Embed Size (px)
Citation preview

1
AMINOPEPTIDASE FINGERPRINTS. AN INTEGRATED APPROACH FOR IDENTIFICATION OF GOOD SUBSTRATES AND OPTIMAL INHIBITORS.
Marcin Drag*,1,2, Matthew Bogyo3, Jonathan A. Ellman4 and Guy S. Salvesen1
From 1Apoptosis and Cell Death Research, The Burnham Institute for Medical Research, La Jolla, CA 92037, USA; 2Division of Medicinal Chemistry and Microbiology, Faculty of Chemistry, Wroclaw University of Technology, Wybrzeze Wyspianskiego 27, 50-370
Wroclaw, Poland; 3 Department of Pathology, Stanford School of Medicine, 300 Pasteur Drive, Stanford, CA 94305, USA; 4Department of Chemistry, University of California,
Berkeley, California 94720, USA Running title: Substrate specificity of aminopeptidases
Address correspondence to: Marcin Drag, Ph.D., Guy Salvesen, Ph.D., Apoptosis and Cell Death Research, The Burnham Institute for Medical Research, La Jolla, CA 92037, USA; Tel.: 858-646-3100; E-mail: [email protected], [email protected]
Aminopeptidases process the N-terminal amino acids of target substrates by sequential cleavage of one residue at a time. They are found in all cells compartments of prokaryotes and eukaryotes being implicated in the major proteolytic events of cell survival, defense, growth and development. We present a new approach for the fast and reliable evaluation of the substrate specificity of individual aminopeptidases. Using solid phase chemistry with the ACC fluorophore we have synthesized a library of 61 individual natural and unnatural amino acids substrates, chosen to cover a broad spectrum of the possible interactions in the S1 pocket of this type of protease. As proof of concept we determined the substrate specificity of human, pig and rat orthologs of aminopeptidase N (CD13), a highly conserved cell surface protease that inactivates enkephalins and other bioactive peptides. Our data reveal a large and hydrophobic character for the S1 pocket of APN that is conserved with APNs. Our approach, which can be applied in principle to all aminopeptidases, yields useful information for the design of specific inhibitors, and more importantly reveals a relationship between the kinetics of substrate hydrolysis and the kinetics of enzyme inhibition.
The largest group of aminopeptidases, proteases that remove amino acids from the N-terminus of target substrates one residue at a time, are members of the metallopeptidases (1). They are found in most cell compartments of prokaryotes and eukaryotes, and are implicated in several
major cell fates such as maintenance of the differentiated state, defense against pathogens, growth and development (2-4). A classic aminopeptidase activity is the removal of the N-terminal Met after the initiation of translation by methionine aminopeptidase (5,6). In addition to their primary proteolytic function, aminopeptidases also play secondary roles as viral or toxin receptors, transcriptional repressors and vesicular trafficking controllers (7-9). The activity of well known aminopeptidases, and the characterization of new aminopeptidases, is generally determined using synthetic substrates containing one of a few natural amino acids attached to chromogenic or fluorogenic leaving groups (10,11). For example, the M1 clan (Alanine Aminopeptidases) is assayed using fluorogenic peptide substrates containing a P1 Ala, and the M17 clan (Leucine Aminopeptidases) using fluorogenic peptide substrates containing a P1 Leu (12-14). But this type of evaluation of enzyme specificity gives limited information about the profile of the S1 pocket, a primary determinant in the absolute specificity of aminopeptidases for their natural substrates.
A number of general substrate-based library screening methods have been developed for the fast and reliable determination of protease specificity (15-20). Importantly, for the cysteine, serine and threonine proteases, substrate-specificity data provides valuable information that can be directly used for the construction of specific inhibitors (18,21). Although exceptions exist, the general principle is that replacing the
http://www.jbc.org/cgi/doi/10.1074/jbc.M109.060418The latest version is at JBC Papers in Press. Published on November 30, 2009 as Manuscript M109.060418
Copyright 2009 by The American Society for Biochemistry and Molecular Biology, Inc.
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

