Embed Size (px)
Citation preview

h THEO : CHEM
ELSEVIER Journal of Molecular Structure (Theochem) 334 (1995) 933100
Viewpoint 16
A new approach for an HIV docking-inhibitor drug designed on the basis of the dual recognition/binding hypothesis between the CD4
receptor and the envelope glycoprotein of the HIV
Zolth Sztkelya1b3’
aMoleculclr Aspects of Drug Design Section, MSL. ABL-Basic Research Program, NCI-Frederick Cancer Research and Development
Center, Frederick MD 21702-1201, USA
‘Department of Chemistry. University of Toronto, Toronto, Ontario MSS IA1 Canada
Received 28 December 1994; accepted 23 February 1995
Abstract
A structural hypothesis is presented in this paper for dual binding sites between the gp120 of HIV and the T-cell. One of the binding sites involved electrostatic attraction between positively charged sites (LYs~~, LYS~~ and Args9) of the CD4 and the negatively charged sites (As~~~~, G1u3” and Asp4”) of the gp120. It is suggested that the benzene ring of Phe43 also acts as a lock at this site. The other site involves the galactose ending of one of the oligosaccharide antennas of the CD4 that docks close to one of the SS linkages (perhaps at Cys296 ~SSCys3”) of the gp120. It is suggested that the activated free OH at C4 position of the galactose acts as a nucleophile to break this strategically located S-S linkage. Such a structural alteration leads to a dramatic conformational change that may in fact trigger the fusion process. On the basis of this structural hypothesis a general glycopeptide structure is proposed that could act as a “bivalent” docking- inhibitor drug.
Keywords; CD4; Drug design; gpl20; HIV; HIV docking-inhibitor; Structural hypothesis
1. Introduction
CD4 is a T-cell surface receptor molecule [1,2] which consists of over 400 amino acid resi- dues and two complex oligosaccharide chains.
One of the function of the CD4 receptor, in the immune system, is the recognition of the
major histocompatibility complex II (MHC II)
’ Permanent address: Department of Medicinal Chemistry, Albert Szent-Gybrgyi Medical University, Dam t&r 8, H-6720
Szeged, Hungary.
molecules on the surfaces of the antigen presenting cells [3,4].
In addition, the CD4 serves as a receptor site for
the human immundeficiency virus (HIV) envelope glycoprotein (gp120) at the initial step of the HIV infection [5]. Numerous studies deal with the CD4- gp120 interaction [6] involving amino acid alter- ations (site-directed mutagenesis) for mapping of the important amino acid residues on the molecu- lar surfaces of both the CD4 [7] and the gp120 [8]. The CD4 is divided into four domains (Dl, D2, D3 and D4) and the oligosaccharide chains are
0166-1280/95/$09.50 0 1995 Elsevier Science B.V. All rights reserved
SSDI 0166-1280(95)04238-5

associated with the third (D3) and fourth (D4) domains (Fig. 1). Recombinant fragments of CD4 were investigated by X-ray diffraction (e.g. DlD2
fragment [991 I] and D3D4 fragment [12,13]). On the basis of X-ray data, molecular dynamical simulations were also performed [ 14,151.
