Embed Size (px)
Citation preview

Plant Physiol. (1 996) 11 1 : 475-485
Solid-State ' 3C Nuclear Magnetic Resonance Characterization of Cellulose in the Cell Walls of
Arabidopsis thaliana Leaves'
Roger H. Newman, Lynette M. Davies, and Philip 1. Harris*
Industrial Research Limited, P.O. Box 31-310, Lower Hutt, New Zealand (R.H.N.); and School of Biological Sciences, T h e University of Auckland, Private Bag 9201 9, Auckland, New Zealand (L.M.D., P.J.H.)
Solid-state 13C nuclear magnetic resonance was used to charac- terize the molecular ordering of cellulose in a cell-wall preparation containing mostly primary walls obtained from the leaves of Arabi- dopsis thaliana. Proton and 13C spin relaxation time constants showed that the cellulose was in a crystalline rather than a paracrystalline state or amorphous state. Cellulose chains were distributed between the interiors (40%) and surfaces (60%) of crystallites, which is consistent with crystallite cross-sectional di- mensions of about 3 nm. Digital resolution enhancement revealed signals indicative of triclinic and monoclinic crystalline forms of cellulose mixed in similar proportions. Of the five nuclear spin relaxation processes used, proton rotating-frame relaxation pro- vided the clearest distinction between cellulose and other cell-wall components for purposes of editing solid-state 13C nuclear magnetic resonance spectra.
~ ~
The cell walls of higher plants consist of two phases, a cellulose microfibrillar phase and a matrix phase. In pri- mary cell walls, the matrix phase consists mostly of po- lysaccharides with a variety of different structures (Bacic et al., 1988). However, relatively little is known about the spatial relationships of the macromolecules in cell walls and how these macromolecules interact (McCann et al., 1990). For example, there is considerable confusion con- cerning the nature of the molecular ordering within the cellulose microfibrils. Descriptions of the crystallinity of cellulose in primary cell walls range from the early study of Frey-Wyssling (1954), in which he described the crystallin- ity in cellulose prepared from the growing root tips and coleoptiles of corn (Zea mays) as being "astonishingly poor," to a recent study of primary cell walls of apple fruit, in which no amorphous cellulose was detected. It is not yet clear whether the diverse conclusions reflect variations in the crystallinity of cellulose between different plant spe- cies, the evolution of new instrumental techniques, or changes in the use of poorly defined words such as "crys- tallinity ."
Until recently, x-ray diffraction has been the main tech- nique used to study the molecular ordering of cellulose.
This work was funded by the New Zealand Foundation for
* Corresponding author; e-mail [email protected]; fax Research Science and Technology under contract no. UOA303.
64 -9 -373-741 6.
However, NMR spectroscopy has shown considerable promise for discriminating between mobile and immobile elements in plant cell walls, enabling in situ studies of the molecular ordering of cellulose and thus avoiding the dam- age that is inevitably associated with the isolation and purification of cellulose (Jarvis and Apperley, 1990). The NMR technique is sensitive to short-range order and can distinguish whether a cellulose chain is exposed on a sur- face or is surrounded by similar chains. It is not sensitive to molecular ordering beyond the adjacent chains. In contrast, x-ray diffraction is sensitive only to long-range order, pro- viding information about the repetition of cellulose chains at regular intervals through a crystallite. For these reasons, almost all published x-ray studies of cellulose have focused on sources in which cellulose cross-sectional dimensions are relatively large, e.g. algae, bacteria, or secondary walls in cotton and ramie. The relatively narrow crystallites of primary cell walls present more demanding challenges.
The term "NMR covers a number of different experi- mental procedures, and it is important to view these in the context of a steady technical development of NMR equip- ment and associated computer software. MacKay et al. (1988) used overlapping proton NMR signals in a study of bean (Phaseolus vulgnuis) primary cell walls. Jarvis and Ap- perley (1990) showed that signals from cellulose, pectic, and other matrix polysaccharides could be resolved by using the superior signal dispersion of I3C NMR. Cross- polarization NMR experiments combine the best features of both nuclei, allowing indirect measurement of proton NMR parameters through variations in the strength of selected 13C NMR signals (Tekely et al., 1989). Cross- polarization NMR was recently used in an investigation of the cellulose in primary cell walls of apple fruit (Newman et al., 1994), and in the present study we have extended this approach.
Nuclear spin relaxation processes can be sensitive to molecular dynamics, and therefore the associated time constants provide convenient criteria for discrimination between mobile and immobile elements in cell walls, i.e. noncrystalline (amorphous) and crystalline domains,
Abbreviations: I, and I,, triclinic and monoclinic crystalline forms of cellulose, respectively; ppm, parts per million; PSRE, proton spin relaxation editing; T,(H) or (C), spin-lattice relaxation time constants; T,,(H) or (C), rotating-frame relaxation time con- stants; T,(H) or (C), spin-spin relaxation time constants.
475
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

476 Newman et al. Plant Physiol. Vol. 11 1, 1996
and the intermediate states known as "paracrystalline" lsolation of Cell Walls or "mesomorphous" domains. The relaxation processes all involve changes in nuclear magnetization, from a perturbed condition toward an equilibrium condition. The processes are complicated by spin diffusion, in which spin information is exchanged between pairs of neighboring nuclei. Proton spin information can diffuse through dimensions of nanometers over time scales of milliseconds (Zumbulyadis, 1983). The experimental val- ues of proton spin relaxation time constants therefore reflect mean values for a11 protons within a finite vol- ume, regardless of which 13C signal is used to monitor proton relaxation. I3C spins diffuse on a longer time scale (VanderHart, 1987); therefore, it is possible to mea- sure different experimental values for different carbon atoms within a molecule. The spin relaxation processes that may be exploited include the following: (a) time constant T,(H) or T,(C), in which spin magnetization approaches an equilibrium value parallel to the static magnetic field; (b) time constant T,,(H) or T,,(C), in which a radiofrequency transmitter is used to "spin- lock" magnetization in a plane perpendicular to the static field; (c) time constant T,(H) or T2(C), in which magnetization decays in a plane perpendicular to the static field while a11 transmitters are switched off.
We exploited a11 of the above time constants, except T,,(C), in the present investigation of the cellulose in a cell-wall preparation containing mostly primary walls ob- tained from the leaves of Arabidopsis thaliana. This dicoty- ledonous plant is increasingly being used as a model plant and it is intended that this study should form a baseline for a future comparison of celluloses in different plant species.
