Embed Size (px)
Citation preview

Apoptosis inhibitor of macrophage (AIM) is requiredfor obesity-associated recruitment of inflammatorymacrophages into adipose tissueJun Kurokawaa, Hiromichi Naganoa, Osamu Oharab, Naoto Kubotac, Takashi Kadowakic, Satoko Araia,and Toru Miyazakia,1
aLaboratory of Molecular Biomedicine for Pathogenesis, Center for Disease Biology and Integrative Medicine, Faculty of Medicine, University of Tokyo,Tokyo 113-0033, Japan; bDepartment of Human Genome Research, Kazusa DNA Research Institute, Kisarazu, Chiba 292-0818, Japan; and cDepartment ofInternal Medicine, Graduate School of Medicine, University of Tokyo, Tokyo 113-0033, Japan
Edited* by Tadatsugu Taniguchi, University of Tokyo, Tokyo, Japan, and approved June 14, 2011 (received for review February 3, 2011)
Infiltration of inflammatory macrophages into adipose tissues withthe progression of obesity triggers insulin resistance and obesity-relatedmetabolic diseases.We recently reported that macrophage-derived apoptosis inhibitor of macrophage (AIM) protein is in-creased inblood in linewithobesityprogression and is incorporatedinto adipocytes, thereby inducing lipolysis in adipose tissue. Herewe show that such a response is required for the recruitment ofadipose tissue macrophages. In vitro, AIM-dependent lipolysisinduced an efflux of palmitic and stearic acids from 3T3-L1 adipo-cytes, thereby stimulating chemokine production in adipocytes viaactivation of toll-like receptor 4 (TLR4). In vivo administration ofrecombinant AIM to TLR4-deficient (TLR4−/−) mice resulted in induc-tion of lipolysis without chemokine production in adipose tissues.Consistently, mRNA levels for the chemokines that affect macro-phages were far lower in AIM-deficient (AIM−/−) than in wild-type(AIM+/+) obese adipose tissue. This reduction in chemokine produc-tion resulted in a marked prevention of inflammatory macrophageinfiltration into adipose tissue in obeseAIM−/−mice, although thesemice showedmore advanced obesity thanAIM+/+mice on a high-fatdiet. Diminished macrophage infiltration resulted in decreased in-flammation locally and systemically in obese AIM−/− mice, therebyprotecting them from insulin resistance and glucose intolerance.These results indicate that the increase in blood AIM is a criticalevent for the initiation of macrophage recruitment into adiposetissue, which is followed by insulin resistance. Thus, AIM sup-pression might be therapeutically applicable for the prevention ofobesity-related metabolic disorders.
diabetes | fatty acid synthase | CD36 | knockout mouse
Chronic, low-grade inflammation observed in adipose tissues ischaracteristic of obesity. Such a subclinical inflammatory state
of adipose tissues is highly associated with insulin resistance bothin adipose tissue and systemically and thus contributes to thedevelopment of multiple obesity-induced metabolic and cardio-vascular diseases (1–4). Evidence has shown that infiltration ofa large number of classically activated inflammatory macrophages(M1 macrophages) into adipose tissue is responsible for obesity-associated inflammation (5–7). Lean adipose tissue contains aresident population of alternatively activated macrophages (M2macrophages), which can suppress the inflammation of both adi-pocytes and macrophages partly via the secretion of interleukin(IL)-10. Hence, obesity induces a switch in macrophage activationstate in adipose tissue toward M1 polarization, which leads toinflammation (8–12). However, the mechanism that promotesinfiltration of inflammatory macrophages into obese adipose tis-sue is as yet unknown.We recently reported that the apoptosis inhibitor of macro-
phage (AIM) protein (13) is incorporated into adipocytes viaCD36-mediated endocytosis, and induces lipolysis by suppressingthe activity of fatty acid synthase (FAS) (14). AIM is a member ofthe scavenger receptor cysteine-rich superfamily and was initiallyidentified as an apoptosis inhibitor that supports the survival of
macrophages against different types of apoptosis-inducing stimuli(13). AIM is a direct target for regulation by nuclear receptor liverX receptor/retinoid X receptor (LXR/RXR) heterodimers (15,16) and is solely produced by tissue macrophages. As a secretedmolecule, AIM is detected in both human and mouse blood atvarious levels (13, 16–19) and increases in blood with the pro-gression of obesity in mice fed a high-fat diet (HFD) (14). Theaugmented blood AIM induced lipolysis, as evident by the factthat the increase of free fatty acids (FFAs) and glycerol in bloodwas suppressed in AIM−/− mice (14). Owing to less lipolysis, ad-ipocyte hypertrophy was more advanced and the overall mass ofvisceral adipose tissues was greater in AIM−/− than in AIM+/+
mice fed a HFD (14). All these observations imply that AIM-in-duced lipolysis might be responsible for the obesity-associatedrecruitment of adipose tissue macrophages.In the present study, we assessed whether AIM affects mac-
rophage accumulation in adipose tissues in obese mice. In ad-dition, we determined the molecular mechanism of how AIM-dependent lipolysis results in the production of chemokines byadipocytes for the effective recruitment of adipose tissue mac-rophages. Finally, we investigated how the absence of AIMinfluences the local and systemic inflammatory state and insulinresistance in mice. On the basis of these results, we discuss theputative role of AIM in the initiation of obesity-associatedchronic inflammation and subsequent metabolic diseases.
