Embed Size (px)
Citation preview

Aaalytita Chikta At:a Elsavicr Puhlbldng Company, Amsterdam Printad in Tho Nethcrlmds
ACTIVATION ANALYSIS OF IWIIE EA’ICTHS
PART XII. THE DETERMINATION OF RARE EARTHS IN MINERALS l3Y TWE SINGLE CONIPARATOR TECHNIQUE
The determination of the rare-earth elements in rocks, meteorites, minerals and similar materials has been described by several authorsr-6. The present paper deals with the application of the so-called single comparator technique to the deter- mination of rare earths in minerals, in this case gadolinite and a tantalocolutnbite. The single comparator technique was proposed by GIRARDI~ who discussed its merits and its application for the determination of chromium in iron, chloride in water, and gallium and copper in aluminium. In our laboratory, the technique has been used by ISSELEE 7 for the determination of As, Sb, Cu and Au in lead. The technique consists essentially in standardising the activity of the rare-earth standards obtained after irradiation in a fixed position and during a constant period against the activity of a flux monitor. The principal advantage of this method is in its applicability to routine analysis since ideally the 16 rare-earth standards can be replaced by a single measurement of the comparator. In fact only xz were standardised according to this procedure for various reasons.
As flux monitor cobalt was chosen, because it is activated easily (a=36 barn) to tbe.oQCo isotope which has a long lifetime (T+ - -5.3 years) and possesses characteristic y-emitting energies (1.17 and 1.33 MeV). The single comparator method requires that for identical irradiation times, always the same ratio between the activities of cobalt and the different rare earths will be obtained. Since the excitation functions (i.e., the activation cross-section as a function of neutron energy) of the rare earths and cobalt are not identical, this can only be the case if the neutron spectrum is constant over the whole series of irradiations.
The errors induced by shifts in the neutron spectrum depend on the fraction of total activation due to epithermal activation of flux monitor and standard. The ratio of the specific activities of the rare earths and cobalt depends on the ratio of the effective activation cross-sections, bnct, defined as:
bact. =ath(I -_fr) +arfr
where r stands for resonance and th for thermal.
baat./br = (ath/ar) (I -fr) +fc
and since (c~)~.~./(o~)e,, is a constant l Rcscarch associate of I.1.K.W. (Belgium).
Anal. Cl&r. A&a, 42 (rg68) 21-28

22 1). 1.. ~lASSA1~1’, J.. HOS’I’E
f+-om the cac.hiutn r~~~io of colxdt and the values of the resonance integral and tile tltcrtnal netiwttion cross-section, one can calculate Jr. For the irradiations in MC-I a value of 0.128 was found. 17rom the cadmium ratios of the rare earths, one can Ck?hTIIhC? Qt)l/Qr, if fr is known. With UI h/Glr the VZdUC CJf (Ua(!t)it.I~./(Qcrot)co WU
calculated for Jr=o.I18 Und fr =0.X38. The perccntagc diffcrcncc bctwccn these values is given in ‘I’ahtc I (Ll’y” talc.).
-___- . Iall ‘l’ill 15r I-10 Tb Gil ITU
.-~“--__._- 8.62
2.38 3.25 2.0s
1.80
1.23
21.5
t 000
?‘*7 t9K4
t3t ttb tt9
20$3p
lislrernc ~ulrrcs cl’l/o OUCY .j clclns.
_-._L.-- ._---._--_.
!iq9 8.0 .[.fb . G.8
17.8 14.0 .1.8
Sill 1.7s Ntl 5.00 1% I r.r<* CC t I.08 I.& 11.7 Co 6.1
_-.._-.---_ --
( 516509 I zGb3 7.5 8 344 0.85 7,s 0
3-e 82.2 5.C) 0. I 7Gr 27.9 I I.0 0. I
25578 598 5.9 0.2
7305 5.3 0
_____,_.,____.___.-__ __- _.__. --_.“._. ._ -..__._ _---_--m_I-
TIIll Y-IENITHtiIXS Slbl.lSCTtSl’)
--- ___.. _.._- -___--.--_ - .._. - ._ . . . _..-. ___-_-.-_ .-. ._. --
Isotope Half-lifi sctcctld cm.!rgy f sotopc Ndf-!ifc .%!leclcd CmYgy _. -______---__-._ -._-..._
1771.u 6.8 d 208 ItcV 16lfllr~u 9.2 h 837 -I- 961 kcV 1 “I*~~,~ rjo tl 8.t I<CV ’ wh 47 11 70 -i- toz kcV 17113. 7.8 h 296 -t- 308 hv 1 .i%l’r 19.2 h 1.57 MCV
t~~f-10 27.3 11 I .3G MCV i.mcc 33 h 294 IceV I “fl’~~, 72 cl sso -t 970 kcV ’ “ %n 40.2 h t .60 MoV ‘ J”GtI 18 11 #L& lrcV fI”Co .5.3 Y I.17 -t x.33 hIcV
_I_.__-_-____.~__ -_-__ --
y-Spectrametric nxxasurernents were carried out with a multichannel analyser. For each rare earth a characteristic y-energy was selcctcd and the counts under the corresponding peak were summed. Sometimes the sum of more than one peak was used. Table II summarises the selected y-energies ‘and the half-lives of the measured isotopes. The clloice of the isotopes for Lu, Tm, I$, Ho, Tb, Eu, Sm, Pr, La and Co is obvious, no other isotopes of the element being formed with a sufficient activity.
