Embed Size (px)
Citation preview

Acute Myeloid Leukemia

Acute Myeloid Leukemia
- Also known as
• Acute myelocytic leukemia• Acute myelogenous leukemia• Acute nonlymphocytic leukemia
Stem cell disorder characterized by Clonal expansion of myeloid precursor cells with reduced capacity to differentiate i.e, MATURATION ARREST.
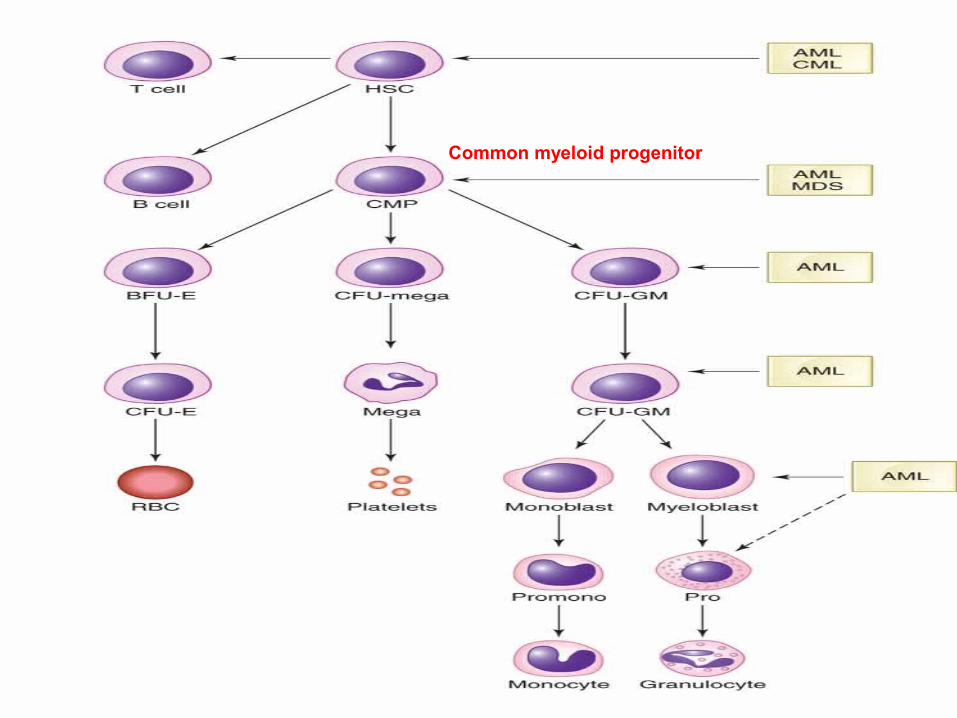
Common myeloid progenitor

Predisposing factors
Congenital factors :-
• Down syndrome.• Bloom syndrome.• Monosomy 7 syndrome• Klinefelter syndrome
• Turner syndrome
• Neurofibromatosis • Congential dysmorphic syndrome

Predisposing factors Marrow failure syndrome:-
• Fanconi anemia
• Dyskeratosis congenita.
• Schwchman Diamond Syndrome• Amegakaryocytic thrombocytopenia• Blackfan Diamond syndrome• Kostmann Agranulocytosis
• Familial aplastic anemia
• Familial platelet disorder.

Predisposing factors
Environmental factors:-
• Solvents ( benzene )• Smoking • Ionizing radiation• Non ionizing radiation
• Chemotherapy
Alkylating agent
Topoisomerase II inhibitor

Biological features
• Leukemogenesis- result from block of differentiation as well as altered proliferation and impaired apoptosis through genetic dysregulation.

Genetic Associations
• Research states that AML is caused by genetic aberrations such as translocations between chromosomes that alter the function of transcriptory regulatory factors
• These translocations are a direct result of chimeric fusion proteins which are caused by the abnormal cells and its inability to allow further growth, proliferation, maturation and differentiation.
• Class 1 and 2: mutations responsible for the development of the neoplastic process of myeloproliferation and de-differentiation

Genetic Associations
• Class 1: mutations that give rise to proliferation and/or differentiation.
• Class 2: mutations that interfere with terminal differentiation and apoptosis thereby providing survival advantage for the mutated cells.


Classification of AML
• WHO classification.
• FAB classification.

Differences between FAB and WHO
FAB-classification:1) Heavily used “Morphologic Findings”2) Special staining (SBB, MPO, NSE, etc), if required
WHO-classification:1) Morphologic findings 2) Special staining (decreased role)3) Immunophenotyping (in the form of FC and IHC) heavily used.4) Cytogentics and Molecular genetics studies frequently used.

WHO Classification of Acute Myelocytic Leukemias

FAB Classification of AML• M0 undifferentiated acute myeloblastic leukemia (5%)• M1 AML with minimal maturation (20%)• M2 AML with maturation (30%)
– t(8;21)• M3 Acute promyelocytic leukemia (5%)
– t(15;17)• M4 Acute myelomonocytic leukemia (20%)• M4 eos Acute myelomonocytic leukemia with eosinophilia (5%)
– inv (16)• M5 Acute monocytic leukemia (10%)
– t(9;11)• M6 Acute erythroid leukemia (3%) (DiGuglielmo's disease)• M7 Acute megakaryoblastic leukemia (3%)

Clinical features
Due to Bone Marrow FailureDue to Organ InfilterationOthers

History from patient with leukemia
Increasing fatigue or decreased exercise tolerance (anemia)
Excess bleeding or bleeding from unusual sites (DIC,
thrombocytopenia)
Fevers or recurrent infections (granulocytopenia)
Headache, vision changes, nonfocal neurologic abnormalities
(CNS leukemia or bleed)
Early satiety (splenomegaly)
Family history of AML (Fanconi, Bloom or Kostmann
syndromes or ataxia telangiectasia)
History of cancer (exposure to alkylating agents, radiation,
topoisomerase II inhibitors)
Occupational exposures (radiation, benzene, petroleum
products, paint, smoking, pesticides)
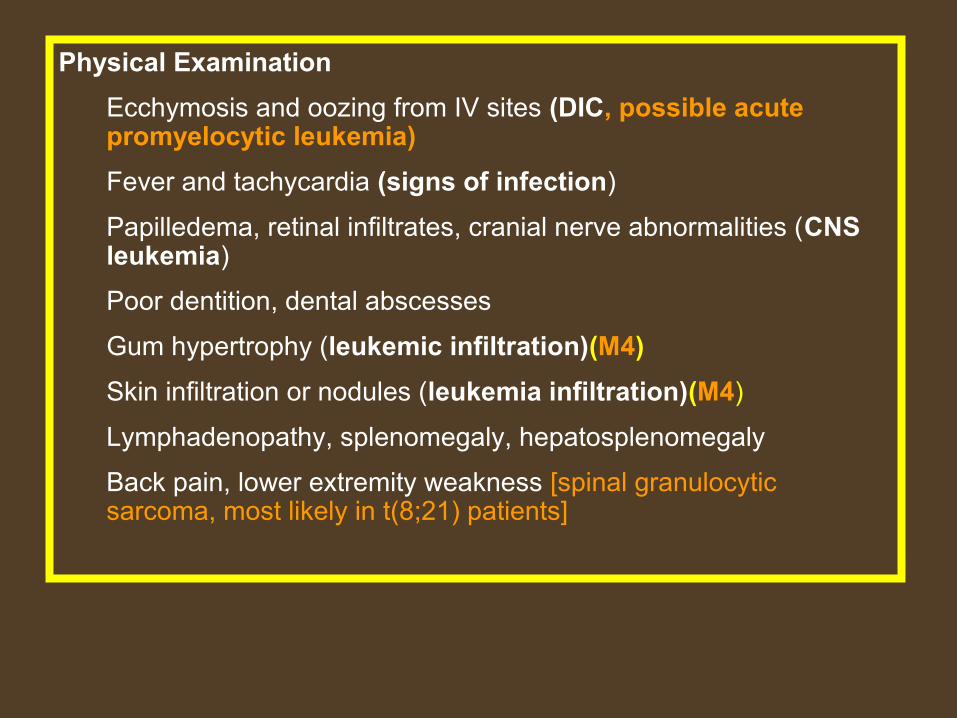
Physical Examination
Ecchymosis and oozing from IV sites (DIC, possible acute promyelocytic leukemia)
Fever and tachycardia (signs of infection)
Papilledema, retinal infiltrates, cranial nerve abnormalities (CNS leukemia)
Poor dentition, dental abscesses
Gum hypertrophy (leukemic infiltration)(M4)
Skin infiltration or nodules (leukemia infiltration)(M4)
Lymphadenopathy, splenomegaly, hepatosplenomegaly
Back pain, lower extremity weakness [spinal granulocytic sarcoma, most likely in t(8;21) patients]

Acute Myeloid Leukemia: Diagnostic Steps
1. Evaluation of an abnormal CBC for possible AML
• Confirm bone marrow failure, assess for blasts/blast equivalents and dysplasia• WBC: non-specific; in AML can be low, normal, or high• ANC: severe neutropenia characteristic of HP failure; typical in AML, but exceptions
occur• Circulating blasts: variable number and percent in AML, but key feature to assess
in blood• RBC features: severe anemia characteristic of HP failure, an expected feature of AML• Polychromasia: reduced, since anemia is result of bone marrow production failure• Other RBC pathology: non-specific• Platelets: severe thrombocytopenia characteristic of HP failure
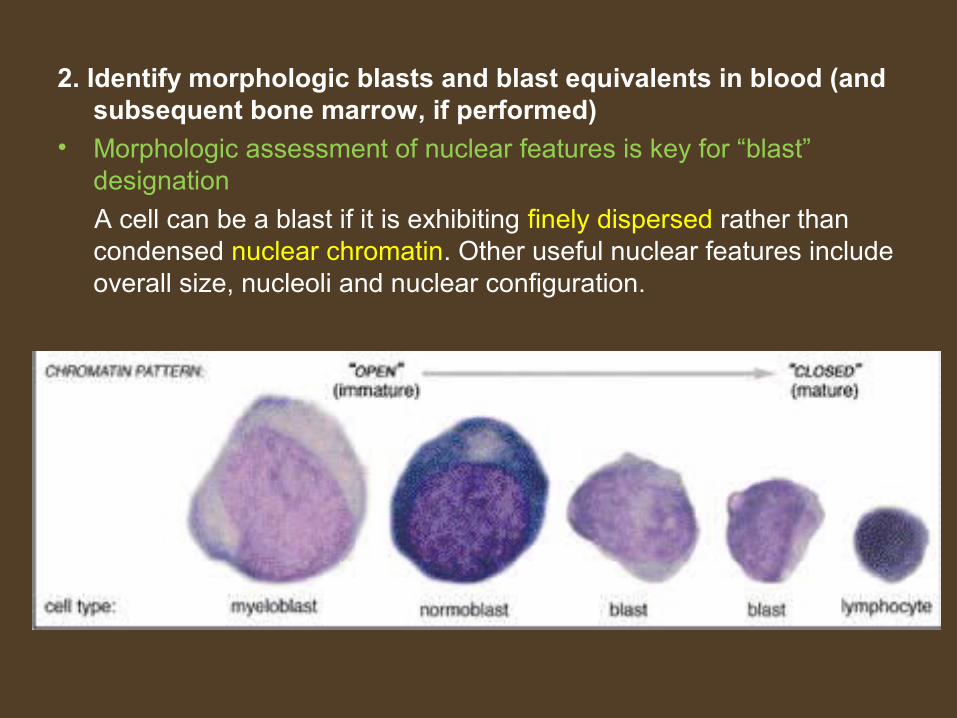
2. Identify morphologic blasts and blast equivalents in blood (and subsequent bone marrow, if performed)
• Morphologic assessment of nuclear features is key for “blast” designation
A cell can be a blast if it is exhibiting finely dispersed rather than condensed nuclear chromatin. Other useful nuclear features include overall size, nucleoli and nuclear configuration.

