Embed Size (px)
Citation preview

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
3.320: Lecture 22 (4/28/05)
Ab-Initio Thermodynamics and Structure Prediction: Time Coarse-graining, Effective
Hamiltonians and Cluster Expansions

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Methods with multiple time scales:Coarse-grain fast one away
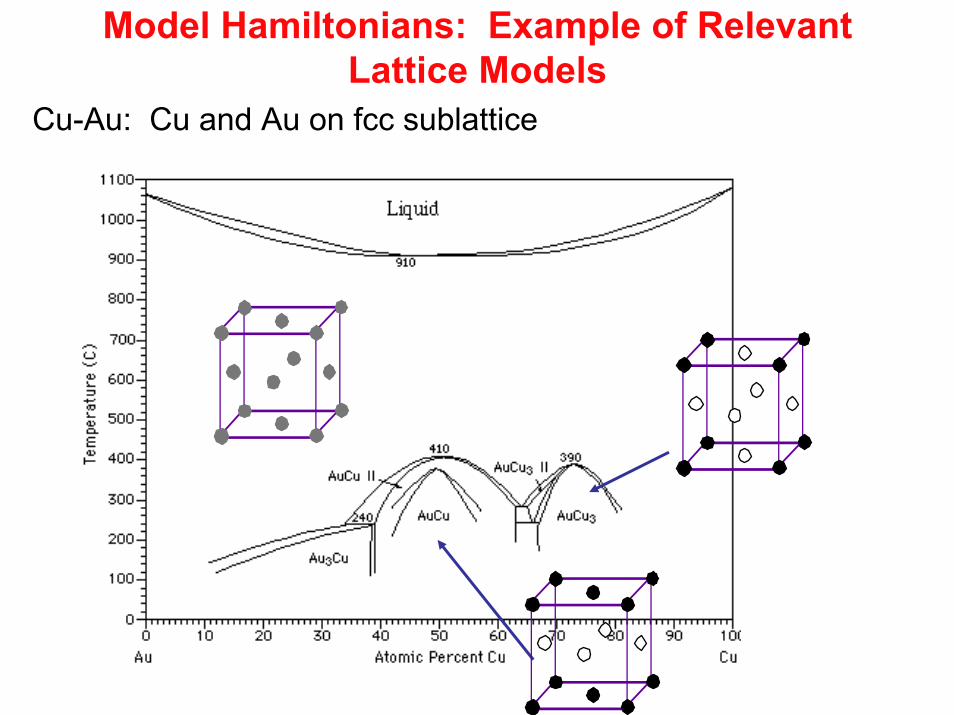
Model Hamiltonians: Example of Relevant Lattice Models
Cu-Au: Cu and Au on fcc sublattice

Configurational Disorder in Fixed HostLiCoO2: Li and vacancies
Triangular lattice of Li and vacancies
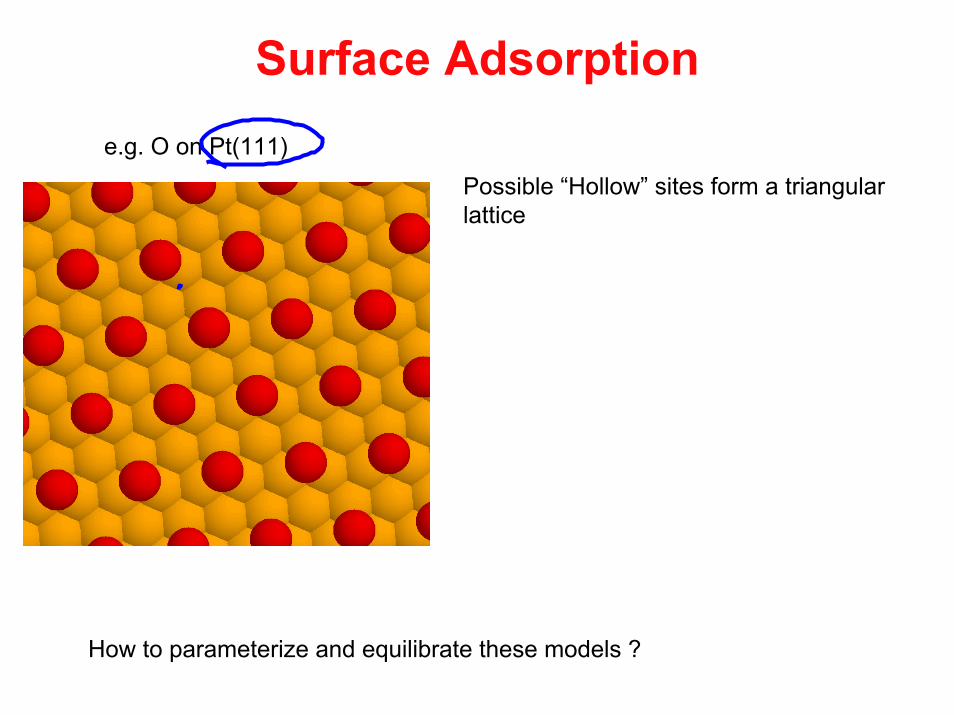
Surface Adsorptione.g. O on Pt(111)
Possible “Hollow” sites form a triangular lattice
How to parameterize and equilibrate these models ?