2
leaving group of a good substrate with an inhibitor “warhead” should yield a potent and specific inhibitor (22-24). There is a presumptive direct relationship between optimal synthetic substrates and potent synthetic inhibitors. The transposition of substrate data to inhibitor design of the two protease groups represented by the metallo- and aspartyl proteases has not been used as often, however good correlations have also been observed between substrate cleavage efficiency and inhibition for inhibitors incorporating phosphonamidate transition-state analogs (25).
We present an approach for the fast and reliable evaluation of the substrate specificity of individual aminopeptidases. Using solid phase chemistry with the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore we synthesized a library of around sixty individual fluorogenic substrates, chosen to cover a broad spectrum of possible interactions within the specificity-determining S1 pocket of this type of protease. This library has utility in determining substrate specificity fingerprints that can be used to predict the shape of the S1 pocket or to design specific inhibitors. Furthermore, in the absence of structural information, such data can be used for in silico construction of specific interaction maps of the binding pockets of the aminopeptidases. The approach can also yield information about of the species specificity of aminopeptidases. Problems with generating inhibitors, especially in the synthesis of enantiomers and diastereomers, makes this approach a method of choice to select the best candidates for further inhibitor synthesis without the need of tedious generation of inhibitors libraries. We provide here substrate specificity data obtained for three mammalian aminopeptidases (human, rat and pig aminopeptidase N, E.C. 3.4.11.2.). We also demonstrate a different relationship between substrate kinetic parameters and inhibitor potency than the generally accepted one reported for covalent serine and cysteine and non-covalent phosphonyl-based metallo endopeptidase inhibitors. The general outline of the methodology is shown in Figure 1.
Experimental Procedures Materials and methods: All chemicals and solvents were obtained from commercial suppliers
and used without further purification, unless otherwise stated. Anhydrous N,N-dimethyl formamide (DMF) was from Sigma-Aldrich. Rink amide resin was purchased from Novabiochem. 1H-NMR spectra were obtained with the aid of the Burnham Structural Biology facility using a Varian 300 spectrometer in dimethyl sulfoxide-d6 (Aldrich). 1H-NMR (300 MHz) spectra are reported as follows: chemical shifts in ppm downfield from TMS, the internal standard; resonance signal description (s, singlet; d, doublet; t, triplet; m, multiplet), integration, and coupling constant (Hz). Analytical high performance liquid chromatography (HPLC) analysis used a Beckman-Coulter System Gold 125 solvent delivery module equipped with a Beckman-Coulter System Gold 166 Detector system by using a Varian Microsorb-MV C18 (250 x 4.8 mm) column. Preparative HPLC analysis used a Beckman-Coulter System Gold 126P solvent delivery module equipped with a Beckman-Coulter System Gold 168 Detector system with a Kromasil 100-10 C18 (20 mm ID) column (Richard Scientific). Solvent composition system A (water/0.1%TFA) and system B (acetonitrile/water 80%/20% with 0.1% of TFA). LC-MS data were recorded with the aid of the Burnham Medicinal Chemistry facility using Shimadzu LCMS-2010EV system. The solid phase substrate library was synthesized using a semiautomatic FlexChem Peptide Synthesis System (Model 202). Enzymatic kinetic studies were performed using Spectra MAX Gemini EM fluorimeter (Molecular Devices) operating in the kinetic mode in 96-well plates. Native human, pig and rat aminopeptidase N (CD13) were purchased from Calbiochem (La Jolla). Synthesis of the substrate library. Fmoc-ACC fluorophore was coupled to Rink Amide Resin (Novabiochem) using a procedure described earlier(26). The Fmoc protecting group was removed using 20% piperidine / DMF (4min + 25min). The resin was washed 6 times with DMF, 3 times with dichloromethane, 3 times with tetrahydrofurane and 3 times with MeOH and dried for 24h in vacuum. The resin containing fluorophore was distributed to separate wells (100 mg per well, 0.039mmol, 1eq.) of a 96 well cartridge in the semi-automatic FlexChem synthesizer and swelled in DMF for 4h. Coupling of the Fmoc or Boc N-terminally protected amino acids (3eq., 0.117 mmol) was carried out in DMF
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

3
(1.5 mL) as described earlier using HATU (3eq., 0.117 mmol) as coupling reagent and in the presence of the 2,4,6-collidine (3eq., 0.117 mmol) base for 24h at r.t. Subsequently, the resin was washed with DMF (5 x 2 mL) and the wells containing Fmoc N-terminally protected amino acids was treated with 20% piperidine / DMF (4min + 25min) and washed with DMF (3 x 2 mL). The resin was washed with dichloromethane (3 x 2 mL), tetrahydrofurane (3 x 2 mL) and MeOH (3 x 2 mL) and dried for 24h under vacuum. Cleavage of the substrates was carried out for 1.5h at r.t. using a mixture of 92.5% TFA, 2.5% water, 2.5% triisopropyl silane and 2.5% phenol (1.5 mL per well). After cleavage, the substrates were precipitated for 10 min at 0°C using tert-butyl-methyl ether (12 mL) and centrifuged. This operation was performed once more. The remaining residues containing fluorogenic substrates were purified using reverse-phase HPLC preparatory chromatography (CH3CN/H2O-0.1% TFA, mode: 0-80% for 45 min and 80-100% for 15 min, 10 mL/min, 210 and 254 nm detection). The purity of the ACC substrates was confirmed by analytical reverse-phase HPLC analysis (CH3CN/H2O-0.1% TFA, 0-90% for 25 min, 1.0 mL/min, 210 nm detection). All the compounds were lyophilized for 48h and analyzed using 1H NMR and LC-MS methods. Subsequently all ACC substrates were dissolved as stock solution at concentration of 50 mM in biological purity DMSO and frozen at -80°C. See table in Supplementary Materials for detailed analysis details. α-Aminoalkylphosphonic acid inhibitors. The appropriate α-aminoalkylphosphonic acids were obtained as described earlier (10,11). The purity of the inhibitors was confirmed by the 1H and 31P NMR spectra. All compounds were racemic mixtures. Assay of the substrate library. Each APN was assayed in a 100 mM Tris-HCl, pH 7.5. The buffer was made at 23°C and assays were performed at 37°C. All enzymes were preincubated for 30 min at 37°C before adding to the wells containing substrate. Final screening of the library was carried out at 1µM substrate concentration and enzymes were between 0.2-5 nM. Release of fluorophore was monitored continuously with excitation at 355 nm and emission at 460 nm.
Total assay time was generally 15-30 min minutes and the linear portion of the progress curve was used to calculate velocity. All experiments were repeated at least twice and the data presented are the average with the bars describing the data ranges. Analysis of the results was based on total RFU (Relative Fluorescence Unit) for each substrate setting the highest value to 100% and adjusting the other results accordingly.
Kinetic parameters (kcat, Km and kcat/Km) determination of individual substrates. Substrates were screened against APNs at 37°C in the above assay buffer. Buffers were prepared at 22°C. Enzymes were preincubated for 30 min at 37°C before adding substrate to the wells of a 96-well plate reader operating in the kinetic mode. Absolute ACC concentrations were calculated by the hydrolysis of three independent ACC coupled substrates at known concentration and average value was determined(27). Enzyme assay conditions were as follows: (100 µL reaction), eight different substrate concentration and enzymes at 0.2 - 5 nM. Release of ACC fluorophore was monitored as above. Each experiment was repeated at least three times and the results presented as an average with the error bars describing the standard deviation. Final substrate concentrations for of kcat/Km determination ranged from 0.25 to 500 µM. Concentration of DMSO in the assay was less than 1% (v/v).
Assay procedure for phosphonate inhibitors. Inhibitors were screened against appropriate APNs at 37°C in the above assay buffer. Buffers were prepared at 22°C. Enzymes were preincubated for 30 min at 37°C with selected inhibitor before adding substrate (Ala-ACC, final concentration equal to 50µM) to the wells of a 96-well plate reader operating in the kinetic mode. Enzyme assay conditions were as follows: (100 µL reaction), eight different inhibitor concentrations and enzymes at 0.2 - 5 nM. Release of ACC fluorophore was monitored as above. Each experiment was repeated at least three times and the results presented as an average. Ki values was calculated from Cheng-Prusoff equation (28): Ki = IC50 / [1 + (S / Km)], where IC50 is the concentration of inhibitor that delivers 50% inhibition, S is the substrate concentration used in the assay, and Km is the Michaelis constant
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