To the best of our present understanding [ 161 the site recognition and docking mechanism of the CD4 and the gp120 consists of four steps:
A. Long-range interaction 1. Electrostatic preorientation
B. Short-range interaction 2. Electrostatic attachment 3. “Locking-in” (via aromatic ring orientation)
C. Internal interaction 4. Induced fit (conformational readjustment)
CD4
A
433 amina acid
residues
Dl
D3
04
I I
INTRb&LLULAR
gP 120
c5 v5
TY~
TOP
In step 1, the positively charged Dl domain is attracted to the negatively charged cavity of the
gp120 (cf. Fig. 1). This long-range electrostatic attraction is completed in step 2, which is the
final phase of the orientation resulting from the short-range electrostatic interaction. In step 3 the aromatic ring of the Phe’” may act as a lock. involving aromatic stacking perhaps, in between the aromatic side chains of Trp427 and Tyr4j5 of the gp120 or perhaps by being involved in the formation an aromatic hydrogen bond ((Ph.. .HO-Tyr4s5) or aromatic interaction with Trp4” and hydrogen-bonded interaction with Tyr4j5 (Trp4" Phe HO-Tyr435) as illustrated in Fig. 2. Step 4 is a conformational readjustment that takes about 30min at 37’C [15,17] (while the
QGIU QAsp
331
v3
296
v2
Vi
Cl
Y
275 511
c2 aa2y
residues
Fig. 1. A stretched out schematic representation of the CD4 and gp120. The four domains (Dl,. ,D4) of the CD4 and the five conserved (Cl,. ,C5) and five variable (Vl,. ,V5) regions of gp120 are also shown. The location of the most important three
positively charged amino acids of CD4 and the negatively charged amino acids of gp120 are clearly marked together with the location
of the amino acids with aromatic side chain which are also involved in the first step of binding. The glycosilation sites (271 and 300) of the
human CD4 are also indicated.

95
Fig. 2. A schematic illustration of a possible system of interactions between the CD4 and gpl20 in the first three steps of binding (cf. Al.
B2 and B3 steps in text). The ionic long-range electrostatic preorientation (Al) and short range electrostatic attachment (B2) while the
aromatic moieties are responsible for the short-range “locking in” (B3) are all included in this figure. It should be emphasized that the
pairing of the charged moieties (e.g. Arg” and AsP’~‘) is purely arbitrary.
previous steps are nearly instantaneous). This pro- cess is normally referred to as “induced fit”.
In addition to the CD4 positive T-cells, other human cells may also be infected by the HIV. Such cells are the neural cells [ 18,191 as well as colonic epithelial cells [20]. In these cases the cells have alternative receptor molecules: galactosylcer- amide (GalCer) or its substituted derivatives at the U-OH position of galactose (the sulfatide contains a sulfate group and the GM4 contains an acetyl- neuraminic group linked to the C3-OH group of the galactose). The recognition [19] and subsequent dock- ing of the above-mentioned glycolipid molecules and the gp120 is illustrated schematically in Fig. 3.
Several studies were published to design small molecule mimetics of CD4 [21] or GalCer [22,23] to inhibit the gpl20-host cell interactions. The present paper proposes a substantial extension of the previous work in the most general sense.
2. Structural hypothesis
Each of the two biantennary complex carbohy-
drate chains of the native CD4 (cf. Fig. 1) has the following structures:
Since the structure has been determined by mass spectrometry [24] it is not certain which glyco- sidic linkage is o and which is p. However, the Q and B configurations given above and below the glycosidic linkages in Scheme 1 are in agreement with numerous NMR experimental results of naturally occurring carbohydrates in general and in particular with the NMR results obtained for a diantennary oligosaccharide of the same composition [25].
While in Scheme 1 the two galactose residues (at the left-hand side), in the biantennary structure, are terminated with N-acetyl-neuraminic acid (NeuAc) an appreciable percentage of molecules have no such termination if n + m = 1. Conse- quently, under these conditions there is at least one free galactose ending with free OH groups at positions C2, C4 and C6 as well as at C3. In such a case the structure of the left-hand side of the biantennary carbohydrate chain (Scheme 1) is ana- logous to the structure of the GalCer and GM4 molecules shown in Fig. 3. However, the C3 hydro- xyl group can tolerate considerable substitution. From this, one may conclude that the free C4
hydroxyl is the structure that is essential for binding. Furthermore, the C4-OH has to be axial.