MATERIALS A N D METHODS
Plant Material
Plants of Arabidopsis thaliana (L.) Heynh. ecotype Colum- bia were grown from seed in compost (three plants per 10-cm-diameter pot) in a controlled environment growth cabinet. Lighting was provided by Sylvania warm-white fluorescent tubes supplemented by incandescent lamps to give an irradiance of 215 pE spl m-' (400-700 nm) at plant level, and 1R:total irradiation of 1:3. The daylength was 19 h, and the temperature was 23°C (day) and 19°C (night). The rosette leaves were harvested when at least two of the terminal flowers were open but before peta1 abscission occurred. The plants were kept in the dark by enclosing them in a black plastic bag for 24 h before harvesting. After this treatment, transverse sections from sample leaves were cut by hand using a razor blade. These sections were treated with a solution of iodine in potassium iodide (0.2 g of iodine and 2 g of potassium iodide in 100 mL of water) to stain starch granules, but none were detected.
L.M. Davies (unpublished data) has shown histochemi- cally that, except for the xylem tracheary elements, all of the cell types in the rosette leaves of A. thaliana have unlignified primary walls. Cell-wall preparations from these leaves thus contained mostly unlignified primary walls, but some walls from xylem tracheary elements were present.
Rosette leaves (including the petioles) were harvested in dim light. They were then weighed, quickly washed with water at 4"C, cut into pieces no larger than 3 x 3 mm, frozen in liquid nitrogen, and ground to a fine powder in a pestle and mortar in the presence of liquid nitrogen. This powder was stored at -70°C until it was required. Portions (5 g) of the frozen powder were added to a mortar at 4°C. Hepes-KOH buffer (20 mM, pH 7.2, 20 mL, at 4"C), con- taining 10 mM 2-mercaptoethanol was added, and the leaf tissue was ground with a pestle for 10 min. The suspension was divided into five equal portions, and each portion was added to a glass tube (150 mm long, 22 mm in diameter) to which was added 20 mL of the buffer containing 10 mM 2-mercaptoethanol at 4°C. The contents of each tube were homogenized on ice at full speed for 4 min with a Polytron homogenizer (model PT 10-35, Kinematica, Kriens-Luzern, Switzerland) fitted with a 12-mm-diameter generator. The cell walls were filtered onto a nylon mesh (pore size 11 pm) and washed with water until the filtrate was clear (500 mL). All of the cell walls were collected and then washed three times in 80% (v/v) ethanol by centrifuging (4508, 5 min). Aliquots were taken of the combined cell-wall material to determine its dry matter content.
Before the cell-wall preparation was washed with 80% (v/v) ethanol, it was examined using bright-field micros- copy after staining with Ponceau 2R (0.2% [w/v] aqueous solution, containing two drops of 18 M H,SO, per 100 mL). This protein stain was used to detect unbroken cells, and small numbers of such cells were found.
N M R Spectroscopy
The sample was filtered from 80% (v/v) ethanol and partially dried in room air for 2 h, until the moisture content decreased to 54% by weight (as determined by oven drying after the NMR experiments). Earlier experi- ments showed that moisture is essential for good resolu- tion of signals from cellulose crystal surfaces (Newman et al., 1994). The crumbs of solid matter were packed in a 7-mm-diameter cylindrical sapphire rotor and retained with Vespel (DuPont) end caps. The rotor was spun at 4 kHz in a Doty Scientific (Columbia, SC) magic-angle spin- ning probe for l3C solid-state NMR spectroscopy at 50.3 MHz on an XL-200 spectrometer (Varian, Harbor City, CA).
"Normal" spectra were obtained with a 6-ps proton preparation pulse, a 1-ms cross-polarization contact time, 30 ms of data acquisition, and a delay of 0.6 s before the sequence was repeated. The proton transmitter power level was increased to provide a field strength of yB/ ( 2 ~ ) = 56 kHz during data acquisition. Transient signals from at least 39,000 contacts were averaged.
T,(H) was investigated by a pulse sequence in which each 90" proton preparation pulse was preceded by a 12-ps 180" pulse and a recovery interval, set at 20 ms for the spectra shown in Figures 1 and 2. T,,(H) was investigated by a pulse sequence in which a proton spin-locking pulse, set at 4 ms for the spectra shown in Figures 3 and 4, was inserted between each 90" proton preparation pulse and
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

13C NMR of Cellulose in Arabidopsis thaliana Cell Walls 477
the cross-polarization contact time. T,(H) was investigated by a pulse sequence in which the proton transmitter was switched off for an interval, set at 15 ps for the spectra shown in Figures 5 and 6, inserted between each 90" proton preparation pulse and the cross-polarization contact time. T,(C) was investigated by a pulse sequence described by Torchia (1978), with a relaxation interval set at 4 s for the spectrum shown in Figure 7. T,,,(C) was not used in the present study because the 13C transmitter could not attain the power output required to ensure that T,,(C) values reflected molecular dynamics rather than static dipole-di- pole interactions (Akasaka et al., 1983). T2(C) was investi- gated by a spin-echo sequence in which each contact was followed by n full-rotor periods, a I3C 180" refocusing pulse, and n more rotor periods before data acquisition commenced. The proton decoupler was left switched on during the spin-echo interval, with power offset from the Hartmann-Hahn condition to prevent cross-polarization during the 13C 180" pulse. The spin-echo interval was set at 8 ms (n = 16) for the spectrum shown in Figure 8.
A resolution-enhanced spectrum was generated from data averaged over a period of 117 h. The NMR free in- duction decay was convoluted with a function of the form (Newman, 1994):
(1)
Values of Tx = 6 ms and T, = 9 ms were chosen by trial and error for the spectrum shown in Figure 9. Other com- binations produced spectra with poorer resolution, poorer noise suppression, and/ or lineshapes distorted by over- compensation for natural broadening.
A sample of commercial microcrystalline cellulose (Avi- cel) was purchased from E. Merck (Darmstadt, Germany). The Avicel was moistened to 48% (w/w) water and a resolution-enhanced spectrum was obtained as described above, except that the recovery delay was extended to 4 s and data averaging was limited to 61 h.
PSRE was also carried out. The theoretical basis for PSRE NMR was described by Newman and Hemmingson (1990) and was applied in a study of the cellulose in primary cell walls of apple fruit (Newman et al., 1994). The theory is summarized here. Suppose a sample comprises a mixture of two types of domains, with different proton spin relax- ation time constants in each. The normal solid-state NMR spectrum is the sum of the two components,
f(t)=exp[(t / Tx)2/ 2- (t /TYl3/ 31.
S=A+B, (2)
where S, A, and B are arrays of data specifying signal heights measured at intervals along the chemical shift scale. If the proton magnetization is perturbed by spin relaxation prior to cross-polarization, then the resultant spectrum S' will be:
S'=faA+fbB, (3)
where f, and fb are factors reflecting the consequences of spin relaxation. Equations 2 and 3 can be solved for A and B:
A=kS+k'S' (4a)
B = ( l - k)S- k'S', (4b)
where
In the present study, the initial estimates of fa and fb were calculated from preliminary estimates of the relevant pro- ton spin relaxation time constants for cellulose and noncel- lulosic components, respectively. These estimates were not regarded as reliable, in view of the degree of overlap between adjacent NMR peaks assigned to different chem- ical components. The values of fa and fb were therefore adjusted until the peak at 89 ppm (assigned to C-4 in crystalline cellulose) was eliminated from subspectrum B and peaks assigned to noncellulosic substances were sup- pressed in subspectrum A, without allowing any peak to become inverted. The final values of fa and fb were then used to calculate improved values of the proton spin re- laxation time constants, as shown in the relevant figure legends.
