Embed Size (px)
Citation preview

CASE OF THE MONTH ABSTRACT: Adult polyglucosan body disease (APBD) is characterized bythe accumulation of insoluble glucose polymers within the central and pe-ripheral nervous systems. A common missense mutation in the glycogenbranching enzyme (GBE1) gene has been identified in Ashkenazi patientswith APBD. We report on a non-Jewish patient with APBD on whom weperformed proton magnetic resonance spectroscopic imaging of the brain.GBE activity in fibroblasts was markedly reduced, and a novel heterozygousmutation was identified in the GBE1 gene. Our findings widen the spectrumof APBD genotypes, underline the importance of performing GBE analysis inall APBD patients, and suggest that brain white matter degeneration inAPBD may result from tissue damage involving axons and myelin.
Muscle Nerve 37: 530–536, 2008
ADULT POLYGLUCOSAN BODY DISEASE: PROTONMAGNETIC RESONANCE SPECTROSCOPY OF THEBRAIN AND NOVEL MUTATION IN THE GBE1 GENE
ROBERTO MASSA, MD,1 CLAUDIO BRUNO, MD,2 ALESSANDRO MARTORANA, MD, PhD1
NICOLA DE STEFANO, MD,3 OTTO P. VAN DIGGELEN, PhD,4 and ANTONIO FEDERICO, MD3
1 Dipartimento di Neuroscienze, Universita di Roma Tor Vergata and Fondazione Santa Lucia,Roma, Italy
2 Unita Operativa Malattie Neuromuscolari, Dipartimento di Pediatria, Universita di Genova andDipartimento di Neuroscienze e Riabilitazione, Istituto G. Gaslini, Genova,Italy
3 Unita Operativa Malattie Neurometaboliche, Istituto di Scienze Neurologiche,Universita di Siena, Siena, Italy
4 Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, The Netherlands
Accepted 17 September 2007
Adult polyglucosan body disease (APBD) is a raredisorder involving both the central and peripheralnervous systems. Patients with APBD usually presentbetween the fourth and the sixth decade of life witha variable combination of symptoms and signs com-monly including corticospinal involvement, periph-eral neuropathy, bladder dysfunction, and, in abouthalf of the patients, cognitive impairment.10,21,23,33 Afew magnetic resonance imaging (MRI) studies haveshown nonspecific, leukodystrophy-like changes incerebral white matter (WM), along with brain andspinal cord atrophy.5,22,23,33 Most cases are appar-
ently sporadic but a few familial clusterings involvingsiblings have been reported.6,12,23,30
The diagnosis of APBD is generally made by ex-clusion of other clinical-pathological entities, andmost cases have been defined by examination ofautopsy material. However, since a peripheral neu-ropathy is invariably present, a diagnosis in vivo ispossible by means of nerve biopsy studies. Thepathological hallmark of this condition is the ratherdiffuse accumulation of polyglucosan bodies (PBs)in the brain and peripheral nerves. PBs consist ofdiastase-resistant glucose polymers that stain withperiodic acid–Schiff (PAS) and, ultrastructurally,they are composed of filamentous and finely granu-lar material.30 Axonal damage, induced by PB dep-osition, is generally considered the pathogenicmechanism of peripheral neuropathy.
The accumulation of this amylopectin-like poly-glucosan is usually ascribed to the deficiency of gly-cogen branching enzyme (GBE), although in theinitial reports the activity of this enzyme was eithernot assayed or, in a few cases, found to be nor-mal.10,12,21,30 However, APBD is presently considereda clinical variant of GBE deficiency, which is charac-
Abbreviations: APBD, adult polyglucosan body disease; Cho, choline; Cr,creatine; FLAIR, fluid-attenuated inversion recovery; GBE, glycogen branch-ing enzyme; 1H-MRSI, proton magnetic resonance spectroscopic imaging;Lac, lactate; MRI, magnetic resonance imaging; NAA, N-acetylaspartate;PAS, periodic acid–Schiff; PB, polyglucosan body; PCR, polymerase chainreaction; PRESS, point-resolved spectroscopy; VOI, volume of interest; WM,white matter.Key words: adult polyglucosan body disease; glycogen branching enzyme;GBE1 gene mutation; nerve pathology; proton magnetic resonance spectro-scopic imagingCorrespondence to: R. Massa; e-mail: [email protected]
© 2007 Wiley Periodicals, Inc.Published online 9 November 2007 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/mus.20916
530 Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008

terized, in its classic form, by liver or neuromuscularinvolvement with onset in infancy.11,27
PB deposition is not peculiar of APBD, since itcan be found in other disorders, such as phospho-fructokinase deficiency and Lafora’s disease, or innormal aging.1,13,25 A recognized mechanism lead-ing to PB formation is an increase in the normalratio between glycogen synthase and branching en-zyme.1,28
In APBD patients from Ashkenazi Jewish families,genetic analysis of the GBE1 gene has identified ahomozygous missense mutation (Tyr329Ser).24 Onlyone patient with reduced GBE activity and GBE1mutations has been reported so far among the non-Ashkenazi population.35 Here we report on an Ital-ian non-Jewish woman with APBD and a likely famil-ial presentation. In order to detect possible diseasemarkers in vivo we performed proton magnetic res-onance spectroscopic imaging (1H-MRSI) of thebrain. Biochemical and genetic studies revealed GBEdeficiency produced by a novel mutation in theGBE1 gene.