• Cytoplasmic features are very helpful in lineage determination, ie, sparse fine granules and Auer rods in myeloblasts, cytoplasmic blebbing in megakaryoblasts, and deeply basophilic, vacuolated cytoplasm in erythroblasts
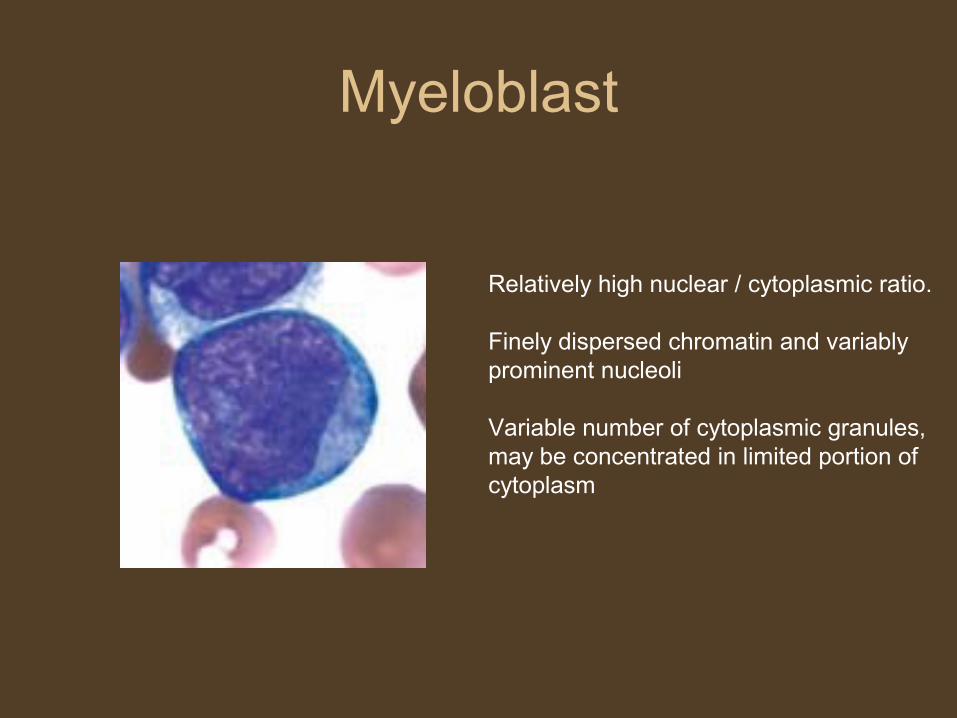
Myeloblast
Relatively high nuclear / cytoplasmic ratio.
Finely dispersed chromatin and variably prominent nucleoli
Variable number of cytoplasmic granules, may be concentrated in limited portion of cytoplasm

Promyelocyte
Nuclear chromatin slightly condensed;
Nucleoli variably prominent;
Nucleus often eccentric, and Golgi zone may be apparent
Numerous cytoplasmic granules that may be more dispersed throughout cytoplasmBlast equivalent in APL only
In APL, intense cytoplasmic granularity usually present .Nuclear configuration variable, but nuclear folding and lobulation characteristic of microgranular variant of APL
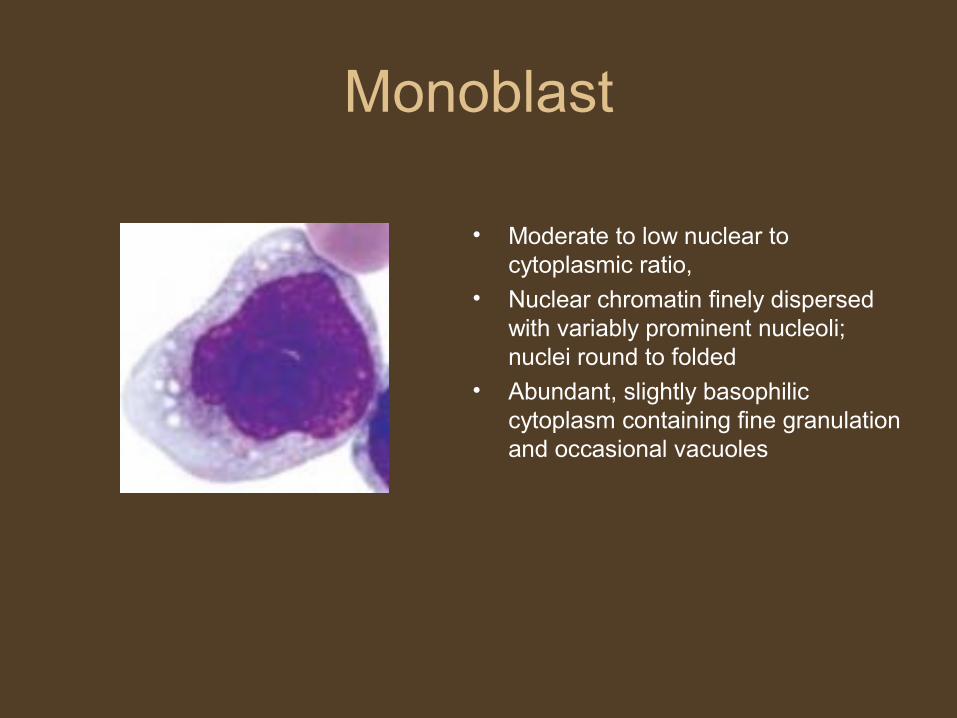
Monoblast
• Moderate to low nuclear to cytoplasmic ratio,
• Nuclear chromatin finely dispersed with variably prominent nucleoli; nuclei round to folded
• Abundant, slightly basophilic cytoplasm containing fine granulation and occasional vacuoles
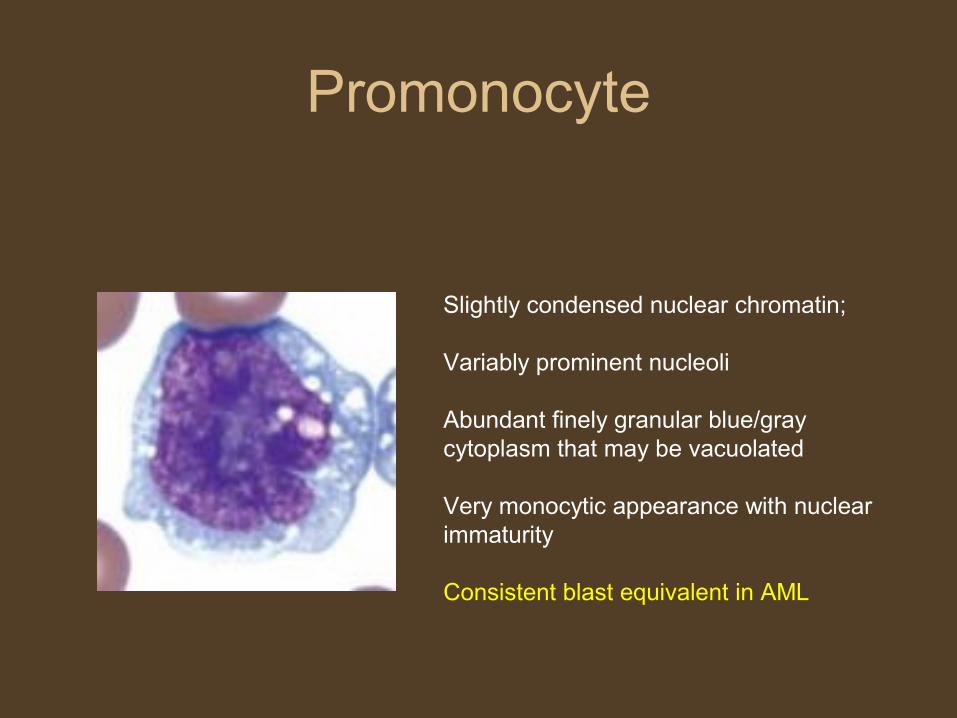
Promonocyte
Slightly condensed nuclear chromatin;
Variably prominent nucleoli
Abundant finely granular blue/gray cytoplasm that may be vacuolated
Very monocytic appearance with nuclear immaturity
Consistent blast equivalent in AML
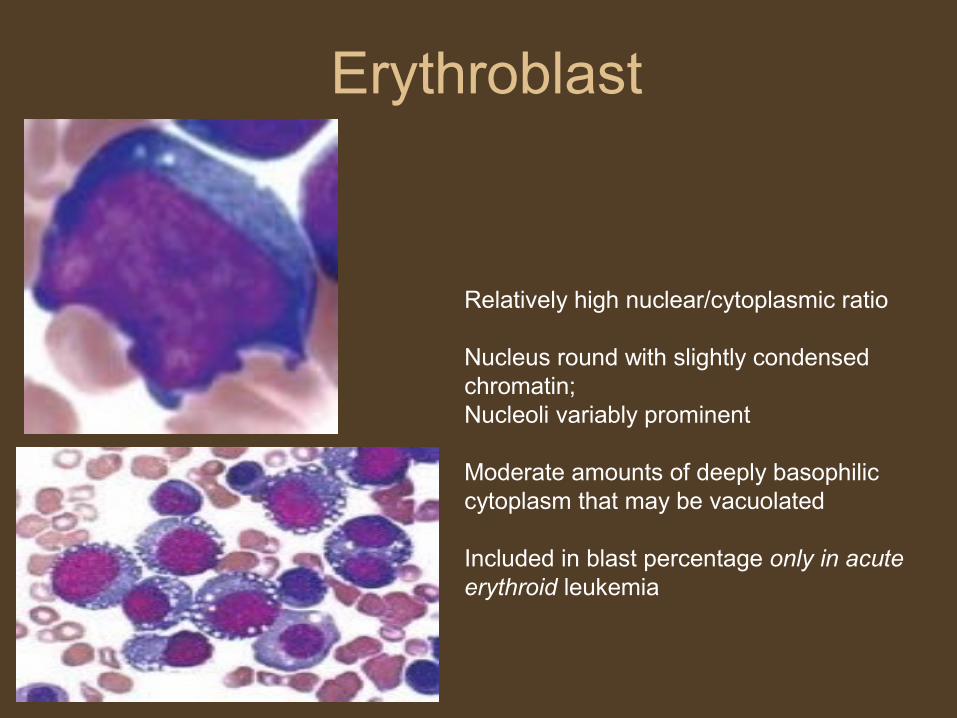
Erythroblast
Relatively high nuclear/cytoplasmic ratio
Nucleus round with slightly condensed chromatin; Nucleoli variably prominent
Moderate amounts of deeply basophilic cytoplasm that may be vacuolated
Included in blast percentage only in acute erythroid leukemia

Megakaryoblast
Highly variable morphologic features;
May be lymphoid-appearing with high nuclear to cytoplasmic ratio
Nuclear chromatin fine to variably condensed
Cytoplasm may be scant to moderate, is usually agranular or contains a few granules;
Blebbing or budding of cytoplasm may be evident
Blasts may form cohesive clumps

MorphologyLymphoblast Myeloblast
Nuclear chromatin
Coarse Fine
Nucleoli 1-2 3-5
N:C ratio High High
Auer rod Absent Present
Accompanying cells
Lymphocytes Myeloid precursor

3. Bone marrow examination often performed to address differential diagnoses from blood assessment or for protocol requirements.
4. Enumeration of blasts/blast equivalents by morphology and differential cell count
• Previously >30% blasts on BM aspirate (per FAB criteria)• As per recent WHO criteria, AML is defined by greater than 20% blasts on
BM aspirate. – patients with certain cytogenetic abnormalities are considered to have
AML regardless of blast percentage• t(8;21)(q22;q22), inversion (16)(p13q22)• t(16;16)(p13;q22), and t(15;17)(q22;q12)
Unique situations compromising blast count:Fibrosis and/or necrosisPredominance (≥50%) of erythroid lineageMarked hypocellularityTechnically poor specimen

