Embed Size (px)
Citation preview

Understand the effects of catalysts in the epitaxial III-V nanowire growth
Mun Teng Soo
Master of Science (Materials Engineering)
A thesis submitted for the degree of Doctor of Philosophy at
The University of Queensland in 2017
School of Mechanical and Mining Engineering

Abstract
Over the past decades, one-dimensional (1D) semiconductor nanostructures have drawn
great interest since the proposed of vapour-liquid-solid (VLS) growth mechanism into the
synthesis of 1D nanostructures. III-V compound semiconductor nanostructures exhibit
exceptional electrical and optical properties. With a broad range of composition and band
structure selection, III-V semiconductor nanostructures are the critical components in a
wide range of potential nanoscale device applications. To fully exploit these 1D III-V
semiconductor nanostructures, particularly nanowires, extensive research has been made
on growth control of 1D nanoscale building blocks, and integration of these nanowire
elements into complex functional architectures in past decades. Nevertheless, there are
still challenges in III-V nanowires control, such as size, shape and crystal quality.
Metal-organic chemical vapour deposition (MOCVD) is one of the most precise and growth
techniques to grow III-V semiconductor nanowires. There are a series of growth
parameters that can be altered in a MOCVD growth chamber, such as temperature and
absolute flow rate of both group-III and group-V precursors (corresponding to V/III ratio).
By tuning these parameters carefully, nanowires with controlled morphologies and crystal
structures can be obtained. Due to the dimensionality of nanowires, advanced electron
microscopy characterisation is demanded to better understand the growth mechanism of
nanowires. Most of the research works have been done focusing on the effects of growth
parameters on the growth of III-V nanowires. However, there is insufficient attention has
been paid to explore the effects of catalysts, in which the catalyst play an important role in
the nucleation and growth of nanowires. Besides, most of the works dedicated to the Au-
catalysed III-V nanowires growth via VLS or vapour-solid-solid (VSS) mechanisms. Little
work has been done on the nanowires growth mediated by non-Au metal catalysts.
The objective of this thesis is to understand the effects of catalysts in the epitaxial III-V
nanowire growth. A systematic study of controlled growth of III-V nanowires using both Au
and non-Au metal catalysts has been conducted. The morphological, structural and
chemical characteristics of the III-V nanowires and their respective catalysts were
investigated by advanced electron microscopy. The findings of this thesis will provide
insights to elucidate the significant role of catalysts and the growth mechanism involved,
which is extremely crucial to design reliable and high-quality III-V nanowires-based
devices.

Declaration by author
This thesis is composed of my original work, and contains no material previously published
or written by another person except where due reference has been made in the text. I
have clearly stated the contribution by others to jointly-authored works that I have included
in my thesis.
I have clearly stated the contribution of others to my thesis as a whole, including statistical
assistance, survey design, data analysis, significant technical procedures, professional
editorial advice, and any other original research work used or reported in my thesis. The
content of my thesis is the result of work I have carried out since the commencement of
my research higher degree candidature and does not include a substantial part of work
that has been submitted to qualify for the award of any other degree or diploma in any
university or other tertiary institution. I have clearly stated which parts of my thesis, if any,
have been submitted to qualify for another award.
I acknowledge that an electronic copy of my thesis must be lodged with the University
Library and, subject to the policy and procedures of The University of Queensland, the
thesis be made available for research and study in accordance with the Copyright Act
1968 unless a period of embargo has been approved by the Dean of the Graduate School.
I acknowledge that copyright of all material contained in my thesis resides with the
copyright holder(s) of that material. Where appropriate I have obtained copyright
permission from the copyright holder to reproduce material in this thesis.

Publications during candidature
Published papers included in this thesis
1. M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Temperature-
dependent side-facets of GaAs nanopillars, Semiconductor Science and
Technology, 31 (9), 094004, 2016.
2. M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Growth of catalyst-
free epitaxial InAs nanowires on Si wafers using metallic masks, Nano Letters, 16
(7), 4189-4193, 2016.
3. M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Mirror-twin induced
bicrystalline InAs nanoleaves, Nano Research, 9 (3), 766-773, 2016.
Published co-authored papers
1. Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M.T. Soo, M. Hong, X. Yan,
G. Qian, J. Zou, A. Dun and X. Yao, A heterostructure coupling of exfoliated Ni–Fe
hydroxide nanosheet and defective graphene as a bifunctional electrocatalyst for
overall water splitting, Advanced Materials, 29 (17), 1700017, 2017.
2. L. Huang, Z. Liu, W. Guo, S. Chen, Q. Duan, C. Zhou and M.T. Soo, Microstructure
and tensile properties of oxide strengthened ferritic steel fabricated using as-milled
powders with dissolved oxygen, Fusion Engineering and Design, 95, 1-6, 2015.

Conference abstracts
1. M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Growth of Ni-
catalysed Epitaxial GaAs Nanowires, International Conference on Nanoscience and
Nanotechnology (ICONN 2016), Canberra, Australia, 2016. (Poster presentation)
2. M.T. Soo, K. Zheng, C. Jagadish and J. Zou, Effect of growth temperature and V/III
ratio on the morphology and crystal structure of nickel-catalysed GaAs nanowires,
2015 EAIT Postgraduate Conference, Brisbane, Australia, 2015. (Oral presentation)
3. M.T. Soo, Q. Gao, Y. Guo, H.H. Tan, C. Jagadish and J. Zou, Low V/III ratio growth
of 1D InAs semiconductor nanostructures by MOCVD method, The 8th International
Conference on Advanced Materials Processing (ICAMP-8), Gold Coast, Australia,
2014 (Oral presentation)
4. M.T. Soo, C. Jagadish and J. Zou, Growth of 1D InAs Semiconductor
Nanostructures on Different Substrates Using Au and Ni Catalysts by MOCVD
Method, 2014 EAIT Postgraduate Conference, Brisbane, Australia, 2014 (Oral
presentation)

Publications included in this thesis
M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Mirror-twin induced
bicrystalline InAs nanoleaves, Nano Research, 9 (3), 766-773, 2016 – incorporated as
Chapter 4.
Contributor Statement of contribution
Mun Teng Soo (Candidate) Designed experiments (70%)
Carried out nanowires growth (50%)
Carried out characterization (100%)
Data analysis (70%)
Prepared the paper (70%)
Kun Zheng Data analysis (10%)
Prepared the paper (10%)
Supervision (20%)
Qiang Gao Designed experiments (10%)
Carried out nanowires growth (40%)
Hark Hoe Tan Designed experiments (10%)
Carried out nanowires growth (10%)
Prepared the paper (5%)
Chennupati Jagadish Prepared the paper (5%)
Supervision (10%)
Jin Zou Designed experiments (10%)
Data analysis (20%)
Prepared the paper (10%)
Supervision (70%)

M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Growth of catalyst-free
epitaxial InAs nanowires on Si wafers using metallic masks, Nano Letters, 16 (7), 4189-
4193, 2016 – incorporated as Chapter 6.
Contributor Statement of contribution
Mun Teng Soo (Candidate) Designed experiments (70%)
Carried out nanowires growth (50%)
Carried out characterization (100%)
Data analysis (70%)
Prepared the paper (70%)
Kun Zheng Data analysis (10%)
Prepared the paper (10%)
Supervision (20%)
Qiang Gao Designed experiments (10%)
Carried out nanowires growth (40%)
Hark Hoe Tan Designed experiments (10%)
Carried out nanowires growth (10%)
Prepared the paper (5%)
Chennupati Jagadish Prepared the paper (5%)
Supervision (10%)
Jin Zou Designed experiments (10%)
Data analysis (20%)
Prepared the paper (10%)
Supervision (70%)

M.T. Soo, K. Zheng, Q. Gao, H.H. Tan, C. Jagadish and J. Zou, Temperature-dependent
side-facets of GaAs nanopillars, Semiconductor Science and Technology, 31 (9), 094004,
2016 – incorporated as Chapter 5.
Contributor Statement of contribution
Mun Teng Soo (Candidate) Designed experiments (70%)
Carried out nanowires growth (50%)
Carried out characterization (100%)
Data analysis (70%)
Prepared the paper (70%)
Kun Zheng Data analysis (10%)
Prepared the paper (10%)
Supervision (20%)
Qiang Gao Designed experiments (10%)
Carried out nanowires growth (40%)
Hark Hoe Tan Designed experiments (10%)
Carried out nanowires growth (10%)
Prepared the paper (5%)
Chennupati Jagadish Prepared the paper (5%)
Supervision (10%)
Jin Zou Designed experiments (10%)
Data analysis (20%)
Prepared the paper (10%)
Supervision (70%)

Contributions by others to the thesis
No contributions by others.
Statement of parts of the thesis submitted to qualify for the award of another degree
None.

Acknowledgements
First and foremost, I would like to thank God for granting me the knowledge, strength,
ability and opportunity to undertake this research study and to preserve and complete it
satisfactorily. This achievement would not have been possible without His blessings.
In my PhD journey, I am blessed enough to have found a teacher, a friend, an inspiration,
and a role model – my principle supervisor, Professor Jin Zou. He has been there
providing his exemplary guidance, creative suggestions, motivation and patience
throughout my research. Besides, I am heartily grateful to my co-supervisors – Professor
Chennupati Jagadish from the Australian National University (ANU) and Dr. Kun Zheng,
for their constant supports, encouragements, and sustained enthusiasm at all times.
I take pride in acknowledging Professor Hark Hoe Tan and Dr. Michael Qiang Gao from
the ANU for their excellent supports and discussions on my nanowires MOCVD growth
experiments. Their expertise in the growth of nanowires by MOCVD has indeed polished
my learning skills. I would like to thank Dr. Yanan Guo and Dr. Jenny Nian Jiang for
guiding my research in III-V nanowires growth.
I have great pleasure in acknowledging my colleagues and fellow research scholars at The
University of Queensland – Dr. Yang Huang, Dr. Justin Hongyi Xu, Dr. Wen Sun, Dr. Zhi
Zhang, Miss Chen Zhou, Mr. Zhiming Liao, Dr. Zhigang Chen, Dr. Lei Yang, Dr. Hong Min,
and Dr. Yichao Zou. Our weekly group meeting and other random discussion have been a
platform for me to learn from each other.
I am also truly indebted and thankful for Dr. Graeme Auchterlonie for helping me develop
my skill or knowledge in operating TEM. I offer my profound gratitude to all staffs at the
Centre for Microscopy and Microanalysis (CMM) in The University of Queensland who
always patiently teach me the knowledge in microscopy analysis.
I would like to acknowledge Australian Government Research Training Program
Scholarship funded by the Australian Government and Commonwealth Government for
financially support my PhD research. Without the scholarship, I would not have this
precious opportunity to do my research in Australia, and meeting many great brains.
Besides, the Australian National Fabrication Facility and Australian Microscopy &

Microanalysis Research Facility, both established under the Australian Government’s
National Collaborative Research Infrastructure Strategy, are gratefully acknowledged for
proving access to the facilities used during my PhD research.
I truthfully thank to The University of Queensland Graduate School for providing me
Graduate School International Travel Award (GSITA) for my research travel to the Institute
of Microstructure and Properties of Advanced Materials, Beijing University of Technology,
China. Besides, the funding support from Overseas Travel Fellowship (OTF) provided by
the Australian Nanotechnology Network (ANN) for my research travel to The
Ultramicroscopy Research Center, Kyushu University, Japan is greatly acknowledged.
These two overseas travel grants enabled me to extend my microscopy skills and to
develop networking skills which enriched my research experience and helped me in
navigating my future career.
I cherish all the friendship I had in Australia. My friends have been an encouragement. The
ladies Bible study group and International students Bible study group – FOCUS at The
University of Queensland always have been a great place that I found friends to support
and pray for each other. The community of St Lucia Bible Church is my family in Christ.
My acknowledgement without be complete without thanking my husband and family for
their unwavering love and support give to me. I thank my husband, Victor Khong, for
putting up with me in difficult moments where I nearly gave up and for encouraging me to
follow my dream of completing my PhD research.
Not to us, O LORD, but to Your name give glory,
for the sake of Your steadfast love and Your faithfulness!
(Psalm 115:1)
Mun Teng Soo (Abby)
25th March, 2017

Keywords
III-V nanowires, indium arsenide, gallium arsenide, metal-organic chemical vapour
deposition, catalyst, zinc-blende, wurtzite, scanning electron microscopy, transmission
electron microscopy
Australian and New Zealand Standard Research Classifications (ANZSRC)
ANZSRC code: 100712, Nanoscale Characterization, 60%
ANZSRC code: 100716, Nanofabrication, Growth and Self Assembly, 20%
ANZSRC code: 091203, Compound Semiconductor, 20%
Fields of Research (FoR) Classification
FoR code: 0912, Materials Engineering, 50%
FoR code: 1007, Nanotechnology, 50%

Table of contents
1. Introduction
1.1. Background introduction .……………………………………………………...
1.2. Objective and scopes .……………………………………………..……….....
1.3. Thesis outline .……………………………………………………….…….…...
1
1
2
2
2. Literature review
2.1. Introduction .………………………………………………………..…………...
2.2. The structural features of epitaxial III-V nanowires …………………...……
2.2.1. Polytypism in III-V nanowires …………………………………...……..
2.2.2. Defects in III-V nanowires ………………………………………..…….
2.2.3. Morphology of III-V nanowires ………………………………..……….
2.2.4. Surface diffusion effects ……………………………………….……….
2.2.5. Growth modes: axial and radial ……………………………………….
2.2.6. Growth direction of III-V nanowires …………………………..……….
2.2.7. Tuning growth parameters – An approach to control nanowire
growth …………………………………………………………………….
2.3. Catalysts – important role in epitaxial III-V nanowire growth ………...……
2.3.1. Nucleation ………………………………………………………………..
2.3.2. Nanowire growth ………………………………………………..………
2.3.3. Effects of catalyst on the nanowires quality ………………………….
2.3.4. Examples of non-Au metal catalysts in III-V nanowires grown …….
2.4. Growth mechanism …………………………………………………….………
2.4.1. Vapour-liquid-solid (VLS) ………………………………………...…….
2.4.2. Vapour-solid-solid (VSS) ………………………………………….…...
2.4.3. Preferential interface nucleation (PIN) ………………………………..
2.4.4. Vapour-solid (VS) …………………………………………………..…...
2.5. Growth techniques ………………………………………………………….….
2.6. Summary ………………………………………………………………..………
References ……………………………………………………………………….……...
4
4
7
7
9
11
13
15
17
18
23
24
26
28
32
35
36
39
40
42
43
49
50
3. Experimental techniques
3.1. Introduction ………………………………………………………………..……
3.2. III-V nanowires epitaxial growth ………………………………………………
3.2.1. Metal-organic chemical vapour deposition (MOCVD) ………………
3.2.1.1. MOCVD design ……………………………………………..…..
65
65
65
65
66

3.2.1.2. Precursors for III-V nanowires growth ………………………..
3.2.1.3. Growth procedures ……………………………………………..
3.2.2. Deposition of catalysts ……………………………….……………….
3.3. Electron microscopy …………………………………………………………...
3.3.1. Interaction between electrons and specimen ………..………………
3.3.2. Scanning electron microscope (SEM) …………………...…………
3.3.3. Transmission electron microscope (TEM) ……………………………
3.4. Electron microscopy samples preparation ………………………..…………
3.4.1. SEM samples …………………………………………………..………..
3.4.2. TEM samples ……………………………………………………..……..
References ……………………………………………………………...……………….
67
69
70
71
72
75
79
83
83
83
84
4. Understanding the growth of different InAs 1D nanostructures
catalysed by Au nanoparticles
4.1. Introduction ………………………………………………………………….….
4.2. Journal publication and manuscript ..….……………………………………..
4.2.1. Mirror-twin induced bicrystalline InAs nanoleaves …………………..
4.2.2. Controlled growth of defect-free zinc-blende -oriented InAs
nanobelts ………………………………………………………………..
87
87
87
88
104
5. Understanding the growth of GaAs 1D nanostructures catalysed by Au
nanoparticles
5.1. Introduction ……………………………………………………………………..
5.2. Journal publication ……………………………………………………………..
5.2.1. Temperature-dependent side-facets of GaAs nanopillars ………….
117
117
117
118
6. Exploring the growth of III-V nanowires using non-Au metals
6.1. Introduction …………………………………………………………………..…
6.2. Journal publication and manuscript ..….……………………………………..
6.2.1. Towards epitaxial growth of defect-free GaAs nanowires catalysed
by Ni ……………….……………………………………………………...
6.2.2. Growth of catalyst-free epitaxial InAs nanowires on Si wafers using
metallic masks …………………………………………………………...
131
131
132
144
7. Conclusions and recommendations
7.1. Conclusions ...………………………………………………………………..…
7.2. Recommendations ..….………………………………………………………..
163
163
165

List of Figures
Figure 2.1 Schematic illustration of nanowire growth (a) induced by metal catalyst, and (b)
by selective-area. Major factors that control the morphological and structural of
epitaxial III-V nanowires growth is described in the diagram.
Figure 2.2 Ball-and-stick models of the observed crystal structures of the polytypes for III-
V nanowires (a) ZB, (b) WZ, (c) 4H, and (d) 6H, including their respective
stacking sequences. Yellow circles in each bilayer represent group-III atoms,
and red circles stand for group-V atoms. Representations of the shortest
repeating stacking unit for each polytype are highlighted in red. (e) TEM and
(f,g) HRTEM images of hetero-structured GaAs/GaAsSb nanowire show ZB,
WZ and 4H segments. (h,i,j) Fast Fourier transform (FFT) of HRTEM images of
the GaAs 4H polytype, the GaAsSb ZB, and the GaAs WZ phases,
respectively. Thick circle in (h) indicates the ¼(0002) reflection characteristic
for a 4H, whereas the two thin grey circles show the and
reflections characteristics for a WZ phases. HRTEM images along the
zone axis of (k) InSb nanowire with segments of ZB, 4H and WZ, and (l) GaAs
nanowires grown at 690°C with segments of 6H.
Figure 2.3 HRTEM images viewed along zone axis show the examples of (a)
rotational twin plane in ZB; and (b) switching from WZ into ZB. The white arrow
shows the length axis or growth direction of the nanowire. The colour coding
indicates the layering of the different crystal structures
Figure 2.4 (a) Schematic illustration of hexagonal prism with diameter D and certain
length L showing dangling bond at the surface. (b) Schematic drawing of
nanowire with either six or sidewall planes for ZB structure; or six
or sidewalls for WZ counterparts. (c) Truncated triangular prism
with three longer {112}B facets and three shorter {112}A facets. (d) Transition of
a hexagonal cross-section into a truncated triangle due to different lateral
growth rate along different facets. (e) Effect of a single twin plane on
nanowire morphology. The triangular segment above the twin plane is rotated
60° about the growth axis, relative to the triangular segment below the twin. (f)
SEM image of GaAs nanowires. (g,h) top and 10°-tilted views of a nanowire
showing the truncated triangular cross-section and overlapping truncated
triangle segments. (i) A slightly tilted view of a nanowire focused at the lower

and top (inset) region. (j) TEM image of a GaAs nanowire with corresponding
HRTEM images (l and m) taken from the boundary of two adjacent segments
and the concave (as marked by arrows). (k) Corresponding SAED pattern of
(j). (n) Octahedral segments with and faces. The dotted lines
with arrows indicate twin planes. (o,p) SEM and TEM image of morphology of
GaAs twinning superlattice structure. (q) SEM image of GaAs nanowire after
annealing at 750°C for 2 min. Red and blue dotted lines in (q) show the
location where the corss-sections in (r) and (s) were taken, respectively.
Figure 2.5 Schematic illustration of adatom contributions to (a) axial gowth and (b) radial
growth. The adatoms contributions include those (1) directly impinging on the
metal catalyst; and those adsorbed on the (2) substrate, and (3) nanowire
sidewalls, which (4) diffuse along the concentration gradient toward the
catalyst/nanowire interface.
Figure 2.6 Schematic illustration of the possible <111> directions on substrate: one
and three <111>A directions.
Figure 2.7 FESEM images of InAs nanowires grown at temperatures range between 400–
525°C with V/III ratio maintained at 46. Scale bar is 1 µm. Samples are tilted at
40°.
Figure 2.8 FESEM images of GaAs nanowires grown at constant flow rate of TMGa
precursors but varying the flow rate of AsH3 precursors, resulting in a series of
V/III ratios. Axis shown is logarithmic. Scale bars are 1 µm. samples are tilted
at 40°.
Figure 2.9 Schematic 3D construction of the GaP nanowire morphologies. (a, b, c, e) are
nanowires with ZB structure, while (d) has the mixture of WZ and ZB structures
(the shaded parts represent the WZ segment). Rotational twin planes for (a, c–
e) do not affect the morphology.
Figure 2.10 SEM images of the growth of InP nanowires on InP (011) substrate: (a) top-
view and (c) view of nanowires after tilting the substrate at 30°. (b) is magnified
image of (a) shows the rectangular shape of nanowires formed by stepped
{110} side facets of the [001]-oriented nanowires. (d) is the magnified image of
a single InP nanowire. (c) Schematic of the two types of growth obtained. Type
I refers to the cases where a direct crystallographic relation between the
nanowire growth and the direct crystallographic relation between the nanowire
growth direction and the substrate in found and Type II when 3D twinning is

found. The drawing shows two nanowires grow from the same point but this
does not have to be the case for Type II.
Figure 2.11 Schematic diagrams of (a) Gibbs-Thompson relationship for the liquid-phase
catalyst formation; and (b) two supersaturated interfaces direct the nucleation
of nanowire growth. Schematic drawing of nanowire growth having (c)
cylindrical morphology; and (d) tapered morphology, and have variations of
contact angle of catalyst (c) larger than 90°, (e) 90°, and (f) smaller than 90°.
SEM images showing catalyst having contact angle (g) larger than 90° and (h)
90°. Scale bar is 100 nm for SEM images.
Figure 2.12 (a–f) ZB structured nanowires grown in different directions. (g) Modelling at the
catalyst/nanowire interface of the nanowires grown in different directions.
Figure 2.13 (a)Time-lapse TEM images and model build to describe kinking nanowire. (b)
Schematic illustration of the key stages of kink formation. The inset is SEM
image of a multiply kinked nanowire. (c) Steady VLS growth modes proposed
by Schwarz and Tersoff.
Figure 2.14 Schematic illustration of VLS mechanism to describe Au-mediated InAs
nanowire growth, assisting by Au-In binary phase diagram.
Figure 2.15 Schematic illustration of VSS mechanism, assisting by Al-Si binary phase
diagram.
Figure 2.16 A schematic diagram describing PIN mechanism.
Figure 2.17 (a) SEM side-view image of InAs nanowires grown on Si (111). The inset is
top-view image of the hexagonal cross-section of nanowires with six side
facets. The scale bar is 1 µm for (a) and 200 nm for the inset. (b) Schematic
illustration of the growth mechanism for self-induced InAs nanowires. (c,d)
Drawing and SEM image of InAs nanomembranes.
Figure 2.18 (a) Schematic diagram summaries the most important chemical reactions
involved in MOCVD growth (thin-film growth is described here; the growth of
nanowires has the same concepts): (1) gas phase reaction; (2) transport to
surface and diffusion through boundary layer; (3) adsorption of precursor; (4)
surface reactions and diffusion; (5) desorption of precursor; (6) nucleation and
island growth; (7) atomic step growth; (8) desorption of volatile surface reaction
products. (b) A schematic Arrhenius plot of the growth rate, R, showing three
distinct growth regimes of a typical III-V semiconductor growth in MOCVD
systems.
Figure 2.19 Schematic drawing of different growth techniques. (a) A typical CVD reactor.

(b) A typical LCG growth chamber. (c) A MOCVD reactor used in this thesis.
(d) A simple MBE growth chamber with the essential growth sources, shutters,
beam flux detector and the RHEED system for monitoring structure during
growth. (e) A typical CBE equipment.
Figure 3.1 (a) AIXTRON 200/4 MOCVD reactor and (b) schematic illustration of a
MOCVD system.
Figure 3.2 (a) Temscal BJD-2000 e-beam evaporator system and (b) schematic diagram
of a typical e-beam evaporator
Figure 3.3 A schematic diagram showing the interaction of electron beam with a thin
specimen
Figure 3.4 (a) A schematic diagram showing the incident electron beam dislodges an
electron from the K shell of a Si atom. An electron from the L shell fills the
vacancy and a Si KαX-ray is emitted. The energy of the X-ray is equal to the
difference between the ionisation energy of both K shell and L shell. (b)
Illustration of the interaction when a SEM electron beam hits on the surface of
a specimen.
Figure 3.5 (a) JEOL JSM-7800F SEM alongside (b) schematic diagram of a SEM column
Figure 3.6 A schematic diagram of the influence of surface topography: (a) flat surface
and (b) sharp edge on the escape volume of SEs. The dotted line represents
the escape depth and the shaded area represents the escape volume.
Figure 3.7 (a) An example of a modern TEM instrument alongside (b) a generalised cut-
away diagram of the internal structure (©
http://www.ammrf.org.au/myscope/tem/introduction/)
Figure 3.8 Ray diagrams for a TEM in BF imaging and selective area diffraction modes.

List of Tables
Table 2.1 Surface energy for different low-index sidewall planes for ZB and WZ
structured GaAs and InAs nanowires.
Table 2.2 Growth directions and catalyst/NW interface dependent on orientation of
substrate.
Table 2.3 Growth directions dependent on the catalyst size.
Table 2.4 Summary of non-Au metal catalysed III-V nanowires growth that has been
reported.
Table 2.5 Differences between CVD and PVD.
Table 2.6 Important processes during MOCVD growth.
Table 3.1 Detailed growth procedures of III-V nanowires by MOCVD.
Table 3.2 Operational parameters of SEMs for III-V nanowires analysis in this thesis.
Table 3.3 List of microanalysis techniques in TEM used in this thesis.
Table 3.4 Purpose and preparation process for different types of nanowire TEM sample.

List of Abbreviations
As arsenic
AsH3 arsine
BF bright-field
BSE back-scattered electrons
CBE chemical beam epitaxy
CBED convergent beam electron diffraction
DI deionised
EBL electron beam lithography
EDS energy dispersive X-ray spectroscopy
EELS electron energy loss spectroscopy
FCC face-centred-cubic
FE-SEM dield-emission scanning electron microscopy
FFT Fast Fourier Transform
HAADF high-angular annular dark-field
HCP hexagonal-close-packed
HR-TEM high-resolution transmission electron microscopy
MBE molecular beam epitaxy
MOCVD metal-organic chemical vapour deposition
PLL poly-L-lysine
SAED selected area electron diffraction
SE secondary electrons
SEM scanning electron microscopy
TEM transmission electron microscopy
TMGa Trimethylgallium, Ga(CH3)3
TMIn Trimethylindium, In(CH3)3
VLS vapour-liquid-solid
VSS vapour-solid-solid
WZ wurtzite
ZB zinc-blende
1D one-dimensional

1
CHAPTER 1
Introduction
1.1 Background introduction
Over the last few decades, major advances have occurred in both understanding and
controlling the growth of semiconductor nanostructures (i.e. structures with at least one
dimension in the range of 1–100 nm) due to their intriguing properties, as well as their
distinctive applications complementary to bulk materials. The ability to generate such small
structures is essential to the advance of various areas in latest science and technology.
One-dimensional (1D) nanostructures (such as wires, belts, tubes, and rods) offer a model
system for exploring the dependence of electronic transport, optical and mechanical
properties on size confinement and dimensionality. III-V semiconductor nanowires, in
particular, should play an important role as both interconnects and active components in
the production of nanoscale electronic and optoelectronic devices. Furthermore, the
controllable growth of III-V nanowires with well-defined size and shape, phase purity,
crystallinity and chemical composition is critical. Therefore, a fundamental, detailed
understanding and good control of nucleation and growth processes at the nanometre
scale are required. This is important in order to fully exploit their potential in fabricating
new types of functional devices. Besides, fascinating physics associated with III-V
nanowires has also begun to be unfolded. These opportunities and challenges have
inspired many groups to launch researches specifically focused on the growth,
characterisation, and utilization of nanowires.
Metal-organic chemical vapour deposition (MOCVD) is one of the accurate methods
to grow epitaxial III-V nanowires, catalysing by metallic catalysts, commonly Au
nanoparticles, via vapour-liquid-solid (VLS) or vapour-solid-solid (VSS) mechanism. In a
MOCVD reactor, there are few main growth parameters can be precisely tuned to grow
nanowires with desired morphologies and crystal structures, such as growth temperature,
the absolute flow rates of group III and group V precursors, and the V/III ratio. Since the
growth of III-V nanowires is mediated by the metallic catalysts, it is in fact a complicated
process. For this reason, it is crucial to understand the role of catalysts in the nanowire

2
growth. Until now, little attention on the role of these metallic catalysts has been paid
sufficiently, both Au- and non-Au metallic catalysts. It is believed that by altering the
chemical composition, morphology and distribution density of the catalyst is also the key
factor in manipulating the nanowire growth. A deeper and thorough investigation is much
needed so that some representative and exciting snapshots regarding the effects of the
catalysts can be provided. It is also hope that readers will enjoy the topics presented in this
thesis and perhaps find inspiration to push this field a step further toward commercial
importance.
1.2 Objective and scopes
The objective of my PhD research is to study and understand the effects of catalyst in the
epitaxial III-V nanowire growth. The correlations between the metal catalyst and the
morphological, structural and chemical characteristics of the III-V nanowires are examined
by advanced electron microscopy. Catalysts are altered by changing their choices of
material and ways of deposition. The growth environment in MOCVD chamber is
controlled so that the influence of metal catalyst on the nanowires quality can be studied.
After detailed studies of these III-V nanowires, growth model will be proposed so that it can
provide the insights on the growth mechanisms of nanowires owing different morphology
and crystal quality, which may lead to new and innovative discovery in III-V nanowires
research field.
1.3 Thesis outline
This thesis first included Chapter 2, which is an up-to-date literatures review on the
structural features of epitaxial grown III-V nanowires. Besides, the nanowire growth
techniques and mechanisms are discussed. The effects of metal catalysts in III-V
nanowires growth are also depicted. A summary is given in the end of this chapter. In
Chapter 3, experimental techniques used in this report are presented. The metal catalysts
deposition methods and MOCVD process for epitaxial grown III-V nanowires are
discussed. Characterisation methods, mainly two types of electron microscopy – scanning
electron microscopy (SEM) and transmission electron microscopy (TEM), are described in
terms of their history background and their respective operation principles. Moreover, few
different sample preparation methods are given.

3
Understanding on the growth of different InAs 1D nanostructures catalysed by Au
nanoparticles is depicted in Chapter 4. The insights into the growth mechanism of
symmetrical leaf-like 1D InAs nanostructures catalysed by Au nanoparticles are described.
These nanoleaves contain relatively low-energy of { } and { } mirror twins acting as
their midribs. The mirror twins lead to identical lateral growth of twinned structures in terms
of crystallography and polarity. Besides, defect-free zinc-blended [ ̅]-nanobelt is
investigated and described.
Chapter 5 investigates the growth behaviour of Au-catalysed GaAs 1D
nanostructures. The effect of the growth temperature on the side-facets of GaAs
nanopillars with hexagonal base was examined. We found that the side-facets of zinc-
blende GaAs nanopillars transform from { } to { } with increasing the growth
temperature as a result of competition between kinetically and thermodynamically
dominated process. Besides, diffusion-induced nanopillar foundations always present the
same { } edge side-facets regardless of the temperature.
The growth of III-V nanowires using non-Au metals have been explored and
included in Chapter 6. The study on the epitaxial growth of defect-free GaAs nanowires
induced by Ni catalyst is demonstrated. Besides, a proof-of-concept technique to grow
catalyst-free InAs nanowires using thick metallic mask is demonstrated. Successful growth
of hetero-epitaxial InAs nanowires on Si ( ) and ( ) wafers were investigated. Ultra-
long defect-free wurtzite InAs nanowires can be grown at 550°C using Ni metallic mask.
Finally, Chapter 7 provides conclusion and recommendations for future works in III-
V nanowires research field using both Au and non-Au metals.

4
CHAPTER 2
Literature Review
2.1 Introduction
In the burgeoning field of semiconductor nanoscience, a major ambition is to synthesize
nanoscale semiconductor building blocks of arbitrary dimensions, morphologies, and
materials of increasing complexity.1 Low-dimensional semiconductor nanostructures are
semiconductors in which at least one-dimension is smaller than 100 nm. Based on this
definition and the fact that bulk semiconductors are three-dimensional (3D) materials, the
dimensions of bulk semiconductors can be reduced in three dimensions. Therefore, one-
dimensional (1D) semiconductor nanostructures are obtained if two dimensions of the
semiconductors are restricted and the restricted one dimension has diameter below ~100
nm. Few examples of 1D nanostructure are nanowires, nanotubes, nanobelts and
nanopillars. The ability to control the size, structure, composition and morphology of these
semiconductor nanostructures, nanowires in particular, makes them ideal 1D building
blocks for a wide-range of potential applications in high-performance nanoelectronics,2-7
nanophotonics,8-15 biotechnology16, 17 and large-area, flexible electronics.18-24 Besides,
they provide experimental models to study size- and shape-dependent thermoelectric,25, 26
and mechanical properties.27 Uniquely, these intriguing semiconductor nanowires can be
readily assembled on various substrates, hence, making them compatible with
complementary metal-oxide-semiconductor (CMOS) processing while avoiding the lattice
mismatch and single-crystalline growth challenges often encountered for epitaxial, planar
thin films.28-30 Besides, due to the large design space made possible by the relaxation of
many constraints of typical thin film growth, nanotechnology based on semiconductor
nanowires has opened new directions in bandgap engineering of optoelectronic devices.
In the past decades, theoretical and experimental studies of III-V semiconductor
nanowires have developed rapidly as they show encouraging signs of being used as
building blocks of future advanced electronic and optoelectronic devices.31 In fact, devices
with outstanding properties such as room temperature UV nanolasers,32 light-emitting
diodes (LEDs),33 and optical detectors,34, 35 proof-of-concept transistors,36 highly sensitive

5
biological and chemical sensors,17, 37 logic gates, and computation devices2 have been
reported widely. Besides, III-V semiconductor nanowires are beneficial in exploring the
fundamental physics problems.38-41 These III-V semiconductor nanowires with 1D
confinement are possible to integrate on silicon (Si).42-44 As a result, advanced
functionalities hybrid electronics consisting of “top-down” Si CMOS technology and
“bottom-up” nanowires may be envisaged.45
In particular, gallium arsenide (GaAs) and indium arsenide (InAs) nanowires, among
those first developed III-V semiconductor nanowires, have been widely explored. Both
GaAs nanowires possess an intrinsically high electron mobility, direct bandgap, and high
photoluminescence efficiency.9, 15, 46-49 InAs as the channel material for high performance
transistors owing to their high electron mobility, small bandgap and the intrinsic surface
charge accumulation layer, which make them readily form near-ohmic, transparent metal
source/drain contacts.24, 50, 51 Additionally, InAs has a large bulk exciton Bohr radius (~34
nm) which is on the order of the radial size of nanowires, resulting in 1D quantum
confinement of the carriers with potentially interesting carrier transport characteristics.52
In order to explore such exciting and diverse potentials of 1D III-V nanowires,
tremendous efforts have been devoted to their fabrication with improved crystallographic
qualities, morphologies using various growth methods.53-55 It is well-known that III-V
nanowires can adopt a range of crystal structures. The two main crystal structures are the
cubic zinc-blende (ZB) and the hexagonal wurtzite (WZ) structure. Besides, III-V
nanowires exhibit side-facets of different orientations. These sidewall facets are usually
those low-index planes because they have fewer dangling bonds, which mean lower
surface energy. The word epitaxy derives from the Greek prefix, in which epi meaning
“upon” or “over” and taxis meaning “arrangement” or “order”. Therefore, epitaxial III-V
nanowires have a registry relative to the underlying substrate’s crystal structure.
Homoepitaxy refers to the growth nanowires are made up of same material as the
substrate whereas in heteroepitaxy the growth nanowires are of different material from the
substrate.
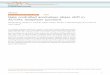
Generally, there are two ways to induce controllable epitaxial growth of III-V
nanowires: (1) growth induced by metal catalyst, in which gold (Au) is the common catalyst
used; and (2) selective-area growth, which involves no catalyst but nano-openings to
confine the growth of 1D nanowire (refer Fig. 2.1). Various growth mechanisms have been

6
proposed based on these two growth approaches, for example vapour-liquid-solid (VLS),
vapour-solid-solid (VSS) and vapour-solid (VS). To better manipulate the morphological
and crystal structural of III-V nanowires, there are two main controlled factors, which are
growth environment in growth chamber56-58 and choice of substrate.59 The details of these
two factors are listed in Fig. 2.1. In nanowires growth induced by metal catalyst, efforts
have been dedicated in tuning the types and sizes of metal catalysts to control nanowires
growth60-62 (refer Fig. 2.1a). For nanowire growth using selective-area epitaxy, the mask
layer is important, in which the nano-openings size, the distance between the nano-
openings and the thickness of mask layer can be altered (refer Fig. 2.1b). Based on the
aforementioned two ways – with or without metal catalyst, there are few common
techniques have been reported to grow epitaxial III-V nanowires, namely metal-organic
chemical vapour deposition (MOCVD),63, 64 molecular beam epitaxy (MBE)65, 66 chemical
vapour deposition (CVD), chemical beam epitaxy (CBE)67-69 and laser-assisted catalytic
growth (LCG),70 principally based on physical or chemical vapour deposition of materials.
Figure 2.1. Schematic illustration of nanowire growth (a) induced by metal catalyst, and
(b) by selective-area. Major factors that control the morphological and structural of
epitaxial III-V nanowires growth is described in the diagram.

7
2.2 The structural features of epitaxial III-V nanowires
Epitaxial III-V nanowires have different chemical potentials from the substrate, which
arises in two fundamental means: (1) the III-V nanowires and the substrate may vary in the
nature and strength of their chemical bonds; (2) the III-V nanowires and substrate may
vary in their lattice structures.71 Epitaxy may be performed close to thermodynamic
equilibrium and the growth is then well described in terms of thermodynamic properties of
the system. Besides, epitaxy can be described as a non-equilibrium process in terms of
kinetics, in which the driving force is proportional to the supersaturation, σ. This results in a
transfer of material towards, and incorporation of material into the crystalline phase. Eqn.
2.1 shows a simplified equation for the flux, J, of material:
(2.1)
where k is the mass transport coefficient. In addition to transfer of material towards the
surface of the crystal, kinetically activated reactions can limit the growth rate. Besides, an
extremely high growth rate can result in a poor crystalline quality.
2.2.1 Polytypism in III-V nanowires
Polytypism is a range of crystal structures in a material resulted from differences in the
stacking periodicity of the identical planes, all with the same atomic composition. All III-V
semiconductors in bulk, except nitrides, have cubic ZB crystal structure, or 3C using the
Ramsdell notation.72 However, when these materials are grown as nanowires, they often
display features of polytypism. This is attributed to the atomic layers in nanowires have the
same in-plane structure but different stacking sequence along the common ⟨ ⟩
⟨ ⟩ growth directions. Two common polytypes formed in III-V nanowires are ZB and
hexagonal WZ structures (2H polytype). The { } planes in the cubic ZB structure and
the { } planes in the hexagonal WZ structure are the closest packed plane, with every
atom having six in-plane neighbours. These directions are the preferential growth
directions of nanowires. The WZ structure has been reported, for instance, in MOCVD-
grown InAs nanowires73 and in MBE-grown GaAs nanowires.74 The ZB structure is
common in MOCVD-grown GaAs and GaP nanowires.75, 76 In several III-V material
systems, segments of polytypes with longer repeating periods, namely hexagonal 4H and

8
6H, have also been reported.77-81 Usually, the crystal structure is most easily recognised
as a random mixture of cubic and hexagonal structures.
Observing from ⟨ ̅⟩ zone axis, ZB structures consist of an ABCABC… stacking
sequence along ⟨ ⟩ direction (Fig. 2.2a). Such stacking sequence is the reason of the
polarity of ZB structure in certain directions, such as in [ ̅ ̅ ̅] direction, in which each ( ̅ ̅ ̅)
plane is terminated with ‘B’ type group V atoms. These directions are termed as ⟨ ⟩
directions. On the other hand, for the most common hexagonal polytype, WZ structure is
generated by stacking of close-packed { } planes, consist of an ABAB… sequence
(Fig. 2.2b). The stacking sequence is ABACABAC… for 4H polytype ABCBACABCBAC
for 6H polytype (Fig. 2.2c,d). Each of the letters A, B and C corresponds to a bilayer of
atoms, consisting of one layer with group-III and one with group-V atoms.
Since these polytypes are only different in their stacking, therefore switching
between these polytypes can happen within a nanowire if there are stacking faults or twin
planes in one or the other structures82 and the stacking fault energy is not extremely
high.83 Hence a mixed structure in III-V nanowires is common (Fig. 2.2e–f). 4H segments
were observed “sandwiched” between ZB and WZ. These effects have an impact on the
nanoscale optoelectronic devices10 and electron scattering at stacking faults or twin planes
can be detrimental to nanoelectronic devices.84, 85 Besides, the possibility to controllable
grow nanowire with same material in different polytypes enables bandgap engineering
using a single material. For all these reasons, careful tuning of the structure of III-V
nanowires is exceptionally important in providing an additional degree of freedom for
device fabrication and functionalization of nanowires.58 The crystal structures of nanowires
can be adjusted by controlling the types and sizes of catalyst, growth temperatures,
source-material flux or impurities. The exact tuning of ZB and WZ crystal structures in
nanowires has been reported in references.78, 83, 86, 87 Various models have been proposed
for crystal structure selection, which emphasize the catalyst geometry, role of
supersaturation and catalyst/nanowire interfacial energies74, 88, 89. Recently, Jacobsson et
al.90 reported the direct observation of the dynamic processes that take place during GaAs
nanowire growth for each crystal phase and during the switch between phases using in
situ TEM.

