Embed Size (px)
Citation preview

doi:10.1182/blood-2013-06-508267Prepublished online October 2, 2013;2013 122: 3713-3722
Frank Ruemmele and Alain FischerRieux-Laucat, Nicole Brousse, Frederic Davi, Véronique Baud, Christoph Klein, Bertrand Nadel,Jean-Laurent Casanova, Stéphane Blanche, Capucine Picard, Olivier Hermine, Frederic Radford-Weiss, Vahid Asnafi, Dhaarini Murugan, Christine Bole, Patrick Nitschke, Olivier Goulet,Suarez, Julien Masliah-Planchon, Katy Billot, Danielle Canioni, Pierre Frange, Isabelle Bénédicte Neven, Emilie Mamessier, Julie Bruneau, Sophie Kaltenbach, Daniel Kotlarz, Felipe deficiencyA Mendelian predisposition to B-cell lymphoma caused by IL-10R
http://bloodjournal.hematologylibrary.org/content/122/23/3713.full.htmlUpdated information and services can be found at:
(396 articles)Plenary Papers � (290 articles)Pediatric Hematology �
(1579 articles)Lymphoid Neoplasia � (5128 articles)Immunobiology �
Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Washington DC 20036.by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

Plenary Paper
LYMPHOID NEOPLASIA
A Mendelian predisposition to B-cell lymphoma caused byIL-10R deficiencyBenedicte Neven,1-4 Emilie Mamessier,5 Julie Bruneau,3,6 Sophie Kaltenbach,2-4,7 Daniel Kotlarz,8 Felipe Suarez,9
Julien Masliah-Planchon,7 Katy Billot,3,10,11 Danielle Canioni,6 Pierre Frange,1,3 Isabelle Radford-Weiss,7 Vahid Asnafi,12
Dhaarini Murugan,8 Christine Bole,3,13 Patrick Nitschke,3,14 Olivier Goulet,15 Jean-Laurent Casanova,1,3,4,16
Stephane Blanche,1,3 Capucine Picard,1,3,4,16,17 Olivier Hermine,3,8,18 Frederic Rieux-Laucat,2-4 Nicole Brousse,5
Frederic Davi,19 Veronique Baud,3,10,11 Christoph Klein,8 Bertrand Nadel,5 Frank Ruemmele,3,4,15,20 and Alain Fischer1-4
1Unite d’immuno-hematologie pediatrique, Hopital Necker-Enfant Malades, Assistance Publique des Hopitaux de Paris (APHP), Paris, France;2Developpement normal et pathologique du systeme immunitaire, Institut National de la Sante et de la Recherche Medicale (INSERM) U768, Paris, France;3Universite Descartes, Sorbonne Paris Cite, Paris, France; 4Institut Imagine, Paris, France; 5Centre d’immunologie de Marseille Luminy, Universite
Aix-Marseille, INSERM U1104, Marseille, France; 6Service d’anatomopathologie, and 7Service de cytogenetique, Hopital Necker-Enfants Malades, APHP,
Paris, France; 8Department of Pediatrics, Dr. von Hauner Children’s Hospital, Ludwig Maximilian University, Munich, Germany; 9Service d’hematologie
adulte, Hopital Necker-Enfants Malades, APHP, Paris, France; 10INSERM, U1016, Institut Cochin, Paris, France; 11Centre National de la Recherche
Scientifique (CNRS), UMR 8104, Paris, France; 12Laboratoire d’hematologie, Hopital Necker-Enfants Malades, APHP, Paris, France; 13Plateforme
Genomique and 14Plateforme de bioinformatique de l’Institut Imagine, Hopital Necker, Paris, France; 15Service de gastro-enterologie pediatrique, Hopital
Necker-Enfants Malades, APHP, Paris, France; 16Laboratoire de genetique humaine des maladies infectieuses, INSERM U980, Faculte Necker, Paris,
France; 17Centre d’Etude des Deficits Immunitaires, APHP, Paris, France; 18Cytokines, hematopoıese et reponses immunes, CNRS UMR 8147, Paris,
France; 19Laboratoire d’hematologie, Hopital de la Pitie Salpetriere, APHP, Paris, France; and 20Interaction de l’epithelium intestinal et du systeme
immunitaire, INSERM U989, Hopital Necker-Enfants Malades, Paris, France
Key Points
• Human inherited IL-10receptor deficiency isassociated with a very highrisk of non-EBV–relateddiffuse large B-celllymphoma.
• IL-10 signaling may beinvolved in the immunecontrol of germinal centerB-cell lymphoma.
Monogenic interleukin-10 (IL-10) and IL-10 receptor (IL-10R) deficiencies cause very early
onset severe inflammatory bowel disease. Here, we report that 5 patients with an IL-10R1
(n5 1) or IL-10R2 (n5 4) deficiency developed B-cell non-Hodgkin lymphoma between the
ages of 5 and 6 years (whichwas recurrent in 1 patient). These lymphomas had someof the
characteristics of diffuse large B-cell lymphomas and containedmonoclonal, Epstein-Barr
virus–negative germinal center B cells. The tumors displayed a remarkably homogeneous
signature, with original activation of the nuclear factor kB pathway and a decrease in
intratumor T-cell infiltration. Hence, IL-10R deficiency is associated with a high risk of
developing B-cell lymphoma. Our results revealed an unexpected role of the IL-10R
pathway in lymphomagenesis. (Blood. 2013;122(23):3713-3722)
Introduction
Inflammatory bowel disease (IBD) encompasses a heterogeneousgroup of diseases that are characterized by chronic intestinal in-flammation with a complex etiology.1-3 Very-early-onset IBDs(VEO-IBDs) are particularly severe, treatment-resistant conditions.4,5
Loss-of-function mutations in one or another of the interleukin-10(IL-10) receptor’s two chains (IL-10R1 and IL-10R2) or in IL-10itself are detected in about 20% of VEO-IBD patients.6-10 Studies ofIL-10/IL-10R knockout mice have shown that this cytokine is a keycheckpoint for maintaining immune homeostasis toward intes-tinal microbiota.11,12
Chronic intestinal inflammation is a known risk factor for thedevelopment of malignancies.13 Infection with Epstein-Barr virus(EBV) and long-term administration of immunosuppressive medi-cation are also associated with a slight increase in the risk of lym-phoma in young children with colitis14 and in adults with IBD.15 AMendelian predisposition to B-cell lymphoma16 has been observed insome primary immunodeficiencies related to DNA repair (such asataxia-telangiectasia17) and dominant-negative STAT3 mutations18,19
but not previously in the monogenic form of IBD. Here, we report onthe occurrence and recurrence of diffuse large B-cell lymphomas
Submitted June 12, 2013; accepted August 14, 2013. Prepublished online as
Blood First Edition paper, October 2, 2013; DOI 10.1182/blood-2013-06-
508267.
E.M., J.B., and S.K. contributed equally to this work.
B.N. and F.R. contributed equally to this work.
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge
payment. Therefore, and solely to indicate this fact, this article is hereby
marked “advertisement” in accordance with 18 USC section 1734.