NeuAc,,2 : 3Gall 2 4GlcNAcl 52Manl 36 4 Man1 3 4GlcNAcl 3 4GlcNAcl 2 Asn’ 171 or 300
P NeuAc,2 -+ 3Gall+ 4GlcNAc 1 + 2Man 1 + 3 R ;j: .i 0
Scheme 1

96 Z. Sz&kely/Journal of Molecular Structure (Theochem) 334 (1995) 93-100
Fig. 3. A schematic illustration of the docking of GalCer in the receptor site of gp120. The structural analogy between the “lipid head” in
GalCer and the Galpi-4GlcNAc$-. moiety of the biantennary carbohydrate chain (Scheme I) is illustrated by the broken line
structural feature of a pyranose ring.
as it is in galactose, because glucose, in which that hydroxyl is equatorial, is inactive for gp120 binding
P91. In view of the foregoing, it appears that one of the
carbohydrate side chains of the CD4 may, in fact, be involved in docking to the gp120. The site of this carbohydrate docking is different from the docking
site of the positively charged amino acid side chains of the CD4 which in turn orients itself towards the negatively charged amino acid side chains of the gp120, as discussed above (Fig. 2). The positively charged amino acid residue side chains of the Dl domain of the CD4 receptor have their docking site in the gp120 molecule at about the amino acid residues 368-457.
The structural analogy between the “lipid head” in GalCer and the Galpl-4GclNAcPl-. moiety of the biantennary carbohydrate chain (Scheme 1) is illustrated by the broken line structural feature of a pyranose ring in Fig. 3.
Recently, it has been suggested [26] that the galactose ending of the biantennary structure has its docking site of the gp120 between amino acid residues number 206-275 although others [27] have
questioned the validity of the experimental basis of that assignment for the galactose binding site. More recently Cys296-Cys331 has been suggested [28] as a likely site for initial attack to break up the S-S link- age. Consequently one may assume that this point may in fact be the docking site of the galactose ending of the biantennary structure of the CD4.
In view of the foregoing the two sites may be separated from each other by several tens of angstroms. Owing to the flexibility of the D2-D3 linkage and the biantennary carbohydrate chain it is not easy to determine the distance between the two docking sites. The average distance between the a-carbon atoms of amino acids 35, 46, 59 and
that the a-carbon atom of amino acid 271 (which holds one of the carbohydrate moieties) is 77A. This value may be modified by the flexibilities discussed above. Thus the best estimate for the separation of the two docking sites is 80 f 10 A [16] which is the geometrical basis for the dual recognition or dual binding hypothesis. This dual recognition or dual binding between the CD4 and gp120 is illustrated schematically in Fig. 4.
It may well be that the second docking (involving the free C4 hydroxyl of the galactose ending of the biantennary carbohydrate) is an unimportant component of the overall recognition/binding process. However it may turn out to be essential. In such a hypothetical case one may ask two important questions. (1) Which one of the two carbohydrate antennas is involved in the binding?
(2) What is the role of the C4 hydroxyl group of galactose at he second docking site?
In connection with the first question, one should emphasize that in most species (e.g. human, monkey, dog, cat and mouse etc.) two oligosaccharide anten- nas at Asn 27’ in the D3 domain and one at Asn300 in the D4 domain of CD4 are always present and only