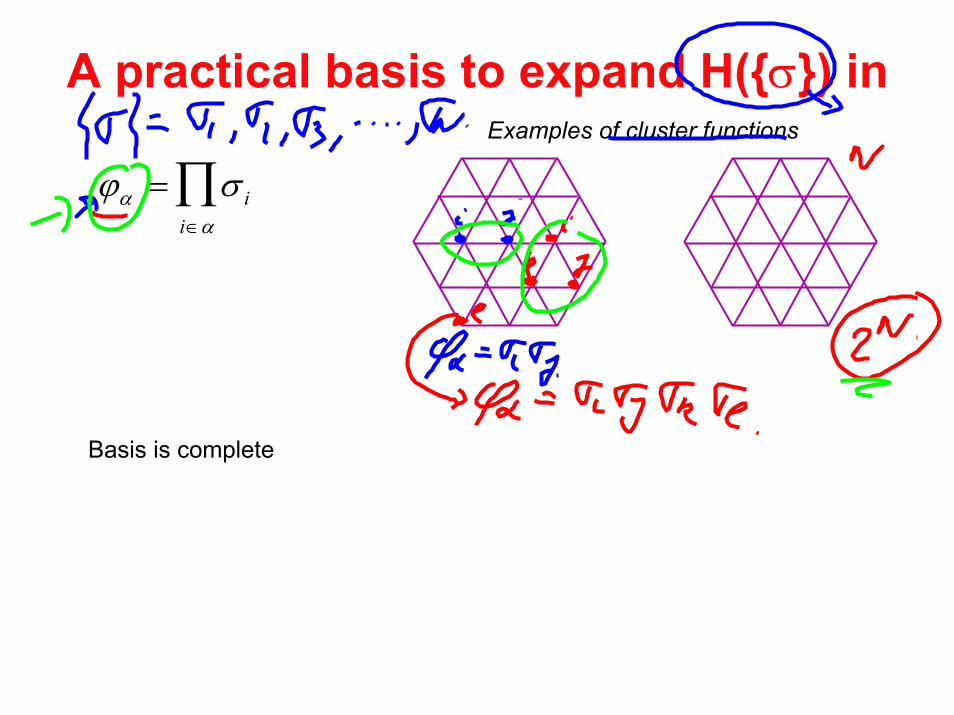
A practical basis to expand H(σ) in
ϕα = σ ii∈α∏
Examples of cluster functions
Basis is complete

Cluster function basis is orthogonalf σ( ) g σ( ) =
12N f σ( )
σ ∑ g σ( )
12N
suitable scalar product
ϕα = 0σ ∑Note that for any basis function
ϕα ϕβ =1
2N ϕασ ∑ ϕβ
orthogonality proof
ϕα ϕβ = σ i2
i∈α∩β∏ σ i
i∈αi∉β
∏ σ ii∈βi∉α
∏ = δαβ
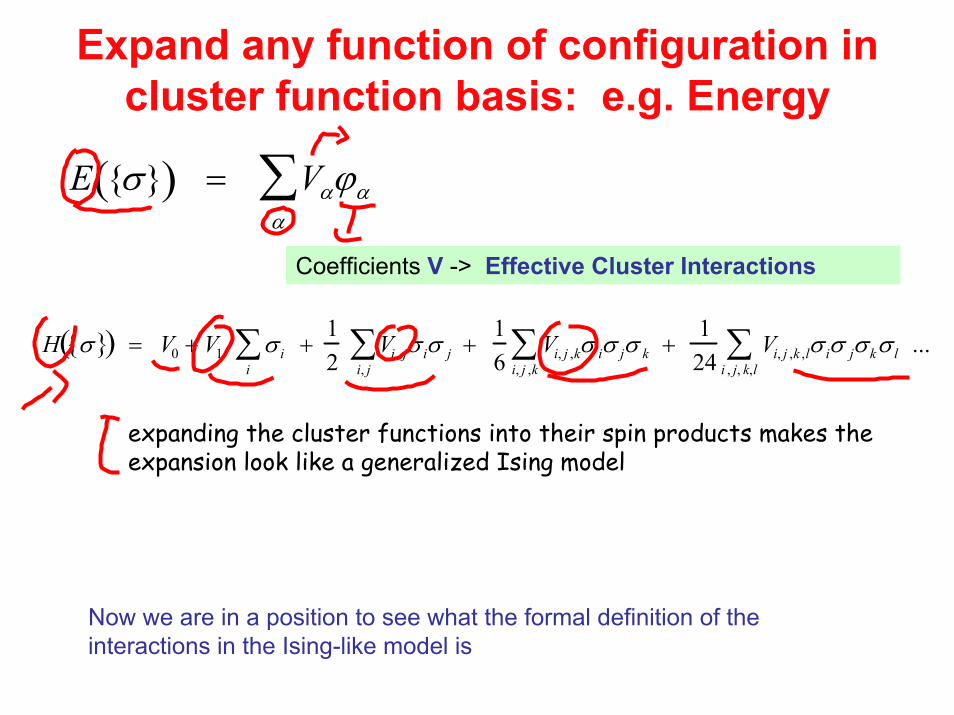
Expand any function of configuration in cluster function basis: e.g. Energy
E σ( ) = Vαϕαα∑
Coefficients V -> Effective Cluster Interactions
H σ ( ) = V0 + V1 σ i + 12
i
∑ Vi , jσ ii, j∑ σ j +
16
Vi, j ,kσ ii, j ,k∑ σ jσ k +
124
Vi, j ,k ,lσ ii , j, k,l∑ σ jσkσ l ...
