Embed Size (px)
Citation preview

A RECOMBINANT MYELOID-BINDING ADENOVIRUS FOR TARGETED PULMONARY GENE THERAPY
by
MICHAEL O. ALBERTI
ZDENEK HEL, COMMITTEE CHAIR GENE P. SIEGAL, COMMITTEE CO-CHAIR
DAVID T. CURIEL, MENTOR JUSTIN C. ROTH, CO-MENTOR
J. EDWIN BLALOCK DAVID D. CHAPLIN
LOUIS B. JUSTEMENT CLAUDE H. STEELE
A DISSERTATION
Submitted to the graduate faculty of The University of Alabama at Birmingham, in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
BIRMINGHAM, ALABAMA
2011

Copyright by Michael O. Alberti
2011

A RECOMBINANT MYELOID-BINDING ADENOVIRUS FOR TARGETED PULMONARY GENE THERAPY
MICHAEL O. ALBERTI
MOLECULAR AND CELLULAR PATHOLOGY
ABSTRACT
Inflammation and airway destruction are hallmarks of many debilitating lung dis-
eases such as chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute
lung injury (ALI), and cancer. Gene-based therapeutic interventions that modulate this
pathologic inflammatory response are likely to reduce the progressive destruction to lung
airways. In this regard, a number of strategies have been evaluated for targeting the pul-
monary vasculature; particularly those based on serotype 5 Adenovirus (Ad5). The ad-
vantages of Ad over other vector systems include: in vivo stability, low oncogenic poten-
tial, and large packaging capacity. Yet, specific and efficient gene delivery to the lung
has been hampered by a number of barriers in vivo; mainly the degree by which Ad5 vec-
tors are sequestered in the liver. The complexity of Ad5 liver tropism has largely been
unraveled, permitting improved efficacy of Ad5 gene delivery. However, Kupffer cell
(KC) scavenging and elimination of Ad5 still represent major obstacles to lung delivery
strategies since KC uptake substantially reduces Ad5 bioavailability for target tissues and
compensatory dose escalation leads to hepatotoxicity.
Efficient targeting of cell types within the lung may preclude the need for viral
modifications that ablate liver sequestration mechanisms. Leukocytes represent an ideal
target for delivery of therapeutics to the pulmonary vasculature as venous blood flow is
first directed through the pulmonary vasculature, and circulating leukocytes accumulate
within the lung due to reduced transit rates through the extensive microvasculature net-
iii

work. The contributing role of leukocytes in the pathogenesis of lung diseases, further
highlights this population as an ideal target for therapeutic intervention of inflammatory
lung disease.
Given the size and location of the lung leukocyte pool, we designed a novel lung-
targeting approach based on modulation of Ad5 vector tropism to myeloid leukocytes.
We demonstrate that this leukocyte-targeting approach specifically localizes Ad5 virions
and gene transfer to the lung microvasculature and prevents KC uptake and hepatocyte
transduction. This strategy resulted in a 165,000-fold enhanced lung-targeting, compared
to unmodified Ad5. This work demonstrates that this novel myeloid-targeting approach,
for the first time, results in significantly enhanced lung gene transfer and elimination of
liver tropism in the absence of further capsid modifications.
Keywords: Adenovirus, Inflammation, Kupffer cells, Lung, Myeloid, Targeting
iv

DEDICATION
To the women first: my magnificent MOMMA, my loving sister Danielle, and my awesomely wonderful girlfriend Kimberly.
And then the men: my Dad and my Bub.
Oh and Barry.
Thank you for all of your support while you have patiently waited for me to return to civilization!
v

ACKNOWLEDGEMENTS
I would first like to thank all of my family and friends in Birmingham, California,
and Chicago for their support. Secondly, I am extremely grateful to all of the people who
“loaned” me reagents or helped me with experiments: Kim Thomas, R. Glenn King, Jes
Werner, Brian Dizon, Jessy Deshane, Svetlana Komarova, Miho Murakami, John
McAuley, Susan Hedley, Pamela Powell, Dezhi Wang, Robert Snelgrove, Matt Hardison,
Heidi Coppersmith, Robert Flynn, Shanrun Liu, Melissa Chimento, Larry Gartland and
Marion Spell. My Coffee Pushers: Forest Perk, Lucy’s, and O’Henry’s.
I am also fortunate to have had great lab members, some of them yet to be named,
both old and new: Hideyo Ugai, Igor Dmitriev, Elena Kashentseva, Alexander Pereboev,
Larisa Pereboeva, Joel Glasgow, Justin Barnes, Meredith Preuss, Travis Lewis, David
Gaston, Ute Saunders, Robert Brunzel, and Jennifer Coleman. Importantly, Jim Markert,
Jackie Parker, and Rich Whitley provided me with much needed space at the end for and
the UAB MSTP, particularly Dr. Robin Lorenz and Dr. Lou Justement, has been ex-
tremely supportive of me throughout; all of which I am extremely thankful for.
I would also like to point out the invaluable experiences I had working with Dr.
Tom Ryan and Dr. Randall Davis here at UAB and Dr. Dennis Revie at California Lu-
theran University, who sparked my curiosity of science and interest in research. I also
want to acknowledge each of my committee members: Dr. Ed Blalock, Dr. David Chap-
lin, Dr. Zdenek Hel, Dr. Lou Justement, Dr. Gene Siegal, and Dr. Chad Steele. You all
vi

have been wonderful and understanding and very helpful throughout whenever I needed
your expertise or advice.
Lastly, my mentors, Dr. David Curiel and Dr. Justin Roth, for all of the knowl-
edge they bestowed and wisdom they shared with me. Their passion for what they do is
infectious… but not in an Adenovirus kind of way. Although they performed different
roles, they both inspired confidence in me to continue forward no matter what the chal-
lenge. Although he keeps himself busy, Dr. Curiel was always just a “Now” email away
from answering any questions or meeting requests I may have had. And aside from being
one of my bosses, Justin has also been a very good friend that I hope to always have since
he has somehow been extremely resilient to my never-ending questioning, heckling, agi-
tating, and sleepless hunger for chicken wings.
vii

TABLE OF CONTENTS
Page
ABSTRACT....................................................................................................................... iii
DEDICATION.....................................................................................................................v
ACKNOWLEDGEMENTS............................................................................................... vi
LIST OF TABLES............................................................................................................. xi
LIST OF FIGURES .......................................................................................................... xii
LIST OF ABREVIATIONS ............................................................................................ xiv
INTRODUCTION ...............................................................................................................1
The Inflammatory Cascade ..........................................................................................1 Nonresolving Inflammation in Pulmonary Disease .....................................................1 Therapeutic Options for Pulmonary Inflammatory Diseases .......................................4 Gene Delivery Vectors and Strategies for Pulmonary Disease ....................................5 Nonviral Strategies................................................................................................6 Viral Strategies......................................................................................................8 Adenovirus .................................................................................................................12 Ad Vectors for Pulmonary Airway Gene Therapy.....................................................17 Intravenous Delivery of Ad Vectors for Pulmonary Gene Therapy...........................21 Barriers to Intravenous Delivery of Ad ..............................................................22 Strategies to Reduce Liver Targeting .................................................................25 Fiber Modification Strategies to Enhance Targeting of Pulmonary Endothelium .....................................................................................28 The Marginated Leukocyte Pool ................................................................................30 Experimental Aims.....................................................................................................31 METHODS ........................................................................................................................32
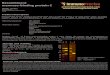
Cell Lines ...................................................................................................................32 Animals ......................................................................................................................34 Phage Display.............................................................................................................34 Myeloid-binding Peptide (MBP) Synthesis ...............................................................35
viii

M13-MBP Mutant Phage Vector Construction..........................................................36 Ad Vector Construction..............................................................................................36 Fiber Shuttle and Expression Vectors.................................................................36 Adenovirus Genome Vectors..............................................................................37 Adenoviruses ..............................................................................................................37 Recombinant Fiber Expression System......................................................................38 Western Blotting.........................................................................................................39 Phage Preparation.......................................................................................................39 In vitro Binding Experiments .....................................................................................40 Transmission Electron Microscopy (TEM)................................................................41 Dihydrorhodamine 123 (DHR123) Assay..................................................................42 In vitro Transduction Experiments.............................................................................42 Luciferase Biodistribution Experiments.....................................................................43 Virus Particle Localization Experiments....................................................................43 Ad.MBP.CMV-GFP in vivo Experiments .................................................................44 Lung Immunohistochemistry .....................................................................................44 Ad-bound Lung Cell Cytospins..................................................................................45 Flow Cytometry..........................................................................................................45 Statistical Analysis .....................................................................................................46 DEVELOPMENT AND CHARACTERIZATION OF A LEUKOCYTE- TARGETED ADENOVIRUS ...........................................................................................51 Introduction ................................................................................................................51 Characterization of a Myeloid-binding Phage............................................................52 Stable Incorporation of a Myeloid-binding Peptide (MBP) into Ad5........................59 Functional Characterization of Ad.MBP....................................................................64 LEUKOCYTE-MEDIATED HAND-OFF OF ADENOVIRUS FOR SPECIFIC AND EFFICIENT LUNG TARGETING..........................................................................69 Introduction ................................................................................................................69 Ad.MBP Particle and Gene Transfer Biodistribution in vivo ....................................70 Assessment of Lung Milieu Following Ad.MBP Gene Transfer ...............................72 Identification of Ad.MBP Transduced Lung Cell Populations ..................................79 Ad.MBP Binding and Transduction of ECs...............................................................83 DISCUSSION....................................................................................................................88
Phage Display Library Screening...............................................................................88 Reduction in Ad5 Hepatocyte Gene Transfer ............................................................90 Abrogation of Acute Liver Sequestration of Ad5 ......................................................92 Leukocyte-mediated Hand-off of Ad.MBP to Pulmonary Vasculature .....................92 Summary and Future Directions.................................................................................94
ix

LIST OF REFERENCES...................................................................................................97
APPENDICES
A IACUC APPROVAL FORM (2006-2010).......................................................121
B IACUC APPROVAL FORM (2011) ................................................................123
C VECTOR MAPS...............................................................................................125
D LUNG GATING SCHEMA FOR FLOW CYTOMETRY ..............................134
x

LIST OF TABLES
Table Page
1 Human Adenovirus (HAdV) Serotypes .....................................................................13
2 List of Oligonucleotides .............................................................................................47
3 Adenoviral Vectors Generated by Homologous Recombination ...............................49
4 Titers of Adenoviruses Generated..............................................................................50
5 Phage Peptide Candidate List.....................................................................................53
xi

LIST OF FIGURES
Figure Page
1 Percent of trials using various vector platforms to date ...............................................7
2 A cartoon section through the HAdV capsid and core...............................................15
3 Mechanism for HAdV cellular entry..........................................................................16
4 HAdV genome organization.......................................................................................17
5 Adenoviral vectors for gene therapy ..........................................................................18
6 Characterization of MiPMVECs ................................................................................33
7 Phage display panning strategies................................................................................54
8 The WTLDRGY phage clone binds BMCs ...............................................................55
9 The WTLDRGY peptide binds broadly but specifically to myeloid BMCs..............56
10 Scanning alanine mutagenesis of the MBP sequence reveals critical residues for binding ..................................................................................................................58 11 MBP-binding is not dependent on MHC class I or II (MHC-I, -II) ...........................60
12 Retargeting of Ad5 .....................................................................................................61
13 Assessment of FF-MBP viability by Western blot.....................................................62
14 FF-MBP retains myeloid-binding specificity.............................................................63
15 MBP fibers are efficiently incorporated into the Ad capsid of purified virions.........63
16 Incorporation of FF-MBP into Ad5 (Ad.MBP) retains myeloid-binding specificity in vitro.......................................................................................................64 17 Expression of hCAR permits efficient Ad5 binding of BMCs ..................................65
xii

18 Ad.MBP binds myeloid cells from various tissues in vitro........................................65
19 Ad.MBP-binding to neutrophils by TEM...................................................................66
20 Ad.MBP-binding to neutrophils does not induce oxidative burst ..............................67
21 Transduction of CD11b+ cells is enhanced with Ad.MBP.........................................68
22 Systemically administered Ad.MBP enhances lung and diminishes liver gene transfer .......................................................................................................73 23 Systemically administered Ad.MBP eliminates liver virus particle sequestration ....74
24 Ad.MBP particles are not removed from the lung with perfusion of the pulmonary circulation.................................................................................................75 25 Ad.MBP lung gene transfer is retained at low doses of systemically administered virus ......................................................................................................76 26 Ad.MBP lung:liver ratio is retained at low doses of systemically administered virus ......................................................................................................77 27 Ad.MBP lung gene transfer does not result in leukocyte recruitment .......................78
28 Ad.MBP lung gene transfer does not alter lung cellular composition .......................80
29 Ad.MBP targets gene transfer to the pulmonary endothelium...................................81
30 Ad.MBP lung gene transfer is localized to alveolar capillaries .................................84
31 ECs are refractive to Ad.MBP binding ......................................................................85
32 ECs are refractive to Ad.MBP transduction...............................................................87
xiii

LIST OF ABBREVIATIONS
α-MEM alpha minimum essential medium
A1AT alpha1-antitrypsin
AAV adeno-associated virus
AAV# AAV serotype #
ACE angiotensin-converting enzyme
Ad adenovirus
Ad# adenovirus serotype #
Ad.MBP Myeloid-targeted Ad5
ALI acute lung injury
AM alveolar macrophage
APC antigen presenting cell
ARDS acute respiratory distress syndrome
bp base pair
β2m β2 microglobulin
BMC bone marrow cell
C4BP C4-binding protein
CAR coxsackie and adenovirus receptor
CF cystic fibrosis
CFTR cystic fibrosis transmembrane conductance regulator
xiv

CIITA MHC class II transactivator
CMV cytomegalovirus
COPD chronic obstructive pulmonary disorder
CTL cytotoxic T lymphocytes
DC dendritic cell
DHR123 dihydrorhodamine 123
DMEM Dulbecco's modified Eagle's medium
EC endothelial cell
FACS fluorescent-activated cell sorting
FCS fetal calf serum
FF fiber-fibritin
FIV feline immunodeficiency virus
FLT fms-related tyrosine kinase
FX factor X
GAP Rho-GTPase-activating protein
GdCl3 gadolinium chloride
GFP enhanced green fluorescent protein
GvHD graft versus host disease
HAdV human adenoviruses
HBSS Hank’s Balanced Salt Solution
hCAR human Coxsackie and Adenovirus Receptor
HDAd helper-dependent Ad
HIV human immunodeficiency virus
xv

HSPG heparin sulfate proteoglycan
HVR hypervariable region
IFN interferon
IHC immunohistochemistry
IL interleukin
i.m. intramuscular
iMBP inverse or inverted MBP
IP-10 IFN-inducible protein 10
i.t. intratracheal
ITR inverted terminal repeat
i.v., IV intravenous
kb kilobase
KC Kupffer cell
KKTK Lys-Lys-Thr-Lys
linMBP linearized MBP
LT leukotriene
LV lentivirus
LUC luciferase
MBP Myeloid-Binding Peptide
MCEC mouse cardiac endothelial cells
MCP monocyte chemotactic protein
mHag minor histocompatibility antigen
MHC-I MHC class I
xvi

MHC-II MHC class II
MIP macrophage inflammatory protein
MiPMVEC mouse induced pulmonary microvascular endothelial cells
MLP major late promoter
MMP matrix metalloproteinase
MOI multiplicity of infection
MoMLV Moloney murine leukemia virus
MW molecular weight
NAb neutralizing antibody
NBF neutral-buffered formalin
NE neutrophil elastase
NK natural killer (cell)
OTC ornithine transcarbamylase
PBS phosphate buffered saline
PEG polyethylene glycol
pfu, PFU plaque forming unit
PMA phorbol myristate acetate
poly-HPMA poly-N-[2-hydroxypropyl]methacrylamide
qPCR quantitative PCR
R123 Rhodamine 123
RES reticuloendothelial system
RGD Arg-Gly-Asp
ROS reactive oxygen species
xvii

RLU relative luciferase units
RV retrovirus
SCID-X1 X-linked severe combined immunodeficiency disorder
SIN self-inactivating
SIRS systemic inflammatory response syndrome
SR scavenger receptor
TEM transmission electron microscopy
TNF tumor necrosis factor
TP (adenovirus) terminal protein
TBS Tris-buffered saline
t.v. tail vein injected
v.p., VP virus particle
VSV-G vesicular stomatitis virus G-protein
vWF von Willebrand factor
xviii

INTRODUCTION
The Inflammatory Cascade
Acute inflammation is an important physiological response to tissue injury or in-
fection (reviewed in Kumar et al., 2005; Medzhitov, 2008). Resident cells, such as ma-
crophages, mast cells and dendritic cells (DCs), respond to stress or injury and release
cytokines that promote vasodilation, increased vascular permeability and leukocyte re-
cruitment. Infiltrating leukocytes, mainly neutrophils and some mononuclear cells (mo-
nocytes and macrophages), release additional inflammatory mediators that propagate and
mature the inflammatory response. Normally this is an organized process that resolves
quickly after the offending insult is removed and levels of anti-inflammatory mediators
increase. However, when an insult persists or when the response is dysregulated, a nonre-
solving pathological state ensues, which can lead to extensive fibrosis, tissue damage, and
organ dysfunction, resulting in significant morbidity and mortality (Nathan and Ding,
2010).
Nonresolving Inflammation in Pulmonary Disease
Nonresolving inflammation is a significant contributor to the pathogenesis of a
wide variety of disease processes, some of which include: atherosclerosis, inflammatory
bowel disease, rheumatoid arthritis, neurodegenerative disease, and many cancers (Na-
than and Ding, 2010). Of particular interest are a number of debilitating inflammatory
lung diseases such as COPD, CF, and ALI. In each of these lung diseases, the pathogene-
sis is characterized by a neutrophilic inflammation associated with excessive release of
1

proteases, including neutrophil elastase (NE), which can cause extensive pulmonary tis-
sue damage (Ohbayashi, 2002). Thus, the inflammatory cells themselves (both neutro-
phils and macrophages) are directly implicated in the initiation and propagation of the
self-amplifying inflammatory loop leading to the pathological state of dysfunction (Beeh
and Beier, 2006; Conese et al., 2003; Nathan and Ding, 2010).
COPD is debilitating condition that is the fourth leading cause of death worldwide
(World Health Organization, 2000). Although the majority of cases appear to be ac-
quired, ~2% of patients have a genetic deficiency in the NE inhibitor, alpha1-antitrypsin
(A1AT) (DeMeo and Silverman, 2004). COPD is generally characterized by
an irreversible and progressive limitation in airflow, usually associated with exposure to
inhaled noxious agents (e.g. smoking or pollution) (Barnes, 2000; MacNee and Tuder,
2009). The chronic inflammatory process involves both the peripheral airways and lung
parenchyma (Barnes, 2000) and is driven by macrophage activation and the local release
of chemokines such as leukotriene (LT)B4 and interleukin (IL)-8 that recruit neutrophils.
In this milieu, both neutrophils and macrophages release multiple proteases (e.g. NE, pro-
teinase 3, matrix metalloproteinases [MMPs]) (Beeh and Beier, 2006; Barnes, 2000) lead-
ing to a proposed protease/antiprotease imbalance (Stockley, 1999 and 2002) that breaks
down connective tissue in the lung parenchyma. These breakdown products recruit addi-
tional inflammatory cells and activate mediators that lead to the amplification and per-
petuation of the chronic inflammatory process and further pulmonary dysfunction (Beeh
and Beier, 2006; Barnes, 2000).
This self-propagating inflammatory loop has also been implicated in other chronic
inflammatory lung diseases, mainly CF (Conese et al., 2003; Birrer et al., 1994; Gaggar
2

et al., 2007). Unlike COPD, CF is a common autosomal recessive disorder caused by mu-
tations in a single gene, the cystic fibrosis transmembrane conductance regulator (CFTR),
which leads to abnormal mucous secretion, chronic bacterial infections, and significant
neutrophil-rich inflammation in the airway (Ramsey, 1996). Failing to undergo apoptosis,
these infiltrating neutrophils accumulate, become necrotic, and release genomic DNA,
proteases, and reactive oxidative products. The DNA viscosity further reduces mucocil-
liary bacterial clearance mechanisms, and the protease and oxidative tissue damage re-
cruits additional neutrophils (Amitani et al., 1991; Donaldson et al., 2006; Hartl et al.,
2007; Perks and Shute, 2000).
While COPD and CF are chronic inflammatory diseases, ALI or its more severe
form, acute respiratory distress syndrome (ARDS), is characterized by a rapidly progres-
sive inflammatory process that leads to hypoxemic respiratory failure and up to 40%
mortality (Rubenfeld et al., 2005; Ware and Matthay, 2000). Any number of direct or in-
direct assaults against the pulmonary vascular endothelium can lead to acute non-
cardiogenic pulmonary edema and release of cytokines and proteases by macrophages
and infiltrating neutrophils (Ware and Matthay, 2000). Despite the fact that the prote-
ase/antiprotease imbalance theory is not as striking (Ohbayashi, 2002) as the evidence
supporting an imbalance in angiogenic growth factors (Gallagher et al., 2008; Karm-
paliotis et al., 2002; Mura et al., 2004; van der Heijden et al., 2008 and 2009), a hallmark
of the disorder is characterized by marked accumulation of neutrophils at both the site of
endothelial injury and within the pulmonary edema fluid.
Thus, in each of these pulmonary afflictions, the capacity of these inflammatory
cells to generate a complex network of pro-inflammatory cytokines and proteases indi-
3

cates that they directly contribute to this positive feedback or feed-forward loop, which
acts to perpetuate inflammatory damage to the point of organ dysfunction. Moreover, the-
rapeutic interventions that modulate this nonresolving inflammatory response, especially
if tailored to the unique pathophysiology in each of these cases, are likely to reduce the
progressive destruction to lung airways (Ramsey, 1996).
Therapeutic Options for Pulmonary Inflammatory Diseases
COPD, CF, and ARDS are all progressive and frequently fatal disorders that have
few (or none in the case of ARDS) pharmacologic strategies that treat the cause of the
disorder, but rather are aimed at treating the symptoms. Thus, existing therapeutics are
mostly directed at either improving lung function, primarily with beta-adrenergic bron-
chodilators (Ramsey, 1996; Fanta, 2009; Sutherland and Cherniack, 2004), or at broadly
limiting inflammation with the use of inhaled corticosteroids (Chung et al., 2009; Falk et
al., 2008; Fanta, 2009). While the use of bronchodilators is important for symptom relief,
anti-inflammatory therapies are more likely to slow the progressive airway destruction in
these diseases (Ramsey, 1996).
Although corticosteroids have a plethora of anti-inflammatory behaviors (van der
Velden, 1998; Barnes, 2005) for the treatment of asthma, their use, particularly in asth-
matic children, has been a concern due to the potential systemic side effects (Barnes,
1995). Furthermore, usage of corticosteroids is less effective for treating neutrophilic in-
flammatory diseases, such as COPD (Sutherland and Cherniack, 2004), since airway neu-
trophilia is less likely to respond to this type of treatment, compared to eosinophilic in-
flammation associated with asthma (Fanta, 2009).
4

Thus, there has been a pressing need to develop new classes of therapeutics that
act directly on inflammatory pathways. To this end, several new drugs are currently in
development, including IL-8 antagonists, protease inhibitors and antioxidants (Barnes,
2002). Since leukocyte subsets are at the heart of many of these inflammatory disorders
and are contributing to the initiation, propagation, and pathology of these diseases, treat-
ment strategies should aim at specifically tailoring therapy to the unique pathophysiology
of each disease to modulate these cells and their mediators. However, the complex nature
of these diseases may require more complex or gene-based products for molecular speci-
ficity and therapeutic efficacy. Therefore, much work has focused on developing a vector
or gene delivery system in order to mediate efficient and specific targeting of lung tissue,
as well as to sustain long term therapeutic gene expression, which at this point has been
unattainable.
Gene Delivery Vectors and Strategies for Pulmonary Disease
The rationale for gene therapy stems from our understanding of disease patho-
genesis, particularly in cases where a known dysfunctional gene or set of genes leads to
the phenotype of the disease, which if rescued with a normal version of the gene(s) leads
to amelioration of the disease phenotype. Gene therapy aims to introduce exogenous
therapeutic genes or proteins into pathologic or malignant cells or tissues to treat disease
in which specific genes or pathways are either abnormal or perturbed. While there are a
number of vector delivery platforms to achieve this end, each system should address a
number of key considerations. Administration should: (i) occur via a non-invasive route,
(ii) specifically and efficiently target the appropriate organ, tissue, or cell types of inter-
5

est, (iii) allow for therapeutic levels of transgene expression for an appropriate length of
time, and (iv) do so in a manner that absolutely minimizes adverse reactions (such as a
severe immune response) and toxicity to the recipient (Kay et al., 2001).
In addition to treating various metabolic diseases (Fischer et al., 2006), inherited
immune deficiency disorders (Kohn, 2008; Qasim et al., 2009), and cancer (Waxman and
Schwartz, 2003; Liu and Kirn, 2008; Brenner and Heslop, 2010), gene based medicines
have long been evaluated for treatment of lung disease and offer potential for gene re-
placement or augmentation (corrective strategies), and even sustained targeting of a spe-
cific pathologic pathway (Friedmann, 1989). Both heritable and acquired forms of lung
disease, particularly CF and cancer, are amenable to genetic amelioration (Albelda et al.,
2000; Kolb et al., 2006), but non-specific and inefficient gene delivery still remains as a
key limitation to this treatment approach, as demonstrated by the extensive time and ef-
fort that has been diverted to unsuccessful CFTR gene delivery to airway epithelial cells
in CF patients (Sueblinvong et al., 2007).
Nonviral Strategies
Several strategies have been employed to enhance lung gene transfer (particularly
to epithelial cells), including nonviral- and viral-based methodologies. Due to growing
safety concerns (discussed below) and the comparatively smaller DNA packaging capac-
ity of viral vectors as a whole, a number of nonviral DNA delivery strategies have been
developed. In fact, since 2004 there has been an increase in the percentage of human clin-
ical trials that utilize these nonviral strategies compared to viral vector applications
(Edelstein et al., 2004 and 2007; http://www.wiley.co.uk/genmed/clinical) (Figure 1).
6