© 2013 by The American Society of Hematology
BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23 3713
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

(DLBCLs) in 5 children with IL-10R1 (n5 1) or IL-10R2 deficiencies(n5 4). None of the DLBCLs was related to EBV infection. Our datastrongly suggest the existence of a direct relationship between IL-10Rdeficiency and the development of B-cell lymphomas.
Patients and methods
Of 18 children being monitored for VEO-IBD at Necker Children’s Hospital(Paris, France), 7 were diagnosed with a defect in the IL-10 pathway (IL-10R1, n 5 1; IL-10R2, n 5 6). Similarly, IL-10 pathway defects werediagnosed in 18 children (IL-10R1, n5 7; IL-10R2, n5 8; IL-10, n5 3) of60 with VEO-IBD treated at Munich Children’s Hospital (Kotlarz et al9 andpersonal data). Four of the 14 patients with IL-10R2 deficiency and 1 of the 8patients with IL-10R1 deficiency developed lymphomas. The clinical char-acteristics of the 5 patients with VEO-IBD who subsequently developedlymphoma (P1 through P5) are summarized in supplemental Table 1. Thefirst signs of colitis (bloody diarrhea) were observed between 2 and 12 weeksof age. The patients subsequently developed recurrent stomatitis and perinealinflammation. In P1, P2, P3, and P5, several episodes of perianal abscesseswere observed. All of the patients except P5 had recurrent flare-ups ofcutaneous folliculitis, which had also started during the first year of life.
Immunophenotype, T-cell function, immunoglobulin levels, and antibodyfunctions of these patients were normal. In view of the persistent colitis, fourpatients were treated with steroids and various immunosuppressive drugs(azathioprine, n 5 4; cyclosporine, n 5 2; mycophenolate mofetil andanti–tumor necrosis factor a monoclonal antibodies [infliximab]) atbetween 1.5 and 5 years of age. P5 did not receive any immunosuppressivedrugs prior to lymphoma onset. In view of the poor observed long-termremission, colectomy was performed in P1, P3, and P4 at between 3 and 4years of age (Figure 1). Furthermore, P4 had brachysyndactyly of bothhands. All participants or their parents/guardians gave their writteninformed consent to participation in accordance with the Declaration ofHelsinki. The study was approved by the Comite Consultatif Pour laRecherche Biologique.
Details of the IL10RA and IL10RB gene mutations found in these 5patients and the functional validation of these mutations (ie, the absence ofIL-10R expression or an in vitro response to IL-10) are given in Figure 1and supplemental Data. We further characterized these lymphomas by per-forming immunohistochemical studies, genome-wide array-based comparativegenomic hybridization (aCGH) (n5 4), cytogenetic tests, fluorescence in situhybridization (FISH) analysis (n5 4), and gene expression profiling (n5 3) ontumor samples. Furthermore, whole-exome sequencing of tumor DNA andcounterpart genomic DNA was performed in 3 tumors from 2 patients. Themethods are described in the supplemental Data. The study was conducted inaccordance with the Declaration of Helsinki.
Figure 1. Genetic characteristics, time course of
lymphoma development, and treatment in patients
with IL-10R deficiency. (A) Pedigrees of the 5 patients
with IL-10R1– and IL-10R2–deficient patients. Consan-
guinity (double horizontal bars), affected individuals
(black boxes and circles), carriers (half-filled boxes and
circles), and subjects not available for participation in the
study (asterisks) are indicated. P1 carried a homozygous
missense mutation in exon 2 (p.Y59C) of the IL-10RB
gene. Thismutationwas absent from genome databases
(including HGMD, Ensembl, and 1000 Genomes). P2
was a heterozygous composite with a frameshift muta-
tion in exon 7 (F269fsX275) and a missense mutation in
exon 5 (p.W204C) of IL-10RB. P3 carried an homozy-
gous nonsensemutation in exon 3 of IL-10RB (p. E141X,
as previously reported by Begue et al8). P4 displayed
homozygous deletion (g.11930-17413 del) in IL-10RB.
P5 carried a homozygous c.368-10 C.G mutation in
intron 3 of IL-10RA. (B) Positions of the IL-10RA and IL-
10RB mutations within the gene sequence. (C) Time
course of lymphoma development and treatment. Immu-
nosuppressive treatment with azathioprine is repre-
sented as a gray rectangle, and administration of an
anti–tumor necrosis factor antibody is shown in blue.
Maintenance therapy with 6-mercaptopurine and
methotrexate was given in P4 after remission of L3
and is represented by a yellow rectangle. P1 (IL-10R2)
died as a result of the progression of EBV-positive
Hodgkin-like lymphoproliferative disease (LPD) (de-
spite chemotherapy); P2 (IL-10R2) died of meningo-
encephalitis (of undetermined etiology) that occurred
during chemotherapy; P3 (IL-10R2) was in remission
from lymphoma when he received a transplant from
a mismatched family donor and is now alive and well
12 months after HSCT; P4 (IL-10R2) was in remission
from L4 when he received a transplant from a geno-
identical sibling. He is alive and well 18 months after
HSCT. P5 (IL-10R1) is in remission from lymphoma
and is alive and well 3.4 years after completion of
chemotherapy (HSCT is pending). wt, wild-type; yrs,
years; occurrence shown by arrows (↓).
3714 NEVEN et al BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom
Results
Characteristics of lymphomas in 5 IL-10R–deficient patients
P1 through P5 all developed high-grade B-cell non-Hodgkin lym-phoma (NHL) at between 5.5 and 6.5 years of age (Figure 1). Threefurther lymphomas occurred in P4 at 9.5, 12.1, and 16.5 years of age(referred to as P4-L2, P4-L3, and P4-L4, respectively) (Figure 1).The B-cell lymphomas were found at various locations withinabdominal lymph nodes (n 5 7), thoracic lymph nodes (n 5 3),spleen (n 5 5), liver (n 5 3), and bone (n 5 2) (Table 1). Lym-phoproliferative mucosal lesions were not documented. All lym-phomas were treated with chemotherapy, which was combined in allbut one case (P1-L1) with administration of an anti-CD20 mono-clonal antibody (Table 1). P1 was in long-term remission when anultimately fatal EBV-related lymphoproliferative disease occurred4.5 years after chemotherapy. P2 died during chemotherapy asa result of meningoencephalitis of unknown etiology. P3underwent hematopoietic stem cell transplantation (HSCT) froma mismatched family donor 12 months after the completion ofeffective chemotherapy and is currently (12 months post-HSCT)in remission. Two months after remission of his fourth lymphoma,P4 underwent HSCT from a geno-identical sibling and is currently(18 months post-HSCT) in remission. P5 is currently in remission
(3.3 years after the completion of chemotherapy), and HSCT is beingconsidered. Data on immunoglobulin H (IgH) sequences, cytogenetictest results, and aCGH data on tumor cells from P4 confirmed that thefirst 3 lymphomas (at least) were distinct and had occurred sequentially.