expanding the cluster functions into their spin products makes the expansion look like a generalized Ising model
Now we are in a position to see what the formal definition of the interactions in the Ising-like model is

Definition of the InteractionsE σ( ) = Vαϕα
α∑
take scalar product with ϕβ
Vβ =1
2Nσ ∑ ϕβ E σ( ) =
12nβ
i= ±1i∈β
∑ ϕβ1
2N−nβσ −β∑ E σ( )
Work out example for pair interaction
Many direct and indirect methods have been developed for calculating the Vα
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
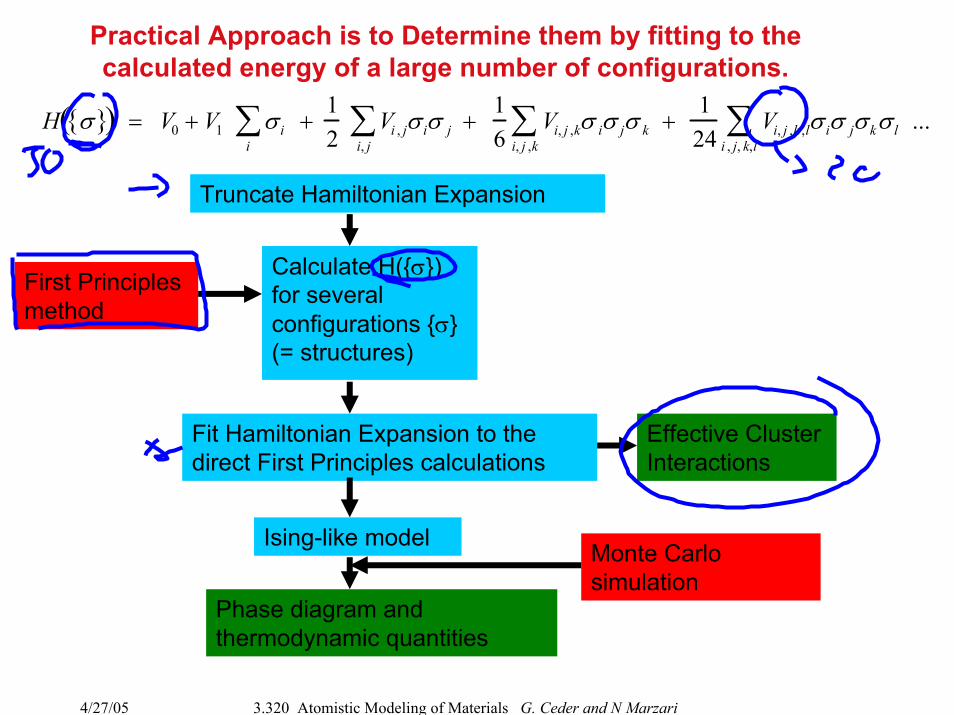
Practical Approach is to Determine them by fitting to the calculated energy of a large number of configurations.
Truncate Hamiltonian Expansion
Calculate H(σ) for several configurations σ (= structures)