CASE REPORT
A 44-year-old woman, the fourth and youngest childof unrelated parents, was admitted to the hospitalwith complaints of urinary urgency for 2 years, dys-phagia for 4 months, and slurred speech and dizzi-ness for several weeks. On examination she had dys-arthria and dysphagia for liquids. Gait was ataxic;dysmetria and dysdiadochokinesia were present inthe upper extremities, especially the left. Strengthand sensation were normal and deep tendon re-flexes were brisk but symmetrical. Plantar responseswere flexor.
Needle electromyography showed diffuse neuro-genic changes, with motor unit potentials of in-creased amplitude and duration, and a reduced in-terference pattern during maximal voluntarycontraction. Nerve conduction studies revealed anaxonal sensory-motor neuropathy in the lower limbs,with markedly decreased amplitude of compoundmuscle action potentials in both tibial nerves andabsent sensory action potentials in both sural nerves.Neuropsychological testing failed to detect any cog-nitive deficit and cerebrospinal fluid examinationwas normal (lactate values were not measured). Abrain MRI showed diffuse hyperintensity of hemi-spheric WM in T2-weighted images. Cardiac func-tion, as assessed by electrocardiography and echo-cardiography, was normal.
A sural nerve biopsy was performed. Light mi-croscopy of cryostat sections revealed a conspicuous
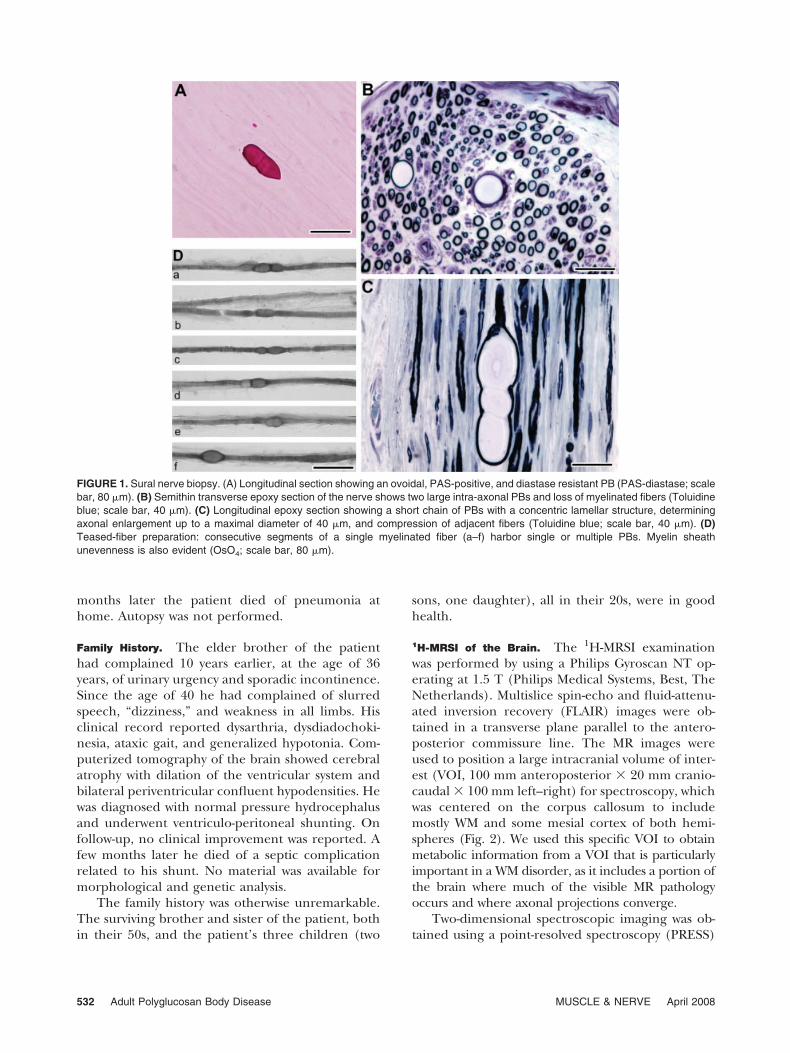
loss of myelinated fibers, together with the presenceof fiber enlargements, corresponding to PBs. Thesewere intra-axonal, PAS-positive, and diastase-resis-tant spherical or ovoidal bodies (Fig. 1A). In resinsemithin sections, PBs appeared as concentricallylaminated structures with a targetoid aspect deter-mining an enlargement of the axon that reached amaximum diameter of 40 �m (Fig. 1B,C). In somecases, PBs were located at paranodal sites, whereasothers were distributed in short chains, with a bead-work appearance. Degenerating axons were rare, butnumerous clusters of axonal regeneration were ob-served. Some remyelinated axons were present, aswell as a few irregular and thickened myelin sheathsresembling tomacula. There was no inflammationbut mast cells were widespread in all nerve compart-ments and sometimes appeared to be close to nervefibers. Teased-fiber preparations showed sporadicdemyelinated and remyelinated internodes. Interest-ingly, only a few (6 of 100) myelinated fibers showedfocal thickenings produced by PBs but in these fibersPBs were disseminated along the fiber length (Fig.1D). Electron microscopy showed the presence ofPBs