Embed Size (px)
Citation preview

Application of molecular dynamics to the evaluation ofdielectric properties of model compounds with ¯exible side
groups of acrylic polymer chains
Enrique Saiz a,*, Evaristo Riande b
a Departamento de Qu�õmica F�õsica, Universidad de Alcal�a, Alcal�a de Henares, 28871 Madrid, Spainb Instituto de Ciencia y Tecnolog�õa de Pol�õmeros (CSIC), 28006 Madrid, Spain
Abstract
Some examples of application of molecular dynamics simulations (MD) to the evaluation of dielectric properties of
model compounds of polymeric chains are presented. Thus, a typical equilibrium property such as the mean squared
dipole moment, hl2i; is calculated for 2-(acetyloxy) ethyl-2-(2-naphthyl)acetate (ANA), model compound of the side
group of poly(2-{[2-(2-naphthyl)-acetyl]oxy}ethyl acrylate), and compared with experimental results. This calculation
illustrates some of the di�culties of MD simulations since the hl2i � 5:8 D2; obtained over a time span of 10 ns by em-
ploying the Tripos force ®eld, is in agreement with the experimental result of 5.3 D2, and yet, the analysis of probability
distributions of the rotational angles of the molecule proves that it does not statistically sample the whole conforma-
tional space of the molecule within the rather long time allowed for the simulation. On the contrary, the Amber force
®eld, with the same trajectory length of 10 ns, provides results that satis®es all conceivable tests. Dynamic properties
such as life times of di�erent conformations and dielectric relaxation times are also computed for 2-chlorocyclohexyl
isobutyrate (CCHI), model compound of the repeating unit of poly(2-cyclohexyl acrylate) (PCCHA). Agreement be-
tween theoretical and experimental results is excellent. Ó 1998 Elsevier Science B.V. All rights reserved.
1. Introduction
Molecular dynamics (MD) procedures allowthe evaluation of the trajectories of the atoms ofa molecular system within a force ®eld. The forcesacting on the atoms come from van der Waals andCoulombic interactions, bond stretching, anglebending, and rotational barriers. These methodsprovide a model of the actual properties of a sys-tem, and can consequently be used to computethe magnitudes of most conceivable macroscopic
properties [1]. Both dynamic and equilibriumproperties can thus be obtained by MD simula-tions, and their magnitudes correlated with micro-scopic structures of samples. However, MDcalculations also present some inconveniences,probably the best known being the computer pow-er required. An appropriate choice of the parame-ters de®ning the force ®eld for a given systemcould also be a problem, although there are somemultipurpose force ®elds that can be employedfor the most common molecules [2±6].
This paper presents MD computations of themean-square dipole moment, hl2i; of 2-(acetyloxy)ethyl-2-(2-naphthyl)acetate (ANA), model com-pound of the side group of poly(2-{[2-(2-naphthyl)-
Journal of Non-Crystalline Solids 235±237 (1998) 353±361
* Corresponding author. Tel.: +34 1 8854 664; fax: +34 1 8854
763; e-mail: [email protected].
0022-3093/98/$19.00 Ó 1998 Elsevier Science B.V. All rights reserved.
PII: S 0 0 2 2 - 3 0 9 3 ( 9 8 ) 0 0 6 5 1 - 6

acetyl]oxy}ethyl acrylate) [7]. The strategy used in-volves monitoring the time dependence of the di-pole moment of the compounds along thetrajectories and the evaluation of the temperaturedependence of the times required to overcome bar-rier energies between low energy conformations.To asses the reliability of the molecular dynamicssimulations, the hl2is calculated from the trajecto-ries are compared with the experimental results ob-tained from dielectric measurements for thiscompound.
The capability of MD simulations to predict thedistribution of dielectric relaxation times for the arelaxation of ¯exible molecular compounds isstudied in 2-chlorocyclohexyl isobutyrate (CCHI),model compound of the repeating unit of poly(2-cyclohexyl acrylate) (PCCHA) [8,9]. In this case,the time dependence of the dipolar correlationfunction, g(t), for the a relaxation process of thiscompound is evaluated [9]. Further transforma-tion of g(t) from the time to the frequency domainwill allow us to evaluate the frequency dependenceof the components of the complex dielectric per-mittivity, e�; of CCHI and the results will be com-pared with the experimental ones [9,10].