Fit Hamiltonian Expansion to the direct First Principles calculations
Ising-like model
Phase diagram and thermodynamic quantities
First Principlesmethod
Monte Carlosimulation
Effective Cluster Interactions
H σ ( ) = V0 + V1 σ i + 12
i
∑ Vi , jσ ii, j∑ σ j +
16
Vi, j ,kσ ii, j ,k∑ σ jσ k +
124
Vi, j ,k ,lσ ii , j, k,l∑ σ jσkσ l ...
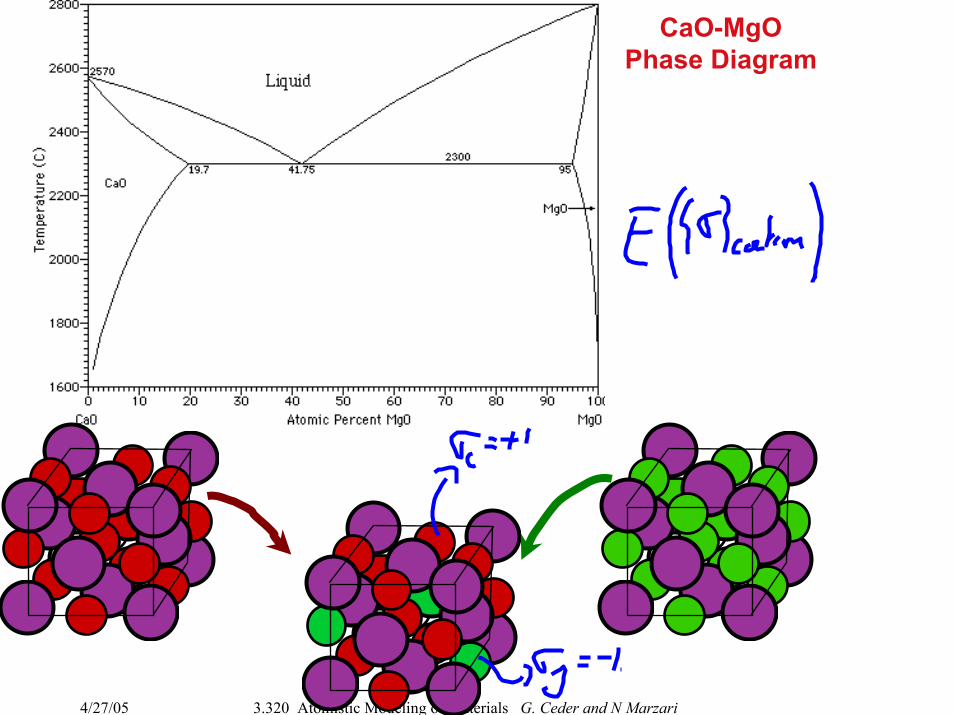
CaO-MgOPhase Diagram
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
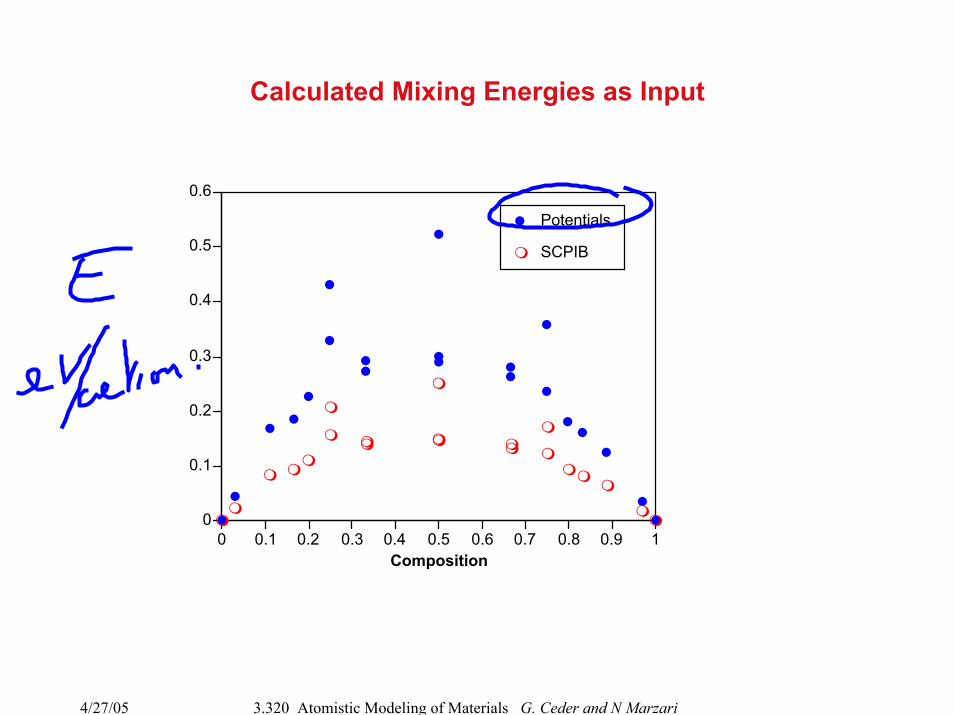
Calculated Mixing Energies as Input
0
0.1
0.2
0.3
0.4
0.5
0.6
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1Composition
Potentials
SCPIB

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Effective Cluster Interactions for CaO-MgO
1 2 3 4 5 6-0.05
-0.04
-0.03
-0.02