Gaclolinium yielcls 3 measurable isotopes: 16JGd and 1soGd by (n,?) reactions and lal’J?b by an (n,y ; /I-) reaction. 163Gd could not be used since it was added as chemical yield monitor (see Part I of this series*). looTb formed by the (n,y) reaction on terbium interferes with the measurement of IalTb as the former gives rise to higher y-energy. Thus 1sQGd was chosen for the determination of gadolinium.

AC,TIVATlON ANALYSIS OF RARE ISA RTIiS. It I “3
Cerium gives rise to one isotope besides the already mentioned ~4Ce, This isotope, i~iCc, has a half-life of 33 days, i4XZe was chosen because there is no intcr- ferencc with its y-energy of 294 keV by 14iCe (highest y-energy r45 keV). *4lCe can be used but its standardisation must be carried out after cotnplete decay of i4Gz There is an interference with the spectrum of i7sYh by the longer-lived i&QYb, hence ytterbium was determined in the mineral by direct comparison with an ytterbium standard. 15*Pm and rd*Pm formed by (n,?; p-} reactions interfere with 14rNd, hence neoclymium W;LS separated from promethium on a cation exchanger witfr cs-hydroxy- isobutyrate (a-HIBA) as eluant. The neodymium was collcctccl in a 50-ml flask ancl tnea~ured under the same conditions as the other rare earths. The 533-keV peak was selected. The dysprosium isotopes are too short-livccl to be measured since txtwccn the end of irradiation and the beginning of the measurements tltere is a time lapse of 20 11. QoY is a pure p-emitter and is not included in the single comparator techniclue.
Table I gives the results of the stancfardisation as well as the measured cadmium ratios. As can lx expected, the range on the results is higfrcst for tfje lower cadmium ratios (‘I’m, Ce and Nd are not active enough to be compared with the others).
The rare earths were separated as a group hefore irracli~~tion and after addition of LJeGd, which has a high specific activity, as a chemical yield monitor (see Part IN), After irradiation, the rare earths were separated by cation exchange with 0.5 M cu-WIBA as the eluting agent. The speed with which the elution was achieved, was mainly determined by the short half-lives of i7lEr and 1G~mEu. Different procedures were tested always at a flow-rate of I[ mlfmin*cm~.
(3) A linear gradient was used with the volume of the two communicating vessels T/t =soo ml, an initial ligand concentration co =0.03 M and a ligand concen- tration in the reservoir ei ,=o.33 M. With this gradient one can coilect the following fractions: Np+Lu +Yb+part of the Tm activity: part of the Tm activity-l-l+; Ho. Subsequently the lighter rare earths were separated quantitatively. In the second fraction Tm does not interfere with the measurement of 171%. After the decay of 171Er, 17eTm can be counted without any interference. In the first fraction, 17’Lu and l?bYb can be determined easily, but interfere with 170Tm, as their half- lives are too long. If required, a subsequent complete separation of Lu-Yb-Tm can be applied on another column.
(2) Elution of lutetium and ytterbium at constant &and concentration was followed by a linear gradient to separate the other rare earths. A quantitative separation was obtained but the procedure was too long to allow determinations of erbium and europium because zo 11 were lost during transportation.
(3) A linear gradient was tested with V,=~OO ml, co=o.03 M, c1,=o.r65 M. This gradient permitted the collection of the following fractions: Np; Lu+Yb; ‘I’m ; 3% followed by the other individual rare earths. This gradient was not as steep as the first but still permitted the determination of erbium. After the clution of yttrium, the concentration CI, in the reservoir was increased to 0.33 M. This procedure was finally adopted for the separation of the rare-earth fraction of gadolinite (Fig. I) and tantalocolumbite (Fig. 2). .