RESULTS AND DISCUSSION
Normal NMR Spectrum
This spectrum is shown in Figure 1 and the assignments of signals follow those suggested by Jarvis (1990) and Jarvis
I . # * * 1 I . I ~ ~ " " ' " ' ' 1
200 150 100 50 O 6 (ppm)
Figure 1. Solid-state 13C NMR spectrum of cell walls of A. thaliana. S, Normal spectrum; S', spectrum obtained with a proton inversion- recovery pulse sequence, in which T,(H) occurred during a 20-ms interval prior to cross-polarization. Carbon numbers refer to cellu- lose. Contributions from proteins and polymethylene chains are labeled "p" and "w," respectively. A band at 102 ppm, assigned to C-l in pectic galacturonans and other noncellulosic polysaccharides, is labeled with an asterisk (*).
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

47% Newman et al. Plant Physiol. Vol. 1 1 1, 1996
and Apperley (1990). The spectrum is dominated by peaks assigned to cellulose, at 62 and 65 ppm (C-6), 73 and 75 ppm (C-2, C-3, C-5), 84 and 89 ppm (C-4), and 105 ppm (C-1). Bands from 15 to 40,50 to 60, and 165 to 180 ppm are assigned primarily to proteins. A peak at 33 ppm is as- signed to polymethylene chains, e.g. in cutin or waxes associated with the cuticle. A band centered on 102 ppm is assigned to C-1 in pectic galacturonans and other noncel- lulosic polysaccharides. The corresponding bands for C-2, C-3, C-4, and C-5 form a broad background under peaks assigned to cellulose, and the band for pectic galacturonans C-6 (expected at 172-175 ppm) is overwhelmed by a band assigned primarily to amide carbon in proteins.
T,(H)
Figure 1 shows the spectrum obtained with a proton spin-lattice relaxation pulse sequence. The inversion-recov- ery curve can be described by a function f:
f=l-2Qexp[ - t/T,(H)], (6)
where Q (O < Q < 1) describes the efficiency of the invert- ing pulse. In the present study, Q = 0.87. The relaxation interval was arbitrarily set at t = 20 ms for spectrum S‘ in Figure 1. Only one signal (from polymethylene chains at 32 ppm) remained inverted. To enhance the discrimination between cellulose and other substances, PSRE subspectra were obtained by linear combinations of S and S’ (Fig. 2). Peaks assigned to cellulose are all confined to subspectrum A, with T,(H) = 13 ms. The band at 102 ppm, characteristic of pectic galacturonans and other noncellulosic polysaccha- rides, is also confined to this subspectrum. This observa-
I 1 I I I I I . . I I I . . I 1 1 1 1 1 I
200 150 1 O0 50 O
6 ( p p m l Figure 2 . PSRE NMR subspectra obtained by taking linear combina- tions of spectra in Figure 1 . A = 0.30s + 1.15s’ and B = 0.70s - 1.1 SS’, corresponding to T,(H) = 13 and 60 ms, respectively. See Figure 1 for symbols and abbreviations.
L ~ ~ ~ ~ I ~ ~ ~ ~ I ~ ~ , I I ~ , I I J
200 150 1 O0 50 O 6 (ppml
Figure 3. Solid-state 13C NMR spectrum of cell walls of A. thaliana. S, Normal spectrum; S’, spectrum obtained with a T,,(H) pulse sequence, in which T,,(H) occurred during a 20-ms interval prior to cross-polarization. See Figure 1 for symbols and other abbreviations.
tion is consistent with proton spin diffusion between cel- lulose microfibrils and the surrounding cell-wall matrix during the relaxation interval, resulting in the averaging of the apparent value of T,(H) for the cell-wall components (Newman et al., 1994). The observation of a distinctly dif- ferent time constant for the polymethylene chains, T,(H) = 60 ms, is consistent with spin-diffusion path lengths of at least tens of nanometers between this material and the cell walls. This is consistent with the polymethylene chains being in the cutin or waxes associated with the cuticle on the outside surface of the leaf epidermal cell walls. Bands assigned to proteins are split between subspectra A and B in Figure 2. This is interpreted in terms of an intermediate time constant, 13 ms < T,(H) < 60 ms, consistent with the deposition of cytoplasmic proteins onto cell-wall surfaces in unbroken cells and inefficient spin diffusion between the proteins and cell-wall components. We conclude that pro- ton spin diffusion during a time constant as long as T,(H) limits the usefulness of T,(H) for resolving details as fine as cellulose crystallites.
Figure 3 shows the spectrum obtained with a proton rotating-frame relaxation pulse sequence. The rotating- frame relaxation curve can be described by a function f:
f =exp[ - t / Tlp(H)]. (7)
An interval of t = 4 ms was adequate for considerable attenuation of signals assigned to noncellulosic substances, but the distinction between T,,(H) values was not suffi- ciently pronounced for separation of subspectra without resorting to PSRE. The PSRE subspectra A and B (Fig. 4)
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

I3C NMR of Cellulose in Arabidopsis thaliana Cell Walls 479
I I c-6
S (ppm) Figure 4. PSRE NMR subspectra obtained by taking linear combina- tions of spectra in Figure 3. A = -0.92s + 4.32s' and B = 1.92s - 4.32S', corresponding to T,,,(H) = 4.9 and 2.6 ms, respectively. See Figure 1 for symbols and abbreviations.
show a clear separation into signals assigned to cellulose and polymethylene chains [subspectrum A, T,,(H) = 4.9 ms] and other substances [subspectrum B, T,,(H) = 2.6 ms]. The observation of distinct values of T,,(H) for cellu- lose and disordered polysaccharides is consistent with the relatively short spin-diffusion path lengths traversed dur- ing T,,(H) relaxation, compared to T,(H) relaxation (Zum- bulyadis, 1983). In other words, T,,(H) can be used to resolve finer detail than T, (H). The observation of indistin- guishable values of T,,(H) for proteins and disordered polysaccharides is attributed to coincidence: the distinct values of T,(H), discussed above, prove that these two categories of molecules are not intimately mixed.
T,(H)
T, relaxation processes differ from T, and TI, processes, in that short values of T, indicate rigidity rather than mobility. Figure 5 shows the spectrum obtained with a spin-spin relaxation pulse sequence. The spin-spin relax- ation curve can be described by a function:
f =exp[ - t/TZ(H)l2/2 (8 )
for rigid solids, or a function:
f =exp[ - t/T,(H)] (9)
for more mobile material. A preliminary experiment showed that Equation 8 was more appropriate for cellulose associated with the peak at 89 ppm and provided an esti- mate of T,(H) = 8.0 t 0.5 ps. This is very similar to a value of T,(H) = 8.8 2 0.3 p s obtained for the sample of Avicel microcrystalline cellulose and provides evidence for a rigid crystalline lattice in the cellulose of the cell walls of A. thaliana leaves. Both values of T,(H) are similar to values reported for crystalline forms of other polymers, e.g. poly- ethylene [T,(H) = 7 ps] (Cheung and Gerstein, 1981).