Immunohistochemical characteristics of the B-cell lymphomas
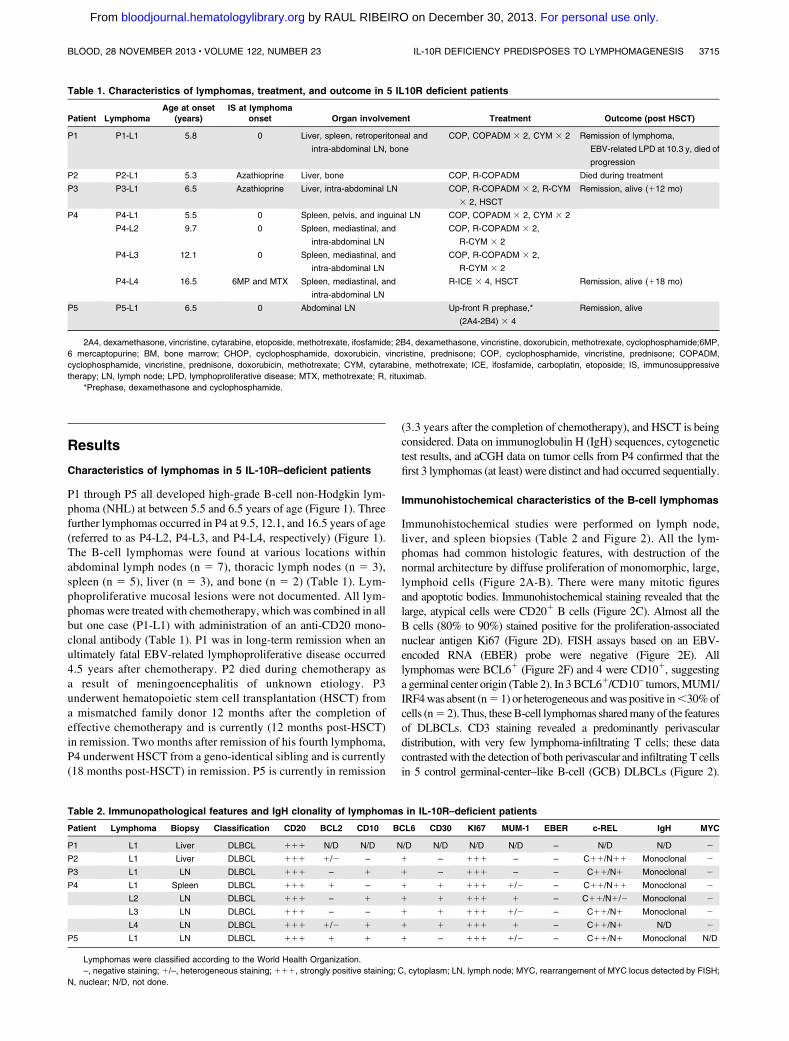
Immunohistochemical studies were performed on lymph node,liver, and spleen biopsies (Table 2 and Figure 2). All the lym-phomas had common histologic features, with destruction of thenormal architecture by diffuse proliferation of monomorphic, large,lymphoid cells (Figure 2A-B). There were many mitotic figuresand apoptotic bodies. Immunohistochemical staining revealed that thelarge, atypical cells were CD201 B cells (Figure 2C). Almost all theB cells (80% to 90%) stained positive for the proliferation-associatednuclear antigen Ki67 (Figure 2D). FISH assays based on an EBV-encoded RNA (EBER) probe were negative (Figure 2E). Alllymphomas were BCL61 (Figure 2F) and 4 were CD101, suggestinga germinal center origin (Table 2). In 3BCL61/CD10– tumors,MUM1/IRF4was absent (n5 1) or heterogeneous andwas positive in,30%ofcells (n5 2). Thus, theseB-cell lymphomas sharedmany of the featuresof DLBCLs. CD3 staining revealed a predominantly perivasculardistribution, with very few lymphoma-infiltrating T cells; these datacontrasted with the detection of both perivascular and infiltrating T cellsin 5 control germinal-center–like B-cell (GCB) DLBCLs (Figure 2).
Table 1. Characteristics of lymphomas, treatment, and outcome in 5 IL10R deficient patients
Patient LymphomaAge at onset
(years)IS at lymphoma
onset Organ involvement Treatment Outcome (post HSCT)
P1 P1-L1 5.8 0 Liver, spleen, retroperitoneal and
intra-abdominal LN, bone
COP, COPADM 3 2, CYM 3 2 Remission of lymphoma,
EBV-related LPD at 10.3 y, died of
progression
P2 P2-L1 5.3 Azathioprine Liver, bone COP, R-COPADM Died during treatment
P3 P3-L1 6.5 Azathioprine Liver, intra-abdominal LN COP, R-COPADM 3 2, R-CYM
3 2, HSCT
Remission, alive (112 mo)
P4 P4-L1 5.5 0 Spleen, pelvis, and inguinal LN COP, COPADM 3 2, CYM 3 2
P4-L2 9.7 0 Spleen, mediastinal, and
intra-abdominal LN
COP, R-COPADM 3 2,
R-CYM 3 2
P4-L3 12.1 0 Spleen, mediastinal, and
intra-abdominal LN
COP, R-COPADM 3 2,
R-CYM 3 2
P4-L4 16.5 6MP and MTX Spleen, mediastinal, and
intra-abdominal LN
R-ICE 3 4, HSCT Remission, alive (118 mo)
P5 P5-L1 6.5 0 Abdominal LN Up-front R prephase,*
(2A4-2B4) 3 4
Remission, alive
2A4, dexamethasone, vincristine, cytarabine, etoposide, methotrexate, ifosfamide; 2B4, dexamethasone, vincristine, doxorubicin, methotrexate, cyclophosphamide;6MP,
6 mercaptopurine; BM, bone marrow; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisone; COP, cyclophosphamide, vincristine, prednisone; COPADM,
cyclophosphamide, vincristine, prednisone, doxorubicin, methotrexate; CYM, cytarabine, methotrexate; ICE, ifosfamide, carboplatin, etoposide; IS, immunosuppressive
therapy; LN, lymph node; LPD, lymphoproliferative disease; MTX, methotrexate; R, rituximab.
*Prephase, dexamethasone and cyclophosphamide.
Table 2. Immunopathological features and IgH clonality of lymphomas in IL-10R–deficient patients
Patient Lymphoma Biopsy Classification CD20 BCL2 CD10 BCL6 CD30 KI67 MUM-1 EBER c-REL IgH MYC
P1 L1 Liver DLBCL 111 N/D N/D N/D N/D N/D N/D – N/D N/D 2
P2 L1 Liver DLBCL 111 1/2 – 1 – 111 – – C11/N11 Monoclonal 2
P3 L1 LN DLBCL 111 – 1 1 – 111 – – C11/N1 Monoclonal 2
P4 L1 Spleen DLBCL 111 1 – 1 1 111 1/2 – C11/N11 Monoclonal 2
L2 LN DLBCL 111 – 1 1 1 111 1 – C11/N1/2 Monoclonal 2
L3 LN DLBCL 111 – – 1 1 111 1/2 – C11/N1 Monoclonal 2
L4 LN DLBCL 111 1/2 1 1 1 111 1 – C11/N1 N/D 2
P5 L1 LN DLBCL 111 1 1 1 – 111 1/2 – C11/N1 Monoclonal N/D
Lymphomas were classified according to the World Health Organization.