And. Ckinr. ncra, 42 (rgG8) 2X-23
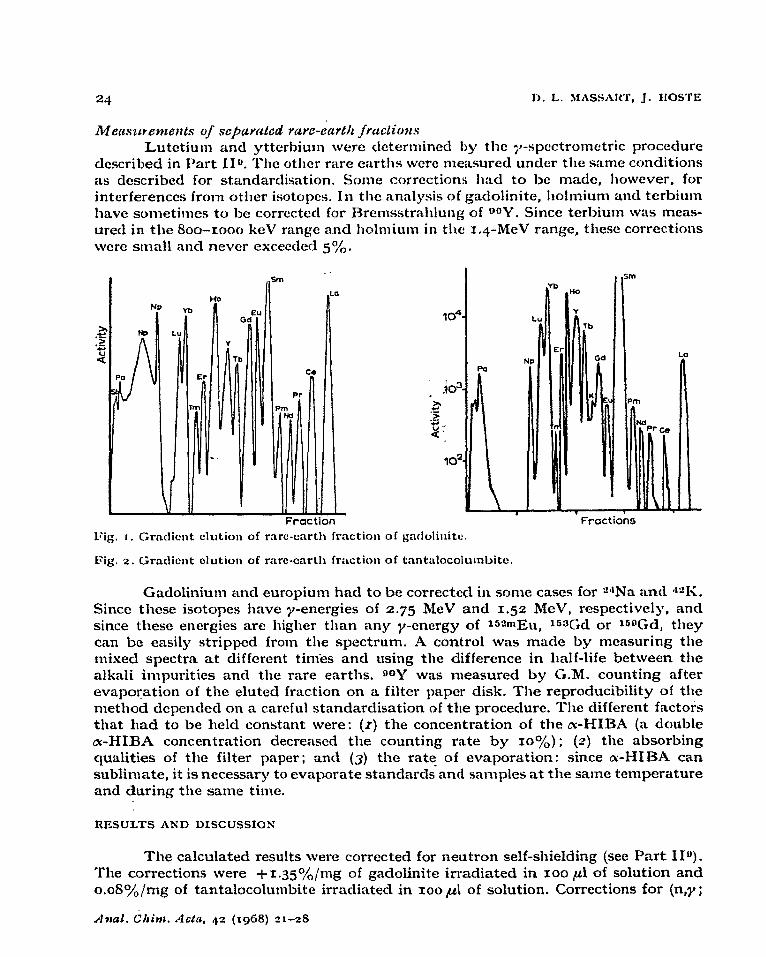
Lutetium and ytterbium were determined by the ;+spcctromctric proceclure described in Part 110. Tllc otllcr rare earths wcrc measured under tile same conditions as &scribed for standardisation. Some corrections tlacl to be ma&, tlowcver, for interferences from other isotopes. In the analysis of gadolinite, tlolmium and terbium have sometimes to be corrected for Brcmsstrailtung of OQY. Since terbium was meas- ured in the 8oo-rooo keV range and holmium in the x.4-MeV range, tilcsc corrections were small and never exceeded so)&.
Fractions 1Gg. I. Crnclicnt clution of mrc-cart11 fraction of gatlolinitc.
Fig. 2. Grncliont olution of rilrc-cart11 frilcti011 of tnntalocolumbitc.
Cadolinium and europium bad to be corrected in some cases for ‘*dNa and &IX. Since these isotopes have y-energies of 2.75 MeV and 1.52 MeV, respectively, and since these energies are higher ttlan any y-energy of 1~~mEu, 153Gd or lfiOGd, they can be easily stripped from the spectrum. A control was macle by measuring the mised spectra at different tim’es and using the difference in half-life between the alkali impurities and tile rare earths. OOY was measured by G.M. counting after evapoqation of the eluted fraction on a filter paper disk. The reproducibility of the method depended on a cnrcful standardisation of the procedure. The different factors that had to be held constant were: (I) the concentration of the a-HIBA (a double c+HIBA concentration decreased the counting rate by IoO/~); (2) the absorbing qualities of the filter paper; and (3) the rate of evaporation: since a-HIBA can sublimate, it is necessary to evaporate standards ancl samples at the same temperature and during the same time.