4
determined for each enzyme/substrate pair. The concentration of DMSO in the assays was less than 1% (v/v).
Intensity Heat Map Analysis. The results from the substrate library assays for APN were analyzed using CLUSTER software. Maximum activity rates were set at 100%, and amino acids that showed no activity were assigned as 0%. The results were analyzed with the program CLUSTER and were visualized using TreeView software as heat map diagrams showing 100% activity as red and 0% activity as black.
RESULTS Design and synthesis of the library. Initially, we synthesized substrates containing all the natural amino acids (except cysteine, because of its susceptibility to oxidation) as well as a diverse set of unnatural amino acids (Supplementary data Table 1). The synthesis was carried out according to previously described methodology using a semiautomatic FlexChem synthesizer. We utilized 7-amino-4-carbamoylmethylcoumarin (ACC) as the leaving group since peptides containing this fluorescent reporter can be synthesized using solid phase methods as described previously (26). Most of the compounds contain an unblocked α-amino group to satisfy the primary specificity of the APNs. However, we also synthesized a few substrates with diverse functionalities (for example, an α-hydroxy group, secondary amine derivatives, or an amine group in other than the α position), to determine how this would influence substrate recognition by the APNs. Following synthesis, all of the compounds were purified using reverse-phase preparative HPLC and analyzed by spectroscopic methods (1H NMR and LC-MS).
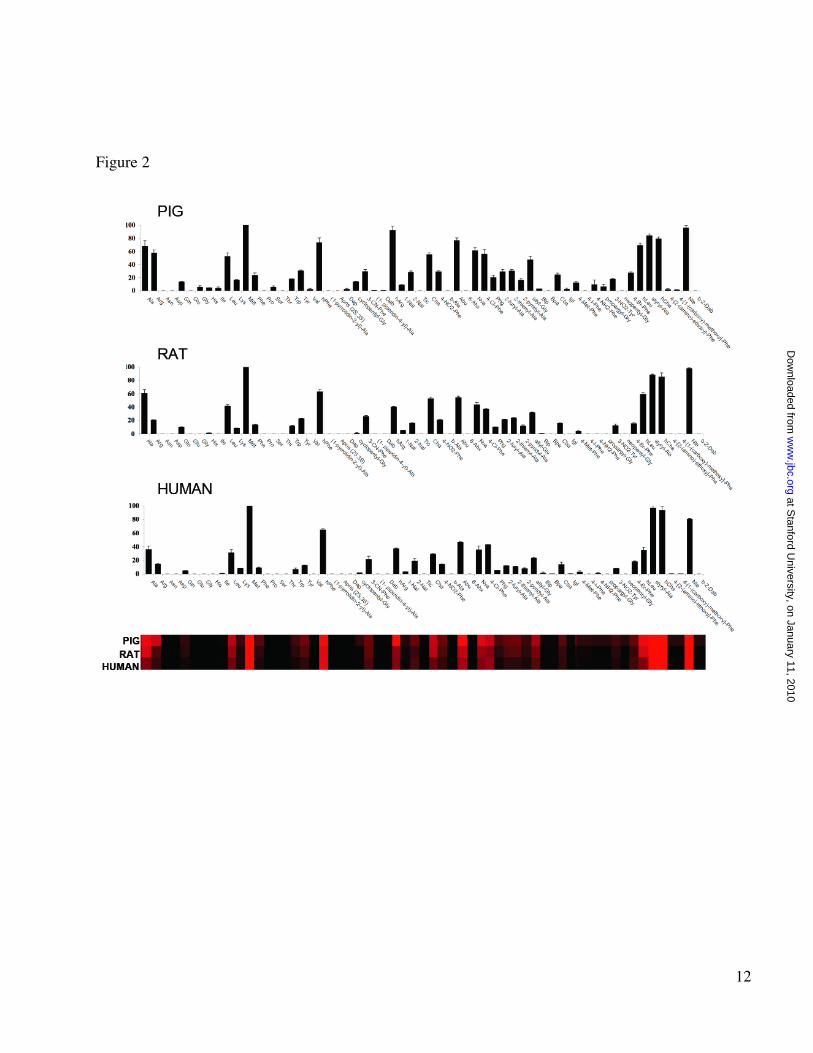
Screening of the substrate library. We performed a preliminary screen of the library to obtain an initial data set with which to establish optimal experimental conditions. Next, we calculated Km values for each substrate using two representative APNs (pig and human), primarily to establish conditions where substrate saturation of the enzymes is not a factor. Subsequently, we screened the library against each enzyme using substrate concentrations that were at least two-fold
lower than the lowest Km for the substrate in the library. Final screening of the library was carried out at 1µM final substrate concentration, which is sufficiently below Km to ensure that velocity data are proportional to kcat/ Km. Every substrate in the library is dissolved in DMSO at a predetermined concentration. Before screening we load an equal volume of each substrate at a given concentration. Subsequent addition of the enzyme in the buffer solution to each of 61 wells allows us to obtain the same concentration of each substrate during screening. Data for the three tested APNs are shown in Figure 2. The enzymes displayed a broad tolerance for several natural and un-natural amino acids, and the overall substrate selectivity was similar for all three enzymes, suggesting a conserved level of structure organization. The most preferred natural amino acids were Met, Ala and Leu, while a striking preference was observed for the non-natural amino acids Nle, Abu, hCha, styrylAla, hArg and hPhe. With the exception of Ala, which has previously been used in substrates of APNs (an alternate name of the enzymes is Alanine Aminopeptidase), the most favored amino acids have rather large hydrophobic side chains. This suggests that the S1 pocket of the enzyme is likely to be open and fits with reports describing potent inhibitors (primarily for pig APN) with bulky P1 residues (11,29-31). The basic amino acids Arg and hArg are also among the preferred amino acids. This is not surprising given recent structural analysis of leukotriene A4 hydrolase/aminopeptidase, which belongs to the same family as APN (32). LTA4H almost equally processed Ala and Arg substrates coupled to the pNA chromophore. Finally, as expected based on the predicted specificity of these enzymes, only substrates with the free α-amino group were cleaved. Comparison of inhibitor and substrate preferences. Recent work with non-peptide, drug like substrates has show that specificity information from substrate libraries can effectively be translated into the design of potent and selective inhibitors (23,24). This method involves conversion of substrates to inhibitors by swapping out the fluorogenic leaving group for a reactive inhibitor “warhead”. For some classes of mechanism-based inhibitor warheads, the velocity (kcat/Km) values can be used to predict the best scaffolds for design of inhibitors. Therefore, we compared our substrate preference data with
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

