Embed Size (px)
Citation preview

This paper is published as part of a PCCP Themed Issue on: Coarse-grained modeling of soft condensed matter
Guest Editor: Roland Faller (UC Davis)
Editorial
Coarse-grained modeling of soft condensed matter Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b903229c
Perspective
Multiscale modeling of emergent materials: biological and soft matter Teemu Murtola, Alex Bunker, Ilpo Vattulainen, Markus Deserno and Mikko Karttunen, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818051b
Communication
Dissipative particle dynamics simulation of quaternary bolaamphiphiles: multi-colour tiling in hexagonal columnar phases Martin A. Bates and Martin Walker, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818926a
Papers
Effective control of the transport coefficients of a coarse-grained liquid and polymer models using the dissipative particle dynamics and Lowe–Andersen equations of motion Hu-Jun Qian, Chee Chin Liew and Florian Müller-Plathe, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b817584e
Adsorption of peptides (A3, Flg, Pd2, Pd4) on gold and palladium surfaces by a coarse-grained Monte Carlo simulation R. B. Pandey, Hendrik Heinz, Jie Feng, Barry L. Farmer, Joseph M. Slocik, Lawrence F. Drummy and Rajesh R. Naik, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b816187a
A coarse-graining procedure for polymer melts applied to 1,4-polybutadiene T. Strauch, L. Yelash and W. Paul, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818271j
Anomalous waterlike behavior in spherically-symmetric water models optimized with the relative entropy Aviel Chaimovich and M. Scott Shell, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818512c
Coarse-graining dipolar interactions in simple fluids and polymer solutions: Monte Carlo studies of the phase behavior B. M. Mognetti, P. Virnau, L. Yelash, W. Paul, K. Binder, M. Müller and L. G. MacDowell, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818020m
Beyond amphiphiles: coarse-grained simulations of star-polyphile liquid crystalline assemblies Jacob Judas Kain Kirkensgaard and Stephen Hyde, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818032f
Salt exclusion in charged porous media: a coarse-graining strategy in the case of montmorillonite clays Marie Jardat, Jean-François Dufrêche, Virginie Marry, Benjamin Rotenberg and Pierre Turq, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818055e
Improved simulations of lattice peptide adsorption Adam D. Swetnam and Michael P. Allen, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818067a
Curvature effects on lipid packing and dynamics in liposomes revealed by coarse grained molecular dynamics simulations
H. Jelger Risselada and Siewert J. Marrink, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818782g
Self-assembling dipeptides: conformational sampling in solvent-free coarse-grained simulation Alessandra Villa, Christine Peter and Nico F. A. van der Vegt, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818144f
Self-assembling dipeptides: including solvent degrees of freedom in a coarse-grained model Alessandra Villa, Nico F. A. van der Vegt and Christine Peter, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818146m
Computing free energies of interfaces in self-assembling systems Marcus Müller, Kostas Ch. Daoulas and Yuki Norizoe, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818111j
Anomalous ductility in thermoset/thermoplastic polymer alloys Debashish Mukherji and Cameron F. Abrams, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818039c
A coarse-grained simulation study of mesophase formation in a series of rod–coil multiblock copolymers Juho S. Lintuvuori and Mark R. Wilson, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818616b
Simulations of rigid bodies in an angle-axis framework Dwaipayan Chakrabarti and David J. Wales, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818054g
Effective force coarse-graining Yanting Wang, W. G. Noid, Pu Liu and Gregory A. Voth, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b819182d
Backmapping coarse-grained polymer models under sheared nonequilibrium conditions Xiaoyu Chen, Paola Carbone, Giuseppe Santangelo, Andrea Di Matteo, Giuseppe Milano and Florian Müller-Plathe, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b817895j
Energy landscapes for shells assembled from pentagonal and hexagonal pyramids Szilard N. Fejer, Tim R. James, Javier Hernández-Rojas and David J. Wales, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818062h
Molecular structure and phase behaviour of hairy-rod polymers David L. Cheung and Alessandro Troisi, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818428c
Molecular dynamics study of the effect of cholesterol on the properties of lipid monolayers at low surface tensions Cameron Laing, Svetlana Baoukina and D. Peter Tieleman, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b819767a
On using a too large integration time step in molecular dynamics simulations of coarse-grained molecular models Moritz Winger, Daniel Trzesniak, Riccardo Baron and Wilfred F. van Gunsteren, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818713d
The influence of polymer architecture on the assembly of poly(ethylene oxide) grafted C60 fullerene clusters in aqueous solution: a molecular dynamics simulation study Justin B. Hooper, Dmitry Bedrov and Grant D. Smith, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b818971d
Determination of pair-wise inter-residue interaction forces from folding pathways and their implementation in coarse-grained folding prediction Sefer Baday, Burak Erman and Yaman Arkun, Phys. Chem. Chem. Phys., 2009 DOI: 10.1039/b820801h
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online / Journal Homepage / Table of Contents for this issue

Adsorption of peptides (A3, Flg, Pd2, Pd4) on gold and palladium
surfaces by a coarse-grained Monte Carlo simulation
R. B. Pandey,*a Hendrik Heinz,b Jie Feng,b Barry L. Farmer,c Joseph M. Slocik,c
Lawrence F. Drummycand Rajesh R. Naik
c
Received 16th September 2008, Accepted 16th December 2008
First published as an Advance Article on the web 26th January 2009
DOI: 10.1039/b816187a
Monte Carlo simulations are performed to study adsorption and desorption of coarse-grained
peptide chains on generic gold and palladium surfaces in the presence of solvent. The atomistic
structural details are ignored within the amino acid residues; however, their specificity and
hydrophobicity are incorporated via an interaction matrix guided by atomistic simulation.
Adsorption probabilities of the peptides A3, Flg, Pd2, Pd4, Gly10, Pro10 on gold and palladium
surfaces are studied via analysis of the mobility of each residue, the interaction energy with the
surface, profiles of the proximity to the surface, the radius of gyration, and comparisons to
homopolymers. In contrast to the desorption of Gly10 and Pro10 (with faster global dynamics),
peptides Pd2, Pd4, Flg, and A3 exhibit various degrees of adsorption on gold and palladium
surfaces (with relatively slower dynamics). Adsorption on both gold and palladium occurs
through aromatic anchoring residues Tyr2 and Phe12 in A3, Tyr2 in Flg, Phe2, His10 and His12 in
Pd2, and His6 and His11 in Pd4. A lower (more negative) surface-interaction energy of these
residues and lower mobility on palladium lead us to conclude that they are slightly more likely to
be adsorbed on palladium surfaces than on gold.