RESULTS AND DISCUSSION
The calculated results were corrected for neutron self-shielding (see Part II@). The corrections were -+x.35°~fn~g of gadolinite irradiated in IOO pl of solution and o.08°/O/mg of tantalocolumbite irradiated in IOO ,ul of solution. Corrections for (n,y ;

p-j, (n,y; P--i n,y) and (n,y ; n,y ; /I-) reactions wcrc calculated. In all cases, these corrections were negligible escept for the iraYb(n,y; p-)r77Lu reaction which has already been discussed in Part II@.
An important correction was required by the reactivation of 153Gd used as chemical yield monitor because it was not carrier-free. This error was determined by reactivating 153Gd samples of known activity and counting the resultant r6*Gd, .The correction never exceeded 25(j/o. The presence of 23QNp activity indicated the presence of uranium in the irradiated sample. By fission this could give rise to positive errors in the lanthanum and cerium cleterminations. Therefore, a determination was carried out on a rare-earth fraction of gadolinite from which uranium had been eiiminatcd before irradiation.
The separation of uranium was achieved by paper chromatography with 5% nitric acid in ether as eluant. This method h,as already been described for the eliminn- tion of uranium on cellulose columnsl0; uranium migrates close to the front and the rare earths remain on the starting line. After activation of the isolated rare-earth fraction and ;L similarly treated blank, lanthanum was cletermined. The result was o.r3z0/0, which indicates that there wcas no positive error caused .by the fission of uranium.
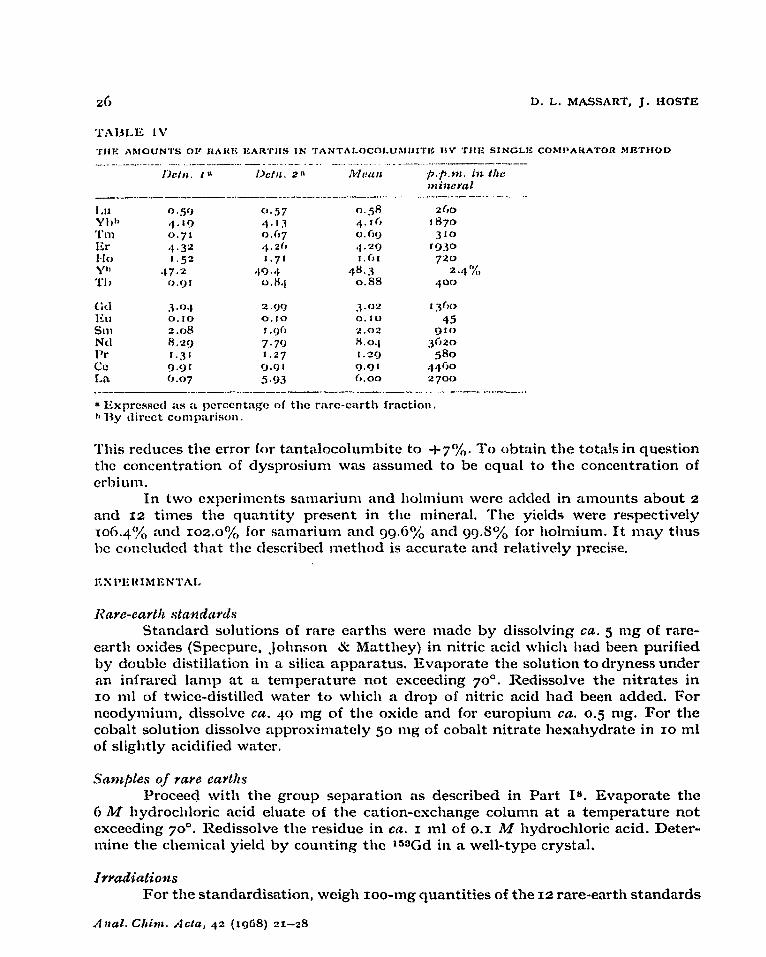
The results obtained for gadolinite by the normal standard procedure and the single comparator method are summarised in Table 1x1. The results obtained by the single comparator method for tantalocolumbite are summarisecl in Table IV.