2. Force ®elds and equilibrium dielectric properties
The force ®eld represents the keystone of MDsimulations since it must represent the forces act-ing over all the atoms in a sample realistically. Ob-viously, if the force ®eld fails, the MD simulationsbecome meaningless. Modern force ®elds are de-signed for a wide variety of molecules; however
they fail in many cases, and what is even worse,it is di�cult to predict the cases where they failand the reason for failure, besides the trivial casesin which the molecule contains atoms, bonds orangles that have not yet been parameterized. Onlya detailed analysis of the results can guarantee anadequate operation of the force ®eld. However,data ®les containing positions and velocities ofthe atoms of the sample are often obtained whosesize, of the order of hundreds of megabyte, rendersdi�cult an exhaustive analysis of the informationin the ®les. Under these circumstances, if propercare is not taken, important issues related to theproperties of the molecules may be overlooked.
Usually, the suitability of MD simulations forthe evaluation of physical properties of moleculesis tested by comparing calculated and experimentalresults. However, agreement between theory andexperiment does not mean that an appropriateforce ®eld is used in the simulations, as the calcu-lation of the mean-square dipole momentsof 2-(acetyloxy)ethyl-2-(2-naphthyl)acetate (ANA),performed in this work, suggests. A scheme of thismolecule in all trans conformations is shown inFig. 1. MD calculations [11] with a simulated timespan of 10 ns employing the Tripos force ®eld [2,3]gives hl2i � 5:8 D2 for this molecule at 30°C, whilethe experimental magnitude of this quantity at thesame temperature is 5.3 D2 [7]. In spite of thisagreement between calculated and experimentalresults for the mean-square dipole moment ofANA, a routine analysis of the results shows thatthe g� and gÿ populations about CH2±O bondsdi�er, a result that, due to the symmetry of themolecule, is obviously wrong. The evolution of
Fig. 1. An schematic sketch of the 2-(acetyloxy)ethyl-2-(2-naphthyl)acetate (ANA) molecule shown in its planar all trans conformation
for which the rotatable bonds on the acyclic residue were set to be /� 180°. Dipole moments of the two ester groups, employed only
for RIS calculations, are represented by arrows pointing from negative to positive centers of charges. Values l1 � l2 � 1:75 D with an
orientation de®ned by s� 121° were used for these dipoles.
354 E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361
the rotational angles about CH2±O bonds suggestthat the cause of this discrepancy lies in the di�-culties found by the molecule in overcoming therotational barriers of these bonds which remainfor very long times in the same state so that thewhole conformational space is not statisticallysampled even with simulations such as the presentones. The analysis of the CH2±CH2 bond is lessconclusive, since it can be argued on whether transor gauche should be the preferred conformationsand if the di�erences obtained in the calculationare of signi®cance.