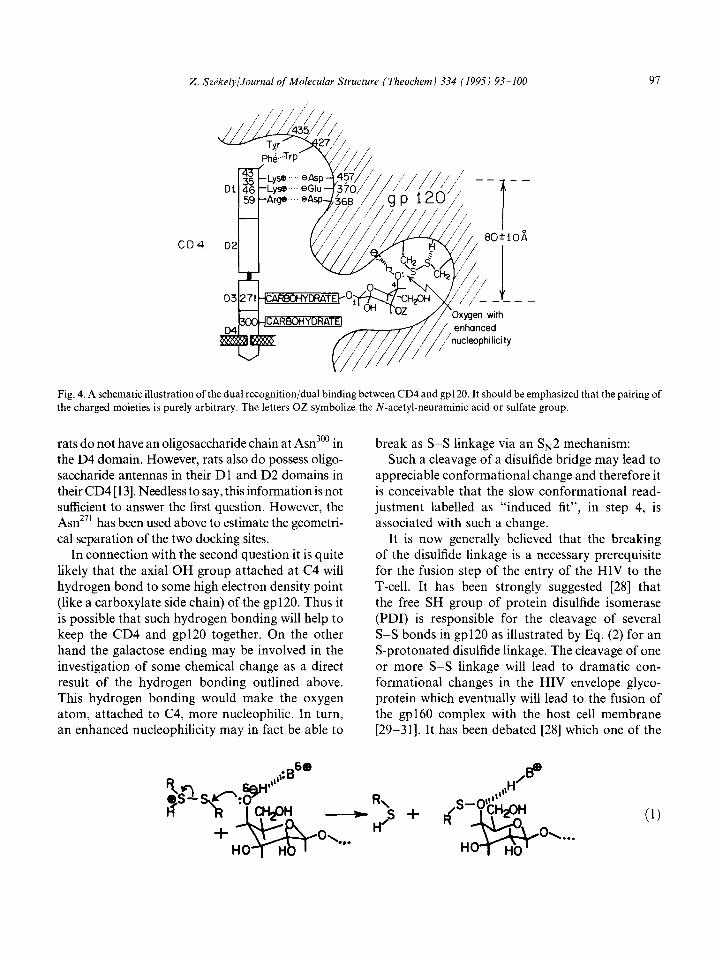
2. SzPkely/Journal of Molecular Struclure (Theochem) 334 (1995) 93-100 91
Fig. 4. A schematic illustration of the dual recognition/dual binding between CD4 and gp120. It should be emphasized that the pairing of
the charged moieties is purely arbitrary. The letters OZ symbolize the N-acetyl-neuraminic acid or sulfate group.
rats do not have an oligosaccharide chain at Asn300 in the D4 domain. However, rats also do possess oligo- saccharide antennas in their Dl and D2 domains in their CD4 [ 131. Needless to say, this information is not sufficient to answer the first question. However, the Asn271 has been used above to estimate the geometri- cal separation of the two docking sites.
In connection with the second question it is quite
likely that the axial OH group attached at C4 will hydrogen bond to some high electron density point (like a carboxylate side chain) of the gp120. Thus it
is possible that such hydrogen bonding will help to keep the CD4 and gp120 together. On the other hand the galactose ending may be involved in the
investigation of some chemical change as a direct result of the hydrogen bonding outlined above. This hydrogen bonding would make the oxygen atom, attached to C4, more nucleophilic. In turn, an enhanced nucleophilicity may in fact be able to
break as S-S linkage via an $2 mechanism: Such a cleavage of a disulfide bridge may lead to
appreciable conformational change and therefore it is conceivable that the slow conformational read- justment labelled as “induced fit”, in step 4, is associated with such a change.
It is now generally believed that the breaking of the disulfide linkage is a necessary prerequisite for the fusion step of the entry of the HIV to the T-cell. It has been strongly suggested [28] that the free SH group of protein disulfide isomerase (PDI) is responsible for the cleavage of several S-S bonds in gp120 as illustrated by Eq. (2) for an S-protonated disulfide linkage. The cleavage of one or more S-S linkage will lead to dramatic con- formational changes in the HIV envelope glyco- protein which eventually will lead to the fusion of the gp160 complex with the host cell membrane [29-311. It has been debated [28] which one of the
R\ - s+
H’ 4 (11
Oh. OX...

98
__t H S-gp 120~-, I^,
!aPQO
nine disulfide linkages [32] is involved. Eventually, the number of SS linkages were narrowed down to four at the following positions: 119-225, 296- 331, 378-445 and 385-418. The last two of these (378-445 and 384-418) were shown to be involved only in the later steps.
Thus the 296-331 disulfide linkage that is at the base of the V3 loop was regarded as the most likely site for the initial binding between the gp120 and the CD4. However, all of these presuppose that the 296-33 1 disulfide bridge is sufficiently exposed for the PDI to attack. Since more than half of the mass
of the gp120 is tied up in surface carbohydrate antennas, one cannot be all that certain that the PDI can make the initial attack at any one of the SSS linkages (like the one at 296633 1) and initiate the cleavage of that particular disulfide linkage according to Eq. (2). After the first disulfide bridge is somehow broken, the gp160 may undergo suffi- cient conformational changes so that the PDI may be able to access the other S-S bonds. In this sense the carbohydrate antenna of the CD4 with its galactose ending may act like an “ignition key” that will initiate the fusion process.