TflK PISRCENTAGlE AMOUSTS OF RARI; ISARTIIS IN GADOLINITE XII’ THIS NORMAL STAZNDARD AND
SINGLE COMPARATOR SIBTIfODS
-__-.. _ _...... _---- -___ _...__ _. ..__. __._..._. .._.c._.. -. .._ -._..... . . . . . ._. .__ ^.. . ._
Giemartf Sittgle SfCtttditTXt DirccC .sfudmd
colirparritor iltwiatioir co?ripaYiso?r dcviiitima f%) (%J
__. .._. .- .._ ..-_.__ _._-.._. _- _.._ _.” . . . . . . ..-... “. . . ..-.-_. _._ . _ _ _..- ---.. I.11 0.201 3 0.200 4
Vb Not cletrl. -- 1.33 . 4 s 5 Till 0. rg’
z o..Jsn 3
15r I.75 1.78 _- HO o.5s7 0-S 0.56 5.5
Y Sot clcttl. -- 3g -- Tit O-53 4 0‘5.5 2
Cd 2.OG 8.5 2.01 13.5 Iizu o.oor3~ .-- 0.0013” .-_ Sill I.293 4 r.qG 7 1%. 0.17’ 0.18~ 6.5
0.59 z 0.59 - 0.13~ 3 0.138 3.5
The sum of the rare earths found by activation analysis was 52.4 (as the osides) whereas gravimetry gave 50.05~/~ for gadolinite. For tantalocolumbite rare- earth contents of 5.27% (activation analysis) and 4.50*/~ (gravimetric method”) were found. The error was -l-4.8% for gadolinite and +13.7% for tantalocolumbite. The gravimetric method for the determination of rare earths, however, employs a homo- geneous precipitation technique for which a loss of 7yo can be predicted (Part 18).
~?JraZ. Cllinr. Ada. 42 (x968) 21-28
ZG D. L. MASSART, J. HOSTE
. __.. .__. __.. ._., _._ _.... ..__---_....... I. ----_-_-
IA$il. ia i3Cfli. 2” Mf!Wl fi.p+ni. ist Ihc: mimwd
____-___.-_-._ __..-__ - ___. ___,__-... ._...._._f --_- .---- t-.-. -. IA 0.59 a.57 n..e 2fm
YII’, at.19 4.1.3 *l-r6 1 H7# ‘I’ll1 0.7’ o.C,7 0.6!, 310 Kr 4t.32 .$.2(J ,l.‘rQ rc)iw HO r.gz 1.71 I.flI 720 ?I
;.1’, 47*2 aI9.4 4H.3 24%
f>.<)f 0.H‘) 0.88 .;oo
(A 3.04 2 .gq J.02 f3C)C-t
Ku 0.10 0.10 0. IO 45 Sill 2.08 I.90 2.02 rj t Cl
Ntl 8.20 7.79 8.0.1 $i20
Pr r.31 1.27 r.aq 580 Co 9*9r rJ.01 Q.91 i)i)fiO
1.n 0.07 5.93 0.00 2700 _____--_._._~ . ..___ __ ----.-- - _.._ -_.. -.-I_
1% I.Cxprcssctf iis a, pcrccntnge of the rare-cnrth frnction. 11 Ry direct conqmrism.
This reduces the error for tantalocolumbite to i-7%. To obtain the totals in question the concentration of dysprosium was assumed to be qua1 to the concentration of erbium.
In two experiments samarium and holmium wcrc aclclccl in amounts about 2
and x2 times the quantity present in the mineral. The yields were respectively 1o(i.4~/~ and roz.o(fl, for samarium ancl gc~.i,~/~ and gq,S”/O for holmium. It may thus bc concluded that the described method is accurate and relatively precise.
Standard solutions of rare earths were maclc by dissolving cu. 5 mg of rare- earth oxides (Specpure, Johnson & Matthey) in nitric acid which had been purified by double clistiliation in a silica apparatus. Evaporate the solution to dryness under an infrared lamp at a temperature not exceeding 70”. Redissolve the nitrates in JO ml of twice-distilled water to which a clrop of nitric acid had been added. For neodymium, dissolve cu. 40 mg of the oxicle and for europium ca. 0.5 mg. For the cobalt solution dissolve approximately 50 mg of cobalt nitrate hexahydrate in IO ml of slightly acidified water.
Sam$es of *we cartlts Proceed with the group separation as described in Part 18. Evaporate the
G M hydrochloric acid eluate of the cation-exchange column at a temperature not exceeding 70”. Redissolve the residue in cn. I ml of 0.1 M hydrochloric acid. Deter- mine the chemical yield by counting the la%d in a well-type crystal.