Equation 9 was found to be more appropriate for the band centered on 173 ppm, and a value of T,(H) = 17 ps was estimated. This was shorter than values in the range 22 to 35 ps, typical of noncrystalline synthetic polymers (Cheung and Gerstein, 1981), but sufficiently long for the material to be labeled "mobile" or "semirigid" rather than "rigid crystalline."
The values of T,(H) were sufficiently distinct for partia1 discrimination between rigid and mobile components (Fig. 5), and the discrimination was improved by PSRE (Fig. 6). The final values of T,(H), selected for cleanest separation of subspectra, were 8.5 ps (subspectrum A) and 19 p s (sub- spectrum B). Values of T,(H) are generally so short that this parameter could, in principle, be used to resolve extremely fine detail (Zumbulyadis, 1983). The resolving power was limited, in the present study, by the use of a 1-ms contact time during which spin information could diffuse over dimensions of nanometers. The resolving power was there- fore similar to that associated with T,,(H).
(C)
Figure 7 shows the spectrum obtained with a 13C spin- lattice relaxation pulse sequence. The pulse sequence de- scribed by Torchia (1978) leads to exponential decay curves rather than the inversion-recovery curves described above for T,(H). Signals from CH and CH, carbon in noncellu- losic substances were considerably attenuated during a relaxation interval of 4 s (Fig. 7). The vertical scale of this spectrum has been increased, relative to Figures 1 to 6, to offset the decay of signals associated with cellulose. Com- parisons with the signal heights in the normal spectrum (not shown) provide the following values: T,(C) = 9 s for
C-2,3,5 1 c-I I I
l u 1 I ~ ~ " ~ ' " ' ~ ' " ' I 20 o 150 100 50 O
6 ( p p m ) Figure 5. Solid-state 13C NMR spectrum of cell walls of A. thaliana. S, Normal spectrum; S', spectrum obtained with a TJH) pulse se- quence, in which T,(H) occurred during a 20-ms interval prior to cross-polarization. See Figure 1 for symbols and other abbreviations.
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

480 Newman et al. Plant Physiol. Vol. 1 1 1, 1996
I * # @ I I I t * * I I I I I I I I I , I
200 150 1 O0 50 O 6 (ppm)
Figure 6. PSRE NMR subspectra obtained by taking linear combina- tions of spectra in Figure 5. A = 1.88s - 4.1 7s' and B = -0.8% + 4.17S', corresponding to TJH) = 8.5 and 19 ps, respectively. See Figure 1 for symbols and abbreviations.
C-4 in crystallite interiors (89 ppm), 6 s for C-4 on crystal- lite surfaces (84 ppm), 5 s for C-6 in crystallite interiors (65 ppm), and <1 s for C-6 on crystallite surfaces (62 ppm). These signal assignments follow Newman et al. (1994). The values of T,(C) fall below the range 15 to 25 s observed for C-4 of cellulose in secondary cell walls of the wood of the dicotyledon Eucalyptus delegatensis and the gymnosperm Pinus radiata (Newman, 1992). The difference is attributed to smaller crystallite dimensions (see below), which results in more efficient I3C spin diffusion mixing between the surface chains, where relaxation is more efficient, and crys- tallite interiors, which are too rigid for efficient T,(C) pro- cesses. The very short value of T,(C) for the peak at 62 ppm is consistent with freedom for rotation of -CH,OH groups around the bond between C-5 and C-6, for chains exposed on crystallite surfaces. Very short values of T,(C) would be expected for C-6 in noncrystalline cellulose also, but other evidence (discussed below) suggests that the A. thaliana cell walls did not contain detectable levels of noncrystalline cellulose.
The clear distinction between values of T,(C) for C-6 in crystallite interiors and on surfaces is relevant to the dis- tinction between crystalline and paracrystalline cellulose. Paracrystalline cellulose, as envisaged by Frey-Wyssling (1954), would consist of bundles of cellulose chains with long axes in approximately parallel orientations but with little molecular ordering along transverse axes. Some inter- chain hydrogen bonds would be stretched and others would be compressed. It seems reasonable to expect that such a loose structure would allow torsional oscillations of -CH,OH groups, if not complete rotation about the bond between C-5 and C-6. In other words, the distinction be- tween interior and surface would be less obvious in a paracrystalline structure. The experimental results provide evidence for a rigid crystalline structure.
The clear distinction between values of T,(C) for C-4 and C-6 on crystallite surfaces is also relevant to the discussion.
The distinction indicates rigid polymer backbones for chains on crystallite surfaces, motion being mostly con- fined to rotation of -CH,OH groups projecting from those backbones. An earlier study of primary cell walls of apple fruit (Newman et al., 1994) could not eliminate the possi- bility of partly ordered material clinging to crystallite sur- faces. That study was based on proton relaxation experi- ments, and the results were ambiguous because proton spins could have diffused between crystallite interiors and partly ordered material during the measurements. The 13C spin relaxation experiments described here are less ambig- uous, since 13C spin diffusion processes are much slower (VanderHart, 1987).
The discussion of T,(C) results is necessarily qualitative, since it is not yet possible to compare the T,(C) values for A. thaliana cell walls with values for pure cellulose in crystalline form. Commercial crystalline celluloses are known to contain noncrystalline material (Sterk et al., 1987) and are therefore not suitable for comparative studies.
Figure 8 shows the spectrum obtained with a I3C spin- spin relaxation pulse sequence. Protons spins were de- coupled during the relaxation interval, and this is known to result in relatively rapid T2(C) decay processes when the frequencies associated with molecular motion are similar to those associated with decoupling (Haw and Johnson, 1986). Persistence of signals assigned to cellulose and polymeth- ylene chains (Fig. 8) provides further evidence for rigid lattices. Persistence of signals assigned to protein side chains (15-25 ppm) and methoxyl carbon in a-D-galactosy- luronate residues of pectic polysaccharides (54 ppm) is attributed to thermal motion at frequencies higher than those associated with decoupling, i.e. 67 kHz in the present study. The signal at 102 ppm, assigned to noncellulosic
I i- I
I C-6i
L I ~ . * ~ I I I n I I t I I I I I I I I I
200 150 1 O0 50 O
6 ( p p m ) Figure 7. Solid-state 13C NMR spectrum of cell walls of A. thaliana obtained with a T,(C) pulse sequence, in which T , ( C ) occurred during a 4-s interval after cross-polarization. "i" and "s" refer to crystallite interiors and surfaces, respectively.