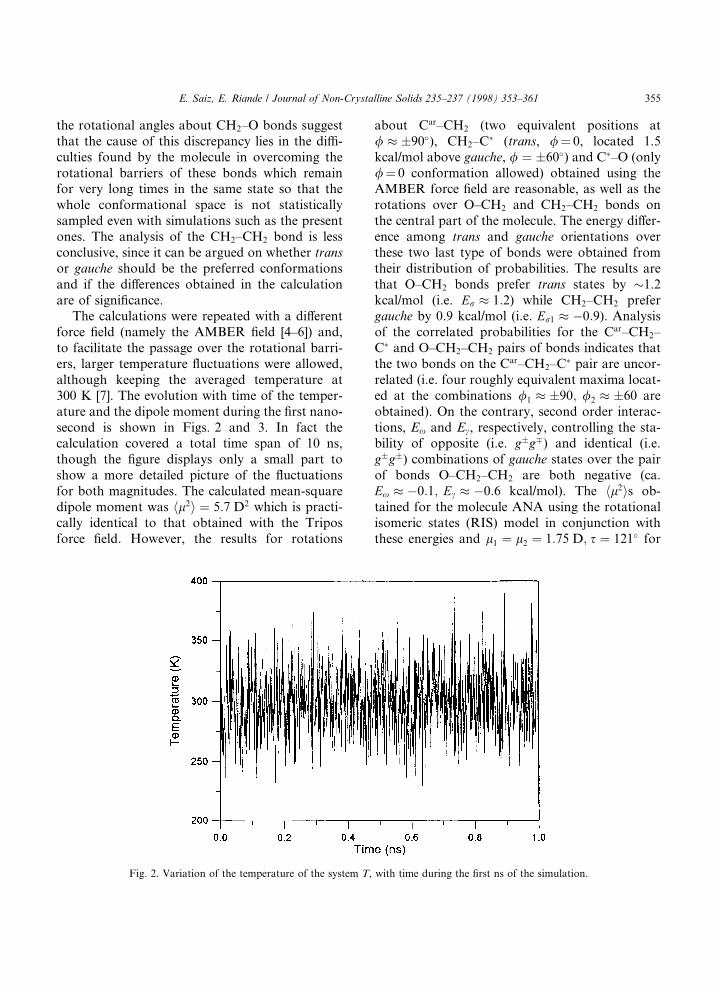
The calculations were repeated with a di�erentforce ®eld (namely the AMBER ®eld [4±6]) and,to facilitate the passage over the rotational barri-ers, larger temperature ¯uctuations were allowed,although keeping the averaged temperature at300 K [7]. The evolution with time of the temper-ature and the dipole moment during the ®rst nano-second is shown in Figs. 2 and 3. In fact thecalculation covered a total time span of 10 ns,though the ®gure displays only a small part toshow a more detailed picture of the ¯uctuationsfor both magnitudes. The calculated mean-squaredipole moment was hl2i � 5:7 D2 which is practi-cally identical to that obtained with the Triposforce ®eld. However, the results for rotations
about Car±CH2 (two equivalent positions at/ � �90�), CH2±C� (trans, /� 0, located 1.5kcal/mol above gauche, / � �60�) and C�±O (only/� 0 conformation allowed) obtained using theAMBER force ®eld are reasonable, as well as therotations over O±CH2 and CH2±CH2 bonds onthe central part of the molecule. The energy di�er-ence among trans and gauche orientations overthese two last type of bonds were obtained fromtheir distribution of probabilities. The results arethat O±CH2 bonds prefer trans states by �1.2kcal/mol (i.e. Er � 1:2) while CH2±CH2 prefergauche by 0.9 kcal/mol (i.e. Er1 � ÿ0:9). Analysisof the correlated probabilities for the Car±CH2±C� and O±CH2±CH2 pairs of bonds indicates thatthe two bonds on the Car±CH2±C� pair are uncor-related (i.e. four roughly equivalent maxima locat-ed at the combinations /1 � �90; /2 � �60 areobtained). On the contrary, second order interac-tions, Ex and Ec, respectively, controlling the sta-bility of opposite (i.e. g�g�) and identical (i.e.g�g�) combinations of gauche states over the pairof bonds O±CH2±CH2 are both negative (ca.Ex � ÿ0:1; Ec � ÿ0:6 kcal/mol). The hl2is ob-tained for the molecule ANA using the rotationalisomeric states (RIS) model in conjunction withthese energies and l1 � l2 � 1:75 D; s � 121� for
Fig. 2. Variation of the temperature of the system T, with time during the ®rst ns of the simulation.
E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361 355
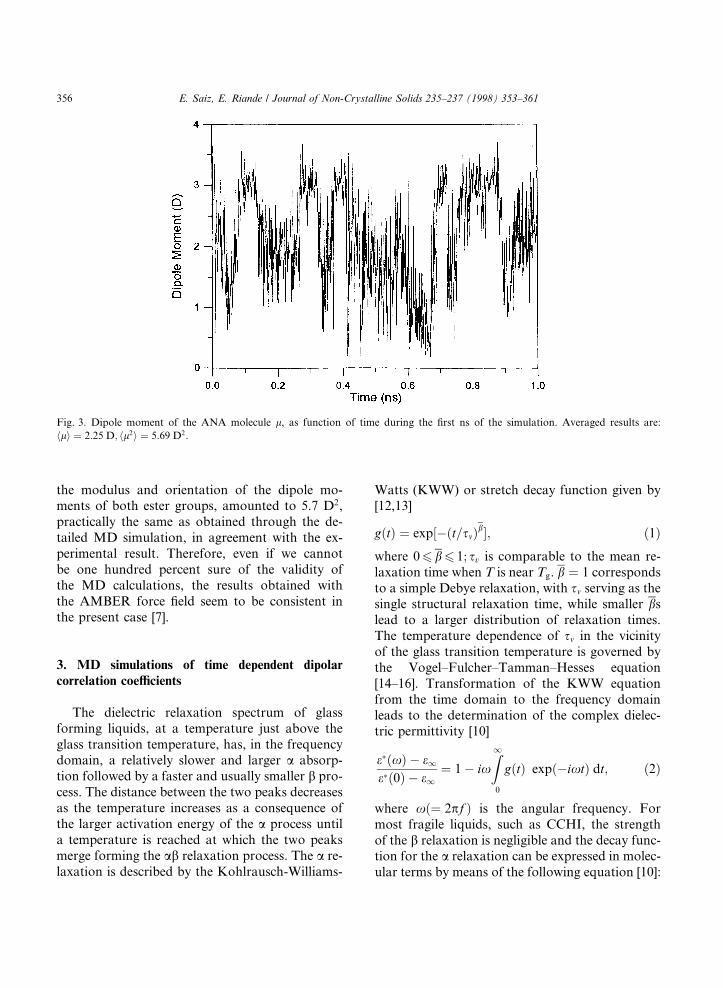
the modulus and orientation of the dipole mo-ments of both ester groups, amounted to 5.7 D2,practically the same as obtained through the de-tailed MD simulation, in agreement with the ex-perimental result. Therefore, even if we cannotbe one hundred percent sure of the validity ofthe MD calculations, the results obtained withthe AMBER force ®eld seem to be consistent inthe present case [7].