-0.01
0
0.01
0.02
SCPIB
Potentials
1 32 1 2
Pairs Triplets Quadruplets
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
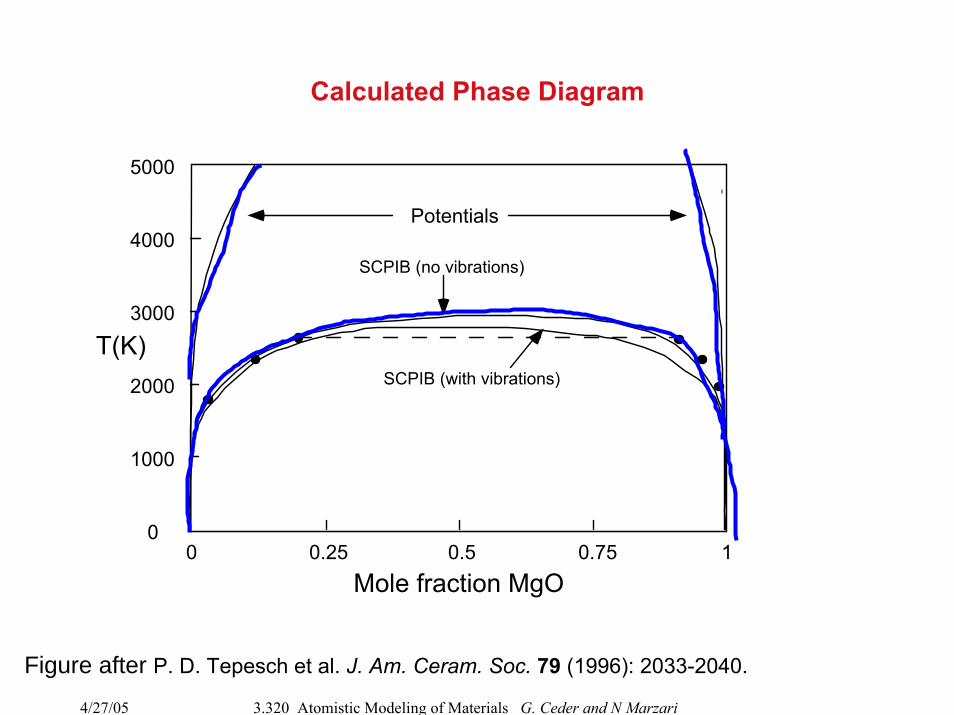
Calculated Phase Diagram
0 0.25 0.5 0.75 1
5000
4000
3000
2000
1000
0
Mole fraction MgO
T(K)
Potentials
SCPIB (no vibrations)
SCPIB (with vibrations)
Figure after P. D. Tepesch et al. J. Am. Ceram. Soc. 79 (1996): 2033-2040.
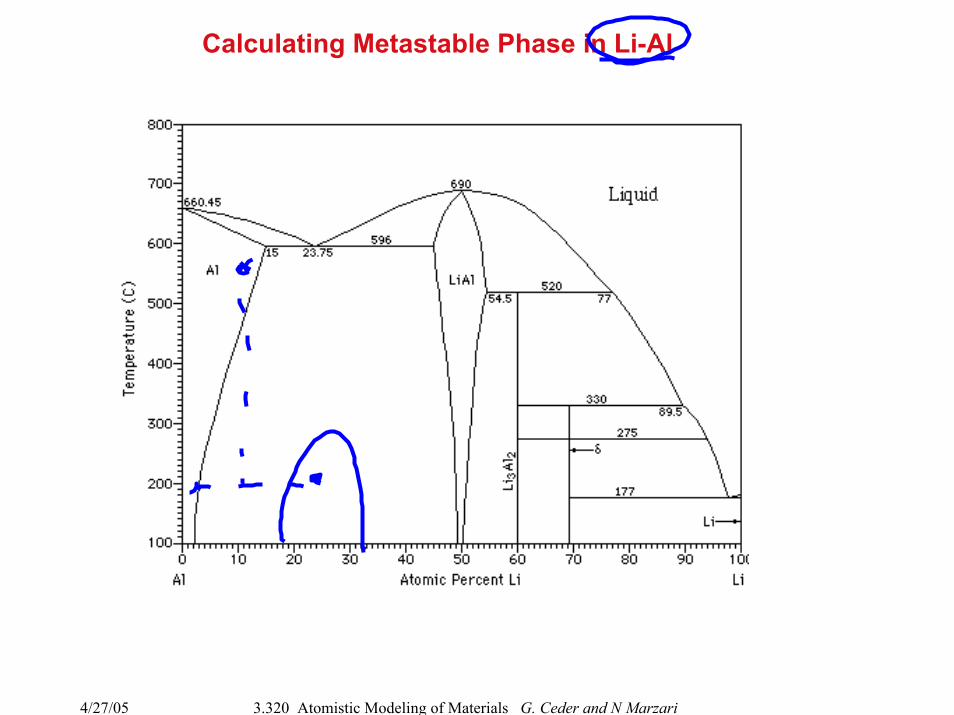
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Calculating Metastable Phase in Li-Al
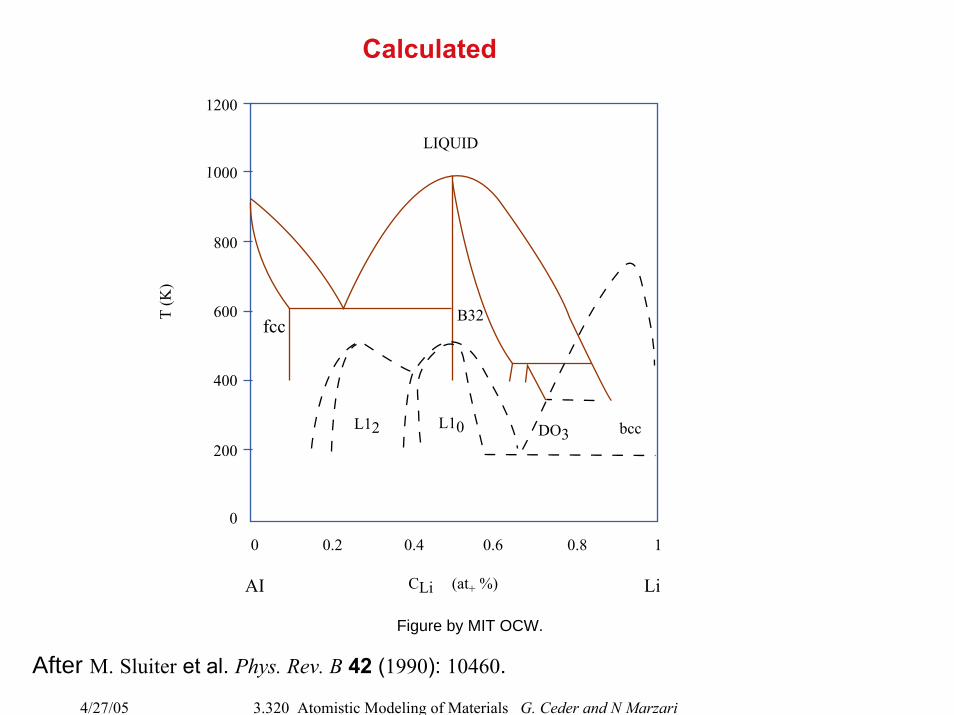
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Calculated
After M. Sluiter et al. Phys. Rev. B 42 (1990): 10460.
1000
800
600
400
200
0
1200
T (K
)
0 0.2 0.4 0.6 0.8 1
CLi
LIQUID
B32
L12 L10 DO3 bcc
