACIL COMPLEXES 40R
charge distribution on the purine and pyrimidine rings. It is known, for example, that the alkylated purines and pyrimidines have slightly different pK values for their ring nitrogens and also react differently to alkylating agents compared with unsubstituted purines and pyrimidines (P. Brookes, personal communication). Another possibility is that lattice forces are important in determining base pairing configurations in the solid state, although, because of the pseudo-symmetry of 1-methylthymine, as discussed above, this seems less likely. Semi-empirical molecular orbital calculations involve significant approximation and it is possible that certain effects may not be apparent using these methods. For example, the consistently short N-H . . . N interactions observed in these studies between the purine and pyrimidine rings (240 A), coupled with the observation that significant ultraviolet hypochromism accompanies complex formation (Thomas & Kyogoku, 1967), may indicate that a sizable amount of quantum mechanical exchange occurs between the purine and pyrimidine rings. Factors such as molecular polarizability and group polarizability are important parameters determining the magnitude of London forces between molecules, and the presence or the absence of heavy halogens on the uracil ring almost certainly alters these parameters. More complete quantum mechanical calculations are required and these almost certainly will lead to a deeper under- standing of hydrogen bonding in nucleic acid structures.
This work has been supported in part by the National Institutes of Health, U.S. Public Healt’h Service, the National Science Foundation, the American Cancer Society, and an institutional grant to the University of Rochester from the American Cancer Societ,y. Facilities for this work were supplied in part from the U.S. Atomic Energy Commission at the University of Rochester Atomic Energy Project and this paper has been assigned Report No. UR-49-1095. We are indebted to Dr David Harker for his interest and for making facilities available to us to do a portion of this work at the Roswell Memorial Park Institute, Center for X-Ray Crystallographic Research. One of us (F.M.) gratefully acknowledges a travel grant from the Italian Minister0 degli Esteri and Consiglio Nazionale delle Ricerche (Rome) to come to the United States. Two of us (H.M.S. and G.K.) are recipients of Research Career Development Awards from the National Institutes of Healt,h, U.S. Public Health Service.
REFERENCES
Baklagine, Y. G., Volkenshtein, M. V. & Kondrashev, Y. D. (1966). Zhurnal Strukurnoi Khimii, 7, 399.
Blow, D. M. (1960). Acta Cryst. 13, 168. Haschemeyer, A. E. V. & Sobell, H. M. (1963). Proc. Nut. Acud. Sci., Wash. 50, 872. Haschemeyer, A. E. V. & Sobell, H. M. (1965). Acta G-y&. 18, 525. Hoogsteen, K. (1959). Acta Cry&. 12, 822. Hoogsteen, K. (1963). Actu Cqst. 16, 907. Hoogsteen, K. (1968). Molecular Associations irk Biology, ed. by B. Pullman. New York:
Academic Press. Katz, L., Tomita, K. & Rich, A. (1965). J. Mol. Biol. 13, 340. Katz, L., Tomita, K. & Rich, A. (1967). Actu Cryst. 21, 754. Kyogoku, Y., Lord, R. C. & Rich, A. (1967). Proc. Nat. Acd Sci., Wash. 57, 250. Labana, L. L. & Sobell, H. M. (1967). Proc. Nut. Acad. Sci., Wash. 57, 459. Mathews, F. S. & Rich, A. (1964). ,J. Mol. BioZ. 8, 89. Miller, J. H., & Sobell, H. M. (1967). J. MOE. BioZ. 24, 345. Pauling, L. (1960). The Nature of the Chemical Bond. Ithaca, New York : Cornell University
Press. Pullman, B. & Caillet, J. (1967). Theoret. Chim. Acta, 8, 223. Sakore, T. D., Tavale, S. S. & Sobell, H. M. (1969). J. Mol. BioZ. 43, 361.

406 T. D. SAKORE, H. M. SOBELL, F. MAZZA AND G. KARTHA
Sobell, H. M. (1969). In Genetic Organization, ed. by A. W. Ravin 8.z E. Caspari, vol. 1. New York: Academic Press. In the press.
Stout, G. H. & Jensen, L. H. (1968). X-ray Structure Determination, A Practical Guide. New York: The Macmillan Company.
Tavale, S. S., Sakore, T. D. & SobeIl, H. M. (1969). J. Mol. BioE. 43, 375. Thomas, G. J., Jr. & Kyogoku, Y. (1967). J. Amer. Chem. Sot. 89, 4170. Tomita, K., Katz, L. & Rich, A. (1967). J. Mol. Biol. 30, 545. Voet, D. & Rich, A. (1969). In Progress in Nucleic Acid Research and Molecular Biology,
ed. by J. N. Davidson & W. E. Cohn. New York: Academic Press. In the press.