For the standardisation, weigh roo-mg quantities of the 12 rare-earth standards
Artal. Cl&u. Ada, 42 (r$Xi) 21-_=8

ACT~VATIOS AKALYSI~ OF rmm EARTHS. xrf 27
and 2 cobalt standards in silica ampoules, Seal and pack in z alurniniu~l~ capsules, each one containing at least one cobalt monitor. For the activation annlysis,,weigh the sample solution, transfer about xoo ,ul to a silica ampoule and weigh agai{i. Seal the ampoule and irradiate together with two cobalt monitors, one ytterbium standard and one yttrium standard.
All irradiations took place in RR-r at Mel (z h at a flus of 4*1ol* n *cm-e*sec-1).
After irradiation, break tbc ampoules and transfer tbc contents to so-ml graduated flasks. Do not fill to the calibration mark, but to the beginning of the neck to obtain a more repro(l~lciblc geometry.
All me~asurcmcnts were carried out on a 3 x 3” flat NaX(T1) crystal coupled to a 4oo-channel annlyscr.
Satttple se#4vution After irradiation break the ampoules and wash the contents with 0.1 N
hyclrochloric acicl into a column of Dowex SOW-X8 (zoo-400 mesl~) of size r4 cm x 0.4 cm”. Elutc with a 0.5 M a-HIDA solution with a lignnd gradient (I/t =soo ml; co = 0.03 N; CL =0.x65 M) and follow the elution by counting the eluted fractions (2 ml) in a well-type crystal. After the elution of yttrium, increase the l&and con- centration in the reservoir to 0.33 M by addition of concentrated ammonia. Collect ca. 200 fractions for the complete elution of tbc rare earths. The clution peaks are identified by y-spectrometry.
Collect the individual separated rare earths in so-ml graduated flasks and also transfer the standards to so-ml graduated flasks with o.i: M hydrochloric acid. For yttrium add first a quantity of LX-HIBA equal to the quantity in the cluted yttrium fraction. Make the measurements under the same conditions as described for the standardisation and for ytterbium as described in Part II”.
The measurement of yttrium is done by G. M. counting. Pipette from both the standard and the eluted yttrium fraction three r-ml portions onto aluminium cups fitted with a filter-paper disk. Place the cups in a ring at cqunl distances from an infrared lamp, evaporate and count.
After the precipitation step with ammonia in the group separation (see Part Is), dissolve the precipitate in a minimal quantity of hydrochloric acid and spot on three Whatman 3MM papers (30 x 15 cm) in a x2-cm long and z-cm wide band. After elution (front zo cm), cut out the starting line, ash, ignite and redissolve in hot 12 N hydrochloric acid. Dilute to 2 N hydrochloric acid and proceed as described in Part 18 with the cation-exchange step.
SunfnfARY
The determination of rare earths in minerals by activation analysis is described. The rare earths are separated as a group from the bulk of the material before irradia-
/far&t. CkiOl. Rein. 42 (r@q 2T-28

tion. After irradiation tllc rare cartlls arc scparatcd from each other by gradient clution with ammonium a-l~ydroxyisol~utyrate on a cation-exchange column. The clcmcnts arc determined 1,~ the single comparator technique. This method permits a practical application of activation analysis to the routine determination of rat-c earths in complex matrices.
On dbcrit un tlosagc clc tcrrcs rares tlans dcs mincrnis. Ides tcrrcs rnres sont s6parf5cs cn un groupc avant irradiation. Aprh irradiation lcs tcrrcs x-arcs sonf” s+ar&s lcs uncs dcs nutrcs par blution shctive B l’aide d’a-hydroxyisobutyrate d’ammonium sur unc colonnc d’&zliangc cle cations. Cllacluc Blrhcnt cst ensuitc closf5 11~1 1ii0yc1~ clc la tccliniclue clu “~oml~iratcur simple”. Cc proc&U pcrmet unc applicn-’ tion pratique dc l’analysc pat- activation au tlosngc clc routine dcs tcrrcs rarcs clans dcs matrices complcscs.
Die Bcstimmung dcr Seltenen Erdcn in Minct-alien mit dcr Aktivierungs- analyst wird besclirieben. Die Scltcncn IQ-den werdcn als Gruppe vor der Bestralilung aus clcm Material abgetrcnnt. Nach der 13estrahlung werden sie mit eincm Kationen- austuuscher getrennt und mit dcr sogcnannten Single-Kompcrator-Teclmik be- stimmt. Die Metlloclc crlaubt einc praktisclle Anwendung dcr Aktivierungsnnalyse bci der ICoutinehcstimlllullg Seltcner Erdcn in komplcxen Matrices.





