Results and DiscussionPrevention of M1 Macrophage Recruitment into Adipose Tissues inObese AIM−/− Mice. In AIM−/− mice, adipocyte hypertrophy wasmore advanced than in AIM+/+ mice, and the overall mass ofvisceral fat as well as body weight was markedly greater com-pared with that of AIM+/+ mice (14). Interestingly, however, farfewer macrophages stained with a pan-macrophage antibodyF4/80 were observed in epididymal adipose tissue in AIM−/− micethan in AIM+/+ mice fed a HFD for 12 wk (Fig. 1A). The numberof IL-6 stained inflammatory type (M1) macrophages in obeseAIM−/− mice was markedly lower than in obese AIM+/+ mice(Fig. 1A). In addition, almost no M1 macrophage clustersforming crown-like structures (CLS) were observed in obeseAIM−/− mice (Fig. 1A). In contrast, the number of M2 adiposetissue macrophages stained for mannose receptor (MR) was notincreased in AIM+/+ or AIM−/− mice after a 12-wk HFD (Fig.1B). Furthermore, the stromal-vascular cell fraction (SVF)containing macrophages was isolated from the epididymal fattissue of lean and obese mice by collagenase treatment and
Author contributions: T.M. designed research; J.K., O.O., N.K., and S.A. performed re-search; H.N. contributed new reagents/analytic tools; J.K., O.O., T.K., and T.M. analyzeddata; and S.A. and T.M. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1101841108/-/DCSupplemental.
12072–12077 | PNAS | July 19, 2011 | vol. 108 | no. 29 www.pnas.org/cgi/doi/10.1073/pnas.1101841108
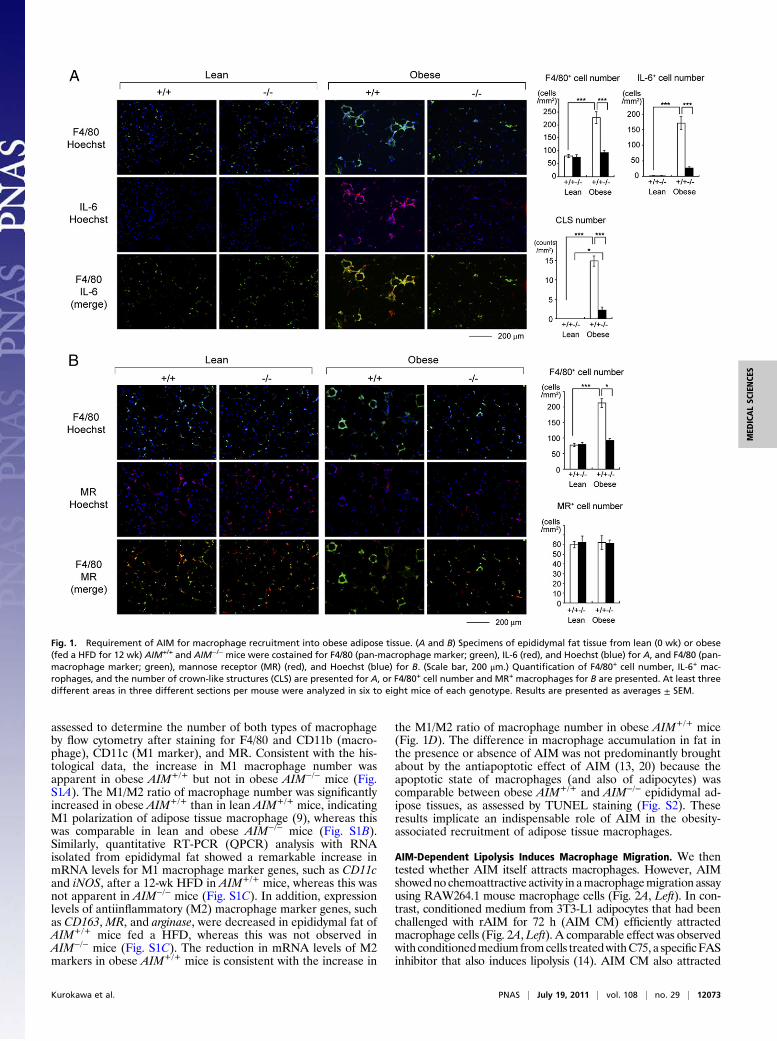
assessed to determine the number of both types of macrophageby flow cytometry after staining for F4/80 and CD11b (macro-phage), CD11c (M1 marker), and MR. Consistent with the his-tological data, the increase in M1 macrophage number wasapparent in obese AIM+/+ but not in obese AIM−/− mice (Fig.S1A). The M1/M2 ratio of macrophage number was significantlyincreased in obese AIM+/+ than in lean AIM+/+ mice, indicatingM1 polarization of adipose tissue macrophage (9), whereas thiswas comparable in lean and obese AIM−/− mice (Fig. S1B).Similarly, quantitative RT-PCR (QPCR) analysis with RNAisolated from epididymal fat showed a remarkable increase inmRNA levels for M1 macrophage marker genes, such as CD11cand iNOS, after a 12-wk HFD in AIM+/+ mice, whereas this wasnot apparent in AIM−/− mice (Fig. S1C). In addition, expressionlevels of antiinflammatory (M2) macrophage marker genes, suchas CD163, MR, and arginase, were decreased in epididymal fat ofAIM+/+ mice fed a HFD, whereas this was not observed inAIM−/− mice (Fig. S1C). The reduction in mRNA levels of M2markers in obese AIM+/+ mice is consistent with the increase in
the M1/M2 ratio of macrophage number in obese AIM+/+ mice(Fig. 1D). The difference in macrophage accumulation in fat inthe presence or absence of AIM was not predominantly broughtabout by the antiapoptotic effect of AIM (13, 20) because theapoptotic state of macrophages (and also of adipocytes) wascomparable between obese AIM+/+ and AIM−/− epididymal ad-ipose tissues, as assessed by TUNEL staining (Fig. S2). Theseresults implicate an indispensable role of AIM in the obesity-associated recruitment of adipose tissue macrophages.