–, negative staining; 1/–, heterogeneous staining;111, strongly positive staining; C, cytoplasm; LN, lymph node; MYC, rearrangement of MYC locus detected by FISH;
N, nuclear; N/D, not done.
BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23 IL-10R DEFICIENCY PREDISPOSES TO LYMPHOMAGENESIS 3715
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom
Granzyme B staining was negative or scattered in all the tested IL-10R–deficient samples (DLBCLs, n 5 7) and was positive in 5lymphoma-infiltrating T-cell control GCB DLBCLs (Figure 2).
Clonality
All tested specimens (6 of 6) displayed monoclonal rearrangementsof the IgH gene locus (Table 2 and data not shown). Somaticmutations in IgV regions were detected in 2 of 3 cases studied(supplemental Table 2).
Tumor gene expression profiling
In an attempt to better characterize the lymphomas, gene expressionprofiles were determined for tumors P3-L1, P4-L2, and P5-L1 andcompared with those in well-defined B-cell lymphoma entities andnormal GCB cells from tonsils. Microarrays signatures from thesevarious entities were downloaded from the open-access GeneExpression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/;identifier, GSE 39503) and compared with samples from ourpatients in an unsupervised analysis. Remarkably, lymphomasP3-L1, P4-L2, and P5-L1 displayed similar profiles (R. 0.650) andclustered in the branch containing all the DLBCL (despite the
latter’s known heterogeneity) and follicular lymphoma samples(R . 0.125) (Figure 3A). This clustering fits with the initialdiagnosis and the immunohistochemical phenotype of the tumors.In view of the heterogeneity of DLBCLs, we sought to determinewhether the patients’ lymphomas were primarily activated B-cell–likeor GCB-like.16,20 In fact, they all clustered in the GCB branch,confirming a major enrichment in the GCB signature (Figure 3B)characterized by markers of germinal center differentiation (MME,BCL6, BCL7, LMO2, CD21 (CR2), CD22, CD27, CD86, XBP1,LRMP, SERPINA9, FAM3C, RGS13, LCK, and IRS1; P , .0001).Furthermore, the 3 lymphomas had additional functional pathways incommonwith GCBDLBCLs, such as upregulation of the neurotrophinsignaling pathway (P , .0003), unsaturated fatty acid biosynthesis(P , .0005), the MAPK-signaling pathway (P , .003), and down-regulation of the BCR signaling pathway (P , .02).
Although GCB DLBCLs were the closest malignant entitiesto lymphomas P3-L1, P4-L2, and P5-L1, a gene set enrichmentanalysis and a pathway analysis revealed that the latter also sharedsome features distinct fromGCB lymphomas (supplemental Figure 2and supplemental Table 3) and nonmalignant, proliferative tonsilB cells (supplemental Table 4 and supplemental Figure 3). Whencompared with GCB DLBCLs, lymphomas P3-L1, P4-L2, and
Figure 2. Histochemical characteristics of lympho-
mas. (A-B) Hematoxylin-eosin staining (original magni-
fication, 3400) of P3-L1 (intra-abdominal lymph node
biopsy) and P4-L2 (inguinal lymph node biopsy), showing
diffuse, massive infiltration by monomorphic large lym-
phoid cells. (C-F) Immunohistochemical studies on P3-L1
biopsy showing (C) CD20 staining, (D) Ki67 staining, (E)
FISH for EBV-encoded RNA, and (F) BCL6 staining. (G-L)
CD3, CD8, and granzyme B immunohistochemical stud-
ies: in control GCB DLBCL biopsy, (G) CD3 and (H) CD8
T cells were detected at perivascular and intratumor sites.
(I) Granzyme B was detected in many T cells. In the
IL-10R–deficient DLBCL (P4-L3) biopsy, (J-K) CD3/CD8
T cells were found in the perivascular region, whereas (L)
no granzyme B was detected within the tumor (3200
magnification). (M) Counts of granzyme-B-positive cells
in 5 control GCB DLBCLs and 7 lymphomas from
IL-10R–deficient patients are shown. Counts per optic
field (3400 magnification). Horizontal bars represent
mean values.
3716 NEVEN et al BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

Figure 3. Gene expression profiles. (A) Unsupervised, hierarchical clustering of IL-10R lymphoma samples and other lymphoma samples. The dendrogram is based on
expression of the most variable genes. Genes whose median expression was above background and whose standard deviation across all samples was .2 in at least 1
lymphoma entity were used for unsupervised hierarchical clustering (n 5 3364). Samples from P3-L1, P4-L2, and P5L1 are shown in white and are indicated by arrows. (B)
Hierarchical clustering of IL-10R lymphoma samples and activated B-cell–like (ABC) (light pink) andGCB (dark pink) DLBCL samples. Samples fromP3-L1, P4-L2, and P5L1 are
shown in white and are indicated by arrows. A list of 227 genes used to discriminate between ABC and GCB DLBCL was extracted via a nonparametric test implemented in the
LIMMA software package (P , .05). Gene lists were submitted to the Cluster program for calculation of the Pearson correlation coefficient (as a similarity metric) and centroid
linkage clustering. The results were visualized with TreeView software. BL, Burkitt’s lymphoma; CB, centroblasts; CC, centrocytes; cHL, classic Hodgkin’s lymphoma; FL,
follicular lymphoma; MCL, mantle cell lymphoma; NPLHL, nodular predominance lymphocytic Hodgkin lymphoma; PMBL, primary mediastinal B-cell lymphoma; THRBL, T-cell/
histiocyte-rich large B-cell lymphoma; Ton, tonsils.
BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23 IL-10R DEFICIENCY PREDISPOSES TO LYMPHOMAGENESIS 3717
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom
P5-L1 showed greater expression of the spliceosome pathway(P, 23 10212) and ubiquitin-mediated proteolysis (P, 23 10211).These characteristics fit with the observed active cell cycling andfrozen GC phenotype. The activation of many pathways involved incancer (such as NOTCH, mTOR, ERBB2, and nuclear factor kB[NF-kB]) (P , 2 3 1025) is consistent with the high proliferationrate observed in these lymphomas. Strikingly, expression ofmolecules related to cytotoxicity and dendritic cell function was lowand thus agreed with the immunohistologic data on T cells. Acomparison with the nonmalignant, proliferating B cells found insecondary lymphoid organs (supplemental Figure 3 and supple-mental Table 4) confirmed the monoclonality of the lymphomas(relative to the polyclonal response observed in tonsils;P, 43 1026)and, most importantly, revealed surprisingly low levels of AICDA,BACH2, andAFF3, 3molecules that are usually upregulated in GCBs.Last, it is noteworthy that the axon guidance pathway was alsoenriched (P , .2 3 1025).
Overall, this signature shows that the lymphomas had high pro-liferative and survival capacities. The low numbers of antitumorimmune cells in the patients’ samples (as assessed by immunohis-tochemical staining and relative to conventional DLBCLs) wasconfirmed by the results of a transcriptome analysis.