9
Figure 2.2. Ball-and-stick models of the observed crystal structures of the polytypes for III-
V nanowires (a) ZB, (b) WZ, (c) 4H, and (d) 6H, including their respective stacking
sequences. Yellow circles in each bilayer represent group-III atoms, and red circles stand
for group-V atoms. Representations of the shortest repeating stacking unit for each
polytype are highlighted in red.81 (e) TEM and (f,g) HRTEM images of hetero-structured
GaAs/GaAsSb nanowire show ZB, WZ and 4H segments.78 (h,i,j) Fast Fourier transform
(FFT) of HRTEM images of the GaAs 4H polytype, the GaAsSb ZB, and the GaAs WZ
phases, respectively. Thick circle in (h) indicates the ⁄ ( ) reflection characteristic for
a 4H, whereas the two thin grey circles show the ( ̅ ) and ( ̅) reflections
characteristics for a WZ phases. HRTEM images along the ⟨ ̅⟩ zone axis of (k) InSb
nanowire with segments of ZB, 4H and WZ,80 and (l) GaAs nanowires grown at 690°C with
segments of 6H.81
2.2.2 Defects in III-V nanowires
Defects are the imperfections in crystal structures and can appear in different varieties,
which are (i) point defects: vacancies, interstitials; (ii) line defects: screw and edge
dislocations; and (iii) planar defects: grain boundaries, anti-phase boundaries, stacking
faults and twins. Twin planes and stacking faults are two widespread defects happen in
nanowires.91 These ‘fault’ layer or twin plane interrupts the perfect crystal phase and often
is affected by the growth environment. As a result, these defects can affect the material,
optical and electrical properties of the nanowires.58. Twinning has been revealed as a
significant issue, as it can lead to crystal structure change and kinking.10, 92, 93 The
consequences for the electronic transport of the existence of twinning and a mixture in
crystal structure have been demonstrated.66 The existence of crystallographically
equivalent twin boundaries with opposite polar bonding across the interface would lead to
even stronger perturbations of electronic structure of the material (in the case where the

10
polarity is conserved, the twin is known as “orthotwin”. When it is not conserved, one
speaks of “paratwin”. See 94 for all the details). However, the issue of polarity conservation
across the twin has only been raised occasionally, mainly due to the difficulties in its
determination.94
Twin plane is defined as a mirror plane between two segments and it happens when
a single bilayer is imperfectly stacked in a ZB structure, which reverses the stacking
sequence. For example, for an ABCABC ZB layering, the sequence becomes ABCACBA,
in which A is the twin plane. The structure on one side of C is rotated 60° along the [ ̅ ̅ ̅]
growth axis relative to the structure on the opposite side. A single twin is insufficient to
produce a stacking fault, and a ZB nanowire containing only single twin plane do not have
WZ segment. In requires at least two sequential twin planes of ABCA|C|A (with the two
fault lines stated). Therefore, the smallest possible WZ segment in ZB requires four
bilayers. On the other hand, in WZ structures with an ABAB sequence, stacking faults
occur when a single bilayer is out-of-place, giving the sequence of ABAB|CBCB, with the
fault line between B and C. This spontaneously generates a single unit of ZB structure
(ABC). Therefore, a WZ structure is corresponding to a ZB structure with a twin plane in
each bilayer. Twin plane and stacking fault in both ZB and WZ occur mainly perpendicular
to the [ ̅ ̅ ̅] [ ̅] direction. Nanowire crystal structure is routinely determined by tilting
the nanowire to the ⟨ ̅ ⟩ zone axis observing using HRTEM. Figure 2.3 are two HRTEM
images of III-V nanowires with twin planes and stacking faults.
Figure 2.3. HRTEM images viewed along ⟨ ̅ ⟩ zone axis show the examples of (a)
rotational twin plane in ZB; and (b) switching from WZ into ZB. The white arrow shows the
length axis or growth direction of the nanowire. The colour coding indicates the layering of
the different crystal structures.91

11
2.2.3 Morphology of III-V nanowires
The surface structure and morphology, down to the atomic scale, of free-standing of III-V
nanowires is central in determining electrical, optical and chemical properties of the wires.
The formation of lateral facets on nanowire surfaces is an attention-grabbing phenomenon
and is closely related with their growth conditions. This is frequently observed in nanowire
growths and is comprehensively investigated in conjunction with their respective crystal
structures.73, 76 Sawtooth morphology is proved at the facets of Si nanowires.95-97
Systematic surface studies of nanowire sidewall growth are especially relevant as the
limited size of their facets can result in behaviour not found for “infinite” planar surfaces.
For most of the envisaged applications based on nanowires, surfaces are the locations
where most of the device activity takes place.98 Through a model that highlights the
contributions to the nanowire formation energy of each structural element (bulk-like core,
sidewalls, and ridges), it has been found that the nanowire stability can be explained in
terms of the surface energy of the sidewalls.99
III-V nanowires have side-facets of different crystallographic planes, which reflects
the symmetry of crystal structure of nanowires. In a crystal system, dangling bond is a
chemical bond associated with an atom in the surface layer of a solid that does not join the
atom with a second atom but extends in the direction of the solid’s exterior (Fig. 2.4a). The
surface energy of a crystal plane comes from the unsatisfied or dangling bonds that are
left on the surface. The bonds that are left un-connected to another atom increase the
energy of the crystal system. Therefore, different crystal planes, with different number of
atoms per unit area, have different numbers of dangling bonds per unit area. In general,
sidewall facets are those low-index planes, because they have fewer dangling bonds,
which mean lower surface energy. Among the nanowires having the same structure, and
same sidewall plane, the nanowires with smallest formation energy are those whose
surface atoms have the smallest number of dangling bonds at the edges.99
Besides, the surface energy depends on the crystal structure. In almost all bulk III-V
materials, bulk cohesive energy is larger for WZ than for ZB structure. For instance, the
bulk energy between WZ and ZB of GaAs is ~24 meV per III-V pair, which is substantial
gap that can be overpassed by applying a huge pressure by applying a huge pressure of
the order of several tens of GPa. However, the WZ structure has been reported for most
⟨ ⟩-oriented III-V nanowires. WZ is energetically preferable for nanowires with small

12
diameters of a few nanometres. The only physical reason for the WZ structure
transformation in nanowires could be a lower surface energy of the WZ sidewalls. These
surface energies cannot be measured directly, while different theoretical calculations give
discrepant results.99, 100 Table 2.1 is the surface energy for different low-index sidewall
planes for ZB and WZ structure in GaAs and InAs nanowires.
Table 2.1. Surface energy for different low-index sidewall planes for ZB and WZ structured
GaAs and InAs nanowires.101, 102
Material Structure Sidewall plane Surface energy, γ (J/m2)
GaAs ZB ( ) 2.1–2.2
( ) 1.5
( ) 1.3 ( ) 1.8
WZ ( ̅ ) 1.5
( ̅ ) 1.3
InAs ZB ( ) 1.4
( ) 1.0–1.1 ( ) 0.8–0.9 ( ) 1.2
WZ ( ̅ ) 1.1
( ̅ ) 0.9
Regular hexahedral nanowire growing in ⟨ ⟩ ⟨ ⟩ direction is restricted by
either six { ̅ } or { ̅} sidewall planes for ZB structure; or six { ̅ } or { ̅ } sidewalls
for WZ counterparts (Fig. 2.4b). The { ̅ } , { ̅ } and { ̅ } surfaces are non-polar
whereas { ̅} facets are polar, which causes the ZB structured nanowires demonstrate a
wide range of morphologies. The six { ̅} facets can be sub-divided into three { } and
three { } facets. Due to the differences in surface stoichiometry, group-III atoms attach
favourably to { } surfaces.76 Under high group-V overpressure growth environment, the
{ } facets, which has lower surface energy are more stable and resulted in a slower
growth rate compared with the { } facets.103 Consequently, the morphology of
nanowire adopts a truncated triangular shape with elongated { } facets and shorter
{ } side-facets, as illustrated in Fig. 2.4c,d, is formed.76, 103 If under low group-V
overpressure growth environment, it can be deduced that elongated { } facets can be
obtained.103
The presence of twin planes changes the overall morphology in the presence of
{ ̅} faceted triangular nanowires. The crystal structure is rotated 60° about [ ̅ ̅ ̅] growth
axis when a twin plan exists.11 Therefore, the section below and above this twin plane will

13
exhibit orientation in an opposite way (Fig. 2.4e). As a result the nanowire structure
resembles a stack of truncated triangles as observed in GaAs nanowires grown at 450°C
using SEM and TEM (Fig. 2.4f–m).76
Interestingly, ZB structured nanowires can be composed of truncated octahedral
segments, each with three { } and three { } side facets and bounded by a twin
plane above and a twin plane below.75 Each octahedral segment has a crystal structure
rotated 60° about the [ ̅ ̅ ̅] growth axis compared to the adjacent segments as illustrated
in Fig. 2.4n. Figure 2o,p shows the SEM and TEM images of this type of morphology,
which results in twinning superlattice formation in ZB structured GaAs nanowires with
larger diameter at higher growth temperature, i.e. 575°C.104
Surface energy of { ̅ } planes is lower than that of { ̅} planes. However, most of
the ZB structured III-V nanowires are terminated by { ̅} facets. From thermodynamically
point-of-view, this is unlikely. Therefore, the formation of { ̅} facets may be kinetically
driven. Transformation of { ̅} facets into { ̅ } facets were observed when high
annealing temperature of 750°C for 2 minutes was applied for a free-standing GaAs
nanowire (Fig. 2q–s).105
2.2.4 Surface diffusion effects
The metal-catalyst-assisted elongating growth of the nanowire is termed as axial growth.
There are two major contributions to axial growth, as illustrated in Fig. 2.5a. Firstly, the
growth is resulting in the direct impingement of reaction species on the catalyst. The other
contribution is due to the absorbance of species on the substrate or nanowire sidewalls.
These adatoms (adsorbed atoms) diffuse along the concentration gradient towards the
metal catalyst, which they are incorporated into axial growth. Surface diffusion is more
significant for group III adatoms, since they have large diffusion lengths (λ); whereas group
V adatoms have much smaller diffusion lengths. In the work of Johansson et al.,106 a
theoretical model describing the dependence of the axial growth rate on surface diffusion
is reported. In regions of high nanowire density, adjacent nanowires spaced within a
diffusion length compete for diffusion adatoms. Thus, nanowires in high density regions
have slower axial growth rates than those in low density regions.

14
Figure 2.4. (a) Schematic illustration of hexagonal prism with diameter D and certain
length L showing dangling bond at the surface.107 (b) Schematic drawing of nanowire with
either six { ̅ } or { ̅} sidewall planes for ZB structure; or six { ̅ } or { ̅ } sidewalls
for WZ counterparts. (c) Truncated triangular prism with three longer { } facets and
three shorter { } facets. (d) Transition of a hexagonal cross-section into a truncated
triangle due to different lateral growth rate along different { ̅} facets. (e) Effect of a
single twin plane on nanowire morphology. The triangular segment above the twin plane is
rotated 60° about the growth axis, relative to the triangular segment below the twin. (f)
SEM image of GaAs nanowires. (g,h) top and 10°-tilted views of a nanowire showing the
truncated triangular cross-section and overlapping truncated triangle segments.76 (i) A
slightly tilted view of a nanowire focused at the lower and top (inset) region. (j) TEM image
of a GaAs nanowire with corresponding HRTEM images (l and m) taken from the boundary
of two adjacent segments and the concave (as marked by arrows). (k) Corresponding
SAED pattern of (j). (n) Octahedral segments with { ̅ ̅} and { ̅ } faces. The dotted
lines with arrows indicate twin planes. (o,p) SEM and TEM image of morphology of GaAs

15
twinning superlattice structure.104 (q) SEM image of GaAs nanowire after annealing at
750°C for 2 min. Red and blue dotted lines in (q) show the location where the corss-
sections in (r) and (s) were taken, respectively.105
2.2.5 Growth modes: axial and radial
There are two major growth modes taking place during metal-catalysed nanowire growth
by MOCVD, which are axial growth and radial growth. The axial growth rate is the
incremental increase in nanowire length, l, per growth time interval, t:
⁄ ⁄ (2.2)
where L is the total length from the base to the catalyst/nanowire interface, and tg is the
total growth time.
Radial growth, sometimes is called as lateral or conformal growth, is the deposition
of material on the nanowire sidewalls. It follows a simple VS growth mechanism, and does
not directly involve in the metal catalyst. A high supersaturation (or concentration) of
growth species in the vapour phase may promote the lateral growth on the side surface of
nanowires with the VS mechanism. As illustrated in Fig. 2.5b, radial growth occurs when
species adsorbed on the substrates and nanowire sidewalls, diffuse and incorporate on
the nanowire sidewalls. Furthermore, a high supersaturation may initiate homogenous
nucleation in the gas phase or secondary nucleation at the surface of nanowires.
Therefore, a conical structure may be developed instead of uniformly sized nanowires. In
this manner, radial growth competes with axial growth.
Tapered nanowire morphologies, whereby nanowires exhibit wider bases and taper
to narrower catalyst-capped tips, are a consequence of radial growth, as explained here.
Nanowire bases are grown first, and hence are exposed to reactants for longer time than
the more recently grown catalyst-capped tip. Besides, owing to their proximity to the
substrate, the nanowire bases receive a greater fraction of precursor materials collected
on and diffusing from the substrate. Thus, lower sections of the nanowire experience more
radial growth, which leads to tapered nanowire morphologies with wider bases and
narrower tips as illustrate in Fig. 2.5b. The radial growth rate is calculated as the increase
in nanowire radius, r, per time interval, t:

16
⁄ ( )
⁄ (2.3)
where rb is the radius at the nanowire base and rt is the radius at the nanowire tip (the
interface of catalyst/nanowire).
In this report, the “tapering parameter” is used to quantify the degree of tapering.
The tapering diameter (nm/µm) is defined as the increase in nanowire radius (nm) per unit
nanowire length (µm) from the catalyst/nanowire interface, l. this is equivalent to the ratio
of radial to axial growth rates.
⁄
⁄ (2.4)
The tapering diameter is thus a figure of merit, with a low tapering parameter signifying
less undesirable tapering.
A further growth process is the deposition of planar or two-dimensional (2D) layers
on the substrate surface. This occurs when adatoms incorporate onto the substrate, rather
than onto the nanowire sidewalls or nanowire tips.
Figure 2.5. Schematic illustration of adatom contributions to (a) axial gowth and (b) radial
growth. The adatoms contributions include those (1) directly impinging on the metal
catalyst; and those adsorbed on the (2) substrate, and (3) nanowire sidewalls, which (4)
diffuse along the concentration gradient toward the catalyst/nanowire interface.

17
2.2.6 Growth direction of III-V nanowires
Control of both the growth orientation and alignment are crucial issues in nanowires
growth.108 Growth orientation can have a strong effect on anisotropic properties of the
semiconductor nanowires. On the other hand, crystallographic alignment occurred
presumably due to a close symmetry and lattice match between the substrates and
observed nanowire growth directions.
By and large, metal-catalyst-assisted nanowire growth takes place in the group-V-
terminated [ ̅ ̅ ̅] crystallographic direction. The catalyst/nanowire interface forms at a
{ } plane of the growing nanowire; this plane is perpendicular to the [ ̅ ̅ ̅] growth
direction. This growth direction is favoured because atoms precipitating on the { }
surface produce the largest decrease in Gibbs free energy. The { } planes have the
smallest separation and the largest density of surface atoms. The { } growth plane has
the lowest interfacial energy, and consequently the [ ̅ ̅ ̅] growth direction minimises the
interfacial energy of the catalyst/nanowire interface. This crystallographic related growth of
nanowires with the substrate is called as epitaxial growth. In III-V materials, there are four
equivalent ⟨ ⟩ directions in total. On a ( ̅ ̅ ̅) surface, there is only one ⟨ ⟩ direction
possible, which is directed outwards perpendicular to the substrate surface (Fig. 2.6).
There are three possible ⟨ ⟩ directions on a ( ̅ ̅ ̅) surface. This means that when
nanowires are grown on { } substrates, the nanowires grow vertical to the substrate
surface. Moreover, the preferential growth directions are totally different when non-( ̅ ̅ ̅)
substrates are used.
Figure 2.6. Schematic illustration of the possible ⟨ ⟩ directions on substrate: one ⟨ ̅ ̅ ̅⟩
and three ⟨ ⟩ directions.

18
Growth directions other than ⟨ ⟩ of epitaxy grown nanowires, such as ⟨ ⟩,109-114
⟨ ⟩111, 113-115 and ⟨ ⟩113, 116, 117 can be induced depending on few factors: diameter of
nanowires,110, 111, 114 side-facets,113 catalyst/nanowire interface, catalyst type, catalyst size
and others.
For large diameters, the direction with the lowest interfacial energy is dominant,
whereas for small diameters the surface energy of the nanowire determines the
preferential growth.111 The interplay of the liquid-solid interface energy with the nanowire
surface energy expressed in terms of an edge tension is responsible for the change of the
growth direction – from ⟨ ⟩ to ⟨ ⟩.
Growth direction of ⟨ ⟩ is possible when the nanwoires are thinner, or the
interface is smaller.115 In these conditions, it seems that the solid-liquid interface becomes
unstable. Thus, the interface may be lying on not only { } plane, but also the other
planes whenever the interface energies are sufficiently low such as the { } plane.
Furthermore, nanowires grown along ⟨ ⟩ can be bounded by surfaces of low energy,
i.e., { }, { } and { }.118
In the work of Wu et al.,114 the smallest-diameter nanowires growing primarily along
⟨ ⟩ and the larger nanowires growing along ⟨ ⟩. They suggested that ⟨ ⟩ direction
is a “translational” direction between the ⟨ ⟩ and ⟨ ⟩ growth directions because the
( ) plane is a stepped plane between the ( ) and ( ) planes.
2.2.7 Tuning growth parameters – An approach to control nanowire growth
The growth of nanowires (focus in MOCVD system) is a complicated process involving
many parameters, for instances, annealing and growth temperatures, V/III ratios, total
pressure, precursor partial pressures, nature of substrates; and types and concentrations
of catalyst used. This is essential to understand the effects of these inter-related
parameters on the crystal structure, morphology and defect density of the III-V nanowires.
Basically, nanowire diameters and lengths are determined by the size of the metal
catalysts and the growth duration, respectively; however, their shape is strongly
determined by growth conditions such as group V/III flux ratio and substrate temperature.
Herein, some of the parameters will be described.

19
Annealing temperature. An anneal step prior to growth in necessary for the growth
of III-V nanowires in MOCVD system to desorb surface contaminants. The substrate is
heated at a relatively high temperature compared to the growth temperature. High
pressure for group V precursor is used to avoid decomposition of the III-V substrates at
this high annealing temperature. This annealing process results in alloying of the catalyst
nanoparticles with the substrate and a good interaction is secured. When Au catalyst is
used, the common annealing temperature applied is 600°C. However, if other metal
catalysts are used, a higher annealing temperature is required to reach the metastable
alloying phase of catalyst and the growing materials. In addition, this thermal anneal can
affect chemistry of the substrate surface. Specifically, a surface oxide can be evaporated,
which affects the surface diffusion kinetics of the precursor molecules.
Growth temperature. This parameter is one of the key factors in controlling the
quality of nanowires. In the work of Joyce et al.,119 a series of Au-catalysed InAs
nanowires were grown at different temperatures (Fig. 2.7). The optimum growth
temperatures are between 400°C and 500°C, in which well-aligned free-standing
nanowires are grown. In the growth window between 400°C and 500°C, the length of
nanowires is longest at the lowest (400°C) and highest (500°C) temperatures, and shorter
at the intermediate temperatures. At 425°C, nanowires are shorter with tapering and kinks.
By increasing 25°C to 425°C, nanowires become shorter with heavy tapering and short
length are induced. Further increasing the temperatures (475°C and 500°C), the tapering
is reduced and the length of nanowires increases. This is due to the increase in In adatom
diffusion length as temperature raises. At the highest temperature of 525°C, the nanowire
growth rate drops sharply. Most of the nucleation at the Au catalysts is not success.
Instead, formation of island-like structures or planar growth on the substrate is observed.
On the other hand, at a much lower temperature (375°C), irregular and kinked
nanostructures are formed. This observation can be explained by taking consideration of
the Au-In alloy eutectic temperature (454.3°C). At this temperature, the catalyst alloy is
expected to be completely solid so that no growth of nanowires can be catalysed by Au.
Irregularly, ultrathin nanowires (~5 nm) are found. It is believed that In is the catalyst,
which is derived from TMIn precursors and segregates into pure In droplets on the surface
of substrate. At 375°C, pure In is in liquid form and these In droplets can induce nanowires
growth via VLS mechanism.

20
Figure 2.7. FESEM images of InAs nanowires grown at temperatures range between 400–
525°C with V/III ratio maintained at 46. Scale bar is 1 µm. Samples are tilted at 40°.119
V/III ratio of precursors. By keeping the flow rate of group III precursors
unchanged, V/III ratio can be altered by monitoring the flow rate of group V precursors. A
set of FESEM images on GaAs nanowires grown at different V/III ratio is displayed in Fig.
2.8. At low V/III ratios (12, 23 and 46), epitaxial nanowires grow in the vertical [ ̅ ̅ ̅]
direction. At a critical V/III ratio of ~90, small number of nanowires growing in non-vertical
direction is observed. The non-vertical growing nanowires increase in amount as the V/III
ratio (190) increases. Besides, the tapering of nanowires increases slightly with V/III ratio.
This is due to the drop of activation energy for 2D planar growth with increasing V/III
ratio.120 The decrease of this 2D growth activation energy causes the radial growth rate
increases,64, 121 resulting in more tapered nanowires. In addition, at V/III ratio of and below
46, the vertical growth rate increase as V/III ratio increases, resulting in a longer nanowire.
This is expected since at low V/III ratio, AsH3 reaction species enhances the
decomposition of TMGa,122 which drives a higher growth rate of GaAs nanowires.
However, at V/III ratio of and above 46, the opposite trend is observed. The axial growth
rate decreases with increasing V/III ratio. This may be attributed to the competition
between axial growth and radial growth for Ga adatoms.123 Besides, at high AsH3 flows,
stable As trimmers form on As-terminated ( ) surfaces,124 which will hinder growth of
vertical nanowires on the ( ) surface.
In the works of Verheijen et al.,103 relationship of the morphology of GaP nanowires
with the growth temperature and V/III ratio is described by giving different 3D models.
Figure 2.9 is the schematic 3D construction of the different morphologies of GaP
nanowires grown at various temperature and V/III ratio.

21
Figure 2.8. FESEM images of GaAs nanowires grown at constant flow rate of TMGa
precursors but varying the flow rate of AsH3 precursors, resulting in a series of V/III ratios.
Axis shown is logarithmic. Scale bars are 1 µm. samples are tilted at 40°.48
Figure 2.9. Schematic 3D construction of the GaP nanowire morphologies. (a, b, c, e) are
nanowires with ZB structure, while (d) has the mixture of WZ and ZB structures (the
shaded parts represent the WZ segment). Rotational twin planes for (a, c–e) do not affect
the morphology.103
Nature of substrates. Generally, the growth direction of nanowire is determined by
the substrate.125 The ( ) substrate are usually used to grow nanowires so that epitaxial
nanowires can be grown since ⟨ ⟩ is the preferential growth direction of nanowires.
Substrate with the identical material as the grown nanowires is selected to minimise the
lattice mismatch between substrate and the nanowires, for instance, InAs nanowires were
grown on InAs ( ) substrate (homoepitaxy). Besides, by using substrates with different
orientations other than ( ) surface, nanowires grow at other directions can be
obtained. In the work of Krishnamachari et al.,126 they reported the growth of InP

22
nanowires in ⟨ ⟩ direction on InP ( ) substrate (Fig. 2.10). The growth is done without
prior annealing, which is believed the reason for the vertical growth of InP in ⟨ ⟩
direction.
To integrate high performance of III-V nanowires with mainstream Si technology,
the study on epitaxial growth of III-V nanowires on Si substrate is significant. It is
understood that the small cross-section of nanowires is able to accommodate and relax
strain from the large lattice mismatch between III-V semiconductor and Si.15, 127 Hetero-
epitaxial growth of GaAs and InP nanowires on Si ( ) substrates; and GaP nanowires
on both Si ( ) and Si ( ) substrates have been achieved (lattice mismatch: GaAs and
Si is 4.1%; InP and Si is 8.1%; GaP and Si is 0.4%).128 However, there are still challenges
in this hetero-epitaxial nanowires growth since the growth on heterogeneous surfaces is
less energetically favourable than on homogeneous surfaces.129 Consequently, nucleation
and growth on a heterogeneous substrate is less likely than on the homogeneous surfaces
and interfaces of the nanowires.
Orthodox theories assume that the initial stage of VLS growth of nanowires takes
place at the liquid–solid interface forming a monocrystalline solid.130 This phenomenon is
consistent with the growth of nanowires epitaxial relation with the substrate. However,
nanowires grown in other directions always appear, especially III-V materials are grown on
group IV materials such as Si or Ge.128, 131 Following the orthodox theory, for catalysed
nanowires growing always in the ( ) direction, only two types of growth orientations
should be observed when grown on group IV ( ) surfaces. For a single seed with B
polarity, i.e., a group-V atom terminated ( ) surface, the nanowires should always grow
vertically at 90° with respect to the substrate surface (Fig. 2.6). For a single seed with A
polarity, i.e. group-III atom terminated ( ) surface, nanowire should turn into growth
direction of 19.47° from the substrate. Uccelli et al.132 introduced a new paradigm for
nanowire growth that explains the unwanted appearance of parasitic non-vertical
nanowires. With a crystal structure polarization analysis of the initial stages of GaAs
nanowire growth on Si substrates, the secondary seeds form due to a three-dimensional
(3D) twinning phenomenon was demonstrated. This 3D twinning causes the manifold of
growth directions, which is typically found to occur and not in direct epitaxial relations with
the substrate (Fig. 2.10c).

23
Figure 2.10. SEM images of the growth of InP nanowires on InP ( ) substrate: (a) top-
view and (c) view of nanowires after tilting the substrate at 30°. (b) is magnified image of
(a) shows the rectangular shape of nanowires formed by stepped { } side facets of the
[ ] -oriented nanowires. (d) is the magnified image of a single InP nanowire.126 (c)
Schematic of the two types of growth obtained. Type I refers to the cases where a direct
crystallographic relation between the nanowire growth and the direct crystallographic
relation between the nanowire growth direction and the substrate in found and Type II
when 3D twinning is found. The drawing shows two nanowires grow from the same point
but this does not have to be the case for Type II.132
2.3 Catalysts – important role in epitaxial III-V nanowire growth
Catalyst is a substance that modifies the transition state to lower the activation energy. It is
important to note that a catalyst increases the rate of reaction without being consumed by
it. In addition, it does not change the energies of the original reactants or products. Rather,
the reactant energy and the product energy remain the same and only the activation
energy is altered (lowered). Catalytic growth is a powerful tool to form a variety of
nanowire-like structures with diameters ranging from just a few nanometres to the
millimetre range.133 There are many characteristics of the catalyst properties that influence
the nanowire growth vividly. Two main stages will be considered in considering a metal-
catalysed nanowire growth: (1) stage of nucleation; and (2) stage of nanowire growth.
Therefore, the effects of catalyst will be reviewed in both two stages. It is crucial to design

24
the growth of nanowires with the combination of catalyst selection and growth condition
choices to grow tip-led nanowires. Joyce and her colleagues63 developed a good work in
understanding these two stages for III-V nanowires growth. A two-temperature method is
applied which allows the Au catalysts to nucleate with growth material precursors at a
relatively higher temperature, and then the growth temperature is reduced to the
conventional growth temperature. Thus, single crystal GaAs nanowires free of planar
defects were grown. Metal catalyst used to induce nanowires growth can be categorized
into (i) foreign metal catalyst – material used as catalyst is different from III-V material, for
example, Au, Ni, Pd and Ag; and (ii) self-catalyst – material used as catalyst is the group-
III material, namely In or Ga.
2.3.1 Nucleation
Nucleation is one of the two main mechanisms of the first order phase transition, the
process of generating a new phase from an old phase whose energy has become higher
than that of the emerging new phase. Another prominent feature of nucleation is
metastability of the old phase, i.e., the transformation requires passage over a free energy
barrier. This is easily understood by considering the free energy changes associated with
the formation of the nucleus. The statement that the free energy per molecule of the new
phase is less than that of the solvated phase only applied to the bulk of the new phase.134
The surface is a different matter. As the surface molecules are less well bound to the
neighbours than are those in the bulk, their contribution to the free energy of the new
phase is greater. The difference between the free energy per molecule of the bulk and that
of the surface is referred to as the interfacial free energy. (It is sometimes called the
surface free energy, but strictly speaking, this term should be reserved for surfaces in
contact with vacuum.) The interfacial energy is always positive term and acts to destabilise
the nucleus. Consequently, at very small size when many of the molecules reside at the
surface the nucleus is unstable. Adding even one molecule just increases the free energy
of the system. On average, such a nucleus will dissolve rather than grow. But once the
nucleus gets large enough, the drop in free energy associated with formation of the bulk
phase becomes sufficiently high that the surface free energy is unimportant, and every
addition of a molecule to the lattice lowers the free energy of the system. There is an
intermediate size at which the free energy of the system is decreased whether the nucleus
grows or dissolves, and this is known as the critical size. This phenomenon is referred to
as the Gibbs-Thomson effect. Obviously, if the supersaturation is high enough, the critical

25
size can be reduced to less than one growth unit. Then the barrier vanishes and the old
phase becomes unstable so that an infinitesimal fluctuation of an order parameter, such as
density, can lead to the appearance of a new phase. The rate of generation and growth of
the new phase is then only limited by the rate of transport or energy.
The existence of a critical size has several implications. First and foremost, because
nucleation of the new phase is the result of fluctuations that bring together sufficient
numbers of molecules to exceed the critical size, the probability of nucleation will be
strongly affected by the value of the critical size. This means that nucleation can be
controlled, to some extent, by modulating the critical size, which is in turn a function of the
interfacial energy. The smaller the interfacial energy, the smaller the critical size and the
more likely nucleation becomes for any given supersaturation. Therefore, by varying either
the solution composition or the supersaturation, the probability of nucleation can be
manipulated. Besides, if a solution is supersaturated, then regardless of the critical size or
the presence of a foreign surface, the solution will eventually crystallize. What is significant
about the critical size is that it controls the probability of a nucleus forming on any given
timescale. In other words, it determines the kinetics of nucleation. The nucleation rate
depends on the supersaturation and the interfacial energy strongly.
The presence of a foreign surface can be used to exert even greater control over
nucleation because, quite often, the interfacial energy between a crystal nucleus and a
solid substrate is lower than that of the crystal in contact with the solution.134 This is
because the molecules in the crystal can form bonds with those in the substrate that are
stronger than the bonds of solvation. Because the enthalpic contribution to the free energy
comes primarily from chemical bonding, stronger bonds lead to a smaller interfacial free
energy. Of course, the strength of bonding at the interface is strongly dependent on the
structure and chemistry of the substrate surface. If the atomic structure of the substrate
surface closely matches a particular plane of the nucleating phase so that lattice strain is
minimized and, in addition, the substrate presents a set of chemical functionalities that
promote strong bonding to the nucleus, then the enthalpy contribution to the interfacial free
energy becomes small, and nucleation occurs preferentially on that crystal plane.
For an understanding on the stage of nucleation, the thermodynamic model is
introduced. The formation of the catalyst droplets in liquid phase can be elucidated by the
minimisation of their free energy. In accordance to the Gibbs-Thompson relationship, the

26
difference in chemical potential (μ) between a curved surface (radius, r) and planar surface
(radius, r = ∞) can be obtained, as given in Eqn. 2.5:135
⁄ (2.5)
where γ is the surface energy of the droplet; and Ω is the molar volume of the species
within the nuclei. The Gibbs-Thompson relationship (Fig. 2.11a) is appropriate to describe
the critical diameter of pure phase nuclei and the vapour-liquid equilibrium differences for
spherical droplet and the planar surface. Gibbs suggested that the shape of the nuclei is
the one ensuring the lowest free energy of the nuclei, i.e. the sphere.136 On the other hand,
this relationship can be applied to explain the formation of liquid phase catalyst droplets in
thermodynamic point of view. Nucleation process of a solid-phase nanowire material will
start after these liquid droplets are saturated by the dissolved precursors.137 In addition,
the shape of these spherical droplets is preserved by the supersaturating interfaces
between the vapour phase precursors and the liquid droplet surfaces.138, 139 Thus, during
the early stage of nanowire growth, there are two supersaturated interfaces that co-exist
(Fig. 2.11b). This is important to annotate that there are several modes of nucleation which
depends on the solubility, critical diameter and Gibbs-Thompson relationship of the
catalyst droplets, supplying precursors and substrates.
For a metal-solute system with reasonably solubility, the critical nucleus size is
expected to be larger than the metal droplet size and hence leads to a tip-led nanowire
growth instead of multiple nucleation.135 On the other hand, considering a system with a
fairly low solubility, spontaneous nucleation, in which the concentration of the dissolved
solute in the relatively large catalyst droplet reaches a supersaturating limit very fast, is
most likely. When this happens, multiple nanowires may grow out from a single catalyst
into different directions.140, 141
2.3.2 Nanowire growth
Once the nucleation initiated, nanowire will start to grow in a tip-led formation manner, in
which the tip is a liquid droplet contains only catalyst metal or catalyst metal and growth
material for VLS mechanism. In VSS mechanism, on the other hand, the catalyst alloy at
the nanowire tip either remains in solid phase or quasi-liquid phase. In all the cases,
despite their phase, catalyst plays similar roles in stimulating nanowire growth. By using

27
metal catalyst, the enhanced growth rate of nanowires can be partly due to the fact that
the condensation surface area for the growth species (on catalyst, especially in liquid form)
is larger than the surface area of the crystal growth. While the growth surface is the
interface between the catalyst and the solid crystal surface, the condensation surface is
the interface between the catalyst and vapour phase. Depending on the contact angle, the
liquid surface area can be several times of the growth surface.
Investigating from the thermodynamic point of view, interfacing energy is the driving
force of nanowires growth. The catalyst droplet, which is defined by the composition of the
materials and the droplet-substrate contact angle, directs the interfacial energy of the
droplet-nanowire (or liquid-solid) interface.142-147 In the interim, the shape and stability of
the droplet are controlled by the combination of different interfacial energies and line
tensions.
Besides, new facet formation occurs with the minimisation of Gibbs free energy.129
The wetting of the surface of the newly grown nanowire tip is the prerequisite for the
growth. The thermodynamic inequality, which determine the possible range of contact
angle, θ, can be expressed using Eqn. 2.6: 135
(2.6)
where αS-V is the solid-vapour interface tension, αL-V is the liquid-vapour interface tension,
θ is the contact angle and δ is the conical angle (Fig. 2.11c). In accordance to this
relationship, catalyst and nanowire material having larger liquid-vapour interface tension,
αL-V, favour stable growth.148 This criterion can be considered for the selection of suitable
catalyst/nanowire systems for a stable tip-led nanowire growth. Nevertheless, the
composition of catalyst is often an alloy with a significant percentage of solute which has
much difference from the pure metal catalyst.
Additionally, the contact angle of the catalyst was found to be related to the growth
rate and crystal quality of the nanowires grown.149-153 This angle can be less than, equal or
larger than 90° (Fig. 2.11c–h). It was found that with the contact angle, θ, being larger than
90°, the growth rate of the nanowires is relatively slower compared to the other two
cases.56, 154 Extending from this observation, nanowires with contact angle larger than 90°

28
exhibit a better crystal quality compared with those having contact angles equal to or less
than 90°.155-159
Figure 2.11. Schematic diagrams of (a) Gibbs-Thompson relationship for the liquid-phase
catalyst formation;135 and (b) two supersaturated interfaces direct the nucleation of
nanowire growth. Schematic drawing of nanowire growth having (c) cylindrical
morphology; and (d) tapered morphology,143 and have variations of contact angle of
catalyst (c) larger than 90°, (e) 90°, and (f) smaller than 90°. SEM images showing catalyst
having contact angle (g) larger than 90° and (h) 90°. Scale bar is 100 nm for SEM
images.160
2.3.3 Effects of catalyst on the nanowires quality
Due to the presence of catalysts in the growth of III-V nanowires growth, the crystal
structure and geometry of the catalyst–nanowire interface has a strong effect on the
nanowire growth, such as the crystal orientation and the defects formation. For example,
for a nanowire with ZB crystal structure, the main growth direction is in ⟨ ⟩ directions.73,
75 Nevertheless, it is possible for the growth of ZB nanowire in ⟨ ⟩ and ⟨ ⟩
directions.59, 110, 161 Wang et al.162 reviewed on the relationships between the growth
direction of nanowires and the catalyst/nanowire interface. For the nanowires grown in

29
⟨ ⟩ directions, the interface underneath the catalyst is perpendicular to the nanowire
growth direction (Fig. 2.12a). Two possible interfaces have been observed when the
nanowire grown in [ ] direction, which are: (i) a straight ( ) plane interface, which are
not perpendicular with the growth axis (Fig. 2.12b); and (ii) a zigzag interface consists of
{ } planes (Fig. 2.12e). On the other hand, nanowires grown in [ ] direction has two
different interfaces, which are: (i) a straight ( ) plane interface (Fig. 2.12c); and (ii) a
zigzag interface consists of { } planes and a small fraction of { } planes (Fig. 2.12d).
Finally, for the nanowires grown in [ ] direction are mostly { } planes (Fig. 2.12f).
Figure 2.12g shows the interfaces models for a variety of growth directions.
Figure 2.12. (a–f) ZB structured nanowires grown in different directions. (g) Modelling at
the catalyst/nanowire interface of the nanowires grown in different directions.162
Lenrick et al.163 observed live of the growth of straight and kinked InAs nanowire
using in situ TEM. They explained that the temperature changes, change of partial
pressure of growth species rapidly and increase flux of In and As species induced the
inclined { } planes in the nanowire. Figure 2.13a shows the time-lapse TEM image at
the catalyst-nanowire interface and the corresponding model build to explain the kinking
phenomena. The catalyst/nanowire interface of the first twin segment was pinned as it
reached the opposing nanowire side-facets. The larger second twin became leading for
the growth, forming an increasing tilted catalyst/nanowire interface with respect to the

30
original interface. Tian et al. also designed multiply kinked nanowires, which was also
altered by changing of the catalyst-nanowire interface as shown in Fig. 2.13b.164 Besides,
Schwarz and Tersoff165 developed a model for steady VLS growth modes for 2D crystals
by considering the catalyst-nanowire interface as illustrated in Fig. 2.13c. The steady
modes proposed for nanowire growth are single-facet (1f), symmetric two-facet (2fs),
asymmetric two-facet (2fa), symmetric inverted two-facet (2fis), and asymmetric inverted
two-facet (2fia).
Figure 2.13. (a)Time-lapse TEM images and model build to describe kinking nanowire. 163
(b) Schematic illustration of the key stages of kink formation. The inset is SEM image of a
multiply kinked nanowire.164 (c) Steady VLS growth modes proposed by Schwarz and
Tersoff.165
Catalyst size effect. As the size of catalyst droplets was reduced, the solubility
would increase. For the growth of very thin nanowires, a very small droplet is required.
However, a convex surface with a very small radius would have a very high solubility.

31
Hence, a high supersaturation of growth species in the vapour phase must be generated.
A high supersaturation in the vapour phase may promote the lateral growth on the side
surface of nanowires with the VS mechanism. Therefore, a conical structure may be
developed instead of uniformly sized nanowires. Further, a high superstation in the vapour
phase may initiate homogeneous nucleation in the gas phase or secondary nucleation at
the surface of nanowires.
Another characteristic in the VLS method should be noted. In accordance to the
Kelvin equation, an equilibrium solubility and supersaturation of growth species in larger
liquid catalyst droplets can be obtained easily than that in smaller droplets. The growth of
nanowires will proceed only when the concentration of growth species is above the
equilibrium solubility. When the concentration or supersaturation in the vapour phase is
appropriately controlled, vapour pressure could be kept below the equilibrium solubility in
small liquid droplets, and the growth of nanowires of thinnest nanowires would terminate.
When the growth proceeds at high temperatures and the grown nanowires are very thin,
radial size instability is often observed. Such instability is explained by the oscillation of the
size of the liquid droplet on the growth tip and the concentration of the growth species in
the liquid droplet. Such instability could be another barrier for the synthesis of very thin
nanowires, which may require high deposition temperatures.
Cai et al.166 demonstrated that the initial growth condition of a ZnSe nanowire is
mainly determined by the size of the Au-Ga binary alloy catalyst, which formed during
annealing of Au nanoparticle on GaAs substrate. Table 2.2 and 2.3 summarises the
growth-direction of ZnSe nanowires dependent on the orientation of GaAs susbtrate and
catalyst size.
Table 2.2. Growth directions and catalyst/NW interface dependent on orientation of
substrate. 166
GaAs substrate Growth directions Catalyst/NW interface
( ) Four ⟨ ⟩, ⟨ ⟩, ⟨ ⟩ { } ( ) [ ] { } ( ) [ ], four ⟨ ⟩, ⟨ ⟩ { }

32
Table 2.3. Growth directions dependent on the catalyst size. 166
Diameter of catalyst Growth directions Structure quality
< 20 nm major ⟨ ⟩, minor ⟨ ⟩ Few defects
> 20 nm ⟨ ⟩ Always have stacking faults
2.3.4 Examples of non-Au metal catalysts in III-V nanowires grown
Among all metal catalyst, Au has been dominated as the primary choice of catalyst to
induce epitaxial growth of III-V nanowires due to its chemical inertness and ability to form
eutectic alloys at relatively low temperatures with various semiconductor elements.31 Au is
also thermally stable.167 There are many excellent works that have been done to grow III-V
nanowires mediated by Au catalysts. Meanwhile, much less attention has been paid to the
effects of other different non-Au metal catalysts on the growth of epitaxial III-V
nanowires.31 Non-Au catalyst, which is generally cheaper in price than Au, is considered a
less mature system to grow III-V nanowires due to the lack of understanding on the effects
of these non-Au metal catalysts with the growing materials. Besides, the controllable
growth environment for this non-Au mediated nanowires growth yet to obtain. Therefore,
an intense study is needed to have a thorough understanding on the non-Au catalysed
nanowires growth. An executive summary table is given which incorporates growth of non-
Au catalysed III-V nanowires that have been reported hitherto (Table 2.4).