3. MD simulations of time dependent dipolar
correlation coe�cients
The dielectric relaxation spectrum of glassforming liquids, at a temperature just above theglass transition temperature, has, in the frequencydomain, a relatively slower and larger a absorp-tion followed by a faster and usually smaller b pro-cess. The distance between the two peaks decreasesas the temperature increases as a consequence ofthe larger activation energy of the a process untila temperature is reached at which the two peaksmerge forming the ab relaxation process. The a re-laxation is described by the Kohlrausch-Williams-
Watts (KWW) or stretch decay function given by[12,13]
g�t� � exp�ÿ�t=sm�b�; �1�where 06 b6 1; sv is comparable to the mean re-laxation time when T is near Tg. b � 1 correspondsto a simple Debye relaxation, with sm serving as thesingle structural relaxation time, while smaller bslead to a larger distribution of relaxation times.The temperature dependence of sm in the vicinityof the glass transition temperature is governed bythe Vogel±Fulcher±Tamman±Hesses equation[14±16]. Transformation of the KWW equationfrom the time domain to the frequency domainleads to the determination of the complex dielec-tric permittivity [10]
e��x� ÿ e1e��0� ÿ e1
� 1ÿ ixZ1
0
g�t� exp�ÿixt� dt; �2�
where x�� 2pf � is the angular frequency. Formost fragile liquids, such as CCHI, the strengthof the b relaxation is negligible and the decay func-tion for the a relaxation can be expressed in molec-ular terms by means of the following equation [10]:
Fig. 3. Dipole moment of the ANA molecule l, as function of time during the ®rst ns of the simulation. Averaged results are:
hli � 2:25 D; hl2i � 5:69 D2:
356 E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361

g�t� � hli�0�li�t�i �P
i6�jhli�0�lj�t�ihli�0�li�0�i �
Pi6�jhli�0�lj�0�i
; �3�
where hli�0�li�t�i is the non-normalized autocor-relation function for the dipole associated withthe molecule i and hli�0�lj�t�i is the non-normal-ized cross correlation function for reorientationalmotions of the dipoles associated with moleculesi and j. In spite of the fact that the cross correla-tion terms in Eq. (3) will have appreciable magni-tude and their signs may be positive or negative,theoretical reasoning and the critical interpretationof experimental dielectric results suggest that theauto- and the cross-correlation functions may havethe same time-dependence. Consequently, the de-cay function can be expressed by the autocorrela-tion function [10]
g�t� � hli�0�li�t�ihl2i ; �4�
where hl2i is the mean-square dipole moment ofthe molecule.