The feasibility of breaking the S-S linkage by nucleophiles including -SH and -OH functional- ities, which is activated by hydrogen bonding, was studied recently by computational methods [33].
3. Theoretical and experimental verification of the structural hypothesis
A theoretical approach via molecular com- putations could shed some light on the role of
CBS- PDI d
galactose after docking which is envisaged to take place via nucleophilic cleavage of the S-S bond
(cf. Eq. (1)). One could evaluate the nucleophili- city of the C4-OH of galactose on its own, with (I) and without (II) internal hydrogen bond- ing, as well as in complexation with an acetate ion (III) that in turn may be regarded as a suitable model for an aspartate or glutamate side chain.
Such an assessment of oxygen nucleophilicity
can be carried out, as has been done previously on pentafuranose [34], using the Bader-type elec- tron density analysis. In addition, the energy differ- ences for reaction (1) where the nucleophilic base denoted by :B. computed for the above three nucleophilic bases (I, II and 111) must also indicate to what extent the nucleophilicity of the galactose C4-OH is enhanced. Whether the reaction is favored, in the thermodynamic sense, depends on the relative stability of the RS-O(H:B)-Gal reac- tion product, where in the case III, H:B is acetic acid (CHs-COOH).
It is perhaps more difficult to establish which carbohydrate antenna of the CD4 is the reactive one. It could be the one at the As?” glycosylation site, it could be the one at the Asn300 glycosylation
site or both may be equally effective. A definitive answer to this question could be
obtained if by mutation one or both of the two sites were to be replaced by alanine so that only one or both of these sites might be free from glycosylation [3,4]. In this way three possibilities might exist in addition to the wild type as shown in Table 1. The generation of these point mutants
(3)

Z. SzPkel.v,‘Journal qf Molecular Structure iTheochenz,i 334 (1995) 93-100 99
Table 1
The wild type and possible point mutants of CD4
CD4
Wild type
Mutants
271
Asn
Asn
Ala
Ala
300 No. of sugar chains
Asn 2
Ala 1
Asn 1
Ala 0
seems to be absolutely essential in finding the answer to the question posed earlier.
4. Practical application of the binding hypothesis
A knowledge of the geometrical parameters of such a dual docking as illustrated in Fig. 4 is essen- tial for designing a drug that would aim to block both receptor sites; the ionic one as well as the
second one with the carbohydrate (i.e. galactose) ending.
It is proposed therefore that a cyclopeptide may be synthesized that contains two lysine and one
arginine residues as well as one phenylalanine strategically located in the cyclopeptide. The size of the ring is crucial so that it may fit into the negatively charged cavity of the gp120. The location of these four amino acids in the cyclo- peptide is also crucial because it should mimic the geometrical arrangements of these four amino acid side chains in the CD4 molecule. It is not likely
that the size of the cyclopeptide could be smaller than that of a cyclohexapeptide. A linear peptide chain of Ser-(Gly),- should also be attached to this cyclopeptide so that a galactose could be linked to the serine side chain in the form of an 0-glycoside. The structure for such a molecule, ful- filling the dual binding hypothesis, is shown sche- matically in Fig. 5. The size of this molecule (i.e. the values of i,j, k, 1, m and n) should come from a detailed structural analysis of the CD4 molecule.
If the dual recognition/binding hypothesis is valid and the dimensions of the proposed molecule and the size and location of the gp120 cavities are comparable then it could be an effective dock- ing inhibition. However, even if dual binding is
not necessary for docking, the above molecule (Fig. 5) could be an effective drug either because it binds with the positively charged phenylalanine-
I I
c=o
Fig. 5. A proposed general structure for a docking-inhibitor
drug based on the dual binding hypothesis between CD4 and
gp 120.
containing cyclopeptide at the appropriate binding site of the gp120 or because it binds with its galac- tose tail at the other receptor site of the gp120.