33
Table 2.4. Summary of non-Au metal catalysed III-V nanowires growth that has been reported.
References Nanowires Non-Au catalyst
Substrates Type
Deposition method
Type Size of
thickness Deposition method
168 GaN MOCVD Au, Fe, Ni thin
film 2–10 nm Thermal evaporation
Si; c-sapphire; a-sapphire
169 GaN AlN
MOCVD Ni NA -- Al2O3
7
InAs GaP GaAs InP
CVD Bi NA -- --
170 GaAs Solid source MBE Mn thin film 1 nm e-beam evaporator SiO2; GaAs ( )
171 GaN MOCVD Ni thin film 2 nm e-beam evaporator Al2O3;
r-sapphire 172 GaN CVD Au-Pd film NA Sputter deposition Si
108 GaN MOCVD Ni drops NA
Dropped diluted nickel nitrate (hexahydrate)
(Ni(NO3)2.6H2O) onto different areas of wafer
(1-102) r-plane sapphire
173 GaAs MOCVD Fe nanoparticles 10 nm
Thermal decomposition of iron pentacarbonyl (IPC) in a
low-pressure microwave plasma reactor
GaAs ( )
18 InAs Solid source CVD Ni thin film 0.5 nm Thermal evaporation Si/ amorphous
SiO2
174 GaN MOCVD Ni thin film NA e-beam evaporator ( ̅ ) r-plane
sapphire
175 InP MOCVD Au thin film Ag thin film
0.6 nm 1 nm
e-beam evaporator Si ( )
176 GaN MOCVD Ni thin film 1–5 nm Sputter deposition c-plane sapphire
120 GaN MOCVD Ni thin film 1–5 nm Sputter deposition ( ) c-plane
sapphire

34
177 InAs Solid source MBE Mn thin film 1–2 nm e-beam evaporator SiO2; GaAs ( );
GaAs ( ) 178 InAs CBE Pd thin film NA Spin-coating InAs ( )
167 InAs CBE Pd thin film
(patterned by EBL)
NA Spin-coating InAs ( )
179 InP MOCVD Cu thin film ≈ 0.6 nm Thermal evaporation InP ( )
180 GaP UHV-TEM (in-situ) Au, Cu, Au-Ga or
Au-Al NA Thermal evaporation Si ( )
181 GaAs Solid source CVD Ni thin film 0.5 nm Thermal evaporation Amorphous
Si/SiO2 182 GaAs Solid source CVD Ni thin film 0.5 nm Thermal evaporation Si/SiO2 (~/50 nm) 68 * Vertically aligned
InSb CBE Ag thin film NA Thermal evaporation InSb ( ̅ ̅ ̅) ;
InAs ( ̅ ̅ ̅)
183 InP MOCVD Cu thin film ≈ 0.6 nm Thermal evaporation InP ( ) 31 InAs MOCVD Pd thin film 2 nm e-beam evaporation GaAs (( ) 184 InAs MOCVD Pd thin film 2 nm e-beam evaporation GaAs (( ) 185 InP-InAs MOCVD Cu thin film ≈ 0.6 nm Thermal evaporation InP ( ) 186 GaN CVD Ni thin film 5 nm NA Si ( )
43 InAs MBE Ag thin film 0.2–2 nm
(optimum: 0.5 nm)
Ag effusion cell Si ( )
187 InAs MBE Pd thin film 0.5–5 nm
(optimum: 1.5 nm)
e-beam evaporation GaAs (( )
188, 189 GaN CVD Ga, Au drops;
Ni thin film 5 nm NA Si ( )
190 GaAs MOCVD Sn droplets NA High flow of TESn GaAs (( ) 191 GaAs MOCVD Pd nanoparticles NA SDG GaAs (( )
192 GaAs MBE Ag thin film 1–2 nm e-beam evaporation GaAs (( )
GaAs ( )

35
2.4 Growth mechanism
“Why do nanowires grow?” “Why would a crystal grow much faster in one direction than
all others?” These questions are important to consider on the growth mechanism of
nanowires. To put this question into perspective one must remember that most of the
nanowire crystal structures do not inherently have one-dimensional symmetry, meaning
that there are equivalent crystallographic planes distributed at precise angles from each
other.129 The equilibrium shape of these crystals is not 1D and thus, unidirectional crystal
growth must be the result of a symmetry breaking element in the crystal growth. There are
few mechanisms proposed to describe the growth of unidirectional crystal growth (why
nanowires grow faster in one direction than in all others).
Vapour-liquid-solid (VLS) mechanism193-198 is the most commonly cited nanowire
growth mode. However, there are other mechanisms have been proposed,67, 199-206
including vapour-solid-solid (VSS) mechanism in which the catalyst is in solid form.205, 207
Both VLS and VSS mechanisms have been observed via in-situ TEM growth studies of Ge
semiconductor nanowires.208 There are still more works need to be done to investigate the
role and nature of the metallic catalyst. Besides, the process of reactant diffusion to the
growth interface has long been questioned. In accordance to the conventional VLS
mechanism, reactant species dissolves into the supersaturated liquid-phase catalyst, and
then readily diffuse through the liquid catalyst to the growth interface. In contrast, the VSS
mechanism proposes solid-phase diffusion, involving dissolution of reactants into the solid
metal catalyst, followed by their diffusion through the solid catalyst to the growth
interface.207 In accordance to both of these mechanisms, reaction species are transported
to the catalyst/nanowire interface via bulk diffusion, despite the phase of catalyst. Instead,
Cheyssac et al.209 proposed that surface diffusion, rather than bulk diffusion, is the main
transport mechanism. In accordance to their model, the reactants adsorb and diffuse along
the outer surface of the catalyst, instead of penetrating it.
On a different note, oxide-assisted growth (OAG) mechanism that governed the Si
nanowires growth (rather than VLS mechanism) was proposed by Lee et al..210-212 This
mechanism does not require a metal catalyst but the growth is induced by the oxide. At the
initial nucleation stage, Si oxide (SiO) vapour condenses on the substrates and forms Si
nanoparticles, which become the nuclei of nanowires. Each Si nanowire nucleus consists
of a polycrystalline Si core containing a high density of defects and a SiO shell. The

36
vapour phase in the reaction chamber mainly consist SiO due to the reaction of Si and
SiO2. This mechanism will not be discussed in detail here.
Besides, the theory of preferential interface nucleation (PIN) has been proposed as
the combining mechanism of nanowire growth, and the fundamental mechanism
supporting the VLS mechanism and variations such as the VSS mechanism.129 In
accordance to the PIN theory, the probability of nucleation at the catalyst/semiconductor
interface is highest and thermodynamically favourable compared with other interfaces in
the system. Preferential nucleation at the catalyst/semiconductor interface induces
nanowire growth. Recent progress toward understanding the mechanism of Au-catalysed
nanowire growth has been reviewed in literature213. In general, all the proposed models of
metal-catalysed nanowire growth suggest that the nucleation rate is the highest at the
catalyst/semiconductor interface, which drives nanowire growth. All in all, of utmost
importance to improve electronic and optical properties of III-V semiconductor nanowires
for future applications, a thorough and sharp understanding on growth mechanisms of III-V
nanowires is imperative.
2.4.1 Vapour-liquid-solid (VLS)
In the VLS growth, a second phase material, commonly referred to as either impurity or
catalyst, is purposely introduced to direct and confine the crystal growth on to a specific
orientation and within a confined area. A catalyst forms a liquid droplet by itself or by
alloying with growth material during the growth, which acts as a trap of growth species.
Enrich growth species in the catalyst droplets subsequently precipitates at the growth
surface resulting in the one-directional growth.
In 1964, Wagner and Ellis193 reported that the metal impurities could mediate the
growth of highly anisotropic Si whiskers by a vapour-liquid-solid (VLS) mechanism. This
mechanism suggested that growth of a material occurs from a liquid alloy zone. This zone
is exposed to at high temperatures to the vapour of a compound of a material to be grown.
Decomposition of the compound causes the liquid to become supersaturated with the
required material and growth occurs by precipitation from the alloy. There are many
important observations pertinent to an understanding in the growth of Si whiskers in the
early 60s:194 (1) Si whiskers are axial screw dislocation-free; (2) certain impurity is
essential for whisker growth. Without such impurities, the Si deposit is in the form of a film

37
or consists of discrete polyhedral crystals; (3) whisker growth is a two-stage process: a
fast-initial extension in length (leader growth) followed by a slow increase in thickness
(layer growth). The impurity concentration in the leader is higher than in the layer growth;
(4) the rapid initial lengthening occurs by addition of material to the tip of the whisker; (5)
the main growth direction of a Si whisker is ⟨ ⟩, known to be the direction of slow growth
from the vapour and from the liquid; (6) Si whiskers can be regrown from partly evaporated
whiskers. The regrowth occurs without change in morphology and without introduction of
defect structure.
During the investigation described in 194, whisker crystals of micron diameter were
observed occasionally to exhibit during regrowth a small “liquid-like” globule at the tip (a
small globule is present at the tip of the whisker during growth). The liquid layers or
globules were considered to be metastable phases, molecular complexes, or intermediate
polymers originating from condensation of the vapour phase. This initial study focused on
Au seed particles at temperatures far above the Au-Si eutectic temperature. For further
explanation, the growth of InAs nanowire via VLS mechanism is depicted in Fig. 2.14 by
referring to Au-In binary phase diagram (In this example, the growth temperature, Tg, is
500°C). In phase (i), solid Au catalyst is deposited on the substrate and loaded into growth
chamber. Vapour state of In atoms are supplied, and absorbed at the Au catalyst sites to
form Au-In alloys via solid-state diffusion. When the composition of In reaches above 28
at.% in the Au-In alloys, the state of the alloys will transform from solid to liquid as
indicated in the Au-In phase diagram. In phase (ii), In atoms continue absorbing into the
Au-In liquid alloys while the temperature in the chamber being kept at 500°C. When the In
concentration reaches about 45 at.% in the Au-In alloys, the composition of the catalyst
droplet reaches the liquidus line. (iii) The continuing supply on In will lead to super-
saturation of In in the Au-In alloy and lead to In precipitate out from the alloy. This
precipitated out In reacts with the As atoms and the interface becomes the nucleation site
of InAs NW. (iv) As this continues, InAs continues to grow vertically. Generally, for VLS
mechanism to be valid, the growth temperature of the nanowires is higher than the eutectic
temperature of the catalyst/nanowire alloy. As a side note, the non-metal As has very low
solid solubility in Au. The typical nanowires growth temperatures are too low to form a
eutectic Au-As alloy. Therefore, the description of the InAs nanowires via VLS growth
mechanism will only take Au-In phase diagram into consideration. Orthodox theories
assume that the initial stage of VLS growth of nanowires takes place at the liquid/solid
interface forming a monocrystalline solid.130

38
The consequences of the VLS growth mechanism have attracted many attentions.
For instance, by assuming that a liquid droplet is critical to nanowire growth, it was
predicted that there would exist a minimum critical size under which nanowires or whiskers
would not grow.150 This is ascribed to the Gibbs-Thomson effect, i.e. as the diameter of
nanowire or whisker decreases, the supersaturation on the growth interface is lowered.
This effect well describes how the effective pressure in a liquid droplet is related to the
curvature of the droplet. For high curvatures (correspond to small sizes of the liquid droplet
in the VLS mechanism) the effective pressure would be so high that the liquid would
evaporate and then unidirectional growth would cease. This argument was expanded to
predict and describe how the growth rates of nanowires depend on their diameters.214-217
Besides, the crystal structures of nanowires are calculated as a functions of
supersaturation and their respective radii.157 The growth dynamics are highly dependent
on changes in morphology of the liquid phase,218 which determines the crystal structure 88
and the occurrence of kinking and crawling phenomena.75, 219, 220
Figure 2.14. Schematic illustration of VLS mechanism193 to describe Au-mediated InAs
nanowire growth, assisting by Au-In binary phase diagram.

39
2.4.2 Vapour-solid-solid (VSS)
The vapour-solid-solid (VSS) mechanism is a lately developed growth mechanism for the
explanation of nanowire grown below the bulk eutectic temperature of the
catalyst/precursor alloy. It has the similar concept as VLS growth mechanism except that
the catalyst/precursor alloy remains in solid phase during the growth of nanowires, which
is first proposed by Persson et al.207 in the growth of Au-catalysed GaAs nanowires by
CBE method. In 2006, the successful growth of Si nanowires by aluminium (Al) catalyst at
450°C (well below the eutectic temperature 577°C of Si-Al alloy) by Wang et al. 221 turned
the spotlight on VSS mechanism. The size of the Al nanoparticles ranges from 20–70 nm,
and the explanation of melting-point depression due to size 222 were excluded in the
explained mechanism.
Besides, observing by in situ TEM, Kodambaka et al.208 showed that Ge nanowires
catalysed by Au nanoparticles can grow below the eutectic temperature with either liquid
or solid catalyst at the same temperature. They related the state of catalyst with the growth
pressure and thermal history. These phenomena may be due to kinetic enrichment of the
eutectic alloy composition and the size of catalyst. In the work of Campos et al.223, they
demonstrated the growth of ZnO nanwoires catalysed by Au particle at low temperature
and showed that the growth was driven by epitaxial match between solid AuZn alloy
catalyst particles and ZnO nanowire via VSS mechanism.
Figure 2.15 depicts the fundamental of VSS mechanism for Al-mediated Si
nanowires. There are some uncertain issues during this mechanism: the alloy is whether in
the state of solid or chemicophysical quasi-liquid.224, 225 In this example, the growth
temperature, Tg, is 550°C (below eutectic temperature, 577°C, of Al-Si alloy). At phase (i),
suitable substrate deposited with Al catalyst is loaded into reaction chamber. Si precursor
is supplied when Tg is reached. By holding at Tg for a given growth time, tg, Si atoms start
to defuse into Al catalyst and solid Al-Si alloy is formed. During phase (ii), Si atoms begin
to precipitate out when supersaturation is reached in the solid Al-Si alloy; hence this
results in nucleation and growth of Si nanowire.
The growth rate of VSS mechanism is not significantly slower than that of the VLS
mechanism. This is interesting since liquid-state diffusion is usually much more favourable
against solid-state diffusion.226, 227 In comparison with VLS mechanism, due to the

40
dependence of nanowire direction and growth rate of the details of the catalyst shape and
orientation relation, nanowires grown by VSS may require more stringent process
control.228 Therefore, additional experiments are necessary to shed light in clarifying all
these unclear issues.
Figure 2.15. Schematic illustration of VSS mechanism, assisting by Al-Si binary phase
diagram.
2.4.3 Preferential interface nucleation (PIN)
As an extension of the VLS growth mechanism, Wacaser et al.129 presented a more
general and fundamental mechanism: preferential interface nucleation (PIN). At large, the
growth characteristics and properties of the nanowires will be dependent on the transport
and diffusion of material in the supply chamber, in the catalyst (or recognised as collector),
and on the crystalline surfaces; the temperature and pressure difference in the system; the
chemical environment and reaction kinetics; the thermodynamic behaviour of the catalyst;
the geometry and design of growth chamber; and the combination of materials involved.
The only constant in these different growth systems is that the growth rate at interface of
catalyst/crystal is faster than the growth rates at all the other interfaces. Based on the
observations of the previous VLS, VSS or other proposed growth paradigms of nanowires,
this research group suggested that a supersaturation is possible for almost any phase of a
catalyst, whether it is a liquid solution, an alloy, or an adsorbed layer.

41
There are few new terminologies introduced for this VLS-like system, as illustrated
in Fig. 2.16a. Collector (c) is named to represent the liquid phase in VLS, which is more
appropriately to emphasize its collection ability of the growth material rather than
emphasize on the particular phase. Supply (s) represents vapour phase in VLS; while the
1D growing material will be called the wire (whether it is a whisker, column, tube or pillar).
The solid material will be generically names as crystal (k). In a three-phase (vapour, liquid
and solid) system, the significant surfaces and interfaces are the supply-collector,
collector-crystal, supply-crystal, and supply-collector-crystal interfaces. The supply-
collector-crystal interface is the line connecting all three phases and is recognised as the
three phase boundary (TPB). Besides, the term collector suggests that for various growth
systems, the phase of this collector can collect material so that the relative concentration
of the growth species can be varied in the collector than in the supply. This is attributed to
the difference of the equilibrium concentration in the collector and that in the supply. For
crystal growth process, the rate of nuclei formation (which is important in initiating the
growth of 1D nanostructure) is dependent on the concentration. By keeping all other
parameters constant, an increase in the concentration results in an increase in the
nucleation rate. However, in order for growth of 1D nanostructures to grow on an
atomically flat surface, the nuclei that are formed during this nucleation must stay steady
long enough for them to initiate growth. Therefore, it explains the other concept: the steady
state nucleation rate is dependent on the critical nucleus size. Figure 2.16b further
explains on the nucleation at different interfaces in a three-phase system. By measuring
the wetting angle (θ) (Fig. 2.16c), one can correlate the different surface free energies. For
a strong interaction between the collector and crystal, the wetting angle is small (θ < 90°)
and the collector-crystal specific surface energy will be smaller than the supply-crystal (σck
< σsk) and vice versa. Figure 2.16d illustrates the nucleation at either the TPB or the
collector-crystal interface and steps exist at the edges of the nuclei in both cases. This
means that these interfaces are no longer atomically flat. The propagation of these steps
along the other surfaces and interfaces in the system results in a birth and spread or layer-
by-layer growth to form a wire (Fig. 2.16e–j).

42
Figure 2.16. A schematic diagram describing PIN mechanism.129
2.4.4 Vapour-solid (VS)
Vapour-solid (VS) mechanism is described for the formation of whiskers or nanowires on a
crystal which is growing by vapour deposition, in which diffusion of either material or heat
is the limiting factor for crystal growth.229 For this mechanism, surface energy, defects, and
growth kinetics are often considered for one-dimensional material formation.230 A vapour of
source materials with certain partial pressure is required for the growth in these vapour-
related processes. VS mechanism is the growth mode proposed in the self-induced III-V
nanowires. In the work of Herttenberger et al.,231 self-induced InAs nanowires were grown
on Si (111) substrate covered with 1–4 nm thick SiOx layer with nanometer-sized opening,
at 480°C in MBE chamber. Abrupt morphology transition and in-plane strain relaxation
revealed that InAs nanowires nucleate without any significant delay and under the
absence of In droplet. These results emphasize the absence of VLS growth by claiming
that there is no sufficient time for In droplets to form due to the fast nucleation of InAs on
Si substrate once after the initiation of growth process.
Dimakis et al.232 investigated carefully the nucleation and growth of InAs nanowires
on bare Si (111) substrate at 450°C by MBE. The nanowires grown are non-tapered with
high aspect ratio (Fig. 2.17a). The nucleation of InAs takes place in In-rich areas forming
spontaneously on the substrate as the growth initiated. The local stoichiometry on the

43
growth interface shifts from In-rich to As-rich, and the growth follows a VS mechanism.
First, In-rich areas formed on Si substrate. Then, InAs nucleated due to In in excess. The
continuous formation of new InAs nuclei on Si caused the decreasing flux of In adatoms at
the growth interface. This results in the fast transition to As-rich conditions. At the final
stage, the growth of InAs by VS mode, and vertical sidewalls developed under As-rich
conditions. VS mechanism was also reported to be the driving force for the growth of V-
shaped nanomembranes as shown in Fig. 2.17c,d.233
Figure 2.17. (a) SEM side-view image of InAs nanowires grown on Si (111). The inset is
top-view image of the hexagonal cross-section of nanowires with six { ̅ } side facets. The
scale bar is 1 µm for (a) and 200 nm for the inset. (b) Schematic illustration of the growth
mechanism for self-induced InAs nanowires.232 (c,d) Drawing and SEM image of InAs
nanomembranes.233
2.5 Growth techniques
Well-developed growth system is vital and many fabrication techniques have been
successfully developed. The growth techniques primarily differ in type of source materials
and the way the source material is brought into the gas phase, which can be classified into
CVD and physical vapour deposition (PVD). Table 2.5 listed the differences between CVD

44
and PVD. Few major III-V nanowires growth techniques, which are CVD, MOCVD, MBE,
CBE and laser-assisted catalytic growth (LCG), will be discussed.
Table 2.5. Differences between CVD and PVD.
Characteristics CVD PVD
Source material Mixed with a volatile precursor
that acts as a carrier Pure
Process to obtain gaseous reactant atoms/molecules
Chemical processes – decomposition of precursor containing source material
Physical processes – gasified of source materials via evaporation
By-product Yes No
Additional enhancements
Heat, plasma High power electricity, laser
ablation
Examples CVD, MOCVD, CBE MBE, LCG
Molecular beam epitaxy. First invented in the late 1960s at Bell Telephone
Laboratories by J.R. Arthur and co-workers,234-239 MBE is a term used to denote the
epitaxial growth of a wide variety of materials, ranging from oxides to semiconductors to
metal (thin films or nanowires) by a process involving the reaction of one or more thermal
molecular beams with a crystalline surface under ultra-high vacuum (UHV) conditions.240
The UHV chamber and the pressure is kept at about 10-9 Torr by using ion pump, turbo-
molecular pump and titanium sublimation pump. It was first applied to the growth of
compound semiconductors. MBE is related to vacuum evaporation, but offers much
improved control over the incident atoms or molecular fluxes so that sticking coefficient
difference may be considered, and allows rapid changing of beam species. In the UHV
environment, the localised beams of atoms or molecules are emitted from the evaporation
cells strike the substrate and promote epitaxial growths. These depositing materials are
significant because the composition can be rapidly changed and the deposition rate is
slow, producing crystalline interface that are almost atomically abrupt, resulting in precise
control of composition during growth.241 Due to the cleanliness of the growth environment
and the precise control over depositing material, MBE structures closely approximate the
idealised models used in solid-state theory. A simple drawing of MBE growth chamber is
shown in Fig. 2.19d.
Chemical vapour deposition. CVD has been extensively used in semiconductor
industries and research facilities.18, 112, 149, 181, 188, 195, 196, 242-245 CVD equipment can
schematically be divided into different sub-units (Fig. 2.19a). The substrate is exposed to

45
one or more volatile precursors in either gas or solid phase, which react and decompose
on the substrate surface to produce desired deposit. Commonly, volatile by-products are
also produced, which are removed by gas flow through the reaction chamber. For an
accurate supply of precursors, it contains a sophisticated gas delivery system, including
precursors together with a distribution system of carrier gas (ultra-pure H2 or N2). The
precursors are individually transported into a mixing chamber before they enter the reactor
cell, so that a laminar gas flow above the substrate can be achieved. The reactor is either
cold-wall or hot-wall. In a cold-wall reactor, only the substrate is heated, typical for
MOCVD conditions, in which the chemical reactions are endothermic. In a hot-wall reactor,
both the substrate and the gas phase are heated, which is typical for hydride vapour phase
epitaxy (HVPE), in which the chemical reactions are exothermic. A number of variations of
CVD techniques have been developed in the past years. These techniques can be
categorized by operating pressure, physical characteristics of vapour, plasma methods
and the use of additional enhancements.
Metal-organic chemical vapour deposition. MOCVD, also known as metal-
organic vapour phase epitaxy (MOVPE) or organometallic vapour phase epitaxy
(OMVPE), is a vapour phase deposition process capable of producing atomically abrupt
interfaces and high purity materials. In particular, MOCVD is a special case of CVD
technique using metal-organic compound as growth precursors with more precise controls
over growth temperature, precursor supply rates and precursor purities. Manasevit and
colleagues246, 247 pioneered the development of MOCVD, initially for growing single-crystal
GaAs on a number of single-crystal insulating oxide substrates. A detailed schematic
diagram of the MOCVD reactor used in this project is shown as Fig. 2.19c. Compared with
other growth methods, MOCVD is flexible, accurate and readily scalable for industrial
mass production. Besides, it is the most economical technique for large-scale commercial
production of almost all III-V and II-VI semiconductor materials.155, 248, 249 At present, it is
the dominant epitaxial technology for photonic devices such as laser diode, LED as well as
electronic device such as heterojunction bipolar transistor (HBT). The most important
processes during MOCVD growth have been summarised by Stringfellow249 as shown in
Table 2.6.

46
Table 2.6. Important processes during MOCVD growth.
Process Details
Mass transport
The carrier gas transports the reactants to reactor cell. A concentration gradient layer (boundary layer) above the growing surface is formed by the laminar flow of the vapour in the reactor cell. Molecules diffuse through the concentration gradient layer towards the surface before surface reactions can take place. The growth pressure and velocity of the carrier gas define the thickness of the concentration gradient layer.
Chemical reactions
The growth by MOCVD is the result of a complex sequence of chemical reactions, which involves surface chemical processes and gas phase chemical reactions. Types of chemical reactions that have been recognised as playing a role in MOCVD are schematically summarised in Fig. 2.18a.250
Thermodynamics
The growth rate is affected by thermodynamic properties since these define the deviation from equilibrium and thus the driving force for growth. The incorporation of native defects is influenced by thermodynamics. Furthermore, the reason to selective growth can be explained by thermodynamics.
Physical processes
Adsorption of molecules and radicals at the substrate surface, heterogeneous deposition of molecules and radicals at the surface, surface diffusion of species at the surface, incorporation of atoms into appropriate lattice positions at kinks or steps, or desorption of reaction products that enter into the vapour phase and then are transported away by the carrier gas are other significant processes for the growth mechanism.
In MOCVD system, crystal growth process is directed by law of thermodynamic and
kinetic processes. Thermodynamics determines the driving force for the entire growth
reaction whereas kinetic defines the rates at which various steps occur. Group III
precursors – gallium (Ga) and indium (In) are provided by metal-organic, MO
(organometallic or metal alkyl) sources trimethylgallium [Ga(CH3)3, TMGa] or
trimethylindium [In(CH3)3, TMIn], respectively during the growth of materials using this
MOCVD reactor. Group V precursors, arsenic (As) in particular, is provided by hydride
sources – arsine (AsH3). The gas-phase AsH3 is stored in high pressure cylinders and
directly supplied to the reactor chamber. TMGa is volatile liquid whereas TMIn is in solid
phase. These volatile MO sources are stored under a constant temperature and pressure
in sealed stainless cylinders. Hydrogen (H2) is used as a carrier gas for the growth. The
flow rate of each precursor can be precisely controlled together with the pressure and
temperature of reactor chamber. Binary III-V compound is the simplest case which
involves a reaction if the vapours of a volatile MO compound and a gaseous hydride, as
shown in Eqn. 2.7:

47
(2.7)
where R is an organic material (tributyl-, trimethyl- or ethyl- radical), M and X are the
constituent Group III and V species respectively for the deposited solid.
During the epitaxial growth, the group V concentration is always maintained in
excess compared to group III concentration due to its higher volatility. Consequently, the
growth rate is limited and controlled by the concentration of group III alkyl elements.
Identical with most of the CVD process, Fig. 2.19b illustrates three growth regimes of the
MOCVD process.251 The slope of the graph is a measure of the activation energy. At low
temperature, the growth is reaction controlled. In particular, surface reactions control the
growth rate. Nanowires are often grown in this range of low temperatures since lateral
growth is suppressed in this region. At intermediate temperature, mass transport governs
the growth rate and stays constant with temperature. At higher temperature, parasitic
spontaneous nucleation or pre-reaction happens. In addition, when temperature
approaches the congruent temperature of III-V compound semiconductors, the
semiconductors start to decompose into their constituent gas phase. As a result, the
growth rate drops. An increased vapour pressure from the constituent source is required to
keep smooth surface morphology.
The surface morphology of III-V semiconductor related closely to V/III ratio during
MOCVD process.252 As a rule of thumb, crystal morphology becomes smoother with a
higher V/III ratio. Nevertheless, by increasing V/III ratio, the surface mobility of group III
reduces. This gives rise to non-stoichiometric structure and more defects are found.
Besides, the decomposed efficiencies of alkyl metals and hydrides depend strongly on the
growth temperature profile, therefore the optimal V/III ratio is a function of the substrate
temperature.

48
Figure 2.18. (a) Schematic diagram summaries the most important chemical reactions
involved in MOCVD growth (thin-film growth is described here; the growth of nanowires
has the same concepts): (1) gas phase reaction; (2) transport to surface and diffusion
through boundary layer; (3) adsorption of precursor; (4) surface reactions and diffusion; (5)
desorption of precursor; (6) nucleation and island growth; (7) atomic step growth; (8)
desorption of volatile surface reaction products.250 (b) A schematic Arrhenius plot of the
growth rate, R, showing three distinct growth regimes of a typical III-V semiconductor
growth in MOCVD systems.251
Chemical beam epitaxy. CBE was first reported by Tsang in 1984.253, 254 This
technology combines the advantages of both MOCVD and MBE. CBE is performed in an
ultrahigh vacuum system. The reactants are in the form of molecular beams of reactive
gases, typically MO or hydride. The precursors used in CBD are thermally cracked so that
molecules are obtained before transporting into the growth chamber. Group III elements
are derived from the pyrolysis of the precursors on the surface of substrate. On the other
hand, by bringing in contact the precursors containing group V elements with heated
tantalum (Ta) or molybdenum (Mo) at 950 – 1200°C, decomposition will occur. Figure
2.19e shows a schematic drawing of CBE equipment. Often, the term CBE is used
interchangeably with metal-organic molecular beam epitaxy (MOMBE). However, these
two nomenclatures have a slight different in terms of the precursors used. When both
group III and group V precursors are in gaseous form, CBE is referred; whereas MOMBE
refers to the process in which the group III precursor is in gaseous form and the group V
precursor is solid.255
Laser-assisted catalytic growth. LCG, or laser ablation, uses a pulsed laser to
vaporise a solid target containing desired material and a catalyst. As a result, catalytic

49
nanoclusters are generated.70, 256-260 These nanoclusters define the size and control the
growth of the semiconductor nanowires by a vapour-liquid-solid (VLS) mechanism. In the
work of Duan et al.,259 they selected Au, silver (Ag) and copper (Cu) as catalysts to grow
GaAs nanowires. High yields of ZB GaAs nanowires were obtained. They recognised that
catalysts for LCG can be selected in the absence of detailed phase diagrams as long as
metals in which the nanowire component elements are soluble in the liquid phase but that
do not form solid compounds more stable than the desired nanowire phase were identified.
It means the ideal metal catalyst should be physical active but chemically stable. From this
standpoint, Au, which is a noble metal, should represent a good starting point for many
materials.
Figure 2.19b shows a typical LCG setup.256-258 A heated flow tube, in which the
pressure and temperature are tuned, is used. The condensation and cluster size can be
controlled by background pressure. On the other hand, catalyst cluster in the liquid state
can be kept by varying the temperature. In accordance to VLS mechanism, the liquid
catalyst alloy (consists of elements from both catalyst and growth nanowire) serves as a
preferential site for absorption of reactant. There is a much higher sticking probability on
liquid vs. liquid surface. When supersaturation reaches, nucleation site for crystallisation
will form. Preferential 1D growth happens in the presence of the reactant if the catalyst
remains liquid.
2.6 Summary
In conclusion, this chapter reviewed the structural features of III-V nanowires, in terms of
their morphology and crystal structures. Growth parameters that can be tuned in a growth
chamber were elucidated. Besides, the important of catalyst in controlling the quality of
nanowires were explained. Growth mechanism of nanowires was also reviewed. Lastly,
available common growth techniques for III-V nanowires were listed.

50
Figure 2.19. Schematic drawing of different growth techniques. (a) A typical CVD reactor.
(b) A typical LCG growth chamber.256-258 (c) A MOCVD reactor used in this thesis.119 (d) A
simple MBE growth chamber with the essential growth sources, shutters, beam flux
detector and the RHEED system for monitoring structure during growth.241 (e) A typical
CBE equipment.261
REFERENCES
1. Bierman, M. J.; Lau, Y. K. A.; Kvit, A. V.; Schmitt, A. L.; Jin, S. Science 2008, 320,
(5879), 1060-1063.
2. Huang, Y.; Duan, X.; Cui, Y.; Lauhon, L. J.; Kim, K.-H.; Lieber, C. M. Science 2001,
294, (5545), 1313-1317.
3. Li, Y.; Qian, F.; Xiang, J.; Lieber, C. M. Mater Today 2006, 9, (10), 18-27.
4. Fan, Z.; Ho, J. C.; Takahashi, T.; Yerushalmi, R.; Takei, K.; Ford, A. C.; Chueh, Y.-
L.; Javey, A. Advanced Materials 2009, 21, (37), 3730-3743.
5. Schroer, M. D.; Petta, J. R. Nano Lett 2010, 10, (5), 1618-1622.

51
6. Halpern, E.; Elias, G.; Kretinin, A. V.; Shtrikman, H.; Rosenwaks, Y. Appl Phys Lett
2012, 100, (26), 262105.
7. Fanfair, D. D.; Korgel, B. A. Cryst Growth Des 2005, 5, (5), 1971-1976.
8. Mokkapati, S.; Saxena, D.; Jiang, N.; Parkinson, P.; Wong-Leung, J.; Gao, Q.; Tan,
H. H.; Jagadish, C. Nano Lett 2012, 12, (12), 6428-6431.
9. Yan, R.; Gargas, D.; Yang, P. Nat Photon 2009, 3, (10), 569-576.
10. Bao, J.; Bell, D. C.; Capasso, F.; Wagner, J. B.; Mårtensson, T.; Trägårdh, J.;
Samuelson, L. Nano Lett 2008, 8, (3), 836-841.
11. Joyce, H. J.; Gao, Q.; Hoe Tan, H.; Jagadish, C.; Kim, Y.; Zou, J.; Smith, L. M.;
Jackson, H. E.; Yarrison-Rice, J. M.; Parkinson, P.; Johnston, M. B. Prog Quant
Electron 2011, 35, (2–3), 23-75.
12. Saxena, D.; Mokkapati, S.; Parkinson, P.; Jiang, N.; Gao, Q.; Tan, H. H.; Jagadish,
C. Nat Photon 2013, 7, (12), 963-968.
13. Xia, H.; Lu, Z.-Y.; Li, T.-X.; Parkinson, P.; Liao, Z.-M.; Liu, F.-H.; Lu, W.; Hu, W.-D.;
Chen, P.-P.; Xu, H.-Y.; Zou, J.; Jagadish, C. ACS Nano 2012, 6, (7), 6005-6013.
14. Wallentin, J.; Anttu, N.; Asoli, D.; Huffman, M.; Åberg, I.; Magnusson, M. H.; Siefer,
G.; Fuss-Kailuweit, P.; Dimroth, F.; Witzigmann, B.; Xu, H. Q.; Samuelson, L.;
Deppert, K.; Borgström, M. T. Science 2013, 339, (6123), 1057-1060.
15. Gudiksen, M. S.; Lauhon, L. J.; Wang, J.; Smith, D. C.; Lieber, C. M. Nature 2002,
415, (6872), 617-620.
16. Fan, G.; Wang, C.; Fang, J. Nano Today 2014, 9, (1), 69-84.
17. Cui, Y.; Wei, Q.; Park, H.; Lieber, C. M. Science 2001, 293, (5533), 1289-1292.
18. Ford, A.; Ho, J.; Fan, Z.; Ergen, O.; Altoe, V.; Aloni, S.; Razavi, H.; Javey, A. Nano
Res. 2008, 1, (1), 32-39.
19. Lieber, C. M.; Wang, Z. L. MRS Bulletin 2007, 32, (02), 99-108.
20. Friedman, R. S.; McAlpine, M. C.; Ricketts, D. S.; Ham, D.; Lieber, C. M. Nature
2005, 434, (7037), 1085-1085.
21. Wang, X.; Song, J.; Liu, J.; Wang, Z. L. Science 2007, 316, (5821), 102-105.
22. Xiang, J.; Lu, W.; Hu, Y.; Wu, Y.; Yan, H.; Lieber, C. M. Nature 2006, 441, (7092),
489-493.
23. McAlpine, M. C.; Ahmad, H.; Wang, D.; Heath, J. R. Nat Mater 2007, 6, (5), 379-
384.
24. Bryllert, T.; Wernersson, L. E.; Froberg, L. E.; Samuelson, L. IEEE Electr Device L
2006, 27, (5), 323-325.

52
25. Lin, Y.-M.; Sun, X.; Dresselhaus, M. S. Physical Review B 2000, 62, (7), 4610-
4623.
26. Lin, Y.-M.; Dresselhaus, M. S. Physical Review B 2003, 68, (7), 075304.
27. Jin, Z. Q.; Ding, Y.; Wang, Z. L. J Appl Phys 2005, 97, (7), 074309.
28. Yerushalmi, R.; Jacobson, Z. A.; Ho, J. C.; Fan, Z.; Javey, A. Appl Phys Lett 2007,
91, (20), 203104.
29. Javey, A.; Nam; Friedman, R. S.; Yan, H.; Lieber, C. M. Nano Lett 2007, 7, (3), 773-
777.
30. Fan, Z.; Ho, J. C.; Jacobson, Z. A.; Yerushalmi, R.; Alley, R. L.; Razavi, H.; Javey,
A. Nano Lett 2008, 8, (1), 20-25.
31. Xu, H.; Wang, Y.; Guo, Y.; Liao, Z.; Gao, Q.; Tan, H. H.; Jagadish, C.; Zou, J. Nano
Lett 2012, 12, (11), 5744-5749.
32. Huang, M. H.; Mao, S.; Feick, H.; Yan, H.; Wu, Y.; Kind, H.; Weber, E.; Russo, R.;
Yang, P. Science 2001, 292, (5523), 1897-1899.
33. Tomioka, K.; Tanaka, T.; Hara, S.; Hiruma, K.; Fukui, T. IEEE Journal of Selected
Topics in Quantum Electronics 2011, 17, (4), 1112-1129.
34. Liu, Z.; Luo, T.; Liang, B.; Chen, G.; Yu, G.; Xie, X.; Chen, D.; Shen, G. Nano Res.
2013, 6, (11), 775-783.
35. Miao, J.; Hu, W.; Guo, N.; Lu, Z.; Zou, X.; Liao, L.; Shi, S.; Chen, P.; Fan, Z.; Ho, J.
C.; Li, T.-X.; Chen, X. S.; Lu, W. ACS Nano 2014, 8, (4), 3628-3635.
36. Nah, J.; Fang, H.; Wang, C.; Takei, K.; Lee, M. H.; Plis, E.; Krishna, S.; Javey, A.
Nano Lett 2012, 12, (7), 3592-3595.
37. Offermans, P.; Crego-Calama, M.; Brongersma, S. H. Nano Lett 2010, 10, (7),
2412-2415.
38. Fasth, C.; Fuhrer, A.; Samuelson, L.; Golovach, V. N.; Loss, D. Physical Review
Letters 2007, 98, (26), 266801.
39. Doh, Y.-J.; van Dam, J. A.; Roest, A. L.; Bakkers, E. P. A. M.; Kouwenhoven, L. P.;
De Franceschi, S. Science 2005, 309, (5732), 272-275.
40. Sand-Jespersen, T.; Paaske, J.; Andersen, B. M.; Grove-Rasmussen, K.;
Jørgensen, H. I.; Aagesen, M.; Sørensen, C. B.; Lindelof, P. E.; Flensberg, K.;
Nygård, J. Physical Review Letters 2007, 99, (12), 126603.
41. Fuhrer, A.; Fasth, C.; Samuelson, L. Appl Phys Lett 2007, 91, (5), 052109.
42. Wei, W.; Bao, X.-Y.; Soci, C.; Ding, Y.; Wang, Z.-L.; Wang, D. Nano Lett 2009, 9,
(8), 2926-2934.

53
43. Pan, D.; Fu, M.; Yu, X.; Wang, X.; Zhu, L.; Nie, S.; Wang, S.; Chen, Q.; Xiong, P.;
von Molnár, S.; Zhao, J. Nano Lett 2014, 14, (3), 1214-1220.
44. Tomioka, K.; Yoshimura, M.; Fukui, T. Nature 2012, 488, (7410), 189-192.
45. Tseng, Y.-C.; Xuan, P.; Javey, A.; Malloy, R.; Wang, Q.; Bokor, J.; Dai, H. Nano
Lett 2004, 4, (1), 123-127.
46. Woodall, J. M. Science 1980, 208, (4446), 908-915.
47. Yoon, J.; Jo, S.; Chun, I. S.; Jung, I.; Kim, H.-S.; Meitl, M.; Menard, E.; Li, X.;
Coleman, J. J.; Paik, U.; Rogers, J. A. Nature 2010, 465, (7296), 329-333.
48. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Fickenscher, M. A.; Perera,
S.; Hoang, T. B.; Smith, L. M.; Jackson, H. E.; Yarrison-Rice, J. M.; Zhang, X.; Zou,
J. Adv Funct Mater 2008, 18, (23), 3794-3800.
49. Parkinson, P.; Lloyd-Hughes, J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Johnston, M. B.;
Herz, L. M. Nano Lett 2007, 7, (7), 2162-2165.
50. Lind, E.; Persson, A. I.; Samuelson, L.; Wernersson, L.-E. Nano Lett 2006, 6, (9),
1842-1846.
51. Dayeh, S. A.; Aplin, D. P. R.; Zhou, X.; Yu, P. K. L.; Yu, E. T.; Wang, D. Small 2007,
3, (2), 326-332.
52. Bleszynski, A. C.; Zwanenburg, F. A.; Westervelt, R. M.; Roest, A. L.; Bakkers, E.
P. A. M.; Kouwenhoven, L. P. Nano Lett 2007, 7, (9), 2559-2562.
53. Guo, Y.-N.; Xu, H.-Y.; Auchterlonie, G. J.; Burgess, T.; Joyce, H. J.; Gao, Q.; Tan,
H. H.; Jagadish, C.; Shu, H.-B.; Chen, X.-S.; Lu, W.; Kim, Y.; Zou, J. Nano Lett
2013, 13, (2), 643-650.
54. Paladugu, M.; Zou, J.; Guo, Y.-N.; Zhang, X.; Kim, Y.; Joyce, H. J.; Gao, Q.; Tan, H.
H.; Jagadish, C. Appl Phys Lett 2008, 93, (10), 101911.
55. Paiman, S.; Gao, Q.; Tan, H. H.; Jagadish, C.; Pemasiri, K.; Montazeri, M.;
Jackson, H. E.; Smith, L. M.; Yarrison-Rice, J. M.; Zhang, X.; Zou, J.
Nanotechnology 2009, 20, (22), 225606.
56. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Fickenscher, M. A.; Perera,
S.; Hoang, T. B.; Smith, L. M.; Jackson, H. E.; Yarrison-Rice, J. M.; Zhang, X.; Zou,
J. Nano Lett 2009, 9, (2), 695-701.
57. Dick, K. A.; Caroff, P.; Bolinsson, J.; Messing, M. E.; Johansson, J.; Deppert, K.;
Wallenberg, L. R.; Samuelson, L. Semiconductor Science and Technology 2010,
25, (2), 024009.
58. Caroff, P.; Dick, K. A.; JohanssonJ; Messing, M. E.; DeppertK; SamuelsonL. Nat
Nano 2009, 4, (1), 50-55.

54
59. Fonseka, H. A.; Caroff, P.; Wong-Leung, J.; Ameruddin, A. S.; Tan, H. H.; Jagadish,
C. ACS Nano 2014.
60. Heo, H.; Kang, K.; Lee, D.; Jin, L.-H.; Back, H.-J.; Hwang, I.; Kim, M.; Lee, H.-S.;
Lee, B.-J.; Yi, G.-C.; Cho, Y.-H.; Jo, M.-H. Nano Lett 2012, 12, (2), 855-860.
61. Kratzer, P.; Sakong, S.; Pankoke, V. Nano Lett 2012, 12, (2), 943-948.
62. Shao, Y. M.; Nie, T. X.; Jiang, Z. M.; Zou, J. Appl Phys Lett 2012, 101, (5), 053104.
63. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Zhang, X.; Guo, Y.; Zou, J.
Nano Lett 2007, 7, (4), 921-926.
64. Dayeh, S. A.; Yu, E. T.; Wang, D. Nano Lett 2007, 7, (8), 2486-2490.
65. Liao, Z.-M.; Chen, Z.-G.; Lu, Z.-Y.; Xu, H.-Y.; Guo, Y.-N.; Sun, W.; Zhang, Z.; Yang,
L.; Chen, P.-P.; Lu, W.; Zou, J. Appl Phys Lett 2013, 102, (6), 063106.
66. Thelander, C.; Caroff, P.; Plissard, S. b.; Dey, A. W.; Dick, K. A. Nano Lett 2011,
11, (6), 2424-2429.
67. Persson, A. I.; Ohlsson, B. J.; Jeppesen, S.; Samuelson, L. Journal of Crystal
Growth 2004, 272, (1–4), 167-174.
68. Vogel, A. T.; de Boor, J.; Becker, M.; Wittemann, J. V.; Mensah, S. L.; Werner, P.;
Schmidt, V. Nanotechnology 2011, 22, (1), 015605.
69. Ryan, A. M.; Mark, A. R., Chemical Beam Epitaxy of Gallium Nitride Nanowires. In
The Science and Function of Nanomaterials: From Synthesis to Application,
American Chemical Society: 2014; Vol. 1183, pp 13-39.
70. Duan, X.; Lieber, C. M. Journal of the American Chemical Society 2000, 122, (1),
188-189.
71. Borgström, M. Epitaxial Growth, Processing and Characterization of Semiconductor
Nanostructures. Lund University, 2003.
72. Ramsdell, L. S. Am Mineral 1947, 32, (1), 64-82.
73. Joyce, H. J.; Wong-Leung, J.; Gao, Q.; Tan, H. H.; Jagadish, C. Nano Lett 2010,
10, (3), 908-915.
74. Glas, F.; Harmand, J.-C.; Patriarche, G. Physical Review Letters 2007, 99, (14),
146101.
75. Johansson, J.; Karlsson, L. S.; Svensson, C. P. T.; Mårtensson, T.; Wacaser, B. A.;
Deppert, K.; Samuelson, L.; Seifert, W. Nature Materials 2006, 5, (7), 574-580.
76. Zou, J.; Paladugu, M.; Wang, H.; Auchterlonie, G. J.; Guo, Y.-N.; Kim, Y.; Gao, Q.;
Joyce, H. J.; Tan, H. H.; Jagadish, C. Small 2007, 3, (3), 389-393.
77. Mariager, S. O.; Sørensen, C. B.; Aagesen, M.; Nygård, J.; Feidenhans’l, R.;
Willmott, P. R. Appl Phys Lett 2007, 91, (8), 083106.