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

13C NMR of Cellulose in Arabidopsis thaliana Cell Walls 48 1
200 150 100 50 O
6 ( p p m ) Figure 8. Solid-state 13C NMR spectrum of cell walls of A. thaliana obtained with a T,(C) pulse sequence, in which T,(C) occurred during an 8-ms interval after cross-polarization. Protons were de- coupled during dephasing and refocusing. A band at 102 ppm, assigned to C-1 in pectic galacturonans and other noncellulosic polysaccharides, is labeled with an asterisk (*).
polysaccharides, is not fully suppressed. It is not clear whether this reflects relatively rigid or relatively mobile molecules.
Crystal Forms
Resolution-enhanced spectra were used to identify the crystal forms of cellulose in the cell walls. Experiments with model substances have shown that disordered non- cellulosic polysaccharides contribute broad peaks that do not respond to resolution enhancement (Kim and New- man, 1995).
The resolution-enhanced NMR spectrum of the cell walls (Fig. 9a) showed a peak within +0.2 ppm of each peak in the spectrum of Avicel microcrystalline cellulose (Fig. 9b). This particular brand of microcrystalline cellulose was cho- sen because it was used as an example of cellulose I in a particularly thorough NMR and diffraction study of cellu- lose polymorphs (Isogai et al., 1989). We therefore inter- preted our spectrum of the A. thaliana cell walls in terms of cellulose I as the dominant form of cellulose. Data for other forms of cellulose showed poor matches, as follows.
Cellulose I1 can be distinguished by one C-1 signal, which occurs at a chemical shift sufficiently large (107.7 ppm) to be clearly resolved from resolution-enhanced sig- nals assigned to C-1 in cellulose I (Hemmingson and Newman, 1995). Figure 9a shows a peak at 107.7 ppm (labeled *), but it is so weak that it could perhaps be dismissed as an artifact of resolution enhancement. This observation places a very low upper limit on the amount of cellulose I1 present.
Cellulose 111 can be distinguished by its relatively simple spectrum (Isogai et al., 1989). In particular, it shows no peaks in the range 63 to 70 ppm and can
therefore not account for peaks observed at 65.4 and 66.4 ppm in Figure 9a.
Cellulose IV contributes peaks at chemical shifts similar to those for cellulose I, although the patterns of relative signal strengths are different (Isogai et al., 1989). The pub- lished data for cellulose IV cannot account for a peak at 62.0 ppm in Figure 9a. The match with cellulose I therefore remains better than a match with cellulose IV. Chanzy et al. (1974) reported electron-diffraction evidence for cellulose IV in the primary cell walls of cotton, but a more recent x-ray diffraction study (Boylston and Hebert, 1995) has shown that the crystal form is cellulose I and has explained why the earlier results indicated another form.
Two distinct crystalline forms of cellulose, I, and I,, have been distinguished in native celluloses using resolution- enhanced solid-state 13C NMR (Cael et al., 1985; Belton et al., 1989; Debzi et al., 1991), electron diffraction (Sugiyama et al., 1991), and IR spectroscopy (Debzi et al., 1991). In the present NMR study, the band at 89 ppm, assigned to C-4 in crystallite interiors, was split into three components by digital resolution enhancement (Fig. 10). The pattern is interpreted as a sum of two overlapping doublets, assigned to the two distinct crystal forms (Atalla and VanderHart, 1984; Newman et al., 1994). The peaks at 90.4 and 88.5 ppm are assigned to I, and I cr stal forms, respectively, and the peak at 89.4 ppm is assigned to a combination of the two. The peak at 90.4 ppm (Fig. 10a) is shifted about 0.2 ppm from the position of the corresponding peak in a spectrum of microcrystalline cellulose (Fig. 10b). We tentatively at- tributed this to a difference in crystallite dimensions. French et al. (1993) suggested that crystal planes are spaced
p .
100 80
:-2,3,5
C- 6
- 60
S ( p p m ) Figure 9. Normal 13C NMR spectra of cell walls of A. thaliana (a) and microcrystalline cellulose (b) processed with digital resolution en- hancement. Carbon numbers refer to cellulose. A signal at 107.7 ppm is labeled with an asterisk (*).
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

482 Newman et al. Plant Physiol. Vol. 1 1 1 , 1996
IS s
I\ s I A
95 9 0 85 80 6 (ppm)
Figure 10. Portion of the normal 13C NMR spectra of cell walls of A. thaliana (a) and microcrystalline cellulose (b) processed with digital resolution enhancement. Peaks are assigned to C-4 in crystallite interiors (labeled " I a " or ''Ip'') or crystallite surfaces (labeled "s"). An unidentified peak is labeled "?."
relatively far apart in cellulose crystals with relatively small cross-sectional dimensions.
The similarities of the peak heights at 90.4 and 88.5 ppm indicate that the I, and I, crystal forms of cellulose in the cell walls of A. thaliana leaves are mixed in similar propor- tions. A similar finding was reported for the cellulose in the primary cell walls of the fruit of apple, another dicotyledon (Newman et al., 1994). It would be interesting to know whether the similarities extend to the cellulose in the pri- mary cell walls of other angiosperms. In contrast to the cellulose in the primary cell walls of dicotyledons, Atalla and VanderHart (1984) reported that the cellulose in the secondary cell walls of the dicotyledons cotton and ramie contained more of the I, form than the I, form. Newman (1994) likewise found the I, form predominant in the sec- ondary cell walls of the angiosperms Castanea sativa, Beilschmiedia tawa, E. delegatensis, and Quercus robur but found the I, form predominant in the secondary cell walls of the gymnosperms P. radiata, Pseudotsuga menziesii, and Agathis australis. It is too early for formulation of general rules concerning proportions of I, and I, celluloses from different sources.
The resolution-enhanced spectrum of A. thaliana cell walls (Fig. 10a) showed a pair of peaks 84.0 and 85.1 ppm, assigned to C-4 in chains exposed on crystallite surfaces. These peaks were resolved in earlier studies of cellulose (Newman, 1994; Newman et al., 1994), and the relative heights were found to be independent of the relative pro- portions of I, and I, forms (Newman, 1994). The pair of peaks has therefore been assigned to pairs of crystallo-
graphically nonequivalent sites on well-ordered crystallite surfaces, regardless of whether the interior is in the I, or I, form (Newman et al., 1994). Surfaces coated with disor- dered cellulose chains, or encrusted in noncellulosic po- lysaccharides, would not be expected to show such a clearly resolved pair of peaks. The heights of the peaks at 84.0 and 85.1 ppm, relative to those at 88.5, 89.4, and 90.4 ppm, are consistent with cellulose crystallites containing so few chains that most of the chains are exposed on crystal- lite surfaces. Newman et al. (1994) illustrated this point by drawing a cross-section of a crystallite containing just 23 chains.