AIM-Dependent Lipolysis Induces Macrophage Migration. We thentested whether AIM itself attracts macrophages. However, AIMshowedno chemoattractive activity in amacrophagemigration assayusing RAW264.1 mouse macrophage cells (Fig. 2A, Left). In con-trast, conditioned medium from 3T3-L1 adipocytes that had beenchallenged with rAIM for 72 h (AIM CM) efficiently attractedmacrophage cells (Fig. 2A,Left). A comparable effect was observedwith conditionedmedium fromcells treatedwithC75, a specificFASinhibitor that also induces lipolysis (14). AIM CM also attracted
Fig. 1. Requirement of AIM for macrophage recruitment into obese adipose tissue. (A and B) Specimens of epididymal fat tissue from lean (0 wk) or obese(fed a HFD for 12 wk) AIM+/+ and AIM−/− mice were costained for F4/80 (pan-macrophage marker; green), IL-6 (red), and Hoechst (blue) for A, and F4/80 (pan-macrophage marker; green), mannose receptor (MR) (red), and Hoechst (blue) for B. (Scale bar, 200 μm.) Quantification of F4/80+ cell number, IL-6+ mac-rophages, and the number of crown-like structures (CLS) are presented for A, or F4/80+ cell number and MR+ macrophages for B are presented. At least threedifferent areas in three different sections per mouse were analyzed in six to eight mice of each genotype. Results are presented as averages ± SEM.
Kurokawa et al. PNAS | July 19, 2011 | vol. 108 | no. 29 | 12073
MED
ICALSC
IENCE
S

J774.1 mouse monocyte cells (Fig. S3A). Furthermore, 3T3-L1adipocytes were treated with rAIM in the presence of a CD36-neutralizing antibody (mouse IgA), which inhibits AIM-dependentlipolysis by disturbing the endocytosis of AIM into adipocytes (14),and the conditioned medium (AIM+αCD36 CM) was assessed inthe macrophage migration assay. The AIM+αCD36 CM did notefficiently attract macrophages (Fig. 2A, Right), suggesting thatAIM-induced lipolysis in adipocytes appears to be responsible formacrophage recruitment. The CD36-neutralizing antibody itselfhad no direct effect on the macrophage migration (Fig. S3B).
Fatty Acids Effluxed from Adipocytes in Response to AIM-DependentLipolysis Stimulated TLR Signaling Pathway and Induced ChemokineProduction in Adipocytes. Accumulating evidence has demon-
strated that saturated fatty acids activate TLR signaling cascadeand that this response is tightly associated with obesity-inducedinflammation (21–25). Thus, it is plausible that an increase inblood AIMmay induce vigorous lipolysis in obese adipose tissues,and saturated fatty acids effluxed from adipocytes as a result oflipolysis might activate chemokine production in adipocytes viathe stimulation of TLR(s) in a paracrine/autocrine fashion (26–28). Indeed, palmitic and stearic acids, the major fatty acidscomprising triglyceride droplets (29) and well known as stim-ulators of TLR4 and TLR2 (21, 25, 30, 31), were identified as thecomponents released by adipocytes in response to lipolysis in-duced by AIM or C75 when the profile of fatty acids in AIM CMand C75 CM was evaluated by gas-chromatography mass-spectrometry analysis.