also in unmyelinated axons and in perineurialand endoneurial cells, the latter being fibroblasts or,less commonly, Schwann cells. PBs appeared as cy-toplasmic, non–membrane-bound, granulo-filamen-tous structures displacing normal axoplasm to themargins of the axon. Other frequent alterations weredemyelination and remyelination, with sporadicclusters of supernumerary Schwann cells formingonion bulbs. Mitochondria had a consistently nor-mal appearance. In view of these findings a diagnosisof APBD was made.
The patient was clinically reexamined 1 year laterand her status was unchanged. After 3 years, how-ever, dysarthria, dysphagia, and ataxia had wors-ened. Dysmetria and dysdiadochokinesia of upperand lower extremities and urinary incontinencewere also evident. Extensor plantar reflexes and pal-momental reflexes were present bilaterally. At thistime neuropsychological tests showed a severe cog-nitive impairment affecting both cortical and subcor-tical functions. A new brain and spinal MRI showedan extension of the WM changes, which affectedmost of the cerebral hemispheres, together withmoderate cortical atrophy and severe atrophy of thecorpus callosum, cerebellum, brainstem, and spinalcord. In order to investigate changes in cerebralmetabolism we performed 1H-MRSI of the brain. Inaddition, GBE activity was measured in cultured fi-broblasts obtained from a skin biopsy and geneticstudies of the GBE1 gene were performed. Nine
Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008 531
months later the patient died of pneumonia athome. Autopsy was not performed.
Family History. The elder brother of the patienthad complained 10 years earlier, at the age of 36years, of urinary urgency and sporadic incontinence.Since the age of 40 he had complained of slurredspeech, “dizziness,” and weakness in all limbs. Hisclinical record reported dysarthria, dysdiadochoki-nesia, ataxic gait, and generalized hypotonia. Com-puterized tomography of the brain showed cerebralatrophy with dilation of the ventricular system andbilateral periventricular confluent hypodensities. Hewas diagnosed with normal pressure hydrocephalusand underwent ventriculo-peritoneal shunting. Onfollow-up, no clinical improvement was reported. Afew months later he died of a septic complicationrelated to his shunt. No material was available formorphological and genetic analysis.
The family history was otherwise unremarkable.The surviving brother and sister of the patient, bothin their 50s, and the patient’s three children (two
sons, one daughter), all in their 20s, were in goodhealth.