(at+ %)AI
fcc
Li
Figure by MIT OCW.
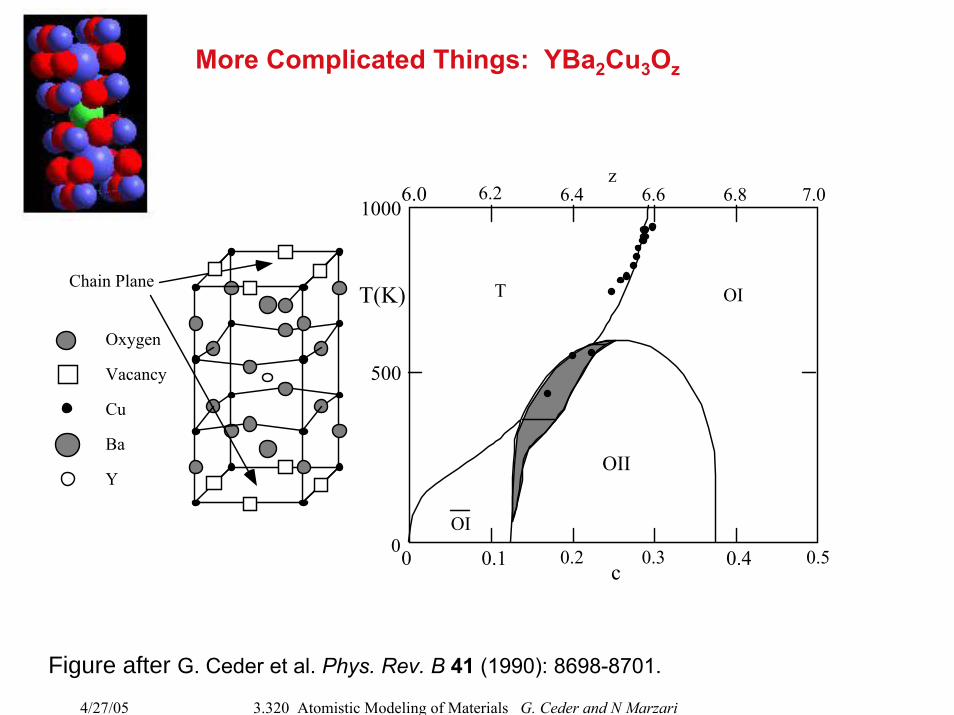
More Complicated Things: YBa2Cu3Oz
Oxygen
Vacancy
Cu
Ba
Y
Chain Plane
0.2 0.3 0.4 0.50 0.1
6.0 6.2 6.4 6.6 6.8 7.0z
1000
500
0
T(K)
OII
T OI
OI
c
Figure after G. Ceder et al. Phys. Rev. B 41 (1990): 8698-8701.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
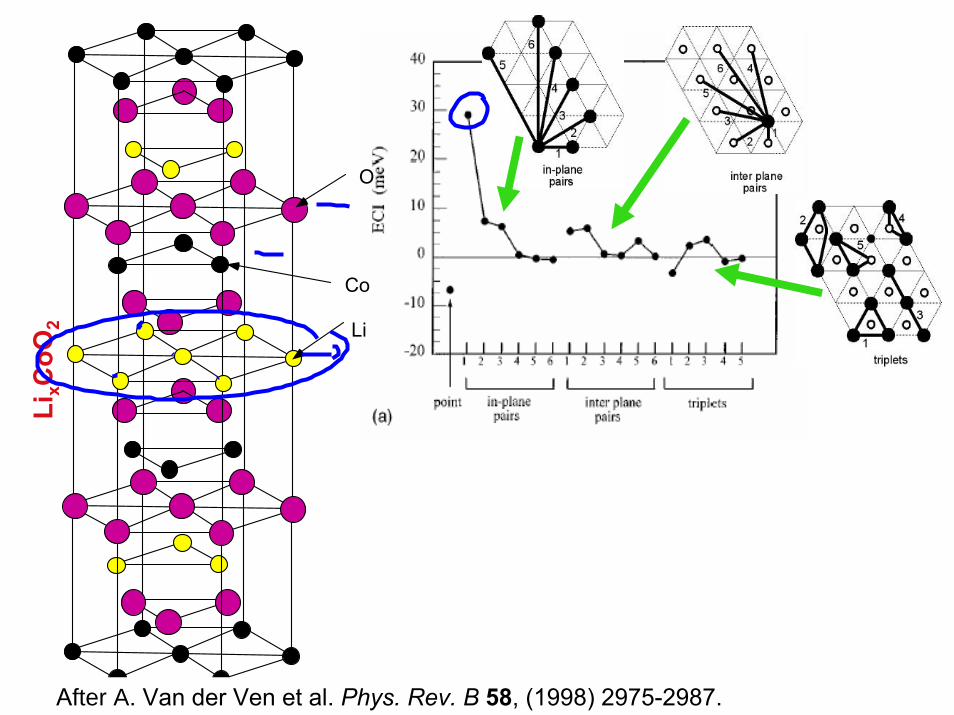
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
LixC
oO2
O
Co
Li
After A. Van der Ven et al. Phys. Rev. B 58, (1998) 2975-2987.
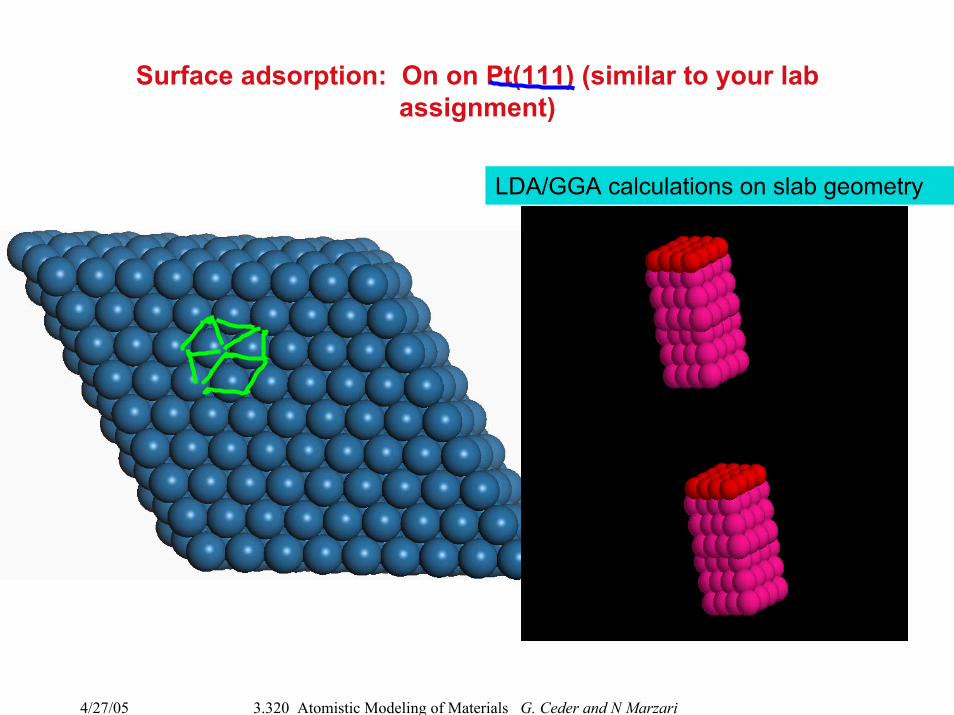
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Surface adsorption: On on Pt(111) (similar to your lab assignment)
LDA/GGA calculations on slab geometry
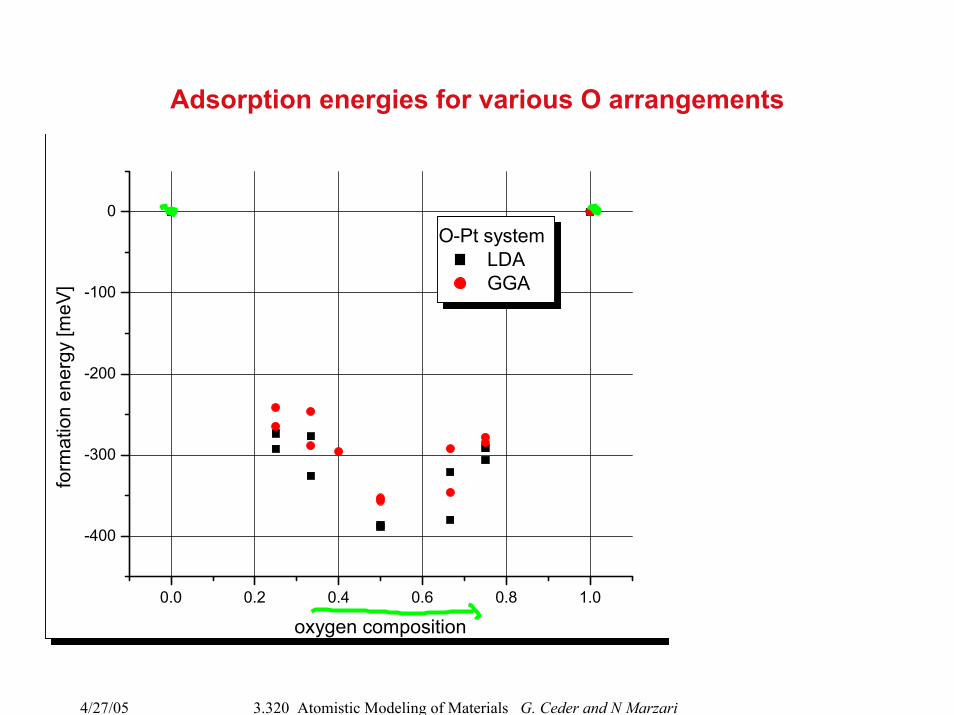