Fig. 2. AIM-dependent lipolysis induces chemokine production in adipocytes via TLR4 stimulation. (A) Chemotaxis of RAW 264.1 cells in response to specifiedstimulant.Attractants: rAIM (25 μg/mL), C75 (25μM),AIMCM/C75CM: conditionedmediumfrom3T3-L1adipocytes treated for 3dwith rAIM (25μg/mL) orC75 (25μM), respectively; AIM+αCD36 CM/AIM+IgA CM: conditionedmedium from 3T3-L1 adipocytes treated for 3 d with rAIM (25 μg/mL) in the presence of anti-CD36Ab or mouse IgA (10 μg/mL each), respectively; none CM, control CM: treated without rAIM or C75; and FM: fresh DMEM culture medium containing 10% FBS.Averages from n = 3 ± SEM.MCP-1 (100 ng/mL) was used as a positive control. (B) Degradation of IkBα in 3T3-L1 adipoctytes in response to specified stimulant intheabsence (−) or presence (+) of aTIRAP inhibitor (100 μM). LPS (100ng/mL)was usedas apositive control. Representative immunoblotting results arepresented.The density of the signal was quantified using National Institutes of Health Image J image analysis software and presented as values relative to those of presti-mulation (Lower twopanels). n = 3. Error bar: SEM. *, versus the value at prestimulation (0min). (C) QPCR analysis ofmRNA levels forMCP-1, CCL5/RANTES,MCP-2,and MCP-3 using RNA isolated from 3T3-L1 adipocytes treated with specified stimulant for 24 h in the absence (white bars) or presence (black bars) of a TIRAPinhibitor. Values were presented as relative expression to those without stimulation (none). n = 3 for each. Error bar: SEM. (D and E) No degradation of IkBα orexpression induction of mRNA for chemokine genes in 3T3-L1 adipoctytes in response to rAIM alone (25 μg/mL) (D) or AIM+αCD36 CM (E).
12074 | www.pnas.org/cgi/doi/10.1073/pnas.1101841108 Kurokawa et al.
Consistent with this result, both AIM CM and C75 CM effi-ciently stimulated the TLR signaling cascade and chemokineproduction in 3T3-L1 adipocytes, as assessed by degradation ofIkBα (Fig. 2B) and mRNA expression of chemokines such asMCP-1, CCL5/RANTES, MCP-2, and MCP-3, which affectsmacrophages (Fig. 2C). AIM CM induced substantial levels ofprotein of these chemokines as assessed by ELISA (Fig. S4A).These responses diminished when adipocytes were treated withAIM CM or C75 CM in the presence of a toll–interleukin-1receptor domain containing adapter protein (TIRAP) inhibitor,which specifically interferes with the interaction of TLR4 (as wellas TLR2) and the adapter protein TIRAP/Mal, resulting in at-
tenuation of TLR signaling (Fig. 2 B and C) (32). Furthermore,we confirmed that similar effects of TLR activation and che-mokine production were observed when 3T3-L1 adipocytes weretreated with palmitic acid (PA) or stearic acid (SA) and that theresponses induced by each fatty acid were reduced when sub-jected to the TIRAP inhibitor (Fig. S5). Consistent with theresults from macrophage migration assay presented in Fig. 2A,neither rAIM alone (Fig. 2D and Fig. S4B) nor AIM+αCD36CM (Fig. 2E and Fig. S4C) stimulated IkBα degradation orchemokine mRNA and protein expression in adipocytes. Thesefindings clearly indicate the necessity of the lipolytic process inthe overall activation of TLR signaling cascade by AIM.