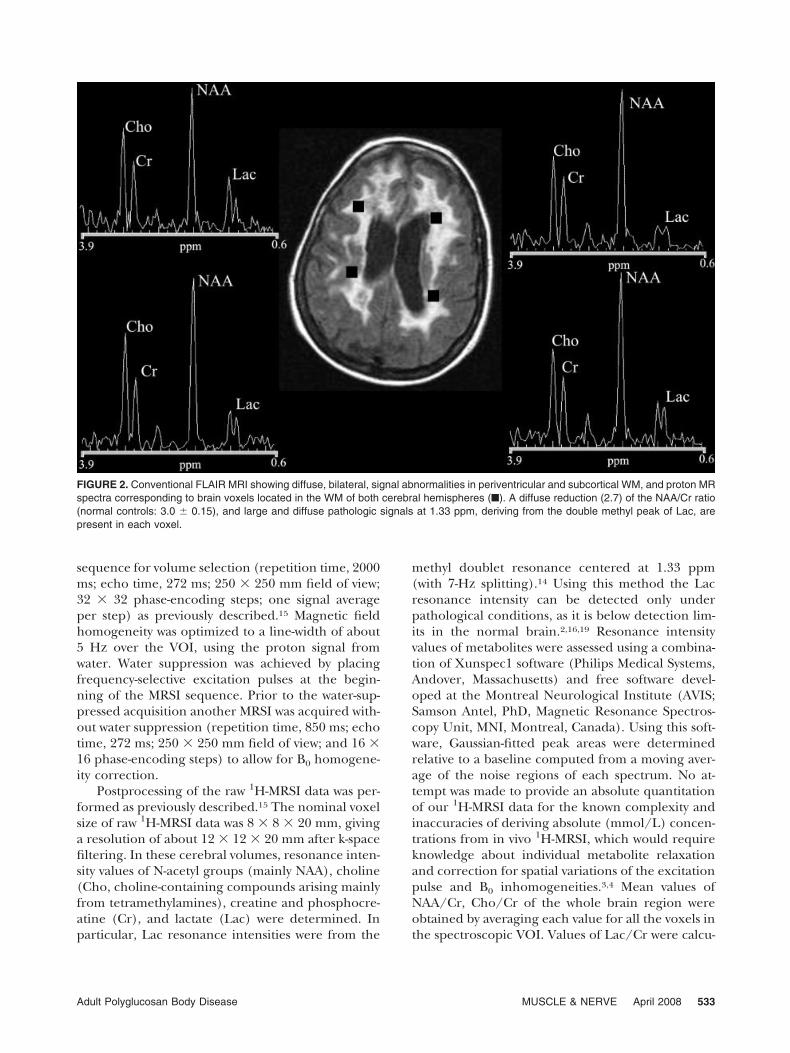
1H-MRSI of the Brain. The 1H-MRSI examinationwas performed by using a Philips Gyroscan NT op-erating at 1.5 T (Philips Medical Systems, Best, TheNetherlands). Multislice spin-echo and fluid-attenu-ated inversion recovery (FLAIR) images were ob-tained in a transverse plane parallel to the antero-posterior commissure line. The MR images wereused to position a large intracranial volume of inter-est (VOI, 100 mm anteroposterior � 20 mm cranio-caudal � 100 mm left–right) for spectroscopy, whichwas centered on the corpus callosum to includemostly WM and some mesial cortex of both hemi-spheres (Fig. 2). We used this specific VOI to obtainmetabolic information from a VOI that is particularlyimportant in a WM disorder, as it includes a portion ofthe brain where much of the visible MR pathologyoccurs and where axonal projections converge.
Two-dimensional spectroscopic imaging was ob-tained using a point-resolved spectroscopy (PRESS)
FIGURE 1. Sural nerve biopsy. (A) Longitudinal section showing an ovoidal, PAS-positive, and diastase resistant PB (PAS-diastase; scalebar, 80 �m). (B) Semithin transverse epoxy section of the nerve shows two large intra-axonal PBs and loss of myelinated fibers (Toluidineblue; scale bar, 40 �m). (C) Longitudinal epoxy section showing a short chain of PBs with a concentric lamellar structure, determiningaxonal enlargement up to a maximal diameter of 40 �m, and compression of adjacent fibers (Toluidine blue; scale bar, 40 �m). (D)Teased-fiber preparation: consecutive segments of a single myelinated fiber (a–f) harbor single or multiple PBs. Myelin sheathunevenness is also evident (OsO4; scale bar, 80 �m).
532 Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008
sequence for volume selection (repetition time, 2000ms; echo time, 272 ms; 250 � 250 mm field of view;32 � 32 phase-encoding steps; one signal averageper step) as previously described.15 Magnetic fieldhomogeneity was optimized to a line-width of about5 Hz over the VOI, using the proton signal fromwater. Water suppression was achieved by placingfrequency-selective excitation pulses at the begin-ning of the MRSI sequence. Prior to the water-sup-pressed acquisition another MRSI was acquired with-out water suppression (repetition time, 850 ms; echotime, 272 ms; 250 � 250 mm field of view; and 16 �16 phase-encoding steps) to allow for B0 homogene-ity correction.
