Embed Size (px)
Citation preview

Aualylica CkimLca Acta EIacvier Publishing Company, Amsterdam Printed in Tho Ncthcrlands
ACTIVATION ANALYSIS OF RARE EARTHS
PART II. DETERMINATION OF LUTETIUM IN GADOLINITE
D. L, MASSART* AND J’. HOSTE
Instifute for Nuclear Sciences. Gke?rt University, Gkenl (GTelgitrnr)
(Rcccivcct Deccmbor :8th, rg67)
Lutetium-x76 is a naturally occurring long-lived isotope with a half-life of 3 *1or0 years, Hence, it can be used for age determinations of rock formationsr. This paper describes the determination in rocks by neutron activation analysis. The methods were tested on gadolinite mineral.
As discussed in Part I*, the rare earths are separated as a group from the rest of the mineral before irradiation. 1sGd is added as chemical yield monitor. After irradiation, lutetium is separated from the other rare earths, by cation exchange with or-hydroxyisobutyrate as the eluting agent. Since lutetium is the heaviest of all rare-earth elements, it leaves the column before all the others. The only possible contaminating rare-earth element is ytterbium,
According to whether the analyst possesses the instrulnentation for y-spectro- metry or not, two methods are applicable:
(a) lutetium and ytterbium are counted integrally as they come from the column and corrections are made for the overlap of the two fractions;
(b) a more rapid separation with a larger ytterbium contamination is carried out. Lutetium and part of the ytterbium are collected and differentiated by y-spectro- metry.
NUCLEAR l3ATA
The nuclear data of lutetium and ytterbium isotopes are given in Table I. The y-energies cited comprise also X-rays and sum peaks. Lutetium-r76 yields on neutron activation also 17@Lu, but owing to its very long half-life, the activity of this isotope is entirely negligible. Ytterbium-r77 decays to r77Lu and will interfere with the determination of lutetium.
From these data it is apparent that differentiation by half-life of 177Lu from r?sYb is barely possible and furthermore would necessitate measurements during at least 3 weeks. However, since the biggest activity of ytterbium is due to 176Yb, ytterbium will interfere with the zo8-keV peak of 177Lu only to a small extent and it is possible to use this y-peak for the determination of lutetium.
* Rcseqh asqxiate of 1.IX.W. (Belgium).
Anal. Chinr. Ada, 42 (1968) 15-20
XG 11. t. MASSART, J. HOSTE
Mofkcr 0 n is0 fo pc ____-.___. ._ . -._-. _ I ..- ------- ..-r... I
‘onI% 0.135 1 I ,c>oo
‘74YIJ 3 1 .f-Q 60
“OYL) 12.73 5-c) ‘7”Lu 97.4 I 35 ‘7@Ltr 2.59 4,000 _.________ . . . . ._. .__. .-. --. _..._.- - __
7’4
. . .._ - ..,.._ -..._ -.._--._ 32 clays
‘1.2 days
I .Q hours 3 *7 llours 0.8 days
. .._. . . .._ .-- ._.
21, 50, 110, 131. 177, 199, 240.2GI 53, 1’4, 137, 1450 283, 396 Not measured Not nxwiurctl 5s. ~13*208,2G3* 321
.-. . . . .__ . . . _...._. ._.__ ______ _.__._
The 6 M hydrochloric acid solution clutcd from the cation-exchange column (see Part I”) was evaporated at a temperature not exceeding 70’. The residue was redissolved in x ml of 0.x M nitric acid. The yield ws determined by counting the 1aaGd in a well-type crystal.
The standard solution was prepared by dissolving 5 mg of lutetium (as LuzO3 sycc pure, johnson and Matthey) in strong nitric acid which had been purified by double distillation in a silica apparatus. The solution was evaporated to dryness at a temperature not exceeding 70~. The lutetium nitrate was redissolved in IO ml of slightly acidified twice-distilled water.
Aliquqts (100 ,ul) of both the sample and the standard solution were pipetted into small silica ampoulcs. These were sealed and irradiated in the BR-I reactor of the S.C.K. in Mol for 2 h (flux 4.10 11 n 9 cm-2 * sec-1). A maximum of 5 samples was activated in one irradiation.