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Adsorption energies for various O arrangements
0.0 0.2 0.4 0.6 0.8 1.0
-400
-300
-200
-100
0
form
atio
n en
ergy
[meV
]
oxygen composition
O-Pt system LDA GGA
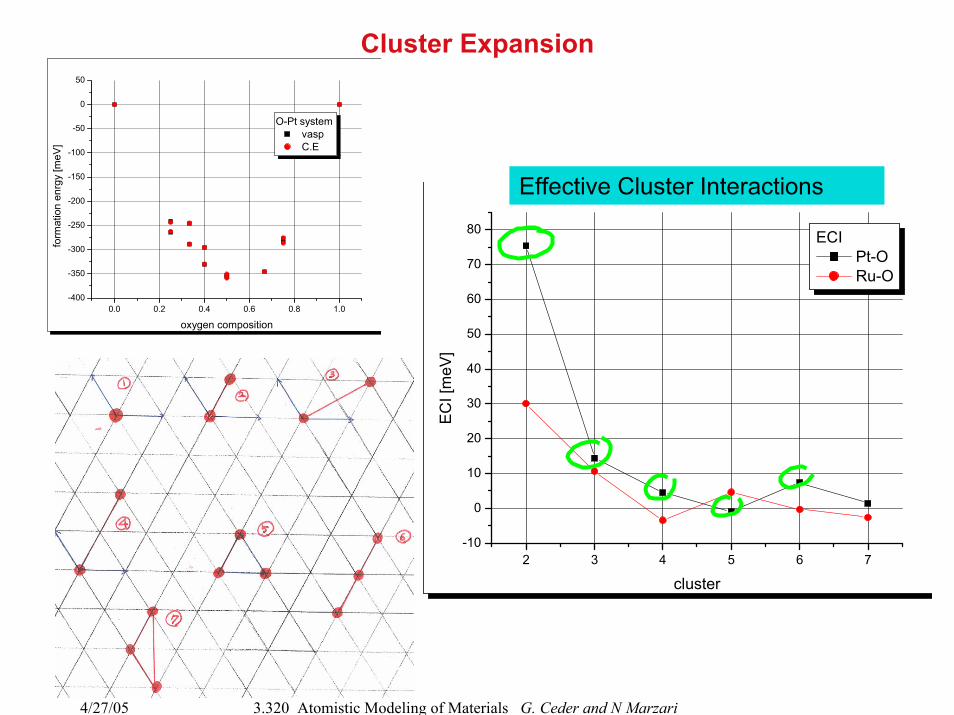
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Cluster Expansion
0.0 0.2 0.4 0.6 0.8 1.0-400
-350
-300
-250
-200
-150
-100
-50
0
50
form
atio
n en
rgy
[meV
]
oxygen composition
O-Pt system vasp C.E
2 3 4 5 6 7-10
0
10
20
30
40
50
60
70
80
ECI [
meV
]
cluster
ECI Pt-O Ru-O
Effective Cluster Interactions
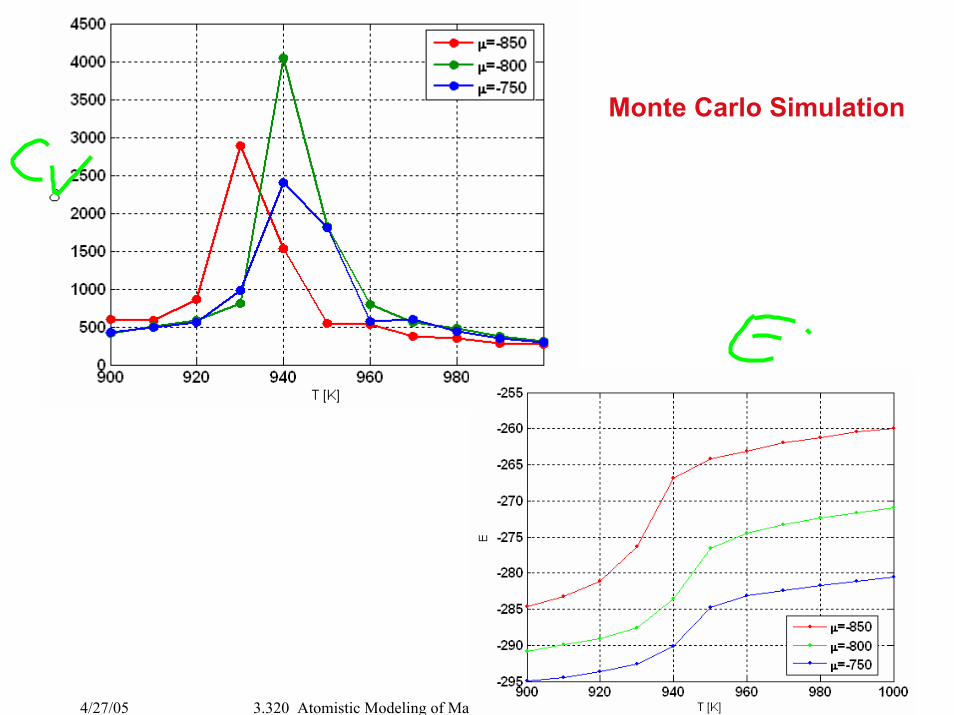
Monte Carlo Simulation
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
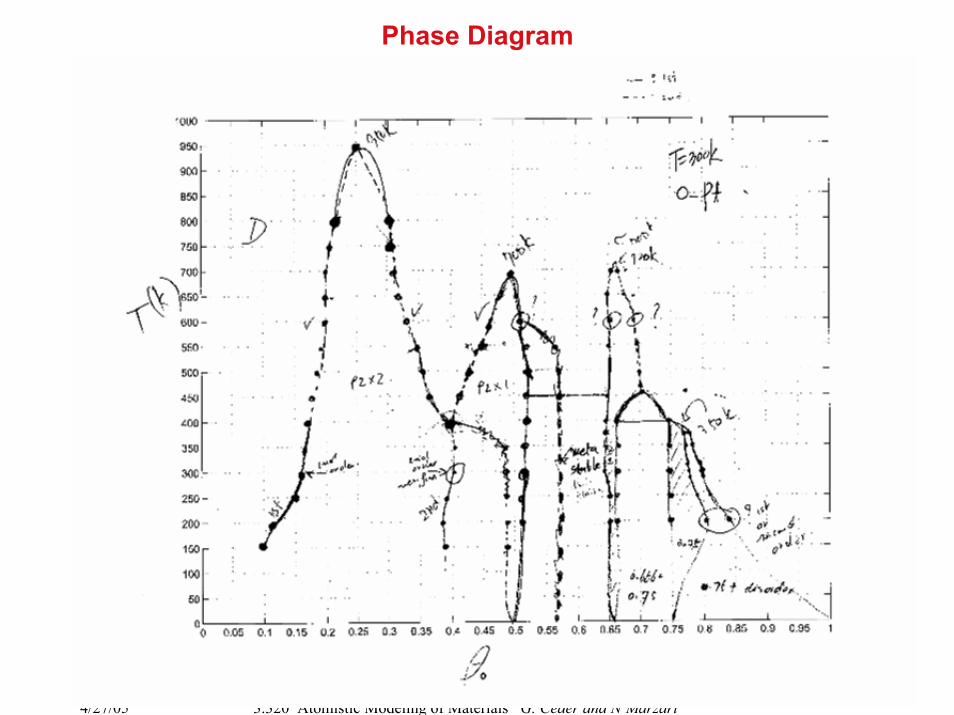
Phase Diagram
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