1. Introduction
Short peptides with specific sequences of residues are increas-
ingly important as a building block in emerging hybrid
materials as well as in understanding ways to incorporate
large proteins at desired targets.1–7 Insight into how to inte-
grate short peptide chains in a matrix of multi-component
systems (inorganic compounds, composites, polymer, and
solvent) is therefore crucial and involves processes such as
thermodynamic equilibration and secondary structure relaxa-
tions. Peptides can also be attached to many surfaces in
biological environments, and preventive measures may be of
interest to enhance the longevity of such surfaces. From a
basic scientific viewpoint, understanding the selective adsorp-
tion of peptides to specific surfaces is desirable within the
broader quest for biomimetic materials.
A number of experiments8–10 are often carried out to assess
the possibility of adsorption of peptides on such surfaces.
Identifying the probability of adsorption or desorption of a
peptide chain with a specific sequence is a laborious experi-
mental task, and it can be difficult to eliminate uncertainties
such as the surface structure or the influence of additional
solutes. It is also challenging to identify how a peptide binds to
specific surfaces, apart from identifying the affinity of certain
residues inside a longer chain. Computer simulation experi-
ments would be valuable not only to complement the experi-
mental efforts but also in predicting the probability of
adsorption of peptides or peptide subunits at different length
scales.
Designing advanced materials from functionalized nano-
particles with specific properties has been the subject of intense
interest in recent years,10 primarily due to the potential for
interesting material and medical applications. Using current
technical advances, surfaces of nanoparticles can be modified
with relative ease to tailor their characteristics in order to
assemble them in a desired morphology for optimal perfor-
mance. However, the incorporation of such nanostructured
materials with functionalized nanoparticles in practical devices
is still a challenge10 due to the difficulty of preserving and
exploiting their unique characteristics. Peptides are excellent
candidates to attach to the surface of a nanoparticle to intro-
duce specific characteristics. They possess unique recognition
motifs with well-defined structures controlled by the sequence
of residues, and interactions with constituents of multi-component
hybrid materials can be modulated through choice of specific
solvents. Of particular interest8–10 are bio-functionalized gold
and palladium nanoparticles for a range of applications
such as sensing, catalysis, bio-transport, and bio-recognition.
Understanding of dynamics and structural stability of
appropriate peptides (e.g. A3, Flg, Pd2, Pd4, Gly10, Pro10)
on gold and palladium surfaces is therefore highly desirable.
In regards to computer simulation and modeling, a peptide
is a short protein with a relatively small number of amino acid
groups. There are two main coarse-grained computer simula-
tion approaches to model a protein chain: on- and off-lattice.
aDepartment of Physics and Astronomy, University of SouthernMississippi, Hattiesburg, MS, 39406, USA
bDepartment of Polymer Engineering, University of Akron, Akron,Ohio 44325, USA
cAir Force Research Laboratory, Materials and ManufacturingDirectorate, AFRL/RXBP, Wright-Patterson AFB, Ohio 45433,USA
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1989
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

Off-lattice approaches (including all-atomic methods)11–15 are
often used to probe such specific configurations as a-helices andb-sheets, especially in the folding of small proteins.16–18 Large
degrees of freedom are very important in probing the structural
fluctuations around known stable structures. The multi-scale
dynamics and related relaxation pathways from one structure to
another, however, are severely limited in proteins due to long
relaxation time when atomistic details are included. In order to
reproduce known characteristics (information-based approach),
some constraints are usually invoked, e.g. peptide group dipole
interaction,19 in addition to standard interactions and con-
straints. The bond-fluctuation (BF) model on a discrete lattice
is a further coarse-grained simplification but has been used to
study the conformational relaxation of a protein chain into the
native structure20 for a general HP (Hydrophobic Polar) protein
chain, a HPE (Hydrophobic, Polar, Electrostatic) protein
model,21 and even for a specific protein, sensory rhodopsin I22
without severe constraints. The main advantages of this
approach over the methods used in probing the structural
dynamics with all-atom descriptions are higher computational
efficiency, coverage of large scales, and avoidance of unphysical
constraints beyond the coarse-grained description on a discrete
lattice; the disadvantage is the lack of atomistic details. Attempts
have been made recently to incorporate specificity of each amino
acid in studying the segmental structure and stochastic mobility
of each residue of an HIV-I protease (1DIFA).23 We would like
to employ BF description here to examine the adsorption and
desorption of a set of peptides on gold and palladium surfaces.
The model is presented in section 2, followed by results and
discussion in section 3, and conclusions in section 4.
2. Model
A peptide (small protein) chain of size M is a specific sequence
of amino acid (AA) residues from a set of twenty. A typical
amino acid group has a central carbon atom (Ca) covalently
bonded by an amino group (NH2), a carboxyl group (COOH),
a hydrogen atom, and a side group (RL) (see Fig. 1). Differ-
ences in the side group distinguish one residue from the other.
The consecutive covalent (peptide) bond between the carboxyl
group of one residue and the amino group of the next
constitutes the peptide chain. Twisting and turning around
the central carbon (Ca) along with the segmental rearrange-
ment of the side groups provides stable structures such as
a-helices, b-sheets, and a whole host of mixed secondary struc-
tures in large polypeptides. A part of computational protein
modeling11–15 is focused on understanding such structures.
One of the major challenges with such modeling is to capture
relevant details and extend it to long time scales to draw
meaningful conclusions and gain useful insights into func-
tional characteristics of the protein. In all bonding (covalent
and non-covalent), electronic densities of states and their
distributions are involved in which the quantum descriptions
are particularly crucial in understanding electronic excitations
and related kinetics. However, it is not feasible to incorporate
such electronic structural details in exploring the long-time
physical properties of a peptide chain. Therefore, coarse-
graining is hard to bypass, i.e. ignoring some microscopic
details but capturing its relevant characteristics via approx-
imate interaction potentials has become a common practice in
modeling such a complex system.