55
78. Dheeraj, D. L.; Patriarche, G.; Zhou, H.; Hoang, T. B.; Moses, A. F.; Grønsberg, S.;
van Helvoort, A. T. J.; Fimland, B.-O.; Weman, H. Nano Lett 2008, 8, (12), 4459-
4463.
79. Soshnikov, I. P.; Cirlin, G. E.; Sibirev, N. V.; Dubrovskii, V. G.; Samsonenko, Y. B.;
Litvinov, D.; Gerthsen, D. Tech. Phys. Lett. 2008, 34, (6), 538-541.
80. Kriegner, D.; Panse, C.; Mandl, B.; Dick, K. A.; Keplinger, M.; Persson, J. M.;
Caroff, P.; Ercolani, D.; Sorba, L.; Bechstedt, F.; Stangl, J.; Bauer, G. Nano Lett
2011, 11, (4), 1483-1489.
81. Johansson, J.; Bolinsson, J.; Ek, M.; Caroff, P.; Dick, K. A. ACS Nano 2012, 6, (7),
6142-6149.
82. Hillerich, K. Influence of Seed Particle Material, Preparation, and Dynamics on
Nanowire Growth. PhD, Lund University, Sweden, 2013.
83. Caroff, P.; Bolinsson, J.; Johansson, J. IEEE Journal of Selected Topics in
Quantum Electronics 2011, 17, (4), 829-846.
84. Stiles, M. D.; Hamann, D. R. Physical Review B 1990, 41, (8), 5280-5282.
85. Stiles, M. D.; Hamann, D. R. Physical Review B 1988, 38, (3), 2021-2037.
86. Dick, K. A.; Thelander, C.; Samuelson, L.; Caroff, P. Nano Lett 2010, 10, (9), 3494-
3499.
87. Mohseni, P. K.; LaPierre, R. R. Nanotechnology 2009, 20, (2), 025610.
88. Krogstrup, P.; Curiotto, S.; Johnson, E.; Aagesen, M.; Nygård, J.; Chatain, D.
Physical Review Letters 2011, 106, (12), 125505.
89. Johansson, J.; Karlsson, L. S.; Dick, K. A.; Bolinsson, J.; Wacaser, B. A.; Deppert,
K.; Samuelson, L. Cryst Growth Des 2009, 9, (2), 766-773.
90. Jacobsson, D.; Panciera, F.; Tersoff, J.; Reuter, M. C.; Lehmann, S.; Hofmann, S.;
Dick, K. A.; Ross, F. M. Nature 2016, 531, (7594), 317-322.
91. Algra, R. E. Twinning and morphology of III-V nanowires. Radboud Universiteit
Nijmegen, Amsterdam, 2010.
92. Davidson, F. M.; Lee, D. C.; Fanfair, D. D.; Korgel, B. A. J Phys Chem C 2007, 111,
(7), 2929-2935.
93. Spirkoska, D.; Arbiol, J.; Gustafsson, A.; Conesa-Boj, S.; Glas, F.; Zardo, I.;
Heigoldt, M.; Gass, M. H.; Bleloch, A. L.; Estrade, S.; Kaniber, M.; Rossler, J.;
Peiro, F.; Morante, J. R.; Abstreiter, G.; Samuelson, L.; Fontcuberta i Morral, A.
Physical Review B 2009, 80, (24), 245325.
94. Cohen, D.; McKernan, S.; Carter, C. B. Microscopy and Microanalysis 1999, 5, (03),
173-186.

56
95. Ross, F. M.; Tersoff, J.; Reuter, M. C. Physical Review Letters 2005, 95, (14),
146104.
96. Xu, T.; Nys, J. P.; Addad, A.; Lebedev, O. I.; Urbieta, A.; Salhi, B.; Berthe, M.;
Grandidier, B.; Stiévenard, D. Physical Review B 2010, 81, (11), 115403.
97. Sansoz, F. Nano Lett 2011, 11, (12), 5378-5382.
98. Dayeh, S. A.; Soci, C.; Bao, X.-Y.; Wang, D. Nano Today 2009, 4, (4), 347-358.
99. Rosini, M.; Magri, R. ACS Nano 2010, 4, (10), 6021-6031.
100. Pankoke, V.; Kratzer, P.; Sakong, S. Physical Review B 2011, 84, (7), 075455.
101. Cahn, J. W.; Hanneman, R. E. Surface Science 1964, 1, (4), 387-398.
102. Sibirev, N. V.; Timofeeva, M. A.; Bol’shakov, A. D.; Nazarenko, M. V.; Dubrovskiĭ,
V. G. Phys. Solid State 2010, 52, (7), 1531-1538.
103. Verheijen, M. A.; Algra, R. E.; Borgström, M. T.; Immink, G.; Sourty, E.; van
Enckevort, W. J. P.; Vlieg, E.; Bakkers, E. P. A. M. Nano Lett 2007, 7, (10), 3051-
3055.
104. Burgess, T.; Breuer, S.; Caroff, P.; Wong-Leung, J.; Gao, Q.; Hoe Tan, H.;
Jagadish, C. ACS Nano 2013, 7, (9), 8105-8114.
105. Nian, J.; Wong-Leung, J.; Qiang, G.; Hark Hoe, T.; Jagadish, C. In Sidewall
evolution in VLS grown GaAs nanowires, 2014 Conference on Optoelectronic and
Microelectronic Materials & Devices (COMMAD), 14-17 Dec. 2014, 2014; pp 87-89.
106. Johansson, J.; Svensson, C. P. T.; Mårtensson, T.; Samuelson, L.; Seifert, W. The
Journal of Physical Chemistry B 2005, 109, (28), 13567-13571.
107. Leitsmann, R.; Bechstedt, F. J Appl Phys 2007, 102, (6), -.
108. Wang, G. T.; Talin, A. A.; Werder, D., J.; Creighton, J. R.; Lai, E.; Anderson, R., J.;
Arslan, I. Nanotechnology 2006, 17, (23), 5773.
109. Givargizov, E. I.; Sheftal, N. N. Journal of Crystal Growth 1971, 9, (0), 326-329.
110. Cui, Y.; Lauhon, L. J.; Gudiksen, M. S.; Wang, J.; Lieber, C. M. Appl Phys Lett
2001, 78, (15), 2214-2216.
111. Schmidt, V.; Senz, S.; Gösele, U. Nano Lett 2005, 5, (5), 931-935.
112. Wang, D.; Dai, H. Angewandte Chemie 2002, 114, (24), 4977-4980.
113. Li, C. P.; Lee, C. S.; Ma, X. L.; Wang, N.; Zhang, R. Q.; Lee, S. T. Advanced
materials 2003, 15, (7 ‐8), 607-609.
114. Wu, Y.; Cui, Y.; Huynh, L.; Barrelet, C. J.; Bell, D. C.; Lieber, C. M. Nano Lett 2004,
4, (3), 433-436.
115. Ozaki, N.; Ohno, Y.; Takeda, S. Appl Phys Lett 1998, 73, (25), 3700-3702.
116. Sharma, S.; Sunkara, M. K. Nanotechnology 2004, 15, (1), 130.

57
117. Lew, K.-K.; Reuther, C.; Carim, A. H.; Redwing, J. M.; Martin, B. R. Journal of
Vacuum Science & Technology B 2002, 20, (1), 389-392.
118. Eaglesham, D. J.; White, A. E.; Feldman, L. C.; Moriya, N.; Jacobson, D. C.
Physical Review Letters 1993, 70, (11), 1643-1646.
119. Joyce, H. J. Growth and Characterisation of III-V Semiconductor Nanowires for
Optoelectronic Device Applications. The Australian National University, Canberra,
2009.
120. Burke, R. A.; Lamborn, D. R.; Weng, X.; Redwing, J. M. Journal of Crystal Growth
2009, 311, (13), 3409-3416.
121. Chuang, L. C.; Moewe, M.; Crankshaw, S.; Chang-Hasnain, C. Appl Phys Lett
2008, 92, (1), 013121.
122. Reep, D. H.; Ghandhi, S. K. Journal of The Electrochemical Society 1983, 130, (3),
675-680.
123. Borgström, M.; Deppert, K.; Samuelson, L.; Seifert, W. Journal of Crystal Growth
2004, 260, (1–2), 18-22.
124. Biegelsen, D. K.; Bringans, R. D.; Northrup, J. E.; Swartz, L. E. Physical Review
Letters 1990, 65, (4), 452-455.
125. Fortuna, S. A.; Li, X. Semiconductor Science and Technology 2010, 25, (2),
024005.
126. Krishnamachari, U.; Borgstrom, M.; Ohlsson, B. J.; Panev, N.; Samuelson, L.;
Seifert, W.; Larsson, M. W.; Wallenberg, L. R. Appl Phys Lett 2004, 85, (11), 2077-
2079.
127. Björk, M. T.; Ohlsson, B. J.; Sass, T.; Persson, A. I.; Thelander, C.; Magnusson, M.
H.; Deppert, K.; Wallenberg, L. R.; Samuelson, L. Appl Phys Lett 2002, 80, (6),
1058-1060.
128. Mårtensson, T.; Svensson, C. P. T.; Wacaser, B. A.; Larsson, M. W.; Seifert, W.;
Deppert, K.; Gustafsson, A.; Wallenberg, L. R.; Samuelson, L. Nano Lett 2004, 4,
(10), 1987-1990.
129. Wacaser, B. A.; Dick, K. A.; Johansson, J.; Borgström, M. T.; Deppert, K.;
Samuelson, L. Advanced Materials 2009, 21, (2), 153-165.
130. Wen, C. Y.; Tersoff, J.; Reuter, M. C.; Stach, E. A.; Ross, F. M. Physical Review
Letters 2010, 105, (19), 195502.
131. Bakkers, E. P. A. M.; van Dam, J. A.; De Franceschi, S.; Kouwenhoven, L. P.;
Kaiser, M.; Verheijen, M.; Wondergem, H.; van der Sluis, P. Nat Mater 2004, 3,
(11), 769-773.

58
132. ccelli, .; Arbiol, J.; Magen, C.; rogstrup, .; Russo-Averchi, .; Heiss, M.;
Mugny, .; Morier- enoud, F. o.; Nygård, J.; Morante, J. R.; Fontcuberta i Morral,
A. Nano Lett 2011, 11, (9), 3827-3832.
133. Kolasinski, K. W. Current Opinion in Solid State and Materials Science 2006, 10,
(3–4), 182-191.
134. De Yoreo, J. J.; Vekilov, P. G. Reviews in mineralogy and geochemistry 2003, 54,
(1), 57-93.
135. Meyyappan, M.; Sunkara, M. K., Inorganic nanowires: applications, properties, and
characterization. CRC Press: 2009.
136. Gibbs, J. W. American Journal of Science 1878, (96), 441-458.
137. Tizei, L. H. G.; Chiaramonte, T.; Ugarte, D.; Cotta, M. A. Nanotechnology 2009, 20,
(27), 275604.
138. Brenner, M. P.; Lohse, D. Physical Review Letters 2008, 101, (21), 214505.
139. Kameda, N.; Nakabayashi, S. Chemical Physics Letters 2008, 461, (1–3), 122-126.
140. Chandrasekaran, H.; Sumanasekara, G. U.; Sunkara, M. K. The Journal of Physical
Chemistry B 2006, 110, (37), 18351-18357.
141. Jang, J.-M.; Kim, C.-R.; Ryu, H.; Razeghi, M.; Jung, W.-G. Journal of Alloys and
Compounds 2008, 463, (1–2), 503-510.
142. Hiruma, K.; Yazawa, M.; Haraguchi, K.; Ogawa, K.; Katsuyama, T.; Koguchi, M.;
Kakibayashi, H. J Appl Phys 1993, 74, (5), 3162-3171.
143. Nebol'sin, V. A.; Shchetinin, A. A. Inorganic Materials 2003, 39, (9), 899-903.
144. Dick, K. A.; Deppert, K.; Karlsson, L. S.; Wallenberg, L. R.; Samuelson, L.; Seifert,
W. Adv Funct Mater 2005, 15, (10), 1603-1610.
145. Mohammad, S. N. Nano Lett 2008, 8, (5), 1532-1538.
146. Clark, T. E.; Nimmatoori, P.; Lew, K.-K.; Pan, L.; Redwing, J. M.; Dickey, E. C.
Nano Lett 2008, 8, (4), 1246-1252.
147. Chung, H.-S.; Jung, Y.; Kim, S. C.; Kim, D. H.; Oh, K. H.; Agarwal, R. Nano Lett
2009, 9, (6), 2395-2401.
148. Pompe, T.; Herminghaus, S. Physical Review Letters 2000, 85, (9), 1930-1933.
149. Wagner, R. S.; Ooherty, C. J. Journal of The Electrochemical Society 1968, 115,
(1), 93-99.
150. Givargizov, E. I. Journal of Crystal Growth 1973, 20, (3), 217-226.
151. Gaydos, J.; Neumann, A. W. Journal of Colloid and Interface Science 1987, 120,
(1), 76-86.
152. Li, D.; Neumann, A. W. Colloids and Surfaces 1990, 43, (2), 195-206.

59
153. Drelich, J.; Miller, J. D.; Hupka, J. Journal of Colloid and Interface Science 1993,
155, (2), 379-385.
154. Kim, Y.; Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Paladugu, M.; Zou, J.;
Suvorova, A. A. Nano Lett 2006, 6, (4), 599-604.
155. Seifert, W.; Borgström, M.; Deppert, K.; Dick, K. A.; Johansson, J.; Larsson, M. W.;
Mårtensson, T.; Sköld, N.; Patrik T. Svensson, C.; Wacaser, B. A.; Reine
Wallenberg, L.; Samuelson, L. Journal of Crystal Growth 2004, 272, (1–4), 211-220.
156. Tchernycheva, M.; Travers, L.; Patriarche, G.; Glas, F.; Harmand, J.-C.; Cirlin, G.
E.; Dubrovskii, V. G. J Appl Phys 2007, 102, (9), 094313.
157. Dubrovskii, V. G.; Sibirev, N. V.; Harmand, J. C.; Glas, F. Physical Review B 2008,
78, (23), 235301.
158. Mohammad, S. N. J Appl Phys 2009, 106, (10), 104311.
159. Jong, E. D.; LaPierre, R. R.; Wen, J. Z. Nanotechnology 2010, 21, (4), 045602.
160. Wallentin, J.; k, M.; Wallenberg, . R.; Samuelson, .; Deppert, .; Borgstr m, M.
T. Nano Lett 2010, 10, (12), 4807-4812.
161. Holmes, J. D.; Johnston, K. P.; Doty, R. C.; Korgel, B. A. Science 2000, 287,
(5457), 1471-1473.
162. Wang, N.; Cai, Y.; Zhang, R. Q. Materials Science and Engineering: R: Reports
2008, 60, (1–6), 1-51.
163. Lenrick, F.; Ek, M.; Deppert, K.; Samuelson, L.; Reine Wallenberg, L. Nano Res.
2014, 7, (8), 1188-1194.
164. Tian, B.; Xie, P.; Kempa, T. J.; Bell, D. C.; Lieber, C. M. Nat Nano 2009, 4, (12),
824-829.
165. Schwarz, K. W.; Tersoff, J. Nano Lett 2012, 12, (3), 1329-1332.
166. Cai, Y.; Chan, S. K.; Sou, I. K.; Chan, Y. F.; Su, D. S.; Wang, N. Advanced
Materials 2006, 18, (1), 109-114.
167. Heun, S.; Radha, B.; Ercolani, D.; Kulkarni, G. U.; Rossi, F.; Grillo, V.; Salviati, G.;
Beltram, F.; Sorba, L. Cryst Growth Des 2010, 10, (9), 4197-4202.
168. Kuykendall, T.; Pauzauskie, P.; Lee, S.; Zhang, Y.; Goldberger, J.; Yang, P. Nano
Lett 2003, 3, (8), 1063-1066.
169. Su, J.; Cui, G.; Gherasimova, M.; Tsukamoto, H.; Han, J.; Ciuparu, D.; Lim, S.;
Pfefferle, L.; He, Y.; Nurmikko, A. V.; Broadbridge, C.; Lehman, A. Appl Phys Lett
2005, 86, (1), 013105.

60
170. Martelli, F.; Rubini, S.; Piccin, M.; Bais, G.; Jabeen, F.; De Franceschi, S.; Grillo, V.;
Carlino, E.; D'Acapito, F.; Boscherini, F.; Cabrini, S.; Lazzarino, M.; Businaro, L.;
Romanato, F.; Franciosi, A. Nano Lett 2006, 6, (9), 2130-2134.
171. Yoo, J.; Hong, Y.-J.; An, S. J.; Yi, G.-C.; Chon, B.; Joo, T.; Kim, J.-W.; Lee, J.-S.
Appl Phys Lett 2006, 89, (4), 043124.
172. Tham, D.; Nam, C. Y.; Fischer, J. E. Adv Funct Mater 2006, 16, (9), 1197-1202.
173. Regolin, I.; Khorenko, V.; Prost, W.; Tegude, F. J.; Sudfeld, D.; Kästner, J.;
Dumpich, G.; Hitzbleck, K.; Wiggers, H. J Appl Phys 2007, 101, (5), 054318.
174. Li, Q.; Wang, G. T. Appl Phys Lett 2008, 93, (4), 043119.
175. Boles, S. T.; Thompson, C. V.; Fitzgerald, E. A. Journal of Crystal Growth 2009,
311, (5), 1446-1450.
176. Weng, X.; Burke, R. A.; Redwing, J. M. Nanotechnology 2009, 20, (8), 085610.
177. Jabeen, F.; Piccin, M.; Felisari, L.; Grillo, V.; Bais, G.; Rubini, S.; Martelli, F.;
d’Acapito, F.; Rovezzi, M.; Boscherini, F. Journal of Vacuum Science &
Technology B 2010, 28, (3), 478-483.
178. Heun, S.; Radha, B.; Ercolani, D.; Kulkarni, G. U.; Rossi, F.; Grillo, V.; Salviati, G.;
Beltram, F.; Sorba, L. Small 2010, 6, (17), 1935-1941.
179. Hillerich, K.; Messing, M. E.; Reine Wallenberg, L.; Deppert, K.; Dick, K. A. Journal
of Crystal Growth 2011, 315, (1), 134-137.
180. Wen, C. Y.; Tersoff, J.; Hillerich, K.; Reuter, M. C.; Park, J. H.; Kodambaka, S.;
Stach, E. A.; Ross, F. M. Physical Review Letters 2011, 107, (2), 025503.
181. Han, N.; Wang, F.; Hui, A. T.; Hou, J. J.; Shan, G.; Xiu, F.; Hung, T.; Ho, J. C.
Nanotechnology 2011, 22, (28), 285607.
182. Han, N.; Hui, A. T.; Wang, F.; Hou, J. J.; Xiu, F.; Hung, T.; Ho, J. C. Appl Phys Lett
2011, 99, (8), 083114.
183. Hillerich, K.; Dick, K. A.; Messing, M. E.; Deppert, K.; Johansson, J. Nano Res.
2012, 5, (5), 297-306.
184. Xu, H.-Y.; Guo, Y.-N.; Liao, Z.-M.; Sun, W.; Gao, Q.; Hoe Tan, H.; Jagadish, C.;
Zou, J. Appl Phys Lett 2013, 102, (20), 203108.
185. Hillerich, K.; Ghidini, D. S.; Dick, K. A.; Deppert, K.; Johansson, J. physica status
solidi (RRL) – Rapid Research Letters 2013, 7, (10), 850-854.
186. Purushothaman, V.; Jeganathan, K. J Nanopart Res 2013, 15, (7), 1-12.
187. Perumal, R.; Zhixin, C.; Patniarche, G.; Jean-Christophe, H.; Kanji, Y.
Semiconductor Science and Technology 2014, 29, (11), 115005.

61
188. Purushothaman, V.; Venkatesh, P. S.; Navamathavan, R.; Jeganathan, K. RSC
Advances 2014, 4, (85), 45100-45108.
189. Purushothaman, V.; Prabhu, S.; Jothivenkatachalam, K.; Parthiban, S.; Kwon, J. Y.;
Jeganathan, K. RSC Advances 2014, 4, (49), 25569-25575.
190. Sun, R.; Jacobsson, D.; Chen, I. J.; Nilsson, M.; Thelander, C.; Lehmann, S.; Dick,
K. A. Nano Lett 2015.
191. Hallberg, R. T.; Lehmann, S.; Messing, M. E.; Dick, K. A. Journal of Materials
Research 2016, 31, (02), 175-185.
192. Lindberg, C.; Whiticar, A.; Dick, K. A.; Sköld, N.; Nygård, J.; Bolinsson, J. Nano Lett
2016, 16, (4), 2181-2188.
193. Wagner, R. S.; Ellis, W. C. Appl Phys Lett 1964, 4, (5), 89-90.
194. Wagner, R. S.; Ellis, W. C.; Jackson, K. A.; Arnold, S. M. J Appl Phys 1964, 35,
(10), 2993-3000.
195. Wagner, R. S.; Ellis, W. C. Trans. Met. Soc. 1965, 233, 1053.
196. Barns, R. L.; Ellis, W. C. J Appl Phys 1965, 36, (7), 2296-2301.
197. James, D. W. F.; Lewis, C. British Journal of Applied Physics 1965, 16, (8), 1089.
198. Wagner, R. S.; Doherty, C. J. Journal of The Electrochemical Society 1966, 113,
(12), 1300-1305.
199. Baker, R. T. K.; Barber, M. A.; Harris, P. S.; Feates, F. S.; Waite, R. J. Journal of
Catalysis 1972, 26, (1), 51-62.
200. Baker, R. T. K.; Harris, P. S.; Thomas, R. B.; Waite, R. J. Journal of Catalysis 1973,
30, (1), 86-95.
201. Baker, R. T. K. Carbon 1989, 27, (3), 315-323.
202. Trentler, T. J.; Hickman, K. M.; Goel, S. C.; Viano, A. M.; Gibbons, P. C.; Buhro, W.
E. Science 1995, 270, (5243), 1791-1794.
203. Markowitz, P. D.; Zach, M. P.; Gibbons, P. C.; Penner, R. M.; Buhro, W. E. Journal
of the American Chemical Society 2001, 123, (19), 4502-4511.
204. Yu, H.; Buhro, W. E. Advanced Materials 2003, 15, (5), 416-419.
205. Dick, K. A.; Deppert, K.; Mårtensson, T.; Mandl, B.; Samuelson, L.; Seifert, W. Nano
Lett 2005, 5, (4), 761-764.
206. Wang, F.; Dong, A.; Sun, J.; Tang, R.; Yu, H.; Buhro, W. E. Inorg Chem 2006, 45,
(19), 7511-7521.
207. Persson, A. I.; Larsson, M. W.; Stenstrom, S.; Ohlsson, B. J.; Samuelson, L.;
Wallenberg, L. R. Nat Mater 2004, 3, (10), 677-681.

62
208. Kodambaka, S.; Tersoff, J.; Reuter, M. C.; Ross, F. M. Science 2007, 316, (5825),
729-732.
209. Cheyssac, P.; Sacilotti, M.; Patriarche, G. J Appl Phys 2006, 100, (4), 044315.
210. Lee, S. T.; Wang, N.; Zhang, Y. F.; Tang, Y. H. MRS Bulletin 1999, 24, (08), 36-42.
211. Shi, W. S.; Zheng, Y. F.; Wang, N.; Lee, C. S.; Lee, S. T. Chemical Physics Letters
2001, 345, (5–6), 377-380.
212. Zhang, R. Q.; Lifshitz, Y.; Lee, S. T. Advanced Materials 2003, 15, (7-8), 635-640.
213. Dick, K. A. Progress in Crystal Growth and Characterization of Materials 2008, 54,
(3–4), 138-173.
214. Dubrovskii, V. G.; Sibirev, N. V.; Cirlin, G. E. Tech. Phys. Lett. 2004, 30, (8), 682-
686.
215. Dubrovskii, V. G.; Sibirev, N. V. Physical Review E 2004, 70, (3), 031604.
216. Chen, Z.; Cao, C. Appl Phys Lett 2006, 88, (14), 143118.
217. Dayeh, S. A.; Picraux, S. T. Nano Lett 2010, 10, (10), 4032-4039.
218. Plissard, S.; Larrieu, G.; Wallart, X.; Caroff, P. Nanotechnology 2011, 22, (27),
275602.
219. Korgel, B. A. Nat Mater 2006, 5, (7), 521-522.
220. Schwarz, K. W.; Tersoff, J. Nano Lett 2011, 11, (2), 316-320.
221. Wang, Y.; Schmidt, V.; Senz, S.; Gosele, U. Nat Nano 2006, 1, (3), 186-189.
222. Buffat, P.; Borel, J. P. Physical Review A 1976, 13, (6), 2287-2298.
223. Campos, L. C.; Tonezzer, M.; Ferlauto, A. S.; Grillo, V.; Magalhães-Paniago, R.;
Oliveira, S.; Ladeira, L. O.; Lacerda, R. G. Advanced Materials 2008, 20, (8), 1499-
1504.
224. Shvartsman, F.; Krongauz, V. Nature 1984, 309, (5969), 608-611.
225. Noor Mohammad, S. The Journal of Chemical Physics 2009, 131, (22), 224702.
226. Ghaisas, S. V.; Das Sarma, S. Physical Review B 1992, 46, (11), 7308-7311.
227. Mohammad, S. N. Journal of Vacuum Science & Technology B 2010, 28, (2),
329-352.
228. Wen, C. Y.; Reuter, M. C.; Tersoff, J.; Stach, E. A.; Ross, F. M. Nano Lett 2010, 10,
(2), 514-519.
229. Brenner, S. S.; Sears, G. W. Acta Metall 1956, 4, (3), 268-270.
230. Zhang, H. Z.; Wang, R. M.; You, L. P.; Yu, J.; Chen, H.; Yu, D. P.; Chen, Y. New
Journal of Physics 2007, 9, (1), 13.
231. Hertenberger, S.; Rudolph, D.; Bolte, S.; Döblinger, M.; Bichler, M.; Spirkoska, D.;
Finley, J. J.; Abstreiter, G.; Koblmüller, G. Appl Phys Lett 2011, 98, (12), 123114.

63
232. Dimakis, .; hnemann, J.; Jahn, .; Breuer, S.; Hilse, M.; eelhaar, .; Riechert,
H. Cryst Growth Des 2011, 11, (9), 4001-4008.
233. Conesa-Boj, S.; Russo-Averchi, E.; Dalmau-Mallorqui, A.; Trevino, J.; Pecora, E. F.;
Forestiere, C.; Handin, A.; Ek, M.; Zweifel, L.; Wallenberg, L. R.; Rüffer, D.; Heiss,
M.; Troadec, D.; Dal Negro, L.; Caroff, P.; Fontcuberta i Morral, A. ACS Nano 2012,
6, (12), 10982-10991.
234. Arthur, J. R. J Appl Phys 1968, 39, (8), 4032-4034.
235. Arthur, J. R.; LePore, J. J. Journal of Vacuum Science & Technology 1969, 6, (4),
545-548.
236. Cho, A. Y. J Appl Phys 1970, 41, (7), 2780-2786.
237. Cho, A. Y. J Appl Phys 1971, 42, (5), 2074-2081.
238. Cho, A. Y.; Hayashi, I. MT 1971, 2, (3), 777-780.
239. Cho, A. Y.; Casey, H. C. Appl Phys Lett 1974, 25, (5), 288-290.
240. Cho, A. Y.; Arthur, J. R. Progress in Solid State Chemistry 1975, 10, Part 3, (0),
157-191.
241. Arthur, J. R. Surface Science 2002, 500, (1–3), 189-217.
242. Givargizov, E. I. Journal of Crystal Growth 1975, 31, (0), 20-30.
243. Chen, C.-C.; Yeh, C.-C.; Chen, C.-H.; Yu, M.-Y.; Liu, H.-L.; Wu, J.-J.; Chen, K.-H.;
Chen, L.-C.; Peng, J.-Y.; Chen, Y.-F. Journal of the American Chemical Society
2001, 123, (12), 2791-2798.
244. Yu, G.; Cao, A.; Lieber, C. M. Nat Nano 2007, 2, (6), 372-377.
245. Chen, J.; Xue, C.; Zhuang, H.; Qin, L.; Li, H.; Yang, Z. Applied Surface Science
2008, 254, (15), 4716-4719.
246. Manasevit, H. M. Appl Phys Lett 1968, 12, (4), 156-159.
247. Manasevit, H. M.; Simpson, W. I. Journal of The Electrochemical Society 1969, 116,
(12), 1725-1732.
248. Hiruma, K.; Katsuyama, T.; Ogawa, K.; Koguchi, M.; Kakibayashi, H.; Morgan, G. P.
Appl Phys Lett 1991, 59, (4), 431-433.
249. Stringfellow, G. B., Organometallic vapor-phase epitaxy: theory and practice. 2nd
ed.; Academic Press: 1999.
250. Jones, A. C.; O'Brien, P., CVD of Compound Semiconductors: Precursor Synthesis,
Development and Applications. John Wiley & Sons: 2008.
251. Smith, D. L., Thin-film deposition: principles and practice. McGraw-hill New York
etc: 1995.

64
252. Zhao, X. Synthesis, Electrical and Optical Characterization of Semiconductor
Nanowires. The Ohio State University, 2010.
253. Tsang, W. T. Appl Phys Lett 1984, 45, (11), 1234-1236.
254. Tsang, W. T. Journal of Crystal Growth 1989, 95, (1–4), 121-131.
255. Kelrich, A.; Dubrovskii, V. G.; Calahorra, Y.; Cohen, S.; Ritter, D. Nanotechnology
2015, 26, (8), 085303.
256. Morales, A. M.; Lieber, C. M. Science 1998, 279, (5348), 208-211.
257. Lieber, C. M. Solid State Communications 1998, 107, (11), 607-616.
258. Hu, J.; Odom, T. W.; Lieber, C. M. Accounts of Chemical Research 1999, 32, (5),
435-445.
259. Duan, X.; Lieber, C. M. Advanced Materials 2000, 12, (4), 298-302.
260. Duan, X.; Huang, Y.; Cui, Y.; Wang, J.; Lieber, C. M. Nature 2001, 409, (6816), 66-
69.
261. Lüth, H. Surface Science 1994, 299–300, (0), 867-877.

65
CHAPTER 3
Experimental Techniques
3.1 Introduction
In this chapter, the operation and principles of the key experimental techniques used to
fabricate and characterise III-V nanowires in this thesis, namely MOCVD and electron
microscopy, are introduced. These techniques are highly complicated; hence the
discussion is restricted to the basic principles and issues relevant to this work. Further
details can be found in the cited references provided for each technique.
The text is arranged as follows: Section 3.2 outlines the central technique of
MOCVD, which was used to grow all the III-V nanowires in this thesis. Electron
microscopy, which is the major characterisation technique, is then discussed in Section
3.3. Two microscopy techniques were pivotal to this research: scanning electron
microscopy (SEM) and transmission electron microscopy (TEM). Lastly, samples
preparation methods for electron microscopy characterisations are given in Section 3.4.
3.2 III-V nanowires epitaxial growth
A series of experiments have been designed to understand the roles of catalyst in III-V
nanowire growth by MOCVD. All III-V nanowire samples were grown at the Australian
National University (ANU) using AIXTRON 200/4 horizontal flow MOCVD reactor (Fig.
3.1a). The process, including growth system and precursors used, is first described.
Subsequently, two types of catalyst deposition methods, which are direct deposition of
colloids and thin-film generated, are explained.
3.2.1 Metal-organic chemical vapour deposition (MOCVD)
MOCVD is the most common and well-established technique to grow epitaxial layers of III-
V and II-VI compound semiconductors in industry due to its high throughput compared with

66
other methods. Therefore, this is the most economically technique for large-scale
commercial production.
Nevertheless, the control of MOCVD process is complex and not yet fully
comprehended (operators often call it a black box).1 The only detailed textbook on
MOCVD is written by G. Stringfellow2 and most of MOCVD-relevant information in this
thesis is referred to this book. There are various parameters that determine the growth,
such as chamber pressure, substrate temperature, precursors’ molar fractions and their
cracking efficiency, and total gas flow. Many of these parameters are inter-dependent.
However, all equipment is unique, which leads to slightly different growth; even so much
development has taken place and results in the high-level of control in this process during
the last forty years.
3.2.1.1 MOCVD design
The essential components of the MOCVD growth system are the reactor, the carrier gas,
the precursors, the heating and the plumbing system, including the valve system as well
as the pressure and the flow control.
The “heart” of the MOCVD system is the reactor. Herein, the substrates are
positioned, the temperature is applied, the precursors are mixed and cracked, and
eventually the growth takes place. The H2 carrier gas transporting the precursor gases
flows over the heated sample holder (susceptor) to the exhaust. At the solid surface of the
reactor (susceptor, sample and wall), the velocity of the gas is zero, since the flow is
laminar.3 Resulting from this boundary condition, a stagnant boundary layer is build up
above the susceptor, in which the velocity of the flow is decreased and the mass transport
happens only by gas phase diffusion 2. There are several types of reactors, for example,
with horizontal or vertical flow direction. In this thesis, a cold wall horizontal reactor is
used. The susceptor consists of inert, high temperature stable silicon carbide-coated
graphite, and is heated by a three-zone infrared lamp arrangement surrounding the
reaction chamber. The susceptor, which accommodates three two-inch wafers on gas foil
rotation plates, capable to rotate both planetary and satellite so that highly uniform
deposition of the growth materials can be achieved. A schematic outline of MOCVD
system is given in Fig. 3.1b.4

67
The system can be operated at atmospheric or at low pressure down to a few tens
of mbar. The total pressure influences the probability of parasitic reactions in the gas
phase, the thermodynamics of the growth, and the diffusion of the growth species. The
standard pressure of MOCVD system is set to 100 mbar. Besides, the total gas flow rates
through the hydride and metal-organic (MO) lines are maintained, at 10 standard litres per
minute (slm) and 5 slm, respectively. This keeps a constant total gas flow rate of 15 slm
through the reactor.
Figure 3.1. (a) AIXTRON 200/4 MOCVD reactor and (b) schematic illustration of a
MOCVD system.4
3.2.1.2 Precursors for III-V nanowires growth
The precursors are transported to the reactor via separate supply lines to avoid pre-
reaction before entering the reactor. Gas sources (often used for group-V materials) are
provided from the gas bottles. MO sources are provided in so-called bubblers, which are
kept at a certain temperature. A carrier gas is led through the liquid or solid source
material, thereby saturated with molecules. The molar fraction fed to the reactor is then
determined by the source flow, which is controlled by mass-flow controllers. In addition, for
MO sources, the molar fraction depends on the amount that is taken up by the carrier gas,
which is controlled by the temperature, and the source pressure. The latter is controlled
using electronic pressure controllers.

68
Two types of precursors are used in MOCVD. MO sources can be used for
providing both group-III and group-V materials. Common group-III sources are sources
with short alkyl groups, such as trimethylgallium (TMGa), triethylgallium (TEGa),
trimethylindium (TMIn) or trimethylaluminium (TMAl), amongst others. Typical MO sources
for group-V are tertiabutylphosphine (TBP) and tertiabutylarsine (TBAs). For group-V
materials, hydrides are often used instead, in which the group-V atom is surrounded by
three hydrogen atoms, such as arsine (AsH3) or phosphine (PH3).
The reason to use quite complicated molecules instead of direct evaporation of
elemental sources is the controllability and handling. The evaporation of pure materials is
difficult to govern, since they mostly have a high vapour pressure and low melting points.
In the other words, the cracking of molecules at a confined place is more convenient, even
though the process if not completely understood.
The cracking, i.e. the dissociation of the precursors, occurs in multi-step reactions,
until the metal or group V atom is set free.2 This process can happen in the gas phase
(homogeneously) or on the substrate surface (heterogeneously). Homogeneous pyrolysis
would cause parasitic reactions and the molecules would not end up in the growing
crystals; thus, heterogeneous pyrolysis is preferred. The cracking efficiency of the
precursors is influenced by the temperature and surfaces, and by the combination of
precursors. For example, the presence of TMIn increase the cracking efficiency of PH3.5 If
the cracking efficiency is low at the desired growth temperatures, a large excess of
precursors has to be fed to the reactor to provide enough material for growth. However,
material will be consumed by parasitic reactions if the desired growth temperatures are so
high that homogeneous pyrolysis takes place. Therefore, it is essential to take account on
the applied growth temperatures for the choice of precursors.
As shown below is the exemplary reaction between MO sources and hydride
precursors for the growth of InAs and GaAs nanowires:
( )
( )

69
3.2.1.3 Growth procedures
The detailed growth of III-V nanowires by MOCVD in this thesis are shown in Table 3.1.
Table 3.1. Detailed growth procedures of III-V nanowires by MOCVD.
Step Procedures Descriptions
1 Selection of substrates
Matching substrates with growth material are selected to eliminate lattice mismatch to grow free-standing nanowire.
Often, GaAs { } and InAs { } substrates are used to grow GaAs and InAs nanowires, respectively. Nanowires are
likely to grown along one of the ⟨ ⟩ directions due to lowest energy state provided.6
Lattice mismatch between different materials of substrate and nanowires may induce defects and strains.
However, when the materials of substrate and nanowire are different, vertical nanowires still can be grown. For example,
vertically standing InAs nanowires can be grown on GaAs { } substrate successfully.
Si substrates and substrates with { } surfaces are selected so that integration with Si electronics is possible.
2 Selection of catalysts
Catalyst selection is important because it can control the growth mechanism, growth rate, growth direction, nanowire morphology and crystal structures (by maintaining other growth parameters such as growth temperature, V/III ratio and precursor flow rates constant).
3 Deposition of catalysts
Au colloid particles in solutions are commercially available.
Thin-film generated by e-beam evaporator followed by annealing is the alternative technique to produce nanoparticles on substrates.
Details of the deposition procedures are included in Section 3.2.2.
4 Selection of growth precursors and sources
Appropriate precursors and sources should be selected.
Before flowing into the reactor chamber, all the precursors will be transformed into vapour form.
Information on the precursors is introduced in Section 3.2.1.2.
5 Substrates annealing
Catalyst-coated substrates are loaded into the low-pressure reactor.
The substrates are heated to selected annealing temperature, Ta (600–800°C) and annealed in situ for 10 min to desorb the surface contaminants, including the surface oxide.
It is performed under group-V overpressure (AsH3 flow of 1.3 x 10-3 mol/min) to prevent decomposition of the substrates.
After annealing, the substrates are cooled to growth temperature, Tg.
6 Growth of nanowires
Tg is typically between 450–600°C.
The group-V flow rate is adjusted for growth.
The group-III precursors are fed to reaction chamber to initiate growth.