5
previously published reports on APN inhibitors. Overall, there was strong correlation between the residues found in some inhibitor compounds (Met or hPhe) with those found in our best substrates (30,31). However, other residues such as Ala, were efficiently processed in our substrates, but are among the worst reported inhibitors for pig APN (30). Therefore, we concluded that good candidate APN inhibitors cannot simply be predicted by substrate velocity (kcat/Km) data. This was quite surprising because comparison of peptide substrates and corresponding inhibitors incorporating alkylating groups and transition-state analogs has shown that the best substrates yielded the best inhibitors (21,23,33). The apparent conflict prompted us to further investigate the relationship.
We hypothesized that instead of the crucial kcat parameter, which describes the turnover speed of substrate processing, we should instead focus on the Km value, which reflects the strength of the binding of the substrate in the active site. Therefore we decided to further evaluate two of the three APNs: pig APN because most of the studies performed to date on inhibitor design have used this enzyme, and human APN because this is the primary pharmaceutical target for inhibitors. When we analyzed substrates for Km (expressed as 1/Km - Figure 3 and Supplementary Material) we observed a substantial difference in preferences compared to the velocity (kcat/Km) data presented in Figure 2. For Pig APN, although a preference for bulky residues (hPhe, Met, Nle or hCha) is evident and matches the velocity data, the Km data shows that residues with small side-chains that score well in the velocity experiments (Ala, Abu, Nva) show overall weak binding constants. Analysis of human APN reveals a similar substrate preference pattern but with strong preference for two amino acids, namely hCha and hPhe.
Detailed relationship between substrates and inhibitors. In light of the differences revealed by velocity (kcat/Km) and binding (Km) data, we wanted to understand which parameters would more closely predict inhibitor behavior. α-Aminoalkilphosphonates are competitive, reverse inhibitors well described in the literature for their activity to efficiently inhibit all the aminopeptidases, thus ideal for our investigations (10,11,34). Previous reports on α-amino
phosphonate activity towards pig APN show that these inhibitors are generally fast binding reversible inhibitors, which we concur with (Supplementary data Figures 4 and 5)(10,11). We therefore selected a series of sequences and converted them into inhibitors using the α-amino phosphonate reactive group to replace the ACC fluorophore of the substrates. We chose one poor substrate (Ala), one average substrate (Nva) and three optimal substrates (hPhe, hCha and Nle) (Figure 4). For these compounds we obtained kcat, Km and kcat/Km values and compared them to Ki values for both APNs (Table 1). A clear linear relationship is apparent between kcat values and the inhibitory constant Ki while the relationship between Ki and kcat/Km is non-linear (Figure 5C). Further analysis revealed that kcat/Km versus Ki more could fit a non-linear model (R = 0.88) for pig APN and (R = 0.92) for human APN (Figure 5D), although the relationship does not fit any simple or even reproducible equation. However, the relationship between kcat and Ki was opposite to that expected, since the best inhibitors (low Ki) had the lowest kcat values (Figure 5B). Instead, we found that the best and most predictive parameter was Km, which showed a linear relationship with Ki and strong correlation coefficients for both pig and human APNs (Figure 5A). Log plots of Km versus Ki (not shown) revealed slopes of 1.00 (R = 0.98) for pig APN and 1.08 (R = 0.99) for human APN, suggesting that binding modes of the inhibitor and substrate are essentially identical, such that the phosphonate group of the inhibitors binds in the same way as the amide of the substrates. Additional comparisons of substrate Km values with a different class of previously published pig APN inhibitors (phosphinic acids) showed the same trend confirming the utility of this type of analysis (data not shown).
We noted that human APN is more than 50-fold less active compared to pig APN in terms of kcat/Km. This lower activity of the human enzyme is mainly due to a reduced kcat, suggesting that the in vivo activity of this enzyme might be differently regulated compared to the pig ortholog. Our results also provide direct comparisons of the Ki data for both enzymes, and reveal that all inhibitors are optimal for the human APN (Table 1). In particular substrates containing hPhe and hCha, are more than ten times more potent for human APN compared with their Ki values for pig APN. To our knowledge this is the first direct
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