In the so-called ‘all-atomic’ model,11–15 one usually consid-
ers all intra-structural details of an amino acid residue (see
Fig. 1) beyond the mesoscopic scales in the framework of a
coarse-grained description; it involves extensive force field
parameters derived from experiment, exploratory simulations
on smaller sub-systems, or a combination thereof. This ap-
proach has enormous advantages in capturing the microscopic
details, especially for short-time properties. However, it is
often not feasible to carry out simulations long enough to
sample the relevant portions of conformational and dynamical
phase space. Therefore, it is highly desirable to look for
alternative approaches that may still be coarse-grained but
computationally efficient to probe long-time properties, in
concert with all-atomic simulation. Such a coarse-grained
modeling approach is presented here with the bond-fluctuation
method for the chain24,25 that incorporates ample degrees of
freedom and the specificity23 of the residues while maintaining
the computational efficiency of a discrete lattice.
2.1 Coarse-grained lattice
We use a three-dimensional discrete (cubic) lattice as the host
matrix. An amino acid residue is represented by a particle or
node, i.e. the internal structural details of the amino acid
(Fig. 1) are ignored but its specific characteristics are in-
corporated (see below). A poly-peptide is a set of residues
(represented by particles or nodes) tethered together in a
flexible chain in a specific sequence. The peptide chain with
the covalently bonded nodes is represented by the bond-
fluctuation model on a lattice with a cubic grid. A node is
represented by a unit cube (by occupying its eight lattice sites)
and the bond length between consecutive nodes can vary
(fluctuate) between 2 and O10 with an exception of O8 in
units of the lattice constant. Such a bond-fluctuation descrip-
tion is known to capture the computational efficiency while
incorporating ample degrees of freedom in complex polymer
systems24,25 and multi-component nanocomposites.26–29 Thus,
a peptide is represented as a chain of unique nodes (amino
acids) in a specific sequence.
Since each node represents a specific residue, it is essential to
capture their specificity or uniqueness. Amino acids can be
divided into three broad categories:21 hydrophobic (H),
hydrophilic or polar (P) and electrostatic (E). An amino acid
within each group is further distinguished23 by its relative
hydrophobicity, polar, or electrostatic strength as shown in
Table 1. In addition to peptide chains, there is a substrate (S)Fig. 1 Amino acid.
1990 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

at the bottom and mobile solvent (water, W) particles each
with the size of a peptide node. This constitutes five main
components, H, P, E, S, and W in which there are three sub-
groups to characterize specificity of each residue: eight hydro-
phobic (H1, H2 . . . H8), eight polar (P1, P2 . . . P8), and four
electrostatic (E1, E2, E3, E4) (see Table 1). A set of pheno-
menological interactions (see below) among these components
are used to execute the stochastic movement of each node and
solvent particle with the Metropolis algorithm.
2.2 Interaction matrix
Interaction among five main components (H, P, E, W, S) can
be represented by a 5 � 5 matrix (see Table 2). The interaction
strength between two elements at sites i and j separated by a
distance rij is represented by the standard Lennard-Jones (LJ)
potential
Uij ¼ f eijsrij
� �12
�"
srij
� �6#; rijorc
where rc is the cut-off range and feij is the interaction strength
with an arbitrary parameter f that can be varied and s= 1. On
a cubic lattice the distance between two sites are discrete and
measured in units of the lattice constant. A variation of the LJ
potential with the distance is shown in Fig. 2 which shows how
it falls off quickly after the minimum distance r between the
two particles (nodes or solvent) is reached. Note that the
minimum distance between two neighboring particles is two
in units of the lattice constant (e.g. r2 = 4) below which the LJ
potential goes to infinity at r o 2. It is worth pointing out that
although the variation of the LJ potential in a continuum host
space is smoother than that on the cubic lattice (Fig. 2), the
general form remains similar. Since the potential falls of very
quickly, we limit the range of the interaction potential to rc2 = 8
which includes as many as 60 neighboring lattice sites. The
depth of the LJ potential is controlled by the magnitude of
the pair interaction strength feij which is relatively large. The
number of the pair-interaction matrix elements is reduced
somewhat due to symmetries, e.g. eij = eji.The magnitude of each interaction element is based on the
insight gained from all-atomistic description and general
characteristics. The structural details from atomic scale in-
cluding covalent bond and non-covalent interaction are gen-
erally considered in an all-atomic description. Ghiringhelli and
Delle Site30 have recently analyzed the structure of an amino
acid, i.e. phenylalanine near inorganic surfaces including
Au(111) and pointed out that both the atomic interaction
and flexibility of the molecule are important in predicting its
stable conformation. Heinz et al. have examined the adsorp-
tion energy of all 20 amino acids (see Table 1) on such surfaces
including Au(111) as shown in Fig. 3.31,32
Note that the amino acids (Phe, Trp, Tyr, His) with
aromatic groups have the largest adsorption energy followed
by the adsorption energy of electrostatic and polar residues.
The adsorption energy of the hydrophobic residues is lowest
among all the amino acids. Accordingly we assume a typical
Table 1 Amino acids and their hydrophobic, polar, and electrostatic strengths; the relative strength is assigned in each group (H, P, E) separately(ref: http://en.wikipedia.org/wiki/Amino_acid)
Amino Acid Hydrophobic/ polar/ electrostatic Relative weight: H/ P/ E
Ile (I): H1 4.5 1.00Val (V): H2 4.2 0.93Leu (L): H3 3.8 0.84Phe (F): H4 2.8 0.62Cys (C): H5 2.5 0.56Met (M): H6 1.9 0.42Ala (A): H7 1.8 0.40Gly (G): H8 �0.4 0.09
Thr (T): P1 �0.7 0.20Ser (S): P2 �0.8 0.23Trp (W): P3 �0.9 0.26Tyr (Y): P4 �1.3 0.37Pro (P): P5 �1.6 0.46His (H): P6 �3.2 0.91Gln (Q): P7 �3.5 1.00Asn (N): P8 �3.5 1.00
Asp (D): E1 �3.5 0.78Glu (E): E2 �3.5 0.78Lys (K): E3 �3.9 0.87Arg (R): E4 �4.5 1.00
Table 2 Interaction matrix among H, P, E, S and W components
H P E W S
H eHH eHP eHE eHW eHS
P ePH ePP ePE ePW ePSE eEH eEP eEE eEW eESW eWH eWP eWE eWW eWS
S eSH eSP eSE eSW eSS
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1991
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

set of values of the interaction matrix presented in Table 3 to
capture the specific characteristics of these amino acids.