1989-2004 1989-2007 1989-Date0
10
20
30 AdRVAAVHSV / Pox / VacciniaNaked/Plasmid DNALipofectionOther or Unknown
Time period
% o
f clin
ical
tria
ls
Figure 1. Percent of trials using various vector platforms to date. Since 2004, the use of retovirus (RV)-based vectors has significantly declined in clinical trials. Adenovirus (Ad) has seen a similar, albeit not as steep, decline. In contrast, nonvi-ral strategies (naked/plasmid DNA and lipofection) have seen an increase in use since 2004. At present, lentiviruses (LV) represent 2.1% of all clinical trials but have been included with "Other or Unknown" instead of "RV" since they were not not specifically mentioned before 2007. Data were pooled from multiple sources: Edelstein et al., 2004 and 2007 and http://www.wiley.co.uk/genmed/clinical.
The least complex but most popular nonviral approach (~18% of all clinical trials;
Edelstein et al., 2007) utilizes naked DNA coding for a therapeutic gene, typically in-
jected by various routes or directly into various tissues for cellular uptake and subsequent
gene expression (Al-Dosari and Gao, 2009). Reporter gene expression from naked DNA
was first demonstrated in muscle cells following intramuscular (i.m.) injection into mice
(Wolff et al., 1990). However, administration of naked DNA, either by aerosolization, or
by intratracheal (i.t.) or intravenous (i.v.) injection does not result in high levels of trans-
gene expression in the lung (Zabner et al., 1997a; Oh et al., 2001) unless encapsulated
with cationic lipid complexes (lipoplexes) (Stribling et al., 1992; Zhu et al., 1993). Yet,
lipoplexes are relatively short lived entities in vivo and can lead to substantial toxicity and
inflammation following administration for lung delivery. In this regard, a number of nov-
7

el strategies such as alternative formulations of lipoplexes as well as DNA-polymer con-
jugates (polyplexes) have been devised to increase both gene delivery and in vivo stabil-
ity, and to decrease toxicity and inflammation that is associated with administration of
these nonviral complexes (reviewed in Kinsey et al., 2005; Al-Dosari and Gao, 2009).
Overall, due to poor lung distribution and low levels of transgene expression, few
of these methods have gone on to show sufficient improvements to lung function in either
animal models or human trials (Caplen et al., 1995; Gill et al., 1997; Konstan et al.,
2004). Thus, virus-based gene delivery approaches currently offer the greatest potential
to achieve both a homogenous distribution of vector and high expression levels within the
lung (Sueblinvong et al., 2007).
Viral Strategies
Viral vectors have the capacity to efficiently enter cells and generate higher levels
of transgene compared to their nonviral counterparts, affording them a long history as
useful therapeutic agents. However, safety concerns raised over a decade ago tempered
much of this early enthusiasm. In 1999, Jesse Gelsinger died from systemic inflammatory
response syndrome (SIRS; cytokine storm) 98 hours after i.v. infusion of a therapeutic
adenovirus vector for ornithine transcarbamylase (OTC) deficiency (Raper et al., 2003).
Then in 2003, it was reported that two children developed leukemia (Hacein-Bey-Abina
S et al., 2003a and 2003b) following treatment for X-linked severe combined immunode-
ficiency disorder (SCID-X1) wtih autologous transplant of retrovirus-corrected CD34+
bone marrow cells (BMCs) (Cavazzana-Calvo M et al., 2000; Hacein-Bey-Abina S et al.,
2002). Despite these setbacks, greater understanding of vector biology and recent cures
8

obtained with viral vectors have demonstrated both the improved safety and efficacy of
virus-based gene delivery platforms (Pearson et al., 2004; Bainbridge et al., 2008; Boztug
et al., 2010) and have piqued renewed interest in these technologies given the limitations
of nonviral gene delivery approaches.
The utility of each vector type has largely been restricted to the native viral entry
and gene expression pathways, and how these pathways complement the therapeutic re-
quirements of the disease. Each vector has a unique set of advantageous and disadvanta-
geous characteristics that must be considered when selecting a viral platform (Verma and
Somia, 1997; Flotte et al., 2007). Although there are many viral vectors that have been
evaluated for pulmonary disease, only those based on retrovirus (RV), lentivirus (LV),
adeno-associated virus (AAV), and adenovirus (Ad) will be presented.
Traditionally, replication-deficient, self-inactivating (SIN) vectors based on RV
(e.g. Moloney murine leukemia virus [MoMLV]) and LV (e.g. human or feline immuno-
deficiency virus [HIV, FIV]) have been favored for therapies requiring long-term stable
gene expression, due to their ability to integrate their genetic payload into the host ge-
nome of many different cell types, including non-dividing cells in the case of LV (Ne-
schadim et al., 2007; Schambach and Baum, 2008). The viral envelope glycoprotein per-
mits both binding and fusion of virus to target cells. As a means of expanding viral tro-
pism and increasing virus stability, RV and LV can be substituted or ‘pseudotyped’ with
glycoproteins from other enveloped viruses (Cronin et al., 2005; Frecha et al., 2008). The
vesicular stomatitis virus G-protein (VSV-G) has been commonly used to pseudotype
both RV (Burns et al., 1993) and LV (Naldini et al., 1996) vectors, thereby broadening
host and tissue tropism, but limiting their use to ex vivo applications in which the target
9

cells are first isolated (typically BMCs or other hematological cell types) before transduc-
tion and re-infusion back into the recipient (Miyoshi et al., 1999; Kohn, 2008). Indeed,
the successful application of this strategy has resulted in cures for a number of hema-
tologic malignancies, as already mentioned (Cavazzana-Calvo M et al., 2000; Boztug et
al., 2010).
Considering the broad non-specific tropism of VSV-G pseudotyped vectors, their
application in vivo has mainly been limited to local delivery methods such as direct injec-
tion into the brain (Naldini et al., 1996) or directly into the airway of the lung (Johnson et
al., 2000). However a number of barriers have limited efficient transduction of lung epi-
thelial cells, including alveolar macrophages (AMs) (McCray et al., 1997; Wilson et al.,
2010; Hirayama et al., 2011), and the need to disrupt epithelial tight junctions in order to
get sufficient expression (Wang et al., 1999; Johnson et al., 2000). Vascular delivery of
LVs with broad tropism properties does not result in efficient or specific targeting of the
pulmonary vasculature (Peng et al., 2001; Follenzi et al., 2002; Pan et al., 2002). Incorpo-
ration of targeting moieties, such as antibodies and ligand or receptor molecules, into mu-
tated envelope glycoproteins from the Sindbis and measles viruses has demonstrated the
ability to target specific cell types in vitro and in vivo, although targeting the pulmonary
vasculature has not yet been demonstrated (Morizono et al., 2005; Yang et al., 2006;
Funke et al., 2008; Anliker et al., 2010). Nevertheless, limiting titers and a number of
safety concerns still limit the immediate usefulness of these vectors for in vivo lung gene
transfer (Verma and Somia, 1997; Kolb et al, 2006; Flotte et al., 2007).
Although other virus platforms, such as Sendai virus (Yonemitsu et al., 2000) and
RSV (Zhang et al., 2002) have received some attention, the lung gene therapy field has
10

mostly favored AAV and Ad vector platforms. Both can be produced at much higher ti-
ters than RV and LV vectors and the wild-type viruses they are derived from cause mi-
nimal disease (or no known disease in the case of AAV) (Verma and Somia, 1997; Kay et
al., 2001). Similar to RV/LV vectors, wild-type AAV can preferentially integrate into
human chromosome 19 at a site designated AAVS1 (Kotin et al., 1992). The small sin-
gle-stranded DNA genome of AAV is flanked by two inverted terminal repeats (ITRs)
and encodes only two genes, rep and cap, which are both removed and replaced with the
transgene in gene therapy vectors (Russell and Kay, 1999). Without rep, AAV vectors
cannot integrate their genome, but can still establish stable episomal latency in target
cells (Russell and Kay, 1999). Early studies demonstrated that AAV serotype 2 (AAV2)
based vectors were capable of stable gene transfer to lung epithelium, however the levels
of transduction were low (Flotte et al., 1993; Halbert et al., 1995 and 1997; Moss et al.,
2004). This is due to a number of factors such as the lack of AAV2 receptor expression at
the epithelial surface and the numerous lung barriers to vector delivery (e.g. innate vector
clearance and the glycocalyx barrier) (Duan et al., 1998 and 2000). Furthermore, it has
been demonstrated that administration of AAV to the airway induces the formation of
neutralizing antibodies (NAbs) which limits the effectiveness of repeated vector delivery
(Halbert et al., 1997 and 1998; Moss et al., 2004).
Given the lack of receptor availability for AAV2, a number of strategies have
been undertaken to increase the efficiency and also the specificity of vector delivery to
the lung. Alternative AAV serotypes have been discovered and some, such as AAV5,
AAV6, AAV8, and AAV9, show enhanced gene transfer to the lung (Zabner et al., 2000;
Halbert et al., 2001; Liqun Wang et al., 2009; Limberis and Wilson, 2006). Some strate-
11

gies aim to develop mutant AAVs that show enhanced lung transduction (Vandenberghe
et al., 2009) while others have genetically incorporated targeting ligands to retarget vi-
ruses to specific receptors and cell types, both i.t. (Liqun Wang et al., 2009) or i.v. (Work
et al., 2006). While use of AAV as a vehicle for lung gene therapy has a promising fu-
ture, AAV vectors are still limited by a few important factors. First, AAV vectors have
traditionally lacked significant DNA packaging capacity (only ~5 kilobases [kb]) (Russell
and Kay, 1999). Although AAVs can now deliver 10 kb of DNA by exploiting the head
to tail viral genome concatamerization that occurs in target cells, this requires the simul-
taneous (and inefficient) delivery of two different viruses each carrying up to 5 kb of the
transgene (Duan et al., 2001). Finally, AAVs are not extremely tolerant to the genetic
manipulations required for improved target cell specificity (Baker, 2003). This is in con-
trast to Ad vectors which can carry up to ~37 kb of exogenous DNA and have been ge-
netically modified to target a plethora of cell types (Waehler et al., 2007).
Adenovirus
Adenoviruses are non-enveloped viruses comprised of an icosahedral capsid har-
boring a linear double-stranded DNA genome complexed into a nucleosome-like core
(Wold and Horwitz, 2007). Following isolation from adenoid tissue in 1953 (Rowe et al.,
1953), human adenoviruses (HAdVs) have since been attributed to self-limiting respira-
tory infections as well as conjunctivitis and gastroenteritis (Wold and Horwitz, 2007).
Within the Mastadenovirus genus there are seven species of HAdVs (groups A to G) to
which more than 50 serotypes belong to (Table 1). HAdV group C serotypes 2 and 5
12

(Ad2, Ad5, respectively) have by far been the most well studied within the entire Adeno-
viridae family.
The HAdV capsid consists of the three major proteins hexon (polypeptide II),
penton base (polypeptide III), and fiber (polypeptide IV) as well as the polypeptides IIIa,
VI, VIII, and IX (Figure 2). Within the core, the 5’ ends of the ~36 kb linear dsDNA ge-
nome are each covalently linked to a single terminal protein (TP), while additional core
polypeptides V, VII, Mu, and IVa2 are non-covalently complexed to the DNA (Russell,
2009).
Table 1
Human Adenovirus (HAdV) Serotypes
Group Serotypes Receptor Binding
A 12, 18, 31, 52 CAR B 3, 4, 7, 11, 14, 16, 21, 34, 35, 50, 55 CD46 C 1, 2, 5, 6, 57 CAR D 8, 9, 10, 13, 15, 17, 19, 20, 22-30, 32, 33, 36-39, 42-49, 51,
53, 54, 56, 58 CAR
E 4, 16 CAR F 40, 41 CAR G 52 ?
There are 240 hexon homotrimers that cover the majority of the icosahedral cap-
sid. The fiber protein forms a homotrimer that projects from the pentameric penton base
at each of the 12 vertices of the capsid and is integral to Ad infection (Philipson et al.,
1968) (Figures 2 and 3). For group C HAdVs such as Ad5, the carboxy-terminal knob
domain of the fiber protein initially tethers the virus particle to the cell surface by binding
13

to the membrane protein coxsackie and adenovirus receptor (CAR), an immunoglobulin
superfamily member (Bergelson et al., 1997; Von Seggern et al., 1999). The cellular in-
tegrins αvβ3 and αvβ5 then bind to the Arg-Gly-Asp (RGD) motif within the penton base,
promoting internalization of the virus (Wickham et al., 1993) into clathrin-coated vesicles
(Wang et al., 1998). Upon cell entry, step-wise uncoating of Ad virions (Greber et al.,
1993) is mediated by the viral encoded protease (Greber et al., 1996). Partially uncoated
virions escape the endosome in a polypeptide VI-dependent process (Weithoff et al.,
2005), and then associate with microtubules for dynein-dependent transport to the nuclear
membrane (Leopold et al., 2000) where additional uncoating occurs and the viral genome
is imported through the nuclear pore into the nucleus (Strunze et al., 2005; Greber et al.,
1996).
After translocation to the nucleus, viral transcription begins in a highly coordi-
nated and temporal manner. There are five early (E) transcriptional units (E1a, E1b, E2,
E3, and E4) that serve various functions (Figure 4). E1a products serve to regulate viral
transcription and stimulate the cell to enter S phase of the cell cycle (Russell, 2000). The
E1b proteins inhibit apoptosis and block host mRNA transport and stabilize and promote
viral mRNA transport (Russell, 2000). E2 encodes for proteins integral to viral DNA rep-
lication and the E3 region products are involved in evasion of the host immune response
(Russell, 2000). The E4 proteins modulate a number of processes including viral DNA
replication, transcriptional regulation, and apoptosis inhibition (Russell, 2000). Three cel-
lular proteins (NFI/CTF, NFII, NFIII/Oct-1) are required for efficient viral DNA replica-
tion to begin from the replication origins that are located within the ~100 base pair (bp)
ITRs (de Jong et al., 2003). Once viral DNA replication ensues, the IX and IVa2 genes
14

Figure 2. A cartoon section through the HAdV capsid and core. The capsid is namely composed of the 240 homotrimeric hexon (polypeptide II) subunits. The pen-ton complex sits at each of the 12 vertices and is composed of a pentameric penton base (III) and homotrimer fiber (IV). There are four additional minor capsid proteins: IIIa, VI, VIII, and IX. Five other proteins are found in the core covalently or noncova-lently bound to the ~36 kb linear double-stranded DNA genome (V, VII, Mu, IVa2, and terminal protein [TP]). Multiple copies of the viral protease are also found within the core.
are highly expressed which then directly mediates activation of the major late promoter
(MLP) (Lutz and Kedinger, 1996; Lutz et al., 1997). The MLP drives transcription of a
single primary transcript of ~30 kb that produces five families of late (L) mRNAs (L1 to
15

L5) through alternative polyadenylation and splicing events (Shaw and Ziff, 1980) (Fig-
ure 4). Late gene expression produces most of the structural proteins of the virus as well
as proteins integral to virus assembly. Nuclear virion assembly proceeds with formation
of the empty capsid, followed by packaging of the viral DNA, and finally viral protease-
mediated maturation of infectious virus particles (Russell, 2000).
Figure 3. Mechanism for HAdV cellular entry. (1) HAdVs (except for subgroup B) bind to their primary receptor, CAR. (2) Clathrin-coated vesicular internalization is mediated by secondary binding interactions between the integrins αvβ3 and αvβ5 and RGD motif within the penton base. (3) After partial uncoating of the capsid, virions escape the endosome and transport to the nuclear membrane. (4) The viral genome, devoid of most capsid proteins, is then imported to the nucleus through the nuclear pore complex.
16

Figure 4. HAdV genome organization. The HAdV double-stranded DNA genome is approximately 36 kb and flanked by inverted terminal repeats (ITRs). The ITRs con-tain the replication origins and the cis-acting packaging recognition signal (Ψ) is lo-cated near the left end of the genome. Shortly after transport to the nucleus, the E1A region is immediately transcribed, followed by temporal expression of the other early (E) regions (E1B, E2, E3, and E4) which occurs from either DNA strands. Following early region transcription, viral replication begins, followed by IX and IVa2 expres-sion, and finally transcription of the late (L) region transcript of ~30 kb from the major late promoter (MLP). This transcript produces five families of genes (L1 to L5) through alternative splicing to the tripartite leader (TPL) sequence.
Ad Vectors for Pulmonary Airway Gene Therapy
Extensive study of Ad5 has allowed Ad5-based vectors a robust history as thera-
peutic agents (Kay et al., 2001). There are many advantages of adenovirus platforms over
other viral systems, such as the stability of the virus in vivo, its low oncogenic potential,
its large packaging capacity (up to ~37 kb), and the ability to produce very high titers of
virus (Verma and Somia, 1997). Yet, like other systems, there are also a number of phy-
siological and immunological barriers that limit effective Ad gene delivery to the lung
(Pickles, 2004). In that regard, various routes of injection have been compared for en-
17

hanced DNA uptake and delivery to specific lung cell subsets (particularly epithelial
cells), including airway delivery (both direct i.t. instillation and aerosolization), trans-
thoracic injection, and i.v. injection (Albelda et al., 2000).
Figure 5. Adenoviral vectors for gene therapy. (a) First generation vectors are de-void of the E1 region, which is replaced with a promoter and transgene of up to ~5 kb. Some vectors also lack the E3 region, increasing the cloning capacity over 8 kb and allowing for alternative or additional transgene placement in the E3 region (not shown). (b) Second generation vectors are typically devoid of both E1 and E3 and some combination of E2A, E2B, E4 resulting in up to 10 kb of available space. (c) The so called 'last generation', or helper-dependent Ad (HDAd) vectors are devoid of all viral coding sequence allowing up to 37 kb cloning capacity. ITR, inverted termi-nal repeat. Ψ, cis-acting packaging recognition signal.
18

Since Ad5 is a common respiratory pathogen, particularly in children (Garnett,
2009), early excitement over the possibility of delivering CFTR to the epithelial surface
of the pulmonary airway helped drive the development of the first generation of Ad vec-
tors. First generation vectors are devoid of the E1 and sometimes the E3 regions, render-
ing them replication-deficient in vivo (Danthinne and Imperiale, 2000) (Figure 5). Per-
missive replication and propagation of first generation viral vectors occurs in trans-
complementing cell lines, such as HEK-293, which contains the E1 region from Ad5 in
its entirety (the E3 region is dispensable for in vitro propagation) (Graham et al., 1977;
Louis et al., 1997). Although early reports demonstrated transduction of bronchial epithe-
lial cells in vivo (Rosenfeld et al., 1992; Mastrangeli et al., 1993), significant problems or
roadblocks to efficient Ad gene transfer were quickly revealed (Grubb et al., 1994; re-
viewed in Pickles, 2004). These include the mucocilliary clearance system, the glycoca-
lyx barrier (Pickles et al., 2000; Walters et al., 2001) (although its effect is controversial),
absence of native Ad receptors on the luminal (apical) epithelial surface (Zabner et al.,
1997b; Pickles et al., 1998), and the slow rate of airway epithelial cell luminal endocyto-
sis (Zabner et al., 1997b; Pickles et al., 1998 and 2000).
In addition, dose related multi-faceted immunological barriers also limit efficient
Ad gene delivery to the epithelium. After instillation into the airway, the majority of Ad
is rapidly sequestered and eliminated by AMs (Worgall et al., 1997a; Zsengeller et al.,
2000) leading to release of cytokines (tumor necrosis factor [TNF]-α] and IL-6) and
chemokines (macrophage inflammatory protein [MIP]-1α and MIP-2). This results in
subsequent neutrophil-mediated clearance of Ad vectors (Otake et al., 1998; Zsengeller et
al., 2000; Cook et al., 1995; Elkon et al., 1997) and additional cytokine (interferon [IFN]-
19

γ) and chemokine (MIP-1α and monocyte chemotactic protein [MCP]-1) release and ac-
cumulation of monocytes/macrophages and natural killer (NK) cells (Zeng et al., 2005).
Furthermore, despite E1/E3 deletion in first generation Ad vectors, low level expression
of remaining viral genes in addition to transgene expression within infected cells leads to
a delayed adaptive immune response in vivo (Yang et al., 1995a and 1996; Tripathy et al.,
1996; Juillard et al., 1995; Yei et al., 1994). First, cytotoxic T lymphocytes (CTLs) medi-
ate clearance of Ad vector-infected cells leading to the abrupt loss of transgene expres-
sion that has been observed in vivo (Yang et al., 1995a and 1996). Second, a humoral
immune response to Ad capsid proteins produces NAbs that severely limits readministra-
tion of the vector (Juillard et al., 1995; Yei et al., 1994).
Additional deletions, in either the E2 or E4 regions (Wang and Finer, 1996), were
engineered into second generation Ad vectors (Figure 5) to further curtail the immune
response to viral gene expression, yet immune-based clearance of transduced cells is still
a significant barrier to efficient gene expression (Engelhardt et al., 1994; Chirmule et al.,
1998). With the advent of the latest helper-dependent Ad (HDAd) or 'gutless' vectors,
safety and efficacy have been significantly improved (Alba et al., 2005; Brunetti-Pierri
and Ng, 2009). HDAds are devoid of all viral coding sequences (permitting insertion of
up to ~37 kb of transgenic DNA) and thus have a complete lack of endogenous viral gene
expression that has previously led to CTL-mediated clearance of Ad-infected cells (Parks
et al., 1999; Toietta et al., 2003). Despite lingering technical difficulties in propagation of
HDAd, it would appear that previous barriers to long-term gene expression with Ad vec-
tors have largely been overcome with HDAds. However, the issues of inefficient Ad de-
livery to airway epithelial cells, due to the physical barriers, remain. Thus, additional me-
20

thods have been devised to enhance gene transfer via perturbing physiological barriers,
such as the glycocalyx, or epithelial tight junctions (Coyne et al., 2000; Johnson et al.,
2003). However, these methods have the potential to induce additional inflammatory re-
sponses (Johnson et al., 2003) and may breach the protective function of these barriers,
demonstrating the need to explore alternative lung gene transfer strategies.
Intravenous Delivery of Ad Vectors for Pulmonary Gene Therapy
Intravenous injection offers an alternative and promising approach for disseminat-
ing Ad vectors throughout the lung when certain factors are considered. First, i.t. delivery
of Ad does not result in a homogenous gene transfer pattern and transduction of alveolar
cells has been particularly difficult. Second, many of these physical and immunological
barriers to vector delivery are enhanced in the context of the diseased lung (e.g. CF and
ARDS), further limiting Ad vector delivery via the airway (McElvaney et al., 1991;
Otake et al., 1998; Sueblinvong et al., 2007). Vector aerosolization can improve alveolar
delivery, however ventilation is often decreased in patients with significant respiratory
disease (McElvaney et al., 1991). In contrast, i.v. vector administration allows for deliv-
ery to the entire pulmonary vascular network which would enable local expression of
therapeutics for diffusion into the alveolar spaces or to act directly on effector cells with-
in the vasculature before they extravasate into the airway.
Barriers to Intravenous Delivery of Ad
Upon i.v. injection, therapeutics first pass through the pulmonary capillary bed.
This allows relatively high amounts of nonviral DNA complexes to be captured (Zhu et
21

al., 1993; Liu et al., 1997), but does not result in substantial accumulation of Ad5 within
the lung. Rather, almost 99% of Ad5 particles are efficiently sequestered within the liver
and spleen (Smith et al., 1993; Huard et al., 1995; Peeters et al., 1996) which not only
limits vector access to target tissues but also leads to acute inflammation and consider-
able hepatotoxicity (Descamps and Benihoud, 2009). Thus, despite its potential, intravas-
cular Ad5-mediated gene delivery to the lung appears to be limited by distinct, yet equal-
ly challenging, barriers to efficacy as airway delivery (Baker et al., 2007; Parker et al.,
2008).
The reticuloendothelial system (RES) quickly eliminates a significant percentage
of i.v. administered Ad vectors from the circulation. First, significant quantities of virus
become trapped within the fenestrated liver sinusoids (Di Paolo et al., 2009) where they
quickly encounter liver macrophages, or KCs. As part of the RES, KCs rapidly eliminate
the bulk (>90%) of i.v. administered Ad5 virions (Lieber et al., 1997; Wolff et al., 1997;
Worgall et al., 1997b; Alemany et al., 2000; Tao et al., 2001). Marginal zone macro-
phages of the spleen also remove circulating virions, albeit on a much smaller scale,
compared to KCs (Zhang et al., 2001). KC uptake of Ad5 occurs, at least in part, through
scavenger receptor interactions with unknown epitopes of the Ad5 capsid (Haisma et al.,
2008 and 2009; Xu et al., 2008). Natural IgM antibodies (from naive mouse serum) and
the complement proteins C3 and C4 opsonize Ad5 in vitro and there is partially reduced
Ad5 uptake by KCs in C3-deficient mice, suggesting that both the classical and an anti-
body-independent complement pathways help mediate clearance of Ad5 by KCs (Kiang
et al., 2006; Xu et al., 2008). Other soluble blood factors, such as C4-binding protein
[C4BP], factor IX, and platelets, have also been implicated in directing KC uptake of Ad
22

through interaction with the fiber knob domain (Shayakhmetov et al., 2005; Stone et al.,
2007), although the biological importance of these effects are questionable and difficult
to interpret in light of more recent reports (Xu et al., 2008; Di Paolo et al., 2009). Never-
theless, virus sequestration by KCs ultimately leads to their death as they rapidly undergo
necrosis (Manickan et al., 2006; Smith et al., 2008).
In addition to KC elimination of Ad5, much of the remaining virus is redirected to
hepatocytes leading to efficient gene transfer in the liver (Smith et al., 1993; Huard et al.,
1995; Peeters et al., 1996). Expression of CAR in the liver (Tomko et al., 1997) and liver
localization of purified recombinant fiber knob after i.v. administration (Zinn et al., 1998)
led to the early hypothesis that Ad5 liver tropsim was attributed to the two-step mecha-
nism of infection for Ad5, in which the Ad fiber knob binds CAR followed by penton
base RGD motif-binding to integrins to promote internalization (Figure 3). However,
failure to demonstrate the relevance of this mechanism in vivo flooded the literature
(Alemany and Curiel, 2001; Mizuguchi et al., 2002; Smith et al., 2003; Nicol et al., 2004)
until it was finally reported that interactions of the Ad5 hexon protein with soluble blood
factors, particularly factor X (FX), actually mediated hepatocyte transduction (Shayak-
hmetov et al., 2005; Parker et al., 2006; Waddington et al., 2008; Kalyuzhniy et al.,
2008). Nevertheless, Ad5 liver tropism has been exploited to deliver a number of thera-
peutics for treatment of various genetic diseases (Smith et al., 1993; Kay et al., 1995;
Bristol et al., 2001) including and culminating with the gene therapy trial that resulted in
the death of Jesse Gelsinger in 1999 (Raper et al., 2003).
Indeed, the death of Jesse Geslinger (which occurred 96 h following i.v. injection
of an Ad5 vector) brought massive attention and focus to the important immunological
23

barriers limiting safe and efficacious intravascular Ad5 delivery. Similar to the innate re-
sponse to Ad5 following airway delivery, i.v. administration of Ad5 induces substantial
release of cytokines and chemokines (e.g. TNF-α, IL-1β, IL-8, IL-6, IL-12, IFN-inducible
protein [IP]-10, MIP-1β, MIP-2, RANTES, MCP-1, IFN-γ) that begins within minutes
after injection and prior to any viral gene expression (Lieber et al., 1997; Muruve et al.,
1999; Zhang et al., 2001; Zaiss et al., 2002; and reviewed in Descamps and Benihoud,
2009). This innate response is mediated primarily through KC and DC uptake of Ad
(Zhang et al., 2001) but can also occur through Ad5 interaction with and activation of
endothelial cells (ECs) directly (Li et al., 2002; Liu et al., 2003). Chemokine release and
local EC activation then help drive the neutrophil inflammation and measurable hepato-
cyte toxicity that follows (Muruve et al., 1999; Li et al., 2002). KC uptake and release of
various effector molecules also leads to systemic EC activation which promotes acute
hemodynamic changes (hypotension, bradycardia, hypothermia) (Schiedner et al., 2003;
Machemer et al., 2005). Subsequent influx of innate cell types, such as mono-
cytes/macrophages and NK cells (Benihoud et al., 2007), perpetuates cytokine production
and stimulates the adaptive immune response which ultimately results in elimination of
transgene-expressing cells (hepatocytes) (Yang et al., 1994a,b and 1995b; Yang and Wil-
son, 1995) and humoral anti-Ad immunity (Juillard et al., 1995; Gahéry-Ségard et al.,
1997; Benihoud et al., 2000).
Hence, vascular delivery of gene therapeutics that specifically and efficiently tar-
get tissues or cells of interest has not yet been fully realized and in order to maximize the
therapeutic potential of Ad for other clinically-relevant tissues, significant rerouting of
Ad5 from the liver will be necessary (Di Paolo and Shayakhmetov, 2009). Fortunately,
24