The study of the 1H nuclear magnetic resonance(NMR) spectrum of the CCHI molecule showsthat the disubstituted cyclohexane ring is in transcon®guration and only the equatorial±equatorialconformation is detected within this con®guration[9]. Fig. 4 shows a schematic representation of
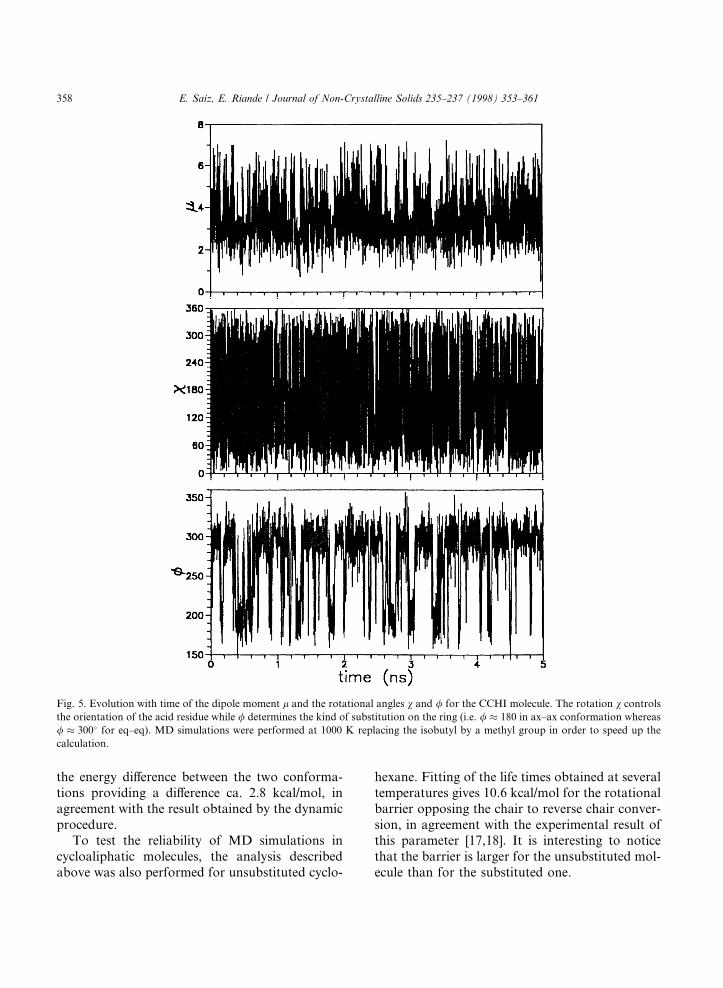
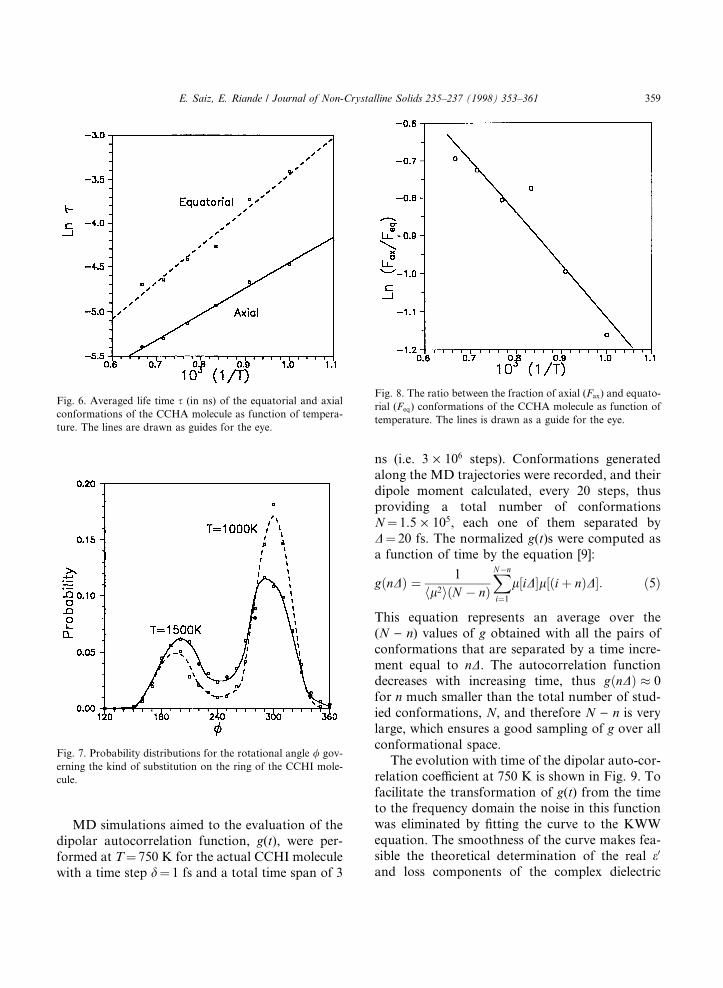
CCHI in the eq±eq conformation (i.e. both substit-uents in the equatorial position). The eq±eq con-formation converts into ax±ax and vice versa bymeans of chair-to-reverse chair transition. Theevolution with time of the dipole moment, the ro-tation, v, that governs the orientation of theisobutyl group, and the rotation, /, that indicatesthe kind of substitution on the ring (i.e./ � 180=300�; respectively, for ax±ax/eq±eq con-formations) are shown in Fig. 5. The calculationswere performed at temperatures, in the range800±1500 K, to facilitate the passage over rota-tional barriers within a reasonable computing time[8,9]. The isobutyl group was replaced by a methylgroup for this calculation, thus, an acetyl acid res-idue was used instead of the actual isobutyl groupof the molecule. This substitution does not a�ectthe features in which we are interested and de-creases the computation time. The results plottedin Fig. 5, obtained at 1000 K, indicate that v is al-most freely rotating at this temperature. The im-portant point is that the life times, s, of axialand equatorial conformations can be evaluatedby averaging the lengths of the visits to those ori-entations. The calculation can be repeated at sev-eral temperatures and represented in anArrhenius plot, shown in Fig. 6, whose slope pro-vides the energy barriers opposing the passage eq® ax and ax ® eq. The di�erence between thesetwo barriers indicates that the eq±eq conformationis preferred over the ax±ax by ca. 2.5 kcal/mol.Moreover, extrapolation to 300 K provides lifetimes, sax � 9:6; seq � 460 ns, respectively, forax±ax and eq±eq conformations [8] with estimateduncertainties of ca. �5%.