6
comparison of the inhibitory profiles for these APNs, as the human analogue was not previously a target of detailed chemical screening with substrates or inhibitors. However, our comparisons indicate that pig APN is a good model for the preliminary design of inhibitors for its human ortholog.
DISCUSSION
Aminopeptidases are key regulators of many intra- and extra-cellular events and several of them are attractive pharmaceutical targets. Clearly, selective inhibitors are required for studies of their function both in vitro and in vivo. A pertinent example of the need for selective inhibitors is the recently characterized malaria parasite neutral aminopeptidases, which have been suggested to be relevant pharmaceutical targets (12,14,35). Since human homologs exist that play essential roles in the host, a highly selective inhibitor for the malarial aminopeptidases will be required. Here we present a library of fluorogenic substrates that can be utilized for fast and reliable determination of individual aminopeptidase substrate preferences, and for the subsequent design of selective inhibitors. One benefit of library screening with a wide variety of natural and synthetic amino acids is that it allows for estimations of the size and shape of the S1 pocket of the investigated enzyme. In the case of APNs, their activity is high on Ala derivatives, which at first glance seems to suggest a restricted pocket. However our data, both in terms of Km and kcat/Km demonstrate that the S1 pocket must be large enough to accommodate bulky hydrophobic residues. Indeed, based on the assumption that Km reflects the ground state binding of the side chains in S1, our data suggests that the APNs bind Ala-ACC weakly, yet show rapid turnover of the substrate. This result could be explained if residues with small side chains are better positioned for catalysis than ones with large side chains, but it is clear that the S1 pocket must be quite large.
Our studies have focused on the S1 pocket, but it is also likely that enhanced interactions with natural peptide and protein substrates utilize interactions to the C-terminal side (S’ side) of the scissile bond. However, libraries designed to scan these positions in the aminopeptidases are likely to
be difficult to design and synthesize. This problem has been overcome for other proteases using internally quenched substrates, but this approach cannot be used because aminopeptidases strictly require only a single free amino acid at the site of hydrolysis. The problem could be solved by the selection of optimal S1 binders using our approach, followed by synthesis of inhibitors, rather than substrates, extended toward S’ pockets. Phosphinate chemistry affords this opportunity, because optimal S1 amino acids derivatized with phopshinates can be extended into the S’ pockets to generate inhibitor libraries (36-39). In addition, information obtained from our substrate library screening data can be also used for the design of specific Activity Based Probes (ABPs). These reagents have attracted considerable interest due to their versatile application for the detection of the enzyme activity both in vitro and in vivo (40-42). In the case of cysteine, serine, and threonine proteases ABPs react covalently with the enzyme and go through the transition state. For these compounds, kcat/Km values of substrates correlate well with their inhibitory constants. Unfortunately metalloproteases are difficult targets for design of ABPs since they cannot be covalently modified in the active site because they do not react with the activated water molecule of the active site, but rather chelate the zinc ion. For this reason enzyme is never found in the transition state and remains in the ground state, and thus the Km of the substrates used to design the inhibitor is dominant. This disadvantage has been overcome using specific photo cross-linking groups that form stable bonds with residues outside the active site. However, this chemistry very often leads to substantial nonspecific labeling. Identification of the optimal binding partners using the substrate-inhibitor approach such as presented here may decrease the amount of these artifacts during labeling as recently demonstrated for several Matrix Metalloproteases (43).
In addition to providing information about substrate specificity and inhibitor design, our approach has highlighted an intriguing relationship between catalysis and inhibitor efficacy. Previous work using other classes of proteases and reactive functional groups has shown that substrates with high kcat/Km (velocity) values can be converted to optimal inhibitors simply by replacing the substrate fluorophore with the desired inhibitor warhead. In fact, the high correlation for metalloproteasess such as
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