Se$amtiolt of lutetium from ytterbiom avid other mre earths* This separation was carried out by means of cation exchange with or-hyclroxy-
idobutyrate as the eluting agent, A column of 14 cm x 0.4 cm” was used. The analytical doncentration of the complex-forming agent was 0.5 M, and the free ligand concen-
Yb
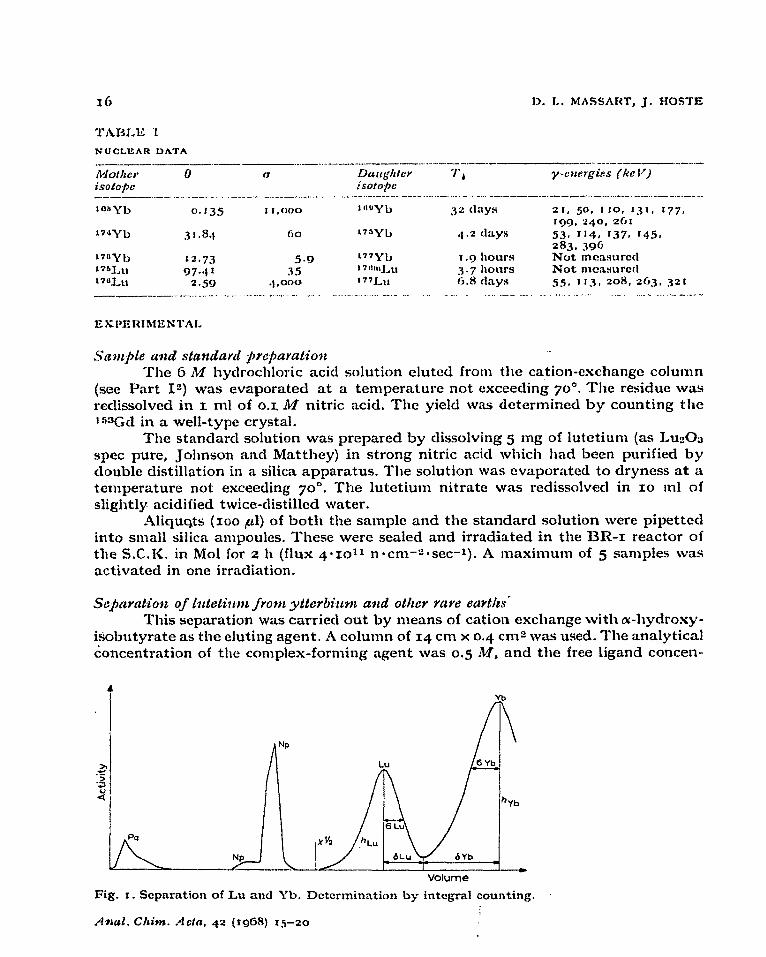
Fig. r. Separation of Lu ancl Yb. Determination by integral counting.
A9iul. CAisn. Actn, 42 (r&8) 15-20
ACTIVATION ANALYSIS OF RARE EARTHS. II 17
tration was o,og M. The eluate was collected in fractions of z ml. A typical separation is sllown in Fig. I. The “3@Np peak stems from ~W_I and the “33Pa peak from 232Th.
Both isotopes were identified by y-spectrometry and half-life determination. ‘fhe presence of thorium and uranium is nof surprising since both elements are very often associated with the rare earths in minerals. These elements accompany the rare earths in tht? group separation.
The separation of lutetium from ytterbium was nearly complete. The cross- contamination never exceeded 5%. For the y-spectrometric determination, the separations were carried out with a ligand concentration”of up to 0.075 &I. In this case, the distribution coefficients were too small to obtain a good separation of lutetium from ytterbium. Ho\vever, 1utetiuIn W;LS completely separated from other rare earths.
The sample was adsorbed quantitatively on the column and eluted. The eluate was collected, the fractions counted in a well-type scintillation counter and the activities plotted as a function of volume. The collected lutetium fraction was corrected for the activity lost under the ytterbium peak and the ytterbium activity present under the lutetium peak according to Fqns. (I) and (2) (see SAID~).