the plasticity of Ad capsid proteins permits significant structural changes aimed at redi-
recting Ad tropism. Thus, many strategies have been evaluated for reduced virus tropism
to the liver as well as enhanced targeting of the pulmonary vasculature (Reynolds, 2011;
Di Paolo and Shayakhmetov, 2009).
Strategies to Reduce Liver Targeting
Early attempts to mitigate Ad5 hepatocyte transduction (‘liver untargeting’) were
based on the hypothesis that Ad5 liver tropsim was a function of Ad fiber knob binding to
CAR on the hepatocytes (Tomko et al., 1997; Zinn et al., 1998). After elucidation of the
critical CAR-binding residues within the various loop motifs within the knob domain
(Roelvink et al., 1999; Jacubczak et al., 2001), Ad vectors harboring these mutations
were generated and, although they all demonstrated ablation of CAR-binding in vitro,
they did not result in reduced liver tropism after i.v. infusion into mice (Alemany and Cu-
riel, 2001; Leissner et al., 2001; Mizuguchi et al., 2002; Smith et al., 2002). In addition,
Ad vectors containing complete removal of the knob domain produced similar results in
vivo (Zinn et al., 2004). Deletion of RGD motifs within the penton base, important for
integrin-mediated endocytosis of Ad vectors, either alone or in conjunction with CAR-
binding ablated mutations also shows no reduction in liver tropism after i.v. infusion of
Ad vectors (Mizuguchi et al., 2002; Koizumi et al., 2003; Smith et al., 2003).
Incorporation of a mutated putative heparin sulfate proteoglycan (HSPG)-binding
motif (Lys-Lys-Thr-Lys or KKTK) within the fiber shaft of Ad5 appeared to drastically
reduce hepatocyte transduction upon i.v. injection (Smith et al., 2003). However, it was
later demonstrated this effect was actually related to the inability of the virus to properly
25

traffic to the nucleus following cellular internalization, rather than its ability to avoid he-
patocyte infection (Kritz et al., 2007). Furthermore, switching of the Ad5 fiber shaft with
those of alternative CAR-binding HAdV serotypes that naturally lack the KKTK motif
(e.g. Ad31 or Ad41) demonstrated efficient liver targeting, indicating mutation of the fi-
ber shaft is not likely to result in mitigation of liver tropism (Di Paolo et al., 2007). In this
regard, switching of the Ad5 fiber knob with those from group B HAdVs (which bind
CD46 instead of CAR) also does not result in significant liver untargeting (Shayakhme-
tov et al., 2004), although replacement of the Ad5 fiber (which contains 2 β-repeat motifs
in its shaft) with shorter fibers (5-7 β-repeats) from alternative HAdV serotypes (e.g.
Ad9, Ad35, Ad40) has been shown to reduce the level of liver transduction (Nakamura et
al., 2003; Koizumi et al., 2003; Shayakhmetov et al., 2004). However, short fiber Ads
still accumulate within KCs and the liver sinusoids shortly after injection, reducing their
ability to target extra-hepatic tissues (Shayakhmetov et al., 2004).
Since the discovery that Ad5 hepatocyte transduction is actually mediated by in-
teractions between hexon and circulating FX, a number of studies have evaluated the ef-
fects mutation of the FX-binding residues within the Ad5 hexon or replacement with
hexons from other HAdV serotypes (e.g. Ad48, Ad26, Ad3) have on liver tropism. These
hexon modification strategies have indeed demonstrated a clear dramatic reduction in he-
patocyte transduction (Waddington et al., 2008; Kalyuzhniy et al., 2008; Alba et al., 2009
and 2010; Short et al., 2010). However, as with Ad short fiber pseudotyping, ablation of
FX-binding does not mitigate KC uptake (Alba et al., 2010). Thus, successful implemen-
tation of strategies to mitigate hepatocyte transduction are important to achieve high lev-
els of target tissue vector delivery, but clearly there are additional mechanisms of Ad
26

liver tropism (i.e. KC uptake and sinusoid trapping) that need to be addressed (Di Paolo
et al., 2009).
While many hepatocyte untargeting strategies have been evaluated, it is important
to remember that over 90% of the administered Ad5 dose is actually sequestered by liver
KCs. Ad-KC interactions also drives a number of undesired processes, such as an innate
immune responses, hepatotoxicity, and altered hemodynamics. Although a variety of KC
untargeting strategies have been evaluated, some rely upon simply removing or depleting
the macrophages prior to Ad delivery via infusion of clodronate liposomes or gadolinium
chloride (GdCl3); both of which are toxic to highly phagocytic cells (Wolff et al., 1997;
Lieber et al., 1997). Other groups have been developing means to shield the virus from in
vivo barriers by chemically attaching various polymers (typically either poly-N-[2-
hydroxypropyl]methacrylamide [poly-HPMA] and polyethylene glycol [PEG]) to the
capsid (Kreppel and Kochanek, 2008). In particular, random incorporation of different
molecular weight (MW) PEG moieties onto the Ad capsid has demonstrated efficient he-
patocyte and KC detargeting (Wortmann et al., 2008), although retargeting these PEGy-
lated vectors has been difficult (Kreppel and Kochanek, 2008). Recently however, di-
rected incorporation of small MW PEG moieties into specific hexon locales of Ad5 al-
lowed for reduced KC uptake and FX-independent transduction of hepatocytes (so called
'hepatocyte retargeting') (Prill et al., 2011). Nevertheless, liver untargeting strategies that
utlize genetic modifications of the capsid instead of chemical modification are still more
favorable at this time due to less complicated and more easily reproducible virus produc-
tion.
27

Genetic capsid modification strategies to mitigate KC sequestration have also
been limited. Although the mechanisms of KC uptake of Ad are still relatively unknown,
the potential importance that scavenger receptor (SR)-A plays in Ad uptake by KCs has
recently been revealed (Xu et al., 2008; Haisma et al., 2009). Since SR-A recognizes
charged structures (Haisma et al., 2008), it was recently speculated that use of less nega-
tively charged (evaluated by adding the number of charged residues within hexon) HAdV
serotypes, such as Ad6, may provide a natural means of KC avoidance (Shashkova et al.,
2009; Weaver et al., 2011). Shortly thereafter, the same group reported that genetic re-
placement of the highly charged hypervariable regions (HVRs) of Ad5 hexon with the
same (less charged) region from Ad6 hexon resulted in significant reduction in KC up-
take of virus (Khare et al., 2011). Yet compared to Ad5, they still observed similar levels
of virus sequestration in the liver (presumably the liver sinusoids) shortly after i.v. injec-
tion which resulted in potent hepatic gene transfer. Thus, it is clear that no single genetic
capsid modification strategy to date has demonstrated the ability to eliminate both hepa-
tocyte and KC tropism which hampers further strategies to retarget Ad vectors to alterna-
tive tissues, such as the lung vasculature.
Fiber Modification Strategies to Enhance Targeting of Pulmonary Endothelium
Although liver untargeting is a major hurdle to any intravascular Ad delivery
strategy, specifically and efficiently targeting of Ad vectors to the pulmonary system is
an equally challenging obstacle. Historically, Ad-mediated gene therapy has been limited
to cell types that express the native receptor, CAR. In vivo, CAR is expressed broadly,
but this expression is mostly localized to inaccessible regions of the cell, such as the ba-
28

solateral surface of epithelial cells (Fechner et al., 1999). Transgenic mice that ubiqui-
tously overexpress a truncated (signaling portion of the cytoplasmic tail was removed)
version of human CAR (hCAR) in the pulmonary vasculature are more efficiently tar-
geted following i.v. infusion of Ad5, demonstrating that pulmonary gene transfer is at-
tainable if the proper molecules are targeted (Tallone et al., 2001; Everts et al., 2005;
Izumi et al., 2005).
In this regard, the plasticity of the Ad capsid for genetic manipulation has allowed
many of the tropism-based restrictions to Ad utility to be overcome. Similar to liver un-
targeting strategies, retargeting approaches are extremely diverse and include genetic
manipulation of many different capsid locales such as hexon, pIX, and fiber (Krasnykh et
al., 2000; Noureddini and Curiel, 2005). However, years of research has revealed what
nature has already optimized. The Ad fiber protein, which normally designates receptor
specificity, is the most optimal retargeting locale (Campos and Barry, 2006). Thus, mul-
tiple fiber targeting approaches, including domain mutation or deletion, pseudotyping of
the shaft and/or knob, and direct ligand insertion, have been evaluated (Krasnykh et al.,
2000; Noureddini and Curiel, 2005; Waehler et al., 2007). To date, a number of these fi-
ber-modified Ad vectors have been created to specifically target a broad range of clini-
cally relevant cell types (reviewed in: Mathis et al., 2006; Waehler et al., 2007), including
pulmonary endothelium (Reynolds, 2011).
In cases where genetic incorporation of targeting ligands into the fiber is not fea-
sible (typically when antibodies are used), two-component adapter molecules have been
used to retarget Ad to alternative receptors (Waehler et al., 2007). In that regard, there are
a number of examples of enhanced endothelial transduction using molecular adapters,
29

although these studies were not limited specifically to lung vasculature and were only
demonstrated in vitro (Wickham et al., 1996; Harari et al., 1999; Trepel et al., 2000; Net-
telbeck et al., 2001). The most efficient in vivo vascular targeting strategy to date has ac-
tually utilized a two-component bispecific antibody system. These bispecific antibodies
bound to the viral capsid, masking Ad5 tropism to its natural receptor CAR, and redi-
rected viral tropism to angiotensin-converting enzyme (ACE) expressed on pulmonary
endothelium (Reynolds et al., 2000). The specificity of gene expression was further im-
proved by incorporating the fms-related tyrosine kinase (FLT)-1 promoter in place of the
ubiquitous cytomegalovirus (CMV) intermediate-early promoter upstream of the viral
reporter gene (Reynolds et al., 2001). However, retargeting was inefficient as large
amount of virions were still observed in the liver with these strategies (Reynolds et al.,
2000 and 2001) indicating that the efficiency in targeting could still be improved upon.
The Marginated Leukocyte Pool
Since venous blood flow is first directed through the pulmonary vasculature, effi-
cient targeting of cell types within the lung may preclude the need for further viral modi-
fications that ablate liver sequestration mechanisms. Consequently, leukocytes may rep-
resent an ideal target for delivery of therapeutics to the pulmonary vasculature. The lung
microvasculature represents a bottleneck that reduces leukocyte transit rate and results in
their net accumulation within this tissue (Downey et al., 1990; Wiggs et al., 1994; Kue-
bler and Goetz, 2002). This often overlooked population of leukocytes is classically re-
ferred to as the marginated pool, and exceeds the circulating pool by an estimated 1.5- to
3-fold (Kuebler and Goetz, 2002). Although neutrophils comprise the majority of this
30

population, considerable numbers of circulating monocytes and lymphocytes also con-
tribute to the pool (Doerschuk et al., 1987 and 1990). Furthermore, leukocytes directly
contribute to the pathogenesis of many debilitating lung diseases making them interesting
and important targets for therapeutic intervention of inflammatory lung disease (Salle-
nave et al., 1997).
Experimental Aims
Given the size and location of this marginated leukocyte pool, and since myeloid
leukocytes naturally home to tissues in response to inflammatory signals, we hypothe-
sized that we could modulate Ad tropism to myeloid cell subsets in the lung for subse-
quent therapeutic intervention of inflammatory lung disorders. In order to study this hy-
pothesis, we first developed a non-replicating Ad vector with a novel myeloid-targeting
peptide genetically incorporated in place of the native CAR-binding domain, knob. Sec-
ondly, we characterized myeloid-targeted Ad (termed Ad.MBP) in vitro to verify target
cell specificity was maintained. Lastly, we investigated the effects our novel targeting
approach would have on the biodistribution of both acute Ad.MBP vector sequestration
and subsequent gene transfer after i.v. administration in vivo. That is, we evaluated
whether Ad.MBP could enhance targeting to pulmonary cell types while also mitigating
liver tropism normally observed with untargeted Ad5.
31

METHODS
Cell Lines
All cells described here were propagated at 37 °C in a humidified atmosphere of
5% CO2. HEK-293 and 293T (Microbix, Toronto, Canada), QBI-293A (Qbiogene, Mont-
real, Canada), and 293-F28 cell lines were all propagated in Dulbecco's modified Eagle's
medium (DMEM)-F12 medium supplemented with 10% (vol/vol) fetal calf serum (FCS),
L-glutamine (2 mM), penicillin (100 I.U. mL–1) and streptomycin (100 μg mL–1). The
293-F28 cell line, a derivative of HEK-293, expresses the Ad5 wild-type fiber for mosaic
virus propagation as described previously (Belousova et al., 2003). 293-F28 cells were
periodically maintained with 600 µg mL–1 Zeocin (Invitrogen, Carlsbad, CA).
The mouse cardiac endothelial cell (MCEC) line (CELLutions Biosystems Inc.,
Burlington, Canada) was propagated in DMEM supplemented with 5% (vol/vol) FCS, L-
glutamine (2 mM), penicillin (100 I.U. mL–1) and streptomycin (100 μg mL–1). The
mouse inducible pulmonary microvascular endothelial cell (MiPMVEC) line was a gen-
erous gift from Dr. Namasivayam Ambalavanan (University of Alabama at Birmingham).
Isolation of these cells is described previously (Frank et al., 2005). Briefly, the cells were
derived from the H-2Kb-tsA58 transgenic mouse (Whitehead et al., 1993; also referred to
as the “Immortomouse”). This strain expresses a temperature-sensitive mutant of the
SV40 large T antigen, so that culturing MiPMVECs at 33 °C allows them to remain im-
mortalized. However, at 37 °C the cells are no longer immortalized and can be used for
experimentation. Prior to experiments, we characterized these cells using a number of
32
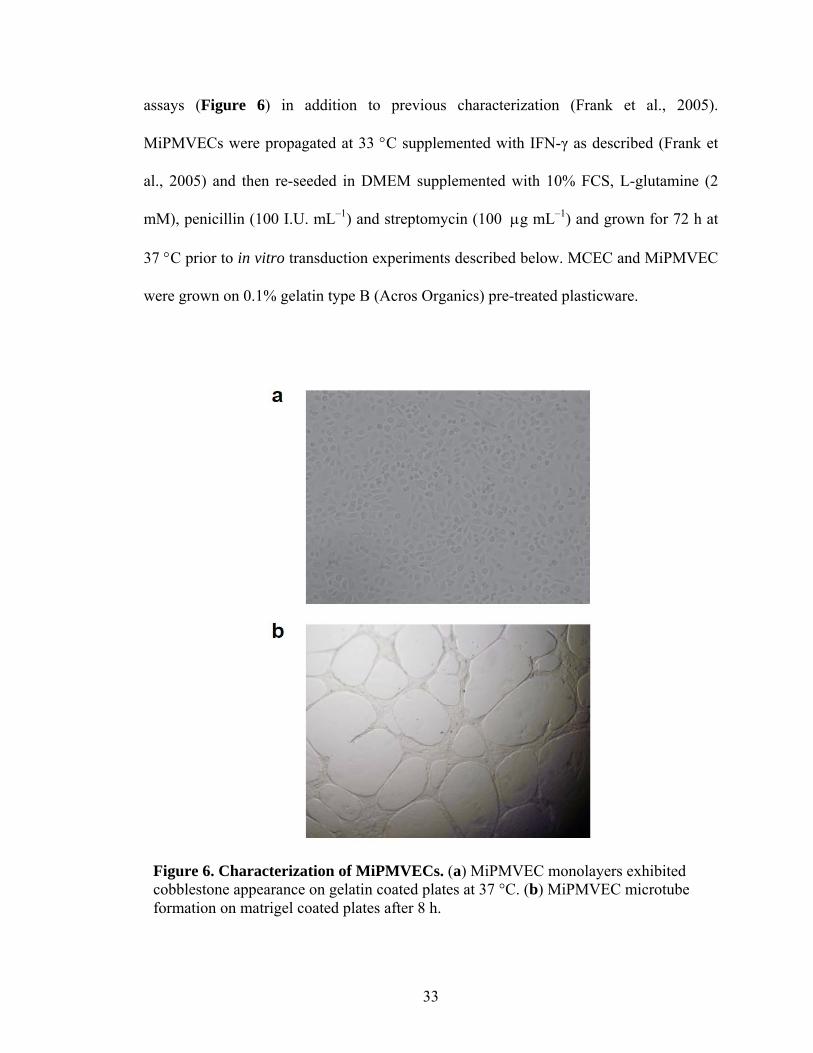
assays (Figure 6) in addition to previous characterization (Frank et al., 2005).
MiPMVECs were propagated at 33 °C supplemented with IFN-γ as described (Frank et
al., 2005) and then re-seeded in DMEM supplemented with 10% FCS, L-glutamine (2
mM), penicillin (100 I.U. mL–1) and streptomycin (100 μg mL–1) and grown for 72 h at
37 °C prior to in vitro transduction experiments described below. MCEC and MiPMVEC
were grown on 0.1% gelatin type B (Acros Organics) pre-treated plasticware.
Figure 6. Characterization of MiPMVECs. (a) MiPMVEC monolayers exhibited cobblestone appearance on gelatin coated plates at 37 °C. (b) MiPMVEC microtube formation on matrigel coated plates after 8 h.
33

Animals
C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) between the ages of 6 to
10 weeks old were used in this study. Transgenic hCAR mice were a gift from Dr. Sven
Pettersson (Karolinska Institute, Stockholm, Sweden). These mice express a truncated
form of hCAR under control of the human ubiquitin-C promoter, allowing hCAR expres-
sion in a variety of tissues (Tallone et al., 2001). β2 microglobulin knockout (β2mnull) and
MHC class II transactivator knockout (CIITAnull) mice were kindly provided by Dr. Ran-
dall S. Davis (University of Alabama at Birmingham). All methods were approved by the
Institutional Animal Care and Use Committee (IACUC) at the University of Alabama at
Birmingham (see Appendices A and B) and performed according to their guidelines.
Phage Display
Phage display was previously performed at Case Western Reserve University
(Roth, 2006). For in vivo phage panning, 2 × 1011 plaque-forming units (pfu) of the
Ph.D.™-C7C M13 Phage Display Library (New England Biolabs, Ipswich, MA) was di-
luted in 200 μL alpha minimum essential medium (α-MEM) and infused into the tail vein
(t.v.) of one mouse. After 10 min, bone marrow cells (BMCs) were harvested from fe-
murs and tibias of mice by flushing marrow cavities with α-MEM supplemented with 2%
FCS (α-MEM/2%). Cells were filtered through 40 µm nylon strainers and centrifuged at
250g. Pellets were resuspended in erythrocyte lysis buffer (155 mM ammonium chloride,
20 mM sodium bicarbonate and 1 mM EDTA, pH 8), centrifuged at 250g and finally re-
suspended in phosphate buffered saline (PBS) supplemented with 2% FCS (FACS buf-
fer). Cells of interest were isolated by fluorescent-activated cell sorting (FACS). Cell-
34

bound phage were eluted with 0.2 M Glycine-HCl (pH 2.2), neutralized with 1 M Tris-
HCl (pH 9.1), then amplified and sequenced according to the library manufacturer’s in-
structions. This process was repeated two additional times with the amplified output
phage at each round used as input for the subsequent panning experiment. For the third
round of panning, two animals were each infused with 2 × 1011 pfu of the output phage
from the second round of panning. However, one mouse was sacrificed after 10 min and
the other mouse was sacrificed after 30 min. For in vitro phage panning, 2 × 1011 pfu of
the same starting library was added to 2.5 × 107 BMCs isolated from naïve mice (as
above) and incubated at 4 °C for 6 h. Excess phage were removed by centrifuging the
cells at 250g and cells of interest were then isolated by FACS. Cell-bound phage were
eluted, amplified and sequenced as described above for a total of three rounds of panning.
Myeloid-binding Peptide (MBP) Synthesis
After identification of a putative myeloid-binding phage clone by in vitro and in
vivo panning, the heptapeptide sequence was used to generate a 14 amino acid (aa) mye-
loid-binding peptide (MBP) with the sequence: CWTLDRGYCSAEKA. The peptide was
engineered to resemble the configuration of the phage-displayed peptide by containing a
putative disulfide bridge (between the bolded residues), three C-terminal flanking resi-
dues based on the M13 phage pIII protein (underlined residues), and a biotinylated lysine
(K) residue at the thirteenth position for detection by flow cytometry. The peptide was
synthesized at the Cleveland Clinic Lerner Research Institute Molecular Biotechnology
Core (Cleveland, OH) on an Omega 396 multiple peptide synthesizer (Advanced Chem-
Tech, Louisville, KY) and dissolved in PBS at 5 mg mL–1.
35

M13-MBP Mutant Phage Vector Construction
M13-MBP bacteriophage mutants were synthesized by substituting an Ala for
each of the seven residues of the MBP sequence (WTLDRGY) and for the flanking cys-
teine (C) residues. An inverse MBP (iMBP) control mutant with the MBP sequence in
reverse (YGRDLTW) was also constructed. Ten different 56 basepair (bp) Acc65I-EagI
oligo linkers (for a list of all oligos, see Table 2 at end of chapter) comprising the MBP,
iMBP, or mutant MBP sequence were cloned into the Acc65I-EagI-linearized M13KE
cloning vector (New England Biolabs) for transformation of E. coli ER2738 without
negative selection according to the New England Biolabs protocol. Phage DNA was iso-
lated according to the New England Biolabs protocol and positive clones were identified
by PCR and verified by sequencing (data not shown).
Ad Vector Construction
Fiber Shuttle and Expression Vectors
The Ad5 fiber shuttle vectors (used for genetic replacement of wild-type Ad5 fi-
ber), pKan-566FF-MBP and pKan-566FF-iMBP, were generated by cloning a 54 bp
BamHI-BclI oligo linker comprising the MBP (or iMBP) sequence into the BamHI-
linearized vector pKan-566FF-BaeI (Noureddini et al., 2006; Hedley et al., 2006).
The mammalian expression vectors, pVS-566FF-MBP and pVS-566FF-iMBP,
containing the modified fiber genes, were then created by replacing the 1.5 kb AgeI frag-
ment of the expression vector pVS-566FF-ΔCd with similar sized AgeI fragments from
the shuttle vectors described above. Plasmid pVS-566FF-ΔCd was constructed by delet-
ing a 0.2 kb BamHI-SwaI fragment, containing the Staph aureus Cd domain, from pVS-
36

566FF-Cd (Noureddini et al., 2006; Hedley et al., 2006) and replacing it with a 58 bp oli-
go linker containing an AgeI site. The expression vector pVSII containing the wild-type
Ad5 fiber gene is described elsewhere (Korokhov et al., 2003).
Adenovirus Genome Vectors
Recombinant, non-replicating, first generation (E1-deleted) Ad genomes contain-
ing the modified MBP or iMBP fiber genes (for a list of constructed recombinants, see
Table 3 at end of chapter) were then derived by homologous DNA recombination in E.
coli BJ5183 between the EcoRI-digested shuttle vectors and SwaI-linearized, pVK700
and pVK900, as previously described (Chartier et al., 1996) The plasmids pVK700 (Be-
lousova et al., 2002) and pVK900 (Murakami et al., 2010) are derivatives of pTG3602
(Chartier et al., 1996) which contain full E1 and fiber gene deletions. In place of the E1
region, pVK700 and pVK900 express the firefly luciferase (Luc) and enhanced green
fluorescent protein (GFP) reporter genes, respectively, from the CMV immediate-early
promoter. Genome size was characterized by restriction digest and gel electrophoresis
and the genomes were sequenced to verify both the fiber and transgene regions.
Schematics and vector maps for all Ad vector cloning and can be found in greater
detail in Appendix C.
Adenoviruses
Ad.MBP viruses were first generated by transfection of 293-F28 cells, which
stably express wild-type Ad5 fiber, with PacI-digested pAd.MBP genomes (from above)
using Lipofectamine™ 2000 (Invitrogen) as described previously (Belousova et al.,
37

2003). Rescued virions at this point are referred to as "mosaic" because they contain both
the wild-type Ad5 fibers and 566FF-MBP fibers. To generate virions with only 566FF-
MBP fibers, we transduced QBI-293A cells with mosaic Ad.5/MBP viruses following
further rounds of amplication in 293-F28 cells. Cells were harvested after CPE was ob-
served and virus was purified by two rounds of cesium chloride density ultracentrifuga-
tion and dialyzed in storage buffer (10% glycerol, 1 mM MgCl2, and 10 mM HEPES, pH
7.8). Isogenic control E1-deleted Ad5.Luc and Ad5.GFP vectors (Krasnykh et al., 2001)
(each expressing transgene from the CMV intermediate-early promoter) were generated
by infection of QBI-293A cells with purified virions. Cells were harvested after CPE was
observed and virus was purified as above. Virus particle (VP) concentration was deter-
mined at 260 nm by using a conversion factor of 1.1 × 1012 VP mL–1 per absorbance unit
as previously described (Maizel et al., 1968) (see Table 4 at end of chapter).
Recombinant Fiber Expression System
293T cells were seeded on 10-cm dishes prior to transfection with each of the ex-
pression plasmids pVS-566FF-MBP, pVS-566FF-iMBP and pVSII (8.5 μg DNA per
dish) using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's protocol.
Cells (including mock transfected control) were harvested after 48 h, washed and then
frozen in PBS (1 mL per 10-cm dish of cells). Protein supernatants (clarified of cell de-
bris) were collected after three freeze/thaw cycles and protein concentrations were deter-
mined by DC Protein Assay (Bio-Rad, Hercules, CA).
38

Western Blotting
Purified Ad5 and Ad.MBP virions (1 × 109 VP) or pVS-transfected 293T protein
supernatants (10 μg total protein) were diluted in Laemmli sample buffer and incubated
at either 95 °C (boiled) or room temperature (unboiled) for 10 min and then separated by
SDS-PAGE (10–12% resolving gels). Proteins were subsequently electroblotted onto
PVDF membranes for western blot using the mouse 4D2 monoclonal antibody to the Ad5
fiber tail domain (Lab Vision, Fremont, CA) followed by horseradish peroxidase-
conjugated rabbit polyclonal secondary antibody to mouse IgG (Dako, Carpinteria, CA)
as previously described (Murakami et al., 2010). Blots were developed using the Amer-
sham™ ECL™ Plus Western Blotting Detection Kit (GE Healthcare, Piscataway, NJ).
Precision Plus Protein™ Kaleidoscope™ Standards (Bio-Rad) were used to estimate mo-
lecular weights of bands.
Phage Preparation
Large scale M13 phage was prepared essentially according to the New England
Biolabs protocol. Briefly, amplified phage were recovered by PEG precipitation of su-
pernatants from 25 mL cultures and resuspended in 1 mL Tris-buffered saline (TBS) with
50% glycerol. Titers were determined by dilution plaque assays on E. coli ER2738. In
order to detect phage binding to BMCs, the original (15 clones identified from phage
panning) M13 phage preps were biotinylated with EZ-Link® Sulfo-NHS-LC-Biotin
(Pierce, Rockford, IL) according to the manufacturer’s protocol. The M13-MBP mutant
phage preps used in subsequent binding experiments were not biotinylated.
39

In vitro Binding Experiments
Prior to binding studies, BMCs (isolated as above), peripheral blood (isolated by
cardiac puncture into heparinized tubes), and spleen cells (filtered through 40 μm strain-
ers) were all centrifuged at 250g. Lungs were removed, chopped into small pieces, resus-
pended in 1 mL Hank’s Balanced Salt Solution (HBSS) supplemented with 0.14 Wünsch
units Liberase™ (Roche, Indianapolis, IN) and 100 units DNase I (Roche) and incubated
for 25 min at 37 °C while shaking (Snelgrove et al., 2010). Lung homogenates were then
passed through 40 μm strainers and centriguged at 250g. Cell pellets from all sources
were resuspended in erythrocyte lysis buffer, centrifuged at 250g, then washed and resus-
pended FACS buffer, for subsequent experimentation. All binding experiments were car-
ried out on ice or at 4 °C.
For phage and peptide binding, 5 × 109 pfu purified phage or 1–100 μg of bioti-
nylated peptide were added to 1 × 106 BMCs. Cell-bound biotinylated phage and peptide
were detected with APC-conjugated streptavidin (BD Pharmingen, San Diego, CA) by
flow cytometry. Unbiotinylated phage-binding was detected with mouse monoclonal an-
tibody to the M13 major coat protein pVIII (GE Healthcare) followed by PE-conjugated
goat polyclonal secondary antibody to mouse IgG (SouthernBiotech, Birmingham, AL).
For fiber protein binding experiments, 575 μg (~100 μL) of 293T protein super-
natant was used to stain 1 × 106 BMCs and cell-bound fiber was detected with mouse
4D2 monoclonal antibody to the Ad5 fiber tail domain (Lab Vision) followed by Alexa
Fluor® 488-conjugated goat polyclonal secondary antibody to mouse IgG (Invitrogen
Molecular Probes, Eugene, OR).
40