Amplification and expression of the cellular homolog of the
reticuloendotheliosis viral oncogene (c-REL) and constitutive
activation of NF-kB
A cytogenetic analysis of the tumor cells revealed multiple chro-mosome rearrangements (supplemental Table 5), and an aCGHanalysis was performed on genomic DNA samples from 4 tumors(P3-L1, P4-L1, P4-L2, and P5-L1). The presence of high copynumber variations in the aCGH analysis of tumor cells agreed infull or in part with the results from a standard karyotype. Of themany genomic variations detected, the only recurrent variation was
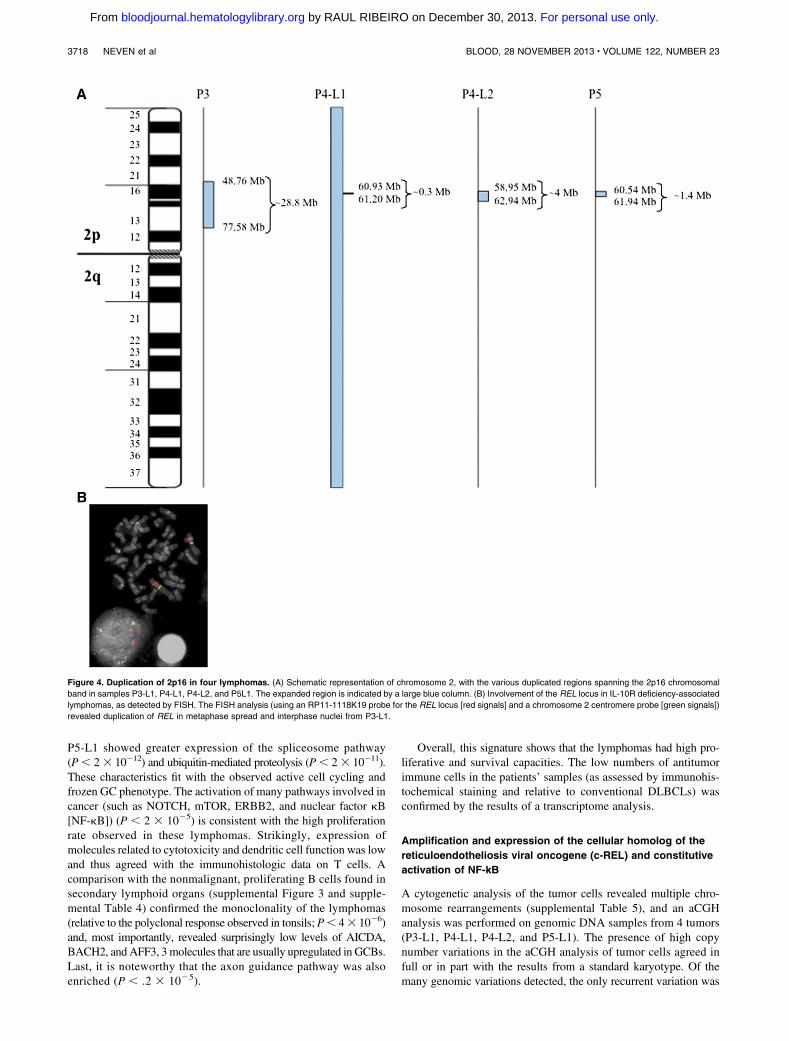
Figure 4. Duplication of 2p16 in four lymphomas. (A) Schematic representation of chromosome 2, with the various duplicated regions spanning the 2p16 chromosomal
band in samples P3-L1, P4-L1, P4-L2, and P5L1. The expanded region is indicated by a large blue column. (B) Involvement of the REL locus in IL-10R deficiency-associated
lymphomas, as detected by FISH. The FISH analysis (using an RP11-1118K19 probe for the REL locus [red signals] and a chromosome 2 centromere probe [green signals])
revealed duplication of REL in metaphase spread and interphase nuclei from P3-L1.
3718 NEVEN et al BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

a gain of the 2p16 chromosomal region (supplemental Table 6 andFigure 4A). The minimum common amplified region defined by thebreakpoint junctions of the P4-L1 sample spanned 0.25 Mb andincluded the PAPOLG, FLJ6341, REL, and PUS10 genes. P4-L2notably exhibited full chromosome 2 trisomy and gain of the 2p16chromosomal region (thus resulting in 2p16 tetraploidy). In P3-L1,gain of the 2p16 locus was confirmed by a FISH analysis with aprobe that flanks the REL locus (Figure 4B).
Given that REL locus amplification was found in all four testedcases, we assayed for expression of the cellular homolog c-REL.The expression levels of c-REL messenger RNA in P3-L1, P4-L2,and (above all) P5-L1 were similar to or greater than those foundin control GCB DLBCL samples (Figure 5A). The expression andcellular localization of c-REL were thus examined by immuno-histochemistry in all specimens but P1-L1. Strikingly, all testedDLBCL specimens stained positive for c-REL in the nucleus(Figure 5B-D and data not shown). Only P4-L2 exhibited mixed,predominantly cytoplasmic c-REL staining. A gene set enrichmentanalysis of P3-L1, P4-L2, and P5-L1 confirmed the upregulation oftranscripts associated with REL functions (such as the cell cycle/proliferation, apoptosis/survival, adhesion/architecture, and innateimmune cell functions). The most activated NF-kB–related path-ways in the 3 lymphomas studied were Toll-like receptor signaling(P5 9.73 10222) and apoptosis (P5 5.093 10217) (supplementalTable 7). The observed overexpression of directly c-REL–responsivegenes (such as CCL5, CXCL9, CCL19, TNFRSF17, TNFRSF9,BCL2A1, and BIRC3) suggested constitutive activation.
To directly assess the status of NF-kB activation, electrophoreticmobility shift assay analysis was performed on protein extractsprepared from P3-L1 and P5-L1. A strong constitutive NF-kBDNA
binding activity composed of two major complexes was observed inboth samples (supplemental Figure 1). The subunit composition ofthe NF-kB DNA-binding complexes was determined by supershiftanalysis. Antibody directed against RelA and p50 supershifted com-plex I almost completely. Complex II was effectively supershiftedwith anti-RelB and p50 antibodies (supplemental Figure 1). Altogether,these results suggest that both the canonical (ie, RelA) andnoncanonical (ie, RelB) NF-kB activation pathways were constitu-tively activated in P3-L1 and P5-L1.
To further characterize the key disease events leading to lym-phomagenesis in our patients, we performed whole-exome sequencingof P3-L1, P4-L1, and P4-L2 DNA and germline DNA in peripheralblood mononuclear cells from P3 and P4. We detected 26, 22, and 9somatic mutations in 24, 21, and 9 genes, respectively, in the threetumors (supplemental Table 8). The numbers of somatic mutationsfound in these lymphomas are in agreement with data on DLBCL.21-23
Some of the somatic mutations previously associated with DLBCLwere found in the patients’ lymphomas, including amutation inMYD88(p.S219C) (P3-L1).24 It is noteworthy that a somatic nonsense mutationin a master NF-kB regulator geneNFKBIA (p. Q68X) was identified inP4-L1, as previously described in classical Hodgkin lymphoma.25
Remarkably, we did not find any somatic mutations in genes involvedin histone and chromatin modifications, in contrast to events describedinDLBCL.26,27 Overall, the atypical but homogeneous gene expressionprofiles, the amplification of 2p16 in all tumors tested, theunconventional localization of c-REL, the constitutive activation ofboth canonical and noncanonical NF-kB pathways, and the presenceof somatic mutations potentially conferring intrinsic activation ofNF-kB suggested that the lymphomas in IL-10R–deficient patientsrepresent a distinct/intermediate subtype of DLBCL.