7n[r --A (bL”/cn.U) 1 YYb =
ALU (2)
.
where A (x) =area under the normal curve of error for afgument .V
liz =l2t\*b~ml.u =hW3!lh
hb, fi1.11, al.ur oYbr hr &b: see Fig. 1
;I.L~ = fractional Lu recovery
?‘Yb = fractional Y b contamination
N(E) d.E
208 kc’/ “‘Lu
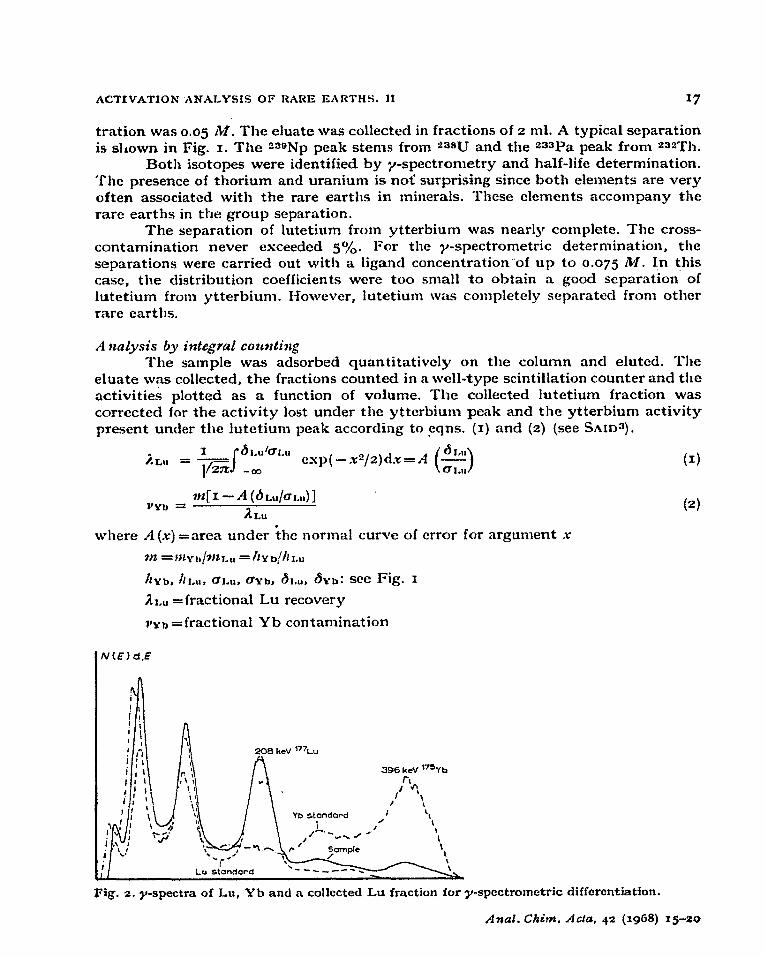
Fig. 2. y-spectra of Lu, Yb and a collected Lu fraction for y-spectrometric differentiation.
Anal. Ckitn, Acta, 42 (xgG8) x5-20

18 D. L. MASSART, J. HOSTE
Rnadysis by y-sfmtrometry This methad required an ytterbium standard as well as a lutetium standard.
After cation exchange, the lutetium activity was collected along with the first quarter of the ytterbium fractions. This ensured a roo”/f recovery of the lutetium activity. The sample and the standards were measured in So-ml ‘calibrated flasks with a 400-channel y-spectrometer.
From the activities in the zoS-keV and the 396-keV energy regions (Fig. z),
two equations were obtained, which were solved for the 177Lu activity under the zoS-keV peak.
PJZSULTS
The experimental results are sumrnariscd in Table II. The first group of 3 results was obtained by integral counting, the second by y-spectrometry and the third by the method used for analysis of all the rare earths described in Part III. Three possible sources of error were investigated (the blank, neutron shadowing and (n,y ; p-) errors) and corrections; have to be made for two of these. These corrections are included in the results of Table II.