Synthetic work is now in progress to produce the above proposed molecule.
Acknowledgments
The financial support of the Hungarian Science Foundation (OTKA F13060) as well as a grant for computational equipment provided by the Higher Educational Development Foundation (FEFA HU-525) are gratefully acknowledged.
The author is grateful for an Eiitvos Fellowship granted by the National Scholarship Board of the Ministry of Culture and Education (Hungary) as well as for a fellowship provided by the Word Bank (W15085).
Thanks are due to Drs. B. Penke, J. Molnir
(A. Szent-Gyorgyi Medical University, Szeged, Hungary) and A. Perczel (Lorand Eotvos Univer- sity, Budapest, Hungary) for helpful discussions

100 2. Szikely/Joumal of Molecular Structure i Theochem) 334 (1995) 93% IO0
and for their moral support. Special thanks are due to Drs. A. Aszalos (Food and Drug Admini- stration, Washington DC, USA), C. Michejda (National Cancer Institute, Frederick, MD, USA) and I.G. Csizmadia (University of Toronto, Tor- onto, Canada) for their helpful discussions as well as for their hospitalities in their respective insti- tutions where the writing of the present paper has been completed.
References
[l] S.C. Harrison, .I. Wang, Y. Yan, T. Garrett, J. Liu, U.
Moebius and E. Reinherz, Cold Spring Harbor Symp.
Quant. Biol., 57 (1992) 541-548.
[2] S.C. Harrison, Act. Chem. Res., 26 (1993) 449-453.
[3] IJ. Moebius, L.K. Clayton, S. Abraham, A. Denier, J.J.
Yunis, S.C. Harrison and E.L. Reinherz, Proc. Natl.
Acad. Sci. USA, 89 (1992) 12008-12012.
[4] U. Moebius, P. Pallai, S.C. Harrison and E.L. Reinherz.
Proc. Natl. Acad. Sci. USA, 90 (1993) 8259-8263.
[5] A.G. Dagleish, P.C.L. Beverly, P.R. Clapham. D.H. Craw-
ford, M.F. Greaves and R.A. Weiss, Nature (London), 312
(1984) 7633767.
[6] J.P. Moore and R.W. Sweet, Perspect. Drug Des. (Suppl.
of Cornput-Aided Drug Des.), 1 (1993) 235-250.
[7] U. Moebius, L.K. Clayton, S. Abraham, S.C. Harrison and
E.L. Reinherz, J. Exp. Med., 176 (1992) 5077517.
[8] U. Olshevsky, E. Helseth, C. Furman, _I. Li, W. Haseltine
and J. Sodroski, J. Virol., 64 (1990) 5701-5707.
[9] J. Wang, Y. Yan, T.P.J. Garrett, J. Liu, D.W. Rodgers,
R.L. Garlick, G.E. Tarr, Y. Hussain, El. Reinherz and
S.C. Harrison, Nature, 348 (1990) 411-418.
[lo] S.-E. Ryu, P.D. Kwong, A. Truneh, T.G. Porter, J. Arthos,
M. Rosemberg, X. Dai. N. Xuong, R. Axel, R.W. Sweet
4$8 W.A. Hendrickson, Nature, 348 (1990) 419-
[I l] T.P.J. Garrett, J. Wang, Y. Yan, J. Liu and S.C. Harrison,
J. Mol. Biol., 234 (1993) 763-778.
[12] R.L. Brady, E.J. Dodson, G.G. Dodson, G. Lange, S.J.
Davis, A.F. Williams and A.N. Barclay, Science, 260
(1993) 9799983.
[13] G. Lange, S.J. Lewis, G.N. Murshudov, G.G. Dodson,
P.C.E. Moody, J.P. Turkenburg, A.N. Barclay and R.L.
Brady, Structure, 2 (1994) 4699481.
[14] Z. Szekely, A. Perczel, B. Penke and J. Molnar, J. Mol.