The resolution-enhanced spectrum of the A. thaliana cell walls (Fig. 10a) also showed a peak at 86.2 ppm, which was not present in the spectrum of apple cell walls (Newman et al., 1994). The narrow lineshape is consistent with a well- ordered (crystalline) environment, but the chemical shift is not consistent with known crystalline forms of cellulose (Isogai et al., 1989). The peak is therefore tentatively as- signed to well-ordered chains of noncellulosic substances, e.g. xyloglucans, adhering to the surfaces of cellulose crys- tallites. Whitney et al. (1995) identified a peak at 99.5 ppm as being characteristic of C-1 of the Xyl residues of xylo- glucans bound to cellulose but did not identify the corre- sponding chemical shift for C-4 of the Glc residues. The similarity in molecular conformation between the back- bones of bound xyloglucans and the underlying cellulose chains could result in similar chemical shifts for Glc resi- dues in both. Whitney et al. (1995) found that the peak at 99.5 ppm was sufficiently sharp to respond to resolution enhancement. At this chemical shift the spectrum of A. thaliana showed only a small peak (Fig. 9) that could not be reliably distinguished from noise and was much smaller than peaks assigned to cellulose on crystal surfaces. The weakness of the signals at 86.2 ppm (possibly xyloglucans) and 99.5 ppm (xyloglucans) indicates that no more than a small proportion of the total surface area of the cellulose crystallites can be involved. This is a surprising finding that requires further work because xyloglucans are a major component of the primary cell walls of dicotyledons, in- cluding A. thaliana (Zablackis et al., 1995), and it is well known that xyloglucans bind to cellulose in vitro (Vincken et al., 1995; Whitney et al., 1995). This binding of xyloglu- cans to cellulose is an important feature of models of dicotyledon primary cell walls (Talbott and Ray, 1992; Carpita and Gibeaut, 1993) in which the xyloglucans are believed to coat and cross-link the cellulose microfibrils. Furthermore, McQueen-Mason and Cosgrove (1994) tenta- tively suggested that expansins (proteins that catalyze the extension of isolated plant cell walls) act by inducing slip- page between the cellulose microfibril and its surface coat of xyloglucans.
The interpretation in terms of a mixture of cellulose I, and I, in thin crystallites, based primarily on signals as- signed to C-4, is supported by evidence found elsewhere in the resolution-enhanced spectrum. The chemical shift range associated with C-1 shows three peaks (Fig. 9a), the outer pair (104.5 and 106.5 ppm) assigned to cellulose I,, the inner one (105.5 ppm) assigned to cellulose Ia, and
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

13C NMR of Cellulose in Arabidopsis thaliana Cell Walls 483
crystallite-surface cellulose (Atalla and VanderHart, 1984). The chemical shift range associated with C-6 shows peaks at 62.0 and 63.1 ppm, assigned to crystallite surfaces, and peaks at 65.4 and 66.4 ppm, assigned to crystallite-interior cellulose. The latter pair of peaks can be interpreted in terms of a contribution from cellulose I,, split into two components, with a contribution from cellulose I, coincid- ing with one component (Atalla and VanderHart, 1984). Relative proportions of signal strength assigned to crystal- lite-surface C-6 and crystallite-interior C-6 are consistent with those observed for C-4, as discussed above.
Cellulose crystallinity
Early studies of the molecular ordering of cellulose in plant cell walls were based on x-ray diffraction or alcohol- ysis techniques. Frey-Wyssling (1954) reported crystallini- ties of 57 and 59% for the cellulose in primary cell walls of roots and coleoptiles of corn (Z. mays) and described these crystallinities as ”astonishingly poor.” Cherno et al. (1982) reported the proportions of amorphous, mesomorphous, and crystalline cellulose in mulberry leaves (excluding veins) as 12.3, 27.4, and 60.3%, respectively. Cherno et al. (1990) reported that apple fruit cellulose could be distin- guished from cotton and wood celluloses by the ”very low degree of order,” i.e. crystallinities of 54 and 55% for pulp and peel, respectively, compared with values of 73% for cotton and 60% for wood.
The results of this NMR investigation suggest that con- cepts of crystallinity require clarification. The “degree of crystallinity” of cellulose has sometimes been estimated from I3C NMR spectra by measuring the area of a band at 89 ppm and dividing that area by the total area assigned to C-4, i.e. the area under bands at 89 and 84 ppm (Horii et al., 1987; Zhbankov et al., 1986; Teeaar et al., 1987). These authors a11 reported good agreement between the NMR results and the degree of crystallinity as determined by x-ray diffraction, although Sterk et al. (1987) reported de- viations for microcrystalline cellulose. The degree of crys- tallinity is commonly assumed to represent the fraction of crystalline cellulose, as distinct from noncrystalline cellu- lose. Newman et al. (1994) exploited T,,(H) to separate a subspectrum of crystalline cellulose from a 13C NMR spec- trum of primary cell walls of apple fruit and concluded that little orno cellulose was contained in amorphous (noncrys- talline) domains. In the current, more-detailed study of cellulose in the cell walls of A. thaliana leaves, more evi- dente has been obtained to support that conclusion. The evidence is as follows:
1. The values of T,(H), T,,(H) and T,(H) are a11 indistin- guishable for bands at 89 and 84 ppm. This is not consistent with the assignment of one band to ordered domains and the other band to disordered domains.
2. The values of T,(C) and T2(C) are similar for the two bands, which confirms the rigidity of the backbone of chains contributing to the band at 84 ppm.
3. Resolution enhancement (Fig. 9) split the band at 84 ppm into two components, assigned to crystallographically inequivalent sites on well-ordered crystal surfaces. Disor-
dered domains would be expected to contribute broad bands not responsive to resolution enhancement.
Published I3C NMR studies of noncrystalline (amor- phous) cellulose have shown only a broad band across the region assigned to C-4, with a nearly flat profile from 80 to 85 ppm and a tail extending to 90 ppm (Atalla et al., 1980). The chemical shift for C-1 remains unchanged at 105 ppm. This combination is not compatible with features separated by a T,,(H) sequence (Fig. 4), since subspectrum .A shows little signal strength at 80 ppm and subspectrum B shows no peak at 105 ppm. It would not be possible for C-1 to contribute to one subspectrum and C-4 to the other, since spin diffusion would mix the spin information on a time scale much shorter than T,,(H). Subspectrum B in Figure 6, separated by exploiting T,(H) and assigned to disordered material, shows a weak peak at 105 ppm. This is too weak to account for more than a trace of noncrystalline cellulose.
We conclude that in discussing the concept of a degree of crystallinity it is important to distinguish between variations in crystallite dimensions and variations in the fraction of crystalline cellulose. The comments of Frey- Wyssling (1954) regarding “astonishingly poor crystal- linity” in primary-wall cellulose may refer to small crys- tallite dimensions rather than a high proportion of noncrystalline cellulose. The small dimensions of the crystallites may have broadened the x-ray diffraction lines to widths approaching those associated with non- crystalline cellulose. Himmelsbach et al. (1986) also com- mented on the difficulty in separating the concepts of crystallinity, morphology, and order used in the context of characterization of cellulose.