• 5. Determine lineage of blasts/blast equivalents (can be performed on blood or bone marrow)
Morphology (nucleus and cytoplasm)CytochemistryImmunophenotype

Myeloperoxidase stain
• Basis- breakdown of hydrogen peroxide by enzyme myeloperoxidase releasing an oxygen radical that reacts with a soluble substrate to form colored precipitate.
• MPO located in peroxisomes of neutrophils and monocytes and specific granules of eosinophils.
• Staining is more pronounced in golgi region.
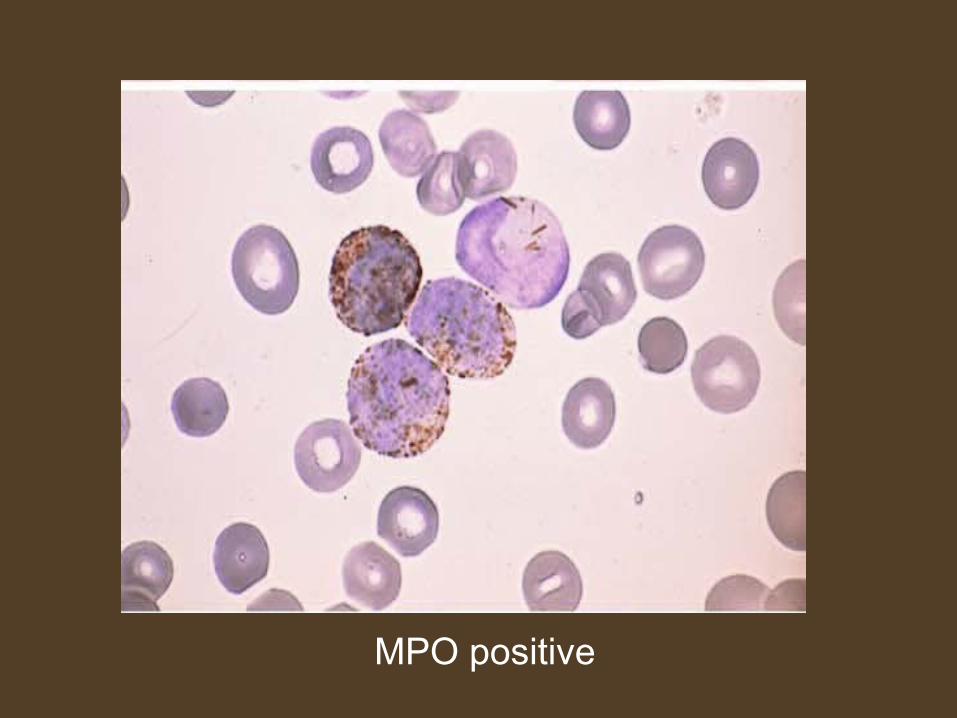
MPO positive

Sudan Black B
• It is a direct stain phospholipid in granular membrane.
• Auer rods are MPO and SBB positive.

Esterase stains
• Non specific esterase reactivity is found in monocytes.
• Basis- Enzymatic release of a side chain from a naphthol ring with subsequent reaction of the free ring with a soluble colour develops to generate a coloured precipitate.
• Most common used substrate for Non specific esterase are Alpha- naphthyl butyrate and Alpha – naphthyl acetate.

NSE positive in MONOCYTIC CELLS
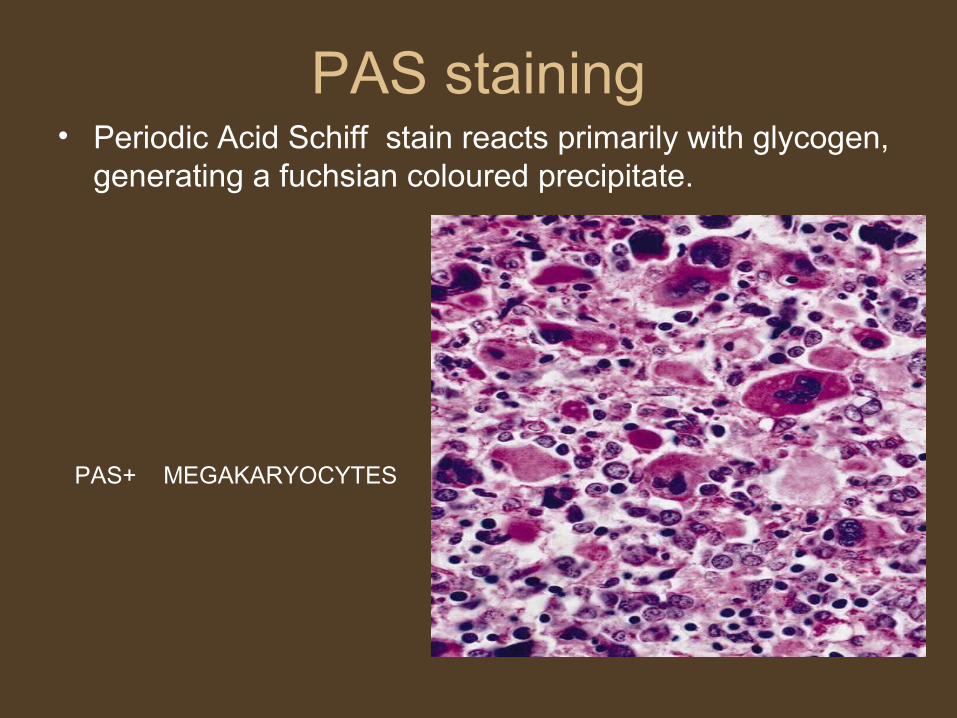
PAS staining• Periodic Acid Schiff stain reacts primarily with glycogen,
generating a fuchsian coloured precipitate.
PAS+ MEGAKARYOCYTES
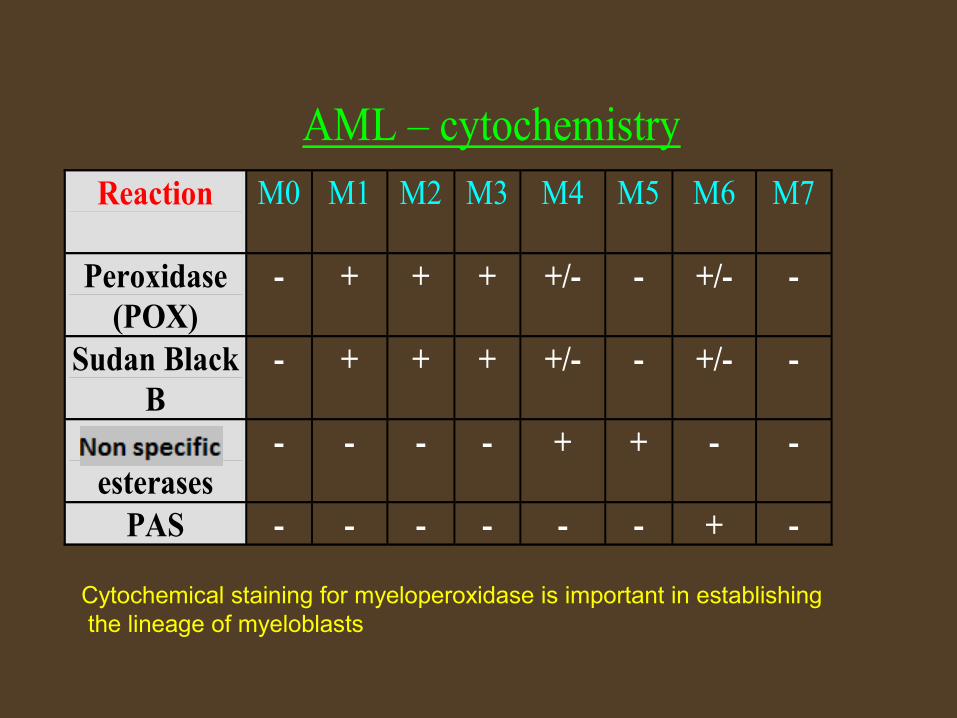
AML – cytochemistry
Reaction
M0
M1 M2 M3 M4 M5 M6 M7
Peroxidase (POX)
- + + + +/- - +/- -
Sudan Black B
- + + + +/- - +/- -
Unspecific esterases
- - - - + + - -
PAS - - - - - - + -
Cytochemical staining for myeloperoxidase is important in establishing the lineage of myeloblasts

Immunophenotyping FAB Immunological marker
AML with minimally differentiated CD13,CD34, HLA-DR, CD33,CD117,CD2,CD7,TdT
AML without maturation CD13,CD14,CD33, CD34
AML with maturation and with t(8;21)
CD34,CD56
Acute promyelocytic leukemia CD13,CD33, HLA-DR absent, CD34 negative
Acute myelomonocytic leukemia with abnormal eosinophils and inversion 16
CD13,CD34,CD11b,CD11c,CD14,CD33
Acute monocytic leukemia and 11q23 abnormalties
CD14,CD4,CD36,CD64
Erythroleukemia Glycophorin 7, Transferrin receptor CD71
Acute Megakaryocytic leukemia cCD41,cCD42b,cCD61

Minimally Differentiated Acute Myeloid Leukemia
• 5% of AML cases• No definite evidence of myeloid differentiation can be given
by morphology & cytochemistry.
• CRITERIA FOR DIAGNOSIS• <3% of blast which are MPO/SBB+(evident on EM)• >20 % of leukemia cells expressing myeloid antigens.
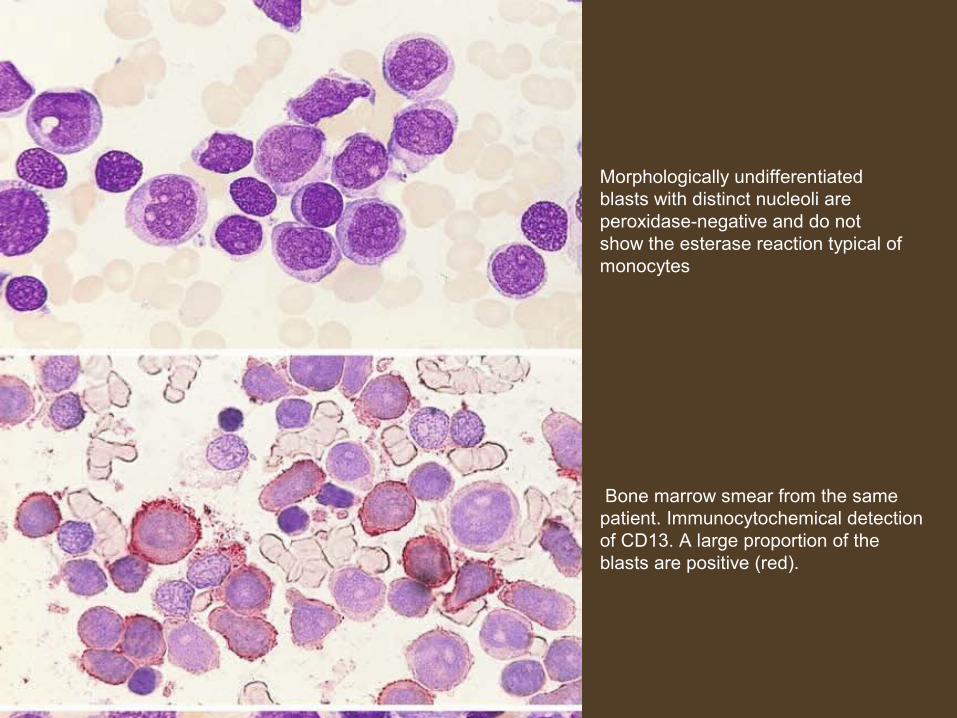
Morphologically undifferentiatedblasts with distinct nucleoli are peroxidase-negative and do not show the esterase reaction typical of monocytes
Bone marrow smear from the samepatient. Immunocytochemical detectionof CD13. A large proportion of the blasts are positive (red).