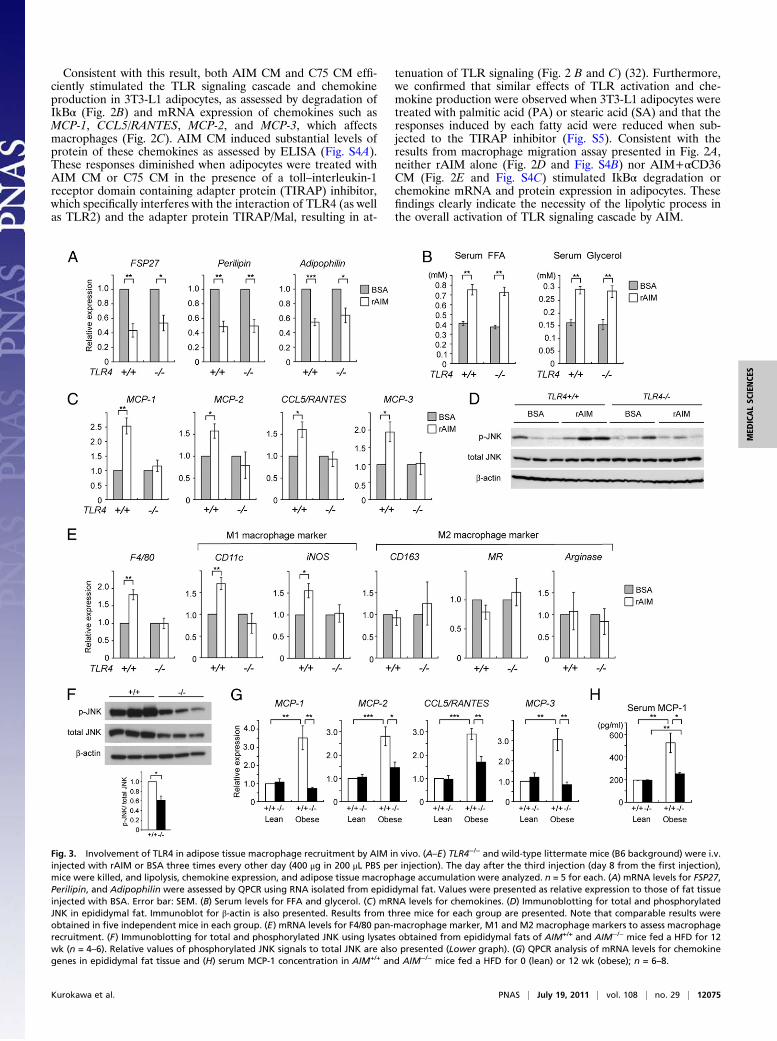
Fig. 3. Involvement of TLR4 in adipose tissue macrophage recruitment by AIM in vivo. (A–E) TLR4−/− and wild-type littermate mice (B6 background) were i.v.injected with rAIM or BSA three times every other day (400 μg in 200 μL PBS per injection). The day after the third injection (day 8 from the first injection),mice were killed, and lipolysis, chemokine expression, and adipose tissue macrophage accumulation were analyzed. n = 5 for each. (A) mRNA levels for FSP27,Perilipin, and Adipophilin were assessed by QPCR using RNA isolated from epididymal fat. Values were presented as relative expression to those of fat tissueinjected with BSA. Error bar: SEM. (B) Serum levels for FFA and glycerol. (C) mRNA levels for chemokines. (D) Immunoblotting for total and phosphorylatedJNK in epididymal fat. Immunoblot for β-actin is also presented. Results from three mice for each group are presented. Note that comparable results wereobtained in five independent mice in each group. (E) mRNA levels for F4/80 pan-macrophage marker, M1 and M2 macrophage markers to assess macrophagerecruitment. (F) Immunoblotting for total and phosphorylated JNK using lysates obtained from epididymal fats of AIM+/+ and AIM−/− mice fed a HFD for 12wk (n = 4–6). Relative values of phosphorylated JNK signals to total JNK are also presented (Lower graph). (G) QPCR analysis of mRNA levels for chemokinegenes in epididymal fat tissue and (H) serum MCP-1 concentration in AIM+/+ and AIM−/− mice fed a HFD for 0 (lean) or 12 wk (obese); n = 6–8.