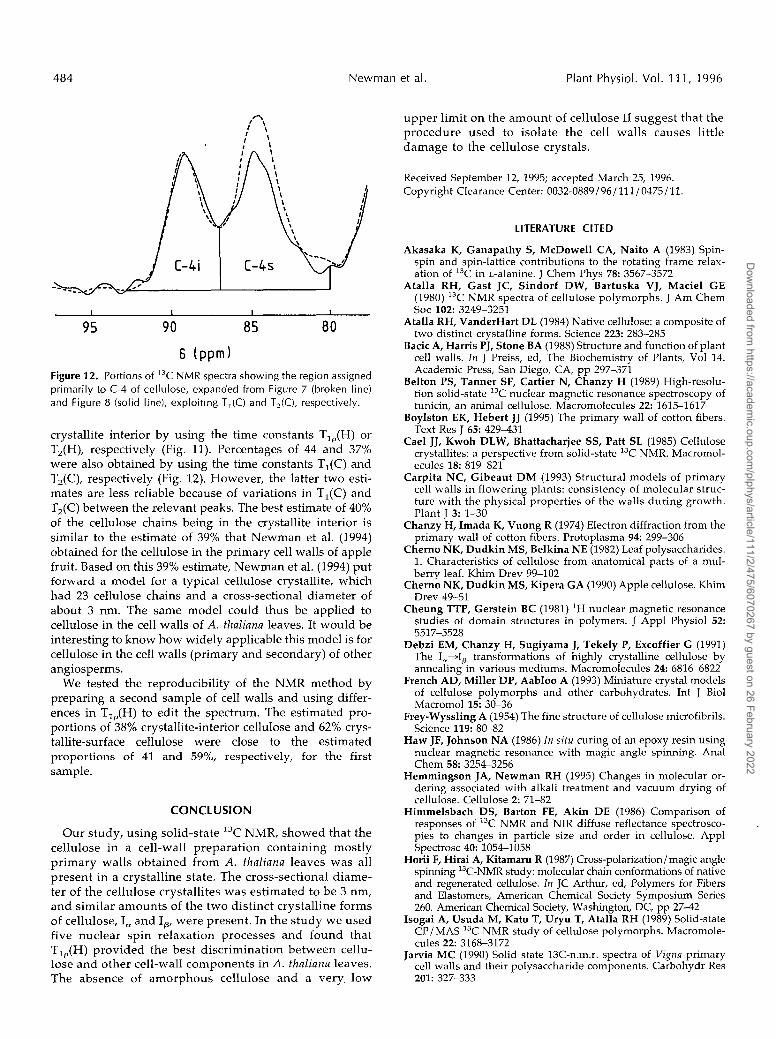
We estimated the proportions of cellulose chains in the crystallite interior and on the crystallite surface from the relative areas of bands at 89 and 84 ppm. Arbitrary bound- aries are shown as vertical lines in Figures 11 and 12. We estimated that 41 or 39% of the cellulose chains were in the
95 90 85 80 6 ( p p m )
Figure 1 1 . Portions of 13C NMR subspectra showing the region assigned to C-4 of cellulose: subspectrum A from Figure 4 (solid line) and subspectrum A from Figure 6 (broken Iine), separated from the total spectrum by exploiting T,,(H) and T,(H), respectively. ” i ” and ”s” refer to interiors and surfaces of crystallites, respectively.
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022
484 Newman et al. Plant Physiol. Vol. 11 1 , 1996
I I I I
95 90 85 80
6 ( p p m ) Figure 12. Portions of 13C NMR spectra showing the region assigned primarily to C-4 of cellulose, expanded from Figure 7 (broken line) and Figure 8 (solid line), exploiting T,(C) and T,(C), respectively.
crystallite interior by using the time constants T,,(H) or T,(H), respectively (Fig. 11). Percentages of 44 and 37% were also obtained by using the time constants T,(C) and T2(C), respectively (Fig. 12). However, the latter two esti- mates are less reliable because of variations in T,(C) and T2(C) between the relevant peaks. The best estimate of 40% of the cellulose chains being in the crystallite interior is similar to the estimate of 39% that Newman et al. (1994) obtained for the cellulose in the primary cell walls of apple fruit. Based on this 39% estimate, Newman et al. (1994) put forward a model for a typical cellulose crystallite, which had 23 cellulose chains and a cross-sectional diameter of about 3 nm. The same model could thus be applied to cellulose in the cell walls of A. thaliana leaves. It would be interesting to know how widely applicable this model is for cellulose in the cell walls (primary and secondary) of other angiosperms.
We tested the reproducibility of the NMR method by preparing a second sample of cell walls and using differ- ences in T,,(H) to edit the spectrum. The estimated pro- portions of 38% crystallite-interior cellulose and 62% crys- tallite-surface cellulose were close to the estimated proportions of 41 and 59%, respectively, for the first sample.
CONCLUSION
Our study, using solid-state I3C NMR, showed that the cellulose in a cell-wall preparation containing mostly primary walls obtained from A. thaliana leaves was a11 present in a crystalline state. The cross-sectional diame- ter of the cellulose crystallites was estimated to be 3 nm, and similar amounts of the two distinct crystalline forms of cellulose, I, and I,, were present. In the study we used five nuclear spin relaxation processes and found that T,,(H) provided the best discrimination between cellu- lose and other cell-wall components in A. thaliana leaves. The absence of amorphous cellulose and a very. low
upper limit on the amount of cellulose I1 suggest that the procedure used to isolate the cell walls causes little damage to the cellulose crystals.