Postprocessing of the raw 1H-MRSI data was per-formed as previously described.15 The nominal voxelsize of raw 1H-MRSI data was 8 � 8 � 20 mm, givinga resolution of about 12 � 12 � 20 mm after k-spacefiltering. In these cerebral volumes, resonance inten-sity values of N-acetyl groups (mainly NAA), choline(Cho, choline-containing compounds arising mainlyfrom tetramethylamines), creatine and phosphocre-atine (Cr), and lactate (Lac) were determined. Inparticular, Lac resonance intensities were from the
methyl doublet resonance centered at 1.33 ppm(with 7-Hz splitting).14 Using this method the Lacresonance intensity can be detected only underpathological conditions, as it is below detection lim-its in the normal brain.2,16,19 Resonance intensityvalues of metabolites were assessed using a combina-tion of Xunspec1 software (Philips Medical Systems,Andover, Massachusetts) and free software devel-oped at the Montreal Neurological Institute (AVIS;Samson Antel, PhD, Magnetic Resonance Spectros-copy Unit, MNI, Montreal, Canada). Using this soft-ware, Gaussian-fitted peak areas were determinedrelative to a baseline computed from a moving aver-age of the noise regions of each spectrum. No at-tempt was made to provide an absolute quantitationof our 1H-MRSI data for the known complexity andinaccuracies of deriving absolute (mmol/L) concen-trations from in vivo 1H-MRSI, which would requireknowledge about individual metabolite relaxationand correction for spatial variations of the excitationpulse and B0 inhomogeneities.3,4 Mean values ofNAA/Cr, Cho/Cr of the whole brain region wereobtained by averaging each value for all the voxels inthe spectroscopic VOI. Values of Lac/Cr were calcu-
FIGURE 2. Conventional FLAIR MRI showing diffuse, bilateral, signal abnormalities in periventricular and subcortical WM, and proton MRspectra corresponding to brain voxels located in the WM of both cerebral hemispheres (■). A diffuse reduction (2.7) of the NAA/Cr ratio(normal controls: 3.0 � 0.15), and large and diffuse pathologic signals at 1.33 ppm, deriving from the double methyl peak of Lac, arepresent in each voxel.
Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008 533

lated by averaging only voxels with detectable Lacresonance intensities. As chemical shift artifacts as-sociated with selective excitation might affect spectraat the edges of the VOI, they were deleted beforeaveraging. 1H-MRSI values of the patient were com-pared to those of a group of 12 age-matched normalcontrols obtained by using the same 1H-MRSI pro-cedure.
The results of the 1H-MRSI examination showeddecreases of the NAA/Cr ratio in the whole VOI(2.7, normal controls � 3.05 � 0.15) and normalvalues of Cho/Cr (1.27, normal controls � 1.3 �0.20). Resonance intensities at 1.33 ppm, derivingfrom the methyl doublet of Lac, were found in eachvoxel of the periventricular WM of both hemi-spheres, with a mean resonance intensity of Lac/Cr � 1.2 (Fig. 2).
Biochemical and Genetic Studies. GBE activity wasassayed in cultured fibroblasts as described.9 Totalcellular RNA and genomic DNA were extracted fromcultured fibroblasts according to standard proce-dures. Six overlapping fragments encompassing theentire coding region of GBE1 were directly amplifiedfrom the patient’s total RNA with the SuperscriptPre-Amplification System kit (Life Technology,Gaithersburg, Maryland), as previously described.11
The promoter region, the entire coding region se-quence, and exon–intron boundaries of the GBE1gene were amplified by polymerase chain reaction(PCR) using a set of intronic primers. The PCR prod-ucts were purified and directly sequenced using theABI Prism Big-Dye terminator cycle sequencing kit andan ABI Prism 3100 Genetic Analyzer (Applied Bio-systems, Foster City, California). To confirm theidentified mutation, specific primer extensiontechniques were employed using minisequencinganalyses (ABI Prism SNaPshot multiplex kit; AppliedBiosystems).
GBE residual activity in cultured fibroblasts wasless than 5% of the normal value. By sequencing theGBE1 gene a heterozygous A-to-G transition in exon12, changing a tyrosine with a cysteine (Tyr535Cys),was identified (Fig. 3). This mutation was absent in apanel of 200 human control chromosomes. Sequenc-ing of the entire coding region and of the promoterregion failed to detect any other variant.
DISCUSSION
Although a “typical” combination of symptoms andsigns may be recognized in most patients with APBD,several clinical variants, mimicking spinocerebellarataxia, extrapyramidal disorders, or motor neuron
disease, have been described.20,26,29 In our patient,clinical presentation at onset lacked some of the“typical” features, such as cognitive impairment andcorticospinal signs, and was characterized by unusualsigns, such as cerebellar dysfunction, dysarthria, anddysphagia that correlated with MRI evidence of cer-ebellar and brainstem atrophy. However, follow-updisclosed the full “typical” clinical setting during thecourse of the disease.