Kurokawa et al. PNAS | July 19, 2011 | vol. 108 | no. 29 | 12075
MED
ICALSC
IENCE
S

Involvement of TLR4. As TIRAP is downstream of not only TLR4but also other TLRs, including TLR2 (32), the precise in-volvement of TLR4 in macrophage recruitment was furtherverified. We first suppressed TLR4 expression by siRNA in 3T3-L1 adipocytes and assessed the induction of MCP-1 by AIM CM.As shown in Fig. S6 A–C, induction of both mRNA and proteinof MCP-1 by AIM CM was significantly reduced in cells trans-fected with siRNA for TLR4. In addition, we injected rAIM i.v.into wild-type and TLR4−/− mice and thereafter assessed thestate of lipolysis and chemokine production in epididymal adi-pose tissue. In both types of mice, the mRNA levels of FSP27(also called Cidec), Perilipin, and Adipophilin, coating elementsfor lipid droplets, were decreased after challenging with rAIM(Fig. 3A), a finding consistent with the progression of lipolysisreported previously (17, 33, 34). Similarly, the increase in bloodFFA and glycerol levels was equivalent in TLR4−/− and wild-typemice (Fig. 3B). In contrast, induction of mRNA for chemokinesby rAIM injection was significantly less efficient in TLR4−/− thanin wild-type mice (Fig. 3C). In line with this, phosphorylationlevels of c-Jun N-terminal kinases (JNKs) in epididymal fat,which represent the state of TLR activation, were up-regulatedin wild-type mice but not in TLR4−/− mice (Fig. 3D). Further-more, the rAIM injection increased mRNA levels for M1 mac-rophage markers in epididymal adipose tissue of wild-type butnot TLR4−/− mice, demonstrating that AIM-induced lipolysiscould not recruit inflammatory macrophages into adipose tissuein the absence of TLR4 (Fig. 3E). There was no significantchange in mRNA levels for M2 macrophage markers in bothTLR4−/− and wild-type mice (Fig. 3E). Histological analysisrevealed the presence of IL-6 expressing M1 macrophages afterthe rAIM injection in epididymal adipose tissue of wild-typemice but not of TLR4−/− mice (Fig. S6D).Consistent results were obtained in obese AIM+/+ and AIM−/−
mice after 12 wk on a HFD. In epididymal fat, phosphorylationlevels of JNKs were decreased in AIM−/− mice compared withAIM+/+ mice (Fig. 3F). In addition, chemokine mRNA levels
were also lower in AIM−/− than in AIM+/+ adipose tissue (Fig.3G). Moreover, the serum level of MCP-1 was lower in AIM−/−
than in AIM+/+ mice (Fig. 3H).It is possible that fatty acids effluxed from adipocytes may
stimulate TLR4 expressed not only on adipocytes but also onresident M2 macrophages within adipose tissue in a paracrinefashion and may induce chemokine expression in macrophages.To assess this possibility, we stained epididymal fat from wild-type AIM+/+ mice fed a HFD for 6 wk for MR, a M2 macro-phage marker, and MCP-1. As shown in Fig. S7, both adipocytesand M2 macrophages stained positive for MCP-1. As expected, inAIM−/−mice, neither adipocytes nor resident macrophages showedobvious MCP-1 expression. Thus, in summary, AIM-induced li-polysis provoked the efflux of saturated fatty acids, includingpalmitic and stearic acids, from adipocytes, and these fatty acidsstimulated chemokine production in both adipocytes and resi-dent macrophages via TLR4 activation, resulting in M1 macro-phage migration.
Prevention of Obesity-Associated Inflammation and Insulin Resistancein AIM−/− Mice. As a consequence of abolished infiltration of in-flammatory macrophages, the progression of obesity-associatedinflammation was prevented both locally and systemically inobese AIM−/− mice. In adipose tissue (Fig. 4A) and the liver (Fig.S8), mRNA levels for proinflammatory cytokines, such as TNFα,IL-6, and IL-1β, were significantly lower in AIM−/− than inAIM+/+ mice after a HFD for 12 wk. Consistent with this finding,serum levels of TNFα and IL-6 were lower in AIM−/− micecompared with AIM+/+mice (Fig. 4B).Having observed decreased inflammation in AIM−/− mice, we
next assessed insulin sensitivity in AIM−/− and AIM+/+ mice fed aHFD for 12 wk. Activation of the insulin signaling pathway after i.v.injection of insulin was studied in adipose tissue, skeletal muscle(gastrocnemius), and liver. As shown in Fig. 4C, substantial insulin-stimulated phosphorylation of AKT and GSK3β protein kinases
Fig. 4. Prevented inflammationand normal insulin sensitivity inobese AIM−/− mice. (A) Local in-flammation. QPCR analysis ofmRNA levels for inflammatorycytokine genes in epididymal fattissue from AIM+/+ or AIM−/− micefed a HFD for 0 (lean) or 12 wk(obese). n = 6–8 for each group.Values were presented as relativeexpression to that in lean AIM+/+
mice. Error bar: SEM. (B) Systemicinflammation. Serum TNFα andIL-6 levels are the same as in A. (C)AIM−/− and AIM+/+ mice fed a HFDfor 12 wk (three mice for each)were fasted for 5 h and treatedwith insulin (10 U/kg body weight)via i.p. injection. Within 15 min,epididymal fat, skeletal muscle(gastrocnemius), and liver wereisolated and examined by im-munoblotting for phosphorylatedAKT (p-AKT), total AKT, phos-phorylated GSK3β (p-GSK3β), to-tal GSK3 (α and β), and β-actin. (D)Glucose tolerance test (GTT) andinsulin tolerance test (ITT) per-formed on AIM+/+ and AIM−/−
mice fed a HFD for 0 (lean) or12 wk (obese); n = 6–8 for eachgroup. For ITT, two panels in-cluding absolute blood glucoselevels (Left) and % of the initial(time 0) glucose level (Right) arepresented.
12076 | www.pnas.org/cgi/doi/10.1073/pnas.1101841108 Kurokawa et al.