Surface interactions with gold and palladium are different:
for gold, eHS = 0.0, ePS = �0.2, eES =�0.2 and for palladium
eHS = 0.0, ePS = �0.27, eES = �0.27. Interactions among the
electrostatic residues are: e11 = e12 = e22 = e33 = e34 = e44 =0.1, e13 = e14 = e23 = e24 = �0.4. Among the hydrophobic
and polar groups, there are four residues (Phe (H4), Trp (P3),
Tyr (P4), His (P6)) with aromatic groups with strong surface
interaction (see Fig. 3) strength eAS = �0.7 with Au and
eAS = �0.9 with Pd. We have used f = 50 as the inter-
action energy factor for gold and palladium to accentuate the
differences in adsorption probabilities for different peptides.
Incorporating the specificity of each amino acid group for a
particular system enhances the size of the interaction matrix.
For example, the interaction energy of the polar group with
the gold surface is given by eP(i)S = Wp(i) � ePS where the
relative weight of each polar residue Wp(1) = 0.20 (Thr),
Wp(2) = 0.23 (Ser), Wp(3) = 0.26 (Trp), . . ., Wp(8) = 1.00
(Asn) (see Table 1). It is worth pointing out that despite these
specified values of the interaction matrix element, there is
ample room for varying the interaction strength including
relative magnitude without altering the qualitative results
significantly. For example, the qualitative result for the prob-
ability of adsorption of these peptides on the substrates can be
distinguished for a range of interaction strength f and a
specific value of f (=50) is selected to see these relative
differences as mentioned above. Thus, the choice of the matrix
elements presented here is primarily to illustrate how to
predict the relative adsorption of a set of peptide groups on
gold and palladium surfaces with a coarse-grained model with
the phenomenological interaction. This approach can be easily
extended to other surfaces and many polypeptides and pro-
teins. Most of our findings seem consistent with experimental
observations8,10 and molecular dynamics simulations with
‘all-atomic’ model32 qualitatively (see below). The main draw-
back is the lack of quantitative comparison. The efficiency of
our coarse-grained approach provides an alternate method to
investigate and predict the relative adsorption of different
polypeptides on gold and palladium substrates.
2.3 Stochastic movement
Each residue and solvent particle executes their stochastic
movement based on the Metropolis algorithm as follows. A
particle (node or solvent) at a site i and one of its 26
neighboring sites, say j, are selected randomly. If site j is
empty, then an attempt is made to move the particle from site
i to site j. If the particle attempted to move is a peptide node, it
is ensured that the length of the bond resulting from such a
move is within the specified limit. Provided the excluded
volume condition is satisfied, the energy in the old (Ei) and
the new configuration (Ej) is compared and the particle is
moved from site i to j with probability exp(�DEij/T), where
DEij = Ej � Ei, the temperature T is in units of the Boltzmann
constant and the interaction energy. Attempts to move each
node and solvent particle once defines one Monte Carlo step
Fig. 2 Variation of the Lennard-Jones potential with r2 (=rij2) with
s = 1, feij = 1 on a cubic lattice in unit of lattice constant.
Fig. 3 Adsorption energy of each amino acid in aqueous solution on an Au {111} surface with an all-atom model (consistent valence force field
(CVFF) with recent parameters for fcc metals, pH = 7, see ref. 31 and 32).
1992 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

(MCS) time. Simulation is performed for a sufficiently long
time to achieve the equilibration. A number of physical
quantities are evaluated during the simulation including the
energy of each residue, their mobility, their neighboring
components (substrate, solvent, and residues), and the
mean square displacement of the center of mass of the peptide
chains. Based on the analysis of these quantities along with
the visual analysis of the snapshots, we predict the
relative probability of adsorption of specific residues and
of the corresponding polypeptides on gold and palladium
substrates.
3. Results and discussion
Simulations are performed on different lattice sizes primarily
to check for severe finite size effects on the qualitative varia-
tion of the physical quantities. All data presented here are
generated on a 643 lattice, as no significant finite size effects are
detected that warrant change in our conclusions. Each simula-
tion consists of 10 copies of each peptide chain brought onto
the substrate with the solvent concentration cw = 0.05. 100
independent samples are used to estimate the average values of
the physical quantities. Animations are used to complement
our conclusions based on data analysis. Initially peptide
chains are dropped on the substrate and their structure,
dynamics, and distributions and dispersion are monitored as
they equilibrate.
In Table 4, coarse-grained descriptions are used to study the
adsorption of following peptide chains on the gold and
palladium surfaces.
Note that the different values of the matrix elements
(eHS, ePS, eES) for the interactions between the residues of
these peptide chains and the substrates distinguishes between
gold and palladium (see section 2).
Snapshots of some of these peptide chains at the end of
simulations are presented in Fig. 4. At a first glance, we see
that A3 and Pd2 are adsorbed on both gold and palladium
while Pro10 is desorbed. Most of the aromatic groups seem to
bind both surfaces. It is difficult to distinguish the degree of
adsorption of A3 and Pd2 on gold and palladium surfaces
from a snapshot at the end of simulation. Therefore, a closer
examination of adsorption is necessary by quantitative
analysis of the energy of each residue, their mobility, and
surrounding profiles as follows.