For characterization of Ad-binding of BMCs, peripheral blood, spleen and lung,
Ad.MBP or Ad5 was incubated with 1 × 106 cells at 500 VP per cell. Cellular-bound Ad
was detected with rabbit polyclonal antiserum to Ad5 (as previously described; Douglas
et al., 1999) followed by either Alexa Fluor® 488-conjugated goat polyclonal or Alexa
Fluor® 647-conjugated chicken polyclonal secondary antibody to rabbit IgG (Invitrogen
Molecular Probes).
For all binding experiments, antibodies to various cell surface markers were in-
cluded with secondary antibody staining to help distinguish various cell populations by
flow cytometry as described below.
Transmission Electron Microscopy (TEM)
BMCs were isolated as above and then neutrophils were purified by negative
magnetic bead-based antibody selection to remove T and B cells, erythrocytes, mono-
cytes and macrophages as described previously (Tsuruta et al., 2007). Ad.MBP binding
was carried out, as described in the in vitro binding studies. Virus-bound cells were fixed
in a modified Karnovsky's fixative (2% Paraformaldehye and 2% Glutaraldehyde in 0.1
M phosphate buffer). After fixation the specimens were rinsed several times with PBS
followed by post-fixation with 1% osmium tetroxide in phosphate buffer for 1 hr. After
rinsing, the tissue specimens were dehydrated through a series of graded ethyl alcohols
from 70 to 100%. After dehydration, the samples were embedded in EMbed 812 resin
(Electron Microscopy Sciences, Hatfield, PA). Images were collected using a FEI Tecnai
F20 FEG transmission electron microscope (FEI, Hillsboro, OR) at 80 KV in the UAB
High Resolution Imaging Facility.
41

Dihydrorhodamine 123 (DHR123) Assay
Peripheral blood was isolated as described above except peripheral blood leuko-
cytes were washed and resuspended in HBSS supplemented with 0.1% bovine serum al-
bumin (BSA) and 1 mM EDTA (Wash Buffer) after red blood cell lysis. The assay was
performed essentially as described previously (Vowells et al., 1995). Briefly, for each
sample, 2 × 105 cells were incubated with 1,000 U mL–1 catalase and 100 mM DHR123
(Invitrogen Molecular Probes) for 5 min at 37 °C followed by an additional 15 min at 37
°C after addition of Wash Buffer, phorbol myristate acetate (PMA; 400 ng mL–1), or
Ad.MBP at various multiplicity of infection (MOI). Conversion of DHR123 to the green
fluorescent analog Rhodamine 123 (R123) was measured by flow cytometry immediately
after incubation.
In vitro Transduction Experiments
For BMC infection, BMCs were harvested and erythrocytes lysed as above.
Ad.MBP.GFP or Ad5.GFP were incubated with 1 × 106 BMCs at various MOI in α-
MEM/2% for 20 min at 4 °C. Cells were washed twice to remove unbound virus and then
plated in 1 mL of α-MEM supplemented with 20% FCS, SCF (50 ng mL–1), IL-3 (20 ng
mL–1), and IL-6 (50 ng mL–1) (all cytokines from R&D Systems, Minneapolis, MN). Af-
ter incubation for 24 h at 37 °C, cells were harvested and stained for flow cytometry.
For MCEC or MiPMVEC infection, 3–4 × 105 trypsinized cells (grown at 37 °C)
were seeded on fresh plates (pre-treated with 0.1% gelatin type B) 24 h prior to addition
of Ad.MBP.GFP or Ad5.GFP at various MOI. After 24 h at 37 °C, cells were harvested
for flow cytometry.
42

Luciferase Biodistribution Experiments
Mice were injected (t.v.) with Ad5.CMV-Luc or Ad.MBP.CMV-Luc diluted in
200 μL α-MEM for gene transfer biodistribution experiments. Tissues were removed 24
h post-injection and homogenized in a Mini-Beadbeater (BioSpec Products, Bartlesville,
OK) for analysis of biodistribution using a luciferase assay system (Promega, Madison,
WI). Protein concentrations of tissue homogenates were determined by DC Protein Assay
(Bio-Rad) in order to normalize relative luciferase units (RLU) values from different tis-
sues and animals.
Virus Particle Localization Experiments
Mice were injected (t.v.) with a cocktail of Ad5 and Ad.MBP at a 1:1 ratio (4 ×
1010 total VP per mouse) in 200 μL HBSS and sacrificed at various time points after in-
jection for organ removal. Some animals were perfused with PBS via the right ventricle
(RV) at time of sacrifice before organ collection. Other mice were first anesthetized with
50 μL pentobarbital i.p. and then the virus cocktail (4 × 1010 total VP per mouse in 50 μL
of HBSS) was injected directly into the left ventricle (LV) upon entry into the chest cav-
ity. At various time points following injection mice were sacrificed for organ removal.
For all samples, whole organs were weighed and PBS was added to each sample at a ratio
of 80 μL per 25 mg tissue and then samples were homogenized in a Mini-Beadbeater
(BioSpec Products). DNA was extracted from 100 μL of homogenized tissue with the
DNeasy Blood and Tissue Kits (Qiagen, Valencia, CA) according to the manufacturer’s
protocol. Standards were prepared for the various tissues by spiking the virus cocktail
into control (uninjected) tissues and extracting DNA in parallel with the experimental
43

samples. An assay for PCR-based detection of viral genomes was designed as a semi-
quantitative measure of VP localization. Since there is a measurable size difference be-
tween the fiber genes of Ad5 and Ad.MBP, a single set of primers (Table 2) was de-
signed such that both products (1127 bp and 1004 bp, respectively) would be amplified in
a single reaction.
Ad.MBP.CMV-GFP in vivo Experiments
Mice were injected (t.v.) with Ad5.CMV-GFP or Ad.MBP.CMV-GFP at 2.5 ×
1010 VP per mouse for both FACS and immunohistochemical analysis. For the former,
lungs were removed 24 h post-injection and a single cell suspension was obtained from
Liberase™ (Roche) digest as described above. Cells were washed in FACS buffer,
counted and stained for flow cytometry. For the latter, mice were anesthetized 24 h post-
injection and then perfused via the right ventricle (RV) with 10 mL PBS supplemented
with heparin (1 U mL–1) followed by 5 mL 10% neutral-buffered formalin (NBF). Lungs
were removed en bloc, inflated with 1–2 mL 10% NBF, then tied off and left at room
temperature overnight submerged in 10% NBF. The next day, lungs were switched to
70% ethanol before trimming and processing for paraffin embedding and sectioning be-
low.
Lung Immunohistochemistry
Paraffin sections (5 μm) were melted on slides at 60 °C for 1 h, deparaffinized in
xylene and then rehydrated in ethanol (100%, 95%, 85%, and 70%). Slides were washed
in PBS before performing antigen retrieval by boiling in Citric Acid Based Antigen Un-
44

masking Solution (Vector Labs, Burlingame, CA) for 12 min in an 800 W microwave.
Slides were washed in PBS, incubated at room temperature in blocking buffer (PBS sup-
plemented with 5% goat serum and 1% bovine serum), followed by incubation with pri-
mary antibodies in fresh blocking buffer overnight at 4 °C in a humidity chamber. Slides
were washed in PBS, incubated at room temperature in blocking buffer, and then with
secondary antibodies for 1 h at room temperature in a humidity chamber. Slides were
rinsed in PBS and mounted in VECTASHIELD Hard Set Mounting Medium with DAPI
(Vector Labs). Image acquisition was performed on a Leica DM6000B epifluorescence
microscope (Leica Micorsystems, Wetzlar, Germany) with SimplePCI Imaging software
(Compix Media, Inc., Irvine, CA). The following antibodies were used: a chicken poly-
clonal antibody to GFP (Abcam, Cambridge, MA), a rabbit antibody to von Willebrand
factor (vWF) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and Alexa Fluor® 488-
and Alexa Fluor® 594-conjugated goat polyclonal secondary antibodies to chicken and
rabbit IgGs, respectively (Invitrogen Molecular Probes).
Ad-bound Lung Cell Cytospins
Ad.MBP-bound lung cells (as prepared above) were isolated by FACS. Cytospins
were prepared on slides by cytocentrifugation at 100g for 5 min. Cells were differentiated
with Diff Quik (Baxter Diagnostics, Inc., Deerfield, IL) following the cytospin.
Flow Cytometry
Antibodies to CD11b (clone M1/70), Gr-1 (RB6-8C5), Ly-6G (1A8), CD11c
(HL3), CD31 (MEC13.3), CD45 (30-F11), CD45.2 (104), were all purchased from BD
45

Pharmingen. Other antibodies were already mentioned. Antibody to CD16/32 (2.4G2;
BD Pharmingen) (Fc Block) was used to block endogenous mouse Fc Receptors before
antibody staining. For binding studies, Fc Block was added after cells had been incubated
with phage, peptide, 293T cell lysate, or virus but before antibody staining. Data were
acquired on a FACSCalibur or LSRII (BD Biosciences, Franklin Lakes, NJ) and analyzed
with FlowJo v.7.6.1 (Tree Star, Inc., Ashland, OR).
Statistical Analysis
Data are presented as means ± s.d. where appropriate. Statistical differences be-
tween groups were determined by analysis of variance (with Bonferroni post-test correc-
tion for comparisons of three or more groups) using Prism 4.0 (GraphPad Software, La
Jolla, CA). Luciferase data were logarithmically transformed before statistical analysis
was performed. Statistical significance was accepted at P < 0.05.
46

Table 2
List of Oligonucleotides
Oligo Name Direction Sequence (5'–3') a
MA-51b,h,i Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTGGACGC-TGGATCGG
MA-52b Reverse cataCGGCCGAACCTCCACCGCAATACCCCCGATCCAGC-GTCCAACA
MA-53c Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTGCTACGC-TGGATCGG
MA-54c Reverse cataCGGCCGAACCTCCACCGCAATACCCCCGATCCAGC-GTAGCACA
MA-55d Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTGGGCAC-TGGATCGG
MA-56d Reverse cataCGGCCGAACCTCCACCGCAATACCCCCGATCCAGT-GCCCAACA
MA-57e Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTGGACGG-CTGATCGG
MA-58e Reverse cataCGGCCGAACCTCCACCGCAATACCCCCGATCAGCC-GTCCAACA
MA-59f Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTGGACGC-TGGCTCGG
MA-60f Reverse cataCGGCCGAACCTCCACCGCAATACCCCCGAGCCAGC-GTCCAACA
MA-61g Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTGGACGC-TGGATGCG
MA-62g Reverse cataCGGCCGAACCTCCACCGCAATACCCCGCATCCAGC-GTCCAACA
MA-63h Reverse cataCGGCCGAACCTCCACCGCAATACGCCCGATCCAGC-GTCCAACA
MA-64i Reverse cataCGGCCGAACCTCCACCGCAAGCCCCCCGATCCAGC-GTCCAACA
MA-65j Forward cataGGTACCTTTCTATTCTCACTCTGCTGCATGGACGC-TGGATCGG
MA-66j Reverse cataCGGCCGAACCTCCACCTGCATACCCCCGATCCAGC-GTCCATGC
MA-67k Forward cataGGTACCTTTCTATTCTCACTCTGCTTGTTATGGGC-GGGATCTG
MA-68k
Reverse cataCGGCCGAACCTCCACCGCACCACGTCAGATCCCGC-
CCATAACA
47

MBP-Fl Forward cataGGATCCCACTCTTGTTGGACGCTGGATCGGGGGTAT MBP-Rl Reverse cataTGATCATTCGGCGGAGCAATACCCCCGATCCAGCGT MA-21m Forward cataGGATCCCACTCTTGTTATGGGCGGGATCTGACGTGG MA-22m Reverse cataTGATCATTCGGCGGAGCACCACGTCAGATCCCGCCC MA-25n Forward cataGGATCCCGGTGGCGGTACCGGTGTATACGGCGGTT-
AATAAAG MA-26n Reverse cataATTTAAATCCCCGGATCTTTATTAACCGCCGTATA-
CACCGG scFv-jxn-5’o Forward TACTGGAGCCTTGGGTTTTG 566FFscrno Reverse GCTATGTGGTGGTGGGGCTA aMany oligos contain a 5’-spacer (cata) followed by a unique restriction site (under-lined). Sequences in bold represent overlapping regions of homology between two oligos for overlap-extension PCR bUsed to generate M13-MBP cUsed to generate M13-MBP[W1A]
dUsed to generate M13-MBP[T2A]
eUsed to generate M13-MBP[L3A]
fUsed to generate M13-MBP[D4A]
gUsed to generate M13-MBP[R5A]
hUsed to generate M13-MBP[G6A]
iUsed to generate M13-MBP[Y7A]
jUsed to generate M13-linMBP
kUsed to generate M13-iMBP
lUsed to generate pKan-566FF-MBP
mUsed to generate pKan-566FF-iMBP
nUsed to generate pVS-566FF-ΔCd
oUsed to amplify a region of the Ad fiber gene that is present in both wild-type Ad5 and fiber-fibritin modified Ad5 genomes
48
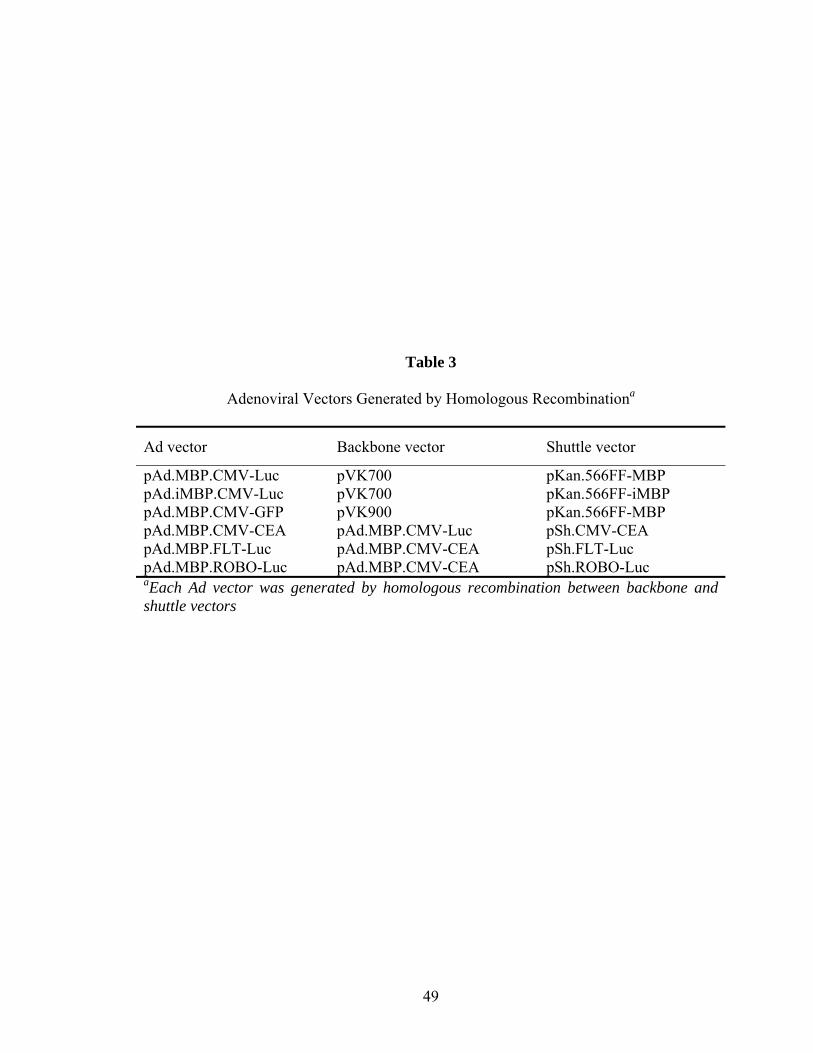
Table 3
Adenoviral Vectors Generated by Homologous Recombinationa
Ad vector Backbone vector Shuttle vector
pAd.MBP.CMV-Luc pVK700 pKan.566FF-MBP pAd.iMBP.CMV-Luc pVK700 pKan.566FF-iMBP pAd.MBP.CMV-GFP pVK900 pKan.566FF-MBP pAd.MBP.CMV-CEA pAd.MBP.CMV-Luc pSh.CMV-CEA pAd.MBP.FLT-Luc pAd.MBP.CMV-CEA pSh.FLT-Luc pAd.MBP.ROBO-Luc pAd.MBP.CMV-CEA pSh.ROBO-Luc aEach Ad vector was generated by homologous recombination between backbone and shuttle vectors
49

Table 4
Titers of Adenoviruses Generated
Titer (×1012 VP mL–1) Virusa
ib ii iii iv Ad.MBP.CMV-Luc 2.60 3.84 3.58 – Ad.iMBP.CMV-Luc 0.31c – – – Ad.MBP.CMV-GFP 1.00 0.47 0.54 – Ad.MBP.CMV-CEA 0.95 – – – Ad.MBP.FLT-Luc Rescuedd – – – Ad.MBP.ROBO-Luc Rescuedd – – – Ad5.CMV-Luc 7.40 – – – Ad5.CMV-GFP 6.50 – – – Ad5.CMV-CEA 7.00 – – – aCMV, human cytomegalovirus intermediate-early promoter; Luc, luciferase; GFP, green fluorescent protein; CEA, human carcinoembryonic antigen; FLT, human fms-related tyrosine kinase-1 promoter; ROBO, human roundabout ho-molog-4 promoter b(i) to (iv) indicate different virus batch preparations cLow titer is resultant of smaller scale preparation and heavy dilution dRescued indicates that only a small amount of unpurified and untitered virus was generated
50

DEVELOPMENT AND CHARACTERIZATION OF A LEUKOCYTE
TARGETED ADENOVIRUS
Introduction
Previously, we (Krasnykh et al., 2001; Noureddini et al., 2006) and others (van
Beusechem et al., 2000; Magnusson et al., 2001) have demonstrated the ability to "untar-
get" Ad5 from its ubiquitously expressed native receptor, CAR, by genetically deleting
the CAR-binding fiber-knob domain. Our group has gone on to show the ability to also
"retarget" Ad5 to an artificial receptor molecule through genetic incorporation of a novel
targeting ligand into knob-less fiber (Hedley e al., 2006) (see Figure 12a), but has yet to
demonstrate retargeting with these vectors in vivo. In order to circumvent many of the
vascular and systemic barriers to systemic adenovirus therapy (Parker et al., 2008; Di
Paolo and Shayakhmetov, 2009), we devised a novel strategy to target adenovirus to the
leukocyte subsets that directly home to sites of inflammation and actively contribute to
disease initiation and propagation.
Given that Ad vectors lack specific tropism to myeloid subsets (Cotter et al.,
2005) and that there have been no reports of a myeloid-targeted adenovirus for systemic
administration, we developed a new vector to overcome these challenges. In this regard,
we employed a technique called phage display library panning in order to identify a tar-
geting ligand for leukocyte subsets. Phage display bio-panning is a powerful tool that has
been used to successfully identify ligands that bind to specific targets in vitro and in vivo
(Pasqualini and Ruoslahti, 1996; Rajotte et al., 1998; Arap et al., 2002).
51

In this chapter, we describe the isolation of a seven amino acid peptide that spe-
cifically labels myeloid-lineage cells from mouse bone marrow. Upon further characteri-
zation, this myeloid-binding peptide (MBP) similarly stained myeloid cells from other
tissues, including the lung. Given the specificity of this novel peptide we genetically in-
corporated it into a knob-less fiber adenovirus (termed Ad.MBP) and demonstrate en-
hanced binding and transduction of myeloid cells in vitro. We further demonstrate that
the myeloid-binding specificity of the MBP sequence is retained in the context of three
independent platforms: phage display, recombinant peptide, and adenovirus.
Characterization of a Myeloid-binding Phage
In order to identify novel myeloid-targeting peptides two complementary phage
display panning strategies (Figure 7) were previously employed at Case Western Reserve
University (Roth, 2006). These unpublished panning data will be summarized below, as
they lay the entire foundation for which this body of work is built on (see Methods for
additional details).
First, a loop-constrained random heptapeptide M13 phage display library (Figure
7) was panned over murine BMCs in vitro. Leukocyte subsets were isolated by FACS
and cell-bound phage were eluted, amplified, and used as input for subsequent pans. A
single peptide, with the sequence WTLDRGY, was identified after three rounds of in vi-
tro panning (Table 5). For the second approach, the phage library was infused into the
tail vein of one mouse. Shortly after injection, BMCs were extracted and leukocyte sub-
sets were fractionated by FACS. Cell-bound phage were eluted, amplified and used as
input for subsequent pans in additional mice. For the third round of panning, two mice
52

were injected with equivalent doses of the library from the second pan. However, one
mouse was sacrificed 10 min post-infusion and the other at 30 min post-infusion. Alto-
gether, three rounds of in vivo panning produced 15 unique peptide sequences (from 32
clones sequenced) but failed to generate a consensus sequence (Table 5). However, the
WTLDRGY peptide identified in vitro was also identified in multiple rounds (second and
third rounds) of in vivo panning.
Table 5
Phage Peptide Candidate List
Frequency In vitro In vivo
Sequence Pan 3a Pan 1 Pan 2 Pan 3.1b Pan 3.2c
WTLDRGY 5 1 2 PPNLKHS 2 PNHSSRT 1 PLVTAQN 1 VPKQTQM 1 LRGETVH 1 PNWPHWP 1 1 1 KFHLGAP 1 2 1 PPFGHTP 1 HPNFLPA 1 SPKTMKH 2 4 LLKDARL 4 SNAGRNA 2 LHNITST 1 TPSTAFQ 1 aOnly clones from the third round of in vitro panning were se-quenced b10 minute post-infusion BM harvest c30 minute post-infusion BM harvest
53

Figure 7. Phage display panning strategies. (a) The New England Biolabs Ph.D.-C7C phage library used in these studies is based on the M13 filamentous phage. The library is composed of approximately 1.28 × 109 unique phage with each expressing, or 'displaying', a different randomized seven amino acid peptide (denoted by varying shapes and colors in cartoon) fused to the N-terminus of the pIII coat protein. (b) The seven amino acid peptides (XXXXXXX) displayed on pIII were engineered to be loop-constrained via disulfide bridge. (c) Schematic representation of the in vitro and in vivo panning strategies used to identify putative myeloid-binding phage peptides from the loop-constrained Ph.D.-C7C library. A complete list of isolated phage pep-tides is presented in Table 5.
54

As a first approach to evaluating binding specificities of the peptides identified in
the screen, BMCs were incubated with clonal stocks of biotinylated phage and binding
was detected by flow cytometry. Compared to other clones, the phage with the WTLDR-
GY sequence demonstrated the highest degree of binding to BMCs (Figure 8). To better
resolve the binding specificity of this clone to individual BMC populations, we synthe-
sized a 14 amino acid (aa) version of the peptide (CWTLDRGYCSAEKA), including a
cysteine bridge, three C-terminal flanking residues based on the M13 phage pIII protein,
with a biotinylated lysine residue at the thirteenth position. The peptide labeled nearly all
of the neutrophils (CD11b+Ly6G+) compared to secondary only controls, as well as the
majority of all CD11b+Ly6G– cells, indicating the binding specificity is primarily re-
stricted to the myeloid (CD11b+) lineage (Figure 9). Thereafter, we designated this pep-
tide: myeloid-binding peptide (MBP).
Inse
rtles
sVP
KQ
TQM
SP
KTM
KHH
PN
FLP
ALL
KD
ARL
PLV
TAQ
NPN
WPH
WP
PN
HSS
RT
SN
AG
RN
AK
FHLG
AP
LRG
ETVH
PPF
GH
TPTP
STA
FQLH
NIT
STW
TLD
RG
Y
0
2
4
50
75
100
Phage Clone
% o
f WTL
DR
GY
phag
e bi
ndin
g
Figure 8. The WTLDRGY phage clone binds BMCs. Flow cytometric detection of binding of individual biotinylated-M13 phage clones to BMCs at 4 °C. Background binding was assessed with insertless M13 control phage from manufacturer. A repre-sentative experiment is shown.
55

CD
11b-
CD
11b+0
25
50
75
100
Lineage
% P
eptid
e-bo
und
cells
Figure 9. The WTLDRGY peptide binds broadly but specifically to myeloid BMCs. The heptapeptide sequence, WTLDRGY, was incorporated into a 14 aa syn-thetic peptide (CWTLDRGYCSAEKA), with a cysteine bridge, 5 C-terminal flanking residues, and a biotinylated lysine residue at position 13, for flow cytometric analysis of binding to CD11b+ (myeloid) and CD11b– BMCs at 4 °C. Gray histograms indicate secondary only controls. A representative experiment is shown.
56

Protein homology searches did not result in any obvious putative binding partners
for the query sequences: WTLDRGY or CWTLDRGYC. Therefore, scanning alanine
mutagenesis was performed for MBP, in an attempt to elucidate the critical myeloid-
binding residues. Each residue within the WTLDRGY sequence was substituted with an
alanine (W1A, T2A, L3A, D4A, R5A, G6A, Y7A) and cloned into the original M13
phage display platform. In order to evaluate whether MBP-binding specificity is depend-
ent on cysteine bridge formation, a double mutant (linMBP) was created in which both
flanking cysteine residues were mutated to alanines. One additional mutant (iMBP) was
generated, in which the MBP sequence was inverted (WTLDRGY to YGRDLTW), to
verify myeloid-binding is sequence specific. A total of 11 phage were generated (includ-
ing recloning of the original MBP phage and amplification of a control phage that lacked
a C7C insert) for evaluation of binding to CD11b+ BMCs in vitro. Scanning alanine mu-
tagenesis revealed that the nature of the fourth (Asp, D) and, to a lesser extent, the fifth
(Arg, R) residues are not important for myeloid-binding specificity (Figure 10). How-
ever, the first (Trp, W), second (Thr, T), third (Leu, L), sixth (Gly, G), and seventh (Tyr,
Y) residues were all absolutely critical for binding. Lack of binding by iMBP demon-
strates that the order of the residues is also important. Furthermore, linearly displayed
MBP (linMBP) shows severely abrogated binding to CD11b+ BMCs. Unfortunately, it is
not clear if abrogated binding is truly due to linearization of MBP or if the flanking cys-
teines are necessary for binding specificity. Individual mutation of each of the Cys resi-
dues will need to be conducted in order to clarify this result. Importantly, none of the mu-
tants demonstrated the ability to bind new cell types, such as CD11b– BMCs.
57