70
Growth time, tg, is selected between 15–30 min, depends on the growth rate and the desired nanowire length.
7 Cooling down under AsH3
Upon completion of growth, each sample is cooled under group-V overpressure (AsH3 flow).
The grown samples are removed from the chamber after the reactor temperature is approximate room temperature.
3.2.2 Deposition of catalysts
Two types of catalyst deposition methods are used to assist III-V nanowires growth. This
treatment aims to distribute nano-sized (~50 nm) catalyst particles on the substrates. First
method applied is direct deposition of colloids. Commercially available monodisperse 50-
nm Au colloidal particles (British Biocell International – Ted Pella, Inc.) are used in this
thesis. This Au-catalysed nanowire growth is used as control parameter for better tuning
and understanding on the behaviour of the nanowires growth. By having negatively
charges, Au colloids do not adhere to III-V substrates. Thus, functionalising of substrate
with poly-L-lysine (PLL), which is a positively charged polyelectrolyte, is necessary. PLL
attracts the negatively-charged Au colloids and immobilised them on the substrate
surface.7 Besides, it helps to prevent the agglomeration of Au colloids during deposition,
hence an even Au colloids distribution is achieved on the substrate.4
The other method of catalyst deposition on substrate is thin-film generated followed
by annealing. Metal films (Ni, Pd and Pt in this thesis) are deposited on the substrate by
Temescal BJD-2000 e-beam evaporator system (Fig. 3.2a) since the commercial colloid
particles for these metals are not available. Fig. 3.2b depicted the schematic diagram of an
e-beam evaporator. The evaporator operates under ultrahigh vacuum condition. The
generation of e-beam by a gun at the bottom of the evaporator is bent by a deflecting
magnet. The curved beam is shined directly on the source material with high purities (>
99.99 %). Then, this high-energy e-beam melts the ingot source and evaporates the
molten source onto the substrate located above the source. The thickness of the deposited
layer is monitored by an accurate sense (next to target substrate).
Subsequently, the coated substrates are loaded into MOCVD reactor to perform in
situ annealing in the reactor. The annealing process will break apart and transform the
metal film into nanoclusters or nanoparticles (the size depends on the annealing
temperature and the density depends on the thickness of film). This method is simple but it
is difficult to obtain a uniform nanoparticle size or distribution. In addition, by supplying

71
group-III precursor, it is suspected that with high annealing temperature the film will melt
and nano-sized liquid droplets of group III/catalyst alloy are formed. These catalyst alloy
droplets will promote the III-V nanowire growth.
Figure 3.2. (a) Temscal BJD-2000 e-beam evaporator system and (b) schematic diagram
of a typical e-beam evaporator.
3.3 Electron microscopy
In early 1878, Ernst Abbé evidenced that the resolution of the optical microscope is
restricted by the wavelength of light,8 in which the smallest detail that can ever be resolved
optically is of the order of 100 millimicrons even when immersion optics and ultraviolet light
are used.9 On the other hand, electrons were first discovered by J.J. Thomson in 1897.10
Two important discoveries were made before the resolving of smaller details by using
electron microscope:
(i) In 1926, Busch11 discovered the analogy between the effect of a convex lens on a light
beam and the effect of magnetic coil on an electron beam.
(ii) In 1942, Louis de Broglie showed theoretically that all particles have an associated
wavelength related to their momentum,12 as shown in the equation below:
(3.1)
where m is the relative mass, v is the relative velocity and h is the Planck’s constant. In his
work, Broglie discovered that electrons have wave-like characteristics with a controllable

72
wavelength substantially less than that of visible light. There were two significant groups
that verified the wave nature of electrons by their experiments of electron diffraction
through crystals, which are Broglie and Reid,13 and Davisson and Germer.14
These discoveries made it plausible that extremely small details can be imagined
with electron lenses and an electron beam due to the smaller wavelength by many orders
of magnitude of the moving electron than the shortest wavelength of light. Few years later,
Knoll and Ruska verified the Busch’s lens formula experimentally and demonstrated the
electron images taken by a TEM.8 In 1936, the first commercial TEM was built. However,
regular production was only started in 1939 by Siemens in Germany. TEM was further
developed when Ardenne added scanning coils to the TEM, in order to build scanning
transmission electron microscope (STEM).8 This invention led to the introduction of a new
class of electron microscope, the SEM. The first used of SEM to examine a solid sample in
1942 by Zworykin et al..15 In 1949, Heidenreich16 thinned metal films to electron
transparency. This was a crucial step for the practical use in TEM, particularly in the field
of materials science. The other main contribution in the development was made when
Hirsch et al.17 at Cambridge developed the theory of transmission electron diffraction
contrast quantitatively. All in all, these developments made electron developments a
distinguished instrument for materials scientists.
3.3.1 Interaction between electrons and specimen
When a beam of electrons hit the surface of a solid, electrons tend to penetrate the solid
and the penetration depth depends upon the energy of the electron beam and the atoms of
a solid. If the specimen is thin (up to several hundred nanometres), and the energy of the
electron beam is sufficiently high (~100keV or higher), the specimen will become
transparent to the electron beam. During electron penetration in the specimen, the
electrons interact with the atoms in the specimen. Thus, many signals are generated.
Some of these signals are schematically summarised in Fig. 3.3. When a high-energy
electron beam hits the surface of a thin specimen, some of the electron beam passes
through the specimen without any interaction (transmitted beam), and some of the
electrons scatter at the surface of the specimen or during their transmission through the
specimen. Those electrons that scatter backwards at the surface of a specimen without
loss of their energy are called backscattered electrons (BSE). The electrons that scatter
during their transmission through the specimen can be divided into elastically scattered

73
and inelastically scattered electrons. The former are the electrons scattered without the
loss of their energy beam is coherent in nature, i.e. the electrons are in phase to each
other, elastically scattered electrons are the electrons which maintain this coherence,
whereas inelastically scattered electrons have no phase relationship with each other.
Figure 3.3. A schematic diagram showing the interaction of electron beam with a thin
specimen.
In TEM, if the sample is crystalline in nature, elastically scattered electrons
generate electron diffraction patterns. On the other hand, inelastically scattered electrons
can be used to analyse the chemical composition of the sample by measuring their energy
loss. From the mathematical point of view, the amplitude of the scattered electrons by an
isolated atom is measured as atomic scattering factor (fθ), given by:18
(
) ( ) (3.3)
where Z is atomic number, fx is atomic scattering factor for X-rays, and θ is the angle made
by the scattering electrons with incident electron beam direction. Generally, this scattering
factor increases with the atomic number, i.e. the heavier the atom, the stronger the
scattering and it decreases with increasing accelerating voltage. This scattering factor is
also a function of the scattering angle (θ), in which the scattering factor decreases at
higher scattering angles.8
In addition to the scattered electrons, other signals are generated from the
specimen, for example, secondary electrons (SEs), Auger electrons and characteristic X-
rays. SEs are those electrons emitted from the surface of the specimen and are ejected

74
due to the bombardment of the incident electron beam. These electrons are emitted due to
the transfer of kinetic energy from the incident electrons to the outer shell electrons of the
specimen atoms. These secondary electrons are used in SEM as the main signals for
image formation. Since an SEM uses bulk specimens, electrons are not transmitted
through specimen, instead, much scattering takes place backwards to the incident beam.
Characteristic X-rays are emitted from the specimen due to the interaction between the
specimen and the incident electron beam. When the high-energy electrons of the incident
electron beam hit the atoms in a specimen, inner-shell electrons of the atoms will be
ejected from their orbitals. This process is due to the transfer of kinetic energy from the
electron beam to the inner shell electrons, leaving empty states in their inner-shell orbitals.
Due to the loss of their inner-shell electrons, the atoms have a high-energy state. To
regain their normal energy state, the electrons from an outer-shell fill the empty states in
the inner-shells. Since the outer-shell electrons fill the inner-shells, a photon, in the form of
X-rays, is emitted due to the energy conservation rule. The energy of the X-ray is equal to
the energy difference between the two shells, which is a characteristic of an element. The
emission of characteristic X-rays is schematically illustrated in Fig. 3.4a. For instance, for
an atom of a specimen, if an empty state in the K-shell is filled by an L-shell electron, a Kα
X-ray will be emitted from the atom. These characteristic X-rays have unique wavelengths
for each element in the periodic table, which allows the detection of the elements
presented in a specimen. A dispersive device made of a semiconductor diode detects
these X-rays and distinguish them on the basis of their energy, which subsequently can be
used to generate an X-ray spectrum as a function of their energy and intensity. This
technique is termed as X-ray energy dispersive spectroscopy (XEDS or EDS). For
quantification of the X-rays, following equation proposed by Cliff and Lorimer19 can be
applied:
[
( )
( )] (
) (3.4)
where nA and nB are the numbers of atoms of A and B elements per unit volume, NA and
NB are the respective number of X-ray photons contributing to their characteristic peaks,
σA and σB are respective ionisation cross-sections of element A and B to create a empty
state in their inner-shells (this ionization cross-section is a measure of the probability of an
atom for ionisation process when it interacts with an electron or a photon, and ωA and ωB
are the X-ray fluorescence yield of respective elements.

75
3.3.2 Scanning electron microscope (SEM)
For thick specimen, a tear-drop shaped interaction volume will be created as shown in Fig.
3.4b. In this interaction volume, SE signals are generated at the surface pf the specimen
(5–50 nm thick), giving the surface information. BSE signals are generated deeper
underneath the surface, which carry mainly compositional information of the specimen,
creating contrast, which is called as a-contrast. Characteristic X-rays are generated at the
entire interaction volume, delivering the analytical signal related with the composition of
the material. The size of the interaction volume depends on the accelerating voltage and
the electrical conductivity of the specimen. In general, with higher accelerating voltage,
lower atomic number, the interaction volume will increase. The typical size of the
interaction volume is about 2 µm (width) by 2 µm (depth).20
Figure 3.4. (a) A schematic diagram showing the incident electron beam dislodges an
electron from the K shell of a Si atom. An electron from the L shell fills the vacancy and a
Si KαX-ray is emitted. The energy of the X-ray is equal to the difference between the
ionisation energy of both K shell and L shell. (b) Illustration of the interaction when a SEM
electron beam hits on the surface of a specimen.20
A SEM can image a specimen by scanning it with a high-energy beam of electrons
(Fig. 3.5). The electrons interact with atoms at or near the surface of the specimen
producing signals that contain information about the specimen’s surface morphology and
composition. In this thesis, several types of SEM instrument were used to image the
morphology of III-V nanowires on the substrates, including JEOL JSM-7100F and JEOL
JSM-7800F.

76
Figure 3.5. (a) JEOL JSM-7800F SEM alongside (b) schematic diagram of a SEM column.
Electrons are extracted form a filament or field-emission tip with a moderate
accelerating voltage, typically in the range of 1–50 kV. The incident electron beam (or
electron probe) has a diameter of 1–10 nm, depending upon the electron source. For
instance, a field-emission gun can produce an electron beam that is narrower in diameter
and has great current density than that can be achieved with conventional thermionic
emitters. The condenser lens controls the spot size by de-magnifying the electron beam
from the gun. The objective lens focuses the electron beam into a small probe, which
controls the focus of the final image. The aperture diameter controls the electron beam
convergence angle, α.
Resolution of a given SEM depends strongly upon the electron beam diameter. The
beam diameter decreases with shorter working distances, which can potentially lead to a
better resolution. Besides, depth-of –field (DOF) is another significant parameter in a SEM.
DOF is defined as the amount of sample (with protrusions) that is acceptably sharp focus.
This parameter limits the amount of sample under focus in an image (the amount of
surface features in focus will increase with a larger DOF). It is important to image those
samples with large protruding surface features, such as nanowires. The DOF can be
increased by decreasing the diameter of aperture, and thereby reducing the electron beam
convergence angle, α. Increasing the DOF can also be achieved by increasing the working
distance. However, increase in working distance reduces the resolution of the image, and
decreases in the aperture size limits the signal strength. Thus, the final aperture size and
the working distance can be chosen by a compromise among signal strength, DOF and
resolution. To generate a SEM image, the specimen is scanned in synchrony with the

77
electron beam of the specimen in a one-to-one relationship between points on the
specimen and points on the image viewing screen.
By and large, a SEM primarily uses the SEs and BSEs as the signals for the
construction of an image. These electrons can be detected using suitable electron
detectors, such as, an Everhart-Thornley detector for SEs and a solid-state or Robinson
detector for BSEs.21
In the SE imaging mode, the SEM can produce very high-resolution images of a
specimen surface. Most SEs are re-absorbed by the sample. It is important to note that
only the electrons produced near the surface (at a depth of less than 5–50 nm) can escape
from the specimen and take part in image formation.22 The number of SEs emitted from
the surface region varies with the topography of the surface. This number increases with
increasing angle between the surface normal and the incident electron beam. This
phenomenon is schematically illustrated in Fig. 3.6, in which the surface is nor planar. The
escape depth of SEs is indicated by the dotted line and the escape volume is shown by
the shaded areas. As the electron beam scans over the protrusion in Fig. 3.6, the escape
volume of SEs of the rough surface is much higher than that of the flat surface. Therefore,
protrusions produce higher counts of SEs, which make these features brighter.
Accordingly, SEs provide details of the surface topography of a sample.
Figure 3.6. A schematic diagram of the influence of surface topography: (a) flat surface
and (b) sharp edge on the escape volume of SEs. The dotted line represents the escape
depth and the shaded area represents the escape volume.20
As it has been described in Section 3.3.1, the number of emitted backscattered
electrons is proportional to the atomic number of the constituting atoms of the specimen.

78
By scanning through the whole specimen, BSE image will be able to give the information
on the compositional of the specimen.
Some SEMs have the X-ray electron dispersive spectroscopy (EDS, EDX or XEDS)
detector installed. EDS uses the X-ray signals generated by the electron-specimen
interaction to investigate the composition information of the specimen, in both quality and
quantity. EDS can be performed at a specific spot or a certain area of the specimen in
SEM. EDS mapping of the specimen can be achieved by using special software. The
interaction volume generated by the X-rays in SEM is 2 µm in size.
Technically, accelerate voltage (kV), spot size (S), aperture (AP) and working
distance (WD) are the key operational parameters which impacts significantly the results of
SEM analysis. Accelerating voltage is the voltage difference between the filament and the
anode which accelerates the electron beam towards the anode. High kV leads to smooth
images with high resolution, while low kV provides detailed surface information. Although
larger spot size contributes to smooth images, smaller spit size is preferable because this
can result in images with high resolution, greater DOF, and less electron beam damage.
Aperture size plays the same role as spot size; therefore, relatively smooth images with
high-resolution conveying the detailed surface information can be obtained by adjusting
these two parameters. Closer WD can improve the resolution but reduce the DOF. It is
important to note that, for nanowires with smaller diameter, the exposure of electron beam
on the tip of nanowires will result in an unwanted cap-like on the tip. This may be due to
the contamination of carbon. Thus, some skills in focusing the nanowires (especially at
higher magnification) are necessary to capture a representative SEM image on a
nanowire. Table 3.2 shows the parameters selected to analyse III-V nanowires using
different SEMs located in the Centre for Microscopy and Microanalysis (CMM), The
University of Queensland.
With the common SEM holder (flat stage), nanowires can be imaged in the direction
perpendicular to the substrate, which is the top-view of nanowires. With a 45° stage
holder, the nanowires can be viewed at this angle so that certain morphology information
of the nanowires can be obtained. With a 90° stage holder, the side-view of nanowires can
be imaged. On top of these, after the holder was loaded into the SEM chamber, the sage
can be rotated 360°, and tilted in a certain range of degrees depending on the working
distance used. Hence, the specimen can be observed from any direction.

79
Table 3.2. Operational parameters of SEMs for III-V nanowires analysis in this thesis.
Operational Parameters kV (kV) S AP WD (mm)
JEOL JSM-7100F 5 – 15 3 – 6 Fixed 2 – 15
JEOL JSM-7800F 5 – 15 2 – 8 Fixed 4 – 10
3.3.3 Transmission electron microscope (TEM)
TEM is a well-established technique for studying the crystallinity and microstructure of
materials. TEM requires a high-energy electron beam, and a thin specimen, up to a few
hundred nanometres in thickness, so that the specimen is transparent to the electron
beam.
As mentioned earlier in Section 3.3.1, electron beam can be forward scattered
during its interaction with a thin specimen in TEM, and these transmitted and/or scattered
beams can be used to form the TEM image of the specimen. A schematic illustration of a
typical TEM is shown in Fig. 3.7. In most cases, electrons in TEM are extracted from a
filament or a field-emission fun (FEG) with an accelerating voltage between 100–300 kV.
FEG emitted electron beam gives a higher spatial coherency and a much higher
brightness, which behaves more monochromatic than that by a thermionic source. This
emitted electron beam is paralleled by the condenser lens before it hits the specimen and
transmits the thin specimen. This transmitted beam is enlarged by the intermediate lens,
transferred through the microscope column, further magnified by the projected lens.
Finally, an image of the specimen will appear on viewing screen.
By having similar imaging principles in the optical system, the objective lens in TEM
forms the image of the specimen on its image plane using these transmitted electrons,
while the electron diffraction (ED) pattern forms at the back focal plane of the objective
lends. In TEM, intermediate lens is used to select the projection of an image or ED pattern
onto viewing screen.
Basically, there are two modes, i.e. imaging mode and diffraction mode, in the TEM,
which can be easily switched between these two modes in modern TEM as illustrated in
Fig. 3.8.

80
Imaging mode. The transmission of the electron beam depends on the mass of the atoms
in the specimen and thus can create the so-called mass-thickness contrast in the TEM
image. After the interaction of incident electron beam with specimen and results in the
transmission of electron beam, it will be focused by the objective lens. The intermediate
lens will then project the intermediate image of the specimen at the image plane onto the
fluorescent viewing screen at the end of the column. These images can be recorded by
negative films or digital camera made of charge-coupled device (CCD). The objective
aperture can also be inserted to increase the diffraction contrast in TEM image.
Diffraction mode. In fact, the major strength of conventional TEM is its ability to generate
diffraction contrast of the specimen using the coherent elastic scattered electrons. If the
specimen is crystalline in nature, the atomic planes of the crystal could diffract some
incident beams due to wave-like characteristic of electrons, as described in Section 3.3.1.
These diffracted electrons will interfere with each other after scattering, leading to
constructive scattering or destructive scattering, defined by the Bragg’s law, as shown in
the following equation:23
(3.5)
where n is an integer, λ is the wavelength of incident wave, d is the spacing between the
planes in the atomic lattice, and θ is the angle between the incident ray and the scattering
planes. Incident electrons that have been diffracted to the same angle are focused at the
same position in the back focal plane of the objective lends, showing the ED pattern. To
make this ED visible of the viewing screen, the intermediate lens is weakened, making the
back focal plane of the objective lens coincide with the objective plane of the projector
lens. Meanwhile, a selective-area-aperture can be inserted in the image plane to confine
physically a specimen area of the specimen that is illuminated by the electron beam. This
ED technique is named as selective-area-electron-diffraction (SAED). The below equation
can be applied to calculate the lattice spacing (d):
(3.6)
where R is the distance of diffraction spot from 000*, L is the camera length, and λ is the
wavelength of electrons.

81
Figure 3.7. (a) An example of a modern TEM instrument alongside (b) a generalised cut-
away diagram of the internal structure. (©
http://www.ammrf.org.au/myscope/tem/introduction/)
Figure 3.8. Ray diagrams for a TEM in BF imaging and selective area diffraction modes.24
TEM is a handy method to perform various microscopy characterisations. Besides
the traditional TEM imaging, there are other techniques that can be used on TEM, such as,
high-resolution transmission electron microscopy (HRTEM), scanning transmission
electron microscopy (STEM), X-ray energy-dispersive spectroscopy (EDS), electron
energy-loss spectroscopy (EELS), and energy-filtered transmission electron microscopy
(EFTEM). In this thesis, HRTEM and SAED techniques are mostly used to determine the
crystal structure of III-V nanowires.

82
Table 3.3 lists the microanalysis techniques that I performed using TEM and their
respective features. In my thesis, few models of TEM that I used, which are FEI Tecnai
F20 (operates at 200kV), FEI Tecnai F30 (operates at 300kV), and FEI Tecnai T12
(operates at 120kV), located in the Centre for Microscopy and Microanalysis (CMM), The
University of Queensland. Besides, I also carried out my TEM analysis using FEI Titan
aberration-corrected TEM operated at 300 kV equipped with an annular dark field (ADF)
detector located at Beijing University of Technology, China.
Table 3.3. List of microanalysis techniques in TEM used in this thesis.
Microanalysis techniques
Characteristics
Conventional imaging
It uses only the transmitted electrons or some of the forward scattered electrons to increase the diffraction contrast in image, in which bright-field (BF) and dark-field (DF) are two standard imaging modes.
In BF images: areas of high scattering appear dark indicating regions of high mass, thickness or strong diffraction effects.
In DF mode: the image is formed with electrons scattered in a specific direction, usually because of diffraction from a particular atomic plane or planes. Therefore, regions of high intensity represent strong scattering.
HRTEM A phase-contrast imaging technique (uses both transmitted and diffracted electrons), which makes it possible to obtain an interfered image with atomic resolution.
It can be used to identify the lattice planes, crystal phases and orientations, and some defects.
The resolution of the TEM needs to be smaller than the planar spacing of the crystal specimen to acquire HRTEM images. Otherwise, the small lattice fringes are beyond the limitation of TEM to resolve.
Thus, the direction of incident electron plane wave must be tuned to the low-index zone axis of the crystalline specimen.
SAED SAED patterns provide crystallographic information from selected regions of the sample from the micron to the ~100 nm scale.
The spacing and orientations of the diffraction spots can be interpreted in terms of the planar spacing and orientation in the sample.
It can be used to identify crystal phases and their orientations (not on such a small spatial scale compared with HRTEM).
EDS It can be used to determine the composition of the sample, and can be conducted from the micro- to the nano-scale.
When the electron beam hits the sample, X-rays are generated which have characteristics energies for each element.
This provides a qualitative analysis of the elements present and, with further analysis, a quantitative composition can be determined.
STEM It involves scanning a nanometre-sized focussed electron beam across the sample with the scanning coils, and then it generates transmitted electron signals (similar SEM). Detectors (rather than CCD camera) count the electron transmitted through the sample, building

83
up an image of the sample point by point as the beam is scanned.
BF images are obtained by collecting electrons scattered through small angles by a small disk-like detector.
High-angle annular dark-field (HAADF) images are obtained by collecting the electrons scattered through large angles by a plate with hole, and provide mass variations information in the sample (also called Z-contrast imaging). Material with higher atomic number, Z, behaves brighter than that with lower Z in a HAADF STEM image.
3.4 Electron microscopy samples preparation
Aiming for a detailed investigation on the III-V nanowires, different techniques of preparing
both SEM and TEM samples are adopted. More information is given in the following sub-
sections.
3.4.1 SEM samples
As the incident electron beams with negative charges will continuously interact with the
sample, it is necessary to get rid of the accumulation of negative charges on the sample.
Therefore, all the SEM samples must be electrically conductive. Usually, a very thin layer
of conductive materials, such as carbon, platinum or gold, will be coated on the surface of
the sample to make it conductive. All the SEM samples in this thesis are III-V 1D
nanostructures, which are semiconductors, all the nanowire samples are not pre-coated
with any conductive layer. Hence, the SEM samples were loaded into the SEM chamber
without any coating. In this thesis, carbon-tape was used to stick at the bottom of the
substrates to the metallic sample stub to obtain top-view and bird-eyes-view of the
samples. On the other hand, facet of the substrate was sticked on the stub to observe the
cross-section of the samples.
3.4.2 TEM samples
For the investigation using TEM, suitable TEM samples preparation is important. In my
thesis, three different methods that I used to prepare samples for TEM analysis: (a)
individual nanowire preparation, (2) Cross-sectional TEM specimen’s preparation, and (3)
cross-section of nanowire sample preparation. The purposes and preparation process for
each sample preparation methods are shown in Table 3.4.

84
Table 3.4. Purpose and preparation process for different types of nanowire TEM sample.
Sample type Purpose Preparation process
Individual nanowire
To investigate crystal structure and composition single nanowire
1. A small substrate with as-grown nanowires are cut and placed in a small container with a little amount of pure ethanol.
2. The container is put into an ultrasonicator for ~10 – 40 min to shake off the nanowires from the substrate.
3. A few drops of this nanowire-ethanol dispersion are dropped onto a holy carbon film coated with copper grid.
Cross-sectional TEM sample
To study the nanowires with the presence of the substrate or the layer growth on substrates
1. A small piece of substrate with as-grown nanowires is cleaved.
2. The substrate is loaded into FEI Scios Dual Beam FIB/SEM, which combines focused ion beam (FIB) and SEM techniques (located at CMM, UQ).
3. The area is selected under observation of SEM and a slice of cross-sectional TEM lamellar specimen is sliced using FIB.
* The thickness of FIB cross-sectional TEM specimen is ~50 nm or thinner for HRTEM imaging.
Nanowire cross-section sample
To observe cross-section of nanowire, especially for those with core-shell structure
1. A small piece of substrate with as-grown nanowires is cut and embedded in Epon resin.
2. Once the resin is set, a razor blade is used to trim the sample to remove the substrate.
3. By removing the substrate, the nanowires are then sliced using a Leica Ultracut UC6 ultramicrotome (located at CMM, UQ).
REFERENCES
1. Hillerich, K. Influence of Seed Particle Material, Preparation, and Dynamics on
Nanowire Growth. PhD, Lund University, Sweden, 2013.
2. Stringfellow, G. B. Organometallic vapor-phase epitaxy: theory and practice. 2nd
ed.; Academic Press: 1999.
3. Smith, D. L. Thin-film deposition: principles and practice. McGraw-hill New York etc:
1995.
4. Joyce, H. J. Growth and Characterisation of III-V Semiconductor Nanowires for
Optoelectronic Device Applications. The Australian National University, Canberra,
2009.
5. Larsen, C.; Buchan, N.; Stringfellow, G. Mass spectrometric studies of phosphine
pyrolysis and OMVPE growth of InP. Journal of Crystal Growth 1987, 85 (1), 148-
153.

85
6. Joyce, H. J.; Paiman, S.; Gao, Q.; Tan, H. H.; Kim, Y.; Smith, L. M.; Jackson, H. E.;
Yarrison-Rice, J. M.; Zhang, X.; Zou, J.; Jagadish, C. In III-V compound
semiconductor nanowires, Nanotechnology Materials and Devices Conference,
2009. NMDC '09. IEEE, 2-5 June 2009; 2009; pp 59-60.
7. (a) Cui, Y.; Lauhon, L. J.; Gudiksen, M. S.; Wang, J.; Lieber, C. M. Diameter-
controlled synthesis of single-crystal silicon nanowires. Appl Phys Lett 2001, 78
(15), 2214-2216; (b) Hochbaum, A. I.; Fan, R.; He, R.; Yang, P. Controlled Growth
of Si Nanowire Arrays for Device Integration. Nano Lett 2005, 5 (3), 457-460.
8. Williams, D. B.; Carter, C. B. The Transmission Electron Microscope. Springer:
1996.
9. Freundlich, M. M. Origin of the electron microscope. Science 1963, 142 (3589),
185-188.
10. Thomson, J. J. XL. Cathode Rays. The London, Edinburgh, and Dublin
Philosophical Magazine and Journal of Science 1897, 44 (269), 293-316.
11. Busch, H. Calculation of the channel of the cathode rays in axial symmetric
electromagnetic fields. Annalen der Physik 1926, 81 (25), 974.
12. Broglie, L. d. A tentative theory of light quanta. Philosophical Magazine Series 6
1924, 47 (278), 446-458.
13. Thomson, G. P.; Reid, A. Diffraction of cathode rays by a thin film. Nature 1927,
119, 890.
14. (a) Davisson, C.; Germer, L. H. Diffraction of Electrons by a Crystal of Nickel.
Physical Review 1927, 30 (6), 705-740; (b) Davisson, C.; Germer, L. H. The
scattering of electrons by a single crystal of nickel. Nature 1927, 119 (2998), 558-
560.
15. Zworykin, V. A. H., J.; Snyder, R. L. A scanning electron microscope. ASTM Bull.
1942, 117, 15-23.
16. Heidenreich, R. D. Electron Microscope and Diffraction Study of Metal Crystal
Textures by Means of Thin Sections. J Appl Phys 1949, 20 (10), 993-1010.
17. Hirsch, P. B.; Howie, A.; Whelan, M. J. A kinematical theory of diffraction contrast of
electron transmission microscope images of dislocations and other defects.
Philosophical Transactions for the Royal Society of London. Series A, Mathematical
and Physical Sciences 1960, 499-529.
18. Edington, J. W. Interpretation of transmission electron micrographs. Macmillan
London: 1975; Vol. 3.

86
19. Cliff, G.; Lorimer, G. The quantitative analysis of thin specimens. Journal of
Microscopy 1975, 103 (2), 203-207.
20. Newbury, D. E.; Joy, D. C.; Echlin, P.; Fiori, C. E.; Goldstein, J. I. Advanced
scanning electron microscopy and X-ray microanalysis. New York.: Plenum Press.
Review by WMS in J. Microsc 1987, 146, 109.
21. Flegler, S. L.; Heckman, J. W.; Klomparens, K. L. Scanning and transmission
electron microscopy : an introduction. Freeman: New York, 1993.
22. Egerton, R. F. Physical Principles of Electron Microscopy : An Introduction to TEM,
SEM, and AEM. Springer Science+Business Media, Inc.: Boston, MA, 2005.
23. Bragg. The diffraction of short electromagnetic waves by a crystal. Proceedings of
the Cambridge Philosophical Society. Mathematical and physical sciences 1913, 17
(43), 4.
24. Fultz, B.; Howe, J. Transmission Electron Microscopy and Diffractometry of
Materials. 4th ed.; Springer Berlin Heidelberg: 2013.

87
CHAPTER 4
Understanding the Growth of Different InAs 1D
Nanostructures Catalysed by Au Nanoparticles
4.1 Introduction
This chapter depicts the growth of different Au-catalysed InAs 1D nanostructures, namely
naoleaf and nanobelt, were studied systematically using electron microscopy. Growth
temperature and V/III ratio used in MOCVD chamber during growth, and different types of
substrate were altered. In Section 4.2.1, insights into the growth mechanism of leaf-like
one-dimensional InAs nanostructures have been examined. Thorough structural
characterization suggests that nanoleaves contain relatively low-energy { } or { }
mirror twins acting as their midribs and narrow sections connecting the nanoleaves and
their underlying bases as petioles. Notably, the mirror twins lead to identical lateral growth
of the twined structures in terms of crystallography and polarity, which is crucial for the
formation of symmetrical nanoleaves. It has been found that the formation of nanoleaves is
energy-minimization-driven of catalyst-induced mirror twined nanostructures. The
successful growth of biomimic semiconductor nanoleaf will be useful in the bio-inspired
nano-architecture. In Section 4.2.2, the growth of defect-free zinc-blende [ ̅]-oriented
nanobelts has been studied. Different types of substrates were used to increase the
density of [ ̅]-oriented nanobelts. The composition of In adsorbed in to Au catalyst was
identified as the factor for the growth of [ ̅]-oriented nanobelts.
4.2 Journal publication and manuscript
Results in Chapter 4 are divided into two sections: Section 4.2.1 is included as it appears
in Nano Research 2016, 9(3), 766–773. https://link.springer.com/article/10.1007/s12274-
015-0955-z; and Section 4.2.2 has been drafted as manuscript.

88
4.2.1 Mirror-twin induced bicrystalline InAs nanoleaves
Mirror-Twin Induced Bicrystalline InAs Nanoleaves
M. Teng Soo1, Kun Zheng2,3,*, Qiang Gao4, H. Hoe Tan4, Chennupati Jagadish4 and Jin
Zou1,2,*
1 Materials Engineering, 2 Centre for Microscopy and Microanalysis, and 3 Australian
Institute for Bioengineering and Nanotechnology, The University of Queensland, St. Lucia,
QLD 4072, Australia
4 Department of Electronic Materials Engineering, Research School of Physics and
Engineering, The Australian National University, Canberra, ACT 2601, Australia
ABSTRACT
In this study, leaf-like one-dimensional InAs nanostructures were grown by metal-organic
chemical vapor deposition method. Detailed structural characterization suggests the
nanoleaves contain relatively low-energy { } or { } mirror twins acting as their midribs
and narrow sections connecting the nanoleaves and their underlying bases as petioles.
Importantly, the mirror twins lead to identical lateral growth of the twined structures in
terms of crystallography and polarity, which is essential for the formation of lateral
symmetrical nanoleaves. It has been found that the formation of nanoleaves is driven by
the catalyst energy minimization. This study provides a biomimic of semiconductor
nanoleaf with that of real leaf in nature.
KEYWORDS: InAs, nanoleaf, twin boundary, mirror twin
Introduction
In the thriving field of nanoscience, a major ambition is to synthesize nanoscale building
blocks of arbitrary dimensions, morphologies, and materials with increasing complexity [1].
Biomimicry is an interdisciplinary study which looks to nature and natural systems for
inspiration in practical engineering design [2]. Discovering a synthetic pathway to artificial
analogs of materials in nature represents a fundamental milestone in the development of
nanomaterials. Studying and replicating these biologically inspired nanostructures may

89
lead to new knowledge for fabrication and design of new and novel nano-scale devices, as
well as providing valuable insight in to how such phenomena can be exploited [3, 4].
III-V semiconductor nanostructures are promising low-dimensional materials with a
wide range of applications, including light-emission, logic, sensing and solar-cell
technology [5-12]. As a main class of III-V semiconductors, InAs one-dimensional (1D)
nanostructures have attracted significant research attention due to their direct and narrow
bandgap, relatively high electron mobility and small electron effective mass [13, 14], which
have made them a potential candidate for applications in resonant tunneling diodes,
Josephson junctions and high-performance transistors [15-17].
Nanoleaf is a type of leaf-like 1D nanostructure, and it was reported mainly for metal
oxides [18-23], and a few III-V materials [24, 25]. In this study, we demonstrated Au-
catalyzed growth of InAs nanoleaves. The structure of these nanoleaves has been
investigated carefully and we found that the axial twin boundary, which acts similar to the
midrib of a real leaf in nature, is necessary in the formation of these nanoleaves. A new
type of twins is identified and their nature is confirmed. The growth pattern of these
nanoleaves is found similar to the growth of dorsiventral leaves in nature [26-28].
Therefore, we believe that by correlating both of the structure of InAs nanoleaves in this
nanoworld and the structure of real dorsiventral leaves in our natural world, a bio-inspired
nano-technology design can be motivated for future electronic or optoelectronic devices.
Experimental Details
InAs nanoleaves were grown via horizontal flow metalorganic chemical vapour deposition
(MOCVD) using trimethylindium (TMIn) as the group III precursor and arsine (AsH3) as the
group V precursor, and Au nanoparticles to drive nanowire growth. InAs ( ̅ ̅ ̅) substrates
were treated with poly-L-lysine (PLL) solution followed by dispersion of a solution
containing 50-nm colloidal Au nanoparticles. Nanoleaves were grown at a pressure of 100
mbar and a total gas flow rate of 15 slm (slm = standard litre per minute). Prior to growth,
each substrate deposited with the Au particles was annealed in situ at 700 °C under AsH3
ambient to desorb surface contaminants. It has been well documented that at high
annealing temperature under AsH3 ambient, Au colloidal nanoparticles can first react with
the InAs substrate to form Au-In alloy droplets [29-31]. AsH3 is typically introduced in the
chamber during temperature ramp-up and annealing to counteract As sublimation and

90
preserve the surface stoichiometry of the InAs substrate. After the substrate was cooled to
the growth temperature of 500 °C, the AsH3 flow was adjusted and TMIn introduced to
initiate nanowire growth. The growth time was 30 min. The absolute flow rate of TMIn and
AsH3 was controlled at 1.2 x 10-5 mol/min and 3.4 x 10-5 mol/min, respectively, to attain a
very low V/III ratio of 2.9 so that a growth environment in the excess of In can be
maintained for promoting the growth of non-vertical 1D nanostructures [32].
Detailed morphology, structure and composition of the InAs nanoleaves were
characterized by field-emission scanning electron microscopy (FE-SEM – JEOL JSM-
7800F, operated at 5 kV) and transmission electron microscopy (TEM – FEI Tecnai F20,
operated at 200 kV; and FEI Tecnai T12, operated at 120 kV). TEM specimens were
prepared by dispersing the nanoleaves in ethanol using an ultrasonic bath for 15 min and
then spreading the drops from the suspension onto holey carbon grids. FE-SEM analysis
was used to identify the general morphology of the nanoleaves such as facet planes and
size. Conventional bright-field and dark-field TEM imaging of the nanoleaves along the
⟨ ⟩ zone axis was used to identify the crystal structures and twin boundary; in
combination with selected-area electron-diffraction (SAED) and lattice imaging.
Nanoleaves were screened for twin boundary over their entire length. The composition of
the Au-In alloy catalyst was studied by energy-dispersive X-ray spectroscopy (EDS)
analysis. Convergent beam electron diffraction (CBED) was employed to determine the
polarity of twin-sections of the nanoleaves through correlating the CBED patterns
simulated using the Bloch wave method in JEMS software [33].
Results and Discussion
Figure 1(a) is a plan-view SEM image and shows the overview of Au-catalyzed InAs
nanostructures grown on an InAs ( ̅ ̅ ̅) substrate, in which inclined leaf-shaped
nanostructures with a density of over 1% can be seen (as arrowed). Figure 1(b) is a
magnified SEM image of such a nanoleaf. Figures 1(c) and 1(d) are a pair of enlarged
SEM images to show the tip and base of the nanoleaf, respectively. As can be seen from
Fig. 1(c), a catalyst is associated with the tip of the nanoleaf, which is asymmetrically
associated with the nanoleaf. On the other hand, at the base region, a line (as marked by
the red arrow) is associated with the axial direction of the nanoleaf that divides the
nanoleaf, starting from the base. In fact, this line acts as a midrib and makes the
nanostructure to look like a plant leaf with a midrib across in the middle. Our extensive

91
SEM investigations indicate that these nanoleaves all have asymmetrical catalysts on their
tips and have straight lines dividing these leaves. By carefully examining these
nanoleaves, unlike those inclined nanowires [32, 34, 35], no specific projected directions
can be found.
Figure 1 (a) Top-view SEM image showing the overview of the Au-catalyzed InAs
nanostructures, in which the red arrows point to the two different nanoleaves. (b) Top-view
image of a single inclined InAs nanoleaf. Enlarged SEM images focused at the (c) tip
(showing the asymmetrical catalyst as indicated by red arrow); and (d) base (showing the
twin boundary located in the middle of nanoleaf starting from the base indicated by red
arrow) of the nanoleaf.
To understand the structural characteristics of these nanoleaves, TEM investigations
were performed, in which individual nanoleaves were prepared lying down on holey carbon
films. In fact, these individual nanoleaves can be characterized by both SEM and TEM.
Figures 2(a) and 2(b) are the typical SEM and TEM images of individual nanoleaves.
Since they are lying flat on the holey carbon films, these SEM and TEM images suggest
that the nanostructure has a leaf-shape indeed. In particular, as shown in Fig. 2(b), a
crystal boundary (as marked by the red arrow) is seen to divide the nanoleaf from the
middle and across the entire leaf from the tip to the bottom, acting as a midrib in a plant
leaf. It is of interest to note that, in both SEM and TEM images, the nanoleaves are
electron transparent, indicating that they are sufficiently thin. Figures 2(c) and 2(d) are
magnified TEM images showing the tip and top regions of the nanoleaf. Interestingly, the
crystal boundary can be clearly seen in both cases. Furthermore, the asymmetrical

92
catalysts can be seen (refer to Fig. 2(c)), which is echoed by the inset in Fig. 2(a). Figures
2(e) and 2(f) are two SAED patterns taken from both sides of the crystal boundary near the
catalyst. These SAED patterns are identical but relatively oriented, and can be respectively
indexed as the [ ] and [ ̅ ̅] zone-axes of the zinc-blende crystal structure, suggesting
that the nanoleaf has the { } surface.
Figure 2 (a) SEM and (b) TEM images of individual InAs nanoleaves showing their flat
nature. Inset in (a) showing the asymmetrical catalyst. Enlarged TEM images from (b) at
the nanoleaf tip (c) showing the asymmetrical catalyst and at the region away from the tip
(d), both have a vertical boundary in the middle of the nanoleaf. (e) and (f) are SAED
patterns taken from left and right of both regions shown in (c) indicating that they have
zinc-blende crystal structure.
To determine the nature of the crystal boundary of our nanoleaves, we employed
high-resolution TEM (HRTEM) and SAED investigations to study a large number of
nanoleaves. Through our extensive TEM investigations, we found that there are two
different kinds of crystal boundaries. Figures 3(a) and 3(b) are HRTEM images taken from
two different cases and their corresponding SAED patterns are shown in Figs. 3(c) and
3(d). Based on both HRTEM images and SAED patterns, twin structures can be clearly
identified. By carefully analyzing the SAED pattern shown in Fig. 3(c), it is found that both
⟨ ⟩ and ⟨ ⟩ diffraction spots are the common diffraction spots for both twinned
structures. Furthermore, by carefully correlating the SAED pattern with the HRTEM image
in Figs. 3(c) and 3(a), the ⟨ ⟩ diffraction spots are perpendicular to the twin boundary
shown in Fig. 3(a), suggesting that the twin boundary is a { } twin. In addition, since the

93
⟨ ⟩ is parallel to the twin boundary and the twin boundary parallel to the axial direction
of the nanoleaf (refer to Fig. (2)), the axial direction of this nanoleaf can be determined to
be along the ⟨ ⟩ direction. Similarly, by correlating the SAED pattern with the HRTEM
image shown in Figs. 3(d) and 3(b), twin boundary shown in Fig. 3(b) can be determined
as the { } twin. Based on the fact that the SAED pattern is taken from ⟨ ⟩ zone-axis
for both twinned structures, the axial direction of this nanoleaf must be perpendicular to the
⟨ ⟩ and ⟨ ⟩, which gives rise to the ⟨ ⟩ direction according to crystallography.
Figure 3 (a) and (b) are typical HRTEM lattice images at the coherent crystal boundary of
two distinct InAs nanoleaves viewed along the [ ] and [ ̅ ̅] zone-axes, with (c) and (d)
corresponding SAED patterns (blue: left region; orange: right region). In (c), common spots
showing the mutual twin boundary of { } plane and growth direction of ⟨ ⟩; and while
in (d), common spots showing the mutual twin boundary of { } plane.
Since there exists polarity in the zinc-blende structure (leading to ( ) ( ̅ ̅ )̅), it is
necessary to determine the nature of the polarity and consequently the nature of the twin
boundaries. Accordingly, CBED was performed on several nanoleaves. Figure 4 shows an

94
example. Figure 4(a) is a bright-field TEM image showing a nanoleaf with a { } twin
boundary. Figures 4(b) and 4(c) are a pair of ⟨ ⟩ zone-axis CBED patterns taken from
both twinned structures, respectively. As can be seen from individual CBED pattern, there
exists a mirror-symmetry, as indicated by their respective dashed lines; and the contrasts
of the two ⟨ ⟩ diffraction disks are different, reflecting the polarity. To determine the
absolute polarity, the theoretical CBED patterns were simulated using the JEMS software
[33]. Figure 4(d) is a simulated InAs [011] zone-axis CBED pattern (where the following
parameters were used for simulation: TEM specimen thickness is 50 nm; incident beam
convergence half-angle is 6.707 mrad), in which the contrasts in both ⟨ ⟩ diffraction
disks are indeed different and there is a vertical mirror symmetry in the simulated CBED
pattern. In fact, such a structural characteristic is demonstrated in Fig. 4(e) that present the
atomic model of zinc-blende structured InAs. By correlating the ⟨ ⟩ contrasts obtained
from experiments (Figs. 4(b) and 4(c)) with the simulation results (Fig. 4(d)), both
experimental ⟨ ⟩ diffraction disks can be unambiguously distinguished, as marked in
Figs. 4(b) and 4(c). Accordingly, the polarities of the both twinned structures are
determined. The comparison of Figs. 4(b) and 4(c) indicates that two CBED patterns are
identical and symmetrical with the twin plane, suggesting that the observed twin belongs to
the mirror twin, so that they have the identical growth direction, which is the essential
component for the formation of the nanoleaf.
To understand the nature of interfaces between the catalyst and twin-structured
nanoleaf, we carefully investigate their crystallographic relationships. Figure 4(f) is a ⟨ ⟩
on-zone TEM image taken from the tip region of the nanoleaf shown in Fig. 4(a), in which
sharp interfaces between the catalyst and two twin-structures can be clearly seen. By
carefully analyzing the CBED pattern shown in Fig. 4(b) and correlating it with the TEM
image shown in Fig. 4(f), the interface between the catalyst and the left twinned structure
is determined to be ( ̅ ̅ ). Using similar analysis, the interface between the catalyst and
right twinned structure is determined to be ( ̅ ̅). According to crystallography, both ( ̅ ̅ )
and ( ̅ ̅) interfaces are A-type { } interfaces, i.e. In terminated interface. Figure 4(g) is
the atomic model representing the crystallographic relationship between the catalyst and
the two twinned structures, in which both interfaces are In terminated interfaces.
Furthermore, according to the crystallographic requirement for the { } twinned nanoleaf,
the angle between these two { } interfaces is 141°, which matches well with our
observation (refer to Fig. 4(f)).