TABLE II
CONCENTRATION OF LUTETXUM IN GADOLINITE (IN “fO OP TIlfJ hIINEllAL)
Method of annlysis
‘Integral counting
Results
o.aoga o.zoG3 O.ZIO~
Mea?l values
0.2076 Y = o.oo2b
y-spcctromctry 0.2033 0.1993
O.ipp H = o.ooj3
0.195”
Sin@ comprrrntor 0.200” 0.2003
0.192” 3 = 0.0083
0.208~
AVClTgC = o.202&o.oo3°/0
A group separation was simulated following the method described in Part 12 but without addition of the rare-earth mineral. Iron (15 mg) was added to carry down possible *rare earths during the hydroxide precipitation step. The iron was purified before the addition by anion exchange in hydrochloric acid. The rare-earth fraction was activated and a separation of lutetium from ytterbium was carried out. Since no activity was found where lutetium and ytterbium should have been eluted, no correction had to be made.
Nez&+on shadowing The presence of gadolinium in the separated rare-earth fractions causes neutron
shadowing and thus a negative error. This error was estimated by the addition of 5ooyg of cobalt to the rare-earth fraction. Cobalt was separated, after irradiation,
Anal. CAim. Acta, 42 (1968) x5--30
ACTIVATION ANALYSIS OF RARE EARTHS. II =9
from the rare earths by anion exchange in hydrochloric acid medium and its activity compared with the activity of a cobalt standard (500 peg Co in zoo ~1 solution) irradiated under the same condition. A mean value of 3.4% for 2.55 mg of gadolinite in a roo-~1 aliquot of solution was obtained. Accordingly, a correction of + x.35%/mg mineral in IOO ~1 of solution was applied.
(12, y ; p-) correction
The formation of 177Lu from 177Yb by the reaction i7~Yb(n,y)r77Yb(P-)177Lu causes a positive error. This error was calculated by means of a general program permitting calculation of all types of secondary order interferences, pro(Frammed in SPS-language and performed on an IBM-1620 computer. For equal quantities of ytterbium and lutetium and a 2-h irradiation, an error of 9.12% was found. Since the ytterbium concentration was 12 times higher (see Part III) than the lutetium concentration, the total error was I.SO/~. When the ytterbium concentration is not known, one can consider that as a general rule it is equal to s-20 times the lutetium concentration.
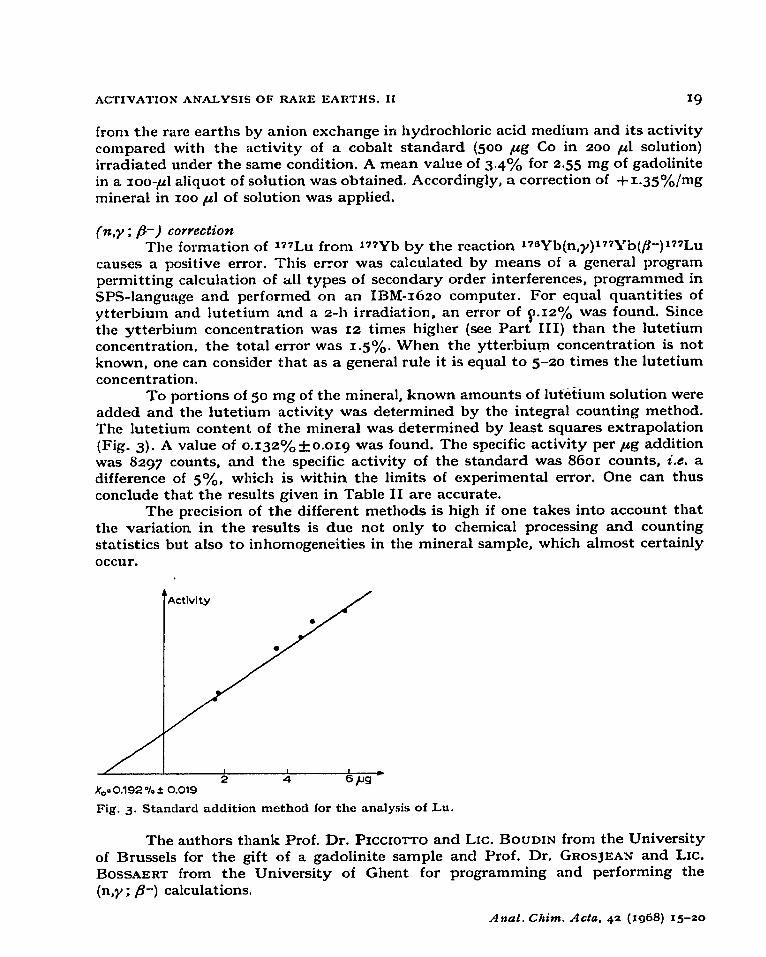
To portions of So mg of the mineral, known amounts of lutetium solution were added and the lutetium activity was determined by the integral counting method. The lutetium content of the mineral was determined by least squares extrapolation (Fig. 3). A value of 0.132%*o.org was found. The specific activity per ,ug addition was 8297 counts, ‘and the specific activity of the standard was 8601 counts, i.e. a difference of soA, which is within the limits of experimental error. One can thus conclude that the results given in Table II are accurate.