This result can also be obtained by computingthe total fraction of time that the molecule expendsin both ax±ax and eq±eq conformations and plot-ting the natural logarithm of the ratio Fax/Feq asfunction of the reciprocal of temperature. Fig. 7shows the probability distribution for the / rota-tion at two temperatures. Integration of the areaunder the peaks at 300 and 180° provide respec-tively the Feq and Fax fractions. Extrapolationto 300 K, represented in Fig. 8, givesFax � 0:02 and Feq � 0:98; in agreement with ex-perimental results obtained by NMR [8]. Further-more, the slope of the plot allows the evaluation of
Fig. 4. The trans con®guration of the 2-chlorocyclohexyl isobu-
tyrate (CCHI) molecule. Dipole moments of chlorine-cyclohex-
yl bond �l1 � 2:2 D� and ester group �l � 1:9 D� are represent
only for illustrative purposes [11].
E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361 357
the energy di�erence between the two conforma-tions providing a di�erence ca. 2.8 kcal/mol, inagreement with the result obtained by the dynamicprocedure.
To test the reliability of MD simulations incycloaliphatic molecules, the analysis describedabove was also performed for unsubstituted cyclo-
hexane. Fitting of the life times obtained at severaltemperatures gives 10.6 kcal/mol for the rotationalbarrier opposing the chair to reverse chair conver-sion, in agreement with the experimental result ofthis parameter [17,18]. It is interesting to noticethat the barrier is larger for the unsubstituted mol-ecule than for the substituted one.
Fig. 5. Evolution with time of the dipole moment l and the rotational angles v and / for the CCHI molecule. The rotation v controls
the orientation of the acid residue while / determines the kind of substitution on the ring (i.e. / � 180 in ax±ax conformation whereas
/ � 300� for eq±eq). MD simulations were performed at 1000 K replacing the isobutyl by a methyl group in order to speed up the
calculation.
358 E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361
MD simulations aimed to the evaluation of thedipolar autocorrelation function, g(t), were per-formed at T� 750 K for the actual CCHI moleculewith a time step d� 1 fs and a total time span of 3
ns (i.e. 3 ´ 106 steps). Conformations generatedalong the MD trajectories were recorded, and theirdipole moment calculated, every 20 steps, thusproviding a total number of conformationsN� 1.5 ´ 105, each one of them separated byD� 20 fs. The normalized g(t)s were computed asa function of time by the equation [9]:
g�nD� � 1
hl2i�N ÿ n�XNÿn
i�1
l�iD�l��i� n�D�: �5�
This equation represents an average over the(N ) n) values of g obtained with all the pairs ofconformations that are separated by a time incre-ment equal to nD. The autocorrelation functiondecreases with increasing time, thus g�nD� � 0for n much smaller than the total number of stud-ied conformations, N, and therefore N ) n is verylarge, which ensures a good sampling of g over allconformational space.
The evolution with time of the dipolar auto-cor-relation coe�cient at 750 K is shown in Fig. 9. Tofacilitate the transformation of g(t) from the timeto the frequency domain the noise in this functionwas eliminated by ®tting the curve to the KWWequation. The smoothness of the curve makes fea-sible the theoretical determination of the real e0
and loss components of the complex dielectric
Fig. 7. Probability distributions for the rotational angle / gov-
erning the kind of substitution on the ring of the CCHI mole-
cule.
Fig. 8. The ratio between the fraction of axial (Fax) and equato-
rial (Feq) conformations of the CCHA molecule as function of
temperature. The lines is drawn as a guide for the eye.