Struct. (Theochem), 286 (1993) 1655182.
1151 L.M. Ptaszek, S. Vijayakumar, G. Ravishanker and D.L.
Beveridge, Biopolymers, 34 (1994) 1145-l 153.
[23] C.R. Bertozzi, P.D. Hoeprich, Jr., and M.D. Bednarski, J.
Org. Chem., 57 (1992) 609226094.
~241
v51
WI
v71
WI
1291
1301
[311
1321
[331
[341
S.A. Carr, M.E. Hemling, G. Folena-Wasserman, R.W.
Sweet, K. Anumula, J.R. Barr, M.J. Huddleston and
P. Taylor, J. Biol. Chem., 264 (1989) 21286621295.
P. de Waard, B.R. Leeflang, J.F.G. Vliegenhart, R. Boe-
lens, G.W. Vuister and R. Kaptein, J. Biomol. NMR, 2
(1992) 21 l-226.
S. Bhat, R.V. Mettus, E.P. Reddy, K.E. Ugen, V. Sri-
kanthan, W.V. Williams and D.B. Weiner, AIDS Res.
Human Retroviruses, 9 (1993) 175-181.
D.G. Cook, J. Fantini, S.L. Spitalnik and F. Gonzalez-
Scarano, Virology, 201 (1994) 2066214.
H.J.-P. Ryser, E.M. Levy, R. Mandel and G.J. DiSciullo,
Proc. Natl. Acad. Sci. USA, 91 (1994) 455994563.
C. Larsen, H. Ellens and J. Bentz, in R.C. Aloia and C.C.
Curtin (Eds.), Membrane Interactions of HIV, Wiley-Liss,
New York, 1992, pp. 143-166.
L.E. Eiden and J.D. Lifson, Immunol. Today, 13 (1992)
201-207.
D.J. Capon and R.H.R. Ward, Annu. Rev. Immunol.. 9
(1991) 6499678.
C.K. Leonard, M.W. Spellman, L. Riddle, R.J. Harris,
J.N. Thomas and T.J. Gregory, J. Biol. Chem., 265
(1990) 10373310382.
W. Viviani, M. Loos, J.-L. Rivail, I. Jalsovszky, A. Perczel
and I.G. Csizmadia, J. Mol. Struct. (Theochem) 1995, in
press.
T.H. Tang, D.M. Whitfield, S.P. Douglas, J.J. Krepinsky
and I.G. Csizmadia, Can. J. Chem., 72 (1994) 180331815.
[16] 2. Szekely, Z. Konya, A. Becskei, A. Perczel, B. Penke,
J. Molnar, A. Aszalos, I.G. Csizmadia, J. Mol. Struct.
(Theochem), in press,
[17] L.A. Lasky, G. Nakamura, D.H. Smith, C. Fennie,
C. Shimasaki, E. Patzer, P. Berman, T. Gregory and
D.J. Capon, Cell, 50 (1987) 9755985.
[18] J.M. Harouse, S. Bhat, S.L. Spitalnik, M. Laughlin,
K. Stefano, D.H. Silberberg and F. Gonzalez-Scarano,
Science, 253 (1991) 320-323.
[19] S. Bhat, S.L. Spitalnik, F. Gonzalez-Scarano and
D.H. Silberberg, Proc. Natl. Acad. Sci. USA, 88 (1991)
7131-7134.
(201 J. Fantini, D.G. Cook, N. Nathalson, S.L. Spitalnik and F. Gonzalez-Scarano, Proc. Natl. Acad. Sci. USA, 90
(1993) 2700-2704.
[21] S. Ma, M.J. McGregor, F.E. Cohen and P.V. Pallai, Bio-
polymers, 34 (1994) 98771000.
[22] C.R. Bertozzi, D.G. Cook, W.R. Kobertz, F. Gonzalez-
Scarano and M.D. Bednarski, J. Am. Chem. Sot., 114
(1992) 10639-10641.





