Figure 10. Scanning alanine mutagenesis of the MBP sequence reveals critical residues for binding. Flow cytometric detection of binding of original MBP phage and mutant MBP phage to CD11b+ (myeloid) BMCs at 4 °C. Each of the seven resi-dues within the MBP sequence, WTLDRGY, was mutated to an alanine (A) to create the mutants. Additional mutants were created such that the MBP sequence was either inverted to YGRDLTW (iMBP) or both flanking cysteine residues were mutated to alanines (linMBP). Background phage binding was assessed by 'insertless' phage (lacking a C7C insert). Gray histograms indicate 'no phage' staining controls. A repre-sentative experiment is shown.
Following these studies, we were able to redefine the MBP sequence and repeat
homology searches using the sequence WTLXRGY or WTLXXGY. From the list of pu-
tative matches for such a small input sequence, the sequence WTLXRGY matched the
minor histocompatibility antigen (mHag) HA-1, which is highly expressed on hemato-
poietic cells (de Bueger et al., 1992). HA-1 is a cytosolic protein with a Rho-GTPase-
58

activating protein (GAP) domain that has been implicated in graft versus host disease
(GvHD), as it is typically processed and displayed by MHC class I (MHC-I) as self anti-
gen (Spierings et al., 2004). To determine if MBP binding is related to the presentation of
HA-1, we evaluated MBP peptide binding to BMCs from MHC-I deficient (β2mnull) ani-
mals (Figure 11). Since some myeloid cell types are antigen presenting cells (APCs), we
also evaluated MBP-binding to MHC-II deficient (CIITAnull) BMCs. However, the data
suggest that MBP does not bind MHC-I or MHC-II.
Nevertheless, additional M13-MBP mutants need to be studied to further charac-
terize the nature of MBP binding to its target ligand or receptor. Ultimately, biochemical
studies (e.g. immunoprecipitation) will most likely need to be conducted to truly isolate
MBP binding partners.
Stable Incorporation of a Myeloid-binding Peptide (MBP) into Ad5
Ad5 cellular entry is mediated by distinct binding and internalization events; the
knob domain of Ad5 fiber initiates attachment through interactions with CAR, while in-
ternalization is mediated by subsequent interactions between integrins and the Ad5 pen-
ton RGD motif (Bergelson et al., 1997; Wickham et al., 1993). Our group has previously
developed a genetic Ad targeting platform, based on key fiber modifications. Specifi-
cally, the knob domain is deleted to ablate its broad tropism to CAR-expressing cells, and
a 95 aa trimerization domain of the T4 phage fibritin protein is substituted to improve
stability and allow display of novel targeting ligands (Krasnykh et al., 2001; Noureddini
et al., 2006). We took advantage of this previously-developed system to develop a mye-
loid-targeted Ad vector (Figure 12).
59
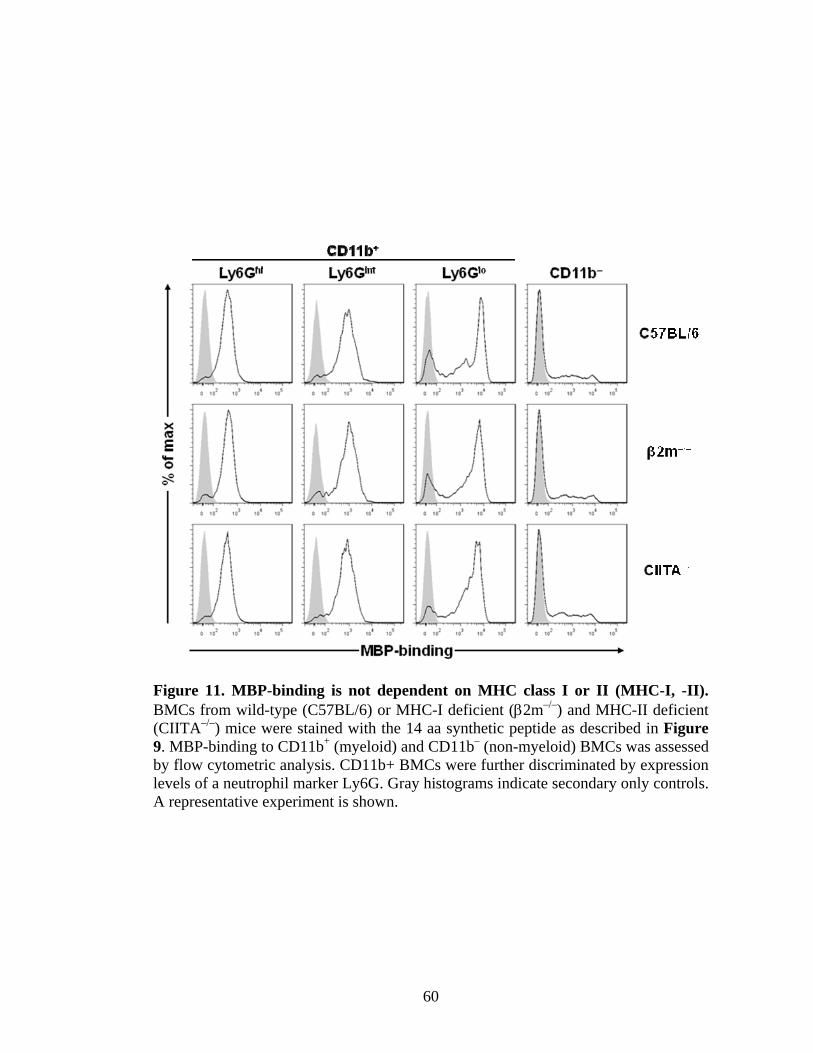
Figure 11. MBP-binding is not dependent on MHC class I or II (MHC-I, -II). BMCs from wild-type (C57BL/6) or MHC-I deficient (β2m–/–) and MHC-II deficient (CIITA–/–) mice were stained with the 14 aa synthetic peptide as described in Figure 9. MBP-binding to CD11b+ (myeloid) and CD11b– (non-myeloid) BMCs was assessed by flow cytometric analysis. CD11b+ BMCs were further discriminated by expression levels of a neutrophil marker Ly6G. Gray histograms indicate secondary only controls. A representative experiment is shown.
60

Figure 12. Retargeting of Ad5. (a) Deletion of the knob domain (red balls) from the Ad5 fiber protein and replacement with a targeting ligand (blue diamonds) ablates binding of Ad5 to CAR and redirects binding to alternative receptors. (b) Diagram comparing wild-type Ad5 and the MBP fibers. The MBP fiber is derived from the 566FF platform that our group has previously developed (Nouredinni et al., 2006). Briefly, the knob domain is removed and replaced with the trimerization region from the T4 phage fibritin protein fused to the MBP (or iMBP) targeting ligand via a flexi-ble linker ([GGGS]4). Numbers indicate amino acid positions.
First, the MBP and control iMBP sequences were inserted into a fiber-fibritin
(FF) shuttle vector (see Methods and Appendix C). Next, FF-MBP and FF-iMBP were
transferred into a mammalian expression vector system (Noureddini et al., 2006; Hedley
et al., 2006; Korokhov et al., 2003) to determine if genetic incorporation of the MBP se-
61

quence into the FF platform produced viable fiber trimers (Figure 13) that also retained
myeloid-binding specificity (Figure 14). The data support the concept that the FF plat-
form can accommodate insertion of both the MBP or iMBP sequences, and that MBP-
binding to myeloid BMCs is retained within the FF context. Additionally, FF-iMBP did
not show substantial binding to any BMC populations, indicating the myeloid-binding
specificity of FF-MBP is mediated entirely by the MBP sequence.
Following this verification, FF-MBP was incorporated into a number of replica-
tion-deficient Ad genomes by homologous recombination in E. coli (see Methods for
complete details). The resultant viruses, all termed Ad.MBP, contained a number of dif-
ferent reporter cassettes for use in various downstream applications (see Table 3 in Me-
thods and Appendix C). Although titers of Ad.MBP viruses were generally much lower
than the corresponding Ad5 viruses (see Table 4 in Methods), FF-MBP incorporation
into purified Ad.MBP particles was similar to wild-type fiber incorporation in Ad5 by
Western blot (Figure 15).
Figure 13. Assessment of FF-MBP viability by Western blot. 293T cells were har-vested 48 h after transfection with expression plasmids for wild-type Ad5, MBP, or iMBP fibers. Protein supernatants from 293T cells were incubated at either 95 °C (boiled, B) or room temperature (unboiled, U) prior to SDS-PAGE separation. Un-boiled samples demonstrate that the majority of fibers are trimerized.
62

Figure 14. FF-MBP retains myeloid-binding specificity. Evaluation of fiber binding to CD11b+ (myeloid) BMCs from C57BL/6 (left panel) and transgenic mice that ex-press human CAR (hCAR) (right panel). 293T protein supernatants (as in Figure 13) were added to BMCs and fiber-bound CD11b+ cells were detected by flow cytometry. A representative experiment is shown.
Figure 15. MBP fibers are efficiently incorporated into the Ad capsid of purified virions. Fiber incorporation into purified virions was assessed by Western blot. Puri-fied virions containing the wild-type Ad5 fiber (Ad5.CMV-Luc), or the smaller MBP fiber (Ad.MBP.CMV-Luc), or both fibers (mosaic Ad5/MBP.CMV-Luc) were incu-bated at 95 °C and then separated by SDS-PAGE.
63

Functional Characterization of Ad.MBP
We next assessed whether incorporation of the MBP sequence into Ad.MBP led
to enhanced binding of specific leukocyte subsets. To do so, Ad5 and Ad.MBP were add-
ed to BMCs at 4 °C, and bound virions were detected by flow cytometric analysis. As
expected, Ad.MBP demonstrated a high degree of binding to CD11b+ myeloid cells (Fig-
ure 16) while Ad5 binding was not detected on any BMC populations except those de-
rived from transgenic mice that express hCAR (Figure 16–18). Additionally, Ad.MBP
and not Ad5 bound to CD11b+ cells in the peripheral blood, spleen, and lung demonstrat-
ing that Ad.MBP has the capacity to bind myeloid cell populations from a variety of tis-
sues ex vivo (Figure 18).
Figure 16. Incorporation of FF-MBP into Ad5 (Ad.MBP) retains myeloid binding specificity in vitro. Assessment of Ad5 or Ad.MBP binding at 4 °C to CD11b+ (mye-loid) or CD11b– (non-myeloid) BMCs (500 VP per cell) by flow cytometry. A repre-sentative experiment is shown.
64

CD11b+ CD11b–
Figure 17. Expression of hCAR permits efficient Ad5 binding of BMCs. Assess-ment of Ad5 binding at 4 °C to myeloid (CD11b+) and non-myeloid (CD11b–) hCAR+ BMCs (500 VP per cell) by flow cytometry. A representative experiment is shown.
0 20 40 60 80 100
Lung CD11b+
Spleen CD11b+
PB CD11b+
BMC CD11b+
BMC CD11b-Ad.MBPAd5
% Ad-bound cells
Ti
ssue
Figure 18. Ad.MBP binds myeloid cells from various tissues in vitro. Assessment of Ad5 or Ad.MBP binding at 4 °C to CD11b+ BMCs, peripheral blood (PB), spleen, and lung cells (500 VP per cell) by flow cytometry. CD11b– BMCs are also shown for comparison. A representative experiment is shown.
65

In addition to flow cytometric binding studies, we utilized TEM to visualize
Ad.MBP binding to BM neutrophils. The transmission electron micrographs demonstrate
numerous virion filled pockets along the neutrophil’s surface (Figure 19). In agreement
with the flow cytometry data, Ad5 was not detected on these populations by TEM (data
not shown). Taken together, these two experiments demonstrate our ability to retarget
virus binding to myeloid populations.
Figure 19. Ad.MBP-binding to neutrophils by TEM. Evaluation of Ad.MBP-binding (arrows) to BM neutrophils was also determined by transmission electron mi-croscopy (TEM). Ad5 was not detected in controls (data not shown). Scale bar, 500 nm.
Upon activation by various inflammatory stimuli, neutrophils generate and release
superoxide anion, hydrogen peroxide, and reactive oxygen species (ROS) through a proc-
ess called an oxidative burst. To determine whether Ad.MBP-binding to neutrophils re-
sults in an oxidative burst, we employed a flow cytometric assay as described previously
(Vowells et al., 1995). Briefly, neutrophils are loaded with intracellular DHR123 and
then incubated with stimulus at 37 °C. Upon activation, DHR123 is oxidized by ROS to
66

the green fluorescent analog R123, which can then be measured by flow cytometry.
While PMA-activated neutrophils resulted in potent conversion of DHR123 to R123, in-
cubation of DHR123-loaded neutrophils with increasing MOIs of Ad.MBP resulted in no
measurable oxidative burst, indicating that Ad.MBP-binding to neutrophils does not lead
to their activation (Figure 20).
Figure 20. Ad.MBP-binding to neutrophils does not induce oxidative burst. Mouse peripheral blood leukocytes, pre-mixed with dihydrorhodamine 123 (DHR123), were incubated with vehicle (red histograms), PMA (assay positive con-trol; green histogram on left), or Ad.MBP (green histogram on right; MOI = 1000 is shown) at increasing MOI at 37 °C. Neutrophils, gated by forward and side scatter, were analyzed by flow cytometry to measure DHR123 oxidation to its green fluores-cent analog rhodamine 123 (R123). A representative experiment is shown.
67
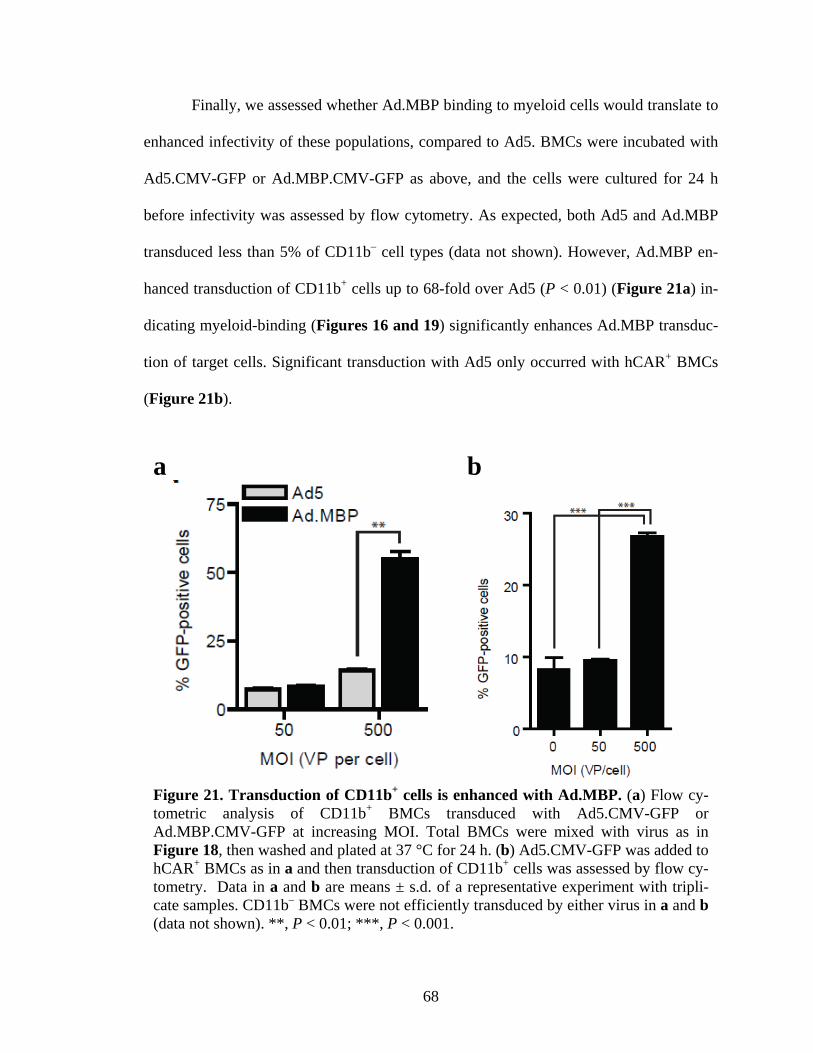
Finally, we assessed whether Ad.MBP binding to myeloid cells would translate to
enhanced infectivity of these populations, compared to Ad5. BMCs were incubated with
Ad5.CMV-GFP or Ad.MBP.CMV-GFP as above, and the cells were cultured for 24 h
before infectivity was assessed by flow cytometry. As expected, both Ad5 and Ad.MBP
transduced less than 5% of CD11b– cell types (data not shown). However, Ad.MBP en-
hanced transduction of CD11b+ cells up to 68-fold over Ad5 (P < 0.01) (Figure 21a) in-
dicating myeloid-binding (Figures 16 and 19) significantly enhances Ad.MBP transduc-
tion of target cells. Significant transduction with Ad5 only occurred with hCAR+ BMCs
(Figure 21b).
a b
Figure 21. Transduction of CD11b+ cells is enhanced with Ad.MBP. (a) Flow cy-tometric analysis of CD11b+ BMCs transduced with Ad5.CMV-GFP or Ad.MBP.CMV-GFP at increasing MOI. Total BMCs were mixed with virus as in Figure 18, then washed and plated at 37 °C for 24 h. (b) Ad5.CMV-GFP was added to hCAR+ BMCs as in a and then transduction of CD11b+ cells was assessed by flow cy-tometry. Data in a and b are means ± s.d. of a representative experiment with tripli-cate samples. CD11b– BMCs were not efficiently transduced by either virus in a and b (data not shown). **, P < 0.01; ***, P < 0.001.
68

LEUKOCYTE-MEDIATED HAND-OFF OF ADENOVIRUS FOR SPECIFIC
AND EFFICIENT LUNG TARGETING
Introduction
Although a number of strategies have been evaluated for targeting the pulmonary
vasculature, systemic use of Ad5 has been limited by a number of barriers in vivo (Parker
et al., 2008; Descamps and Benihoud, 2009). The primary in vivo barrier is the large ex-
tent to which intravenously administered Ad5 virions are sequestered by KCs in the liver
(Lieber et al., 1997; Tao et al., 2001). Much of the remaining virus is redirected to hepa-
tocytes through interactions with soluble blood factors (Shayakhmetov et al., 2005), par-
ticularly FX (Parker et al., 2006; Waddington et al., 2008). Additional mechanisms, at-
tributed to the unique anatomical composition of the liver, also contribute to Ad5 seques-
tration (Di Paolo et al., 2009). Hence, to maximize the therapeutic potential of Ad for
other clinically-relevant tissues, significant rerouting of Ad5 from the liver will be neces-
sary (Di Paolo and Shayakhmetov, 2009).
Efficient targeting of cell types within the lung may preclude the need for viral
modifications that ablate liver sequestration mechanisms. Leukocytes represent an ideal
target for delivery of therapeutics to the pulmonary vasculature as venous blood flow is
first directed through the pulmonary vasculature, and circulating leukocytes accumulate
within the lung due to reduced transit rates through the extensive microvasculature net-
work within this tissue (Wiggs et al., 1994; Kuebler and Goetz, 2002). Given the size and
69

location of the lung leukocyte pool, we designed a novel lung-targeting approach utiliz-
ing our myeloid-targeted Ad (Ad.MBP), described in the previous chapter.
In this chapter, we demonstrate that intravenous administration of Ad.MBP results
in significant rerouting of Ad5 tropism. Specifically, we demonstrate that this leukocyte-
targeting approach specifically localizes Ad5 virions and gene transfer to the lung mi-
crovasculature and prevents KC uptake and hepatocyte transduction. To our knowledge,
this is the first study to demonstrate significantly enhanced lung gene transfer and elimi-
nation of liver tropism in the absence of further capsid modifications. Furthermore, this
work validates the application of future knob-less fiber retargeting strategies.
Ad.MBP Particle and Gene Transfer Biodistribution in vivo
Because circulating leukocytes are concentrated in the lung vasculature (Kuebler
and Goetz, 2002) and venous blood flow passes through the pulmonary system prior to
the liver, we hypothesized that tail vein injection of the myeloid-targeted Ad.MBP vector
would result in improved gene delivery to the leukocyte-rich lung, in contrast to Ad5.
Thus, we compared the biodistribution of Luc gene transfer following i.v. infusion of
Ad5.CMV-Luc or Ad.MBP.CMV-Luc vectors (at 2.5 × 1010 VP per mouse). In accor-
dance with other reports (Smith et al., 1993; Huard et al., 1995; Peeters et al., 1996; Rey-
nolds et al., 2000 and 2001), Ad5 gene expression is primarily localized to the liver and
spleen at 24 h post-administration (Figure 22a). In stark contrast, Ad.MBP gene expres-
sion is localized almost exclusively to the lungs and is essentially absent from the liver
and spleen (Figure 22a), resulting in a 205-fold increase (P < 0.001) in lung gene expres-
sion and 820-fold (P < 0.001) and 260-fold (P < 0.001) reductions in liver and spleen
70

gene expression, respectively. Previous reports have utilized the ratio of lung to liver
gene expression as a measure of targeting efficiency (Reynolds et al., 2001; Everts et al.,
2005; Izumi et al., 2005) Thus, our targeting approach realized 6,700-fold higher trans-
gene expression in the lung than in the liver (P < 0.001). Since, the lung:liver ratio of un-
targeted Ad5 was 0.04 (P < 0.01), our targeting approach resulted in an ~165,000-fold
enhancement in lung targeting of over that of Ad5. Furthermore, Ad.MBP also realized a
>56,000-fold improvement in lung:spleen ratio, compared to Ad5. Lastly, in order to en-
sure that the low gene expression observed in the liver for Ad.MBP.CMV-Luc was not
due to preferential expression in the lung, quantitative PCR (qPCR) was used to measure
the number of Ad.MBP genomes in both the liver and lung at 24 h. The data reveal that
there is 100-fold more Ad.MBP DNA in the lung than in the liver, reaffirming that low
levels of Ad.MBP gene expression in the liver are likely a result of poor transduction of
the liver and not due to expression differences (Figure 22b).
Previous studies have concluded that Ad5 vectors with knob mutations or dele-
tions do not significantly alter reporter gene biodistribution (Alemany and Curiel, 2001;
Zinn et al., 2004). To investigate whether reduced liver tropism was a result of a first-
pass effect through the pulmonary circulation, we evaluated the VP biodistribution of
Ad.MBP immediately after i.v. injection. Both Ad5 and Ad.MBP were co-injected i.v. at
a 1:1 ratio (4 × 1010 total VP per mouse) and the animals were sacrificed 5 min post-
injection for genomic DNA extraction from whole organs and PCR-based detection of
viral genomes. Since there is a measurable size difference between the fiber genes of Ad5
and Ad.MBP, a single set of primers was designed to amplify both products as a semi-
quantitative measure of VP localization. Analogous to gene expression biodistribution
71

(Figure 22a), Ad5 virions were predominantly detected in the liver, while Ad.MBP viri-
ons were detected almost exclusively in the lung (Figure 23a). Additional animals co-
injected with the same virus mixture and perfused either immediately before tissue isola-
tion (Figure 24) or sacrificed 45 min later (Figure 23a) showed similar findings.
Ad.MBP was only detectable in the liver when the Ad5:Ad.MBP mixture was directly
injected into the left ventricle (Figure 23b). In the aggregate, these data support that
Ad.MBP avoids KC and liver sinusoidal VP sequestration and FX-mediated liver trans-
duction due to a strong first-pass effect through the lung microvasculature before the vi-
rus enters systemic circulation.
Since high VP loads are associated with an acute immune response (Lieber et al.,
1997; Raper et al., 2003; and reviewed in Descamps and Benihoud, 2009), we titrated
Ad.MBP gene expression biodistribution down to viral doses of 107 VP per mouse to de-
termine if efficient lung targeting was retained. Even at these doses, lung expression was
significantly higher than any other tissue (P < 0.001) (Figure 25), including the liver and
spleen (Figures 25 and 26), further demonstrating the specificity and efficiency with
which Ad.MBP targets the pulmonary system due to the first-pass effect (Figure 23).
Assessment of Lung Milieu Following Ad.MBP Gene Transfer
Intravenous administration of Ad5 results in vector accumulation within the liver
and induces an acute immune response, characterized by cytokine and chemokine release
and recruitment of leukocytes to the liver (Muruve et al., 1999; Li et al., 2002). Thus far,
we have demonstrated that administration of Ad.MBP by the same route, redirects almost
all virus to the lung (Figures 22–26) suggesting that this innate response and subsequent
72

a b
Nai
ve
Ad.
MB
P
Nai
ve
Ad.
MB
P
100
101
102
103
104
105
LungLiver
Animal Group
Ad.
MB
P ge
nom
es p
er 2
0 ng
gD
NA
Figure 22. Systemically administered Ad.MBP enhances lung and diminishes liv-er gene transfer. (a) Biodistribution of Luc gene transfer in mice injected (i.v.) with 2.5 × 1010 VP of Ad.MBP.CMV-Luc or Ad5.CMV-Luc 24 h prior to assessment. ***, P < 0.001 compared to Ad5; ###, P < 0.001 compared to Ad.MBP Lung. (b) Ad.MBP genome copy number was assessed by qPCR in lung and liver tissues of mice injected with 2.5 × 1010 VP of Ad.MBP.CMV-Luc. Tissues were removed 24 h after injection and genomic DNA (gDNA) was extracted for qPCR. Naive mice were included to show background levels of detection. Data in a and b are means ± s.d. for n = 3 mice per group for each experiment.
hepatotoxicity is largely attenuated in the liver but may have shifted to the lung. Histo-
logical examination of lung tissue sections from animals injected with Ad,MBP 24 h
prior reveals no gross changes in tissue architecture and no observable increase in leuko-
cyte number, compared to both naive and Ad5-injected animals (Figure 27) suggesting
that Ad.MBP lung sequestration does not induce an acute inflammatory response. How-
ever, further analysis of cytokine levels should be undertaken to corroborate these gross
observations.
73

Figure 23. Systemically administered Ad.MBP eliminates liver virus particle se-questration. Biodistribution of VP was assessed to evaluate sequestration and first-pass effect of Ad.MBP. (a) Mice were injected (i.v.) with a 1:1 ratio of Ad5:Ad.MBP (4 × 1010 total VP) and sacrificed at either 5 or 45 min post-injection. Lungs and livers were removed and genomic DNA was extracted for subsequent PCR-based assessment of Ad genome content. (b) Mice were also injected directly into the left ventricle (LV) and sacrificed at 5 min post-injection to compare to i.v. injected (TV) mice. Spiked samples contain the 1:1 viral cocktail added directly to naïve tissues prior to genomic DNA isolation. Gray and black arrowheads indicate positions for Ad5 and Ad.MBP PCR products, respectively. Data are representative of n = 3–5 mice per group.
74

Figure 24. Ad.MBP particles are not removed from the lung with perfusion of the pulmonary circulation. Biodistribution of VP following vascular perfusion was as-sessed to determine if Ad.MBP particles are associated with soluble blood factors or bound to resident structures of the lung microvasculature shortly after injection. Mice were injected (i.v.) with a 1:1 ratio of Ad5:Ad.MBP (4.0 × 1010 total VP) and sacri-ficed at 5 min post-injection. Lungs and livers were removed and genomic DNA was extracted for subsequent PCR-based assessment of Ad genome content. Some animals were perfused with PBS via the right ventricle (Perf) immediately before tissue isola-tion. Non-perfused mice (No Perf). Spiked samples contain either Ad5, or Ad.MBP, or the 1:1 viral cocktail added directly to naïve lung tissue prior to genomic DNA isola-tion. Gray and black arrowheads indicate positions for Ad5 and Ad.MBP PCR prod-ucts, respectively. Data are representative of n = 3 mice per group.
75

Figure 25. Ad.MBP lung gene transfer is retained at low doses of systemically administered virus. Biodistribution of Luc gene transfer in mice injected (i.v.) with 107–1010 VP of Ad.MBP.CMV-Luc 24 h prior to assessment. Data are means ± s.d. of n = 3 mice per group. ***, P < 0.001 for lung compared to all other tissues.
76

Figure 26. Ad.MBP lung:liver ratio is retained at low doses of systemically ad-ministered virus. The lung:liver and lung:spleen Luc gene expression ratios were de-termined for the same mice injected (i.v.) with 107–1010 VP of Ad.MBP.CMV-Luc in Figure 25. Data are means ± s.d. for n = 3 mice per group.
77