95
Figure 4 (a) TEM image of a typical nanoleaf. (b) and (c) are experimental CBED patterns
taken from the left and right twined structures. (d) Simulated [ ] CBED pattern of a 50-
nm thick InAs. (e) Atomic model of InAs zinc-blende structure viewed along the [ ]
zone-axis. (f) HRTEM lattice image and (g) atomic model of bicrystalline InAs nanoleaf at
the growth front viewed along the ⟨ ⟩ zone-axes, which consists of a { } twin
boundary.
Based on our extensive TEM investigations, the surface of the nanoleaves has { }
planes with their midribs being either { } or { } twin boundaries. These twin
boundaries are perpendicular to the leaf surfaces and to their axial directions.
Nevertheless, the interfaces between the catalysts and twinned structures of nanoleaves
are all { } interfaces. These rigid crystallographic relationships require specific angles
(θ) between the two adjacent interfaces. For { } twinned nanoleaves, { } (Fig.
5(a)); while for { } twinned nanoleaves, { } (Fig. 5(b)). Since { } { },
for a spherical catalyst being cut by the two { } planes of a nanoleaf shown in Figs.
5(a) and 5(b), the surface area of the catalyst is larger in the case of { } twinned

96
nanoleaf (Fig. 5(b)), suggesting a higher catalyst surface energy, which may result in the
system being unstable. Interestingly, by carefully studying the catalysts in { } twinned
nanoleaves, we found that the interfaces between the catalysts and nanoleaves are not
simple, as illustrated in Fig. 5(b). As shown in Fig. 2(c), there are two interfaces between
the catalyst and the right twinned section: one is ( ̅ ̅) and the other one is ( ̅ ̅) with the
latter one being parallel to the twin plane of the nanoleaf. The resultant catalyst/nanoleaf
relationship is illustrated in Fig. 5(c). The comparison of the surface energies of the
catalysts in the configurations shown in Figs. 5(a) and 5(c) indicates that the surface
energy becomes lowered for the { } twinned nanoleaves if the dimensions of the two
catalysts are the same. Furthermore, the ( ̅ ̅) interface has been observed in most of the
studied { } twinned nanoleaves, suggesting that such a configuration is energetically
stable. Thus the observed ( ̅ ̅) interface between the catalyst and a twinned structure
must have a relatively low interfacial energy; otherwise, the configuration shown in Fig.
5(b) would be preferred. Based on our detailed TEM investigations, we anticipate that the
complicated interfacial configurations found in the { } twinned nanoleaves is due to the
required minimization of overall catalyst surface/interfacial energies. On the other hand,
the fact that we did not find such a complicated interfacial configuration in { } twinned
nanoleaf indicates that the { } interface might have a relatively higher interfacial energy
when such a configuration is adopted to lower the overall catalyst energy. We found that
most nanoleaves are { } twinned nanoleaves, which is coincidence with the prediction
that { } twins have relatively low energy than { } twins [36, 37]. On this basis, we
anticipate that most { } twinned nanoleaves are attributed to their relatively low energy
required for their formation. It is of interest to note that no { } twinned nanoleaf is
observed, possibly due to the high-energy requirement for forming { } mirror twins
caused by the polarity reversal [38, 39]. Based on these analyses and discussion, we
anticipate that the extraordinary twin boundaries of { } or { } found in the InAs
nanoleaves are formed due to the required formation of { } interfaces between Au
catalysts and their induced InAs nanostructures, in which two { } interfaces are formed
(driven by the catalyst energy minimization). This is very different from the common { }
interface found between Au catalysts and InAs nanowires, in which only one { }
interface are formed between the catalysts and their underlying nanowires leading to the
nanowire growth [40]. In our case, once the mirror twins are formed, the lateral growth of
the twinned nanostructure leads to formation of leaf-shaped nanostructure. Therefore, the
fact that asymmetrical catalytic particle at the tip of a nanoleaf produce a symmetrical
nanoleaf is the rigid crystallographic requirement. On other hand, since the { } surfaces

97
have a relatively low energy for InAs (although higher than the {111} surface energy), the
formation of all nanoleaves with flat { } surfaces is driven by crystallographic necessity,
rather than the absolute energy minimization.
Figure 5 Schematic illustrations at the growth front of nanoleaves with (a) { } twin
boundary, with two { } interfaces between catalyst and nanoleaf; (b) { } twin
boundary, with two { } interfaces between catalyst and nanoleaf; and (c) { } twin
boundary, with two interfaces between the catalyst and the right twinned structure: one is
( ̅ ̅) and the other one is ( ̅ ̅).
According the different growth mechanisms proposed in the literature, our
nanoleaves (containing twins as midribs) should be grown synergistically by three growth
mechanisms, namely via vapor-liquid solid (VLS) [41], twin-plane re-entrant-edge (TPRE)
[42, 43], and vapor-solid (VS) mechanisms [44]. Both VLS and TPRE growth mechanisms
lead to the axial growth of nanoleaves whereas the VS growth mechanism regulates the
lateral growth at sidewalls of nanoleaves. It is important to note that the midribs of our
nanoleaves are mirror twins, leading to growth fronts of sidewalls of both twined structures
are identical in terms of crystallography and polarity. This fact in turn enables the identical
lateral growth of both twined structures in a nanoleaf, thus forming the symmetrical
structure [45, 46]. In general, the lateral growth of one-dimensional nanostructures, such
as nanowires, leads to the formation of tapered nanostructures when these nanostructures
are directly grown from the underlying substrates [45, 47]. In our case, all nanoleaves were
developed from bases with narrow sections between them and these narrow sections act
as “petiole” (refer to Fig. 1(d)), which is critically essential for the formation of nanoleaves.
The physical resemblances between our unique nanoleaf and real leaf are presented in
the Electronic Supplementary Material.

98
Conclusion
In summary, bicrystalline InAs nanoleaves with { } and { } mirror twin boundaries
were, for the first time, grown under an excessive In condition. Detailed electron
microscopy investigations were performed, from which the growth behavior and
fundamental reasons attributed to the growth of nanoleaves are clarified. Specifically, the
necessary conditions for formation of nanoleaves are the formation of mirror twins and the
existence of a narrow connecting section between the nanoleaves and their underlying
bases. These biomimetic nanoleaves are inter-related with natural plant leaves so that
some valuable hints for a bio-inspired nano-technology design may be inspired.
ASSOCIATED CONTENT
Electronic Supplementary Material: Physical resemblances of a real leaf and nanoleaf.
This material is available in the online version of this article.
AUTHOR INFORMATION
Corresponding Author
* E-mail: [email protected], [email protected].
ACKNOWLEDGMENT
This research was supported by the Australian Research Council. The Australian National
Fabrication Facility and Australian Microscopy & Microanalysis Research Facility, both
established under the Australian Government’s National Collaborative Research
Infrastructure Strategy, are gratefully acknowledged for proving access to the facilities
used in this work.

99
ELECTRONIC SUPPLEMENTARY MATERIAL (ESM)
Mirror-twin induced bicrystalline InAs nanoleaves
M. Teng Soo1, Kun Zheng2,3,*, Qiang Gao4, H. Hoe Tan4, Chennupati Jagadish4 and Jin
Zou1,2,*
1 Materials Engineering, 2 Centre for Microscopy and Microanalysis, and 3 Australian
Institute for Bioengineering and Nanotechnology, The University of Queensland, St. Lucia,
QLD 4072, Australia
4 Department of Electronic Materials Engineering, Research School of Physics and
Engineering, The Australian National University, Canberra, ACT 2601, Australia
Physical resemblances of a real leaf and nanoleaf
This nanoleaf resembles a real leaf in both the structure and growth mechanism. The
structures of both are shown in Figure S1. The parts of nanoleaf analogue of the real leaf
are given in Table S1. In nature, leaves of vascular plant have mirror symmetry with the
midrib located at the center along the axial axis and they have two flat surfaces, so-called
blade. The margin is the side of the leaf. The petiole is the base of leaf, which attaches the
leaf blade to the stem. Besides, simple leaves initiate as entire structures at the flanks of
the shoot apical meristem [48] and result in a sharp apex. Thus, the sharp apex and
petiole bound the shape of leaves. In our work, the catalyst and base that attached to the
substrate are the boundary of InAs nanoleaves.
Figure S1. The structures of a real leaf (left) and InAs nanoleaf (right).

100
Table S1. Different parts of an InAs nanoleaf analogue of a real leaf.
True leaf InAs nanoleaf
Apex Tip Primary vein (midrib) Twin boundary
Margin Symmetry sidewall Lamina/Blade { } flat surface
Petiole Base
REFERENCES [1] M. J. Bierman, Y. K. A. Lau, A. V. Kvit, A. L. Schmitt, and S. Jin, "Dislocation-Driven
Nanowire Growth and Eshelby Twist," Science, vol. 320, no. 5879, pp. 1060-1063,
May 23, 2008 2008.
[2] J. F. V. Vincent, O. A. Bogatyreva, N. R. Bogatyrev, A. Bowyer, and A.-K. Pahl,
"Biomimetics: its practice and theory," Journal of the Royal Society Interface,
Journal Article vol. 3, no. 9, pp. 471-482, 2006-08-22 00:00:00 2006.
[3] B. Han et al., "Bio-inspired networks for optoelectronic applications," Nature
Communications, Article vol. 5, 11/28/online 2014.
[4] K. Buhl, Z. Roth, P. Srinivasan, R. Rumpf, and E. Johnson, "Biologically inspired
optics: analog semiconductor model of the beetle exoskeleton," in Proceedings of
the SPIE 7057, 2008, pp. 705707-705707-8: International Society for Optics and
Photonics.
[5] X. Duan, Y. Huang, Y. Cui, J. Wang, and C. M. Lieber, "Indium phosphide
nanowires as building blocks for nanoscale electronic and optoelectronic devices,"
Nature, 10.1038/35051047 vol. 409, no. 6816, pp. 66-69, 2001.
[6] Y. Huang, X. Duan, Y. Cui, L. J. Lauhon, K.-H. Kim, and C. M. Lieber, "Logic Gates
and Computation from Assembled Nanowire Building Blocks," Science, vol. 294, no.
5545, pp. 1313-1317, 2001.
[7] Z. Zhong, D. Wang, Y. Cui, M. W. Bockrath, and C. M. Lieber, "Nanowire Crossbar
Arrays as Address Decoders for Integrated Nanosystems," Science, vol. 302, no.
5649, pp. 1377-1379, November 21, 2003 2003.
[8] P. Parkinson et al., "Transient Terahertz Conductivity of GaAs Nanowires," Nano
Letters, vol. 7, no. 7, pp. 2162-2165, 2007.
[9] H. J. Joyce et al., "III–V semiconductor nanowires for optoelectronic device
applications," Progress in Quantum Electronics, vol. 35, no. 2–3, pp. 23-75, 2011.

101
[10] H. Xia et al., "Distinct Photocurrent Response of Individual GaAs Nanowires
Induced by n-Type Doping," ACS Nano, vol. 6, no. 7, pp. 6005-6013, 2012/07/24
2012.
[11] D. Saxena et al., "Optically pumped room-temperature GaAs nanowire lasers,"
Nature Photonics, Letter vol. 7, no. 12, pp. 963-968, 2013.
[12] R. Chau, B. Doyle, S. Datta, J. Kavalieros, and K. Zhang, "Integrated
nanoelectronics for the future," Nat Mater, 10.1038/nmat2014 vol. 6, no. 11, pp.
810-812, 11//print 2007.
[13] A. G. Milnes and A. Y. Polyakov, "Indium arsenide: a semiconductor for high speed
and electro-optical devices," Materials Science and Engineering: B, vol. 18, no. 3,
pp. 237-259, 4/15/ 1993.
[14] S. A. Dayeh, D. P. R. Aplin, X. Zhou, P. K. L. Yu, E. T. Yu, and D. Wang, "High
Electron Mobility InAs Nanowire Field-Effect Transistors," Small, vol. 3, no. 2, pp.
326-332, 2007.
[15] M. T. Björk et al., "Nanowire resonant tunneling diodes," Applied Physics Letters,
vol. 81, no. 23, pp. 4458-4460, 2002.
[16] J. Miao et al., "Single InAs Nanowire Room-Temperature Near-Infrared
Photodetectors," ACS Nano, vol. 8, no. 4, pp. 3628-3635, 2014.
[17] Y.-J. Doh, J. A. van Dam, A. L. Roest, E. P. A. M. Bakkers, L. P. Kouwenhoven,
and S. De Franceschi, "Tunable Supercurrent Through Semiconductor Nanowires,"
Science, vol. 309, no. 5732, pp. 272-275, 2005.
[18] Z. Chun-Zhi, G. Hong, Z. Di, and Z. Xi-Tian, "Local Homoepitaxial Growth and
Optical Properties of ZnO Polar Nanoleaves," Chinese Physics Letters, vol. 25, no.
1, p. 302, 2008.
[19] Y. Yang, Q. Liao, J. Qi, W. Guo, and Y. Zhang, "Synthesis and transverse
electromechanical characterization of single crystalline ZnO nanoleaves," Physical
Chemistry Chemical Physics, 10.1039/B918326D vol. 12, no. 3, pp. 552-555, 2010.
[20] H. Xu, W. Wang, W. Zhu, L. Zhou, and M. Ruan, "Hierarchical-Oriented Attachment:
From One-Dimensional Cu(OH)2 Nanowires to Two-Dimensional CuO
Nanoleaves," Crystal Growth & Design, vol. 7, no. 12, pp. 2720-2724, 2007/12/01
2007.
[21] X. Xu, M. Zhang, J. Feng, and M. Zhang, "Shape-controlled synthesis of single-
crystalline cupric oxide by microwave heating using an ionic liquid," Materials
Letters, vol. 62, no. 17–18, pp. 2787-2790, 6/30/ 2008.

102
[22] Y. J. He, J. F. Peng, W. Chu, Y. Z. Li, and D. G. Tong, "Black mesoporous anatase
TiO2 nanoleaves: a high capacity and high rate anode for aqueous Al-ion batteries,"
Journal of Materials Chemistry A, 10.1039/C3TA13906A vol. 2, no. 6, pp. 1721-
1731, 2014.
[23] P. K. Samanta, S. Basak, and P. R. Chaudhuri, "Fern leaves: The secret life of zinc
oxide," Materials Today, vol. 14, no. 6, p. 295, 6// 2011.
[24] F. Martelli et al., "Photoluminescence of Mn-catalyzed GaAs nanowires grown by
molecular beam epitaxy," Nanotechnology, vol. 18, no. 12, p. 125603, 2007.
[25] J. Li, J. Liu, L.-S. Wang, and R. P. Chang, "Physical and electrical properties of
chemical vapor grown GaN nano/microstructures," Inorganic Chemistry, vol. 47, no.
22, pp. 10325-10329, 2008.
[26] E. Di Giacomo, M. Iannelli, and G. Frugis, "TALE and Shape: How to Make a Leaf
Different," Plants, vol. 2, no. 2, pp. 317-342, 2013.
[27] M. Nakata and K. Okada, "The Leaf Adaxial-Abaxial Boundary and Lamina
Growth," Plants, vol. 2, no. 2, pp. 174-202, 2013.
[28] M. Tsiantis and J. A. Langdale, "The formation of leaves," Current Opinion in Plant
Biology, vol. 1, no. 1, pp. 43-48, 1998.
[29] K. Hiruma et al., "Growth and optical properties of nanometer‐scale GaAs and InAs
whiskers," Journal of Applied Physics, vol. 77, no. 2, pp. 447-462, 1995.
[30] K. A. Dick, K. Deppert, T. Mårtensson, B. Mandl, L. Samuelson, and W. Seifert,
"Failure of the Vapor−Liquid−Solid Mechanism in Au-Assisted MOVPE Growth of
InAs Nanowires," Nano Letters, vol. 5, no. 4, pp. 761-764, 2005/04/01 2005.
[31] S. A. Dayeh, E. T. Yu, and D. Wang, "III−V Nanowire Growth Mechanism: V/III
Ratio and Temperature Effects," Nano Letters, vol. 7, no. 8, pp. 2486-2490, 2007.
[32] Z. Zhang, Z.-Y. Lu, P.-P. Chen, W. Lu, and J. Zou, "Controlling the crystal phase
and structural quality of epitaxial InAs nanowires by tuning V/III ratio in molecular
beam epitaxy," Acta Materialia, vol. 92, no. 0, pp. 25-32, 6/15/ 2015.
[33] P. A. Stadelmann. JAVA-EMS: JEMS. http://cime.epfl.ch/research/jems.
[34] H. Xu et al., "Defect-free< 110> zinc-blende structured InAs nanowires catalyzed by
palladium," Nano Letters, vol. 12, no. 11, pp. 5744-5749, 2012.
[35] Z. Zhang, K. Zheng, Z.-Y. Lu, P.-P. Chen, W. Lu, and J. Zou, "Catalyst Orientation-
Induced Growth of Defect-Free inc- lende Structured 1 InAs Nanowires,"
Nano Letters, vol. 15, no. 2, pp. 876-882, 2015.

103
[36] D. Wolf, "Atomic-level geometry of crystalline interfaces," in Materials interfaces:
atomic-level structure and properties, D. Wolf and S. Yip, Eds. Cambridge UK:
Chapman & Hall, 1992, pp. 1-57.
[37] D. Wolf and K. L. Merkle, "Correlation between the structure and energy of grain
boundaries in metals," in Materials interfaces: atomic-level structure and properties,
D. Wolf and S. Yip, Eds. Cambridge UK: Chapman & Hall, 1992, pp. 87-150.
[38] B. T. Lee, J. Y. Lee, and E. D. Bourret, "Atomic structure of twins in GaAs," Applied
Physics Letters, vol. 57, no. 4, pp. 346-347, 1990.
[39] L. Jin et al., "{113} Twinned ZnSe Bicrystal Nanobelts Filled with <111> Twinnings,"
The Journal of Physical Chemistry C, vol. 112, no. 13, pp. 4903-4907, 2008/04/01
2008.
[40] Z. Zhang, Z. Lu, H. Xu, P. Chen, W. Lu, and J. Zou, "Structure and quality
controlled growth of InAs nanowires through catalyst engineering," (in English),
Nano Research, vol. 7, pp. 1-10, 2014/08/23 2014.
[41] R. S. Wagner and W. C. Ellis, "Vapor‐Liquid‐Solid Mechanism of Single Crystal
Growth," Applied Physics Letters, vol. 4, no. 5, pp. 89-90, 1964.
[42] J. W. Faust Jr and H. F. John, "The growth of semiconductor crystals from solution
using the twin-plane reentrant-edge mechanism," Journal of Physics and Chemistry
of Solids, vol. 25, no. 12, pp. 1407-1415, 12// 1964.
[43] A. D. Gamalski, P. W. Voorhees, C. Ducati, R. Sharma, and S. Hofmann, "Twin
Plane Re-entrant Mechanism for Catalytic Nanowire Growth," Nano Letters, vol. 14,
no. 3, pp. 1288-1292, 2014/03/12 2014.
[44] S. S. Brenner and G. W. Sears, "Mechanism of whisker growth — III nature of
growth sites," Acta Metallurgica, vol. 4, no. 3, pp. 268-270, 1956.
[45] J. Zou et al., "Growth Mechanism of Truncated Triangular III–V Nanowires," Small,
vol. 3, no. 3, pp. 389-393, 2007.
[46] M. Paladugu et al., "Formation of Hierarchical InAs Nanoring / GaAs Nanowire
Heterostructures," Angewandte Chemie International Edition, vol. 48, no. 4, pp.
780-783, 2009.
[47] M. Paladugu et al., "Polarity driven formation of InAs/GaAs hierarchical nanowire
heterostructures," Applied Physics Letters, vol. 93, no. 20, p. 201908, 2008.
[48] D. Vlad et al., "Leaf Shape Evolution Through Duplication, Regulatory
Diversification, and Loss of a Homeobox Gene," Science, vol. 343, no. 6172, pp.
780-783, February 14, 2014 2014.

104
4.2.2 Controlled growth of defect-free zinc-blende [ ̅]-oriented InAs nanobelts
ABSTRACT
Understanding and controlling the structural properties of defect-free semiconductor one-
dimensional (1D) nanostructures is critical in developing future functional devices based
on 1D nanostructures. In this study, growth of [ ̅]-oriented InAs nanobelts on different
types of substrate are investigated systematically. The density of inclined [ ̅]-oriented
InAs nanobelts is higher when growing heteroepitaxy on GaAs ( ̅ ̅ ̅) substrate compared
with growing homoepitaxy on InAs ( ̅ ̅ ̅) substrate. All the [ ̅]-oriented nanobelts,
despite of the substrates, are all defect-free zinc-blende. The reason to induce defect-free
nanobelt is the lower In composition adsorbed into the Au catalyst during growth. Finally,
successful growth vertical [ ̅]-oriented nanobelt on GaAs (001) substrate has been
realised.
KEYWORDS: InAs nanobelts, [ ̅]-orientation, zinc-blende, defect-free
Introduction
Ever since the first reported observation of catalysed whisker growth by Wagner and Ellis,1
semiconductor nanowires have attracted tremendous attention due to their unique
properties and wide application prospects in nanoelectronics and nanophotonics.2 Among
these, InAs semiconductor nanowires exhibit direct bandgap, high carrier mobility and
small electron effective mass.3 III-V nanowires are often grown on the (111)B surfaces of
the III-V materials because this is the surface that easily obtain epitaxial growth of vertical
[ ̅ ̅ ̅]-oriented nanowires induced by Au catalyst.4 Nanowires growing in non-⟨ ⟩
directions are usually harder to be achieved due to the competition between anisotropy
nanowires growth and two-dimensional planar growth on surfaces. Few reports on the
growth of non-⟨ ⟩-oriented nanowires by applying growing surface with different
orientation,5 altering the size of Au catalysts6 and also using a non-Au catalyst.7
The common III-V one-dimensional nanostructure reported is [ ̅ ̅ ̅]-oriented
nanowire owing six hexagonal { }/{ } sidewalls for zinc-blended structure.8 [ ̅ ̅ ̅]-
oriented nanowires of larger diameter have been observed to have twinning superlattice

105
structure composed of truncated octahedral segments with six { } side-facets.9 This is
interest to note that for non-⟨ ⟩-oriented 1D nanostructure, special morphology with
different sidewall orientation has been observed. For example, [ ]-oriented InAs
nanoplates and InP whisker with four { } sidewalls,10 [ ̅ ̅ ]-oriented InAs nanowires
with two { } and four { } sidewalls, and ⟨ ⟩-oriented bicrystalline InAs nanoleaves
with two { } flat surfaces.11
In this study, the growth of defect-free zinc-blended [ ̅]-oriented InAs nanobelts
on different type of substrates are studied. We observed that heteroepitaxy growth of InAs
on GaAs (111)B substrate can yield higher density of nanobelts compared with
homoepitaxy growth of InAs on InAs (111)B substrate. Therefore, by keeping the same
growth parameters, we successfully grow vertical [ ̅]-oriented InAs nanobelts on GaAs
(001) substrate, which may contribute to the development of III-V 1D nanostructures on
the common used Si (001) industry.
Experimental detail
InAs 1D nanosctructures were grown on different substrates with different orientations,
namely InAs (111)B, GaAs (111)B and GaAs (001) substrates via horizontal flow metal-
organic chemical vapor deposition (MOCVD) using trimethylindium (TMIn) as the group III
precursor and arsine (AsH3) as the group V precursor, and Au nanoparticles to induce
nanowire growth. All the substrates were treated with poly-L-lysine (PLL) solution followed
by dispersion of a solution containing 50-nm colloidal Au nanoparticles before loading into
growth chamber. For the 1D nanostructures comparison of the consistency growth
environment, all substrates were loaded into the chamber simultaneously. Prior to growth,
the substrates deposited with the Au particles were annealed in situ at 700 °C under AsH3
ambient to desorb surface contaminants. It has been well documented that at high
annealing temperature under AsH3 ambient, Au colloidal nanoparticles can first react with
the III-V substrates to form Au-III alloy droplets.12 AsH3 is introduced in the chamber during
temperature ramp-up and annealing to prevent As sublimation and preserve the surface
stoichiometry of the substrates. After the substrates were cooled to growth temperature of
500 °C, the AsH3 flow was adjusted and TMIn introduced to initiate the nanowire growth.
The growth time was set as 30 min. The absolute flow rate of TMIn and AsH3 was
respectively controlled at 1.2 x 10-5 mol/min and 3.4 x 10-5 mol/min to attain a V/III ratio of

106
2.9. The pressure of chamber was 100 mbar and a total gas flow rate of 15 slm was
applied.
Detailed morphology, structure and composition of grown InAs nanostructures were
characterized by field-emission scanning electron microscopy (FE-SEM, JEOL JSM-
7800F, operated at 8 kV) and transmission electron microscopy (TEM, FEI Tecnai F20 and
F30, operated at 200 kV and 300 kV, equipped with X-ray energy dispersive spectroscopy
(EDS) for compositional analysis). Atomic-scale STEM HAADF images were taken using
an FEI Titan aberration-corrected TEM operated at 300 kV equipped with an annular dark
field (ADF) detector. Cross-sectional TEM samples were prepared by focused ion beam
(FIB) (FEI Scios Dual Beam FIB/SEM).
Results and discussion
Figure 1a,b show top-view SEM images of Au-catalysed InAs 1D nanostructures grown on
InAs and GaAs ( ̅ ̅ ̅) substrates, from which two kinds of 1D nanostructures were found:
the inclined and vertical aligned, both with respect to the substrate surfaces. The inclined
nanostructures are projected in certain crystallographic orientations, while vertically grown
nanostructures are represented as “white” dots (note that the electron beam is parallel to
the axial direction of these nanostructures). The side-view SEM images revealed that
these “white” dots are vertical nanowires growing along the [ ̅] [ ̅ ̅ ̅] direction (refer to
Figure S1 in Supporting Information). It is of interest to note that the density of inclined 1D
nanostructures grown on the GaAs substrate is much higher (several tens times) than that
on the InAs substrate (refer to Table S1 in Supporting Information), suggesting that the
inclined 1D nanostructures are much easier to be formed on the GaAs substrate. Using
the fact that the cleavage planes for both the zinc-blende structured substrates are { },
the crystallographic geometry of the projection of 1D nanostructures on the substrate can
be determined. As depicted in the inset of Figure 1a, the projections of the 1D
nanostructures on the ( ̅ ̅ ̅) substrates are along ⟨ ⟩ directions, suggesting that the
general growth directions of the inclined 1D nanostructures are along the ⟨ ⟩ or ⟨ ⟩
directions where n is an integer if these 1D nanostructures have the zinc-blende
structure.7, 13 The plan-view SEM specimen was then tilted to determine the axial directions
of these epitaxial inclined 1D nanostructures when they are aligned with the electron
beam. In this case, both the axial direction and sidewall facets of 1D nanostructures can
be determined unambiguously.7, 14 Figure 1c–e is a series of titled SEM images of a typical

107
inclined 1D nanostructure grown on GaAs (111)B substrate. When the titling angle reaches
to ~55° (refer to Figure 1e), the electron beam becomes parallel to the growth direction of
the 1D nanostructure, suggesting the growth direction is [ ̅]. It can be observed that the
1D nanostructure sidewalls have a rectangular cross-section with four sidewall facets of
{ }, namely the ±( ̅ ) and ±( ) planes. This morphological characteristic has been
confirmed by SEM investigations of several inclined 1D nanostructures. Therefore, our 1D
nanostructures are named as nanobelts.
Figure 1 Top-view SEM images of InAs nanowires grown on ( ̅ ̅ ̅)-type (a) InAs and (b)
GaAs substrates under identical growth conditions. The inset in (a) is the sketch of the
orientation relationship between ⟨ ⟩ and ⟨ ⟩ directions on the ( ̅ ̅ ̅) substrate, in
which red arrows and blue arrows representing ⟨ ⟩ and ⟨ ⟩ directions, respectively.
(c–e) A series of SEM images showing the facet details and growth direction of typical
inclined 1D nanostructure, in which the e-beam is (c) parallel, (d) 30°, and (e) 55°
(magnified) tilted to the normal of the ( ̅ ̅ ̅) substrate. The tilt axis is [ ̅ ̅ ].
To understand the structural and chemical characteristics of our inclined nanobelts,
detailed TEM investigation was performed. Figure 2a is a TEM image of a typical inclined
InAs nanobelt and shows the tapered morphology near the growth front, and Figure 2b is
the zoom-in TEM image focusing at the growth front of nanobelt, in which the catalyst-

108
nanobelt interface is ( ̅) that is perpendicular to the growth direction. Figure 2c,d is a
high-resolution TEM (HRTEM) image and the corresponding FFT pattern taken from the
middle section of the nanobelt, and shows that the perfect zinc-blende structure without
any observable defects. Our extensive HRTEM studies over a dozen of [ ̅] nanobelts
confirmed the nature of defect-free zinc-blende structure along the entire nanobelts. Figure
2e is a [ ̅ ] zone-axis STEM HAADF image taken from the other identical nanobelt and
confirms again the zinc-blende structure, in which individual In-As dumbbells are observed
with the larger/brighter atomic columns being In atoms as described in the line intensity
scan outlined in Figure 2g. Figure 2f illustrated the atomic model. Based on this
crystallography information, the growth direction can be determined as [ ̅] and the four
rectangular sidewall facets are ±( ̅ ) and ±( ) planes. This indicates that the lateral
growth rates of these sidewalls are anisotropy, in which lateral growth along ±( )
sidewall facets are faster than that of ±( ̅ ). This is due to the higher density of dangling
bonds of Ga atoms at the ±( ) steps compared with the Ga atoms at the ±( ̅ ) steps
(refer to Figure S2 in Supporting Information).10c, 15 This results in the shorter length of
±( ) sidewall facets than that of ±( ̅ ), which represented by the schematic drawing in
Figure 2h.
Figure 2 Detailed TEM investigations of [ ̅]-oriented InAs nanobelt: (a) the overview of
typical [ ̅] InAs nanobelt; (b) growth front of nanobelt; (c,d) corresponding HRTEM
image and FFT pattern showing defect-free zinc-blende structure. (e) Aberration-corrected
STEM HAADF image of zinc-blended nanobelt. (f) Line intensity scan of the atoms
crossing the In-As dumbbell highlighted in (e). (g) [ ̅ ] zone-axis atomic model of zinc-
blende structured InAs. (h) Atomic configuration of a [ ̅] nanobelt with four { }
sidewalls.

109
To understand the driving force of the inclined nanobelts formation and why they
are much easier to be formed on the GaAs substrate compared with InAs substrate, we
examined the cross-sectional TEM sample of the post-growth InAs and GaAs ( ̅ ̅ ̅)-type
substrates. Figure 3a,b is the cross-sectional TEM images of InAs and GaAs substrates
and a layer of InAs planar layer grew on both of the substrates. The chemical analysis to
confirm the composition of this layer growth is shown in Figure S3 in Supporting
Information. The heteroepitaxy InAs planar layer on GaAs substrate is thicker suggests
that most of the In adatoms on the substrate have been participated in InAs layer growth
rather than adsorb by the catalyst. Our EDS analysis shows that the catalyst of both
vertical nanowires and inclined nanobelts is Au-In alloy (refer to Figure S4 in Supporting
Information). The In composition is ~51 at.% in nanowire catalyst and ~20 at.% in nanobelt
catalyst during growth. Therefore, we deduce that lower In atoms have been adsorbed into
the Au catalyst is the main reason for the growth of this inclined defect-free zinc-blended
InAs nanobelt.
Figure 3 Cross-sectional TEM of post-growth ( ̅ ̅ ̅)-type (a) InAs, and (b) GaAs
substrates. The red dashed line indicates the interface of the InAs planar growth on the
substrate. The InAs layer is ~80 nm on InAs substrate and ~200 nm on GaAs substrate.
Growth of vertical ⟨ ⟩-nanobelt on (001) orientation substrate. Since we identified
that using GaAs ( ̅ ̅ ̅) substrate is an advantage to yield higher density of defect-free zinc-
blende structured [ ̅]-nanobelts, we repeated this identical growth by using GaAs (001)
substrate so that we can grow vertical ⟨ ⟩-nanobelts on (001) orientation substrate.
Figure 4a,b is a SEM images showing the facet details and growth direction of typical
⟨ ⟩-nanobelt growing vertically on GaAs (001) substrate; the e-beam is (a) parallel, and
(b) 55° tilted to the normal of the (001) substrate. The overview of a typical [ ̅]-nanobelt
is shown in Figure 4c with ( ̅) catalyst-nanobelt interface (refer to Figure 4d) and defect-

110
free zinc-blended nanobelt (refer to Figure 4e). The density of the nanobelts grown on
(001) substrate are lower compared with ( ̅ ̅ ̅) substrate due to the competition of layer
growth on (001) substrate. The successful growth of [ ̅]-nanobelts on GaAs (001)
substrate is expected to give an insight to the direct integration of III-V 1D nanostructures
on Si (001) substrate which is the most common industry used orientation in fabricating
electronic devices.
Figure 4 (a,b) SEM images showing the facet details and growth direction of typical [ ̅]-
nanobelt growing vertically on GaAs (001) substrate; the e-beam is (a) parallel, , and (b)
55° tilted to the normal of the (001) substrate. (c) Overview of typical [ ̅] InAs nanowire
grown on GaAs (001) substrate and (d) its corresponding growth front. (e) Corresponding
SAED image of nanobelt showing defect-free zinc-blende structure.
Conclusion
In conclusion, we demonstrated the controlled growth of defect-free zinc-blended [ ̅]-
oriented InAs nanobelts. [ ̅]-type InAs nanobelts grew preferable on GaAs than InAs
( ̅ ̅ ̅)-type substrate. By identifying the reason of heteroepitaxy growth of [ ̅]-type
nanobelt on GaAs substrate, we successfully grew vertical [ ̅]-type nanobelts on GaAs
(001) substrate.

111
SUPPORTING INFORMATION
Controlled growth of defect-free zinc-blende [ ̅]-oriented InAs nanobelts
1. Morphology analysis by SEM
Side-view SEM images revealed that the “white” dots observed in the top-view SEM
images (Figure S1) are vertical nanowires grown perpendicularly to the substrate,
suggesting that these epitaxial nanowires growing along the [ ̅] ⟨ ⟩ direction on
the substrate. The angle between the inclined nanowires and the substrate normal is ~55°
(refer to Figure S1b).
Figure S1. Side-view SEM images of InAs nanowires grown on (a) InAs and (b) GaAs
{ } substrates under identical growth conditions. The angle between 1D nanostructure
growth direction and [ ̅ ̅ ̅] is ~55° as shown in (b).
2. Density of different 1D nanostructures
The density of both types of 1D nanostructures grown on two different materials was
examined by SEM observation over the area of (240x180) µm2, and the comparison of the
results is displayed in Table S1.

112
Table S1 Comparison of different 1D nanostructures density grown on different substrates.
Substrate
InAs (111)B GaAs (111)B
Vertical nanowires
96.6 % 70.5 %
Inclined nanobelts 3.4 % 29.5 %
3. Cross-sections of ( ) steps and ( ̅ ) steps
In As-rich growth environment, As species impinge onto a growing surface is abundant.
Hence low desorption probability for As species and the growing surface is considered to
be completely covered with As species. Atomic model of InAs cross-section of [ ] and
[ ̅ ] steps under As-rich environment is schematically described in Figure S2. Since the
probability of incorporation into the step sites is higher than that for flat surfaces, according
to classical growth model, migrating In atoms are easily caught in the [ ] and/or [ ̅ ]
steps. The In atom at the the [ ̅ ] step ( [ ̅ ]) is bound with two bonds; both to the step
bottom whereas [ ] is bound with three bonds; two to the step bottom and one to the
step side (refer to Figure S2). Therefore, Ga atoms migration is easier to be incorporated
into the [ ] steps than into the [ ̅ ] steps. As a result, the [ ] growth rate is
essentially higher than the [ ̅ ] one in As-rich environment.
Figure S2. Zinc-blended InAs atomic model description of (a) [ ] step viewed along
[ ̅ ] zone axis, and (b) [ ̅ ] step viewed along [ ] zone axis.

113
4. EDS analysis of the planar layer growth and catalyst
Figure S3 is the EDS area mapping of cross-sectional TEM samples of post-growth InAs
nanobelts on ( ̅ ̅ ̅)-type InAs and GaAs substrates. The EDS mapping shows that the
obvious of InAs planar growth on both InAs and GaAs substrates, in which the layer is
thicker on GaAs substrates.
Figure S3. Cross-sectional sample of InAs planar growth on ( ̅ ̅ ̅)-type (a–e) InAs, and
(f–i) GaAs substrate. (a,e) Dark-field STEM image. (b,f) the combination area mapping of
different elements. Elements mapping of (c,g) Ga. The Ga shown at the upper region is
contributed by the Ga deposition during FIB process, (d,h) In, and (e,i) As.
Figure S4 is the EDS spectra show that both post-growth catalyst of vertical nanowire and
inclined nanobelt is Au-In alloy. The In composition of catalyst is higher in catalyst of
nanowire than that of nanobelt.

114
Figure S4. EDS spectra of post-growth catalyst for (a) vertical nanowire, and (b) inclined
nanobelt.
REFERENCES 1. Wagner, R. S.; Ellis, W. C. Vapor‐Liquid‐Solid Mechanism of Single Crystal Growth.
Appl Phys Lett 1964, 4 (5), 89-90.
2. (a) Yan, R. X.; Gargas, D.; Yang, P. D. Nanowire photonics. Nat Photon 2009, 3; (b)
Akiyama, T.; Yamashita, T.; Nakamura, K.; Ito, T. Band Alignment Tuning in Twin-
Plane Superlattices of Semiconductor Nanowires. Nano Lett 2010, 10 (11), 4614-
4618; (c) Lu, W.; Lieber, C. M. Nanoelectronics from the bottom up. Nature
materials 2007, 6 (11), 841-850.
3. (a) Björk, M. T.; Ohlsson, B. J.; Thelander, C.; Persson, A. I.; Deppert, K.;
Wallenberg, L. R.; Samuelson, L. Nanowire resonant tunneling diodes. Appl Phys
Lett 2002, 81 (23), 4458-4460; (b) Doh, Y.-J.; van Dam, J. A.; Roest, A. L.; Bakkers,
E. P. A. M.; Kouwenhoven, L. P.; De Franceschi, S. Tunable Supercurrent Through
Semiconductor Nanowires. Science 2005, 309 (5732), 272-275; (c) Miao, J.; Hu,
W.; Guo, N.; Lu, Z.; Zou, X.; Liao, L.; Shi, S.; Chen, P.; Fan, Z.; Ho, J. C.; Li, T.-X.;
Chen, X. S.; Lu, W. Single InAs Nanowire Room-Temperature Near-Infrared
Photodetectors. ACS Nano 2014, 8 (4), 3628-3635.
4. (a) Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Zhang, X.; Guo, Y.; Zou,
J. Twin-Free Uniform Epitaxial GaAs Nanowires Grown by a Two-Temperature
Process. Nano Lett 2007, 7 (4), 921-926; (b) Zhang, Z.; Lu, Z.-Y.; Chen, P.-P.; Xu,
H.-Y.; Guo, Y.-N.; Liao, Z.-M.; Shi, S.-X.; Lu, W.; Zou, J. Quality of epitaxial InAs
nanowires controlled by catalyst size in molecular beam epitaxy. Appl Phys Lett
2013, 103 (7), 073109.

115
5. (a) Fonseka, H. A.; Caroff, P.; Wong-Leung, J.; Ameruddin, A. S.; Tan, H. H.;
Jagadish, C. Nanowires Grown on InP (100): Growth Directions, Facets, Crystal
Structures, and Relative Yield Control. ACS Nano 2014; (b) Krishnamachari, U.;
Borgstrom, M.; Ohlsson, B. J.; Panev, N.; Samuelson, L.; Seifert, W.; Larsson, M.
W.; Wallenberg, L. R. Defect-free InP nanowires grown in [001] direction on InP
(001). Appl Phys Lett 2004, 85 (11), 2077-2079.
6. Cai, Y.; Chan, S. K.; Sou, I. K.; Chan, Y. F.; Su, D. S.; Wang, N. The Size-
Dependent Growth Direction of ZnSe Nanowires. Advanced Materials 2006, 18 (1),
109-114.
7. Xu, H.; Wang, Y.; Guo, Y.; Liao, Z.; Gao, Q.; Tan, H. H.; Jagadish, C.; Zou, J.
Defect-free< 110> zinc-blende structured InAs nanowires catalyzed by palladium.
Nano Lett 2012, 12 (11), 5744-5749.
8. (a) Verheijen, M. A.; Algra, R. E.; Borgström, M. T.; Immink, G.; Sourty, E.; van
Enckevort, W. J. P.; Vlieg, E.; Bakkers, E. P. A. M. Three-Dimensional Morphology
of GaP−GaAs Nanowires Revealed by Transmission Electron Microscopy
Tomography. Nano Lett 2007, 7 (10), 3051-3055; (b) Jiang, N.; Wong-Leung, J.;
Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C. Understanding the True Shape of
Au-Catalyzed GaAs Nanowires. Nano Lett 2014, 14 (10), 5865-5872.
9. (a) Algra, R. E.; Verheijen, M. A.; Borgstrom, M. T.; Feiner, L.-F.; Immink, G.; van
Enckevort, W. J. P.; Vlieg, E.; Bakkers, E. P. A. M. Twinning superlattices in indium
phosphide nanowires. Nature 2008, 456 (7220), 369-372; (b) Burgess, T.; Breuer,
S.; Caroff, P.; Wong-Leung, J.; Gao, Q.; Hoe Tan, H.; Jagadish, C. Twinning
Superlattice Formation in GaAs Nanowires. ACS Nano 2013, 7 (9), 8105-8114.
10. (a) Aagesen, M.; Johnson, E.; Sorensen, C. B.; Mariager, S. O.; Feidenhans'l, R.;
Spiecker, E.; Nygard, J.; Lindelof, P. E. Molecular beam epitaxy growth of free-
standing plane-parallel InAs nanoplates. Nat Nano 2007, 2 (12), 761-764; (b)
Seifert, W.; Borgström, M.; Deppert, K.; Dick, K. A.; Johansson, J.; Larsson, M. W.;
Mårtensson, T.; Sköld, N.; Patrik T. Svensson, C.; Wacaser, B. A.; Reine
Wallenberg, L.; Samuelson, L. Growth of one-dimensional nanostructures in
MOVPE. Journal of Crystal Growth 2004, 272 (1–4), 211-220; (c) Zhang, Z.; Zheng,
K.; Lu, Z.-Y.; Chen, P.-P.; Lu, W.; Zou, J. Catalyst Orientation-Induced Growth of
Defect-Free Zinc- lende Structured nAs Nanowires Nano Lett 2015, 15 (2),
876-882.
11. Soo, M. T.; Zheng, K.; Gao, Q.; Tan, H. H.; Jagadish, C.; Zou, J. Mirror-twin induced
bicrystalline InAs nanoleaves. Nano Res. 2016, 9 (3), 766-773.

116
12. (a) Hiruma, K.; Yazawa, M.; Katsuyama, T.; Ogawa, K.; Haraguchi, K.; Koguchi, M.;
Kakibayashi, H. Growth and optical properties of nanometer‐scale GaAs and InAs
whiskers. J Appl Phys 1995, 77 (2), 447-462; (b) Dick, K. A.; Deppert, K.;
Mårtensson, T.; Mandl, B.; Samuelson, L.; Seifert, W. Failure of the
Vapor−Liquid−Solid Mechanism in Au-Assisted MOVPE Growth of InAs Nanowires.
Nano Lett 2005, 5 (4), 761-764; (c) Dayeh, S A ; Yu, E T ; Wang, D −V
Nanowire Growth Mechanism: V/ Ratio and Temperature Effects Nano Lett
2007, 7 (8), 2486-2490.
13. Zhang, Z.; Lu, Z.; Xu, H.; Chen, P.; Lu, W.; Zou, J. Structure and quality controlled
growth of InAs nanowires through catalyst engineering. Nano Res. 2014, 7, 1-10.
14. Zhang, Z.; Han, X.; Zou, J. Direct realizing the growth direction of epitaxial
nanowires by electron microscopy. Sci. China Mater. 2015, 58 (6), 433-440.
15. Asai, H. Anisotropic lateral growth in GaAs MOCVD layers on (001) substrates.
Journal of Crystal Growth 1987, 80 (2), 425-433.