7
thermolysin between peptide substrates and their corresponding noncovalent, phosphonyl-based transition-state inhibitors, suggested that a similar correlation might also have been expected here (25). Instead, we observed an almost opposite correlation where Km dominates the relationship. Indeed, such a relationship has been observed before, with the peptidyl fluoroalkane inhibitors of thermolysin where inhibition robustly correlates with Km of the corresponding substrate (44). Our results strongly suggest that for the aminopeptidases, the amino acyl phosphonates are likely to bind the ground state of the enzyme. Ground state binding is dominated by the dissociation constant Ks which itself is related to the observed quantity Km.
Depending on the kinetic relationship between type of warhead and corresponding
substrate, inhibitors are sometimes classified as kcat-type (for example the fast, irreversible, ketone-based inactivators of serine and cysteine proteases) or kcat/Km-type (for example the phosphonamidate-based metalloprotease inhibitors) (25,45). Because our inhibitor binding efficiency is related to Km, the term “Km-type inhibitors” would be more reasonable for this class of non-covalent effectors (Figure 6). In conclusion, this study provides a rational explanation for the design of ground state-based, inhibitors for specific groups of enzymes and inhibitor pharmacophores using specific kinetic parameters. The results also demonstrate the importance of choosing the appropriate kinetic parameter from a substrate library screen, before applying this to inhibitor design.
REFERENCES 1. Lowther, W. T., and Matthews, B. W. (2002) Chem Rev 102, 4581-4608 2. Taylor, A. (1993) FASEB J 7, 290-298 3. Saiki, I., Fujii, H., Yoneda, J., Abe, F., Nakajima, M., Tsuruo, T., and Azuma, I. (1993) Int J Cancer
54, 137-143 4. Bradshaw, R. A., and Yi, E. (2002) Essays Biochem 38, 65-78 5. Bradshaw, R. A., Brickey, W. W., and Walker, K. W. (1998) Trends Biochem Sci 23, 263-267 6. Li, X., and Chang, Y. H. (1995) Proc Natl Acad Sci U S A 92, 12357-12361 7. Delmas, B., Gelfi, J., L'Haridon, R., Vogel, L. K., Sjostrom, H., Noren, O., and Laude, H. (1992)
Nature 357, 417-420 8. Yeager, C. L., Ashmun, R. A., Williams, R. K., Cardellichio, C. B., Shapiro, L. H., Look, A. T., and
Holmes, K. V. (1992) Nature 357, 420-422 9. Petrovic, N., Schacke, W., Gahagan, J. R., O'Conor, C. A., Winnicka, B., Conway, R. E., Mina-
Osorio, P., and Shapiro, L. H. (2007) Blood 110, 142-150 10. Lejczak, B., Kafarski, P., and Zygmunt, J. (1989) Biochemistry 28, 3549-3555 11. Drag, M., Grembecka, J., Pawelczak, M., and Kafarski, P. (2005) Eur J Med Chem 40, 764-771 12. Skinner-Adams, T. S., Lowther, J., Teuscher, F., Stack, C. M., Grembecka, J., Mucha, A., Kafarski,
P., Trenholme, K. R., Dalton, J. P., and Gardiner, D. L. (2007) J Med Chem 50, 6024-6031 13. Stack, C. M., Lowther, J., Cunningham, E., Donnelly, S., Gardiner, D. L., Trenholme, K. R.,
Skinner-Adams, T. S., Teuscher, F., Grembecka, J., Mucha, A., Kafarski, P., Lua, L., Bell, A., and Dalton, J. P. (2007) J Biol Chem 282, 2069-2080
14. Cunningham, E., Drag, M., Kafarski, P., and Bell, A. (2008) Antimicrob Agents Chemother 15. Backes, B. J., Harris, J. L., Leonetti, F., Craik, C. S., and Ellman, J. A. (2000) Nat Biotechnol 18,
187-193 16. Harris, J. L., Backes, B. J., Leonetti, F., Mahrus, S., Ellman, J. A., and Craik, C. S. (2000) Proc Natl
Acad Sci U S A 97, 7754-7759 17. Harris, J. L., Niles, A., Burdick, K., Maffitt, M., Backes, B. J., Ellman, J. A., Kuntz, I., Haak-
Frendscho, M., and Craik, C. S. (2001) J Biol Chem 276, 34941-34947 18. Choe, Y., Leonetti, F., Greenbaum, D. C., Lecaille, F., Bogyo, M., Bromme, D., Ellman, J. A., and
Craik, C. S. (2006) J Biol Chem 281, 12824-12832
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