Figure 27. Ad.MBP lung gene transfer does not result in leukocyte recruitment. Mice were injected (i.v.) with 2.5 × 1010 VP of Ad.MBP.CMV-GFP or Ad5.CMV-GFP 24 h prior to sacrifice. Panels show H&E staining on formalin-fixed, paraffin-embedded lung tissue. Images are representative of n = 2 mice per group.
Further examination of total lung cells by flow cytometry suggests that the cellu-
lar composition of the lung may be slightly altered at 24 h. Compared to naive animals,
Ad.MBP-injected animals have a higher ratio of myeloid (CD11b+) to non-myeloid
(CD11b–) cell types (Figure 28a) although this result was not significant (P > 0.05).
Even so, we cannot determine whether this trend is due to an overall increase in myeloid
cells or a general decrease in non-myeloid cells in Ad.MBP animals, because lung cell
78

counts (of collagenase-digested whole lung tissue) were unreliable and therefore not ob-
tained. However, we observed no difference in lung weights from the two groups and our
results from gross histological examination (Figure 27) revealed no obvious leukocyte
recruitment. Furthermore, there was no perturbation of the ratio of neutrophils
(CD11b+Ly6G+) to monocytes/macrophages (CD11b+Ly6G–) between the two groups
(Figure 28b). Altogether, these data suggest that there is no change in cellular composi-
tion of the lung 24 h following Ad.MBP injection, although a more detailed analysis of
lung cellularity should be performed that includes absolute cell counts and additional
time-points.
Identification of Ad.MBP Transduced Lung Cell Populations
Given the significant specificity of VP and gene transfer localization in the lung,
we wanted to assess whether or not myeloid cells (particularly marginated leukocytes in
the pulmonary microvasculature) were the main population transduced by the Ad.MBP
virus. Mice were i.v. injected with Ad.MBP.CMV-GFP and the lungs were harvested 24
h later. After collagenase digestion of lung tissue, GFP+ lung cells (Figure 29a) were
analyzed by flow cytometry using different antibody cocktails. A myeloid (CD11b+)
population representing about 25% of GFP+ lung cells was consistently detected in
Ad.MBP injected animals (Figure 29b,c). Less than 5% of these cells were neutrophils
(CD11b+Ly6G+), indicating that the major transduced myeloid population was non-
alveolar macrophages or monocytes (CD11b+CD11c–) (Figure 29b,c). However, about
75% of the GFP+ lung cells lacked myeloid marker (CD11b–Ly6G–CD11c–) expression
(Figure 29b,c). Therefore, a second antibody cocktail was used to distinguish leukocyte
79
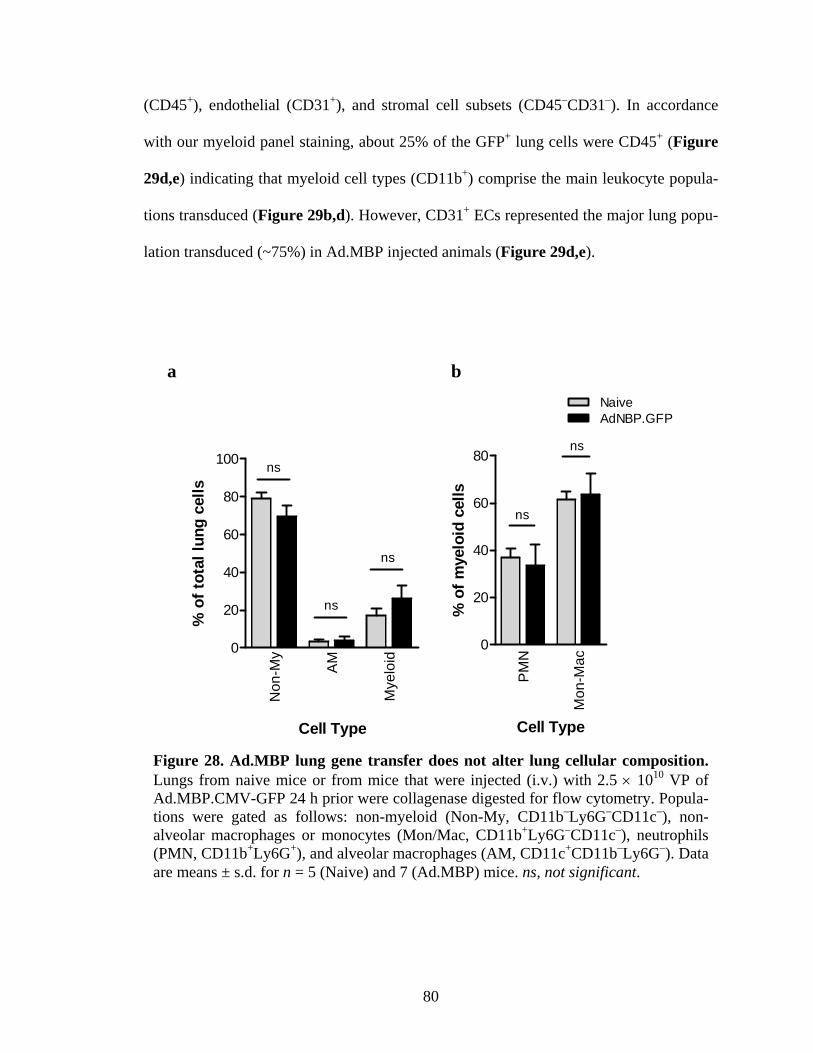
(CD45+), endothelial (CD31+), and stromal cell subsets (CD45–CD31–). In accordance
with our myeloid panel staining, about 25% of the GFP+ lung cells were CD45+ (Figure
29d,e) indicating that myeloid cell types (CD11b+) comprise the main leukocyte popula-
tions transduced (Figure 29b,d). However, CD31+ ECs represented the major lung popu-
lation transduced (~75%) in Ad.MBP injected animals (Figure 29d,e).
a b
Non
-My
AM
Mye
loid
0
20
40
60
80
100
Cell Type
% o
f tot
al lu
ng c
ells
ns
ns
ns
PM
N
Mon
-Mac
0
20
40
60
80
AdNBP.GFPNaive
Cell Type
% o
f mye
loid
cel
ls
ns
ns
Figure 28. Ad.MBP lung gene transfer does not alter lung cellular composition. Lungs from naive mice or from mice that were injected (i.v.) with 2.5 × 1010 VP of Ad.MBP.CMV-GFP 24 h prior were collagenase digested for flow cytometry. Popula-tions were gated as follows: non-myeloid (Non-My, CD11b–Ly6G–CD11c–), non-alveolar macrophages or monocytes (Mon/Mac, CD11b+Ly6G–CD11c–), neutrophils (PMN, CD11b+Ly6G+), and alveolar macrophages (AM, CD11c+CD11b–Ly6G–). Data are means ± s.d. for n = 5 (Naive) and 7 (Ad.MBP) mice. ns, not significant.
80

Naive Ad5 Ad.MBP0
2
4
6
8
10
Virus
% G
FP-p
ositi
ve c
ells
Figure 29 (Part I). Ad.MBP targets gene transfer to the pulmonary endothelium. (a–e) Cell populations transduced by Ad.MBP were evaluated by flow cytometry of collagenase digested lungs from mice injected (i.v.) with 2.5 × 1010 VP of Ad5.CMV-GFP or Ad.MBP.CMV-GFP 24 h prior. (a) Transduced (GFPP
+) cells were gated based on the staining profile of lung cells from uninjected controls and the number of GFP+ lung cells are shown graphically to the right. Representative scatter plots are shown. Data are means ± s.d. for n = 5 (naive), 2 (Ad.5), and 7 (Ad.MBP) mice.
81

Figure 29 (Part II). (b,c) Analysis of GFP+ lung cells gated in a for expression of the markers CD11b, CD11c, and Ly6G. Populations were gated as follows: non-myeloid (Non-My, CD11b–Ly6G–CD11c–), non-alveolar macrophages or monocytes (Mon/Mac, CD11b+Ly6G–CD11c–), neutrophils (PMN, CD11b+Ly6G+), and alveolar macrophages (AM, CD11c+CD11b–Ly6G–). (d,e) Analysis of GFP+ lung cells gated in a for expression of the markers CD45 and CD31. Populations were gated as follows: total leukocuytes (Leuk, CD45+), endothelial cells (EC, CD31+), and other stromal cells (Stromal, CD45–CD31–). Data in b and d are means ± s.d. for n = 5 and 7 mice, respectively. ***, P < 0.001. Representative scatter plots are shown in c and e. Gating schema for b–e were based on lung staining patterns from uninjected controls (see Appendix D).
82

In addition to flow cytometric analysis, some lungs were harvested en bloc from
Ad.MBP.CMV-GFP injected mice for further analysis by immunohistochemistry. Com-
pared to uninjected controls and Ad5.CMV-GFP injected mice, there is substantially
more GFP staining within the lung sections from Ad.MBP.CMV-GFP injected mice
(Figure 30), corroborating our flow cytometric analyses (Figure 29). Furthermore, GFP+
cells from Ad.MBP lungs were predominantly localized to the smaller alveolar capillaries
(tissue lacking von vWF staining) rather than the ECs that comprise the larger blood ves-
sels (vWF+) (Figure 30).
Ad.MBP Binding and Transduction of ECs
To determine whether Ad.MBP also binds and transduces pulmonary ECs, we
decided to reevaluate Ad.MBP binding to various lung cell populations. Lungs from na-
ïve mice were collagenase digested to make a single cell suspension and Ad.MBP was
added ex vivo. Unbound virus was removed and binding to individual cell populations
was evaluated by flow cytometry. In agreement with our earlier data (Figures 16 and 18
from the previous chapter), Ad.MBP bound a large fraction of CD11b+ cells (Figure
31a). However, further investigation revealed Ad.MBP did not show substantial binding
to CD31+ pulmonary ECs (Figure 31a). Furthermore, the majority of total Ad.MBP-
bound lung cells expressed the pan-leukocyte marker CD45 (Figure 31b) and morpho-
logically resembled myeloid cell types (monocytes, macrophages and neutrophils) and
not other cell populations such as ECs (Figure 31c). These data suggest that Ad.MBP
first binds myeloid cells and is subsequently “handed-off” to PECs.
83

Figure 30. Ad.MBP lung gene transfer is localized to alveolar capillaries. Mice were injected (i.v.) with 2.5 × 1010 VP of Ad.MBP.GFP 24 h prior to sacrifice. Panels show staining for the GFP transgene (green fluorescence), von Willebrand factor (red), and nuclei (DAPI, blue) in uninjected controls (a) and mice injected with control Ad5.GFP (b) or Ad.MBP.GFP (c). Scale bar, 40 μm. Images are representative of n = 2 mice per group.
84
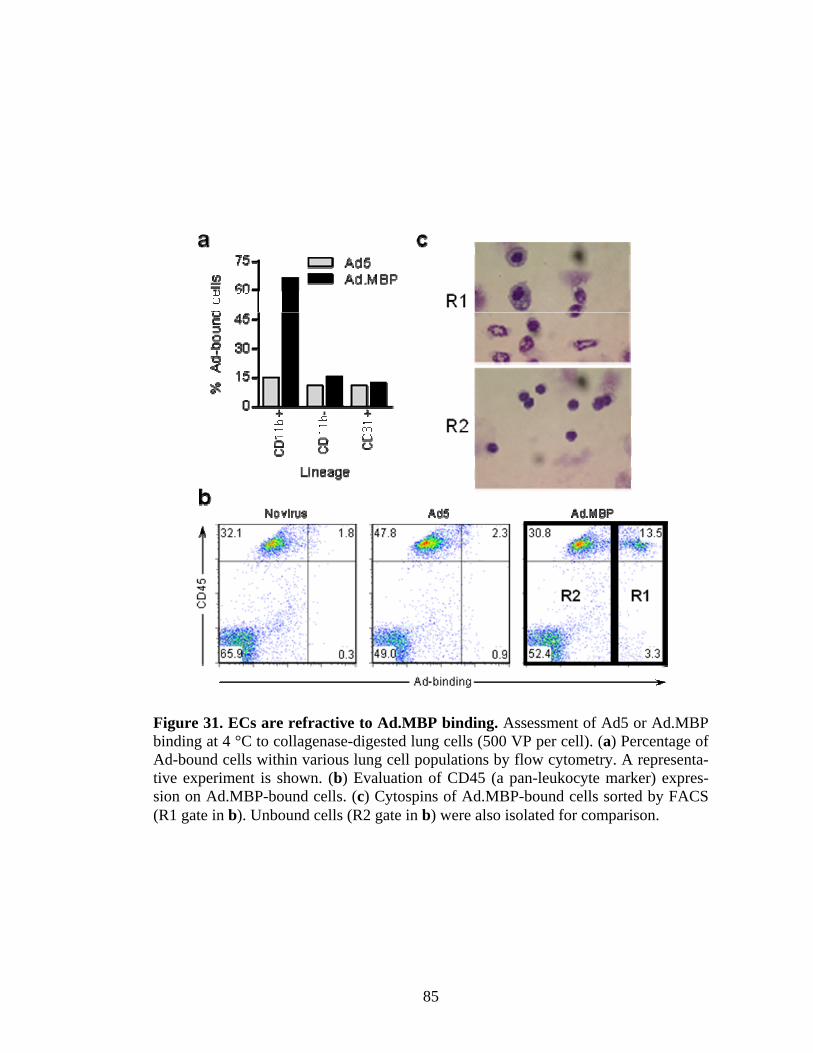
Figure 31. ECs are refractive to Ad.MBP binding. Assessment of Ad5 or Ad.MBP binding at 4 °C to collagenase-digested lung cells (500 VP per cell). (a) Percentage of Ad-bound cells within various lung cell populations by flow cytometry. A representa-tive experiment is shown. (b) Evaluation of CD45 (a pan-leukocyte marker) expres-sion on Ad.MBP-bound cells. (c) Cytospins of Ad.MBP-bound cells sorted by FACS (R1 gate in b). Unbound cells (R2 gate in b) were also isolated for comparison.
85

To assess whether Ad.MBP can transduce ECs directly, two murine EC lines were
obtained for direct in vitro testing. First, an immortalized mouse cardiac endothelial cell
(MCEC) line was purchased. Ad.MBP.CMV-GFP or Ad5.CMV-GFP control vectors
were added to MCEC and gene transfer was assessed by flow cytometry 24 h later. Al-
though Ad.MBP transduced CD11b+ BMCs very effectively (Figure 21); it did not effi-
ciently transduce MCECs, even at a high MOI (Figure 32a). Conversely, Ad5 efficiently
transduced MCECs (Figure 32a). Next, a murine conditionally-immortalized pulmonary
microvascular endothelial cell line, designated MiPMVEC (a generous gift from Dr. Na-
masivayam Ambalavanan, UAB), was previously established by isolating pulmonary ECs
from H-2Kb-tsA58 transgenic mice (Immortomouse) (Frank et al., 2005; Whitehead et
al., 1993). When grown at 37 °C, the cells behave like differentiated ECs (Frank et al.,
2005; also see Figure 6 in Methods). Similar to before, Ad.MBP.CMV-GFP or
Ad5.CMV-GFP control vectors were added to MiPMVEC and gene transfer was assessed
by flow cytometry 24 h later. Ad5, but not Ad.MBP, efficiently transduced MiPMVECs
at all MOIs (Figure 32b). Together, both of these data further suggest that myeloid cell-
mediated hand-off may be required for efficient pulmonary EC transduction in vivo.
86

a b
10 100 10000
2
4
10
40
70
100Ad5AdNBP
MOI (vp/cell)
% G
FP+
MC
EC
*
***
***
10 100 10000
2
4
10
40
70
100Ad5AdNBP
MOI (vp/cell)
% G
FP+
MiP
MVE
C
**
***
***
Ad5 Ad.MBP
Ad5
Ad.MBP Ad5
Ad.MBP
Figure 32. ECs are refractive to Ad.MBP transduction. Transduction of MCEC (a) or MiPMVEC (b) with Ad5.GFP or Ad.MBP.GFP at increasing MOI. After 24 h, the percent of cells transduced were analyzed by flow cytometry. Data are means ± s.d. of a representative experiment with triplicate samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
87

DISCUSSION
The inability to achieve specific and efficient gene transfer in target cells remains
as a key barrier to systemic gene delivery approaches. This lack of specificity reduces the
effective dose accessible to target cells and compensatory dose escalation can lead to tox-
icity. Here we demonstrate, for the first time, a novel genetic capsid modification strategy
that retargets Ad to the lung microvasculature and prevents both KC uptake and hepato-
cyte transduction in the liver. These data establish an important benchmark in vector tar-
geting research, and offers an important first step in validating systemic Ad targeting to
myeloid leukocytes which may have clinical utility for treatment of inflammatory lung
diseases (Sallenave et al., 1997).
Phage Display Library Screening
Pasqualini and Ruoslahti first demonstrated that phage display library screening
could be used to identify peptides that selectively "home", or target to, specific organs in
vivo (Pasqualini and Ruoslahti, 1996). Since then, a number of reports have been pub-
lished that further validate this technique (Pasqualini et al., 1995; Arap et al., 1998;
Tamm et al., 2003). However, studies that identified peptides specific for the lung vascu-
lature were of particular interest to us (Rajotte et al., 1998; Fukuda et al., 2000; Valadon
et al., 2006; Work et al., 2006). While there have been many such peptides identified, a
few of these have been further evaluated in the context of vector retargeting (e.g. AAV,
Ad), and even fewer have demonstrated enhancement in lung tissue targeting in vivo
88

(Work et al., 2006). Thus, we devised a novel lung targeting strategy by attempting to
target the large reservoir of leukocytes within the lung microvasculature rather than the
vasculature itself. These cell types were attractive targets for three reasons: (1) this large
reservoir of leukocytes, referred to as the marginated pool (Doerschuk et al., 1990), ex-
ceeds the circulating pool of leukocytes by up to an estimated threefold due to their net
accumulation within the lung microvascular capillary bed; (2) leukocytes directly con-
tribute to the pathogenesis of many debilitating lung diseases; and (3) in the context of
Ad vector targeting, sequestration of our retargeted Ad by circulating cell types may in-
crease vector bioavailability.
Although the marginated pool is composed of all types of leukocytes, neutrophils,
and to a lesser extent monocytes, comprise the largest fraction of these cells. Therefore,
our phage panning strategy aimed to identify peptides specific for myeloid cell types both
in vitro and in vivo. Interestingly, a single peptide, with the sequence WTLDRGY,
emerged after the third and the final round of in vitro panning and perhaps even moreso,
the same peptide was identified in mutliple rounds of in vivo panning. Extensive charac-
terization of this myeloid-binding peptide, or MBP, demonstrate that the myeloid-binding
specificity of the MBP sequence is retained in the context of three independent platforms:
phage display, recombinant peptide, and adenovirus. This further validates the use of
phage display library screening as a means to identify novel targeting moieties for spe-
cific cell subsets that may be directly incorporated into viral vectors such as Ad.
89

Reduction in Ad5 Hepatocyte Gene Transfer
Historically, specific and efficient systemic targeting of Ad5 has been difficult to
achieve. Retargeting strategies so far have mostly focused on manipulation of the viral
capsid, particularly ligand insertion, domain deletion, or pseudotyping of the fiber, pIX,
and hexon proteins (Krasnykh et al., 2000; Waehler et al., 2007). The most successful
transductional-targeting of Ad to the pulmonary system was obtained nearly ten years ago
using bispecific adapters that redirected Ad5 tropism to ACE expressed on pulmonary
endothelium (Reynolds et al., 2000). This was the first study to report enhanced targeting
of Ad5 to a specific organ after systemic delivery. However, this vector-targeting strategy
failed to reduce liver accumulation (i.e. KC sequestration) of Ad5 particles (Reynolds et
al., 2000), which causes significant hepatotoxicity (Lieber et al., 1997; Descamps and
Benihoud, 2009), suggesting the need for further improvement in liver untargeting ef-
forts.
To improve upon these studies, we conceived a novel targeting approach to local-
ize Ad to the lung by way of the marginated pool of leukocytes, rather than the pulmo-
nary microvasculature itself. After its identification and validation in vitro, the MBP pep-
tide was genetically incorporated into a knob-deleted Ad fiber platform previously de-
scribed by our group (Krasnkh et al., 2001; Noureddini et al., 2006; Hedley et al., 2006).
Although genetic modifications can be more technically challenging to construct, de-
pending on the targeting ligand of interest and capsid locale chosen, they afford advan-
tages over the two-component strategies based on adapters. These include the fact that
genetically modified or “single-component” agents allow for more consistent vector pro-
90

duction and are also less prone to in vivo instability that is associated with two-
component adapter systems (Waehler et al., 2007).
Despite these potential shortcomings, the two-component adapter targeting ap-
proach exploited by Reynolds et al. realized an almost 20-fold increase in lung:liver gene
expression over untargeted Ad5 vector (Reynolds et al., 2000). However, the incorpora-
tion of the MBP targeting ligand into our knob-deleted fiber Ad resulted in a considerably
higher 165,000-fold enhancement in the lung:liver ratio, compared to Ad5. While both
studies demonstrate considerable lung targeting in comparison to Ad5, adapter targeting
realized only 83% reduction in liver transgene expression (Reynolds et al., 2000) com-
pared to the 99.9% reduction achieved with Ad.MBP. In this regard, Reynolds et al. used
an adapter targeting approach coupled to a transcriptional-targeting strategy in which an
EC specific promoter (FLT-1) was utilized to silence transgene expression in non-
pulmonary tissues such as the liver (Reynolds et al., 2001). This was the first such study
to demonstrate that both transductional- (albeit still a two-component adapter system) and
transcriptional-targeting strategies acted synergistically to both improve lung and reduce
liver transgene expression (Reynolds et al., 2001). Indeed, this dual-targeting approach
(Reynolds et al., 2001) revealed a 12,000-fold improvement in liver untargeting, com-
pared to their former study (Reynolds et al., 2000). In this regard, similar transcriptional-
targeting strategies may enhance the specificity of Ad.MBP gene expression to either
myeloid or endothelial subsets in the lung, depending on the therapeutic endpoints de-
sired. Nevertheless, true transductional targeting specificity should preclude the need for
these combined approaches and reduce viral particle dissemination to other tissues.
91

Abrogation of Acute Liver Sequestration of Ad5
Beyond enhanced lung gene targeting, our study also demonstrates that viral par-
ticle localization was efficiently restricted to the lung. In fact, this is the first Ad-targeting
study, to our knowledge, to report complete abrogation of liver tropism (i.e. KC uptake
and hepatocyte transduction) in addition to efficient targeting of gene transfer to a spe-
cific organ (i.e. the lung). The data herein strongly support a mechanism of first-pass lung
sequestration of myeloid-targeted Ad, as direct injection of Ad.MBP into the left ventri-
cle (an injection site distal to the pulmonary circulation) partially restored accumulation
of virions in the liver. However, a substantial amount of particles were still localized to
the lung shortly after injection. This finding is interesting, since previous studies have
implicated the fiber knob domain as an important component for KC sequestration (Sha-
yakhmetov et al., 2005). While our data does not directly demonstrate mitigation of KC
interaction with Ad particles, our study demonstrates that KC sequestration, and likely
hepatotoxicity and the acute immune response typically associated with KC uptake (Lie-
ber et al., 1997; Descamps and Benihoud, 2009), can be minimized through more effi-
cient binding of Ad to other tissues, such as the lung. Thus, liver untargeting via addi-
tional capsid modifications may not be necessary, as long as the route of injection and the
efficiency of targeting or sequestration supercede viral access to the liver.
Leukocyte-mediated Hand-off of Ad.MBP to Pulmonary Vasculature
Finally, our data suggest the marginated pool plays an integral role in the efficient
and specific targeting of Ad.MBP particle binding and gene transfer to the lung. The
marginated pool of neutrophils, monocytes and lymphocytes (Doerschuk et al., 1990)
92

represents the net accumulation of circulating leukocytes from the systemic pool that be-
come transiently sequestered within the much smaller alveolar capillary network (Wiggs
et al., 1994; Kuebler and Goetz, 2002). The marginated pool is in constant exchange with
the circulating pool; these marginated leukocytes adjust their shape to navigate through
the pulmonary microvasculature and eventually reenter the systemic circulation. Thus, we
hypothesize that Ad.MBP virions are first sequestered on leukocytes from the marginated
pool for subsequent viral “hand-off” and gene transfer to pulmonary ECs. Indeed, similar
“hand-off” or “hitchhiking” mechanisms have been described for other viruses in vivo
(Cole et al., 2005). Since we have yet to identify the molecule(s) that interact with the
MBP peptide, we cannot be certain that MBP-binding is indeed specific to myeloid cells
and not other cell types, including pulmonary EC subsets. However, lung cell binding and
MiPMVEC transduction experiments demonstrate the inability of Ad.MBP to both bind
and infect pulmonary ECs directly. Furthermore, the ability of Ad.MBP to bind myeloid
lung cell types ex vivo and the disproportionate number of Ad.MBP transduced alveolar
capillaries compared to larger pulmonary vessels in vivo further supports that Ad.MBP
virions are first sequestered by the marginated pool for subsequent viral “hand-off” and
gene transfer to pulmonary ECs. We cannot rule out the potential that factors in vivo may
contribute to MBP-binding to pulmonary ECs, such as the case with FX-mediated Ad5
transduction of hepatocytes (Waddington et al., 2008). Yet, based on the design of our in
vivo phage panning strategy, which required extensive incubation of target cells with
phage both in vivo and ex vivo, recovery of myeloid cell-bound phage was biased for non-
internalizing clones. This phenomenon may be important in that leukocyte-bound virions
may be in such close contact with alveolar ECs that penton (i.e. RGD)-inegrin interac-
93

tions can occur, allowing virus to be efficiently internalized directly by pulmonary ECs,
bypassing the need for the initial attachment step of cell transduction. RGD-deleted
Ad.MBP and/or infusion of Ad.MBP into integrin knockout mice may validate this hy-
pothesis. Additional studies will be necessary to carefully elucidate the apparent dual-
tropic nature of our targeting approach and whether pulmonary EC gene transfer is truly a
myeloid-dependent process in vivo. Nevertheless, our approach results in efficient and
specific transduction of lung cells indicating this strategy should be evaluated for thera-
peutic delivery in the context of pulmonary disease.
Summary and Future Directions
Overall, these data establish an important benchmark in vector retargeting re-
search. Our innovative myeloid-binding strategy demonstrates that efficient targeting of
appropriate cell types may preclude the need for additional liver untargeting strategies,
such as hexon mutations that abrogate FX-binding (Alba et al., 2009 and 2010) or KC
uptake (Khare et al., 2011). Nevertheless, evaluation of hexon-modified Ad.MBP vectors
may reveal absolute liver untargeting and even further enhance the efficacy of gene ex-
pression in the pulmonary system. In fact, the hexon mutation strategies described to date
have not yet been extensively evaluated in conjunction with vector targeting strategies in
vivo. Additionally, while deletion of the knob domain may or may not directly affect KC
uptake of Ad.MBP virions, knob-deletion strategies may help dampen anti-vector immu-
nity in future clinical applications (Molinier-Frenkel, 2003), as previous knob-deleted
Ads have reported decreased immunogenicity in mouse models (Myhre et al., 2007).
However, the hexon protein, the most abundant Ad capsid protein, is much more immu-
94

nogenic (Sumida et al., 2005), and similar to knob-deleted Ads, hexon substituted Ads
demonstrate a marked decrease in host anti-vector immunity (Roberts et al., 2006). Thus,
all encompassing hexon modification strategies that ablate liver tropism and anti-vector
immunity could be evaluated in conjunction with the Ad.MBP vector to further improve
the vector safety profile and improve targeting of Ad.MBP to leukocytes within non-
pulmonary tissues.
Historically, inefficient gene delivery to target cells has been a major limitation to
the gene therapy field. The vector-targeting studies described in this body of work are an
important first step in validating systemic Ad-targeting strategies of myeloid cell types as
a more efficient approach for gene therapy of acute pulmonary diseases such as ARDS
and even chronic conditions, such as CF, COPD, idiopathic pulmonary fibrosis (IPF) and
cancer. In regard to moving closer to treating these chronic conditions, additional strate-
gies must be employed to ensure long-term gene expression. Although still somewhat
technically challenging to produce, MBP-outfitted HDAd vectors may provide an avenue
to achieve such long-term gene therapy of chronic lung disease, as they have been re-
ported to significantly reduce anti-Ad cellular and humoral immunity (Morral et al.,
1999; Balague et al., 2000). Based on our principle interest in evaluating targeting, the
difficulties of HDAd production, and the systems available in our lab, we used a first
generation Ad vector, which is known to incite an immune response (Liber et al., 1997).
We did not directly evaluate any acute responses following infusion of first generation
Ad.MBP, as these observations are beyond the scope of our targeting aims, and true eval-
uation of Ad-induced innate and acquired immunity should be performed with a clinically
relevant vector platform.
95