117
CHAPTER 5
Understanding the Growth of GaAs 1D
Nanostructures Catalysed by Au Nanoparticles
5.1 Introduction
Growth temperatures exert a significant influence on III-V nanowires growth. It is one of
the most important parameters that have substantial influence on the growth rate and
morphology of resulting nanowires. In this chapter, the effect of the growth temperature on
the side-facets of Au-catalysed GaAs nanopillars with hexagonal base was studied. We
found that the side-facets of zinc-blende GaAs nanopillars transform from { } to { }
with increasing the growth temperature because of competition between kinetically and
thermodynamically dominated process. Besides, diffusion-induced nanopillar foundations
always present the same { } edge side-facets regardless of the temperature.
5.2 Journal publication
Results in Section 5.2.1 are included as it appears in Semiconductor Science and
Technology 2016, 31, 094004. http://iopscience.iop.org/article/10.1088/0268-
1242/31/9/094004/meta

118
5.2.1 Temperature-dependent side-facets of GaAs nanopillars
Temperature-dependent side-facets of GaAs nanopillars
Mun Teng Soo,1 Kun Zheng,2,3,* Qiang Gao,4 Hark Hoe Tan,4 Chennupati Jagadish4 and
Jin Zou1,2 *
1 Materials Engineering, The University of Queensland, Australia, QLD 4072, Australia
2 Centre for Microscopy and Microanalysis, The University of Queensland, QLD 4072,
Australia
3 Australian Institute for Bioengineering and Nanotechnology, The University of
Queensland, QLD 4072, Australia
4 Department of Electronic Materials Engineering, Research School of Physics and
Engineering, The Australian National University, Canberra, ACT 2601, Australia
ABSTRACT
In this study, the effect of growth temperature on the structural properties of Au-catalysed
epitaxial GaAs semiconductor nanopillars grown by metal-organic chemical vapour
deposition is investigated by electron microscopy. It has been found that the growth
temperature plays a significant role on the evolution of side-facets of zinc-blende
structured GaAs nanopillars. At a growth temperature of 550 °C, six { } side-facets are
formed; whereas at a higher growth temperature of 600 °C, six { } side-facets are
observed. It is believed that the formation of { } side-facets is a kinetically dominated
process while the formation of { } side-facets is a thermodynamical process. Besides,
the diffusion-induced nanopillar foundations present the same { } edge side-facets
regardless of the growth temperature.
KEYWORDS: GaAs, side-facets, temperature, kinetic, thermodynamic
1. Introduction
One dimensional (1D) semiconductor nanostructures, such as nanowires and nanopillars,
represent a unique system for exploring phenomena at the nanoscale and have been one
of the most active globally-focused research fields over the past decades due to their

119
critical role as nanoscale building blocks for the next generation electronic and
optoelectronic applications [1-5]. As one of the key class of 1D III-V semiconductor
nanostructures, GaAs have drawn significant attention due to their high electron mobility
and absorption coefficients, wide direct bandgap (~1.4 eV) as well as their possibilities to
fabricate 1D nanostructures and their compatibility with Si technology, which made them a
promising candidate for the extensive applications in nano-photonic and nano-electronic
devices [6-10].
The surface facets of 1D nanostructures are important in determining their
electronic [11-14], thermoelectric [15], and structural [16, 17] properties due to their large
surface-to-volume ratio. Systematic surface investigations of 1D nanostructure side-facets
are especially relevant as the limited size of their facets can result in behaviour not found
for “infinite” planar surfaces and also lead to enhanced fundamental understanding on
shell growth of 1D nanostructure [18]. Besides, for most of the envisaged applications
based on 1D nanostructures, surfaces are the locations where most of the device activity
takes place. For example, 1D nanostructure surfaces play an important role in the
formation of contacts for nano-electronic applications [19, 20], the passivation with
functionalizing groups or dyes for solar cells [21, 22], and molecular detection for nano-
sensors [23-26].
1D III-V nanostructures, which generally grow via the vapour-liquid-solid
mechanism, exhibit side-facets of different orientations. For instance, 1D III-V nanowires
growing epitaxially along [ ̅ ̅ ̅] [ ̅] directions, most of them have hexagonal cross-
section with six { ̅ } or six { ̅} side-facets for zinc-blende structured 1D nanostructures
[27-29] or six { ̅ } or six { ̅ } side-facets for their wurtzite counterparts [30, 31]. In
zinc-blende structured 1D III-V nanostructures containing regular twins, six { } side-
facets have been experimentally observed [32-34]. During the axial growth, the side-facets
of 1D III-V nanostructures can grow radially due to the adsorption of group-III adatoms on
the side-facets [35].
In this study, we examined the effect of the growth temperature on the side-facets
of GaAs nanopillars with hexagonal base. We found that the side-facets of zinc-blende
GaAs nanopillars transform from { } to { } with increasing the growth temperature as
a result of competition between kinetically and thermodynamically dominated process.

120
Besides, diffusion-induced nanopillar foundations always present the same { } edge
side-facets regardless of the temperature.
2. Experimental
GaAs nanopillars were epitaxially grown on GaAs ( ) substrates (pre-treated with poly-
L-lysine) using a horizontal flow metal-organic chemical vapour deposition (MOCVD)
system at a constant pressure of 100 mbar. The total input of gas flow rate was maintained
at 15 slm. Au nanoparticles with a diameter of 50 nm (from commercially available colloidal
Au solution) were used as catalysts for inducing the nanopillar growth. Prior to growth,
each substrate deposited with the Au nanoparticles was annealed in situ at 700 °C under
AsH3 ambient. Trimethylgallium (TMGa) and arsine (AsH3) were used as the precursors for
Ga and As, respectively. The growth time was 15 min and the V/III ratio was 5. The growth
temperature was set to either 550 or 600 °C.
The morphological characteristics of the grown nanostructures were investigated by field-
emission scanning electron microscopy (FE-SEM, JEOL JSM-7800F, 5 kV). Transmission
electron microscopy (Philips Tecnai F20, 200 kV) was also used to determine the crystal
structures of grown nanostructures and it has been found that they have the zinc-blende
structure. The cross-sections of individual nanostructures for TEM observations was
prepared by embedding the nanostructures in resin (EPON) and then sectioning with an
ultramicrotome (Leica EM UC6). The initial section was cut from the bottom of the
nanostructures with the location and thickness of each subsequent cross-sectional slice
carefully controlled and measured (approximately 50 nm per section in this study). The thin
slices of nanostructure cross-sections were then transferred onto Cu mesh grids. Besides,
the cross-sectional TEM specimen of post-annealed sample was prepared by focused ion
beam (FIB) (FEI Scios Dual Beam FIB/SEM).
3. Results and discussion
Figure 1a,b are typical plan-view SEM images taken from both samples, in which the
diameter of the grown nanostructures is smaller in the sample grown at 550 °C, compared
to that grown at 600 °C. To understand the morphological characteristics of these
nanostructures, the SEM specimens were tilted for 45°, and corresponding SEM images
were taken, as shown in Figure 1c,d, respectively. As can be seen, the grown
nanostructures with the nanopillar morphology and hexagonal bases grow vertically from

121
the substrate for both cases. By comparing these two samples, the length of nanopillars
increases with temperature, which is attributed to the lower energy barrier and the
increased surface mobility of group-III adatoms [36, 37]. In addition, radial growth at side-
facets of nanopillars can be witnessed at higher growth temperature leading to significant
tapering [38]. However, upon careful observation, the two samples have different
morphologies. The magnified SEM images of an individual nanopillars from each sample
are shown in Fig. 1e,f, in which different regions can be distinguished by their respective
contrasts. Although both have a hexagonal cross section, the sample grown at 550 °C has
two regions (indicated as R1 and R2), whereas the sample grown at 600 °C has three
different regions (indicated as R1, R1-2 and R2), in which the edges of R1 are rotated by
30° when compared with R1-2 and R2. We can thus conclude that, in both cases, the
sections related to R1 are the nanopillar sections, R2 sections are the nanopillar
foundations, while R1-2 section of nanopillars grown at 600 °C is the connecting section
between R1 and R2. The foundation is a lateral growth at the root of a nanopillar due to
the Ga surface diffusion [39]. It has been reported that the elbow where the nanopillar
meets the substrate surface has a higher growth rate with increasing available diffusion
area and develops into the foundation [40, 41].
Figure 1. SEM images of GaAs nanopillars grown at 550 °C (a, c, e) and 600 °C (b, d, f),
respectively. (a,b) Top-view SEM images. (c,d) Corresponding SEM images taken with the
samples tilted at 45°. Scale bars are 1 µm for (a–d). (e,f) Magnified top-view SEM images
of the GaAs nanopillars, viewed along the nanopillar growth direction.

122
In order to determine the aforementioned side-facets of nanopillars and
foundations of both samples, we applied the knowledge of crystallographic relationship of
epitaxial nanopillars and substrate. It has been well-documented that the cleavage planes
of zinc-blende structured GaAs are along the { } atomic planes [42], which can be used
to determine the crystallographic information of the nanopillar side-facets [43]. Figure 2a,b
are low-magnification plan-view SEM images showing the cleaved edges of the GaAs
substrates, from which the { } planes can be identified. Figure 2c,d are plan-view SEM
images taken from the edges of the sample, from which the side-facets of nanopillars can
be conclusively determined. Accordingly, the edges of nanopillar foundations (namely R2)
can be determined as six { } side-facets. On the other hand, the nanopillars grown at
550 °C have six { } side-facets whereas those grown at 600 °C have six { } side-
facets. The connecting R1-2 section of nanopillars grown at 600 °C has the combination of
both six { } and six{ } side-facets.
Figure 2. SEM images showing the { } cleavage planes to identify the side-facets of
GaAs nanopillars grown at 550 °C (a, c) and 600 °C (b, d). Inset in (b) is the sketch of the
orientation relationship between ⟨ ⟩ and ⟨ ⟩ directions on the ( ) surface, in which
green arrows and orange arrows represent the ⟨ ⟩ and ⟨ ⟩ directions, respectively.
(c,d) Top-view SEM images of GaAs nanopillars taken with the cleaved edges.

123
In addition, the side-facets of the nanopillars and their corresponding foundations
(in particular, nanopillars and their associated foundations grown at 600 °C that consists of
both { } and { } side-facets at the top and bottom region, respectively) are further
verified by TEM investigations of the cross-sections of individual nanopillars. By
investigating the facets of different sections along the nanopillars, the growth behaviour of
nanopillars and their corresponding foundations may be clarified. Figure 3a–c are SEM
images of GaAs nanopillars grown at 600 °C, viewed along different directions, namely
top, tilted (45°) and side view (along the direction that is perpendicular to the cleavage
plane). Figure 3d–f show the bright-field TEM images taken respectively from cross-
sections obtained from the top, middle and bottom regions of GaAs nanopillars grown at
600 °C, as indicated by the green, pink and orange lines marked in SEM images shown in
Fig. 3a–c. Figure 3g–i are the corresponding selected area electron diffraction (SAED)
patterns. By correlating the side-facets found in TEM images and the diffraction spots
indexed in the SAED patterns, the side-facets of different regions can be determined. As
marked in Fig. 3d-f, the side-facets at the top region are { } and the side-facets of the
bottom region is { } whereas the middle region consists of both { } and { }side-
facets, which are in excellent agreement with the determined side-facets using SEM.
Based on our detailed characterization outlined above, we conclude that (1) the
side-facets of GaAs nanopillars changed from { } to { } with increasing growth
temperature; and (2) the edges of nanopillar foundations always retain at { } side-facets
in spite of the temperatures.
To understand the different side-facets of epitaxial nanopillars grown at different
temperatures, we need to compare the surface energy (γ) for different facets. It has been
calculated that, for zinc-blende structured GaAs, { } = 1.79 J/m2 and { } = 1.54 J/m2
[44], so that the fact of { } > { } indicates that the { } surfaces have a lower
energy than the { } surfaces, and thus the { } side-facets are energetically stable
surfaces when compared with { } side-facets. The fact that { } side-facets have been
constantly observed at lower growth temperatures [27, 45] indicates that the formation of
{ } side-facets at relatively low growth temperature is kinetically controlled process. It is
anticipated that there is a kinetic energy barrier for the formation of { } side-facets [18].
At higher growth temperature, say 600 °C in our case, higher thermal energy is available in
the system. In this case, surface atoms can gain higher kinetic energy to overcome the
energy barrier, so that nanopillar sidewalls can adopt the lower surface energy side-facets

124
of { }. Therefore, we believe that the formation of { } side-facets is
thermodynamically controlled.
Figure 3. SEM images of the nanopillars grown at 600 °C: (a) top-view; (b) 45° tilted-view;
and (c) side-view. (d–f) Cross-sectional TEM images from the top, middle and bottom of
GaAs nanopillars as indicated by green, pink and orange lines, respectively, in (a–c). The
{ } and { } facets are indicated. (g–i) Corresponding SAED patterns taken along
[ ̅̅ ̅̅ ̅] zone axis of (d–f), respectively.
It is of interest to note that facet change of GaAs nanopillars from the { } side-
facets to the { } side-facets has been achieved through annealing them at 750 °C [18],
which has been explained as the result of surface atom migration to overcome the kinetic
energy barrier. In our study, { } side-facets of zinc-blende structured GaAs nanopillars
were achieved by setting the growth temperature of 600 °C. These studies suggest that
thermodynamically favoured { } side-facets of GaAs nanopillars can be achieved either
by growing the nanopillars at a relatively high temperature (say 600 °C in our case) or by
annealing the pre-grown nanopillars at high temperature (such as 750 °C [18]). The

125
comparison of both cases indicates that facet switching via annealing needs a higher
temperature, possibly due to the fact that the atomic migration from pre-grown nanopillars
requires a higher thermal energy.
It is well known that the nanopillar and its corresponding foundation are formed
differently [40, 41]. Therefore, we anticipate that two growth modes are involved: (1)
vapour-liquid-solid mechanism which leads to the axial growth of nanopillars. The change
of side-facets from { } to { } is governed by kinetically controlled process (at low
temperature) and thermodynamically controlled process (at high temperature),
respectively; (2) adatoms diffusion-induced process which results in the formation of
nanopillar foundations and the tapering of nanopillars. Figure 4 is the schematic diagram
to illustrate the formation of our nanopillars. (i) Deposition of Au catalysts on the substrate.
(ii) Annealing of the Au deposited substrate. At this stage, Ga adatoms on substrate
diffuse towards the catalyst in which a foundation forms below the catalyst (refer to the
inset, a cross-sectional ⟨ ⟩ TEM image showing the annealed catalyst with its underlying
foundation). (iii) Growth of nanopillars at different temperatures. At this stage, vapour-
liquid-solid mechanism leads to the growth of nanopillars with different side-facets: { }
at 550 °C through a kinetically-controlled process; and { } at 600 °C through a
thermodynamically controlled process. As can be seen from inset of Fig. 4(ii), the GaAs
foundation forms below the catalyst even after the annealing process, agreed with our
previous study [46]. The increasing of size of foundations is attributed to the increased
diffusion length of adatoms on the substrate with increasing the temperature [40], through
a diffusion-induced process.

126
Figure 4. Schematic diagram shows the flow of formation of GaAs nanopillars at two
different temperatures: (i) Deposition of Au catalyst on substrate. (ii) Annealing of Au-
deposited substrate. The inset is the cross-sectional ⟨ ⟩ TEM image of annealed
catalyst with its underlying foundation. (iii) Growth of nanopillars at 550 and 600 °C.
4. Conclusion
In conclusion, we have demonstrated that the transformation of side-facets of zinc-blende
structured GaAs nanopillars from { } to { } by controlling growth temperature. The
formation of { } side-facets which have higher surface energy is a kinetically limited
process. On the other hand, the formation of { } side-facets is a thermodynamically
controlled process. The diffusion-induced nanopillar foundations retain the { } edge
side-facets despite growth temperature. This study verifies the importance of growth
temperature on the formation of the side-facets of GaAs nanopillars.
Acknowledgement
The Australian Research Council is acknowledged for providing financial support to this
project. The Australian National Fabrication Facility and Australian Microscopy and
Microanalysis Research Facility, both established under the Australian Government’s
National Collaborative Research Infrastructure Strategy, are gratefully acknowledged for
providing access to the facilities used in this work.

127
References [1] Q. Gao et al., "Selective-Area Epitaxy of Pure Wurtzite InP Nanowires: High
Quantum Efficiency and Room-Temperature Lasing," Nano Letters, vol. 14, no. 9,
pp. 5206-5211, 2014/09/10 2014.
[2] M. S. Vitiello et al., "Room-Temperature Terahertz Detectors Based on
Semiconductor Nanowire Field-Effect Transistors," Nano Letters, vol. 12, no. 1, pp.
96-101, 2012/01/11 2012.
[3] F. Patolsky et al., "Detection, Stimulation, and Inhibition of Neuronal Signals with
High-Density Nanowire Transistor Arrays," Science, vol. 313, no. 5790, pp. 1100-
1104, 2006-08-25 00:00:00 2006.
[4] M. W. Shao, Y. Y. Shan, N. B. Wong, and S. T. Lee, "Silicon Nanowire Sensors for
Bioanalytical Applications: Glucose and Hydrogen Peroxide Detection," Advanced
Functional Materials, vol. 15, no. 9, pp. 1478-1482, 2005.
[5] P. Krogstrup et al., "Single-nanowire solar cells beyond the Shockley-Queisser
limit," Nat Photon, 10.1038/nphoton.2013.32 vol. 7, no. 4, pp. 306-310, 04//print
2013.
[6] D. Saxena et al., "Optically pumped room-temperature GaAs nanowire lasers,"
Nature Photonics, Letter vol. 7, no. 12, pp. 963-968, 2013.
[7] K. Tomioka, J. Motohisa, S. Hara, K. Hiruma, and T. Fukui, "GaAs/AlGaAs Core
Multishell Nanowire-Based Light-Emitting Diodes on Si," Nano Letters, vol. 10, no.
5, pp. 1639-1644, 2010/05/12 2010.
[8] N. Han et al., "High performance GaAs nanowire solar cells for flexible and
transparent photovoltaics," ACS Applied Materials & Interfaces, vol. 7, no. 36, pp.
20454-20459, 2015/08/18 2015.
[9] S. Perera et al., "Nearly intrinsic exciton lifetimes in single twin-free GaAs∕AlGaAs
core-shell nanowire heterostructures," Applied Physics Letters, vol. 93, no. 5, p.
053110, 2008.
[10] N. Han et al., "GaAs Nanowires: From Manipulation of Defect Formation to
Controllable Electronic Transport Properties," ACS Nano, vol. 7, no. 10, pp. 9138-
9146, 2013/10/22 2013.
[11] D. Migas, A. Filonov, D. Yatsyna, D. Rusli, and C. Soci, "Role of edge facets on
stability and electronic properties of III–V nanowires," (in English), Nano
Convergence, vol. 2, no. 1, pp. 1-6, 2015/07/08 2015, Art. no. 14.

128
[12] M. Rosini and R. Magri, "Surface Effects on the Atomic and Electronic Structure of
Unpassivated GaAs Nanowires," ACS Nano, vol. 4, no. 10, pp. 6021-6031,
2010/10/26 2010.
[13] H. Shu, X. Chen, Z. Ding, R. Dong, and W. Lu, "First-Principles Study of the Doping
of InAs Nanowires: Role of Surface Dangling Bonds," The Journal of Physical
Chemistry C, vol. 115, no. 30, pp. 14449-14454, 2011/08/04 2011.
[14] Y. Zhao and B. I. Yakobson, "What is the Ground-State Structure of the Thinnest Si
Nanowires?," Physical Review Letters, vol. 91, no. 3, p. 035501, 07/17/ 2003.
[15] F. Sansoz, "Surface Faceting Dependence of Thermal Transport in Silicon
Nanowires," Nano Letters, vol. 11, no. 12, pp. 5378-5382, 2011.
[16] F. Li, P. D. Nellist, C. Lang, and D. J. H. Cockayne, "Dependence of Surface Facet
Period on the Diameter of Nanowires," ACS Nano, vol. 4, no. 2, pp. 632-636,
2010/02/23 2010.
[17] X. Yang, H. Shu, P. Liang, D. Cao, and X. Chen, "Crystal Phase and Facet Effects
on the Structural Stability and Electronic Properties of GaP Nanowires," The Journal
of Physical Chemistry C, vol. 119, no. 21, pp. 12030-12036, 2015/05/28 2015.
[18] N. Jiang, J. Wong-Leung, H. J. Joyce, Q. Gao, H. H. Tan, and C. Jagadish,
"Understanding the True Shape of Au-Catalyzed GaAs Nanowires," Nano Letters,
vol. 14, no. 10, pp. 5865-5872, 2014/10/08 2014.
[19] S. A. Dayeh, P. Chen, Y. Jing, E. T. Yu, S. S. Lau, and D. Wang, "Integration of
vertical InAs nanowire arrays on insulator-on-silicon for electrical isolation," Applied
Physics Letters, vol. 93, no. 20, p. 203109, 2008.
[20] S. A. Dayeh, C. Soci, X.-Y. Bao, and D. Wang, "Advances in the synthesis of InAs
and GaAs nanowires for electronic applications," Nano Today, vol. 4, no. 4, pp. 347-
358, 8// 2009.
[21] C. J. Novotny, E. T. Yu, and P. K. L. Yu, "InP Nanowire/Polymer Hybrid
Photodiode," Nano Letters, vol. 8, no. 3, pp. 775-779, 2008/03/01 2008.
[22] S. Ren, N. Zhao, S. C. Crawford, M. Tambe, V. Bulović, and S. Gradečak,
"Heterojunction Photovoltaics Using GaAs Nanowires and Conjugated Polymers,"
Nano Letters, vol. 11, no. 2, pp. 408-413, 2011/02/09 2011.
[23] Y. Cui, Q. Wei, H. Park, and C. M. Lieber, "Nanowire Nanosensors for Highly
Sensitive and Selective Detection of Biological and Chemical Species," Science,
vol. 293, no. 5533, pp. 1289-1292, 2001.

129
[24] A. Artzy Schnirman et al., "Antibody Molecules Discriminate between Crystalline
Facets of a Gallium Arsenide Semiconductor," Nano Letters, vol. 6, no. 9, pp. 1870-
1874, 2006/09/01 2006.
[25] J. Du, D. Liang, H. Tang, and X. P. A. Gao, "InAs Nanowire Transistors as Gas
Sensor and the Response Mechanism," Nano Letters, vol. 9, no. 12, pp. 4348-4351,
2009/12/09 2009.
[26] P. Offermans, M. Crego-Calama, and S. H. Brongersma, "Gas Detection with
Vertical InAs Nanowire Arrays," Nano Letters, vol. 10, no. 7, pp. 2412-2415, 2010.
[27] J. Zou et al., "Growth Mechanism of Truncated Triangular III–V Nanowires," Small,
vol. 3, no. 3, pp. 389-393, 2007.
[28] M. A. Verheijen et al., "Three-Dimensional Morphology of GaP−GaAs Nanowires
Revealed by Transmission Electron Microscopy Tomography," Nano Letters, vol. 7,
no. 10, pp. 3051-3055, 2007.
[29] H. Xu et al., "High-Density, Defect-Free, and Taper-Restrained Epitaxial GaAs
Nanowires Induced from Annealed Au Thin Films," Crystal Growth & Design, vol.
12, no. 4, pp. 2018-2022, 2012.
[30] H. J. Joyce, J. Wong-Leung, Q. Gao, H. H. Tan, and C. Jagadish, "Phase
Perfection in Zinc Blende and Wurtzite III−V Nanowires Using Basic Growth
Parameters," Nano Letters, vol. 10, no. 3, pp. 908-915, 2010.
[31] C. Zhou et al., "Quality Control of GaAs Nanowire Structures by Limiting As Flux in
Molecular Beam Epitaxy," The Journal of Physical Chemistry C, vol. 119, no. 35,
pp. 20721-20727, 2015/08/03 2015.
[32] T. Burgess et al., "Twinning Superlattice Formation in GaAs Nanowires," ACS
Nano, vol. 7, no. 9, pp. 8105-8114, 2013/09/24 2013.
[33] P. Caroff, K. A. Dick, JohanssonJ, M. E. Messing, DeppertK, and SamuelsonL,
"Controlled polytypic and twin-plane superlattices in III-V nanowires," Nat Nano,
10.1038/nnano.2008.359 vol. 4, no. 1, pp. 50-55, 2009.
[34] J. Johansson et al., "Structural properties of <111> B-oriented III–V nanowires,"
Nature Materials, vol. 5, no. 7, pp. 574-580, 2006.
[35] H. J. Joyce, G. Qiang, J. Wong-Leung, Y. Kim, H. H. Tan, and C. Jagadish,
"Tailoring GaAs, InAs, and InGaAs Nanowires for Optoelectronic Device
Applications," Selected Topics in Quantum Electronics, IEEE Journal of, vol. 17, no.
4, pp. 766-778, 2011.
[36] S. A. Dayeh, E. T. Yu, and D. Wang, "III−V Nanowire Growth Mechanism: V/III
Ratio and Temperature Effects," Nano Letters, vol. 7, no. 8, pp. 2486-2490, 2007.

130
[37] S. A. Dayeh, E. T. Yu, and D. Wang, "Surface Diffusion and Substrate−Nanowire
Adatom Exchange in InAs Nanowire Growth," Nano Letters, vol. 9, no. 5, pp. 1967-
1972, 2009/05/13 2009.
[38] H. Kenji, H. Keiichi, Y. Masamitsu, M. Yuuichi, and K. Toshio, "Nanometre-sized
GaAs wires grown by organo-metallic vapour-phase epitaxy," Nanotechnology, vol.
17, no. 11, p. S369, 2006.
[39] J. R. Arthur, "Interaction of Ga and As2 Molecular Beams with GaAs Surfaces,"
Journal of Applied Physics, vol. 39, no. 8, pp. 4032-4034, 1968.
[40] A. I. Persson, B. J. Ohlsson, S. Jeppesen, and L. Samuelson, "Growth mechanisms
for GaAs nanowires grown in CBE," Journal of Crystal Growth, vol. 272, no. 1–4,
pp. 167-174, 12/10/ 2004.
[41] Y. Kim et al., "Influence of Nanowire Density on the Shape and Optical Properties of
Ternary InGaAs Nanowires," Nano Letters, vol. 6, no. 4, pp. 599-604, 2006/04/01
2006.
[42] A. U. M. Rae and G. W. Gobeli, "Low‐Energy Electron‐Diffraction Study of the
Cleaved (110) Surfaces of InSb, InAs, GaAs, and GaSb," Journal of Applied
Physics, vol. 35, no. 5, pp. 1629-1638, 1964.
[43] Z. Zhang, X. Han, and J. Zou, "Direct realizing the growth direction of epitaxial
nanowires by electron microscopy," (in English), Science China Materials, vol. 58,
no. 6, pp. 433-440, 2015/06/06 2015.
[44] V. G. Dubrovskii, "Crystal Structure of III-V Nanowires," in Nucleation Theory and
Growth of Nanostructures: Springer, 2014, pp. 499-571.
[45] Y.-N. Guo, T. Burgess, Q. Gao, H. H. Tan, C. Jagadish, and J. Zou, "Polarity-driven
Nonuniform Composition in InGaAs Nanowires," Nano Letters, vol. 13, no. 11, pp.
5085-5089, 2013.
[46] Z.-M. Liao et al., "Au impact on GaAs epitaxial growth on GaAs (111)B substrates in
molecular beam epitaxy," Applied Physics Letters, vol. 102, no. 6, p. 063106, 2013.

131
CHAPTER 6
Exploring the growth of III-V nanowires using non-
Au metals
6.1 Introduction
Chapter 5 and 6 described the growth of III-V 1D nanostructures catalysed by Au
nanoparticles. In this chapter, the possibility of growth of III-V nanowires using non-Au
metals was explored. Section 6.2.1 reports on the successful growth of GaAs nanowires
using Ni catalyst by tuning the growth temperature and V/III ratio. The defect-free epitaxial
GaAs nanowires have been examined by detailed electron microscopy. On the other hand,
the thickness of non-Au metallic films is crucial in determining whether they can be
transformed into nanoparticles to induce nanowires growth. Section 6.2.2 described a
proof-of-concept technique, which is developed for catalyst-free hetero-epitaxy growth of
InAs nanowires on Si wafers. Before the growth of InAs nanowires, a thick metallic film
was pre-deposited on the Si wafer and then annealed to form nano-scaled openings.
These nano-openings exposed the surface of Si substrate, which allowed subsequent
nucleation and growth of nanowires; simultaneously the small size of the nano-openings
limited the lateral growth of the nucleus but induced the axial growth. Catalyst-free and
hetero-epitaxy nanowires were grown on both Si (111) and (001) wafers successfully at
growth temperatures of 550 and 600°C. This work exhibits a very simple, low cost and
general method to grow catalyst-free hetero-epitaxy III-V nanowires. The simplicity of the
technique opens a new avenue for the growth and integration of catalyst-free
heteroepitaxial III-V nanowires on Si wafers.
6.2 Journal publication and manuscript
Results in Chapter 6 have been sub-categorized into two sections: Section 6.2.1 has been
drafted as manuscript; and Section 6.2.2 is included as it appears in Nano Letters 2016,
16, 4189–4193. http://pubs.acs.org/doi/abs/10.1021/acs.nanolett.6b01064

132
6.2.1 Towards epitaxial growth of defect-free GaAs nanowires catalysed by Ni
ABSTRACT
Deep understanding on non-Au catalysts growth of III-V nanowires is significant for the
exploration of the quality and property of the nanowires which may distinct from the
conventional Au-catalyzed nanowire; and hence provides a new insight into the growth
window and growth mechanism that will set a benchmark for the design of non-Au
catalyzed III-V nanowires growth. In this study, Ni-catalyzed GaAs nanowires have been
successfully grown by metal-organic chemical vapour deposition (MOCVD) by altering the
growth temperature and V/III ratio. A suitable growth window related with growth
temperature and V/III ratio has been explored; and via adjusting these factors, defect-free
zinc-blende GaAs nanowires growing along [ ̅ ̅ ̅] direction or defect-free wurtzite GaAs
nanowires growing along the [ ̅] direction can be achieved. The fundamental
mechanisms for the epitaxial growth of defect-free GaAs nanowires are presented.
KEYWORDS: GaAs nanowires, Ni catalyst, defect-free, zinc-blende, wurtzite
Introduction
In the past decade, significant research efforts have been devoted to the growth of III-V
nanowires for understanding their fundamental physics and for their potential applications
in nanophotonics, nanoelectronics and sensing devices.1, 2 Ongoing efforts have been put
forth into catalyst-induced III-V nanowires growth, particularly GaAs nanowires due to its
excellent carrier mobility and special coupling effect with light, which are promising for
electronic, phonics and solar cells applications.3, 4
Gold (Au) has been widely used to catalyze the growth of epitaxial III-V
nanowires,5, 6 while limited studies have been carried out using non-Au catalysts.7, 8 This is
anticipated that by using non-Au catalyst, in-depth insights related to fundamental issues
and technical issues could be obtained. However, there are less works reporting on non-
Au metals catalyzed III-V nanowires due to the unclear growth mechanism using non-Au
metals, which poses a challenge in identifying the suitable growth window for the growth of
epitaxial defect-free nanowires using different types of non-Au metals. To grow good
quality nanowires using non-Au metals epitaxially is important for integration of nanowires

133
directly onto substrates, which is important for future electronic and opto-electronic
applications.
Nickel (Ni) is anticipated to be one of the prominent catalysts since it is greatly
cheaper than Au, and is in principle compatible with Si electronics.9 Therefore, we believe
that using Ni as catalyst to induce III-V nanowires is promising in the future large-scale
electronic and opto-electronic devices. Han et al.7, 10 is the only group reported on the
growth of Ni-catalyzed GaAs nanowires. They used solid-source CVD methods to grow Ni-
catalyzed GaAs nanowires without any substrates. To the best of our knowledge, there is
still no reported on the success Ni-catalyzed epitaxial growth of GaAs nanowires on the
GaAs (111)B substrate. The lack of consensus on the suitable growth parameters to grow
epitaxial defect-free Ni-catalyzed GaAs nanowires has motivated us to tune the basic
growth parameters, namely growth temperature and V/III ratio, towards epitaxial growth of
defect-free Ni-catalyzed GaAs nanowires using MOCVD. In this study, we demonstrate the
realization of epitaxial growth of GaAs nanowires on GaAs (111)B substrates using Ni as
catalyst by identifying the suitable growth window. The reasons of the epitaxial growth of
defect-free GaAs nanowires are elucidated.
Experimental details
A ~2 nm thick Ni film was firstly deposited onto the GaAs(111)B substrates using an e-
beam evaporator. For better control of epitaxial growth of Ni-induced GaAs nanowires, we
design the experiments using GaAs (111)B substrate so that lattice-mismatch between
nanowires and wafer will be eliminated. We started the growth temperature and V/III ratio
using the optimize growth parameters for our Au-catalyzed GaAs nanowires,11 which is
450°C and V/III ratio of 46. Prior to the nanowire growth, each substrate was annealed in
situ at 600°C under AsH3 ambient to desorb GaAs surface oxides and surface
contaminants in metal-organic chemical vapor deposition (MOCVD) chamber under arsine
(AsH3) ambient The other reason for annealing is to obtain Ni nanoparticles, which play a
role as catalyst to induce GaAs nanowires growth. After the substrate cooled to growth
temperature, which were carried out at 450 and 550°C, the AsH3 flow was adjusted and
trimethylgallium (TMGa) was introduced to initiate GaAs nanowires growth. The flow rate
of TMGa was set at 1.16 x 10-5 mol/min for all growths; and the flow rates of AsH3 were set
respectively as 1.16 x 10-5, 5.79 x 10-5 and 5.36 x 10-4 mol/min (corresponding to V/III
ratios of 1, 5 and 46). Detailed morphology, structure and composition of GaAs nanowires

134
were characterized by field-emission scanning electron microscopy (FE-SEM, JEOL JSM-
7800F, operated at 8 kV) and transmission electron microscopy (TEM, FEI Tecnai F20,
operated at 200 kV). Individual nanowires for TEM observation were prepared by
dispersing the nanowires in ethanol using an ultrasonic bath for 15 min and then spreading
a few drops from the suspension onto holey carbon grids. For some samples have very
short and low density nanowires on substrates, the above conventional sample
preparation could not obtain a suitable TEM sample, focused ion beam (FIB) technique
was carried out to prepare thin cross-sectional TEM samples along ⟨ ⟩ zone-axis (FEI
Scios Dual Beam FIB/SEM).
Results and discussion
Figure 1 shows the (a,c) 45° tilted and (b,d) side-view SEM images of GaAs nanowires
grown on GaAs (111)B substrate at (a,b) 450°C and (c,d) 550°C. Figure 1a shows that
majority nanowires are curly (defined by their frequently changing growth direction and
kinks, refer to Figure 1a inset) and only a few nanowires are straight at 450°C. Moreover,
none of the nanowires grew in vertical [ ] direction, which is perpendicular to substrate
surface (refer to Figure 1b). The nanowire orientations exhibit no apparent relationship
with the substrate. For all the nanowires, we observed a small nanoparticle at the tip of
nanowires as highlighted in Figure 1b inset. At higher growth temperature of 550°C,
although nano-pyramids rather than nanowires were grown because the axial growth was
competing with radial growth of nanowires at this growth environment, which results in the
tapering of most of the nanowires, however, growth direction perpendicular to substrate for
majority nano-pyramids is apparent from Figure 1c,d. That is to say, the temperature is
important to their growth directions.

135
Figure 1. SEM morphologies of GaAs 1D nanostructures obtained at different growth
temperature under a V/III ratio of 46. (a,b) Growth temperature: 450°C; 45° tilted and side-
view SEM images. The inset of (b) is the enlarged SEM image focused at nanowire tip of
the blue box region in (b). (c,d) Growth temperature: 550°C; 45° tilted and side-view SEM
images. All scale-bars are 1 µm.
Based on the facts that axial growth is mass transport limited11 and an increase in
the radial growth rate of the nanowire due to reduce diffusion length of the Ga adatoms
under higher As flux,12 we reduce the AsH3 flow rate during growth to enhance the axial
growth of nanowires at 550°C. Figure 2a,b presents 45° tilted and side-view SEM images
of GaAs nanowires grown under a V/III ratio of 5. As expected, the radial growth of
nanowires was greatly reduced. Both thin vertical nanowires and tapered inclined
nanowires were observed. Due to { } substrate surface, the vertical epitaxial
nanowires should have the [ ̅] [ ̅ ̅ ̅] growth direction. Statistically speaking, the
majority inclined nanowires have an angle with respect to the substrate normal of ~19°. In
this paper, our focus is to investigate the suitable growth window for Ni-catalyzed epitaxial
nanowires vertically grown on the GaAs substrate.
Non-[ ̅ ̅ ̅] nanowires have been reported in Au-catalyzed GaAs13 and InAs14
nanowires as V/III ratio increasing attributed to the formation of stable As-trimer surface
reconstruction on As-terminated { } surface under As-rich conditions.15 The surface

136
reconstruction favors a non-[ ̅ ̅ ̅] growth direction.16 Therefore, we carried out the other
growth under V/III ratio of 1, which As precursors was decreased to reduce the non-[ ̅ ̅ ̅]
type nanowires. Figure 2c,d is the SEM images of 45° tilted and side-view SEM images of
this sample. Inclined nanowires were suppressed and only vertical nanowires were
induced, however the length is shorter and the density is much lower.
Figure 2. SEM morphologies of GaAs 1D nanostructures obtained under different V/III
ratio at growth temperature of 550°C. (a,b) V/III: 5; 45° tilted and side-view SEM images.
(c,d) V/III: 1, 45° tilted and side-view SEM images. All scale-bars are 1 µm.
To understand the structural characteristics of these epitaxial growth of vertical
GaAs nanowires at 550°C, TEM analyses were performed. We realized that it is
impractical to obtain individual vertical nanowires formed under V/III ratio 1 and 46 by
suspension of nanowires-contained ethanol onto holey carbon grids due to a very low
density. Hence, the area consisting of the vertical nanowires was identified and very thin
cross-section TEM sample was milled using FIB along ⟨ ⟩ zone-axis (which can be
recognized by the natural cleavage plane of { } substrate). Figure 3 shows ⟨ ⟩ zone-
axis TEM images of vertical nanowires grown at 550°C under V/III ratios of 46 (a,b), 5
(c,d), and 1(e,f), respectively. Figure 3a is a thin cross-sectional TEM sample viewing from
⟨ ⟩ zone-axis prepared using FIB and shows three vertical nanowires grown under V/III
ratio of 46 on GaAs (111)B substrate. The layer around the nanowires is platinum coating

137
as a protective layer during milling process of FIB. These vertical nanowires are tapered
towards their tip (refer to Figure 3a). Figure 3b is the corresponding HRTEM and FFT
images show that the nanowire has zinc-blende structure with twin planes and the growing
direction of nanowires is [ ̅ ̅ ̅]. Figure 3c is a typical vertical nanowire grew under a low
V/III ratio of 5 viewing along ⟨ ⟩ zone-axis and Figure 3d is the corresponding HRTEM
and images showing a defect-free zinc-blende structure. We performed an extensive
HRTEM study on over a dozen of vertical nanowires and confirmed this defect-free zinc-
blende structure nature along entire nanowires. Figure 3e is a thin cross-sectional TEM
sample viewing from ⟨ ⟩ zone-axis and shows a single vertical nanowires grown under
V/III ratio of 1 on GaAs (111)B substrate. Figure 3f is the corresponding HRTEM image and
SAED pattern show the nanowire has wurtzite structure and the growth direction for these
epitaxial nanowires is [ ̅]. The strong diffraction rings in SAED pattern (refer to Figure
3f inset) is attributed to the strong background of platinum coating. To note that the
seemingly not straight ( ̅ ̅ ̅) planes in the HRTEM image of the nanowire (refer to Figure
3b,f) is due to the uneven thickness of the background platinum coating during FIB
process.