Figure 5. Expression of c-REL messenger RNA and protein. (A) c-REL transcript levels in tumor cells from P3-L1, P4-L2, and P5L1 in comparison with ABC DLBCL and
GCB DLBCL controls. The horizontal bar represents the mean value. (B-E) Immunohistochemical staining of c-REL on tumor biopsies (B,D: original magnification,3400; C,E:
detail from B and D, respectively). (B-C) A P4-L4 sample showing nuclear expression of c-REL in tumor cells and the absence of staining in endothelial cells. (D-E) A P4-L1
sample showing c-REL expression in both the nucleus and cytoplasm of tumor cells.
BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23 IL-10R DEFICIENCY PREDISPOSES TO LYMPHOMAGENESIS 3719
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

Discussion
Here, we reported the occurrence of B-cell lymphomas in 5IL-10R–deficient patients. Strikingly, one patient (P4) developedat least 3 distinct lymphomas. This hitherto unrecognized asso-ciation between IL-10/IL-10R deficiency and lymphoma raises anumber of questions. EBV-negative B-cell lymphomas were mono-clonal. They contained highly proliferative, monomorphic, largeB cells and showed several of the immunohistologic and molecularcharacteristics of DLBCLs. A gene expression analysis confirmed theorigin of the lymphoma germinal center28,29 and revealed a strikinglyhomogeneous phenotype that was similar (but not identical) to that ofGCB DLBCL. A key finding was the amplification of the 2p16chromosomal region in four tested tumors. This feature has beenreported previously in 17% to 26% of GCB DLBCLs16,30 and inHodgkin lymphoma.31-33 The minimal amplified region containedREL but not the closely located oncogene BCL11A. In fact, RELencodes c-REL, a member of the NF-kB transcription factor complexinvolved in driving cell survival and proliferation.34 It has been shownthat mutations in REL lead to constitutive NF-kB activation in B-celllymphomas,29,35 with the detection of NF-kB transcription factors(including c-REL) in the nucleus,30,36,37 as observed here for c-REL.This characteristic also distinguishes the patients’ lymphomas fromtypical GCB DLBCLs.35,38 The observed pattern of gene over-expression is consistent with aberrant triggering of the NF-kB path-way and is further suggested by the observation of somatic mutationsthat potentially lead to gain of function of the NF-kB pathway in 2 ofthe 3 lymphomas tested. In addition, electrophoretic mobility shiftassay combined with supershift analysis performed on 2 samplesshowed constitutive activation of both canonical and noncanonicalNF-kB pathways. This pattern in unusual for GCB DLBCL.
The occurrence of 1 or more B-cell lymphomas in 5 patients withIL-10R deficiency cannot be viewed as a chance event, given thatthe incidence of NHL in childhood is 1 3 1025 per year and thatDLBCLs account for only 15% of childhood NHLs.39 Furthermore,the characteristics shared by the lymphomas of all the patients arguestrongly against random occurrence. This increased risk of lym-phomas might conceivably have been related to the immunosup-pressive therapy that 4 of the 5 patients were receiving as treatmentfor IBD. In adult IBD, a fivefold increase in the risk of lym-phoproliferative disorders has been observed in thiopurine-exposedpatients.15,40 The CESAME study of almost 20 000 IBD patientsreported 23 lymphoproliferative disorders (12 of which were EBV-related).15 However, none of the 53 children with idiopathic VEO-IBD in our series (who were also exposed to thiopurine-basedimmunosuppression for a comparable period) developed lympho-mas.8 Likewise, patients with monogenic IBDs (such as chronicgranulomatous disease or X-linked inhibitor of apoptosis proteindeficiency) do not develop lymphoma more frequently than wouldbe expected.41,42 Thus, in the context of IL-10R deficiency,azathioprine therapy is probably at most a weak risk factor forlymphomagenesis.
Our data therefore point to a direct relationship between theoccurrence of lymphoma and a deficiency in the IL-10 pathway.Taking into account all reported patients with IL-10 (n 5 5),IL-10R1 (n 5 11), or IL-10R2 (n 5 19) deficiencies,6-10,43 thefrequency of developing lymphoma is estimated to be 36% (5 of 14)at the age of 7 years (in the absence of a previous HSCT). It isnoteworthy that lymphomagenesis has not been reported in mice
with impaired IL-10–mediated pathways, despite the onset ofIBD,11,44 suggesting either the existence of marked differencesbetween mice and humans or insufficient follow-up of the micein an adequate environment. A role for the IL-10 pathway inhuman lymphomagenesis has been suggested by several studiesshowing a significant association between genetic variants inIL-10 and IL-10RB on one hand and NHL (and particularlyDLBCL) on the other.45,46 Since disruption of the IL-10/IL-10Raxis in both humans and mice leads to severe inflammatoryenterocolitis,12 one can hypothesize that chronic inflammationmay create a favorable milieu for B-cell lymphomagenesis,perhaps through protracted B-cell activation.47 However, few of thepatients’ nodal lymphomas were in the gut, no mucosa-associatedlymphomas were detected, and an activated B-cell lymphomaphenotype was not observed. Furthermore, VEO-IBD was notfound to be associated with lymphoma when the IL-10/IL-10Rpathway was unaffected. We thus conclude either that gutinflammation is not a main driver of lymphomagenesis or thata distinct pattern of inflammation (related to IL-10/IL-10Rdeficiency) is involved in lymphomagenesis. Future research willhave to evaluate these possibilities.
Hence, two mutually nonexclusive models may account for theoccurrence of B-cell lymphomas in the absence of a fully competentIL-10 signaling pathway. Given that IL-10 controls the proliferationof nonactivated B cells,48 defective signaling may lead to anincrease in DNA replication and, in turn, acquisition of rearrange-ment events and somatic mutations. An alternative mechanismmight involve a local T-cell immunodeficiency caused by impairmentof the IL-10/IL-10R pathway. Surprisingly, in view of the im-munosuppressive role of IL-10, recent studies of murine modelshave found that this cytokine promotes CD8 T-cell infiltration intotumors, local interferon-g secretion, cytotoxicity, antigen pre-sentation, and thus tumor suppression.49 Furthermore, IL-10 candirectly induce cytotoxicity in intratumor CD8 T cells. Overall,these experimental data tend to confer a role of IL-10 inimmunosurveillance. This hypothesis is attractive but remains tobe tested. The scarcity of intratumor infiltrative granzyme-B–positive T cells in lymphomas from IL-10R–deficient patients(relative to control DLBCLs) shown in immunohistochemicalanalyses and confirmed at transcriptomic level is consistent withthe presence of a deficiency in an IL-10–triggered local antitumorimmunity pathway. It is noteworthy that patients with heterozygousSTAT3mutations (resulting in partial loss of function) are also proneto the development of B-cell lymphomas18,19 (albeit to a lesserextent). The observation that IL-10 signaling is STAT3 dependentsupports the hypothesis in which the IL-10R/STAT3 pathway isinvolved in controlling lymphomas. An additional role of the IL-10R2signaling pathway could be suggested by the fact that the IL-10R2chain is common to the IL-22, IL-26, and interferon l (IL-28A, IL-28B,and IL-29) receptors50 However, the occurrence of B-cell lymphomawith the very same characteristics in a patient with IL-10R1 deficiencymakes this hypothesis very unlikely.