AML without maturation
10 – 20% of AML cases CRITERIA FOR DIAGNOSIS
Predominance of myeloblast ( > 90% ) without evidence of maturation ( < 10% promyelocytes or others) in marrow .
IF no auer rods , at least 3% of blast must be MPO OR SBB positive .
Median age : 45-50 yrs. Generally chemosensitive and prognostically favourable
unless hyperleukocytosis or complex karyotype present.
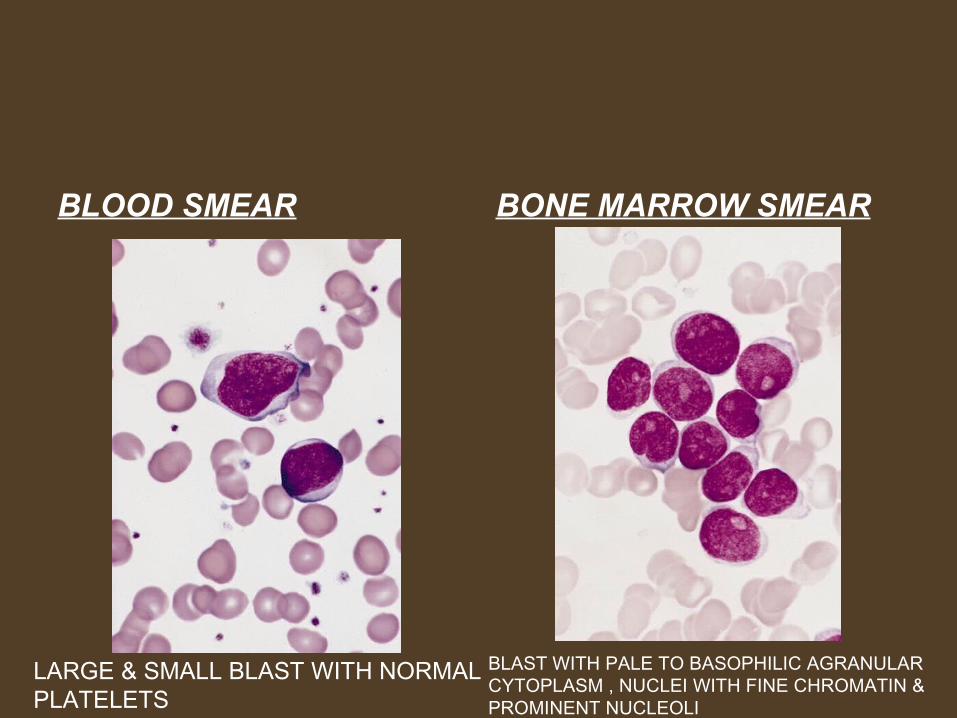
BLOOD SMEAR BONE MARROW SMEAR
LARGE & SMALL BLAST WITH NORMAL PLATELETS
BLAST WITH PALE TO BASOPHILIC AGRANULAR CYTOPLASM , NUCLEI WITH FINE CHROMATIN & PROMINENT NUCLEOLI
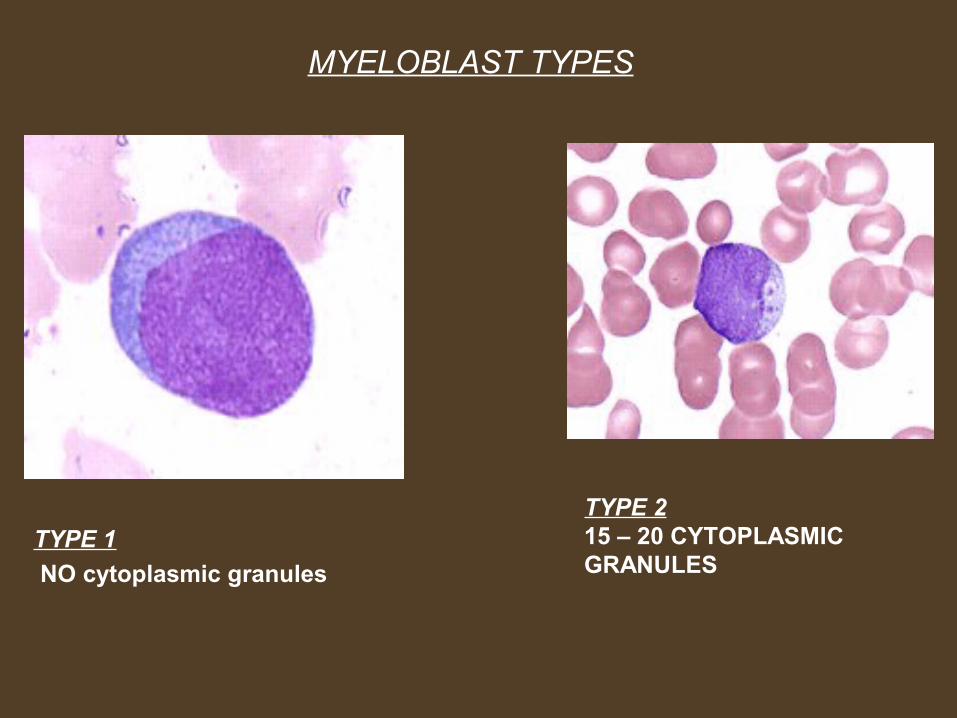
MYELOBLAST TYPES
TYPE 1
NO cytoplasmic granules
TYPE 215 – 20 CYTOPLASMIC GRANULES
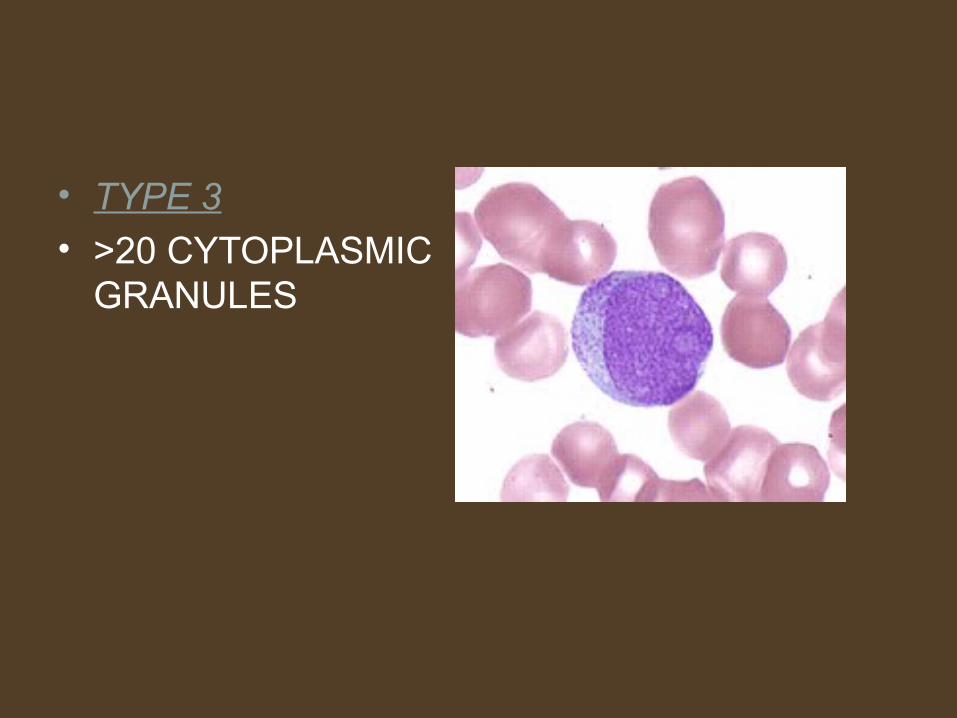
• TYPE 3• >20 CYTOPLASMIC
GRANULES

AML with maturation and with t(8;21)
• 30-45% of AML cases (Most frequent).• Genes involved in t(8;21) are AML1 at 21q22 and ETO
(eight twenty one) at 8q22.• CRITERIA FOR DIAGNOSIS• Blast 20% or more(20-89) of all nucleated cells in bone
marrow• Mature cells (promyelocytes to granulocytes) > 10%• Monocytic cells < 20%.

BONE MARROW SMEAR
BLAST WITH NUCLEOLI AUER RODS

Acute promyelocytic leukemia
• Median age 30-38 yrs (young patient).
• It is generally not preceded by myelodysplastic syndrome.
• Most patient present with hemorrhagic manifestation secondary to DIC.
• Associated with t(15;17).• Retinoic acid receptor (RAR- alpha) gene on
chromosome 17q12.
• Promyelocytic gene (PML gene) on chromosome 15q22.
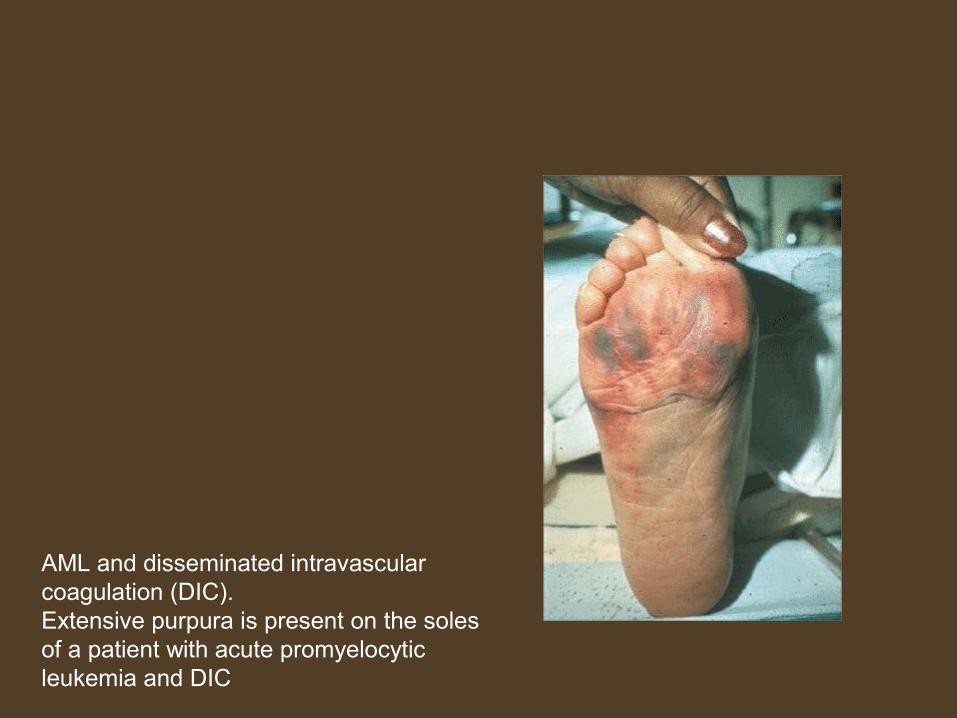
AML and disseminated intravascular coagulation (DIC). Extensive purpura is present on the soles of a patient with acute promyelocytic leukemia and DIC