In the presence of such clinical findings the re-sults of sural nerve biopsy led to the unequivocaldiagnosis of APBD. We observed PBs not only inmyelinated and unmyelinated fibers, but also withinendoneurial fibroblasts and perineurial cells, as pre-viously reported in a juvenile case of polyglucosanbody disease.31 Interestingly, our teased-fiber find-ings suggest that deposition of PBs, rather than oc-curring randomly, affects single nerve fibersthroughout their length, possibly favored by localmetabolic stress. Besides, we detected several exam-ples of segmental demyelination and remyelination,
FIGURE 3. Electropherogram of mutation in exon 12 of the GBE1gene. Pherogram from a normal control (NC) is shown above thatof the patient (Pt). The alteration responsible for mutation isindicated by an arrow. Nucleotide and amino acid sequences ofnormal and mutant alleles are shown next to the correspondingpherograms. In the lower part of the figure the high phylogeneticconservation at amino acid residue 535 is shown.
534 Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008

Schwann cell vacuolation and hyperplasia, and en-doneurial mast cell infiltration. These data supportthe hypothesis that accumulation of PBs, in additionto its deleterious effects on axonal integrity, may alsoinduce direct or indirect myelin damage. Indeed,demyelination accompanies axonal loss in the brain,according to the few available postmortem studies,and has been sporadically reported in peripheralnerves.21,30,32
Brain MRI findings showed severe alterations ofthe hemispheric WM that, although described inAPBD, are not distinctive compared to those seen inthe leukodystrophies.5,22,23,33 In considering our 1H-MRSI findings, a reduction of NAA/Cr should beinterpreted, as in other neurological disorders, as anindex of neuro-axonal damage or loss due to thedecrease of NAA.7,17 The presence of widespreadneuro-axonal damage in postmortem APBD brainsseems to give further support to this hypothe-sis.20,30,32 However, increases in cerebral Cr levels,resulting from gliosis occurring in the cerebral WM,cannot be categorically excluded.
The diffuse presence of a large Lac signal in thealtered brain WM was the most interesting 1H-MRSIfeature in our patient. Lac is the endproduct ofglycolysis and accumulates when oxidative metabo-lism cannot meet energy requirements. Lac reso-nance intensity detected by in vivo spectroscopy cancome from both intra- and extracellular compart-ments and its increases are usually seen in patientswith either focal brain injuries or mitochondrial ab-normalities.18,19 In conditions associated with focalinflammation, Lac accumulation reflects the metab-olism of inflammatory cells.18 In contrast, increasesin brain Lac resonance intensities not localized tovisible MRI lesions are usually due to signals comingfrom the brain parenchyma as a consequence ofmitochondrial disturbance.19 Thus, the large anddiffuse presence of Lac signal in the abnormal WMof our patient could be related to infiltrating mac-rophages recruited by an intense demyelination, asdescribed in postmortem studies.21,30,32 Interest-ingly, this mechanism has been proposed in recent1H-MRSI studies to explain WM accumulation of Lacin different forms of leukodystrophies.8,34 However,an impaired mitochondrial metabolism of the brainparenchyma cannot be definitely ruled out, al-though we did not observe morphological mitochon-drial alterations in the peripheral nerve. Therefore,our 1H-MRSI data, in agreement with pathologicalfindings, suggest that brain WM degeneration inAPBD may result from tissue damage involving bothaxons and myelin.
In our patient we identified a novel missensemutation in the GBE1 gene in a heterozygous con-dition. We consider this single amino acid substitu-tion to be pathogenic because: (1) it is associatedwith a deficiency of GBE activity; (2) it replaces anamino acid that is evolutionarily conserved with aresidue of different conformation; and (3) the mu-tation was absent in a panel of 200 control chromo-somes. To our knowledge, this is the first Italian caseof APBD to be reported, and the first non-Ashkenazipatient with a likely familial presentation. Indeed,examination of the proband’s brother clinicalrecords allowed us to challenge his diagnosis of nor-mal pressure hydrocephalus, suggesting instead thathe had APBD. Age at onset, the rich combination ofsymptoms and signs, very similar to those presentedby his sister, and the lack of clinical improvementafter ventricular shunting support this hypothesis.Moreover, his brain CT scan findings were compa-rable to those in other cases of APBD.5
To date, GBE deficiency and mutations in theGBE1 gene have been found almost exclusively inAPBD patients of Ashkenazi descent.24 The only non-Jewish patient reported so far carried two previouslyunreported heterozygous mutations (Arg515His andArg524Gln).35 Recently, two cases of typical APBDwith “atypical” GBE insufficiency have been de-scribed. The first case was a non-Jewish patient withcomplete GBE deficiency and no mutation in theGBE1 gene.22 The second case was a patient of Ash-kenazi descent with partial GBE deficiency (in theheterozygous range) and the Tyr329Ser mutation inheterozygosity. At variance with other carriers, thispatient displayed the full-blown disease.33 In order toexplain PB deposition in these two cases the occur-rence of posttranscriptional or posttranslational de-fects and the influence of environmental factorshave been suggested.22,33 This hypothesis may applyto other cases with “atypical” GBE insufficiency.