Fig. 6. Averaged life time s (in ns) of the equatorial and axial
conformations of the CCHA molecule as function of tempera-
ture. The lines are drawn as guides for the eye.
E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361 359
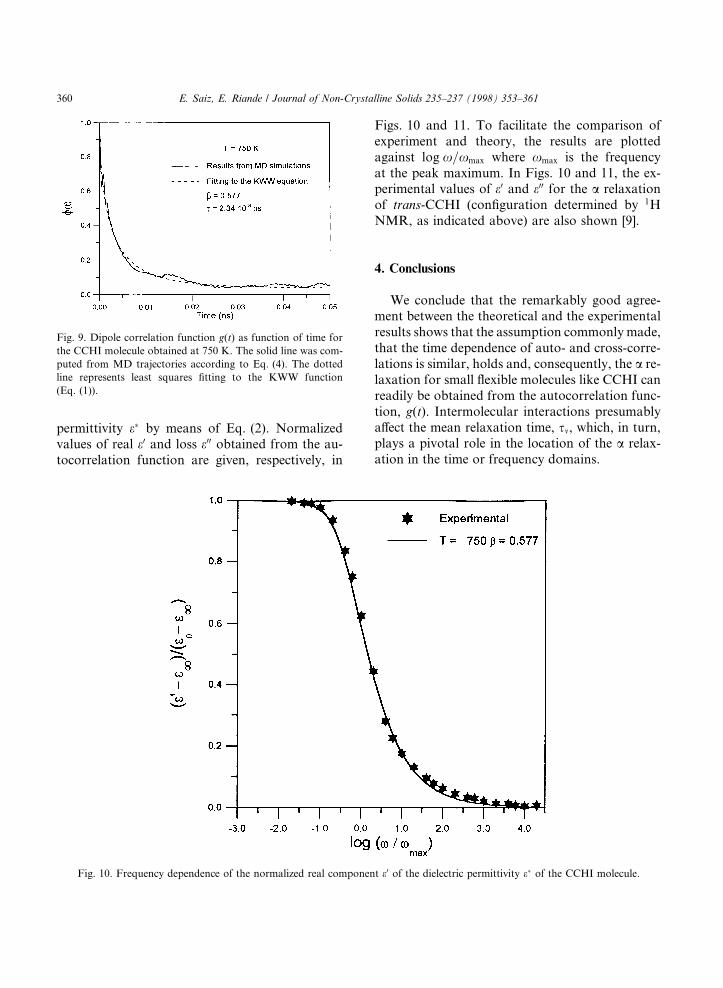
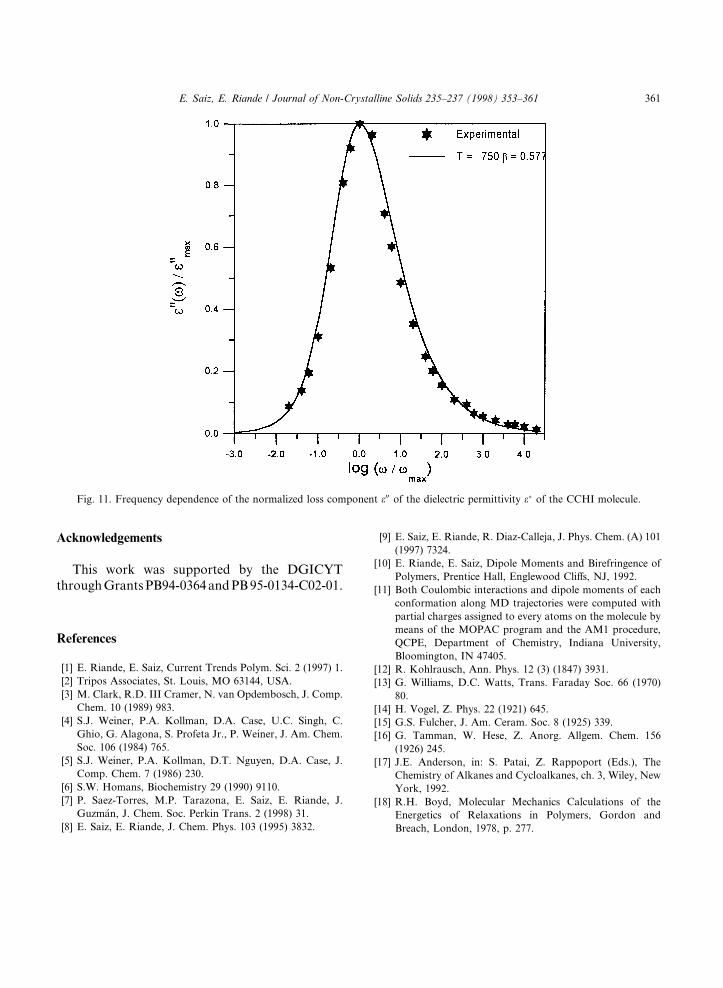
permittivity e� by means of Eq. (2). Normalizedvalues of real e0 and loss e00 obtained from the au-tocorrelation function are given, respectively, in
Figs. 10 and 11. To facilitate the comparison ofexperiment and theory, the results are plottedagainst log x=xmax where xmax is the frequencyat the peak maximum. In Figs. 10 and 11, the ex-perimental values of e0 and e00 for the a relaxationof trans-CCHI (con®guration determined by 1HNMR, as indicated above) are also shown [9].