Acute promyelocytic leukemia
• Either HYPERGRANULAR OR MICROGRANULAR.• Hypergranular type is most common.
• Leukopenia is seen in Hypergranular APL.
• Leukocytosis in Microgranular APL.
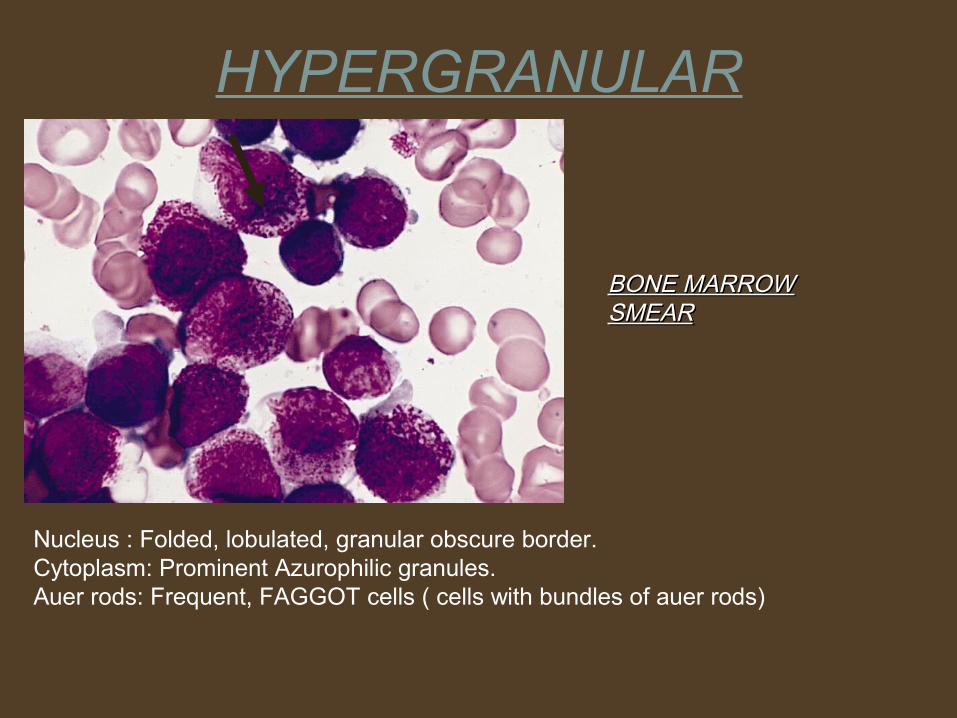
BONE MARROW BONE MARROW SMEARSMEAR
HYPERGRANULAR
Nucleus : Folded, lobulated, granular obscure border.Cytoplasm: Prominent Azurophilic granules.Auer rods: Frequent, FAGGOT cells ( cells with bundles of auer rods)

Variations on the appearance of “faggot cells” in several different cases of APL.

BONE MARROW BIOPSY
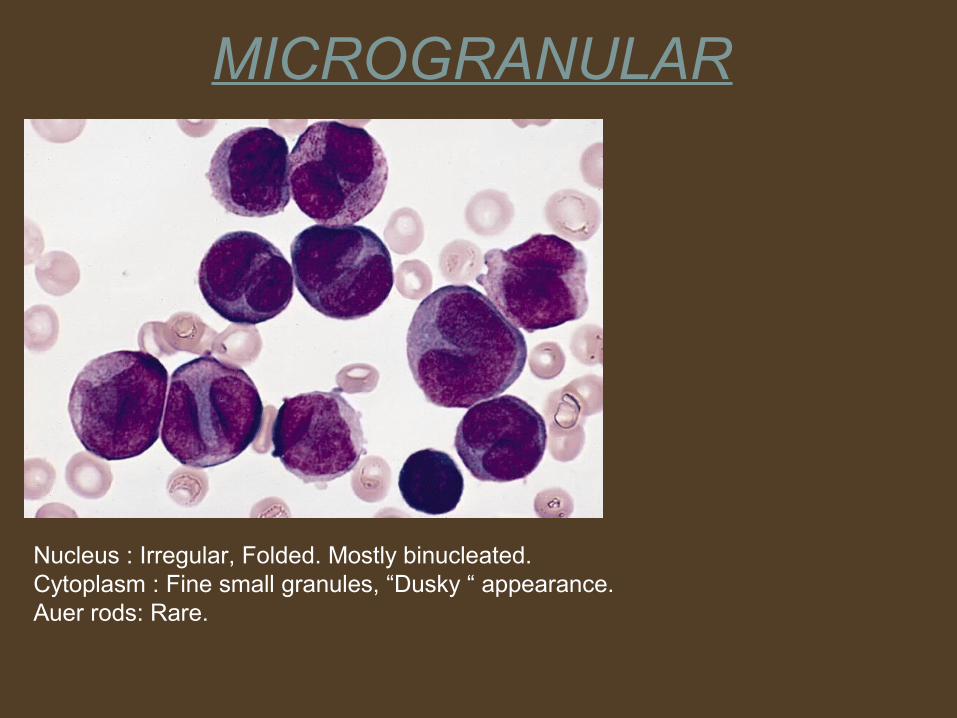
MICROGRANULAR
Nucleus : Irregular, Folded. Mostly binucleated.Cytoplasm : Fine small granules, “Dusky “ appearance.Auer rods: Rare.

ACUTE MYELOMONOCYTIC LEUKEMIA with ABNORMAL EOSINOPHILS and INVERSION Of Chrosome 16
• 15-25% of AML cases• CRITERIA FOR DIAGNOSIS
Blast >20%
Monocytic cells & their precursor
Neutrophil & their precursor• Median age : 40 – 45 yrs.• Leukocytosis is present in most of the patients.
• Prognosis is better than M1, M2, or M3.
>20%
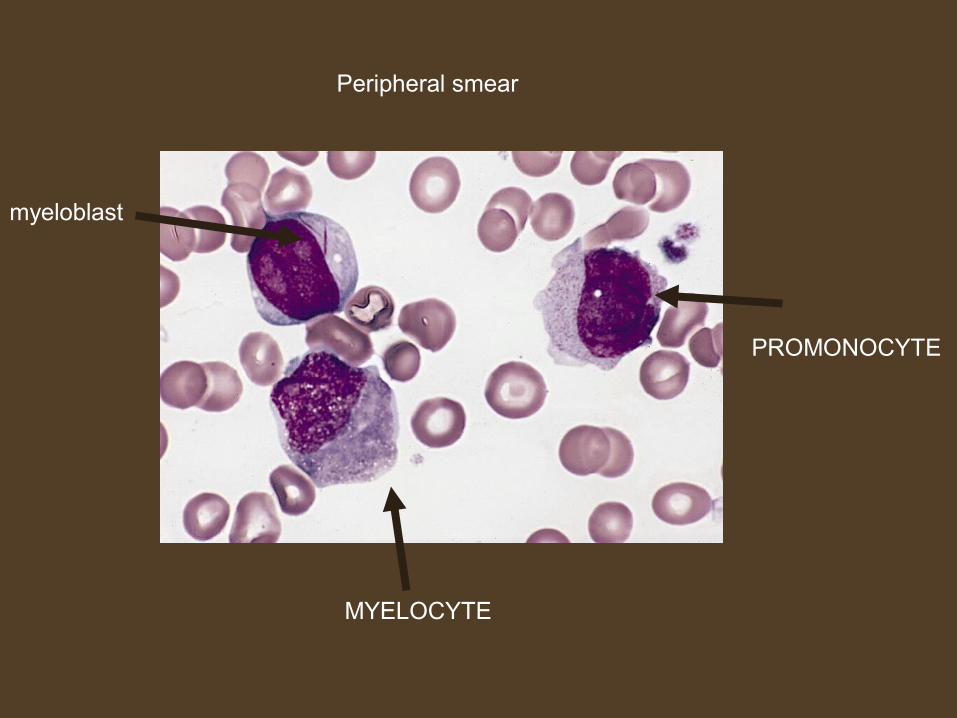
Peripheral smear
myeloblast
MYELOCYTE
PROMONOCYTE

BONE MARROW SMEAR
MONOCYTES & NEUTROPHIL AT VARIOUS STAGES OF MATURATION
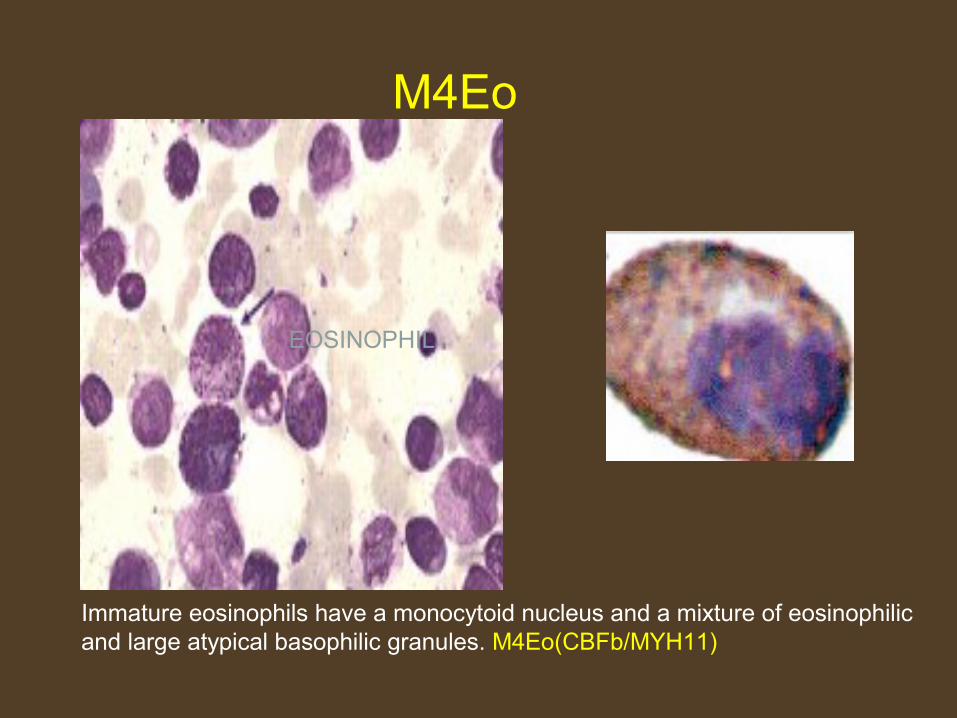
EOSINOPHIL
M4Eo
Immature eosinophils have a monocytoid nucleus and a mixture of eosinophilic and large atypical basophilic granules. M4Eo(CBFb/MYH11)

Acute monocytic leukemia & 11q23 abnormalities
• Two types : M5a and M5b
• M5a :-
Poorly differentiated
Trisomy 8 is most common abnormality seen.• M5b :-
Well differentiated.
FLT3 mutation is most common abnormality seen.• Extramedullary disease occur in > 50% of the patient.• It has a very poor prognosis , 6 -12 months
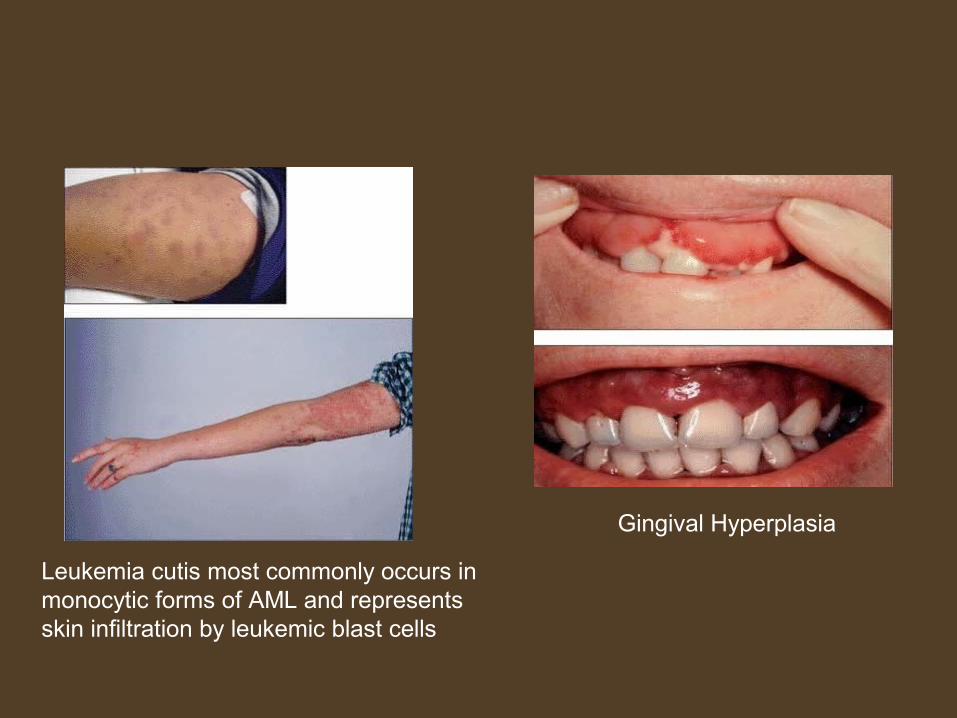
Leukemia cutis most commonly occurs inmonocytic forms of AML and represents skin infiltration by leukemic blast cells
Gingival Hyperplasia