In conclusion, our findings widen the spectrumof APBD genotypes and underline the importance toperform GBE enzymatic assay and GBE1 gene anal-ysis in all APBD patients, independent of their ethnicbackground. Finally, brain 1H-MRSI data may pro-vide a better understanding of the pathogenesis ofAPBD.
Financial support was provided by research grants from Fondazi-one Santa Lucia and Ministero dell’Universita e Ricerca Scienti-fica (COFIN-2005060584) to R.M. The authors thank GiorgioBernardi, MD, for encouragement and criticism, Chiara Terrac-ciano, MD, for help in electron microscopy, Denise Cassandrini,PhD, for help in sequence analysis, and Mr. Graziano Bonelli forphotographic assistance.
Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008 535

REFERENCES
1. Agamanolis DP, Askari AD, Di Mauro S, Hays A, Kumar K,Lipton M, et al. Muscle phosphofructokinase deficiency: twocases with unusual polysaccharide accumulation and immu-nologically active enzyme protein. Muscle Nerve 1980;3:456–467.
2. Arnold DL, Matthews PM. Practical aspects of clinical appli-cations of MRS in the brain. In: Young IR, Charles HC,editors. MR spectroscopy: clinical applications and tech-niques. London: Martin Dunitz; 1996:139–159.
3. Barantin L, Le PA, Akoka S. A new method for absolutequantitation of MRS metabolites. Magn Reson Med 1997;38:179–182.
4. Barker PB, Soher BJ, Blackband SJ, Chatham JC, Mathews VP,Bryan RN. Quantitation of proton NMR spectra of the humanbrain using tissue water as an internal concentration refer-ence. NMR Biomed 1993;6:89–94.
5. Berkhoff M, Weis J, Schroth G, Sturzenegger M. Extensivewhite-matter changes in case of adult polyglucosan body dis-ease. Neuroradiology 2001;43:234–236.
6. Bigio EH, Weiner MF, Bonte FJ, White CL. Familial dementiadue to adult polyglucosan body disease. Clin Neuropathol1997;16:227–234.
7. Bjartmar C, Battistuta J, Terada N, Dupree E, Trapp BD.N-acetylaspartate is an axon-specific marker of maturewhite matter in vivo: a biochemical and immunohistochem-ical study on the rat optic nerve. Ann Neurol 2002;51:51–58.
8. Brockmann K, Dechent P, Meins M, Haupt M, Sperner J,Stephani U, et al. Cerebral proton magnetic resonance spec-troscopy in infantile Alexander disease. J Neurol 2003;250:300–306.
9. Bruno C, DiRocco M, Lamba LD, Bado M, Marino C, TsujinoS, et al. A novel missense mutation in the glycogen branchingenzyme gene in a child with myopathy and hepatopathy.Neuromuscul Disord 1999;9:403–407.
10. Bruno C, Servidei S, Shanske S, Karpati G, Carpenter S,McKee D, et al. Glycogen branching enzyme deficiency inadult polyglucosan body disease. Ann Neurol 1993;33:88 –93.
11. Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M,Giuffre B, Donati MA, et al. Clinical and genetic heterogene-ity of branching enzyme deficiency (glycogenosis type IV).Neurology 2004;63:1053–1058.
12. Cafferty MS, Lovelace RE, Hays AP, Servidei S, DiMauro S,Rowland LP. Polyglucosan body disease. Muscle Nerve 1991;14:102–107.
13. Cavanagh JB. Corpora-amylacea and the family of polyglu-cosan diseases. Brain Res Rev 1999;29:265–295.
14. Danielsen ER, Ross BD. Magnetic resonance spectroscopy:diagnosis of neurological diseases. New York: Marcel Dekker;1999.
15. De Stefano N, Balestri P, Dotti MT, Grosso S, Mortilla M,Morgese G, et al. Severe metabolic abnormalities in the whitematter of patients with vacuolating megalencephalic leukoen-cephalopathy with subcortical cysts. A proton MR spectro-scopic imaging study. J Neurol 2001;248:403–409.
16. De Stefano N, Matthews PM, Antel JP, Preul M, Francis G,Arnold DL. Chemical pathology of acute demyelinating le-sions and its correlation with disability. Ann Neurol 1995;38:901–909.
17. De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tarta-glia MC, Antel JP, et al. Evidence of axonal damage in theearly stages of multiple sclerosis and its relevance to disability.Arch Neurol 2001;58:65–70.