4. Conclusions
We conclude that the remarkably good agree-ment between the theoretical and the experimentalresults shows that the assumption commonly made,that the time dependence of auto- and cross-corre-lations is similar, holds and, consequently, the a re-laxation for small ¯exible molecules like CCHI canreadily be obtained from the autocorrelation func-tion, g(t). Intermolecular interactions presumablya�ect the mean relaxation time, sm, which, in turn,plays a pivotal role in the location of the a relax-ation in the time or frequency domains.
Fig. 10. Frequency dependence of the normalized real component e0 of the dielectric permittivity e� of the CCHI molecule.
Fig. 9. Dipole correlation function g(t) as function of time for
the CCHI molecule obtained at 750 K. The solid line was com-
puted from MD trajectories according to Eq. (4). The dotted
line represents least squares ®tting to the KWW function
(Eq. (1)).
360 E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361
Acknowledgements
This work was supported by the DGICYTthrough Grants PB94-0364 and PB 95-0134-C02-01.
References
[1] E. Riande, E. Saiz, Current Trends Polym. Sci. 2 (1997) 1.
[2] Tripos Associates, St. Louis, MO 63144, USA.
[3] M. Clark, R.D. III Cramer, N. van Opdembosch, J. Comp.
Chem. 10 (1989) 983.
[4] S.J. Weiner, P.A. Kollman, D.A. Case, U.C. Singh, C.
Ghio, G. Alagona, S. Profeta Jr., P. Weiner, J. Am. Chem.
Soc. 106 (1984) 765.
[5] S.J. Weiner, P.A. Kollman, D.T. Nguyen, D.A. Case, J.
Comp. Chem. 7 (1986) 230.
[6] S.W. Homans, Biochemistry 29 (1990) 9110.
[7] P. Saez-Torres, M.P. Tarazona, E. Saiz, E. Riande, J.
Guzm�an, J. Chem. Soc. Perkin Trans. 2 (1998) 31.
[8] E. Saiz, E. Riande, J. Chem. Phys. 103 (1995) 3832.
[9] E. Saiz, E. Riande, R. Diaz-Calleja, J. Phys. Chem. (A) 101
(1997) 7324.
[10] E. Riande, E. Saiz, Dipole Moments and Birefringence of
Polymers, Prentice Hall, Englewood Cli�s, NJ, 1992.
[11] Both Coulombic interactions and dipole moments of each
conformation along MD trajectories were computed with
partial charges assigned to every atoms on the molecule by
means of the MOPAC program and the AM1 procedure,
QCPE, Department of Chemistry, Indiana University,
Bloomington, IN 47405.
[12] R. Kohlrausch, Ann. Phys. 12 (3) (1847) 3931.
[13] G. Williams, D.C. Watts, Trans. Faraday Soc. 66 (1970)
80.
[14] H. Vogel, Z. Phys. 22 (1921) 645.
[15] G.S. Fulcher, J. Am. Ceram. Soc. 8 (1925) 339.
[16] G. Tamman, W. Hese, Z. Anorg. Allgem. Chem. 156
(1926) 245.
[17] J.E. Anderson, in: S. Patai, Z. Rappoport (Eds.), The
Chemistry of Alkanes and Cycloalkanes, ch. 3, Wiley, New
York, 1992.
[18] R.H. Boyd, Molecular Mechanics Calculations of the
Energetics of Relaxations in Polymers, Gordon and
Breach, London, 1978, p. 277.
Fig. 11. Frequency dependence of the normalized loss component e00 of the dielectric permittivity e� of the CCHI molecule.
E. Saiz, E. Riande / Journal of Non-Crystalline Solids 235±237 (1998) 353±361 361





