Alternative vector platforms may achieve similar endpoints. LV vectors stably in-
tegrate their genomes into both dividing and non-dividing cell types, providing non-
episomal (i.e. AAV) long-term expression until the cell is eliminated. This property
makes LV ideal candidates for chronic pulmonary therapy. With the fidelity the MBP
peptide has displayed in the variety of platforms already tested (i.e. phage, peptide, Ad),
and with the growing development of LV-targeting strategies, we are beginning to evalu-
ate the feasibility of LV.MBP-mediated gene delivery to the pulmonary vasculature.
Although leukocytes were transduced less frequently than ECs in the lung, the
dual-tropic nature of Ad.MBP provides the opportunity to restrict gene expression to ei-
ther leukocyte or endothelial cell types through transcriptional-control elements, which
may be useful for targeting therapies for different pulmonary diseases. Lastly, since leu-
kocytes naturally home to and infiltrate tissues in response to inflammatory signals, we
propose that our unique strategy may allow targeting of therapeutics directly to non-
pulmonary sites of inflammation, utilizing a cell vehicle approach (Roth et al., 2008) and
local vascular administration or additional capsid modifications that would allow for
evaluation in non-pulmonary inflammatory disease.
In closing, we have isolated and characterized a seven amino peptide that specifi-
cally binds to myeloid leukocytes. Its myeloid-binding specificity is conferred through
only five of the seven residues and its specificity is retained after incorporation into a
number of vector platforms. Furthermore, we show for the first time, that incorporation of
only this seven amino acid peptide results in near complete retargeting of Ad5 through
efficient sequestration of virions within the lung microvasculature by what we hypothe-
size is leukocyte-mediated “hand-off” to the endothelium.
96

97
LIST OF REFERENCES
1. Al-Dosari MS, Gao X. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 2009;11:671-81.
2. Alba R, Bosch A, Chillon M. Gutless adenovirus: last-generation adenovirus for gene therapy. Gene Ther. 2005;12:S18-27.
3. Alba R, Bradshaw AC, Parker AL, Bhella D, Waddington SN, Nicklin SA, van Rooijen N, Custers J, Goudsmit J, Barouch DH, McVey JH, Baker AH. Identifica-tion of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: ef-fect of mutagenesis on FX interactions and gene transfer. Blood. 2009;114:965-71.
4. Alba R, Bradshaw AC, Coughlan L, Denby L, McDonald RA, Waddington SN, Buckley SM, Greig JA, Parker AL, Miller AM, Wang H, Lieber A, van Rooi-jen N,McVey JH, Nicklin SA, Baker AH. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood. 2010;116:2656-64.
5. Albelda SM, Wiewrodt R, Zuckerman JB. Gene therapy for lung disease: hype or hope? Ann Intern Med. 2000;132:649-660.
6. Alemany R, Suzuki K, Curiel DT. Blood clearance rates of adenovirus type 5 in mice. J Gen Virol. 2000;81:2605-9.
7. Alemany R, Curiel DT. CAR-binding ablation does not change biodistribution and toxicity of adenoviral vectors. Gene Ther. 2001;8:1347-53. Amitani R, Wilson R, Rutman A, et al. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am J Respir Cell Mol Biol. 1991;4:26-32.
8. Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J, Schneider IC, Münch RC, Petznek H, Kontermann RE, Koehl U, Johnston IC, Keinänen K, Müller UC, Hohenadl C, Monyer H, Cichutek K, Buchholz CJ. Specific gene transfer to neu-rons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat Methods. 2010;7:929-35.
9. Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377-80.

98
10. Arap W, Kolonin MG, Trepel M, Lahdenranta J, Cardó-Vila M, Giordano RJ, Mintz PJ, Ardelt PU, Yao VJ, Vidal CI, Chen L, Flamm A, Valtanen H, Weavind LM, Hicks ME, Pollock RE, Botz GH, Bucana CD, Koivunen E, Cahill D, Troncoso P, Baggerly KA, Pentz RD, Do KA, Logothetis CJ, Pasqualini R. Steps toward mapping the human vasculature by phage display. Nat Med. 2002;8:121-7.
11. Baker AH. Targeting AAV vectors. Mol Ther. 2003;7:433-4.
12. Baker AH, Mcvey JH, Waddington SN, Di Paolo NC, Shayakhmetov DM. The in-fluence of blood on in vivo adenovirus bio-distribution and transduction. Mol Ther. 2007;15:1410-6.
13. Balagué C, Zhou J, Dai Y, Alemany R, Josephs SF, Andreason G, Hariharan M, Sethi E, Prokopenko E, Jan HY, Lou YC, Hubert-Leslie D, Ruiz L, Zhang WW. Sustained high-level expression of full-length human factor VIII and restoration of clotting activity in hemophilic mice using a minimal adenovirus vector. Blood. 2000;95:820-8.
14. Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231-9.
15. Barnes PJ. Inhaled glucocorticoids for asthma. N Engl J Med. 1995;332:868-875.
16. Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269-280.
17. Barnes PJ. Advances in chronic obstructive pulmonary disease. Trans Med Soc Lond. 2002;119:41-51.
18. Barnes PJ. Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol Allergy Clin North Am. 2005;25:451-68.
19. Beeh KM, Beier J. Handle with care: targeting neutrophils in chronic obstructive pulmonary disease and severe asthma? Clin Exp Allergy. 2006;36:142-157.
20. Belousova N, Krendelchtchikova V, Curiel DT, Krasnykh V. Modulation of adeno-virus vector tropism via incorporation of polypeptide ligands into the fiber protein. J Virol. 2002;76:8621-31.
21. Belousova N, Korokhov N, Krendelshchikova V, Simonenko V, Mikheeva G, Triozzi PL, Aldrich WA, Banerjee PT, Gillies SD, Curiel DT, Krasnykh V. Geneti-

99
cally targeted adenovirus vector directed to CD40-expressing cells. J Virol. 2003;77:11367-77.
22. Benihoud K, Salone B, Esselin S, Opolon P, Poli V, Di Giovine M, Perricaudet M, Saggio I. The role of IL-6 in the inflammatory and humoral response to adenovi-ral vectors. J Gene Med. 2000;2:194-203.
23. Benihoud K, Esselin S, Descamps D, Jullienne B, Salone B, Bobé P, Bonardelle D, Connault E, Opolon P, Saggio I, Perricaudet M. Respective roles of TNF-alpha and IL-6 in the immune response-elicited by adenovirus-mediated gene transfer in mice. Gene Ther. 2007;14:533-44.
24. Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320-3.
25. Birrer P, McElvaney NG, Rudeberg A, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;150:207-213.
26. Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA, Böhm M, Nowrouzi A, Ball CR, Glimm H, Naundorf S, Kühlcke K, Blasczyk R, Kondratenko I, Maródi L, Orange JS, von Kalle C, Klein C. Stem-cell gene ther-apy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363:1918-27.
27. Brenner MK, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immu-nol. 2010;22:251-7.
28. Bristol JA, Gallo-Penn A, Andrews J, Idamakanti N, Kaleko M, Connelly S. Ade-novirus-mediated factor VIII gene expression results in attenuated anti-factor VIII-specific immunity in hemophilia A mice compared with factor VIII protein infu-sion. Hum Gene Ther. 2001;12:1651-61.
29. Brunetti-Pierri N, Ng P. Progress towards liver and lung-directed gene therapy with helper-dependent adenoviral vectors. Curr Gene Ther. 2009;9:329-40.
30. Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A. 1993;90:8033-7.
31. Campos SK, Barry MA. Comparison of adenovirus fiber, protein IX, and hexon capsomeres as scaffolds for vector purification and cell targeting. Virology. 2006;349:453-62.

100
32. Caplen NJ, Alton EW, Middleton PG, et al. Liposome-mediated CFTR gene trans-fer to the nasal epithelium of patients with cystic fibrosis. Nat Med. 1995;1:39-46.
33. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, Bousso P, Deist FL, Fischer A. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669-72.
34. Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Es-cherichia coli. J Virol. 1996;70:4805-10.
35. Chirmule N, Hughes JV, Gao GP, Raper SE, Wilson JM. Role of E4 in eliciting CD4 T-cell and B-cell responses to adenovirus vectors delivered to murine and nonhuman primate lungs. J Virol. 1998;72:6138-45.
36. Chung KF, Caramori G, Adcock IM. Inhaled corticosteroids as combination therapy with beta-adrenergic agonists in airways disease: present and future. Eur J Clin Pharmacol. 2009;65:853-871.
37. Cole C, Qiao J, Kottke T, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11:1073-1081.
38. Conese M, Copreni E, Di Gioia S, De Rinaldis P, Fumarulo R. Neutrophil recruit-ment and airway epithelial cell involvement in chronic cystic fibrosis lung disease. J Cyst Fibros. 2003;2:129-135.
39. Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science. 1995;269:1583-5.
40. Cotter MJ, Zaiss AK, Muruve DA. Neutrophils interact with adenovirus vectors via Fc receptors and complement receptor 1. J Virol. 2005;79:14622-31.
41. Coyne CB, Kelly MM, Boucher RC, Johnson LG. Enhanced epithelial gene transfer by modulation of tight junctions with sodium caprate. Am J Respir Cell Mol Biol. 2000;23:602-9.
42. Cronin J, Zhang XY, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr Gene Ther. 2005 Aug;5(4):387-98.
43. Danthinne X, Imperiale MJ. Production of first generation adenovirus vectors: a review. Gene Ther. 2000;7:1707-14.

101
44. de Bueger M, Bakker A, Van Rood JJ, Van der Woude F, Goulmy E. Tissue distri-bution of human minor histocompatibility antigens. Ubiquitous versus restricted tis-sue distribution indicates heterogeneity among human cytotoxic T lymphocyte-defined non-MHC antigens. J Immunol. 1992;149:1788-94.
45. de Jong RN, van der Vliet PC, Brenkman AB. Adenovirus DNA replication: protein priming, jumping back and the role of the DNA binding protein DBP. Curr Top Mi-crobiol Immunol. 2003;272:187-211.
46. DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2: genetic aspects of al-pha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004;59:259-64.
47. Descamps D, Benihoud K. Two key challenges for effective adenovirus-mediated liver gene therapy: innate immune responses and hepatocyte-specific transduction. Curr Gene Ther. 2009;9:115-27.
48. Di Paolo NC, Kalyuzhniy O, Shayakhmetov DM. Fiber shaft-chimeric adenovirus vectors lacking the KKTK motif efficiently infect liver cells in vivo. J Virol. 2007;81:12249-59.
49. Di Paolo NC, Shayakhmetov DM. Adenovirus de-targeting from the liver. Curr Opin Mol Ther. 2009;11:523-31.
50. Di Paolo NC, van Rooijen N, Shayakhmetov DM. Redundant and synergistic mechanisms control the sequestration of blood-born adenovirus in the liver. Mol Ther. 2009;17:675-84.
51. Doerschuk CM, Allard MF, Martin BA, MacKenzie A, Autor AP, Hogg JC. Margi-nated pool of neutrophils in rabbit lungs. J Appl Physiol. 1987;63:1806-1815.
52. Doerschuk CM, Downey GP, Doherty DE, et al. Leukocyte and platelet margina-tion within microvasculature of rabbit lungs. J Appl Physiol. 1990;68:1956-1961.
53. Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241-250.
54. Douglas JT, Miller CR, Kim M, Dmitriev I, Mikheeva G, Krasnykh V, Curiel DT. A system for the propagation of adenoviral vectors with genetically modified recep-tor specificities. Nat Biotechnol. 1999;17:470-5.
55. Downey GP, Doherty DE, Schwab B, 3rd, Elson EL, Henson PM, Worthen GS. Re-tention of leukocytes in capillaries: role of cell size and deformability. J Appl Physiol. 1990;69:1767-1778.

102
56. Duan D, Yue Y, Yan Z, McCray PB Jr, Engelhardt JF. Polarity influences the effi-ciency of recombinant adenoassociated virus infection in differentiated airway epi-thelia. Hum Gene Ther. 1998;9:2761-76.
57. Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin In-vest. 2000;105:1573-87.
58. Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4:383-91.
59. Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene therapy clinical trials worldwide 1989-2004-an overview. J Gene Med. 2004;6:597-602.
60. Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007--an update. J Gene Med. 2007;9:833-42.
61. Elkon KB, Liu CC, Gall JG, Trevejo J, Marino MW, Abrahamsen KA, Song X, Zhou JL, Old LJ, Crystal RG, Falck-Pedersen E. Tumor necrosis factor alpha plays a central role in immune-mediated clearance of adenoviral vectors. Proc Natl Acad Sci U S A. 1997;94:9814-9.
62. Engelhardt JF, Litzky L, Wilson JM. Prolonged transgene expression in cotton rat lung with recombinant adenoviruses defective in E2a. Hum Gene Ther. 1994;5:1217-29.
63. Everts M, Kim-Park SA, Preuss MA, Passineau MJ, Glasgow JN, Pereboev AV, Mahasreshti PJ, Grizzle WE, Reynolds PN, Curiel DT. Selective induction of tumor-associated antigens in murine pulmonary vasculature using double-targeted adenoviral vectors. Gene Ther. 2005;12:1042-8.
64. Falk JA, Minai OA, Mosenifar Z. Inhaled and systemic corticosteroids in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:506-512.
65. Fanta CH. Asthma. N Engl J Med. 2009;360:1002-1014.
66. Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy of metabolic diseases. J Inherit Metab Dis. 2006;29:409-12.
67. Fechner H, Haack A, Wang H, Wang X, Eizema K, Pauschinger M, Schoemaker R, Veghel R, Houtsmuller A, Schultheiss HP, Lamers J, Poller W. Expression of coxsackie adenovirus receptor and alphav-integrin does not correlate with ade-novector targeting in vivo indicating anatomical vector barriers. Gene Ther. 1999;6:1520-35.

103
68. Flotte TR, Afione SA, Conrad C, McGrath SA, Solow R, Oka H, Zeitlin PL, Guggino WB, Carter BJ. Stable in vivo expression of the cystic fibrosis trans-membrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci U S A. 1993;90:10613-7.
69. Flotte TR, Ng P, Dylla DE, McCray PB Jr, Wang G, Kolls JK, Hu J. Viral vector-mediated and cell-based therapies for treatment of cystic fibrosis. Mol Ther. 2007;15:229-41.
70. Follenzi A, Sabatino G, Lombardo A, Boccaccio C, Naldini L. Efficient gene deliv-ery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2002;13:243-60.
71. Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE, de Caestecker MP. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res. 2005;97:496-504.
72. Frecha C, Szécsi J, Cosset FL, Verhoeyen E. Strategies for targeting lentiviral vec-tors. Curr Gene Ther. 2008;8:449-60.
73. Friedmann T. Progress toward human gene therapy. Science. 1989;244:1275-81.
74. Fukuda MN, Ohyama C, Lowitz K, Matsuo O, Pasqualini R, Ruoslahti E, Fukuda M. A peptide mimic of E-selectin ligand inhibits sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res. 2000 Jan;60:450-6.
75. Funke S, Maisner A, Mühlebach MD, Koehl U, Grez M, Cattaneo R, Cichutek K, Buchholz CJ. Targeted cell entry of lentiviral vectors. Mol Ther. 2008;16:1427-36.
76. Gaggar A, Li Y, Weathington N, et al. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am J Physiol Lung Cell Mol Physiol. 2007;293:L96-L104.
77. Gahéry-Ségard H, Juillard V, Gaston J, Lengagne R, Pavirani A, Boulanger P, Guillet JG. Humoral immune response to the capsid components of recombinant adenoviruses: routes of immunization modulate virus-induced Ig subclass shifts. Eur J Immunol. 1997;27:653-9.
78. Gallagher DC, Parikh SM, Balonov K, et al. Circulating angiopoietin 2 correlates with mortality in a surgical population with acute lung injury/adult respiratory dis-tress syndrome. Shock. 2008;29:656-661.
79. Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. Latent species C adenoviruses in human tonsil tissues. J Virol. 2009;83:2417-28.

104
80. Gill DR, Southern KW, Mofford KA, et al. A placebo-controlled study of liposome-mediated gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 1997;4:199-209.
81. Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59-74.
82. Greber UF, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75:477-86.
83. Greber UF, Webster P, Weber J, Helenius A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996;15:1766-77.
84. Grubb BR, Pickles RJ, Ye H, Yankaskas JR, Vick RN, Engelhardt JF, Wilson JM, Johnson LG, Boucher RC. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and humans. Nature. 1994;371:802-6.
85. Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R,Dupuis-Girod S, Fischer A, Davies EG, Kuis W, Leiva L, Cavazzana-Calvo M. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185-93.
86. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC,Cavazzana-Calvo M, Fischer A. A serious ad-verse event after successful gene therapy for X-linked severe combined immunode-ficiency. N Engl J Med. 2003a;348:255-6.
87. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R,Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A,Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003b;302:415-9.
88. Haisma HJ, Kamps JA, Kamps GK, Plantinga JA, Rots MG, Bellu AR. Polyinos-inic acid enhances delivery of adenovirus vectors in vivo by preventing sequestra-tion in liver macrophages. J Gen Virol. 2008;89:1097-105.
89. Haisma HJ, Boesjes M, Beerens AM, van der Strate BW, Curiel DT, Plüddemann A, Gordon S, Bellu AR. Scavenger receptor A: a new route for adenovirus 5. Mol Pharm. 2009;6:366-74.

105
90. Halbert CL, Alexander IE, Wolgamot GM, Miller AD. Adeno-associated virus vec-tors transduce primary cells much less efficiently than immortalized cells. J Vi-rol. 1995;69:1473-9.
91. Halbert CL, Standaert TA, Aitken ML, Alexander IE, Russell DW, Miller AD. Transduction by adeno-associated virus vectors in the rabbit airway: efficiency, per-sistence, and readministration. J Virol. 1997;71:5932-41.
92. Halbert CL, Standaert TA, Wilson CB, Miller AD. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosup-pression during the initial exposure. J Virol. 1998;72:9795-805.
93. Halbert CL, Allen JM, Miller AD. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J Virol. 2001;75:6615-24.
94. Harari OA, Wickham TJ, Stocker CJ, Kovesdi I, Segal DM, Huehns TY, Sarraf C, Haskard DO. Targeting an adenoviral gene vector to cytokine-activated vascular endothelium via E-selectin. Gene Ther. 1999;6:801-7.
95. Hartl D, Latzin P, Hordijk P, et al. Cleavage of CXCR1 on neutrophils disables bac-terial killing in cystic fibrosis lung disease. Nat Med. 2007;13:1423-1430.
96. Hedley SJ, Auf der Maur A, Hohn S, Escher D, Barberis A, Glasgow JN, Douglas JT, Korokhov N, Curiel DT. An adenovirus vector with a chimeric fiber incorporat-ing stabilized single chain antibody achieves targeted gene delivery. Gene Ther. 2006;13:88-94.
97. Hirayama S, Sato M, Loisel-Meyer S, Liu M, Yeung JC, Wagnetz D, Cypel M, Zehong G, Medin JA, Keshavjee S. Local Long-term Expression of Lentivirally Delivered IL-10 in the Lung Attenuates Obliteration of Intrapulmonary Allograft Airways. Hum Gene Ther. 2011. [Epub ahead of print]
98. Huard J, Lochmüller H, Acsadi G, Jani A, Massie B, Karpati G. The route of ad-ministration is a major determinant of the transduction efficiency of rat tissues by adenoviral recombinants. Gene Ther. 1995;2:107-15.
99. Izumi M, Kawakami Y, Glasgow JN, Belousova N, Everts M, Kim-Park S, Yamamoto S, Wang M, Le LP, Reynolds PN, Curiel DT. In vivo analysis of a genetically modified adenoviral vector targeted to human CD40 using a novel tran-sient transgenic model. J Gene Med. 2005;7:1517-25.
100. Jakubczak JL, Rollence ML, Stewart DA, Jafari JD, Von Seggern DJ, Nemerow GR, Stevenson SC, Hallenbeck PL. Adenovirus type 5 viral particles pseudotyped with mutagenized fiber proteins show diminished infectivity of coxsackie B-adenovirus receptor-bearing cells. J Virol. 2001;75:2972-81.

106
101. Johnson LG, Olsen JC, Naldini L, Boucher RC. Pseudotyped human lentiviral vec-tor-mediated gene transfer to airway epithelia in vivo. Gene Ther. 2000;7:568-74.
102. Johnson LG, Vanhook MK, Coyne CB, Haykal-Coates N, Gavett SH. Safety and efficiency of modulating paracellular permeability to enhance airway epithelial gene transfer in vivo. Hum Gene Ther. 2003;14:729-47.
103. Juillard V, Villefroy P, Godfrin D, Pavirani A, Venet A, Guillet JG. Long-term hu-moral and cellular immunity induced by a single immunization with replication-defective adenovirus recombinant vector. Eur J Immunol. 1995;25:3467-73.
104. Kalyuzhniy O, Di Paolo NC, Silvestry M, Hofherr SE, Barry MA, Stewart PL, Shayakhmetov DM. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci U S A. 2008;105:5483-8.
105. Karmpaliotis D, Kosmidou I, Ingenito EP, et al. Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L585-595.
106. Kay MA, Graham F, Leland F, Woo SL. Therapeutic serum concentrations of hu-man alpha-1-antitrypsin after adenoviral-mediated gene transfer into mouse hepato-cytes. Hepatology. 1995;21:815-819.
107. Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33-40.
108. Khare R, May SM, Vetrini F, Weaver EA, Palmer D, Rosewell A, Grove N, Ng P, Barry MA. Generation of a Kupffer Cell-evading Adenovirus for Systemic and Liver-directed Gene Transfer. Mol Ther. 2011. [Epub ahead of print]
109. Kiang A, Hartman ZC, Everett RS, Serra D, Jiang H, Frank MM, Amalfitano A. Multiple innate inflammatory responses induced after systemic adenovirus vector delivery depend on a functional complement system. Mol Ther. 2006;14:588-98.
110. Kinsey BM, Densmore CL, Orson FM. Non-viral gene delivery to the lungs. Curr Gene Ther. 2005;5,181-194.
111. Kohn DB. Gene therapy for childhood immunological diseases. Bone Marrow Transplant. 2008;41:199-205.
112. Koizumi N, Mizuguchi H, Sakurai F, Yamaguchi T, Watanabe Y, Hayakawa T. Reduction of natural adenovirus tropism to mouse liver by fiber-shaft exchange in combination with both CAR- and alphav integrin-binding ablation. J Vi-rol. 2003;77:13062-72.

107
113. Kolb M, Martin G, Medina M, Ask K, Gauldie J. Gene therapy for pulmonary dis-eases. Chest. 2006;130:879-884.
114. Konstan MW, Davis PB, Wagener JS, et al. Compacted DNA nanoparticles admin-istered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate par-tial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther. 2004;15:1255-1269.
115. Korokhov N, Mikheeva G, Krendelshchikov A, Belousova N, Simonenko V, Krendelshchikova V, Pereboev A, Kotov A, Kotova O, Triozzi PL, Aldrich WA, Douglas JT, Lo KM, Banerjee PT, Gillies SD, Curiel DT, Krasnykh V. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J Virol. 2003;77:12931-40.
116. Kotin RM, Linden RM, Berns KI. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992;11:5071-8.
117. Krasnykh VN, Douglas JT, van Beusechem VW. Genetic targeting of adenoviral vectors. Mol Ther. 2000;1:391-405.
118. Krasnykh V, Belousova N, Korokhov N, Mikheeva G, Curiel DT. Genetic targeting of an adenovirus vector via replacement of the fiber protein with the phage T4 fi-britin. J Virol. 2001;75:4176-83.
119. Kreppel F, Kochanek S. Modification of adenovirus gene transfer vectors with syn-thetic polymers: a scientific review and technical guide. Mol Ther. 2008;16:16-29.
120. Kritz AB, Nicol CG, Dishart KL, Nelson R, Holbeck S, Von Seggern DJ, Work LM, McVey JH, Nicklin SA, Baker AH. Adenovirus 5 fibers mutated at the puta-tive HSPG-binding site show restricted retargeting with targeting peptides in the HI loop. Mol Ther. 2007;15:741-9.
121. Kuebler WM, Goetz AE. The marginated pool. Eur Surg Res. 2002;34:92-100. 122. Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS. Robbins and Cotran
pathologic basis of disease (ed 7th). Philadelphia: Elsevier Saunders; 2005.
123. Leissner P, Legrand V, Schlesinger Y, Hadji DA, van Raaij M, Cusack S, Pavirani A, Mehtali M. Influence of adenoviral fiber mutations on viral encapsidation, infec-tivity and in vivo tropism. Gene Ther. 2001;8:49-57.
124. Leopold PL, Kreitzer G, Miyazawa N, Rempel S, Pfister KK, Rodriguez-Boulan E, Crystal RG. Dynein- and microtubule-mediated translocation of adenovirus sero-type 5 occurs after endosomal lysis. Hum Gene Ther. 2000;11:151-65.

108
125. Li Y, Muruve DA, Collins RG, Lee SS, Kubes P. The role of selectins and integrins in adenovirus vector-induced neutrophil recruitment to the liver. Eur J Immu-nol. 2002;32:3443-52.
126. Lieber A, He CY, Meuse L, Schowalter D, Kirillova I, Winther B, Kay MA. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J Virol. 1997;71:8798-807.
127. Limberis MP, Wilson JM. Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc Natl Acad Sci U S A. 2006;103:12993-8.
128. Liqun Wang R, McLaughlin T, Cossette T, Tang Q, Foust K, Campbell-Thompson M, Martino A, Cruz P, Loiler S, Mueller C, Flotte TR. Recombinant AAV serotype and capsid mutant comparison for pulmonary gene transfer of alpha-1-antitrypsin using invasive and noninvasive delivery. Mol Ther. 2009;17:81-7.
129. Liu F, Qi H, Huang L, Liu D. Factors controlling the efficiency of cationic lipid-mediated transfection in vivo via intravenous administration. Gene Ther. 1997;4:517-23.
130. Liu Q, Zaiss AK, Colarusso P, Patel K, Haljan G, Wickham TJ, Muruve DA. The role of capsid-endothelial interactions in the innate immune response to adenovirus vectors. Hum Gene Ther. 2003;14:627-43.
131. Liu TC, Kirn D. Gene therapy progress and prospects cancer: oncolytic viruses. Gene Ther. 2008;15:877-84.
132. Louis N, Evelegh C, Graham FL. Cloning and sequencing of the cellular-viral junc-tions from the human adenovirus type 5 transformed 293 cell line. Virology. 1997;233:423-9.
133. Lutz P, Kedinger C. Properties of the adenovirus IVa2 gene product, an effector of late-phase-dependent activation of the major late promoter. J Virol. 1996;70:1396-405.
134. Lutz P, Rosa-Calatrava M, Kedinger C. The product of the adenovirus intermediate gene IX is a transcriptional activator. J Virol. 1997;71:5102-9.
135. Machemer T, Engler H, Tsai V, Lee S, Cannon-Carlson S, Voloch M, Schluep T, Ravindran S, Vellekamp G, Brin E, Cornell D, Sutjipto S, Wen SF, Horn M,Van Rooijen N, Maneval D, Hutchins B, LaFace D. Characterization of hemodynamic events following intravascular infusion of recombinant adenovirus reveals possible solutions for mitigating cardiovascular responses. Mol Ther. 2005 Aug;12(2):254-63.