In conclusion, our results indicate that IL-10R deficiency pre-disposes to the development of a subtype of DLBCLs with germinalcenter origin characterized by original constitutive activation of theNF-kB pathway and a defective local T-cell immune response.Further work will be needed to fully elucidate the IL-10R pathway’sprotective effect. Meanwhile, our data support early HSCT in patientswith an impaired IL-10 pathway; this procedure is able to cure IBDand may well prevent the occurrence or recurrence of lymphoma.
3720 NEVEN et al BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

Acknowledgments
The authors thank the patients and their families for their co-operation; Dr V. Minard, Dr C. Patte, Dr F. Doz, Dr D. Orbach,and Dr G. Couillaut for excellent care of the patients; Dr N. CerfBensussan for critical reading of the manuscript; C. Daussyand J. Pasquet for excellent technical assistance; C. Wouters,C. Janssens, and T. Roeskam for assistance in immunohistochem-ical staining; T. Molina for discussion and critical review of themanuscript; T. Petrella for providing patient samples; and the staff atthe Necker Hospital’s “tumorotheque” biological resource center foranalytical assistance.
This work was funded by grants from the Institut National de laSante et de la Recherche Medicale, the Fondation pour la RechercheMedicale (B. Nadel), the Agence Nationale pour la Recherche(F.R.-L.), The German Research Foundation (SFB1054) (C.K.),and an Advanced Senior Grant from the European ResearchCouncil (PID Immune 249816) (A.F.).
Authorship
Contribution: B. Neven designed the research, collected and analyzeddata, and participated in drafting the paper and clinical care; E.M.
performed and interpreted microarray studies and participated indrafting the manuscript; J.B., D.C., and N.B. performed pathologyand immunohistochemical studies and participated in drafting themanuscript; S.K., J.M.-P. and I.R.-W. performed cytogenetic andmicroarray studies and participated in drafting the manuscript;F.R.-L. andC.P. participated in genetic testing, functional analysis, andcritical review of the manuscript; V.A. and F.D. performed clonalityand somatic mutations in studies of IgV regions and participated incritical review of the manuscript; C.B. and P. N. performed whole-exome sequencing; F.S., P.F., S.B., O.G., J.-L.C., andO.H. participatedin clinical care and critical review of the manuscript; D.K., D.M.,and C.K. performed genetic and functional studies and participatedin clinical care of patients and critical review of the manuscript; K.B.performed analysis of NF-kB activation status; V.B. designed theNF-kB activation study and participated in data analyses anddrafting the manuscript; B. Nadel designed the research andparticipated in data analyses and drafting the manuscript; F.R.participated in data analyses, drafting the paper, and clinical care;and A.F. designed the research and participated in data analyses,drafting the manuscript, and clinical care.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Alain Fischer, Hopital Necker-Enfant Malades,Assistance Publique des Hopitaux de Paris, 149 rue de Sevres, 75015Paris, France; e-mail: [email protected].
References
1. Abraham C, Cho JH. Inflammatory bowel disease.N Engl J Med. 2009;361(21):2066-2078.
2. Cho JH. The genetics and immunopathogenesisof inflammatory bowel disease. Nat Rev Immunol.2008;8(6):458-466.
3. Xavier RJ, Podolsky DK. Unravelling thepathogenesis of inflammatory bowel disease.Nature. 2007;448(7152):427-434.
4. Van Limbergen J, Russell RK, Drummond HE,et al. Definition of phenotypic characteristics ofchildhood-onset inflammatory bowel disease.Gastroenterology. 2008;135(4):1114-1122.
5. Levine A, Griffiths A, Markowitz J, et al. Pediatricmodification of the Montreal classification forinflammatory bowel disease: the Parisclassification. Inflamm Bowel Dis. 2011;17(6):1314-1321.
6. Glocker EO, Kotlarz D, Boztug K, et al.Inflammatory bowel disease and mutationsaffecting the interleukin-10 receptor. N Engl JMed. 2009;361(21):2033-2045.
7. Glocker EO, Frede N, Perro M, Sebire N, ElawadM, Shah N, Grimbacher B. Infant colitis—it’s in thegenes. Lancet. 2010;376(9748):1272.
8. Begue B, Verdier J, Rieux-Laucat F, et al.Defective IL10 signaling defining a subgroup ofpatients with inflammatory bowel disease. Am JGastroenterol. 2011;106(8):1544-1555.
9. Kotlarz D, Beier R, Murugan D, et al. Loss ofinterleukin-10 signaling and infantile inflammatorybowel disease: implications for diagnosis andtherapy. Gastroenterology. 2012;143(2):347-355.
10. Engelhardt KR, Shah N, Faizura-Yeop I, et al.Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoieticstem cell transplantation. J Allergy Clin Immunol.2013;131(3):825-830.
11. Kuhn R, Lohler J, Rennick D, Rajewsky K, MullerW. Interleukin-10-deficient mice develop chronicenterocolitis. Cell. 1993;75(2):263-274.
12. Barnes MJ, Powrie F. Regulatory T cells reinforceintestinal homeostasis. Immunity. 2009;31(3):401-411.
13. Pohl C, Hombach A, Kruis W. Chronicinflammatory bowel disease and cancer.Hepatogastroenterology. 2000;47(31):57-70.
14. Thapar N, Shah N, Ramsay AD, Lindley KJ, MillaPJ. Long-term outcome of intractable ulceratingenterocolitis of infancy. J Pediatr GastroenterolNutr. 2005;40(5):582-588.
15. Beaugerie L, Brousse N, Bouvier AM, et al;CESAME Study Group. Lymphoproliferativedisorders in patients receiving thiopurines forinflammatory bowel disease: a prospectiveobservational cohort study. Lancet. 2009;374(9701):1617-1625.
16. Lenz G, Wright GW, Emre NC, et al. Molecularsubtypes of diffuse large B-cell lymphoma ariseby distinct genetic pathways. Proc Natl Acad SciUSA. 2008;105(36):13520-13525.
17. Kastan MB, Lim DS. The many substrates andfunctions of ATM. Nat Rev Mol Cell Biol. 2000;1(3):179-186.
18. Kumanovics A, Perkins SL, Gilbert H, CessnaMH, Augustine NH, Hill HR. Diffuse large B celllymphoma in hyper-IgE syndrome due to STAT3mutation. J Clin Immunol. 2010;30(6):886-893.