The precision of the different methods is high if one takes into account that the variation in the results is due not only to chemical processing and counting statistics but also to inhomogeneities in the mineral sample, which almost certainly occur.
I I I
2 4 =rJs .
x0= 0.192 % * 0.019
Pig. 3. Standard addition method for the analysis of Lu.
The authors thank Prof. Dr. PICCIOITO and LIC. BOUDIN from the University of Brussels for the gift of a gadolinite sample and Prof. Dr. GROSJEAN and LIC. BOSSAERT from the University of Ghent for programming and performing the (n,r ; fi-) calculations.
Anal. Chim. Ada, 42 (1968) 15-20

20 D. L. MASSART, J. EIOSTE
SUMMARY
Two precise and accurate methods for the determination of lutetium in tile presence of other rare earths and after separation from a mineral, gadolinite, are described. Both methods require separation of lutetium and ytterbium from the other rare earths. A complete separation of Lu and Yb is necessary when integral counting is used. The other method differentiates between the two elements by y-spectrometry.
On dCcrit deux mbthodes prCcises et r&productibles pour le dosage du lut&ium en pr&ence d’autres terres rares et apr&s sCparation d’un minerai, de la gadolinite par exemple. Les deux pro&d& ndcessitent une sdparation du lutetium et de l’ytterbium - d’avec d’autres terres rares. Une s¶tion totale de Lu et Yb est nCccssaire avcc le comptage intdgral. L’autre m&hode distingue les deux c”l6ments par spectromCtrie-y.
ZUSAMMENFASSUNG
Zwei reproduzierbarc und zuverl%liche Methoden zur Bestimmung von Luthe- tium in Gegenwart anderer Seltener Erden und nach Abtrennung clieser aus dem Gadolinit werden beschrieben. Beide Methoden erfordern die Trennung des Luthetiums und Ytterbiums von den anderen Seltenen Erden. Eine vollst%ndige Abtrennung dcs Lu und Yb ist notwendig, wenn eine integrale ZHhlweise angewendet wird. Die andere Methode unterscheidet mit Hilfe der y-Spektrometrie zwischen den beiden Elementen.
REFERENCES
I PICCIOTTO AND A. BOUDIN, personal communicntion. 2 D. L. MASSART AND J. HOSTE, And. Chkn. Acta, 42 (rgG8) 7. 3 A. S. SAID, J. Gas Chomatog., 2 (xgG4) Go.
A*tal. Chinr. Rctn, 42 (x968) x5-20





























![The positions of lanthanum (actinium) and lutetium .... B. Jensen/Reprints/018. La vs Lu.pdfThe Positions of Lanthanum (Actinium] and Lutetium (Lawrencium] in the Periodic Table William](https://img.pdfslide.us/doc/110x75/5ae6424f7f8b9a08778ccf1d/the-positions-of-lanthanum-actinium-and-lutetium-b-jensenreprints018.jpg)