Received September 12, 1995; accepted March 25, 1996. Copyright Clearance Center: 0032-0889/96/ 111/0475/11
LITERATURE ClTED
Akasaka K, Ganapathy S, McDowell CA, Naito A (1983) Spin- spin and spin-lattice contributions to the rotating frame relax- ation of I3C in L-alanine. J Chem Phys 78: 3567-3572
Atalla RH, Gast JC, Sindorf DW, Bartuska VJ, Maciel GE (1980) 13C NMR spectra of cellulose polymorphs. J Am Chem
Atalla RH, VanderHart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223: 283-285
Bacic A, Harris PJ, Stone BA (1988) Structure and function of plant cell walls. In J Preiss, ed, The Biochemistry of Plants, Vol 14. Academic Press, San Diego, CA, pp 297-371
Belton PS, Tanner SF, Cartier N, Chanzy H (1989) High-resolu- tion solid-state 13C nuclear magnetic resonance spectroscopy of tunicin, an animal cellulose. Macromolecules 22: 1615-1617
Boylston EK, Hebert JJ (1995) The primary wall of cotton fibers. Text Res J 65: 429431
Cael JJ, Kwoh DLW, Bhattacharjee SS, Patt SL (1985) Cellulose crystallites: a perspective from solid-state 13C NMR. Macromol- ecules 18: 819-821
Carpita NC, Gibeaut DM (1993) Structural models of primary cell walls in flowering plants: consistency of molecular struc- ture with the physical properties of the walls during growth. Plant J 3: 1-30
Chanzy H, Imada K, Vuong R (1974) Electron diffraction from the primary wall of cotton fibers. Protoplasma 94: 299-306
Cherno NK, Dudkin MS, Belkina NE (1982) Leaf polysaccharides. 1. Characteristics of cellulose from anatomical parts of a mul- berry leaf. Khim Drev 99-102
Cherno NK, Dudkin MS, Kipera GA (1990) Apple cellulose. Khim Drev 49-51
Cheung TTP, Gerstein BC (1981) 'H nuclear magnetic resonance studies of domain structures in polymers. J Appl Physiol 52:
Debzi EM, Chanzy H, Sugiyama J, Tekely P, Excoffier G (1991) The Im+Ip transformations of highly crystalline cellulose by annealing in various mediums. Macromolecules 24: 6816-6822
French AD, Miller DP, Aabloo A (1993) Miniature crystal models of cellulose polymorphs and other carbohydrates. Int J Biol Macromol 15: 30-36
Frey-Wyssling A (1954) The fine structure of cellulose microfibrils. Science 119: 80-82
Haw JF, Johnson NA (1986) In situ curing of an epoxy resin using nuclear magnetic resonance with magic angle spinning. Ana1 Chem 58: 3254-3256
Hemmingson JA, Newman RH (1995) Changes in molecular or- dering associated with alkali treatment and vacuum drying of cellulose. Cellulose 2 71-82
Himmelsbach DS, Barton FE, Akin DE (1986) Comparison of responses of 13C NMR and NIR diffuse reflectance spectrosco- pies to changes in particle size and order in cellulose. Appl Spectrosc 40: 1054-1058
Horii F, Hirai A, Kitamaru R (1987) Cross-polarization/magic angle spinning 13C-NMR study: molecular chain conformations of native and regenerated cellulose. In JC Arthur, ed, Polymers for Fibers and Elastomers, American Chemical Society Symposium Series 260. American Chemical Society, Washington, DC, pp 27-42
Isogai A, Usuda M, Kato T, Uryu T, Atalla RH (1989) Solid-state CP/MAS 13C NMR study of cellulose polymorphs. Macromole- cules 22: 3168-3172
Jarvis MC (1990) Solid state 13C-n.m.r. spectra of Vigna primary cell walls and their polysaccharide components. Carbohydr Res
SOC 102: 3249-3251
5517-5528
201: 327-333
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022

13C NMR of Cellulose in Arabidopsis thaliana Cell Walls 485
Jarvis MC, Apperley DC (1990) Direct observation of cell wall structure in living plant tissues by solid-state I3C NMR spec- troscopy. Plant Physiol 92: 61-65
Kim YS, Newman RH (1995) Solid state I3C NMR study of wood degraded by the brown rot fungus Gloeophyllum tvabeum. Holz- forschung 49: 109-114
MacKay AL, Wallace JC, Sasaki K, Taylor IEP (1988) Investiga- tion of the physical structure of the primary plant wall by proton magnetic resonance. Biochemistry 27: 1467-1473
McCann MC, Wells 8, Roberts K (1990) Direct visualization of cross-links in the primary cell wall. J Cell Sci 96: 323-334
McQueen-Mason S, Cosgrove DJ (1994) Disruption of hydrogen bonding between plant cell wall polymers by proteins that in- duce wall extension. Proc Natl Acad Sci USA 91: 6574-6578
Newman RH (1992) I3C NMR spectroscopy of multiphase bioma- terials. In WG Glasser, H Hatakeyama, eds, Viscoelasticity of Biomaterials, American Chemical Society Symposium Series 489. American Chemical Society, Washington, DC, pp 311-319
Newman RH (1994) Crystalline forms of cellulose in softwoods and hardwoods. J Wood Chem Technol 14: 451466
Newman RH, Ha M-A, Melton LD (1994) Solid-state 13C NMR investigation of molecular ordering in the cellulose of apple cell walls. J Agric Food Chem 42: 1402-1406
Newman RH, Hemmingson JA (1990) Determination of the de- gree of cellulose crystallinity in wood by 13C nuclear magnetic resonance spectroscopy. Holzforschung 44: 351-355
Sterk H, Sattler W, Janosi A, Paul D, Esterbauer H (1987) Einsatz der Festkorper 13C-NMR-Spektroskopie fiir die Bestimmung der Kristallinitat in Cellulosen. Das Papier 41: 664-668
Sugiyama A, Vuong R, Chanzy H (1991) Electron diffraction study on the two crystalline phases occurring in native cellulose from an alga1 cell wall. Macromolecules 2 4 41684175
Talbott LD, Ray I'M (1992) Molecular size and separability fea- tures of pea cell wall polysaccharides. Implications for models of primary wall structure. Plant Physiol 98: 357-368
Teeaar R, Serimaa R, Paakkari T (1987) Crystallinity of cellulose, as determined by CP/MAS NMR and XRD methods. Polymer Bull 17: 231-237
Tekely P, Canet D, Delpuech J-J (1989) Observation of 'H nuclei in heterogeneous solids via cross-polarization I3C N.M.R. Mo1 Physiol 67: 81-96
Torchia DA (1978) The measurement of proton-enhanced car- bon-13 T, values by a method which suppresses artefacts. J Magn Reson 30 613-616
VanderHart DL (1987) Natural-abundance l3C-I3C spin exchange in rigid crystalline organic solids. J Magn Reson 72: 1 3 4 7
Vincken J-I', de Keizer A, Beldman G, Voragen AGJ (1995) Fractionation of xyloglucan fragments and their interaction with cellulose. Plant Physiol 108: 1579-1585
Whitney SEC, Brigham JE, Darke AH, Reid JSG, Gidley MJ (1995) ln vitro assembly of cellulose/xyloglucan networks: ul- trastructural and molecular aspects. Plant J 8: 491-504
Zablackis E, Huang J, Miiller 8, Darvill AG, Albersheim P (1995) Characterization of the cell-wall polysaccharides of Arabidopsis thaliana leaves. Plant Physiol 107: 1129-1138
Zhbankov RG, Ioelovich MYa, Treimanis A, Lippmaa ET, Teejaer R, Kaputskii FN, Grinshpan DD, Lushchik LG (1986) Determination of the degree of crystallinity of cellulose by high- resolution solid-state carbon-13 NMR. Khim Drev 3-6
Zumbulyadis N (1983) Selective carbon excitation and the detec- tion of spatial heterogeneity in cross-polarization magic-angle- spinning NMR. J Magn Reson 53: 486494
Dow
nloaded from https://academ
ic.oup.com/plphys/article/111/2/475/6070267 by guest on 26 February 2022