was observed in all three tissues in AIM−/− mice in contrast to themarkedly diminished phosphorylation levels in AIM+/+ mice.Thus, insulin sensitivity was preserved in obese AIM−/− mice.Consistent with these results, whole-body glucose intolerance andinsulin resistance observed in AIM+/+ mice were found to beameliorated in AIM−/− mice by i.p. glucose and insulin tolerancetests (GTT and ITT, respectively; Fig. 4D). Insulin productionin pancreatic β cells in response to glucose was comparable inAIM−/− and AIM+/+ mice, as assessed in vivo (Fig. S8B) andin vitro using isolated pancreatic Langerhans islets (Fig. S8C).
ConclusionThe present results provide unique and important evidence re-garding the role of AIM in the initiation of chronic inflammationthat connects obesity and insulin resistance. Firstly, macrophagerecruitment into obese adipose tissues requires AIM-induced li-polysis. Augmentation of blood AIM levels may induce vigorouslipolysis in obese adipose tissues, increasing local extracellularfatty acid concentration to a level sufficient for the stimulation ofTLR4, which triggers chemokine production by adipocytes andmacrophage recruitment (summarized in Fig. S9). Although weand others previously reported some related facts underlying thisconclusion, which were observed in a number of different physi-ological and experimental conditions (12, 14, 21–33), we wouldlike to emphasize that this study, which focused on AIM, hasuniquely linked apparently independent elements to a processthat occurs during the progression to obesity.Secondly, adipocyte hypertrophy is not solely sufficient for the
initiation of macrophage infiltration; an increase in blood AIMneeds to be accompanied. In AIM−/− mice, although the level ofAIM-independent lipolysis increases in line with adipocyte hy-pertrophy (14), it may not reach a level sufficient for macrophagerecruitment (Fig. S9). Thirdly, within adipose tissue, crosstalk
between macrophages and adipocytes establishes a vicious cyclethat accelerates inflammation; saturated fatty acids broughtabout by lipolysis activate TLR4 to induce TNFα, which in turnactivates the TNFα receptor to produce inflammatory cytokines/adipokines and chemokines (35). The end point of this responseis further progression of inflammation, lipolysis, and macrophagerecruitment. It is likely that via an increase in lipolysis, AIM maystrengthen this crosstalk, further contributing to the progressionof inflammation (Fig. S9).Thus, this study might not only advance our knowledge about
the events triggering obesity-associated inflammation, but alsoopen a door to the development of next-generation antimeta-bolic therapies via suppression of AIM.
Materials and MethodsMice. AIM−/− mice (13) had been backcrossed to C57BL/6 (B6) for 13 gen-erations before used for experiments. HFD (HFD32, fat kcal: 60%) was pur-chased from CREA. TLR4−/− mice (36) were kindly provided from Drs. S. Akira(Osaka University, Osaka, Japan) and K. Miyake (The Institute of MedicalScience, University of Tokyo, Tokyo, Japan). All mice were maintained undera specific pathogen free condition.
Statistical Analysis. A two-tailed Mann-Whitney test was used to calculateP values. ***P < 0.001, **P < 0.01, *P < 0.05. Error bars: SEM.
Please see SI Materials and Methods for further details.
ACKNOWLEDGMENTS. We thank Genostaff Inc. for technical assistance inhistology. This work was supported by Grants-in-Aid for Scientific Research(B) (Japan Society for the Promotion of Science), the Global Centers ofExcellence (COE) Program (T.M.), Kanae Foundation for the Promotion ofMedical Science, the Astellas Foundation for Research on MetabolicDisorders, and the Ono Medical Research Foundation (S.A.).
1. Hotamisligil GS, Shargill N-S, Spiegelman B-M (1993) Adipose expression of tumornecrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 259:87–91.
2. Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adiposetissue. J Clin Invest 112:1785–1788.
3. Arkan MC, et al. (2005) IKK-beta links inflammation to obesity-induced insulin re-sistance. Nat Med 11:191–198.
4. Shoelson SE, Lee J, Goldfine AB (2006) Inflammation and insulin resistance. J ClinInvest 116:1793–1801.
5. Weisberg SP, et al. (2003) Obesity is associated with macrophage accumulation inadipose tissue. J Clin Invest 112:1796–1808.
6. Xu H, et al. (2003) Chronic inflammation in fat plays a crucial role in the developmentof obesity-related insulin resistance. J Clin Invest 112:1821–1830.
7. Solinas G, et al. (2007) JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab 6:386–397.
8. Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nat Rev Im-munol 5:953–964.
9. Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adi-pose tissue macrophage polarization. J Clin Invest 117:175–184.
10. Ozcan U, et al. (2004) Endoplasmic reticulum stress links obesity, insulin action, andtype 2 diabetes. Science 306:457–461.