8
19. Drag, M., Mikolajczyk, J., Bekes, M., Reyes-Turcu, F., Ellman, J., Wilkinson, K., and Salvesen, G. (2008) Biochem J
20. Drag, M., Mikolajczyk, J., Krishnakumar, I. M., Huang, Z., and Salvesen, G. S. (2008) Biochem J 409, 461-469
21. Rano, T. A., Timkey, T., Peterson, E. P., Rotonda, J., Nicholson, D. W., Becker, J. W., Chapman, K. T., and Thornberry, N. A. (1997) Chem Biol 4, 149-155
22. Coombs, G. S., Rao, M. S., Olson, A. J., Dawson, P. E., and Madison, E. L. (1999) J Biol Chem 274, 24074-24079
23. Wood, W. J., Patterson, A. W., Tsuruoka, H., Jain, R. K., and Ellman, J. A. (2005) J Am Chem Soc 127, 15521-15527
24. Patterson, A. W., Wood, W. J., and Ellman, J. A. (2007) Nat Protoc 2, 424-433 25. Mader, M. M., and Bartlett, P. A. (1997) Chem Rev 97, 1281-1302 26. Maly, D. J., Leonetti, F., Backes, B. J., Dauber, D. S., Harris, J. L., Craik, C. S., and Ellman, J. A.
(2002) J Org Chem 67, 910-915 27. Schneider, E. L., and Craik, C. S. (2009) Methods Mol Biol 539, 59-78 28. Cheng, Y., and Prusoff, W. H. (1973) Biochem Pharmacol 22, 3099-3108 29. Fournie-Zaluski, M. C., Coric, P., Turcaud, S., Bruetschy, L., Lucas, E., Noble, F., and Roques, B.
P. (1992) J Med Chem 35, 1259-1266 30. Chen, H., Roques, B. P., and Fournie-Zaluski, M. C. (1999) Bioorg Med Chem Lett 9, 1511-1516 31. Drag, M., Grzywa, R., and Oleksyszyn, J. (2007) Bioorg Med Chem Lett 17, 1516-1519 32. Tholander, F., Muroya, A., Roques, B. P., Fournie-Zaluski, M. C., Thunnissen, M. M., and
Haeggstrom, J. Z. (2008) Chem Biol 15, 920-929 33. Morgan, B. P., Scholtz, J. M., Ballinger, M. D., Zipkin, I. D., and Bartlett, P. A. (1991) Journal of
the American Chemical Society 113, 297-307 34. Giannousis, P. P., and Bartlett, P. A. (1987) J Med Chem 30, 1603-1609 35. Gardiner, D. L., Trenholme, K. R., Skinner-Adams, T. S., Stack, C. M., and Dalton, J. P. (2006) J
Biol Chem 281, 1741-1745 36. Vassiliou, S., Xeilari, M., Yiotakis, A., Grembecka, J., Pawelczak, M., Kafarski, P., and Mucha, A.
(2007) Bioorg Med Chem 15, 3187-3200 37. Dive, V., Georgiadis, D., Matziari, M., Makaritis, A., Beau, F., Cuniasse, P., and Yiotakis, A. (2004)
Cell Mol Life Sci 61, 2010-2019 38. Drag, M., Dlugosz, K., and Oleksyszyn, J. (2006) Synthetic Commun 36, 2787-2795 39. Drag, M., and Oleksyszyn, J. (2005) Tetrahedron Lett 46, 3359-3362 40. Sieber, S. A., Niessen, S., Hoover, H. S., and Cravatt, B. F. (2006) Nat Chem Biol 2, 274-281 41. Chan, E. W., Chattopadhaya, S., Panicker, R. C., Huang, X., and Yao, S. Q. (2004) J Am Chem Soc
126, 14435-14446 42. Bogyo, M. (2006) Nat Chem Biol 2, 229-230 43. David, A., Steer, D., Bregant, S., Devel, L., Makaritis, A., Beau, F., Yiotakis, A., and Dive, V.
(2007) Angew Chem Int Ed Engl 46, 3275-3277 44. Bartlett, P. A., and Otake, A. (1995) Journal of Organic Chemistry 60, 3107-3111 45. Rando, R. R. (1974) Science 185, 320-324
FOOTNOTES
This work was supported by NIH grants AIG1139 and RR20843 (to GSS) GM054051 (to JAE) and EB005011 (to MB), and by State for Scientific Research Grant 2 P05A 013 30 in Poland. We thank Jozef Oleksyszyn (Division of Medicinal Chemistry and Microbiology, Faculty of Chemistry, Wroclaw University of Technology, Poland) for the gift of the phosphonate inhibitors and Scott Snipas (Burnham Institute for Medical Research) for excellent technical assistances. We thank also Ed Madison (Catalyst Biosciences, San Francisco) and Cristina Pop (Burnham Institute for Medical Research) for stimulating discussions. Marcin Drag is grateful to Foundation for Polish Science for support.
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