M5a( Acute Monoblastic Leukemia )
BLOOD SMEAR BONE MARROW SMEAR
MONOBLAST
80% or more are MONOBLAST Abundant cytoplasm Round nuclei with nucleoli
MONOBLAST WITH ABUNDANT CYTOPLASM WITH FINE GRANULES

M5b( Monocytic Leukemia )
BLOOD SMEAR BONE MARROW SMEAR
PROMONOCYTES
<80% Monoblast
Mature monocytes or promonocytes predominate

ACUTE ERYTHROID LEUKEMIA
M6a (ERYTHROLEUKEMIA)
5% of AML casesMore COMMON THAN pure erythroid leukemia.Bimodal distribution- <20 yrs and >60yrs.
CRITERIA FOR DIAGNOSIS
>50% of nucleated marrow cells are erythroid lineage>20% of nonerythroid cells are myeloblastDyserythropoiesis is prominent
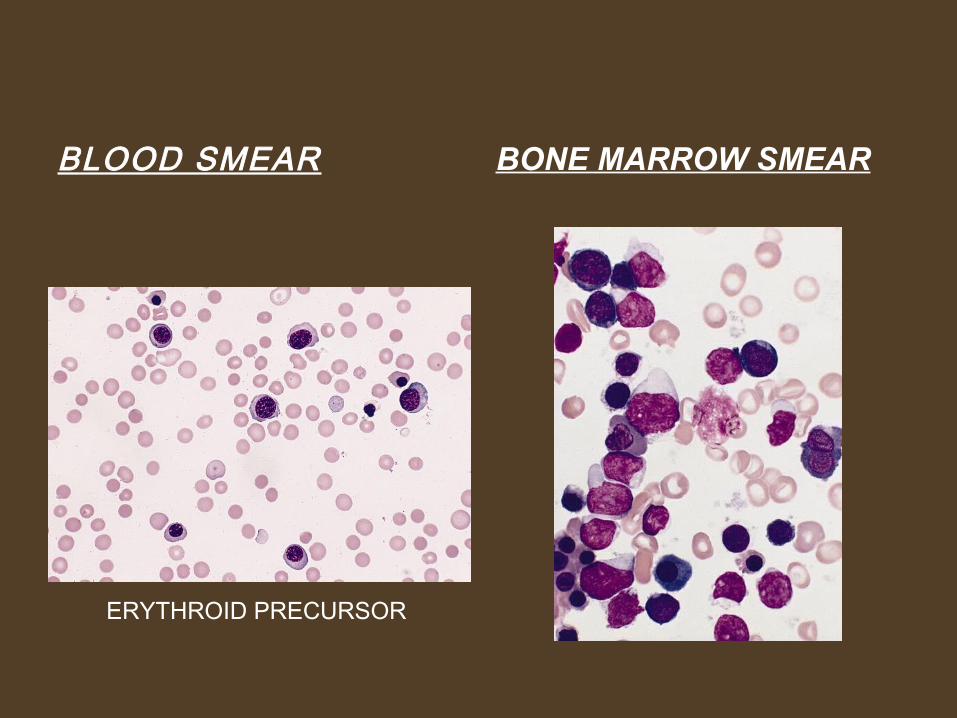
BLOOD SMEAR BONE MARROW SMEAR
ERYTHROID PRECURSOR

M6b (PURE ERYTHROID LEUKEMIA )Very rareAlso called ERYTHEMIC MYELOSIS , ACUTE Di GUGLIELMO SYNDROME>80% of marrow cells are erythroblastNo significant myeloblastic component
ACUTE ERYTHROID LEUKEMIA
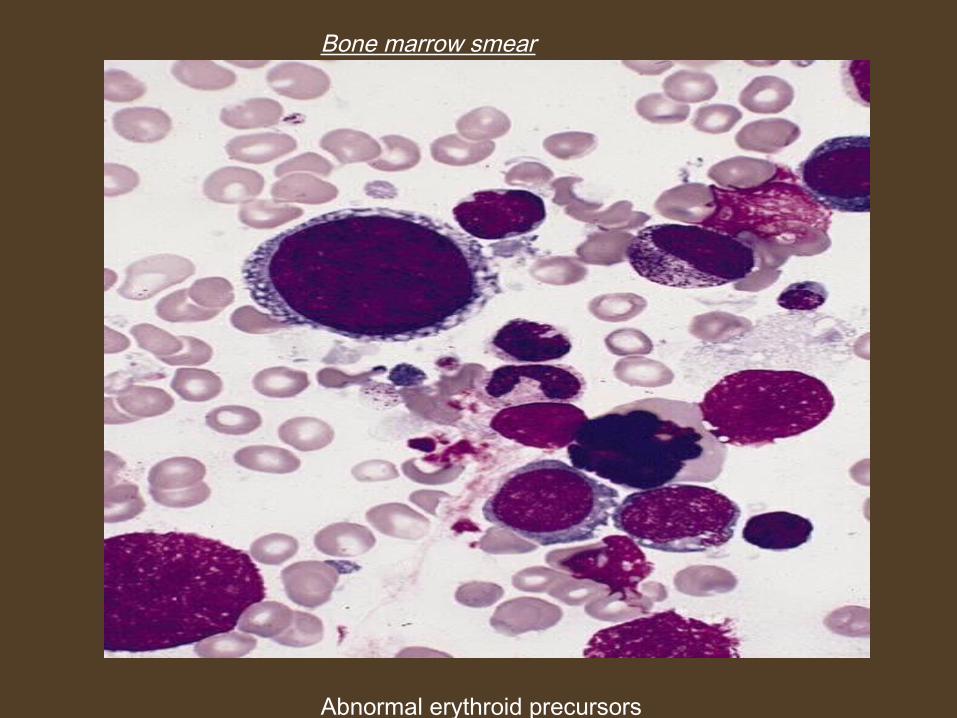
Bone marrow smear
Abnormal erythroid precursors

ACUTE MEGAKARYOBLASTIC LEUKEMIA
• 10% of AML in children & 5% of adult AML• Bimodal distribution- Infancy and elderlyCRITERIA FOR DIAGNOSIS• Megakaryoblast 20% or more in BM• Bone marrow fibrosis Megakaryoblast are either small to round with scanty
cytoplasm & coarse chromatin (resembling lymphoblasts) or medium to large with fine chromatin & prominent 1-3 nucleoli

ACUTE MEGAKARYOBLASTIC LEUKEMIA
• Morphologically confused with
- L2 subtype of ALL
- AML M1.• Diagnosis depends on expression of at least one platelet
antigen ( i.e., CD41,CD42b, CD61 or factor VIII related antigen)
• Most common leukemia seen in Down’s Syndrome.
• Platelet show impaired aggregation response.
• Elevated serum Lactate Dehydrogenase level.
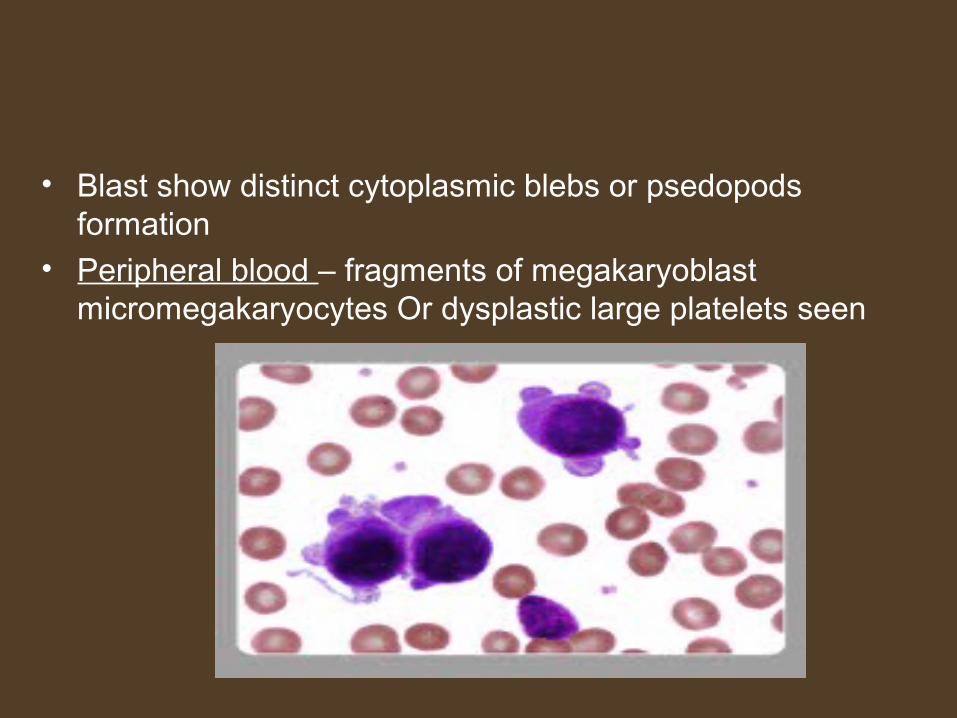
• Blast show distinct cytoplasmic blebs or psedopods formation
• Peripheral blood – fragments of megakaryoblast micromegakaryocytes Or dysplastic large platelets seen
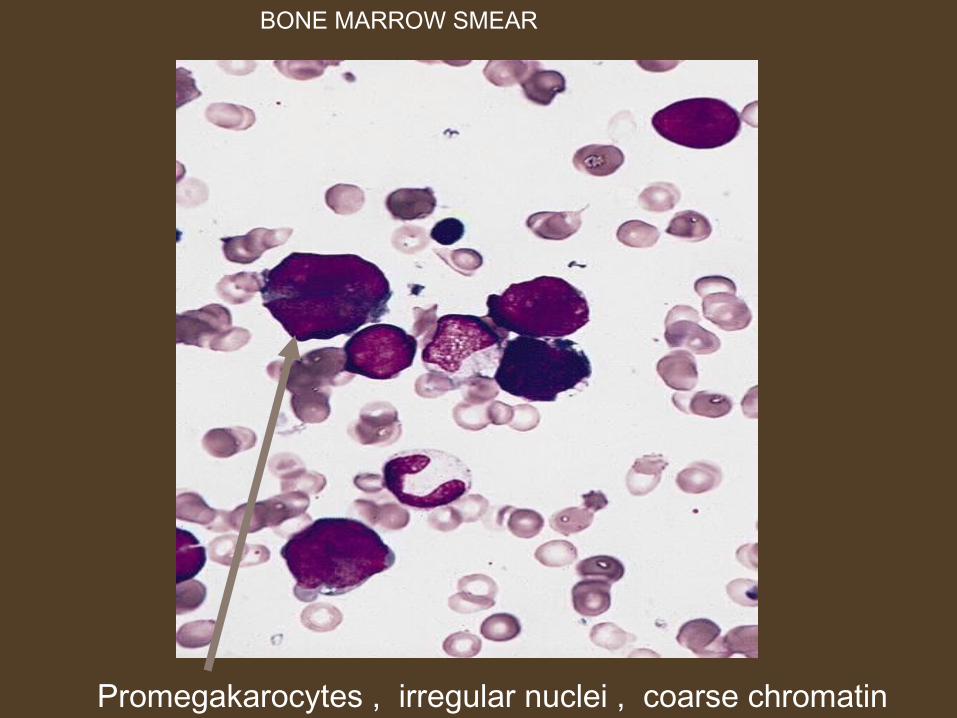
Promegakarocytes , irregular nuclei , coarse chromatin
BONE MARROW SMEAR