138
Figure 3. TEM analysis of vertical GaAs nanowires obtained at 550°C under different V/ III
ratio. (a,b) V/III: 46; Cross-sectional ⟨ ⟩ TEM image of vertical nanowire; and the
corresponding HRTEM and FFT (inset) images showing zinc-blende structure with twin
planes; (c,d) V/III: 5; TEM image of single nanowire grew viewed along ⟨ ⟩ zone axis,
and its corresponding HRTEM and FFT (inset) images showing defect-free zinc-blende
structure; (e,f) V/III: 1; Cross-sectional ⟨ ⟩ TEM image of vertical nanowire and the
corresponding HRTEM image and SAED pattern (inset) of nanowire showing defect-free
wurtzite structure. The diffraction ring is contributed by the amorphous platinum coating on
the growth surface during FIB process.
To understand our observations for the crystal structure change of these vertical
Ni-catalyzed GaAs nanowires with decreasing V/III ratio, we need to clarify two
phenomena: (1) why the twin planes can be eliminated in the epitaxial nanowires when the

139
V/III ratio decreases from 46 to 5? (2) Why the defect-free crystal structure switches from
zinc-blende to wurtzite, and they have the same length under V/III ratio of 1?
To explain the first observation, we consider the energy barrier for ordinary plane
nucleation which can be expressed as5, 17
( )
(
)
⁄ (1)
and for nuclei with twins, we obtain
( )
(
)
⁄ (2)
where h is the height of a nucleus, a is the area per III-V pair on the growth interface,
is the chemical potential difference per III-V pair. The chemical potential is a measure of
the supersaturation of the growth system5; thus and supersaturation can be used
interchangeably. represents the twin formation energy. is the total change in surface
and interfacial energy due to nucleus formation; b and c are coefficients independent of
supersaturation.
Then we define as the ratio between the nucleation barriers with and without
twins from Equations (1) and (2), we get the expression
(3)
Based on Equation (3), for twin nucleation to occur, must be high. The can
be related to the growth parameters as expressed below5
(4)
where is the Boltzmann constant, T is the growth temperature. CGa,eq and CGa are the
concentrations of Ga at equilibrium with the GaAs crystal and in the catalyst during
nanowire growth, respectively; since Ga is expected to be supplied from the catalyst. CAs,eq

140
and CAs refer to the vapor phase As concentrations as As is expected to be supplied to the
growth interface from the vapor phase.
According to Equation (4), a high CAs increases ; and thereby promote twin
nucleation as described in Equation (3). This explains the reason for twin planes formed in
GaAs nanowires grown under the V/III ratio of 46, in which the As concentration in the
growth chamber is much higher and other parameters remain unchanged.
To describe the switching of defect-free structured GaAs nanowires from zinc-
blende to wurzite under the V/III ratio of 1, we consider the nucleation kinetics in catalyst-
induced nanowire growth. Under the identical growth conditions, the nucleation barriers for
the formation of two different crystal structures wurtzite and zinc-blende are fixed but
different; and the ratio between wurtzite and zinc-blende nucleation barriers can be
expressed as18, 19
(5)
where is the extra cohesive energy needed for wurtzite III-V pairs formation, is the
ratio between the change in effective surface energies due to the formation of a wurtzite
and zinc-blende nucleus ( and ) as expressed below19
( ) ( )
( ) ( ) (6)
where , and are the interfacial energies of the vapor-liquid-solid system, is the
areal share of nucleus in contact with the vapor phase ( ), is the contact angle,
and ( ) ( ) is the ratio between the lateral solid-vapor surface energies of
wurtzite and zinc-blende nuclei, which because of less bonding bonds on the
wurtzite surface.
According to Equation (5), both and can affect the crystal structure of
nanowires, namely, lower and is close to 1 favor the formation of zinc-blende
structure. Otherwise, formation of wurtzite will be favored. Based on Equations (5) and (6),
the newly formed monolayer will adopt zinc-blende structure for higher values of .20
Accordingly, we anticipate that a lower , or more precisely an increase in . Such
a change in surface energies would increase the difference of the term ( )

141
in Equation (6), which would increase and , and would promote zinc-
blende formation. It has been reported that arsenic is known to act as surface active agent
that can lower the surface energies.21 In our designed growth, we altered the V/III ratio by
solely changing the AsH3 flow rate. Therefore, under V/III ratio of 5, there is more arsenic
in the growth environment and we speculate that the surface energy of catalyst is
lowered for zinc-blende growth condition. Arsenic can also affect the catalyst/nanowire
interfacial energy , however this effect is expected to be much less significant due to the
rapid consumption of any available arsenic at the growth interface into the nanowire
growth.11 On this basis, the nanowires grown under V/III ratio of 1 (relatively lesser arsine
in the vapor) tend to increase more significantly, and in turn, nanowires prefer for the
wurtzite nucleation.
Alternatively, it is well-documented that higher supersaturation, which results in
the higher values of , will promote wurtzite formation.18 At this very low V/III ratio, Ga
vapor pressure in growth chamber should be relatively higher and catalysts can adsorb
more Ga, resulting in high supersaturation in catalysts. It is of interest to note that an
increase in supersaturation should also corresponds to higher axial growth rate of
nanowires; however, this is not consistent with our observations – the nanowires grown
under V/III ratio of 1 are all have the same short length, and this phenomena has been
reported elsewhere.22, 23 Based on this observation, it is sensible to postulate that Ga
concentration in the catalyst is not the dominating factor for nanowires growth rate in this
growth environment. The role of arsine is therefore considered. At this low V/III ratio,
arsine flow rate is decreased and hence decreasing the chances of arsine atoms reaching
the growth front and incorporating with Ga into the nanowires, which results in the slow
growth rate of nanowires. Perfect crystal structure is favorable to be adopted for nanowires
grown at slow growth rate.
Conclusion
In summary, epitaxial defect-free Ni-catalyzed GaAs nanowires have been successful
developed. The growth temperature for epitaxial vertical [ ̅ ̅ ̅]/[ ̅] nanowires is
relatively higher so that the mobility Ga atoms can be increased to diffuse to the catalyst.
V/III ratio is important to control the crystal quality of nanowires, in which by decreasing
V/III ratio from 46 to 5, twin planes can be eliminated and defect-free zinc-blended GaAs
[ ̅ ̅ ̅] nanowires can be achieved. At very low V/III ratio of 1, defect-free wurtzite [ ̅]

142
nanowires can be obtained. This success epitaxial defect-free nanowires growth is
expected to open a new avenue in III-V research field that intensely looking for alternative
Si-compatible catalyst to replace Au catalyst.
References 1. Yan, R.; Gargas, D.; Yang, P. Nat Photon 2009, 3, (10), 569-576.
2. Offermans, P.; Crego-Calama, M.; Brongersma, S. H. Nano Lett 2010, 10, (7),
2412-2415.
3. Gudiksen, M. S.; Lauhon, L. J.; Wang, J.; Smith, D. C.; Lieber, C. M. Nature 2002,
415, (6872), 617-620.
4. Yoon, J.; Jo, S.; Chun, I. S.; Jung, I.; Kim, H.-S.; Meitl, M.; Menard, E.; Li, X.;
Coleman, J. J.; Paik, U.; Rogers, J. A. Nature 2010, 465, (7296), 329-333.
5. Joyce, H. J.; Wong-Leung, J.; Gao, Q.; Tan, H. H.; Jagadish, C. Nano Lett 2010,
10, (3), 908-915.
6.
Riechert, H. Nano Lett 2011, 11, (3), 1276-1279.
7. Han, N.; Wang, F.; Hui, A. T.; Hou, J. J.; Shan, G.; Xiu, F.; Hung, T.; Ho, J. C.
Nanotechnology 2011, 22, (28), 285607.
8. Xu, H.; Wang, Y.; Guo, Y.; Liao, Z.; Gao, Q.; Tan, H. H.; Jagadish, C.; Zou, J. Nano
Lett 2012, 12, (11), 5744-5749.
9. Dick, K. A.; Caroff, P. Nanoscale 2014.
10. Han, N.; Hui, A. T.; Wang, F.; Hou, J. J.; Xiu, F.; Hung, T.; Ho, J. C. Appl Phys Lett
2011, 99, (8), 083114.
11. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Fickenscher, M. A.; Perera,
S.; Hoang, T. B.; Smith, L. M.; Jackson, H. E.; Yarrison-Rice, J. M.; Zhang, X.; Zou,
J. Nano Lett 2009, 9, (2), 695-701.
12. Dheeraj, D. L.; Munshi, A. M.; Scheffler, M.; Helvoort, A. T. J. v.; Weman, H.;
Fimland, B. O. Nanotechnology 2013, 24, (1), 015601.
13. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Fickenscher, M. A.; Perera,
S.; Hoang, T. B.; Smith, L. M.; Jackson, H. E.; Yarrison-Rice, J. M.; Zhang, X.; Zou,
J. Adv Funct Mater 2008, 18, (23), 3794-3800.
14. Dayeh, S. A.; Yu, E. T.; Wang, D. Nano Lett 2007, 7, (8), 2486-2490.
15. Biegelsen, D. K.; Bringans, R. D.; Northrup, J. E.; Swartz, L. E. Physical Review
Letters 1990, 65, (4), 452-455.

143
16. Ikejiri, K.; Noborisaka, J.; Hara, S.; Motohisa, J.; Fukui, T. Journal of Crystal Growth
2007, 298, (0), 616-619.
17. Johansson, J.; Karlsson, L. S.; Svensson, C. P. T.; Mårtensson, T.; Wacaser, B. A.;
Deppert, K.; Samuelson, L.; Seifert, W. Nature Materials 2006, 5, (7), 574-580.
18. Glas, F.; Harmand, J.-C.; Patriarche, G. Physical Review Letters 2007, 99, (14),
146101.
19.
T. Nano Lett 2010, 10, (12), 4807-4812.
20. Zhang, Z.; Lu, Z.-Y.; Chen, P.-P.; Lu, W.; Zou, J. Acta Mater 2015, 92, (0), 25-32.
21. Copel, M.; Reuter, M. C.; Kaxiras, E.; Tromp, R. M. Physical Review Letters 1989,
63, (6), 632-635.
22. Zhou, C.; Zheng, K.; Lu, Z.; Zhang, Z.; Liao, Z.; Chen, P.; Lu, W.; Zou, J. J Phys
Chem C 2015, 119, (35), 20721-20727.
23. Lehmann, S.; Wallentin, J.; Jacobsson, D.; Deppert, K.; Dick, K. A. Nano Lett 2013,
13, (9), 4099-4105.

144
6.2.2 Growth of catalyst-free epitaxial InAs nanowires on Si wafers using metallic
masks
Growth of Catalyst-free Epitaxial InAs Nanowires on Si
Wafers Using Metallic Masks
M. Teng Soo,† Kun Zheng,*,‡,§ Qiang Gao,∥ H. Hoe Tan,∥ Chennupati Jagadish∥ and Jin
Zou*,†,‡
† Materials Engineering, ‡ Centre for Microscopy and Microanalysis, and § Australian
Institute for Bioengineering and Nanotechnology, The University of Queensland, St. Lucia,
QLD 4072, Australia
∥ Department of Electronic Materials Engineering, Research School of Physics and
Engineering, The Australian National University, Canberra, ACT 2601, Australia
ABSTRACT
Development of heteroepitaxy growth of catalyst-free vertical III-V nanowires on Si wafers
is highly desirable for future nanoscale Si-based electronic and optoelectronic devices. In
this study, a proof-of-concept approach is developed for catalyst-free heteroepitaxy growth
of InAs nanowires on Si wafers. Before the growth of InAs nanowires, a Si-compatibly
metallic film with a thickness of several tens of nanometers was pre-deposited on a Si
wafer, and then annealed to form nano-size openings so as to obtain a metallic mask.
These nano-openings exposed the surface of the Si wafer, which allowed subsequent
nucleation and growth of InAs nanowires directly on the surface of the Si wafer. The small
size of the nano-openings limits the lateral growth of the nanostructures but promotes their
axial growth. Through this approach, catalyst-free InAs nanowires were grown on both Si
(111) and (001) wafers successfully under different growth temperatures. In particular,
ultra-long defect-free InAs nanowires with the wurtzite structure were grown the Si (111)
wafers at 550 °C using the Ni mask. This study offers a simple, cost-effective, and scalable
method to grow catalyst-free III-V nanowires on Si wafers. The simplicity of the approach
opens a new avenue for the growth and integration of catalyst-free high-quality
heteroepitaxial III-V nanowires on Si wafers.
KEYWORDS: InAs nanowires, catalyst-free, hetero-epitaxial, metallic mask, Si wafers

145
Introduction
Recent development in the growth of one-dimensional (1D) semiconductor nanowires has
raised great interests for their unique potential to be used as building blocks for nano-
photonics,1, 2 nano-electronics,3 and bio-sensing devices.4 As one of the technologically
significant III-V semiconductors with exceptionally high electron mobility and narrow
bandgap at room temperature, InAs has attracted much research attention aiming at high-
frequency electronics and long-wavelength photonics.5, 6 To date, much work has been
done to grow InAs nanowires on various substrates.7-11 Among them, InAs nanowires
grown on Si substrates are of particular significance and have been investigated
extensively since it may enable seamless integration of nanowire-based
electronic/optoelectronic devices with the industrially matured Si platform. More recently,
the ability to directly grow epitaxial InAs nanowires on Si wafers has opened new
possibilities for novel integrated device architectures, such as high-speed field-effect-
transistors (FETs)12, 13 or tunnel FETs with steep-slope current-voltage characteristics.14, 15
InAs nanowires can be monolithically integrated on Si complementary metal-oxide-
semiconductor platforms, lifting the lattice misfit restrictions typically present in their thin
film counterparts.16 Besides, in most nanowires-based FET devices, on-chip processing is
the preferred technology,17 in which vertical nanowires can serve as wrap-gated
channels.18 In these devices, a metallic layer is often sputtered onto the wafer to function
as the bottom contact of the nanowires.19
Au catalyst is mostly used for the epitaxial growth of III-V nanowires via the vapor-liquid-
solid mechanism.20, 21 However, for nanowires grown on Si wafers, Au-catalyzed nanowire
growth often result in unwanted effects in Si, such as formation of deep-level defect states
and Au migration on the semiconductor surface,22, 23 which in turn degrade their
corresponding optical and electrical properties.24 As a consequence, it is incompatible with
the Si platform. To solve this problem, the selective-area epitaxy method has been
developed for growing epitaxial III-V nanowires, which offers a catalyst-free approach that
avoids contamination.9, 25 At present, the common method is to deposit a SiO2 layer as a
mask9, 26 or a self-assembled polymer coating27 on Si wafers, followed by patterning the
nano-openings in the SiO2 barrier layer using electron-beam lithography or in the polymer
coating by etching. In particular, the lithography process is relatively costly and it is difficult
to control the crystal quality of III-V nanowires grown on Si wafers by selective area
epitaxy, in which stacking faults are always found in the nanowires.27 So far, InP

146
nanowires grown via the selective-area epitaxy method are the only reported defect-free
III-V nanowires, but they can only be realized on InP { } substrates.28, 29 There is still
no report on the selective-area epitaxy growth of defect-free III-V nanowires
heteroepitaxially on Si wafers directly.
Void formation in aluminum or copper films has been a common problematic issue in Si-
based integrated circuits industry.30-32 These stress-induced voids are caused by the
relaxation of thermal stress generated during thermal processing or thermal aging in chip
processing. Besides, high thermal stress can be generated due to the difference in the
thermal expansion coefficients between the deposited metallic film and Si wafer. In this
study, we turn this disadvantage to develop nano-openings in metallic films deposited on
Si wafers for the direct growth of InAs nanowires within these nano-openings, rather than
using the deposited metallic films to catalyze the nanowire growth. It is found that InAs
nanowires can be epitaxially grown on both Si (111) and (001) wafers masked by different
types of metallic layers at different growth temperatures.
Experimental Details
Si (111) and (001) wafers were treated with diluted hydrofluoric (HF) acid before being
loaded into a Temescal BJD-2000 e-beam evaporator system for the deposition of
different metallic films, i.e. Ni, Pd and Pt with nominal thicknesses of 30, 60 and 90 nm,
respectively. The metal coated wafers were loaded into a horizontal flow metalorganic
chemical vapor deposition (MOCVD) system. Prior to growth, each wafer was annealed in
situ at 700 °C under AsH3 ambient to produce nano-openings that exposed Si surface for
subsequent InAs nanowire growth,32 to desorb Si surface contaminants and to form As-
terminated surface within the nano-openings.9 After that the wafer was cooled to the
temperature (namely 500, 550 and 600 °C in our experiment) and trimethylindium (TMIn)
was introduced to initiate InAs growth. During the InAs growth, the flow rates of TMIn and
AsH3 were maintained at 1.2 x 10-5 mol/min and 3.4 x 10-5 mol/min, respectively, to attain a
V/III ratio of 2.9. Figure 1a schematically illustrates the process flow, and Figure 1b a
scanning electron microscopy (SEM) image taken from the Ni-coated Si (111) wafer at
annealed at 700 °C which shows many nano-openings on the surface. Figure 1c shows
the close-up view of the same sample that confirms the formation of nano-openings.

147
Figure 1. (a) Schematic illustration of the process flow for MOCVD growth of catalyst-free
InAs nanowires on Si wafer pre-coated with metallic film: (i) coating the metallic film by e-
beam evaporation; (ii) annealing the pre-coated wafer to produce a mask layer with
nanoscale openings; (iii) growing InAs nanowires directly from the openings. (b) SEM
image of a Ni-coated Si (111) wafer (annealed at 700 °C) showing many nano-openings
(dark contrast) on the surface. (c) SEM image showing an enlarged nano-opening shown
in (b).
Detailed morphological, structural and compositional characteristics of the InAs
nanostructures were investigated by SEM (JEOL JSM-7800F and JEOL JSM-7100F,
equipped with energy-dispersive X-ray spectroscopy (EDS) for compositional analysis)
and transmission electron microscopy (TEM; FEI Tecnai-F20; operated at 200 kV,
equipped with an EDS system; FEI Tecnai-F30; operated at 300 kV). Individual nanowires
for TEM observation were prepared by dispersing the nanowires in ethanol using an
ultrasonic bath for 15 min and then spreading a few drops from the suspension onto holey
carbon grids. To identify the layer morphology and composition, cross-section TEM
specimens were prepared by focused ion beam (FIB) (FEI Scios Dual Beam FIB/SEM).

148
Results and Discussion
Figure 2 is a set of SEM images (viewed from top and side) taken from InAs grown on Ni-
coated Si (111) wafers at different temperatures. Figure 2a,b show top and side views,
respectively, of InAs grown at 500 °C, in which no obvious nanowire was found. Instead, a
~ 500 nm-thick InAs layer (as indicated by the blue arrows in the inset of Figure 2b) was
grown on the Ni film (as indicated by the orange arrows in the inset of Figure 2b). The
inset in Figure 2a shows a nano-opening, and a short nanowire grown from the nano-
opening. By increasing the growth temperature to 550 °C, the overgrowth of the InAs layer
directly on the metallic film was suppressed significantly and transformed into InAs clusters
(refer to Figure 2c). The formation of these clusters is due to thermal-induced dewetting33
via the Volmer-Weber growth mode,34, 35 in order to minimize the interaction area of InAs
with metal. However, growth of ultra-long but curved thin InAs nanowires from the nano-
openings was observed. These nano-openings that promote these ultra-long and thin
nanowires are the intersections of nano-cracks of the annealed Ni film and the nanowires
almost covered the entire nano-openings (refer to Figure 2c inset). The curved feature of
these ultra-long wires may be due to their large aspect ratio.36 By further increasing the
growth temperature to 600 °C, Figure 2e,f show that vertical InAs nanowires from the
nano-openings were observed, in which small “white dots” shown in Figure 2e are the top-
view of vertical nanowires (confirmed by the slightly tilted SEM image shown in the inset).
Furthermore, the inset of Figure 2e indicates that the nanowire is indeed induced within
the nano-opening. Interestingly, these vertical InAs nanowires have relatively smaller
aspect ratio than those curved wires and are straight. Our detailed SEM investigation
indicates that the nanowires have relatively flat tips without metal catalysts, as shown in
the inset of Figure 2f. The composition of overgrowth InAs layer (500 °C) and InAs clusters
on Ni film (550 and 600 °C) is confirmed by EDS analyses (refer to Figure S1–S2 in
Supporting Information).

149
Figure 2. (a, c, e) Top-view; and (b, d, f) side-view SEM images of InAs grown on Ni-
coated Si (111) wafers. The insets of (a, c, e) showing nanowires grown from nano-
openings. The inset of (b) showing InAs overgrowth layer, and the Ni film. The inset of (f)
showing the flat tip of a typical InAs nanowire. The growth temperatures are (a, b) 500, (c,
d) 550, and (e, f) 600 °C.
To understand the structural characteristics of the nanowires, TEM investigation was
performed on over a dozen of nanowires from each type of nanowires. Figure 3 shows
TEM images of typical nanowires grown at 550 and 600 °C on Ni-coated Si (111) wafers.
Figure 3a is a TEM image of an ultra-long nanowire grown at 550 °C, and shows that the
nanowire is catalyst-free (Figure 3a inset) and slightly tapered toward the tip. Figure 3b is
the HRTEM image showing that the nanowire has a defect-free wurtzite structure with the
epitaxy growth direction of <0001>. Figure 3c is the corresponding SAED pattern, and
confirms the <0001> growth direction. More TEM investigation evidences of defect-free
InAs nanowires are shown in Figure S3 in Supporting Information. Figure 3d is a TEM
image showing a nanowire grown at 600 °C, which has a lateral size of slightly larger than
100 nm and a length of over 6 μm. Figure 3e is a magnified TEM image showing the top

150
section of the nanowire, in which no catalyst can be seen. Figure 3f is the high-resolution
TEM image and confirm the nanowire has the wurtzite structure with the <0001> growth
direction, agreeing with our SEM observations (e.g. Figure 2f). Since it has been reported
that zinc-blende structure is less stable at higher growth temperatures and at low V/III
ratios,37 wurtzite structure would be preferred for the formation of InAs nanowires under
our growth conditions. This experimental result suggests that InAs nanowires can be
indeed be epitaxially grown on the Si wafers in the same manner as homoepitaxial growth
despite the large lattice mismatch between InAs and Si.38 In addition, stacking faults can
be observed (refer to Figure 3f). In fact, the high-density of stacking faults can also be
witnessed by the streaks along the <0001> direction in the corresponding SAED pattern
(Figure 3g inset). Figure 3g shows an EDS spot analysis taken from the nanowire
confirming that it is indeed InAs, which contains no metal contamination.
.
Figure 3. (a) TEM image of a typical ultra-long InAs nanowire grown on Ni-coated Si (111)
wafer at 550 °C. Inset showing the catalyst-free tip of the nanowire. (b, c) Corresponding
HRTEM image and SAED pattern showing defect-free wurtzite structure. (d) TEM image of
a typical vertical InAs nanowire grown on Ni-coated Si (111) wafer at 600 °C. (e) Magnified
TEM image showing the nanowire tip; (f) Corresponding HRTEM image showing wurtzite
structure with stacking faults. (g) EDS spot analysis of nanowire with inset being the
corresponding SAED pattern.

151
To clarify the general availability of using metallic masks to grow catalyst-free InAs
nanowires on Si wafers with different orientations, we grew InAs nanowires respectively on
the Ni-coated Si (001) wafer as well as Pd- and Pt-coated Si (111) and (001) wafers; and
investigated their corresponding growth behaviors. Figure 4 shows the summary of our
extensive SEM investigations, in which InAs nanowires have been successfully grown in
all samples epitaxially. Similar to Figure 2a,b, overgrown InAs layer can be found on all
metal-coated Si wafers grown at 500 °C (refer to Figure S4–S8 in the Supporting
Information). Our detailed analysis of Figure 4 indicates that relatively higher growth
temperature (namely 550 and 600 °C) coupled with a low V/III ratio, which are identical
with the growth conditions for the epitaxial growth of InAs films,39, 40 is essential for
catalyst-free InAs nanowires grown on Si wafers with different orientations.
Figure 4. Cross-sectional SEM images of InAs nanowires on Si (111) and (001) wafers
pre-coated with Ni, Pd or Pt film grown at 550 and 600 °C, respectively. All scale bars are
1 µm.

152
Interestingly, the pre-coated metallic films are commonly used as a source of catalysts
for inducing the nanowire growth.41, 42 However, in this study, we employed the pre-coated
metallic films to realize the direct-epitaxial growth of InAs nanowires on Si wafers. To
understand the different role of the metallic films, we note that the key factor is the
formation of thermal stress-induced voids in metallic films at high annealing
temperatures.32, 43 The voids behave as the nano-openings to expose the underlying Si
surface. The growth species are anticipated to migrate to nano-openings either by surface
diffusion of the growth species from metallic surface or direct deposition from the vapor
phase,44 similar to the growth behaviors of catalyst-free III-V nanowires grown on
lithographically defined Si wafers.45 In this study, our growth environment of high
temperature and low V/III ratio, which favor adatom mobility,40 results in the fast surface
diffusion of growth species to the nano-openings. After the arrival of these growth species
in the openings, they will nucleate on Si and subsequent growth species would be able to
attach to any initially available nucleation site.46 In this way, the continuous supply of
growth species results in the epitaxial growth of InAs nanowires via the vapor-solid (VS)
mechanism.47, 48 The nano-openings limit the lateral growth of the nucleus but promote the
axial growth. In fact, our experimental results support the tentative growth model shown in
Figure 1a, from which InAs nanowires were indeed epitaxially grown on different Si wafers
with nano-openings. Besides, a variety of densities and lengths of nanowires were
observed by using different metallic mask at different growth temperatures (namely 550
and 600 °C) may be attributed to the (1) different diffusion length of growth species on
different metals,49 and (2) different growth rate at different temperature.50
It should be noted that the metallic films selected in this study (namely Ni, Pd and Pt) are
compatible with Si-based device fabrication51, 52 and they can be utilized directly as the
bottom contact in a FET device, which can minimize the damage to nanowires during
device fabrication and/or avoids the additional fabrication procedure, so that this proof-of-
concept approach offers a simple, time-saving and scalable method for growing high-
quality III-V nanowires directly on Si wafers.
Conclusion
In this study, a proof-of-concept approach to grow catalyst-free InAs epitaxial nanowires
on Si wafers is presented. This is a simple, economical, time-effective and scalable
technique to grow III-V nanowires on Si wafers. By pre-coating metallic films with suitable

153
thicknesses on Si wafers, the annealing, via stresses-induced voiding, results in the
formation of nano-openings in the metallic films, which act as the mask for the growth of
nanowires via the VS mechanism. Especially, defect-free wurtzite InAs nanowires have
been grown heteroepitaxially on the Ni-masked Si (111) successfully at 550 °C. This study
provides a practical insight to realize the growth of catalyst-free III-V nanowires on Si
wafers. Specifically, our results also demonstrated that, by tuning the growth parameters,
growth of defect-free InAs nanowires directly on Si wafers are possible, so that this study
opens up an avenue for realizing the epitaxial growth of high quality III-V nanowires on Si
wafers.
ASSOCIATED CONTENT
Supporting Information. 1. Compositional analysis. 2. Structural analysis of defect-free
InAs nanowires. 3. Morphology analysis by SEM. This material is available free of charge
via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author
* E-mail: [email protected], [email protected]
ACKNOWLEDGMENT
This research was supported by the Australian Research Council. The Australian National
Fabrication Facility and Australian Microscopy & Microanalysis Research Facility, both
established under the Australian Government’s National Collaborative Research
Infrastructure Strategy, are gratefully acknowledged for proving access to the facilities
used in this work.

154
SUPPORTING INFORMATION
Growth of Catalyst-free Epitaxial InAs Nanowires on Si Wafers Using Metallic Mask
M. Teng Soo,† Kun Zheng,*,‡,§ Qiang Gao,∥ H. Hoe Tan,∥ Chennupati Jagadish∥ and Jin
Zou*,†,‡
† Materials Engineering, ‡ Centre for Microscopy and Microanalysis, and § Australian
Institute for Bioengineering and Nanotechnology, The University of Queensland, St. Lucia,
QLD 4072, Australia
∥ Department of Electronic Materials Engineering, Research School of Physics and
Engineering, The Australian National University, Canberra, ACT 2601, Australia

155
SUPPORTING INFORMATION
1. Compositional analysis
Figure S1. Top-view SEM images of InAs nanowires grown on Si (111) wafers pre-coated
with Ni film at (a) 500 and (c) 550 °C. (b,d) EDS spot analyses of (a) and (c), respectively,
showing different compositions at different region.
Figure S2. (a) Cross-sectional TEM image (prepared by FIB) of sample grown at 600 °C
on Si (111) wafer pre-coated with Ni film showing the InAs cluster-Ni-Si interface. The
same sample was then ultrasonicated (to remove the InAs nanowires and clusters): (b,d)
BSE images at different areas. The brighter contrast indicates elements with higher atomic
weight and vice versa; (c) EDS spot analyses of (b) showing different compositions at
different region. (e-h) Area mapping of (d) shows the distribution of different elements.

156
2. Structural analysis by TEM
Figure S3. (a, d, g, j) TEM images of various ultra-long InAs nanowires grown on Ni-
coated Si (111) wafer at 550 °C. (b, e, h, k) Corresponding HRTEM images and (c, f, i, l)
SAED or FFT patterns showing defect-free wurtzite structure.

157
3. Morphology analysis by SEM
Figure S4. (a, c, e) Top-view; and (b, d, f) cross-sectional SEM micrographs of InAs
nanowires grown on Si (001) wafers pre-coated with Ni film. The inset of (a) is the
magnified SEM image shows the top-view of two nanowires (indicated by arrows). The
insets of (b) and (f) are the magnified SEM image focuses at the InAs-Ni-Si wafer
interface. The short nanowire was indicated by arrow in inset of (b).The growth
temperatures are (a, b) 500; (c, d) 550 and (e, f) 600 °C.

158
Figure S5. (a, c, e) Top-view; and (b, d, f) cross-sectional SEM micrographs of InAs
nanowires grown on Si (111) wafers pre-coated with Pd film. The growth temperatures are
(a, b) 500; (c, d) 550 and (e, f) 600 °C.
Figure S6. (a, c, e) Top-view; and (b, d, f) cross-sectional SEM micrographs of InAs
nanowires grown on Si (001) wafers pre-coated with Pd film. The growth temperatures are
(a, b) 500; (c, d) 550 and (e, f) 600 °C.

159
Figure S7. (a, c, e) Top-view; and (b, d, f) cross-sectional SEM micrographs of InAs
nanowires grown on Si (111) wafers pre-coated with Pt film. The growth temperatures are
(a, b) 500; (c, d) 550 and (e, f) 600 °C.
Figure S8. (a, c, e) Top-view; and (b, d, f) cross-sectional SEM micrographs of InAs
nanowires grown on Si (001) wafers pre-coated with Pt film. The growth temperatures are
(a, b) 500; (c, d) 550 and (e, f) 600 °C.

160
REFERENCES 1. Duan, X.; Huang, Y.; Agarwal, R.; Lieber, C. M. Nature 2003, 421, (6920), 241-245.
2. Xia, H.; Lu, Z.-Y.; Li, T.-X.; Parkinson, P.; Liao, Z.-M.; Liu, F.-H.; Lu, W.; Hu, W.-D.;
Chen, P.-P.; Xu, H.-Y.; Zou, J.; Jagadish, C. ACS Nano 2012, 6, (7), 6005-6013.
3. Guo, N.; Hu, W.; Liao, L.; Yip, S.; Ho, J. C.; Miao, J.; Zhang, Z.; Zou, J.; Jiang, T.;
Wu, S.; Chen, X.; Lu, W. Advanced Materials 2014, 26, (48), 8203-8209.
4. Cui, Y.; Wei, Q.; Park, H.; Lieber, C. M. Science 2001, 293, (5533), 1289-1292.
5. Chuang, S.; Gao, Q.; Kapadia, R.; Ford, A. C.; Guo, J.; Javey, A. Nano Lett 2012,
13, (2), 555-558.
6. Miao, J.; Hu, W.; Guo, N.; Lu, Z.; Zou, X.; Liao, L.; Shi, S.; Chen, P.; Fan, Z.; Ho, J.
C.; Li, T.-X.; Chen, X. S.; Lu, W. ACS Nano 2014, 8, (4), 3628-3635.
7. Mandl, B.; Stangl, J.; Mårtensson, T.; Mikkelsen, A.; Eriksson, J.; Karlsson, L. S.;
Bauer, G.; uuml; nther; Samuelson, L.; Seifert, W. Nano Lett 2006, 6, (8), 1817-
1821.
8. Dayeh, S. A.; Yu, E. T.; Wang, D. J Phys Chem C 2007, 111, (36), 13331-13336.
9. Tomioka, K.; Motohisa, J.; Hara, S.; Fukui, T. Nano Lett 2008, 8, (10), 3475-3480.
10. Dayeh, S. A.; Yu, E. T.; Wang, D. Nano Lett 2009, 9, (5), 1967-1972.
11. Zhang, Z.; Zheng, K.; Lu, Z.-Y.; Chen, P.-P.; Lu, W.; Zou, J. Nano Lett 2015, 15,
(2), 876-882.
12. Tanaka, T.; Tomioka, K.; Hara, S.; Motohisa, J.; Sano, E.; Fukui, T. Applied Physics
Express 2010, 3, (2), 025003.
13. Johansson, S.; Egard, M.; Ghalamestani, S. G.; Borg, B. M.; Berg, M.; Wernersson,
L. E.; Lind, E. IEEE T Microw Theory 2011, 59, (10), 2733-2738.
14. Ionescu, A. M.; Riel, H. Nature 2011, 479, (7373), 329-337.
15. Tomioka, K.; Fukui, T. Appl Phys Lett 2011, 98, (8), 083114.
16. Bakkers, E. P. A. M.; Borgström, M. T.; Verheijen, M. A. MRS Bulletin 2007, 32,
(02), 117-122.
17. Wernersson, L. E.; Thelander, C.; Lind, E.; Samuelson, L. Proceedings of the IEEE
2010, 98, (12), 2047-2060.
18. Thelander, C.; FrobergFroberg, L. E.; Rehnstedt, C.; Samuelson, L.; Wernersson,
L. E. IEEE Electr Device L 2008, 29, (3), 206-208.
19. Egard, M.; Johansson, S.; Johansson, A. C.; Persson, K. M.; Dey, A. W.; Borg, B.
M.; Thelander, C.; Wernersson, L. E.; Lind, E. Nano Lett 2010, 10, (3), 809-812.

161
20. Joyce, H. J.; Gao, Q.; Tan, H. H.; Jagadish, C.; Kim, Y.; Fickenscher, M. A.; Perera,
S.; Hoang, T. B.; Smith, L. M.; Jackson, H. E.; Yarrison-Rice, J. M.; Zhang, X.; Zou,
J. Nano Lett 2009, 9, (2), 695-701.
21. Guo, Y.-N.; Xu, H.-Y.; Auchterlonie, G. J.; Burgess, T.; Joyce, H. J.; Gao, Q.; Tan,
H. H.; Jagadish, C.; Shu, H.-B.; Chen, X.-S.; Lu, W.; Kim, Y.; Zou, J. Nano Lett
2013, 13, (2), 643-650.
22. Lang, D. V.; Grimmeiss, H. G.; Meijer, E.; Jaros, M. Physical Review B 1980, 22,
(8), 3917-3934.
23. Oh, S. H.; Benthem, K. v.; Molina, S. I.; Borisevich, A. Y.; Luo, W.; Werner, P.;
Zakharov, N. D.; Kumar, D.; Pantelides, S. T.; Pennycook, S. J. Nano Lett 2008, 8,
(4), 1016-1019.
24. Collins, C. B.; Carlson, R. O.; Gallagher, C. J. Physical Review 1957, 105, (4),
1168-1173.
25. Rudolph, ertenberger, olte, aosangthong, pir os a, e
blinger, ichler, Finle , Abstreiter, G oblm ller, G Nano Lett
2011, 11, (9), 3848-3854.
26. Björk, M. T.; Schmid, H.; Breslin, C. M.; Gignac, L.; Riel, H. Journal of Crystal
Growth 2012, 344, (1), 31-37.
27. Huang, Y.; Kim, T. W.; Xiong, S.; Mawst, L. J.; Kuech, T. F.; Nealey, P. F.; Dai, Y.;
Wang, Z.; Guo, W.; Forbes, D.; Hubbard, S. M.; Nesnidal, M. Nano Lett 2013, 13,
(12), 5979-5984.
28. Mohan, P.; Motohisa, J.; Fukui, T. Nanotechnology 2005, 16, (12), 2903.
29. Gao, Q.; Saxena, D.; Wang, F.; Fu, L.; Mokkapati, S.; Guo, Y.; Li, L.; Wong-Leung,
J.; Caroff, P.; Tan, H. H.; Jagadish, C. Nano Lett 2014, 14, (9), 5206-5211.
30. Yue, J. T.; Funsten, W. P.; Taylor, R. V. In Stress Induced Voids in Aluminum
Interconnects During IC Processing, Reliability Physics Symposium, 1985. 23rd
Annual, March 1985, 1985; pp 126-137.
31. Yeo, I. S.; Ho, P. S.; Anderson, S. G. H. J Appl Phys 1995, 78, (2), 945-952.
32. An, J. H. Thermal stress induced voids in nanoscale copper interconnects by in-situ
TEM heating. D.Eng., The University of Texas at Austin, Ann Arbor, 2007.
33. Martin, J.; Plain, J. Journal of Physics D: Applied Physics 2015, 48, (18), 184002.
34. Volmer, M.; Weber, A. Z. phys. Chem 1926, 119, 277-301.
35. Seel, S. C.; Thompson, C. V.; Hearne, S. J.; Floro, J. A. J Appl Phys 2000, 88, (12),
7079-7088.
36. James, D. W. F.; Lewis, C. British Journal of Applied Physics 1965, 16, (8), 1089.

162
37. Joyce, H. J.; Wong-Leung, J.; Gao, Q.; Tan, H. H.; Jagadish, C. Nano Lett 2010,
10, (3), 908-915.
38. Tomioka, K.; Motohisa, J.; Hara, S.; Fukui, T. Japanese Journal of Applied Physics
2007, 46, L1102-L1104.
39. Partin, D. L.; Green, L.; Morelli, D. T.; Heremans, J.; Fuller, B. K.; Thrush, C. M.
Journal of Elec Materi 1991, 20, (12), 1109-1115.
40. Ghalamestani, S. G. The growth of InAs and GaSb layers and nanowires on Si
(111). Lund University, Sweden, 2012.
41. Xu, H.-Y.; Guo, Y.-N.; Liao, Z.-M.; Sun, W.; Gao, Q.; Hoe Tan, H.; Jagadish, C.;
Zou, J. Appl Phys Lett 2013, 102, (20), 203108.
42. Xu, H.; Wang, Y.; Guo, Y.; Liao, Z.; Gao, Q.; Tan, H. H.; Jagadish, C.; Zou, J. Nano
Lett 2012, 12, (11), 5744-5749.
43. Zhang, Z.; Wang, S. J.; Yu, T.; Wu, T. J Phys Chem C 2007, 111, (47), 17500-
17505.
44. Paladugu, M.; Merckling, C.; Loo, R.; Richard, O.; Bender, H.; Dekoster, J.;
Vandervorst, W.; Caymax, M.; Heyns, M. Cryst Growth Des 2012, 12, (10), 4696-
4702.
45. Hertenberger, S.; Rudolph, D.; Bichler, M.; Finley, J. J.; Abstreiter, G.; Koblmüller,
G. J Appl Phys 2010, 108, (11), 114316.
46. Saitoh, T.; Matsubara, S.; Minagawa, S. Thin Solid Films 1978, 48, (3), 339-344.
47. Du, W.; Yang, X.; Pan, H.; Wang, X.; Ji, H.; Luo, S.; Ji, X.; Wang, Z.; Yang, T. Cryst
Growth Des 2015, 15, (5), 2413-2418.
48. Hertenberger, S.; Rudolph, D.; Bolte, S.; Döblinger, M.; Bichler, M.; Spirkoska, D.;
Finley, J. J.; Abstreiter, G.; Koblmüller, G. Appl Phys Lett 2011, 98, (12), 123114.
49. Antczak, G.; Ehrlich, G., Surface diffusion: metals, metal atoms, and clusters.
Cambridge University Press: 2010.
50. Dayeh, S. A.; Yu, E. T.; Wang, D. Nano Lett 2007, 7, (8), 2486-2490.
51. Yearsley, J.; Lin, J.; Hwang, E.; Datta, S.; Mohney, S. J Appl Phys 2012, 112, (5),
054510.
52. Wu, Y.-T.; Huang, C.-W.; Chiu, C.-H.; Chang, C.-F.; Chen, J.-Y.; Lin, T.-Y.; Huang,
Y.-T.; Lu, K.-C.; Yeh, P.-H.; Wu, W.-W. Nano Lett 2016, 16, (2), 1086-1091.

163
CHAPTER 7
Conclusions and Recommendations
7.1 Conclusions
A literature study on the structural features, growth mechanism and growth techniques of
epitaxial III-V nanowires have been reviewed. The role of catalyst in nucleation and
subsequent growth of nanowires is described and a complete list of III-V nanowires growth
catalysed by non-Au metals is compiled. This literature review provides a comprehensive
study on the catalyst-induced III-V nanowires growth.
In this thesis, intriguing results on the III-V 1D nanostructures have been observed,
both using Au and non-Au metals to assist the growth. These findings are summarized
accordingly.
Growth of different InAs 1D nanostructures catalysed by Au nanoparticles
Two different types of InAs 1D nanostructures, namely nanoleaf and nanobelt, have been
investigated in detailed by electron microscopy. Firstly, insights into the growth mechanism
of leaf-like one-dimensional InAs nanostructures is depicted. Structural characterization
suggests that nanoleaves contain relatively low-energy {122} or {133} mirror twins acting
as their midribs and narrow sections connecting the nanoleaves and their underlying
bases as petioles. Notably, the mirror twins lead to identical lateral growth of the twined
structures in terms of crystallography and polarity, which is crucial for the formation of
symmetrical nanoleaves. It has been found that the formation of catalyst-induced mirror
twined nanoleaves is energy-minimization-driven. The successful growth of biomimic
semiconductor nanoleaf will be useful in the bio-inspired nano-architecture.
Growth of [ ̅]-oriented InAs nanobelts on different types of substrate are
investigated systematically. The density of inclined [ ̅]-oriented InAs nanobelts is higher
when growing heteroepitaxy on GaAs ( ̅ ̅ ̅) substrate compared with growing

164
homoepitaxy on InAs ( ̅ ̅ ̅) substrate. All the [ ̅]-oriented nanobelts, despite of the
substrates, are all defect-free zinc-blende. The reason to induce defect-free nanobelt is the
lower In composition adsorbed into the Au catalyst during growth. Finally, successful
growth vertical [ ̅]-oriented nanobelt on GaAs (001) substrate has been realised. This
detailed understanding of the growth of [ ̅]-oriented nanobelt can open up new avenues
in developing future functional devices based on this nanostructure.
Side-facets of GaAs 1D nanostructures catalysed by Au nanoparticles
The effect of growth temperature on the side-facets of Au-catalysed GaAs nanopillars with
hexagonal base was studied. We found that the side-facets of zinc-blende GaAs
nanopillars transform from {112} to {110} with increasing the growth temperature because
of competition between kinetically and thermodynamically dominated process. Besides,
diffusion-induced nanopillar foundations always present the same {112} edge side-facets
regardless of the temperature.
Exploring the growth of III-V nanowires using non-Au metals
Non-Au metals have been rarely used in assisting the growth of epitaxial III-V nanowires
due to the complexity of the reaction between non-Au metals with both group-III and
group-V species. Using Au-free methods for III-V nanowires growth are significant for
fundamental understanding on the nanowires growth mechanism that could not found in
Au-catalysed nanowires; and also of technical important for the development of future
electronic and opto-electronic devices. Success epitaxial growth of defect-free Ni-
catalysed GaAs nanowires has been reported in this thesis. During in situ annealing of a
very thin pre-coated Ni film in MOCVD chamber, the thin film would break into
nanoparticles. The suitable growth window has been identified and the possible reasons
for the epitaxial defect-free GaAs nanowires growth have been given. Nickel is anticipated
to be one of the prominent catalysts since it is greatly cheaper than Au, and is in principle
compatible with Si electronics, which will open up opportunities to grow Au-free GaAs
nanowires on Si.
The thickness of metallic film is critical to determine whether it can be function as
catalyst to induced nanowires growth. As the thickness of this film increases, it will not
break into nanoparticles but forming nano-holes on the metallic film during annealing

165
process. Taking advantage of this phenomenon, a proof-of-concept technique is
developed for catalyst-free hetero-epitaxy growth of InAs nanowires on Si wafers, which is
a very simple, low cost and general method. Before the growth of InAs nanowires, a thick
metallic film was pre-deposited on the Si wafer and then annealed to form nano-scaled
openings. These nano-openings exposed the surface of Si substrate, which allowed
subsequent nucleation and growth of nanowires; simultaneously the small size of the
nano-openings limited the lateral growth of the nucleus but induced the axial growth.
Catalyst-free and hetero-epitaxy nanowires were grown on both Si (111) and (001) wafers
successfully at growth temperatures of 550 and 600°C. One growth condition to be
highlighted is ultra-long defect-free wurtizte InAs nanowires were grown at 550°C using Ni
mask. This development of hetero-epitaxy growth of vertical III-V nanowires without
assistance of metallic catalysts on Si wafers is highly demanded for future Si-based
electronic and optoelectronic devices.
7.2 Recommendations
Indeed, tremendous research has been done to gain the understanding on the morphology
and structural of III-V nanowires. However, there are many issues have yet to be resolved,
for instance, the effects of the catalysts in the growth of epitaxial III-V nanowires. In view of
that, few issues that need pressing considerations are listed.
Composition and structural properties of metal catalyst
The thorough understanding of the composition and crystal structure of the metal catalyst
is urgent to determine whether the classic VLS mechanism is valid or other growth
mechanism is involved.
Catalyst/nanowire interface
Catalyst/nanowire interface has a significant role in determining the morphology and
structural quality of nanowires since the nucleation initiates at the interface. By altering the
metal catalyst, non-{ } catalyst/nanowire interface can be obtained. This will definitely
provide an insight into the correlation between the interface and quality of III-V nanowires.

166
Based on Au-As phase diagram, the solubility of As in Au is less than 1%. In
general, this is still unclear regarding the mechanism of group-V elements incorporating
into nanowires. It is believed that the interface for Au-catalysed nanowires between the
liquid binary Au-based alloy and the nanowire is the growth front for the incorporation of
group-V elements. Therefore, by collecting the analytical data at the catalyst/nanowire
interface using other catalyst system, exciting results are expected to explain the unclear
mechanism involved.
Controllable crystal quality of III-V nanowires by using different metal catalysts
There are strong experimental and theoretical data that support the tuning of crystal
structures, commonly zinc-blende and wurtzite, by changing the diameter of catalyst, the
growth temperatures and V/III ratio. Nevertheless, there is little attention have been paid
on altering the crystal structures of nanowire by changing the properties of catalyst. By
utilizing different metal catalysts, it is anticipated to provide better understanding on the
driving force of common zinc-blende and wurtzite structures or less reported 4H and 6H
structures formed in III-V nanowires.
Application of III-V nanowires or other 1D nanostructures for future electronic and opto-
electronic devices
It is important to test the electronic or opto-electronic performance of these III-V 1D
nanostructures after successful control of their morphology and structural quality. The
design of III-V nanowires assisted by Si-friendly non-Au metals is practical to be integrated
in the future Si-based electronic and opto-electronic devices.