The energy of each residue with gold and palladium sub-
strates is presented in Fig. 5 and 6 respectively. Note that the
total energy of a residue includes its interaction with other
residues of the same peptide and that of its neighboring
peptides, surrounding solvent, and the substrate. There is a
considerable change in energy from one residue to another in
the same peptide chain as well as among different peptide
chains. The comparison of Fig. 5 and 6 shows no change in
pattern in samples with gold and palladium apart from minor
differences in the magnitude. Residues with low energy in both
Figures include Pro11 of A3, Asp4 and Lys8 of Flg, Pro6 and
Arg7 of Pd2, Asn3, Thr8, and His11 of Pd4. Despite the overall
low energy, one cannot tell which peptide has higher adsorp-
tion probability to these substrates (Au, Pd) or which residue
anchors them. Based on purely non-covalent interaction thermo-
dynamics, the stability of equilibrium (and subsequent
structure) is dictated by low energy. The peptide (homopolymer)
chains Gly and Pro possess very little cohesive energy; the
residue energy in these peptides is too large in comparison to
low-energy residues in rest of the peptides, e.g. A3, Flg, Pd2
and Pd4.
In order to assess the adsorption of the peptides and identify
the specific residues that may bind to the substrate, it is
important to analyze their profiles of surface-interaction
(adsorption) energies. Unlike the total energy of each residue
described above (Fig. 5 and 6), only the interaction energy of
each residue with the substrate within the range of interaction
of the surface sites constitutes the surface-interaction energy.
Table 3 A typical interaction matrix among H, P, E, S and W components
H P E W S
H 0.0 0.0 0.0 0.1 eHS
P 0.0 �0.2 �0.2 �0.2 ePSE 0.0 �0.2 eEE �0.3 eESW 0.1 �0.2 �0.3 �0.1 0.0S eSH eSP eSE 0.0 0.0
Table 4 Peptide chain details
Peptide chain Amino acids
A3 Ala–Tyr–Ser–Ser–Gly–Ala–Pro–Pro–Met–Pro–Pro–Phe[H7–P4–P2–P2–H8–H7–P5–P5–H6–P5–P5–H4]
Flg Asp–Tyr–Lys–Asp–Asp–Asp–Asp–Lys[E1–P4–E3–E1–E1–E1–E1–E3]
Pd2 Asn–Phe–Met–Ser–Leu–Pro–Arg–Leu–Gly–His–Met–His[P8–H4–H6–P2–H3–P5–E4–H3–H8–P6–H6–P6]
Pd4 Thr–Ser–Asn–Ala–Val–His–Pro–Thr–Leu–Arg–His–Leu[P1–P2–P8–H7–H2–P6–P5–P1–H3–E4–P6–H3]
Pro10 Pro–Pro–. . .–Pro[P5–P5–. . .– P5]
Gly10 Gly-Gly–. . .–Gly[H8–H8–. . .–H8]
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1993
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

Fig. 7 and 8 show the profiles of the surface-interaction
energy of each residue in all peptide chains with gold and
palladium surfaces, respectively. The residues in each peptide
with low (e.g. large negative) surface-interaction energy can be
easily identified. Peptide A3 has two residues, Tyr2 and Phe12,
with very low surface-interaction energy on both substrates
(Au and Pd). Both residues are aromatic and have potential to
anchor A3 on the substrates. The surface-interaction energy of
these residues is comparatively lower at the palladium surface.
Therefore, the probability of adsorption of A3 on the palla-
dium substrate is higher than on the gold substrate. Similarly,
the peptide Flg is anchored by an aromatic residue Tyr2 on
both surfaces and the probability of adsorption is higher on
palladium. The probability of adsorption of the peptides Pd2
and Pd4 is also higher on the palladium substrate in compar-
ison to gold. Binding residues are Phe2, His10 and His12 for
Pd2 and His10 and His11 for Pd4; these residues are also
aromatic. The peptides Gly and Pro possess only very minor
negative surface interaction energy and do not adsorb on
either substrate.
Since the interaction potential and matrix in our coarse-
grained description (presented above) are phenomenological,
Fig. 4 Snapshots of A3, Pd2 and Pro on gold (Au, top row) and palladium (Pd, bottom row) surfaces are shown at the end of simulations
(105 MCS time) on a 643 lattice. (green (H), gold (P), blue (E), pink (A), grey (W), dirty gold (substrate)).
Fig. 5 Profile of the total energy of each residue of each peptide on a gold substrate. Statistics: sample size 643, 10 peptides in each of 100
independent samples, with 105 time steps.
1994 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

one may question the validity of conclusions based on the
specificity of the residues and their sequences. To test the
sensitivity of the role of amino acids in a specific sequence, we
have examined the physical properties of analogous homo-
polymers of each residue in some of these peptides. As an
example, Flg appears suitable as it contains only three differ-
ent amino acids, i.e. Asp, Tyr, and Lys which lead to three
homopolymer chains25 Asp8, Tyr8, and Lys8, each with eight
nodes of the respective residue. The adsorption energy of each
monomer segment in these three homopolymers on the palla-
dium surface is presented in Fig. 9 along with that of the
peptide Flg. We notice the contrast in adsorption energy
among the homopolymers and the peptide. Despite the same
characteristics of each monomer in a homopolymer (e.g. Asp8
and Lys8), there is a considerable difference in surface-
interaction energy from one node (segment) to another. This
implies that the conformation of the chain at the surface and
steric constraints play a significant role in adsorption and
Fig. 6 Profile of the total energy of each residue of each peptide on a palladium substrate. Statistics: sample size 643, 10 peptides in each of 100
independent samples, with 105 time steps.
Fig. 7 Surface-interaction energy Ens of each residue of each peptide with the gold substrate. Statistics: sample size 643, 10 peptides in each of 100
independent samples, with 105 time steps.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1995
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

anchoring (strong adsorption). The lowest surface-interaction
energy of Tyr2 in Flg, however, is due to its interaction with
the Pd surface. Anchoring of Flg by Tyr2 leads to a different
conformation than that of corresponding homopolymers,
e.g. Tyr8.