BONE MARROW BIOPSY
PROLIFERATION OF MEGAKARYOCYTES

AML WITH MULTILINEAGEDYSPLASIA
• Multilineage dysplasia – dysplasia present >50% of cells in 2 or more myeloid cell lines
• Occur in elderly
• With / without prior h/o MDS
• Poor prognosis
• Chromosomal abnormalities similar to MDS
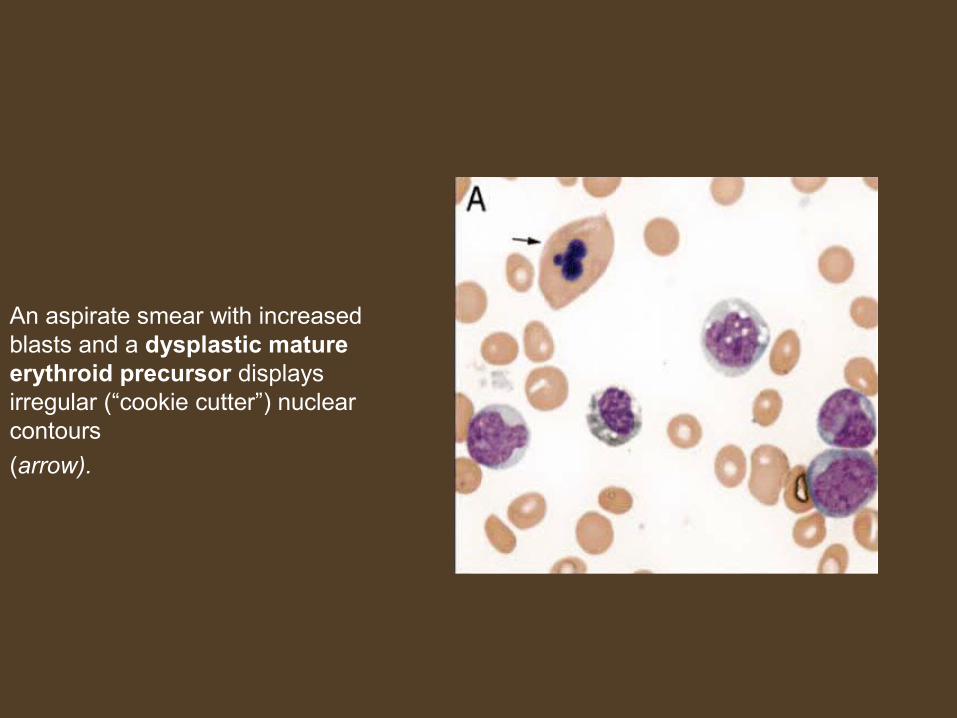
An aspirate smear with increased blasts and a dysplastic mature erythroid precursor displays irregular (“cookie cutter”) nuclear contours
(arrow).
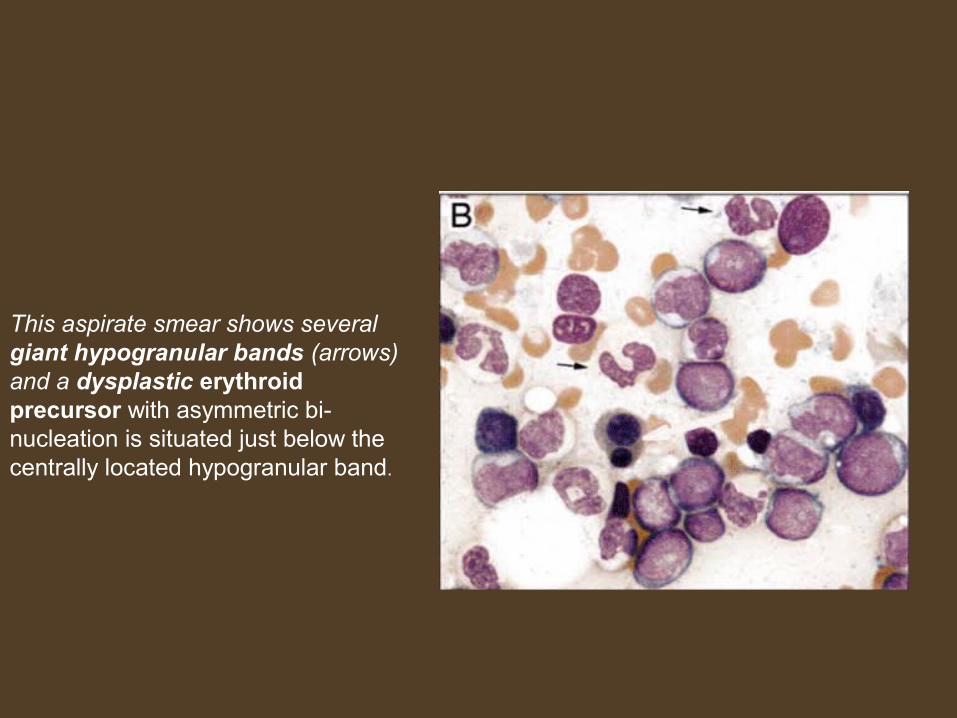
This aspirate smear shows several giant hypogranular bands (arrows) and a dysplastic erythroid precursor with asymmetric bi-nucleation is situated just below the centrally located hypogranular band.
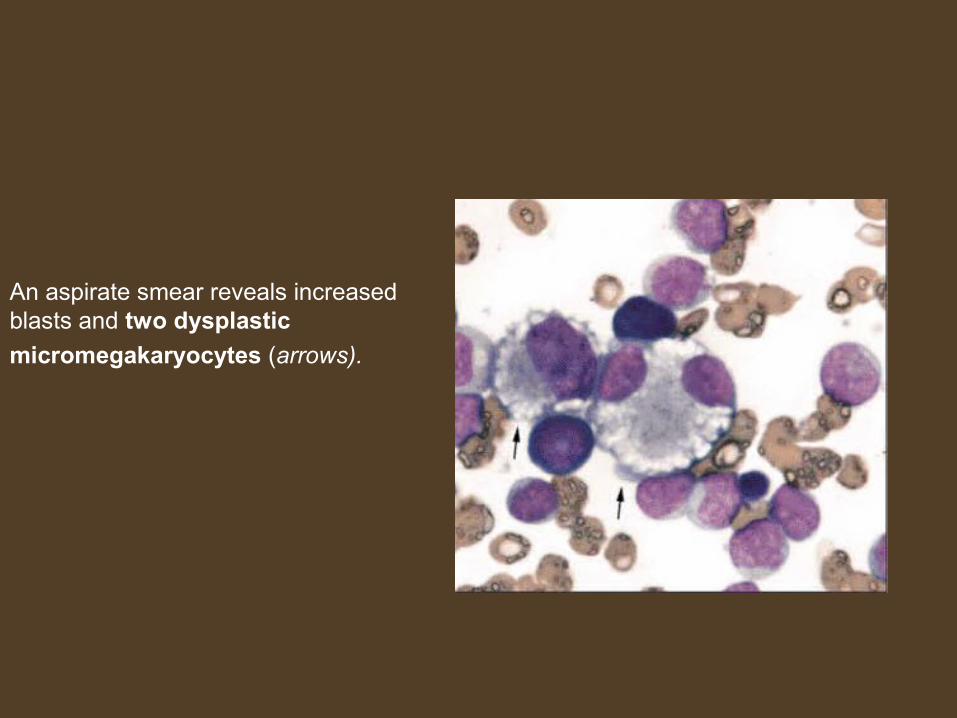
An aspirate smear reveals increased blasts and two dysplastic
micromegakaryocytes (arrows).

AML & MDS therapy related
• Different from denovo AML• Characteristic cytogenetic abnormalities• Multilineage dysplasia• Refractoriness to therapy• Short survival• Follow TOPOISOMERASE II INHIBITOR
( myeloid/lymphoid) OR ALKYLATING AGENTS
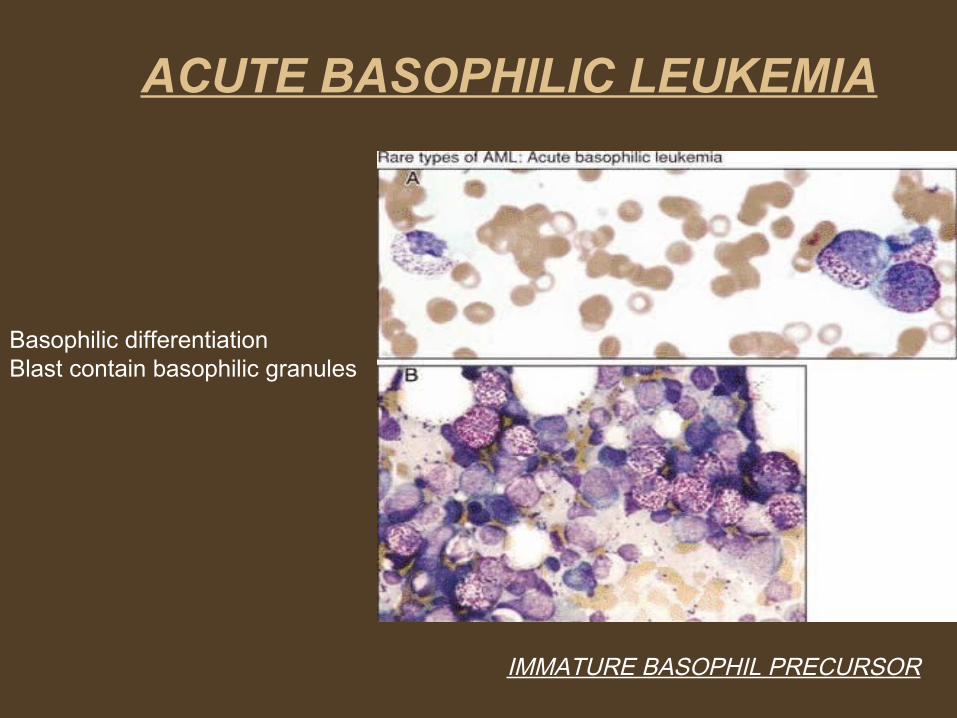
IMMATURE BASOPHIL PRECURSOR
Basophilic differentiationBlast contain basophilic granules
ACUTE BASOPHILIC LEUKEMIA
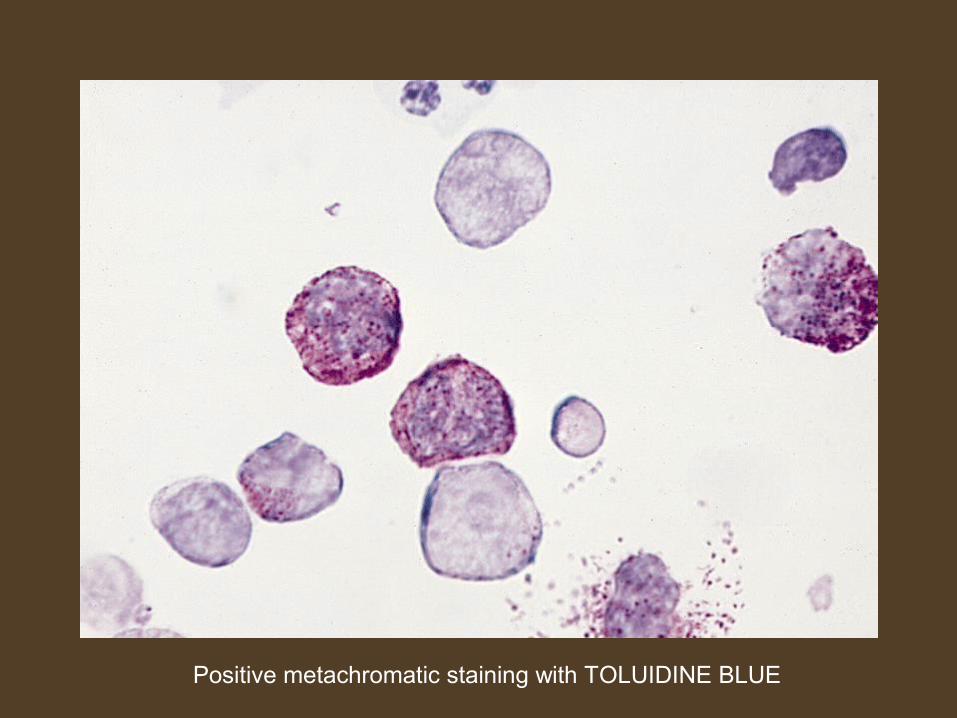
Positive metachromatic staining with TOLUIDINE BLUE