11. Kahn SE, Hull RL, Utzschneider KM (2006) Mechanisms linking obesity to insulin re-sistance and type 2 diabetes. Nature 444:840–846.
12. Kosteli A, et al. (2010) Weight loss and lipolysis promote a dynamic immune responsein murine adipose tissue. J Clin Invest 120:3466–3479.
13. Miyazaki T, Hirokami Y, Matsuhashi N, Takatsuka H, Naito M (1999) Increased sus-ceptibility of thymocytes to apoptosis in mice lacking AIM, a novel murine macrophage-derived soluble factor belonging to the scavenger receptor cysteine-rich domain su-perfamily. J Exp Med 189:413–422.
14. Kurokawa J, et al. (2010) Macrophage-derived AIM is endocytosed into adipocytesand decreases lipid droplets via inhibition of fatty acid synthase activity. Cell Metab11:479–492.
15. Joseph SB, et al. (2004) LXR-dependent gene expression is important for macrophagesurvival and the innate immune response. Cell 119:299–309.
16. Valledor AF, et al. (2004) Activation of liver X receptors and retinoid X receptorsprevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci USA 101:17813–17818.
17. Gebe JA, et al. (1997) Molecular cloning, mapping to human chromosome 1 q21-q23,and cell binding characteristics of Spalpha, a new member of the scavenger receptorcysteine-rich (SRCR) family of proteins. J Biol Chem 272:6151–6158.
18. Kim WK, et al. (2008) Glycoproteomic analysis of plasma from patients with atopicdermatitis: CD5L and ApoE as potential biomarkers. Exp Mol Med 40:677–685.
19. Gray J, et al. (2009) A proteomic strategy to identify novel serum biomarkers for liver cir-rhosis and hepatocellular cancer in individuals with fatty liver disease. BMC Cancer 9:271.
20. Arai S, et al. (2005) A role for the apoptosis inhibitory factor AIM/Spalpha/Api6 inatherosclerosis development. Cell Metab 1:201–213.
21. Shi H, et al. (2006) TLR4 links innate immunity and fatty acid-induced insulin re-sistance. J Clin Invest 116:3015–3025.
22. Suganami T, et al. (2007) Role of the Toll-like receptor 4/NF-kappaB pathway in sat-urated fatty acid-induced inflammatory changes in the interaction between adipo-cytes and macrophages. Arterioscler Thromb Vasc Biol 27:84–91.
23. Poggi M, et al. (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation areprotected against the development of insulin resistance in white adipose tissue inresponse to a high-fat diet. Diabetologia 50:1267–1276.
24. Tsukumo DM, et al. (2007) Loss-of-function mutation in Toll-like receptor 4 preventsdiet-induced obesity and insulin resistance. Diabetes 56:1986–1998.
25. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME (2008) Tlr-4 deficiency selectivelyprotects against obesity induced by diets high in saturated fat. Obesity (Silver Spring)16:1248–1255.
26. Kamei N, et al. (2006) Overexpression of monocyte chemoattractant protein-1 inadipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem281:26602–26614.
27. Kanda H, et al. (2006) MCP-1 contributes to macrophage infiltration into adiposetissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494–1505.
28. Keophiphath M, Rouault C, Divoux A, Clément K, Lacasa D (2010) CCL5 promotesmacrophage recruitment and survival in human adipose tissue. Arterioscler ThrombVasc Biol 30:39–45.
29. Soma MR, Mims MP, Chari MV, Rees D, Morrisett JD (1992) Triglyceride metabolism in3T3-L1 cells. An in vivo 13C NMR study. J Biol Chem 267:11168–11175.
30. Kopp A, et al. (2009) Fatty acids as metabolic mediators in innate immunity. Eur J ClinInvest 39:924–933.
31. Schaeffler A, et al. (2009) Fatty acid-induced induction of Toll-like receptor-4/nuclearfactor-kappaB pathway in adipocytes links nutritional signalling with innate immu-nity. Immunology 126:233–245.
32. Jenkins KA, Mansell A (2010) TIR-containing adaptors in Toll-like receptor signalling.Cytokine 49:237–244.
33. Zechner R, Strauss JG, Haemmerle G, Lass A, Zimmermann R (2005) Lipolysis: Pathwayunder construction. Curr Opin Lipidol 16:333–340.
34. Nishino N, et al. (2008) FSP27 contributes to efficient energy storage in murine whiteadipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest 118:2693–2696.
35. Schäffler A, Schölmerich J, Salzberger B (2007) Adipose tissue as an immunologicalorgan: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol 28:393–399.
36. Hoshino K, et al. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice arehyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product.J Immunol 162:3749–3752.
Kurokawa et al. PNAS | July 19, 2011 | vol. 108 | no. 29 | 12077
MED
ICALSC
IENCE
S





