The mobility of each residue is evaluated from their average
number of successful moves per unit time step. Fig. 10 and 11
show the mobility profiles of each residue in all peptide chains
with gold and palladium surfaces, respectively. The mobility of
Tyr2 is the lowest among all residues of peptide A3 on gold
substrate. Despite the low surface-interaction energy of Phe12
of A3 (Fig. 7), it is more mobile on gold (Fig. 10). We conclude
that the adsorption of A3 on gold is for the most part
mediated by Tyr2 and not by Phe12. The mobility of Tyr2 of
A3 is much lower on palladium than that on gold (see Fig. 10
and 11). Therefore, Tyr2 is the major anchoring residue for A3
on palladium as well. The mobility of Phe12 of A3 on
palladium is higher than Tyr2 but it is still low in comparison
Fig. 8 Surface-interaction energy Ens of each residue of each peptide with the palladium substrate. Statistics: sample size 643, 10 peptides in each
of 100 independent samples, with 105 time steps.
Fig. 9 Surface-interaction energy Ens of each residue in various homopolymers (Asp8, Lys8, Tyr8) and peptide Flg on a palladium substrate.
Statistics: sample size 643, 10 peptides in each of 100 independent samples, with 105 time steps.
1996 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

to Phe12 and comparable to Tyr2 on gold. Thus, Phe12 is an
additional anchor for the adsorption of A3 on palladium due
to both low surface-interaction energy (Fig. 8) and low
mobility (Fig. 11). Residue Tyr2 of Flg is of lowest mobility
on both gold and palladium surfaces (Fig. 10 and 11) and
anchors the adsorption of Flg. It should be pointed out the
contributions of residues Lys3 and Asp5 in anchoring (at least
partially) the peptide Flg for its adsorption cannot be ruled
out due to their relatively low mobility and low surface energy.
Low mobility of Phe2, His10, and His12 of Pd2 and His6 and
His11 of Pd4 along with their low surface-interaction energy on
both gold and palladium substrates (Fig. 7–11), with lower
Fig. 10 Mobility Mn of each residue of all 10 peptide chains on the gold substrate; mobility (y-axis) of each residue per peptide chain should be
divided by 10. Statistics: sample size 643, 10 peptides in each of 100 independent samples, with the 105 time steps.
Fig. 11 MobilityMn of each residue of all 10 peptide chains on the palladium substrate; mobility (y-axis) of each residue per peptide chain should
be divided by 10. Statistics: sample size 643, 10 peptides in each of 100 independent samples, with 105 time steps.
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1997
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

surface-interaction energy on the latter, show that these
residues most strongly anchor the peptides Pd2 and Pd4. All
residues of Gly and Pro are relatively more mobile and offer
little probability of adsorption or anchoring.
Local structural profiles are also examined by monitoring
the number of different components (residue groups, solvent,
substrate) within the range of interaction of each residue in all
peptide with both gold and palladium surfaces. The profile for
the number of surface sites (Ns) within the range of interaction
of each residue may be most relevant, especially for assessing
the adsorption of peptides and their anchoring residues.
Fig. 12 and 13 show such surface-interaction profiles. The
closer a residue is to the substrate, the larger a value Ns would
have and therefore the higher its adsorption probability. High
numbers of surface-interaction sites (Ns) are found for Tyr2
and Phe12 of peptide A3, Tyr2 of peptide Flg, Phe2, His10, and
His12 of peptide Pd2, and His6 and His11 of peptide Pd4 on
both gold and palladium substrates (Fig. 12 and 13). Structural
profiles near surfaces support our above analysis in identifying
the residues that anchor the peptides to the surface and
are consistent with the above analysis based on their low
surface-interaction energy and low mobility.
Note that the molecular weight of all peptide chains is not
the same (the smallest is Flg with eight residues) and the size of
each chain is too small to analyze the scaling of the radius of
gyration with the molecular weight. The radius of gyration
(Rg) can be calculated, nevertheless, and its magnitude can be
compared in the sample with the gold surface to the sample
with the palladium surface. The equilibrium value of the
radius of gyration hRgi of each peptide chain is different in
both samples. Estimates of hRgi for each peptide chain are
presented in Table 5. We see that hRgi is generally lower in the
sample with the palladium substrates. Of particular note is the
difference in the magnitude of hRgi of A3 which is reduced by
about 70% on the palladium surface in comparison to its value
on the gold surface. In general, palladium is found to be more
adsorbing than gold.
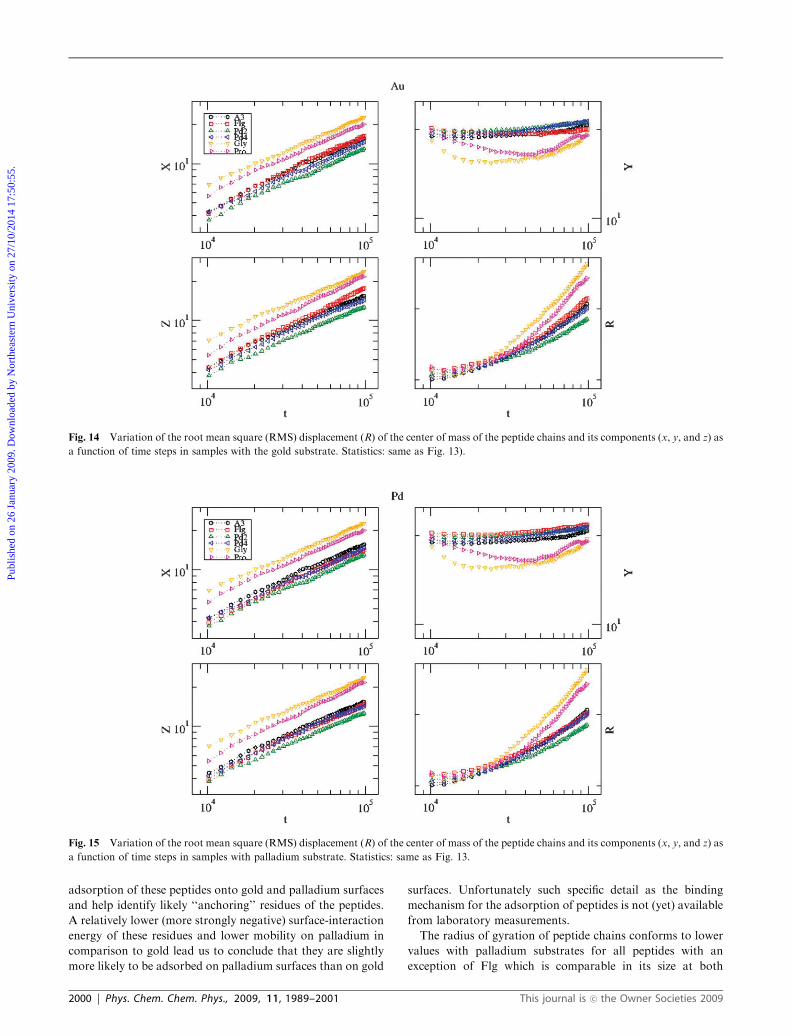
Finally, the overall dynamics of the peptide chains can be
studied by analyzing the variation of the root mean square
(RMS) displacement of the chains with the number of time
steps. Variations of the RMS displacement of the center of
mass of the peptide chains and its components (x, y, z) as a
function of time steps (number of Monte Carlo moves) are
presented in Fig. 14 and 15 with gold and palladium sub-
strates, respectively. The collective dynamics of the peptides
appears very similar in the samples with gold (Fig. 14) and
palladium surfaces (Fig. 15). However, there are marked
differences in the dynamics between the individual peptides
on the surfaces. For example, the rate of change of the RMS
displacement (R) of the peptide Pd2 is the lowest followed by
Pd4, A3, and Flg while that of the peptides Gly and Pro
(homopolymers) is the highest. Peptides Gly and Pro are most
mobile and have the lowest probability of adsorption on both
substrates. The motion of the remaining peptide chains
(A3, Flg, Pd2, Pd4) is considerably slower due to their adsorption
on the substrates. Further the differences in motion of peptides
along different directions (x, y, and z) should be pointed
out. The metallic substrates considered here lie in the
zx-plane normal to y, the vertical direction (see Fig. 4).