ACUTE PANMYELOSIS WITH MYELOFIBROSIS
• Very rare type.• Median age – 57 to 67 yrs.
• Pancytopenia with < 5% blast.
• No history of preceding myeloproliferative disorder.• Proliferation of all major myeloid cell lines• Dyspalstic changes are present along with fibrosis of bone
marrow
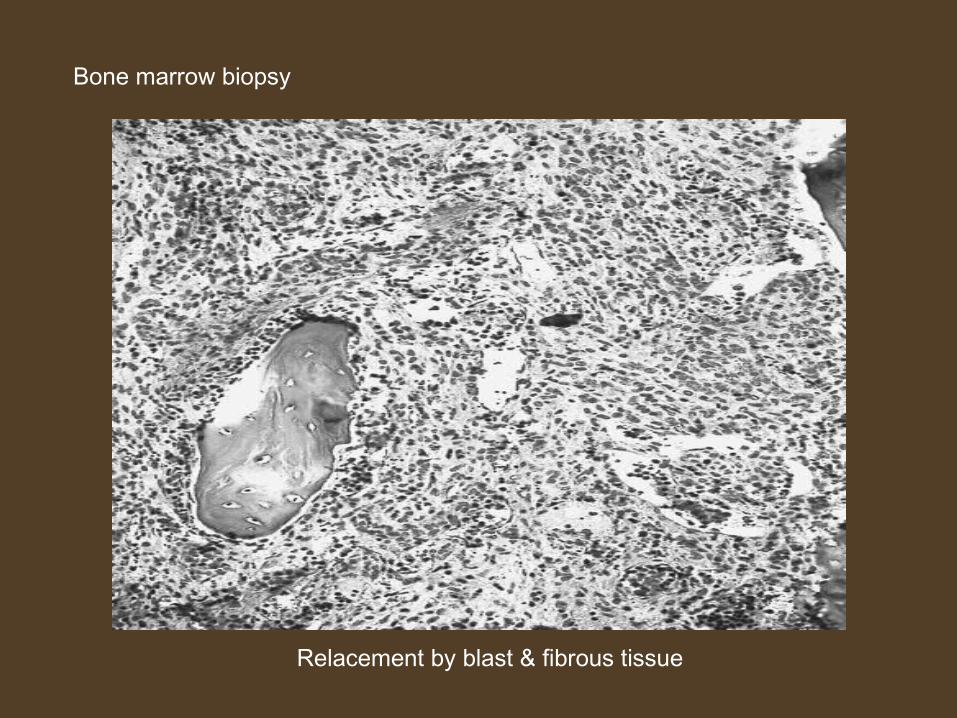
Relacement by blast & fibrous tissue
Bone marrow biopsy

MYELOID (granulocytic) SARCOMA (Myeloblastoma)
• Isolated tumour mass.• Also known as Chloroma because some appear green or
turn green in dilute acid secondary to expression of MPO.
• Composed of myeloblast or immature cells in extramedullary site
• Sign of relapse in a treated case of AML• Common sites – orbits and the paranasal sinuses.• The diagnosis should be suspected if eosinophilic
myelocytes are present in H & E stained biopsy sections.

Differential diagnosis
• 1. Leukaemoid reaction
• 2. Myelodysplastic Syndrome
• 3. Acute Lymphoblastic Leukemia
• 4. Blast crisis of Chronic Myeloid Leukemia

LEUKAEMOID REACTION
• Refers to the presence of markedly increased leucocyte count (>50,000/mm3) and immature white blood cells in peripheral blood resembling leukemia but occurring in non-leukaemic conditions.
• Causes of leukaemoid reaction- Severe bacterial or viral infection. Severe acute haemolysis. Severe haemorrhage Cancer metastatic to bone marrow. Tuberculosis

LEUKAEMOID REACTION
Differentiation from AML is made by following features:
• Clinical presentation.• Presence of underlying disease.• Morphology on blood smear.• % of blasts in bone marrow.
• Correction of leukaemoid blood picture after treatment of underlying disease.

Myelodysplastic syndrome
• Differentiation of AML from MDS depends on proportion of myeloblasts in the bone marrow.
• In AML, myeloblasts are greater than 20%.• In MDS, myeloblasts are less than 20%.• MPO staining may also be useful for diagnosis of MDS
wherein granulocytes may lose MPO reactivity.

ALL Vs AMLALL AML
Age Mainly children Mainly adults
Lymphadenopathy Usually present Usually absent
Gum hypertrophy -ve +ve in M4/M5
Skin infiltration -ve +ve in M4/M5
Granulocytic sarcoma -ve +ve in few cases
Mediastinal mass +ve in T-ALL -
Associated DIC -ve +ve in M3

Blast crisis of CML
• Presence of marked splenomegaly, basophilia and Philadelphia chromosome are suggestive of CML . These features differentiate blast crisis of CML with AML.
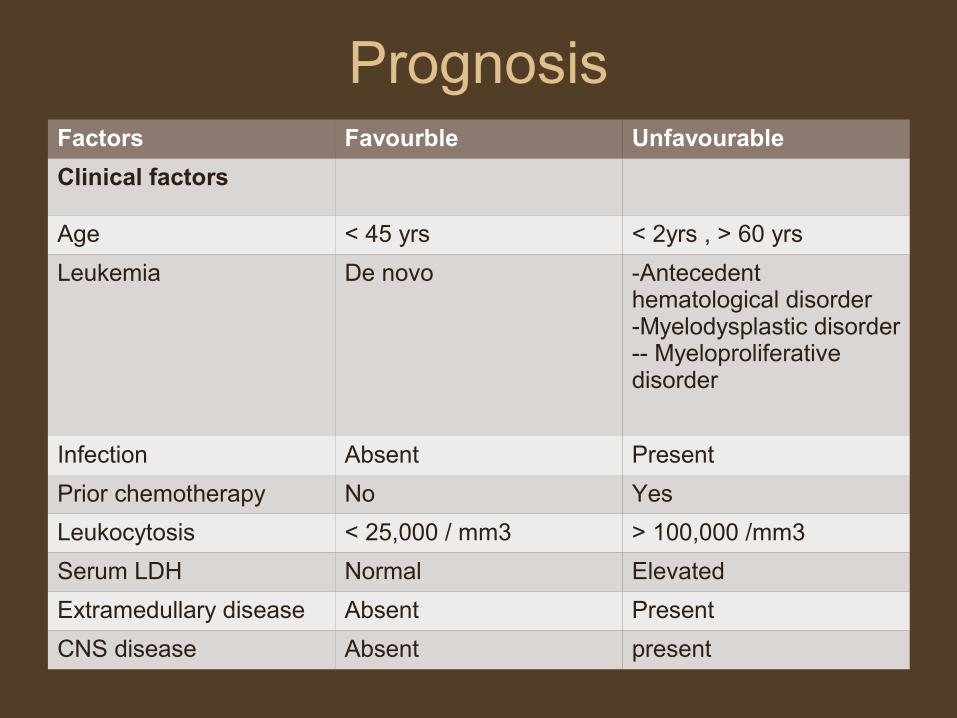
Prognosis Factors Favourble Unfavourable
Clinical factors
Age < 45 yrs < 2yrs , > 60 yrs
Leukemia De novo -Antecedent hematological disorder-Myelodysplastic disorder-- Myeloproliferative disorder
Infection Absent Present
Prior chemotherapy No Yes
Leukocytosis < 25,000 / mm3 > 100,000 /mm3
Serum LDH Normal Elevated
Extramedullary disease Absent Present
CNS disease Absent present

Prognosis
Factors Favourble Unfavourable
Morphology
Auer rods Present Absent
Eosinophils Present Absent
Megaloblastic erythroid Absent Present
Dysplastic megakaryocytes
Absent Present
FAB type M2,M3,M4 M0,M6,M7
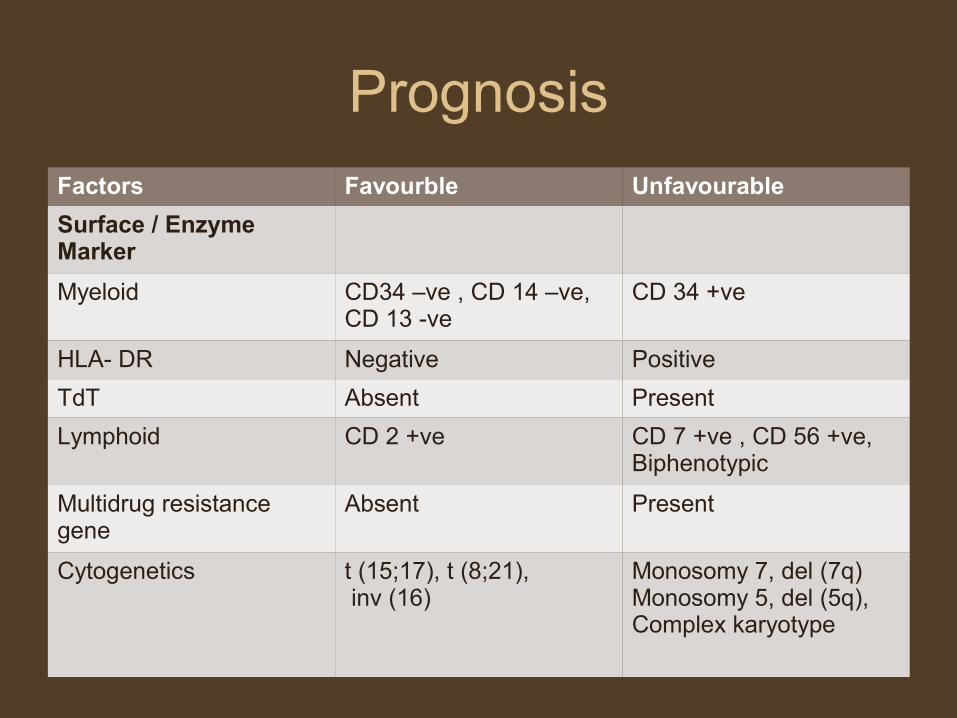
Prognosis
Factors Favourble Unfavourable
Surface / Enzyme Marker
Myeloid CD34 –ve , CD 14 –ve, CD 13 -ve
CD 34 +ve
HLA- DR Negative Positive
TdT Absent Present
Lymphoid CD 2 +ve CD 7 +ve , CD 56 +ve, Biphenotypic
Multidrug resistance gene
Absent Present
Cytogenetics t (15;17), t (8;21), inv (16)
Monosomy 7, del (7q)Monosomy 5, del (5q),Complex karyotype

THANK YOU
Presented by Dr. Monika Nema














![[Ghiduri][Cancer]Acute Myeloid Leukemia](https://img.pdfslide.us/doc/110x75/55cf9686550346d0338c0f55/ghiduricanceracute-myeloid-leukemia.jpg)