109
136. MacNee W, Tuder RM. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc. 2009;6:527-531.
137. Magnusson MK, Hong SS, Boulanger P, Lindholm L. Genetic retargeting of adeno-virus: novel strategy employing "deknobbing" of the fiber. J Virol. 2001;75:7280-9.
138. Maizel JV Jr, White DO, Scharff MD. The polypeptides of adenovirus. I. Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology. 1968;36:115-25.
139. Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, Byrnes AP. Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Mol Ther. 2006;13:108-17.
140. Mastrangeli A, Danel C, Rosenfeld MA, Stratford-Perricaudet L, Perricaudet M, Pavirani A, Lecocq JP, Crystal RG. Diversity of airway epithelial cell targets for in vivo recombinant adenovirus-mediated gene transfer. J Clin Invest. 1993;91:225-34.
141. McCray PB, Jr., Wang G, Kline JN, et al. Alveolar macrophages inhibit retrovirus-mediated gene transfer to airway epithelia. Hum Gene Ther. 1997;8:1087-1093.
142. McElvaney NG, Hubbard RC, Birrer P, Chernick MS, Caplan DB, Frank MM, Crystal RG. Aerosol alpha 1-antitrypsin treatment for cystic fibrosis. Lancet. 1991;337:392-4.
143. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428-435.
144. Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vec-tors. Science. 1999;283:682-6.
145. Mizuguchi H, Koizumi N, Hosono T, Ishii-Watabe A, Uchida E, Utoguchi N, Watanabe Y, Hayakawa T. CAR- or alphav integrin-binding ablated adenovirus vectors, but not fiber-modified vectors containing RGD peptide, do not change the systemic gene transfer properties in mice. Gene Ther. 2002;9:769-76.
146. Molinier-Frenkel V, Prevost-Blondel A, Hong SS, et al. The maturation of murine dendritic cells induced by human adenovirus is mediated by the fiber knob domain. J Biol Chem. 2003;278:37175-37182.
147. Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, Wu L, Chen IS. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through in-travenous injection. Nat Med. 2005;11:346-52.

110
148. Morral N, O'Neal W, Rice K, Leland M, Kaplan J, Piedra PA, Zhou H, Parks RJ, Velji R, Aguilar-Córdova E, Wadsworth S, Graham FL, Kochanek S, Carey KD, Beaudet AL. Administration of helper-dependent adenoviral vectors and se-quential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc Natl Acad Sci U S A. 1999;96:12816-21.
149. Moss RB, Rodman D, Spencer LT, Aitken ML, Zeitlin PL, Waltz D, Milla C, Brody AS, Clancy JP, Ramsey B, Hamblett N, Heald AE. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regula-tor gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest. 2004;125:509-21.
150. Mura M, dos Santos CC, Stewart D, Liu M. Vascular endothelial growth factor and related molecules in acute lung injury. J Appl Physiol. 2004;97:1605-1617.
151. Murakami M, Ugai H, Wang M, Belousova N, Dent P, Fisher PB, Glasgow JN, Everts M, Curiel DT. An adenoviral vector expressing human adenovirus 5 and 3 fiber proteins for targeting heterogeneous cell populations. Virology. 2010;407:196-205.
152. Muruve DA, Barnes MJ, Stillman IE, Libermann TA. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent he-patic injury in vivo. Hum Gene Ther. 1999;10:965-76.
153. Myhre S, Henning P, Granio O, Tylö AS, Nygren PA, Olofsson S, Boulanger P, Lindholm L, Hong SS. Decreased immune reactivity towards a knobless, affi-body-targeted adenovirus type 5 vector. Gene Ther. 2007;14:376-81.
154. Nakamura T, Sato K, Hamada H. Reduction of natural adenovirus tropism to the liver by both ablation of fiber-coxsackievirus and adenovirus receptor interaction and use of replaceable short fiber. J Virol. 2003;77:2512-21.
155. Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263-7.
156. Nathan C, Ding A. Nonresolving inflammation. Cell 2010;140:871-882.
157. Neschadim A, McCart JA, Keating A, Medin JA. A roadmap to safe, efficient, and stable lentivirus-mediated gene therapy with hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2007;13:1407-1416.
158. Nettelbeck DM, Miller DW, Jérôme V, Zuzarte M, Watkins SJ, Hawkins RE, Müller R, Kontermann RE. Targeting of adenovirus to endothelial cells by a bispecific single-chain diabody directed against the adenovirus fiber knob domain and human endoglin (CD105). Mol Ther. 2001;3:882-91.

111
159. Nicol CG, Graham D, Miller WH, White SJ, Smith TA, Nicklin SA, Stevenson SC, Baker AH. Effect of adenovirus serotype 5 fiber and penton modifications on in vivo tropism in rats. Mol Ther. 2004;10:344-54.
160. Noureddini SC, Curiel DT. Genetic targeting strategies for adenovirus. Mol Pharm. 2005;2:341-7.
161. Noureddini SC, Krendelshchikov A, Simonenko V, Hedley SJ, Douglas JT, Curiel DT, Korokhov N. Generation and selection of targeted adenoviruses embodying op-timized vector properties. Virus Res. 2006;116:185-95.
162. Oh YK, Kim JP, Yoon H, Kim JM, Yang JS, Kim CK. Prolonged organ retention and safety of plasmid DNA administered in polyethylenimine complexes. Gene Ther. 2001;8:1587-92.
163. Ohbayashi H. Novel neutrophil elastase inhibitors as a treatment for neutrophil-predominant inflammatory lung diseases. IDrugs. 2002;5:910-923.
164. Otake K, Ennist DL, Harrod K, Trapnell BC. Nonspecific inflammation inhibits adenovirus-mediated pulmonary gene transfer and expression independent of spe-cific acquired immune responses. Hum Gene Ther. 1998;9:2207-22.
165. Pan D, Gunther R, Duan W, Wendell S, Kaemmerer W, Kafri T, Verma IM, Whitley CB. Biodistribution and toxicity studies of VSVG-pseudotyped lentiviral vector after intravenous administration in mice with the observation of in vivo transduction of bone marrow. Mol Ther. 2002;6:19-29.
166. Parker AL, Waddington SN, Nicol CG, et al. Multiple vitamin K-dependent coagu-lation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood. 2006;108:2554-2561.
167. Parker AL, Nicklin SA, Baker AH. Interactions of adenovirus vectors with blood: implications for intravascular gene therapy applications. Curr Opin Mol Ther. 2008;10:439-48.
168. Parks RJ, Bramson JL, Wan Y, Addison CL, Graham FL. Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J Vi-rol. 1999;73:8027-34.
169. Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide li-braries. Nature. 1996;380:364-6.
170. Pasqualini R, Koivunen E, Ruoslahti E. A peptide isolated from phage display li-braries is a structural and functional mimic of an RGD-binding site on integrins. J Cell Biol. 1995;130:1189-96.

112
171. Pearson S, Jia H, Kandachi K. China approves first gene therapy. Nat Biotech-nol. 2004;22:3-4.
172. Peeters MJ, Patijn GA, Lieber A, Meuse L, Kay MA. Adenovirus-mediated hepatic gene transfer in mice: comparison of intravascular and biliary administration. Hum Gene Ther. 1996;7:1693-9.
173. Peng KW, Pham L, Ye H, Zufferey R, Trono D, Cosset FL, Russell SJ. Organ dis-tribution of gene expression after intravenous infusion of targeted and untargeted lentiviral vectors. Gene Ther. 2001;8:1456-63.
174. Perks B, Shute JK. DNA and actin bind and inhibit interleukin-8 function in cystic fibrosis sputa: in vitro effects of mucolytics. Am J Respir Crit Care Med. 2000;162:1767-1772.
175. Philipson L, Lonberg-Holm K, Pettersson U. Virus-receptor interaction in an ade-novirus system. J Virol. 1968;2:1064-75.
176. Pickles RJ, McCarty D, Matsui H, Hart PJ, Randell SH, Boucher RC. Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer. J Virol. 1998;72:6014-6023.
177. Pickles RJ, Fahrner JA, Petrella JM, Boucher RC, Bergelson JM. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J Vi-rol. 2000;74:6050-6057.
178. Pickles RJ. Physical and biological barriers to viral vector-mediated delivery of genes to the airway epithelium. Proc Am Thorac Soc. 2004;1:302-308.
179. Prill JM, Espenlaub S, Samen U, Engler T, Schmidt E, Vetrini F, Rosewell A, Grove N, Palmer D, Ng P, Kochanek S, Kreppel F. Modifications of adenovirus hexon allow for either hepatocyte detargeting or targeting with potential evasion from Kupffer cells. Mol Ther. 2011;19:83-92.
180. Qasim W, Gaspar HB, Thrasher AJ. Progress and prospects: gene therapy for inher-ited immunodeficiencies. Gene Ther. 2009;16:1285-91.
181. Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E. Molecu-lar heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Invest. 1998;102:430-7.
182. Ramsey BW. Management of pulmonary disease in patients with cystic fibrosis. N Engl J Med. 1996;335:179-188.

113
183. Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamy-lase deficient patient following adenoviral gene transfer. Mol Genet Me-tab. 2003;80:148-58.
184. Reynolds PN, Zinn KR, Gavrilyuk VD, Balyasnikova IV, Rogers BE, Buchsbaum DJ, Wang MH, Miletich DJ, Grizzle WE, Douglas JT, Danilov SM, Curiel DT. A targetable, injectable adenoviral vector for selective gene delivery to pulmonary en-dothelium in vivo. Mol Ther. 2000;2:562-78.
185. Reynolds PN, Nicklin SA, Kaliberova L, Boatman BG, Grizzle WE, Balyasnikova IV, Baker AH, Danilov SM, Curiel DT. Combined transductional and transcrip-tional targeting improves the specificity of transgene expression in vivo. Nat Bio-technol. 2001;19:838-42.
186. Reynolds PN. Gene therapy for pulmonary hypertension: prospects and challenges. Expert Opin Biol Ther. 2011;11:133-43.
187. Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, Liu J, Thorner AR, Swanson PE, Gorgone DA, Lifton MA, Lemckert AA, Holterman L,Chen B, Dilraj A, Carville A, Mansfield KG, Goudsmit J, Barouch DH. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immu-nity. Nature. 2006;441:239-43.
188. Roelvink PW, Mi Lee G, Einfeld DA, Kovesdi I, Wickham TJ. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adeno-viridae. Science. 1999;286:1568-71.
189. Rosenfeld MA, Yoshimura K, Trapnell BC, Yoneyama K, Rosenthal ER, Dalemans W, Fukayama M, Bargon J, Stier LE,Stratford-Perricaudet L, et al. In vivo transfer of the human cystic fibrosis transmembrane conductance regulator gene to the air-way epithelium. Cell. 1992;68:143-55.
190. Roth JC. Dual-gene transfer and vector targeting for hematopoietic stem cell gene therapy. Unpublished Ph.D. Thesis. 2006. Case Western Reserve University, Cleve-land, OH. OhioLink ETD Center. Web Accessed June 15, 2011.
191. Rowe WP, Huebner RJ, Gilmore LK, Parrott RH, Ward TG. Isolation of a cytopa-thogenic agent from human adenoids undergoing spontaneous degeneration in tis-sue culture. Proc Soc Exp Biol Med. 1953;84:570-3.
192. Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685-1693.
193. Russell DW, Kay MA. Adeno-associated virus vectors and hematology. Blood. 1999;94:864-74.

114
194. Russell WC. Update on adenovirus and its vectors. J Gen Virol. 2000;81:2573-604.
195. Russell WC. Adenoviruses: update on structure and function. J Gen Vi-rol. 2009;90:1-20.
196. Sallenave JM, Porteous DJ, Haslett C. Gene therapy for lung inflammatory dis-eases: not so far away? Thorax. 1997;52:742-4.
197. Schambach A, Baum C. Clinical application of lentiviral vectors - concepts and practice. Curr Gene Ther. 2008;8:474-482.
198. Schiedner G, Bloch W, Hertel S, Johnston M, Molojavyi A, Dries V, Varga G, Van Rooijen N, Kochanek S. A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum Gene Ther. 2003 Nov 20;14(17):1631-41.
199. Shashkova EV, May SM, Barry MA. Characterization of human adenovirus sero-types 5, 6, 11, and 35 as anticancer agents. Virology. 2009;394:311-20.
200. Shaw AR, Ziff EB. Transcripts from the adenovirus-2 major late promoter yield a single early family of 3' coterminal mRNAs and five late families. Cell. 1980;22:905-16.
201. Shayakhmetov DM, Gaggar A, Ni S, Li ZY, Lieber A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J Virol. 2005;79:7478-7491.
202. Smith JS, Xu Z, Tian J, Stevenson SC, Byrnes AP. Interaction of systemically de-livered adenovirus vectors with Kupffer cells in mouse liver. Hum Gene Ther. 2008;19:547-54.
203. Short JJ, Rivera AA, Wu H, Walter MR, Yamamoto M, Mathis JM, Curiel DT. Substitution of adenovirus serotype 3 hexon onto a serotype 5 oncolytic adenovirus reduces factor X binding, decreases liver tropism, and improves antitumor efficacy. Mol Cancer Ther. 2010;9:2536-44.
204. Smith TA, Mehaffey MG, Kayda DB, Saunders JM, Yei S, Trapnell BC, McClelland A, Kaleko M. Adenovirus mediated expression of therapeutic plasma levels of human factor IX in mice. Nat Genet. 1993;5:397-402.
205. Smith T, Idamakanti N, Kylefjord H, Rollence M, King L, Kaloss M, Kaleko M, Stevenson SC. In vivo hepatic adenoviral gene delivery occurs independently of the coxsackievirus-adenovirus receptor. Mol Ther. 2002;5:770-9.
206. Smith TA, Idamakanti N, Rollence ML, et al. Adenovirus serotype 5 fiber shaft in-fluences in vivo gene transfer in mice. Hum Gene Ther. 2003;14:777-787.

115
207. Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, Shastry S, Rowe SM, Shim YM, Hussell T, Blalock JE. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science. 2010;330:90-4.
208. Spierings E, Wieles B, Goulmy E. Minor histocompatibility antigens--big in tumour therapy. Trends Immunol. 2004 Feb;25(2):56-60.
209. Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160:S49-52.
210. Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121:151S-155S.
211. Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni S, Lieber A. Adenovirus-platelet in-teraction in blood causes virus sequestration to the reticuloendothelial system of the liver. J Virol. 2007;81:4866-4871.
212. Stribling R, Brunette E, Liggitt D, Gaensler K, Debs R. Aerosol gene delivery in vivo. Proc Natl Acad Sci U S A. 1992;89:11277-81.
213. Strunze S, Trotman LC, Boucke K, Greber UF. Nuclear targeting of adenovirus type 2 requires CRM1-mediated nuclear export. Mol Biol Cell. 2005;16:2999-3009.
214. Sueblinvong V, Suratt BT, Weiss DJ. Novel therapies for the treatment of cystic fibrosis: new developments in gene and stem cell therapy. Clin Chest Med. 2007;28:361-379.
215. Sumida SM, Truitt DM, Lemckert AA, Vogels R, Custers JH, Addo MM, Lockman S, Peter T, Peyerl FW, Kishko MG, Jackson SS, Gorgone DA, Lifton MA,Essex M, Walker BD, Goudsmit J, Havenga MJ, Barouch DH. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174:7179-85.
216. Sutherland ER, Cherniack RM. Management of chronic obstructive pulmonary dis-ease. N Engl J Med. 2004;350:2689-2697.
217. Tallone T, Malin S, Samuelsson A, Wilbertz J, Miyahara M, Okamoto K, Poellinger L, Philipson L, Pettersson S. A mouse model for adenovirus gene deliv-ery. Proc Natl Acad Sci U S A. 2001;98:7910-5.
218. Tamm I, Trepel M, Cardó-Vila M, Sun Y, Welsh K, Cabezas E, Swatterthwait A, Arap W, Reed JC, Pasqualini R. Peptides targeting caspase inhibitors. J Biol Chem. 2003;278:14401-5.

116
219. Tao N, Gao GP, Parr M, Johnston J, Baradet T, Wilson JM, Barsoum J, Fawell SE. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose re-sponse of transduction in liver. Mol Ther. 2001;3:28-35.
220. Toietta G, Koehler DR, Finegold MJ, Lee B, Hu J, Beaudet AL. Reduced inflam-mation and improved airway expression using helper-dependent adenoviral vectors with a K18 promoter. Mol Ther. 2003;7:649-58.
221. Tomko RP, Xu R, Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci U S A. 1997;94:3352-6.
222. Trepel M, Grifman M, Weitzman MD, Pasqualini R. Molecular adaptors for vascu-lar-targeted adenoviral gene delivery. Hum Gene Ther. 2000;11:1971-81.
223. Tripathy SK, Black HB, Goldwasser E, Leiden JM. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med. 1996;2:545-50.
224. Tsuruta Y, Park YJ, Siegal GP, Liu G, Abraham E. Involvement of vitronectin in lipopolysaccaride-induced acute lung injury. J Immunol. 2007;179:7079-86.
225. Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc Natl Acad Sci U S A. 2006;103:407-12.
226. van Beusechem VW, van Rijswijk AL, van Es HH, Haisma HJ, Pinedo HM, Gerritsen WR. Recombinant adenovirus vectors with knobless fibers for tar-geted gene transfer. Gene Ther. 2000;7:1940-6.
227. van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VW, Groeneveld AB. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax. 2008;63:903-909.
228. van der Heijden M, van Nieuw Amerongen GP, Chedamni S, van Hinsbergh VW, Johan Groeneveld AB. The angiopoietin-Tie2 system as a therapeutic target in sep-sis and acute lung injury. Expert Opin Ther Targets. 2009;13:39-53.
229. van der Velden VH. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediators Inflamm. 1998;7:229-237.
230. Vandenberghe LH, Wilson JM, Gao G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009;16:311-9.
231. Verma IM, Somia N. Gene therapy -- promises, problems and prospects. Nature. 1997;389:239-42.

117
232. Von Seggern DJ, Chiu CY, Fleck SK, Stewart PL, Nemerow GR. A helper-independent adenovirus vector with E1, E3, and fiber deleted: structure and infec-tivity of fiberless particles. J Virol. 1999;73:1601-8.
233. Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods. 1995;178:89-97.
234. Waddington SN, McVey JH, Bhella D, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397-409.
235. Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene ther-apy. Nat Rev Genet. 2007;8:573-587.
236. Walters RW, van't Hof W, Yi SM, et al. Apical localization of the coxsackie-adenovirus receptor by glycosyl-phosphatidylinositol modification is sufficient for adenovirus-mediated gene transfer through the apical surface of human airway epi-thelia. J Virol. 2001;75:7703-7711.
237. Wang K, Huang S, Kapoor-Munshi A, Nemerow G. Adenovirus internalization and infection require dynamin. J Virol. 1998 Apr;72(4):3455-8.
238. Wang G, Slepushkin V, Zabner J, Keshavjee S, Johnston JC, Sauter SL, Jolly DJ, Dubensky TW Jr, Davidson BL, McCray PB Jr. Feline immunodeficiency virus vectors persistently transduce nondividing airway epithelia and correct the cystic fi-brosis defect. J Clin Invest. 1999;104:R55-62.
239. Wang Q, Finer MH. Second-generation adenovirus vectors. Nat Med. 1996;2:714-6.
240. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334-1349.
241. Waxman DJ, Schwartz PS. Harnessing apoptosis for improved anticancer gene therapy. Cancer Res. 2003;63:8563-72.
242. Weaver EA, Hillestad ML, Khare R, Palmer D, Ng P, Barry MA. Characteriza-tion of species C human adenovirus serotype 6 (Ad6). Virology. 2011;412:19-27.
243. Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A. 1993 Jan 15;90(2):587-91.

118
244. Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309-19.
245. Wickham TJ, Segal DM, Roelvink PW, Carrion ME, Lizonova A, Lee GM, Kovesdi I. Targeted adenovirus gene transfer to endothelial and smooth mus-cle cells by using bispecific antibodies. J Virol. 1996 Oct;70(10):6831-8.
246. Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus protein VI medi-ates membrane disruption following capsid disassembly. J Virol. 2005;79:1992-2000.
247. Wiggs BR, English D, Quinlan WM, Doyle NA, Hogg JC, Doerschuk CM. Contri-butions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J Appl Physiol. 1994;77:463-70.
248. Wilson AA, Murphy GJ, Hamakawa H, Kwok LW, Srinivasan S, Hovav AH, Mulligan RC, Amar S, Suki B, Kotton DN. Amelioration of emphysema in mice through lentiviral transduction of long-lived pulmonary alveolar macrophages. J Clin Invest. 2010;120:379-89.
249. Wold WSM, Horwitz MS. Adenoviruses. In: Fields BN, Knipe DM, Howley PM, eds. Fields Virology. 5th ed. Philadelphia: Lippincott-Raven; 2007: 2395-2436.
250. Wolff G, Worgall S, van Rooijen N, Song WR, Harvey BG, Crystal RG. Enhance-ment of in vivo adenovirus-mediated gene transfer and expression by prior deple-tion of tissue macrophages in the target organ. J Virol. 1997 Jan;71(1):624-9.
251. Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL. Di-rect gene transfer into mouse muscle in vivo. Science. 1990;247:1465-8.
252. Worgall S, Leopold PL, Wolff G, Ferris B, Van Roijen N, Crystal RG. Role of al-veolar macrophages in rapid elimination of adenovirus vectors administered to the epithelial surface of the respiratory tract. Hum Gene Ther. 1997a;8:1675-1684.
253. Worgall S, Wolff G, Falck-Pedersen E, Crystal RG. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum Gene Ther. 1997b;8:37-44.
254. Work LM, Büning H, Hunt E, Nicklin SA, Denby L, Britton N, Leike K, Odenthal M, Drebber U, Hallek M, Baker AH. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol Ther. 2006;13:683-93.
255. World Health Organization. World Health Report 2000. Geneva, World Health Or-ganization, 2000.

119
256. Wortmann A, Vöhringer S, Engler T, Corjon S, Schirmbeck R, Reimann J, Kochanek S, Kreppel F. Fully detargeted polyethylene glycol-coated adenovirus vectors are potent genetic vaccines and escape from pre-existing anti-adenovirus antibodies. Mol Ther. 2008;16:154-62.
257. Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Vi-rol. 2008;82:11705-13.
258. Yang L, Bailey L, Baltimore D, Wang P. Targeting lentiviral vectors to specific cell types in vivo. Proc Natl Acad Sci U S A. 2006;103:11479-84.
259. Yang Y, Ertl HC, Wilson JM. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994a;1:433-42.
260. Yang Y, Nunes FA, Berencsi K, Furth EE, Gönczöl E, Wilson JM. Cellular immu-nity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci U S A. 1994b;91:4407-11.
261. Yang Y, Wilson JM. Clearance of adenovirus-infected hepatocytes by MHC class I-restricted CD4+ CTLs in vivo. J Immunol. 1995;155:2564-70.
262. Yang Y, Li Q, Ertl HC, Wilson JM. Cellular and humoral immune responses to vi-ral antigens create barriers to lung-directed gene therapy with recombinant adenovi-ruses. J Virol. 1995a;69:2004-15.
263. Yang Y, Xiang Z, Ertl HC, Wilson JM. Upregulation of class I major histocompati-bility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc Natl Acad Sci U S A. 1995b;92:7257-61.
264. Yang Y, Su Q, Wilson JM. Role of viral antigens in destructive cellular immune responses to adenovirus vector-transduced cells in mouse lungs. J Vi-rol. 1996;70:7209-12.
265. Yei S, Mittereder N, Tang K, O'Sullivan C, Trapnell BC. Adenovirus-mediated gene transfer for cystic fibrosis: quantitative evaluation of repeated in vivo vector administration to the lung. Gene Ther. 1994;1:192-200.
266. Yonemitsu Y, Kitson C, Ferrari S, et al. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotechnol. 2000;18:970-973.
267. Zabner J, Cheng SH, Meeker D, et al. Comparison of DNA-lipid complexes and DNA alone for gene transfer to cystic fibrosis airway epithelia in vivo. J Clin In-vest. 1997a;100:1529-1537.

120
268. Zabner J, Freimuth P, Puga A, Fabrega A, Welsh MJ. Lack of high affinity fiber receptor activity explains the resistance of ciliated airway epithelia to adenovirus in-fection. J Clin Invest. 1997b;100:1144-1149.
269. Zabner J, Seiler M, Walters R, Kotin RM, Fulgeras W, Davidson BL, Chiorini JA. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J Virol. 2000;74:3852-8.
270. Zaiss AK, Liu Q, Bowen GP, Wong NC, Bartlett JS, Muruve DA. Differential acti-vation of innate immune responses by adenovirus and adeno-associated virus vec-tors. J Virol. 2002;76:4580-90.
271. Zeng X, Moore TA, Newstead MW, Deng JC, Lukacs NW, Standiford TJ. IP-10 mediates selective mononuclear cell accumulation and activation in response to in-trapulmonary transgenic expression and during adenovirus-induced pulmonary in-flammation. J Interferon Cytokine Res. 2005;25:103-12.
272. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. 2002;76:5654-5666.
273. Zhang Y, Chirmule N, Gao GP, Qian R, Croyle M, Joshi B, Tazelaar J, Wilson JM. Acute cytokine response to systemic adenoviral vectors in mice is mediated by den-dritic cells and macrophages. Mol Ther. 2001 May;3(5 Pt 1):697-707.
274. Zhu N, Liggitt D, Liu Y, Debs R. Systemic gene expression after intravenous DNA delivery into adult mice. Science. 1993;261:209-211.
275. Zinn KR, Douglas JT, Smyth CA, Liu HG, Wu Q, Krasnykh VN, Mountz JD, Curiel DT, Mountz JM. Imaging and tissue biodistribution of 99mTc-labeled ade-novirus knob (serotype 5). Gene Ther. 1998;5:798-808.
276. Zinn KR, Szalai AJ, Stargel A, Krasnykh V, Chaudhuri TR. Bioluminescence imag-ing reveals a significant role for complement in liver transduction following intra-venous delivery of adenovirus. Gene Ther. 2004;11:1482-6.
277. Zsengellér Z, Otake K, Hossain SA, Berclaz PY, Trapnell BC. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling dur-ing acute respiratory tract infection. J Virol. 2000;74:9655-67.

APPENDIX A
IACUC APPROVAL FORM (2006-2010)
121

122

APPENDIX B
IACUC APPROVAL FORM (2011)
123

124

APPENDIX C
Vector Maps
125

126

127

128

129

130

131

132

133

APPENDIX D
Lung Gating Schema for Flow Cytometry
134
CD
11b
CD11c
CD
11b
Ly6G
16.5 0.2
81.3
44.1
2.0
CD
45
CD31
22.8
1.1 76.1
a b
Flow cytometry gating schema for characterization of GFP+ lung cells in Ad.MBP.GFP injected mice. Expression of the markers CD11b, CD11c, and Ly6G (a) or CD45 and CD31 (b) was evaluated on collagenase digested lung cells from uninjected (naive) mice in order to establish cell population gates applied in Figure 29. (a) Populations were gated as follows: non-myeloid (Non-My, CD11b–Ly6G–
CD11c–), non-alveolar macrophages or monocytes (Mon/Mac, CD11b+Ly6G–CD11c–
), neutrophils (PMN, CD11b+Ly6G+), and alveolar macrophages (AM, CD11c+CD11b–Ly6G–). (b) Populations were gated as follows: total leukocuytes (Leuk, CD45+), endothelial cells (EC, CD31+), and other stromal cells (Stromal, CD45–CD31–). Representative scatter plots are shown in a and b.
135