9
Abbreviations used: Ac, acetyl; ACC, 7-amino-4-carbamoylmethylcoumarin; APN – aminopeptidase N, DMF - N,N-dimethyl formamide; TFA, trifluoroacetate.
Figure legends Fig. 1. Schematic representation of the synthesis and application of the Fluorogenic Substrate Library for aminopeptidases. The library contains 61 natural and unnatural amino acids. Fig. 2. Individual substrate velocities of human, pig and rat aminopeptidases. Enzyme concentrations were in the range 0.2-5 nM and the final concentration of the substrate in each well was 1µM. ACC production was monitored using an fmax multi-well fluorescence plate reader (Molecular Devices) at excitation wavelength of 355 nm and an emission wavelength of 460 nm. Assay time 15-30 min. The x-axis represents the abbreviated amino acid names (for full name and structure see Suppl. Material). The y-axis represents the average relative activity expressed as a percent of the best amino acid. In the heat map view the most preferred positions are displayed in bright red, whereas a complete lack of activity is in black, with intermediate values represented by intermediate shades of red.
Fig. 3. Individual reciprocal Km values of human, pig and rat aminopeptidases. The enzyme concentration was in the range 0.6-5 nM and the final concentration of the substrate in each well was in the range 0.25-500 µM. ACC production was monitored using a fmax multi-well fluorescence plate reader (Molecular Devices) at excitation wavelength of 355 nm and an emission wavelength of 460 nm. The x-axis represents the abbreviated amino acid names (for full name and structure see Suppl. Material). The y-axis represents the average reciprocal Km expressed as a percent of the best amino acid. In the heat map view the most preferred positions are displayed in bright red, whereas a complete lack of activity is in black, with intermediate values represented by intermediate shades of red.
Fig. 4. Structures of the tested α-aminoalkanephosphonic acids.
Fig. 5. Plot of the kinetic parameters for the fluorogenic substrates versus their appropriate inhibitor Ki value (data from Table 1). (A) Plot of the substrate Km versus corresponding phosphonate inhibitor Ki; (B) Plot of the substrate kcat versus corresponding phosphonate inhibitor Ki; (C) Plot (linear approach) of the substrate kcat/Km versus corresponding phosphonate inhibitor Ki (D) Non-linear plots of the substrate kcat/Km versus corresponding phosphonate inhibitor Ki. Error bars represent the standard deviation of the Ki and kinetic terms of experiments run in triplicate.
Fig. 6. kcat versus Km inhibitors - predicting optimal inhibitors based on substrate screens. Inhibitor potency depends on the mechanism of binding, and we recognize two primary categories. Inhibitors that operate through mechanism-based inactivation, or transition state analogs, can be broadly classified as kcat inhibitors, where inhibitor efficiency parallels the kcat term in the cleavage of equivalent substrates. Relative potency of these inhibitors can be predicted simply by determining the hydrolysis rates of substrates in library screens. On the other hand, inhibitors that bind the ground state do not always follow this relationship, and it is inappropriate to extrapolate substrate cleavage data to inhibitor design. In the later case, the Km term in substrate cleavage predicts the order, and more importantly the potency, of inhibitor efficiency. As the repertoire of inhibitor types available to medicinal chemists increases, distinguishing between these two mechanisms will become an essential early step in the design of specific protease inhibitors.
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

10
Table 1. Comparison of the kinetic properties of the selected substrates (kcat, Km and kcat/Km) and racemic mixtures of phosphonate inhibitors (Ki) for pig and human APN. Data represent the mean and standard deviation of at least triplicate experiments. Note that overall processing of the substrates by human APN is significantly lower than for pig APN.
pig APN human APN
kcat
[s-1]
Km
[µM]
kcat/Km
[s-1 µM-1]
Ki
[µM]
kcat
[s-1]
Km
[µM]
kcat/Km
[s-1 µM-1]
Ki
[µM]
Ala 47.4 ± 4.8 79.7 ± 2.7 0.59 ± 0.042 35.6 ± 5.3 1.15 ± 0.12 134.6 ±11.4 0.0087 ± 0.0011 19.3 ± 2.7
Nva 14.3 ± 1.6 23 ± 1.1 0.62 ± 0.069 14.0 ± 2.1 0.34 ± 0.018 30.2 ± 2.9 0.011 ± 0.0065 6.5 ± 2.2
Nle 12.8 ±1.2 11 ± 2.2 1.2 ± 0.11 4.3 ± 0.8 0.30 ± 0.042 15.2 ± 1.3 0.0196 ± 0.0012 2.9 ± 0.38
hPhe 6.6 ± 0.2 7.4 ± 1.5 0.91 ± 0.065 6.1 ± 0.9 0.09 ± 0.012 3.7 ± 0.3 0.0238 ± 0.0035 0.8 ± 0.1
hCha 10.4 ± 0.4 8.7 ± 1.7 1.2 ± 0.12 4.0 ± 0.6 0.11 ± 0.014 2.1 ± 0.2 0.0509 ± 0.0055 0.45 ± 0.052
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

11
Figure 1
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from
12
Figure 2
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

13
Figure 3
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

14
Figure 4
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

15
Figure 5
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from

16
Figure 6
at Stanford U
niversity, on January 11, 2010w
ww
.jbc.orgD
ownloaded from











![Activity-Based Probes German Edition:DOI:10.1002/ange ...med.stanford.edu/content/dam/sm/bogyolab/documents/...[9a] Selective release of the leaving group adds additional versatility](https://img.pdfslide.us/doc/110x75/5f926c06722aa625ac671fd1/activity-based-probes-german-editiondoi101002ange-med-9a-selective.jpg)

