The motion of peptide chains along the longitudinal (y)
direction is constrained by their adsorption on the substrates
as seen from the y-component of the RMS displacement of
the center of mass of peptide chains. In transverse directions
(z and x), there is no such constraint and chains diffuse freely.
These differences in the dynamics of each peptide along the
longitudinal and transverse directions are clearly seen in
Fig. 12 Number of surface sites (Ns) in the vicinity of each residue of each peptide on the gold substrate. Statistics: sample size 643, 10 peptides in
each of 100 independent samples, with the 105 time steps.
1998 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

Fig. 14 and 15 from the rate of change of corresponding RMS
displacements.
4. Summary and conclusions
A coarse-grained computer simulation model is introduced to
study the adsorption of the peptides A3, Flg, Pd2, Pd4, Gly10,
Pro10 on gold and palladium surfaces. With the simplicity of
the model and computational efficiency it is possible to predict
the probability of adsorption of peptides and their specific
binding (anchoring) residues. A peptide is represented by a
specific set of residues (represented by particles or nodes)
tethered together by fluctuating bonds in a flexible chain of
a specific sequence. Although the internal structural details of
each residue are ignored, their specific characteristics are
captured via an interaction matrix for the coefficients of a LJ
potential; the relative magnitude of the interaction matrix
elements are based on the all-atomic analysis31,32 of each
residue.
The adsorption probability of each peptide chain is deter-
mined by analyzing a number of global and local quantities
including the energy of each peptide chain and that of each
residue in their unique sequence position, the interaction
energy of each residue with the surface, their neighborhood
profiles, their mobility, the radius of gyration of the peptides
on the different surfaces, and the variation of the root mean
square displacement of the peptide chains as a function of the
number of simulation steps. In addition, visual inspection is
also used to assess the degree of adsorption of each peptide.
We find that peptides A3, Flg, Pd2, and Pd4 are more likely
to be adsorbed on both gold and palladium surfaces with
various degrees while Pro10 and Gly10 are desorbed. These
observations, though speculative, seem consistent with the
experimental observation (SEM images of bi-metallic nano-
particles in presence of A3 and Flg and their dimers and
surface coverage of Au and Pd,9 as well as with TEM micro-
graphs10 of the aggregation of spherical gold nanoparticles
with A3 and Flg). There are many residues with lower
(negative) surface-interaction energy and lower mobility in
A3, Flg, Pd2, and Pd4 than those in Gly10 and Pro10. Slightly
negative surface-interaction energy does not necessarily indi-
cate higher adsorption probability. Such residues include Ser4,
Pro8, Pro11 in peptide A3, Asp4 and Lys8 in Flg, Pro6 and Arg7
in Pd2, Asn3, Thr8 and His11 in Pd4 and Pro10 in Pro. Residues
with significantly negative surface-interaction energy and thus
strong adsorption with both gold and palladium surfaces, on
the other hand, are Tyr2 and Phe12 in A3, Tyr2 in Flg, Phe2,
His10, and Phe12 in Pd2, and His6 and His11 in Pd4. Mobility is
also low for these residues on both gold and palladium
surfaces with an exception, i.e. Phe12 in A3 with gold. This
means that the conformational entropy (physical constraints
on the shape) plays a more important role for the peptide than
for a small molecule such as a single amino acid,30 in concert
with the energy in anchoring the peptide chains to the surface.
The number of surrounding substrate sites of residues in the
peptide chains (surface-interaction profiles) confirm the
Fig. 13 Number of surface sites (Ns) in the vicinity of each residue of each peptide on the palladium substrate. Statistics: sample size 643,
10 peptides in each of 100 independent samples, with 105 time steps.
Table 5 Radius of gyration (Rg) of peptides on gold and palladiumsurfaces
Peptide hRgi on Au hRgi on Pd % difference
A3 4.484 � 0.031 2.619 � 0.024 71.2Flg 3.240 � 0.019 3.194 � 0.016 1.4Pd2 4.480 � 0.033 2.804 � 0.027 37.4Pd4 4.486 � 0.032 2.702 � 0.035 39.8Gly 4.086 � 0.025 2.249 � 0.028 44.9Pro 3.951 � 0.020 2.190 � 0.025 44.6
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 1999
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online
adsorption of these peptides onto gold and palladium surfaces
and help identify likely ‘‘anchoring’’ residues of the peptides.