18. De Stefano N, Narayanan S, Matthews PM, Mortilla M, DottiMT, Federico A, et al. Proton MR spectroscopy to assess
axonal damage in multiple sclerosis and other white matterdisorders. J Neurovirol 2000;6(Suppl 2):S121–S129.
19. Dubeau F, De Stefano N, Zifkin BG, Arnold DL, ShoubridgeEA. Oxidative phosphorylation defect in the brains of carriersof the tRNAleu (UUR) A3243G mutation in a MELAS pedi-gree. Ann Neurol 2000;47:179–185.
20. Felice KJ, Grunnet ML, Rao KR, Wolfson LI. Childhood-onsetspinocerebellar syndrome associated with massive polyglu-cosan body deposition. Acta Neurol Scand 1997;95:60–64.
21. Gray F, Gherardi R, Marshall A, Janota I, Poirier J. Adultpolyglucosan body disease (APBD). J Neuropathol Exp Neu-rol 1988;47:459–474.
22. Klein CJ, Boes CJ, Chapin JE, Lynch CD, Campeau NG, DyckPJB, et al. Adult polyglucosan body disease: case descriptionof an expanding genetic and clinical syndrome. Muscle Nerve2004;29:323–328.
23. Lossos A, Barash V, Soffer D, Argov Z, Gomori M, Ben-NariahZ, et al. Hereditary branching enzyme dysfunction in adultpolyglucosan body disease: a possible metabolic cause in twopatients. Ann Neurol 1991;30:655–662.
24. Lossos A, Meiner Z, Barash V, Soffer D, Schlesinger I,Abramsky O, et al. Adult polyglucosan body disease inAshkenazi Jewish patients carrying the Tyr329Ser mutationin the glycogen-branching enzyme gene. Ann Neurol 1998;44:867– 872.
25. Massa R, Lodi R, Barbiroli B, Servidei S, Sancesario G, Man-fredi G, et al. Partial block of glycolysis in late-onset phospho-fructokinase deficiency myopathy. Acta Neuropathol 1996;91:322–329.
26. McDonald TD, Faust PL, Bruno C, DiMauro S, Goldman JE.Polyglucosan body disease simulating amyotrophic lateralsclerosis. Neurology 1993;43:785–790.
27. Moses SW, Parvari R. The variable presentations of glycogenstorage disease type IV: a review of clinical, enzymatic andmolecular studies. Curr Mol Med 2002;2:177–188.
28. Raben N, Danon M, Lu N, Lee E, Shliselfeld L, Skurat AV, etal. Surprises of genetic engineering: a possible model ofpolyglucosan body disease. Neurology 2001;56:1739–1745.
29. Robertson NP, Wharton S, Anderson J, Scolding NJ. Adultpolyglucosan body disease associated with an extrapyrami-dal syndrome. J Neurol Neurosurg Psychiatry 1998;65:788 –790.
30. Robitaille Y, Carpenter S, Karpati G, DiMauro S. A distinctform of adult polyglucosan body disease with massive in-volvement of central and peripheral neuronal processesand astrocytes: a report of four cases and a review of theoccurrence of polyglucosan bodies in other conditionssuch as Lafora’s disease and normal ageing. Brain 1980;103:315–336.
31. Schroder JM, May R, Shin YS, Sigmund M, Nase-HuppmeierS. Juvenile hereditary polyglucosan body disease with com-plete branching enzyme deficiency (type IV glycogenosis).Acta Neuropathol 1993;85:419–430.
32. Sindern E, Ziemssen F, Ziemssen T, Podskarbi T, Shin Y,Brasch F, et al. Adult polyglucosan body disease: a postmor-tem correlation study. Neurology 2003;61:263–265.
33. Ubogu EE, Hong STK, Akman HO, DiMauro S, Katirji B, Pres-ton DC, et al. Adult polyglucosan body disease: a case report ofa manifesting heterozygote. Muscle Nerve 2005;32:675–681.
34. van der Voorn JP, Pouwels PJ, Hart AA, Serrarens J, Willem-sen MA, Kremer HP, et al. Childhood white matter disorders:quantitative MR imaging and spectroscopy. Radiology 2006;241:510–517.
35. Ziemssen F, Sindern E, Schroder JM, Shin YS, Zange J, Kili-mann MW, et al. Novel missense mutations in the glycogen-branching enzyme gene in adult polyglucosan body disease.Ann Neurol 2000;47:536–540.
536 Adult Polyglucosan Body Disease MUSCLE & NERVE April 2008





