19. Chandesris MO, Melki I, Natividad A, et al.Autosomal dominant STAT3 deficiency andhyper-IgE syndrome: molecular, cellular, andclinical features from a French national survey.Medicine (Baltimore). 2012;91(4):e1-e19.
20. Alizadeh AA, Eisen MB, Davis RE, et al. Distincttypes of diffuse large B-cell lymphoma identifiedby gene expression profiling. Nature. 2000;403(6769):503-511.
21. Compagno M, Lim WK, Grunn A, et al. Mutationsof multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature.2009;459(7247):717-721.
22. Pasqualucci L, Trifonov V, Fabbri G, et al.Analysis of the coding genome of diffuse largeB-cell lymphoma. Nat Genet. 2011;43(9):830-837.
23. Lohr JG, Stojanov P, Lawrence MS, et al.Discovery and prioritization of somatic mutationsin diffuse large B-cell lymphoma (DLBCL) by
whole-exome sequencing. Proc Natl Acad SciUSA. 2012;109(10):3879-3884.
24. Ngo VN, Young RM, Schmitz R, et al.Oncogenically active MYD88 mutations in humanlymphoma. Nature. 2011;470(7332):115-119.
25. Lake A, Shield LA, Cordano P, et al. Mutations ofNFKBIA, encoding IkappaB alpha, are a recurrentfinding in classical Hodgkin lymphoma but are nota unifying feature of non-EBV-associated cases.Int J Cancer. 2009;125(6):1334-1342.
26. Morin RD, Mendez-Lago M, Mungall AJ, et al.Frequent mutation of histone-modifying genes innon-Hodgkin lymphoma. Nature. 2011;476(7360):298-303.
27. Pasqualucci L, Dominguez-Sola D, Chiarenza A,et al. Inactivating mutations of acetyltransferasegenes in B-cell lymphoma. Nature. 2011;471(7337):189-195.
28. Rosenwald A, Wright G, Chan WC, et al;Lymphoma/Leukemia Molecular Profiling Project.The use of molecular profiling to predict survivalafter chemotherapy for diffuse large-B-celllymphoma. N Engl J Med. 2002;346(25):1937-1947.
29. Lenz G, Staudt LM. Aggressive lymphomas.N Engl J Med. 2010;362(15):1417-1429.
30. Feuerhake F, Kutok JL, Monti S, et al. NFkappaBactivity, function, and target-gene signatures inprimary mediastinal large B-cell lymphoma anddiffuse large B-cell lymphoma subtypes. Blood.2005;106(4):1392-1399.
31. Martın-Subero JI, Gesk S, Harder L, et al.Recurrent involvement of the REL and BCL11Aloci in classical Hodgkin lymphoma. Blood. 2002;99(4):1474-1477.
32. Toujani S, Dessen P, Ithzar N, et al. Highresolution genome-wide analysis of chromosomalalterations in Burkitt’s lymphoma. PLoS ONE.2009;4(9):e7089.
33. Robledo C, Garcıa JL, Caballero D, et al; SpanishLymphoma/Autologous Bone Marrow TransplantStudy Group (GEL-TAMO). Array comparativegenomic hybridization identifies genetic regionsassociated with outcome in aggressive diffuse
BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23 IL-10R DEFICIENCY PREDISPOSES TO LYMPHOMAGENESIS 3721
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom

large B-cell lymphomas. Cancer. 2009;115(16):3728-3737.
34. Gugasyan R, Grumont R, Grossmann M,Nakamura Y, Pohl T, Nesic D, Gerondakis S. Rel/NF-kappaB transcription factors: key mediators ofB-cell activation. Immunol Rev. 2000;176:134-140.
35. Lim KH, Yang Y, Staudt LM. Pathogeneticimportance and therapeutic implications of NF-kBin lymphoid malignancies. Immunol Rev. 2012;246(1):359-378.
36. Houldsworth J, Olshen AB, Cattoretti G, et al.Relationship between REL amplification, RELfunction, and clinical and biologic features indiffuse large B-cell lymphomas. Blood. 2004;103(5):1862-1868.
37. Barth TF, Martin-Subero JI, Joos S, et al. Gains of2p involving the REL locus correlate with nuclearc-Rel protein accumulation in neoplastic cells ofclassical Hodgkin lymphoma. Blood. 2003;101(9):3681-3686.
38. Shaffer AL III, Young RM, Staudt LM.Pathogenesis of human B cell lymphomas. AnnuRev Immunol. 2012;30:565-610.
39. Izarzugaza MI, Steliarova-Foucher E, MartosMC, Zivkovic S. Non-Hodgkin’s lymphoma
incidence and survival in European children andadolescents (1978-1997): report from theAutomated Childhood Cancer InformationSystem project. Eur J Cancer. 2006;42(13):2050-2063.
40. Kandiel A, Fraser AG, Korelitz BI, Brensinger C,Lewis JD. Increased risk of lymphoma amonginflammatory bowel disease patients treated withazathioprine and 6-mercaptopurine. Gut. 2005;54(8):1121-1125.
41. Holland SM. Chronic granulomatous disease. ClinRev Allergy Immunol. 2010;38(1):3-10.
42. Pachlopnik Schmid J, Canioni D, Moshous D, et al.Clinical similarities and differences of patients withX-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAPdeficiency). Blood. 2011;117(5):1522-1529.
43. Mao H, Yang W, Lee PP, et al. Exomesequencing identifies novel compoundheterozygous mutations of IL-10 receptor 1 inneonatal-onset Crohn’s disease. Genes Immun.2012;13(5):437-442.
44. Spencer SD, Di Marco F, Hooley J, et al. Theorphan receptor CRF2-4 is an essential subunit of
the interleukin 10 receptor. J Exp Med. 1998;187(4):571-578.
45. Rothman N, Skibola CF, Wang SS, et al. Geneticvariation in TNF and IL10 and risk of non-Hodgkinlymphoma: a report from the InterLymphConsortium. Lancet Oncol. 2006;7(1):27-38.
46. Lan Q, Wang SS, Menashe I, et al. Geneticvariation in Th1/Th2 pathway genes and risk ofnon-Hodgkin lymphoma: a pooled analysis ofthree population-based case-control studies. Br JHaematol. 2011;153(3):341-350.
47. Rui L, Schmitz R, Ceribelli M, Staudt LM.Malignant pirates of the immune system. NatImmunol. 2011;12(10):933-940.
48. Moore KW, de Waal Malefyt R, Coffman RL,O’Garra A. Interleukin-10 and the interleukin-10receptor. Annu Rev Immunol. 2001;19:683-765.
49. Mumm JB, Emmerich J, Zhang X, et al.IL-10 elicits IFNg-dependent tumor immunesurveillance. Cancer Cell. 2011;20(6):781-796.
50. Ouyang W, Rutz S, Crellin NK, Valdez PA,Hymowitz SG. Regulation and functions of theIL-10 family of cytokines in inflammation anddisease. Annu Rev Immunol. 2011;29:71-109.
3722 NEVEN et al BLOOD, 28 NOVEMBER 2013 x VOLUME 122, NUMBER 23
For personal use only. by RAUL RIBEIRO on December 30, 2013. bloodjournal.hematologylibrary.orgFrom