A relatively lower (more strongly negative) surface-interaction
energy of these residues and lower mobility on palladium in
comparison to gold lead us to conclude that they are slightly
more likely to be adsorbed on palladium surfaces than on gold
surfaces. Unfortunately such specific detail as the binding
mechanism for the adsorption of peptides is not (yet) available
from laboratory measurements.
The radius of gyration of peptide chains conforms to lower
values with palladium substrates for all peptides with an
exception of Flg which is comparable in its size at both
Fig. 15 Variation of the root mean square (RMS) displacement (R) of the center of mass of the peptide chains and its components (x, y, and z) as
a function of time steps in samples with palladium substrate. Statistics: same as Fig. 13.
Fig. 14 Variation of the root mean square (RMS) displacement (R) of the center of mass of the peptide chains and its components (x, y, and z) as
a function of time steps in samples with the gold substrate. Statistics: same as Fig. 13).
2000 | Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 This journal is �c the Owner Societies 2009
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online

(gold and palladium) surfaces. The global dynamics of peptide
based on the variation of the RMS displacement of peptide
chains show that Gly and Pro move fast after desorption from
the substrates while A3, Flg, Pd2, and Pd4 remains slow.
These findings seem consistent with all-atomic simulations32
and laboratory observations.8–10
The model and the results presented here certainly leave
room for improvement. The interaction matrix selected for the
phenomenological interaction potential is one of our early
attempts which can be fine-tuned in coordination with labora-
tory data. Moreover, the combination with bioinformatics
approaches in the future might provide interesting opportu-
nities for in silico screening of the huge number of potential
peptides to control binding properties to a given surface.
Acknowledgements
Support from the Materials and Manufacturing Directorate of
the Air Force Research Laboratory, the Air Force Office of
Scientific Research (AFOSR) is gratefully acknowledged.
H. H. and J. F. acknowledge further support from the University
of Akron and from the Ohio Supercomputing Center.
References
1 M. Sarikaya, C. Tamerler, D. T. Schwartz and F. Baneyx, AnnualRev. Mater. Res., 2004, 34, 373.
2 C. Tamerler, E. E. Oren, M. Duman, E. Venkatasubramanian andM. Sarikaya, Langmuir, 2006, 22, 7712.
3 G. W.M. Vandermeulen and H.-A. Klok,Macromol. Biosci., 2004,4, 383.
4 Engineered Inorganic-Binding Polypeptides for BiotechnologyBioMEMS and Biomedical Nenotechnolgy, eds. M. Ferrari,A. P. Lee and L. J. Lee, Speinger, US, 2006.
5 R. Fairman and K. S. Akerfeldt, Curr. Opinion in Struct. Biol.,2005, 15, 453.
6 V. P. Raut, M. A. Agashe, S. J. Stuart and R. A. Latour,Langmuir, 2005, 21, 1629.
7 D. Horinek et al., P.A.N.S, 2008, 105, 2842.8 J. M. Slocik, M. O. Stone and R. R. Naik, Small, 2005, 1,1048.
9 J. M. Slocik and R. R. Naik, Adv. Mater., 2006, 18, 1988.10 M. M. Tomczak, J. M. Slocik, M. O. Stone and R. R. Naik, MRS
Bulletin, 2008, 33, 519, and references therein.11 U. H. E. Hansmann, Comput. Sci. Eng., 2003, 5, 64.12 Y. Zhou and M. Karplus, Nature, 1999, 401, 400.13 E. J. Sorin and V. S. Pande, Biophys. J., 2005, 88, 2472.14 M. Shen and K. F. Freed, Proteins, 2002, 49, 439.15 B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, L. Barry,
B. L. Stoddard and D. Baker, Science, 2003, 302, 1364.16 Y. Wei, W. Nadler and U. H. E. Hansmann, J. Chem. Phys., 2008,
128, 025105.17 O. Zimmermann and U. H. E. Hansmann, Biochim. Et Biophys.
Acta, 2008, 1784, 252.18 T. Herges and W. Wenzel, Phys. Rev. Lett., 2005, 94, 018101.19 A. Liwo, M. R. Pincus, R. J. Wawak, S. Rackovsky and
H. A. Scheraga, Prot. Sci., 1993, 2, 1697.20 J. Bjursell and R. B. Pandey, Phys. Rev. E., 2004, 70, 052904.21 R. B. Pandey and B. L. Farmer, Phys. Rev. E, 2008, 77, 031902.22 C.-M. Chen and C.-C. Chen, Biophys. J., 2003, 84, 1902.23 R. B. Pandey and B. L. Farmer, J. Chem. Phys., 2009 in press.24 I. Carmesin and K. Kremer, Macromol., 1988, 21, 2819.25 Monte Carlo and Molecular Dynamics Simulations in Polymer
Science, ed. K. Binder, Oxford University Press, New York,1995.
26 R. B. Pandey, K. L. Anderson, H. Heinz and B. L. Farmer,J. Polym. Sci. Part B: Polym. Phys., 2005, 43, 1041.
27 R. B. Pandey, K. L. Anderson and B. L. Farmer, Phys. Rev. E,2007, 75, 061913.
28 R. B. Pandey and B. L. Farmer, Macromol. Theor. Sim., 2008, 17,208.
29 R. B. Pandey and B. L. Farmer, J. Polym. Sci. Part B: Polym.Phys., 2008, 46, 2696.
30 L. M. Ghiringhelli and L. Delle Site, J. Am. Chem. Soc., 2008, 130,2634.
31 H. Heinz, R. A. Vaia, B. L. Farmer and R. R. Naik, J. Phys. Chem.C, 2008, 112, 17281.
32 H. Heinz et al. (to appear) and J. Feng et al. (to appear).
This journal is �c the Owner Societies 2009 Phys. Chem. Chem. Phys., 2009, 11, 1989–2001 | 2001
Publ
ishe
d on
26
Janu
ary
2009
. Dow
nloa
ded
by N
orth
east
ern
Uni
vers
ity o
n 27
/10/
2014
17:
50:5
5.
View Article Online






















![Case presentation pd2[1]](https://img.pdfslide.us/doc/110x75/548404cfb47959140d8b4a9f/case-presentation-pd21.jpg)