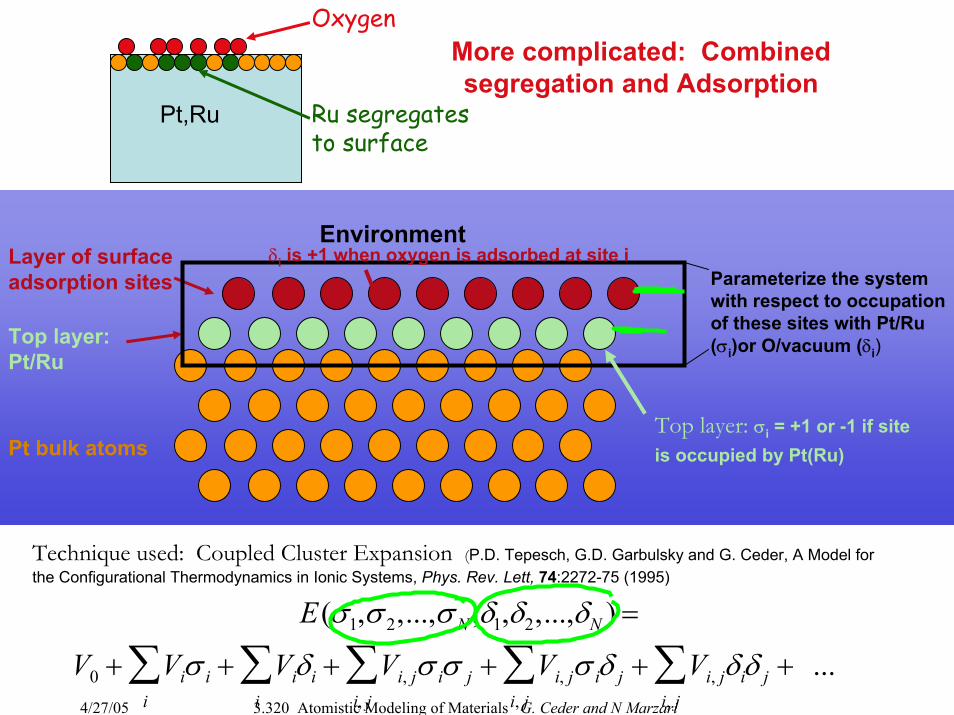
More complicated: Combined segregation and Adsorption
Pt,Ru Ru segregatesto surface
Oxygen
Layer of surface adsorption sites
Pt bulk atoms
Top layer: Pt/Ru
Parameterize the system with respect to occupation of these sites with Pt/Ru(σi)or O/vacuum (δi)
Technique used: Coupled Cluster Expansion (P.D. Tepesch, G.D. Garbulsky and G. Ceder, A Model for the Configurational Thermodynamics in Ionic Systems, Phys. Rev. Lett, 74:2272-75 (1995)
E(σ1,σ 2,...,σ N ,δ1,δ2,...,δN ) =
V0 + Viσ ii
∑ + Viδii
∑ + Vi, jσ iσ ji, j∑ + Vi, jσ iδ j
i, j∑ + Vi, jδiδ j
i, j∑ + ...
δi is +1 when oxygen is adsorbed at site i
Top layer: σi = +1 or -1 if site is occupied by Pt(Ru)
Environment
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
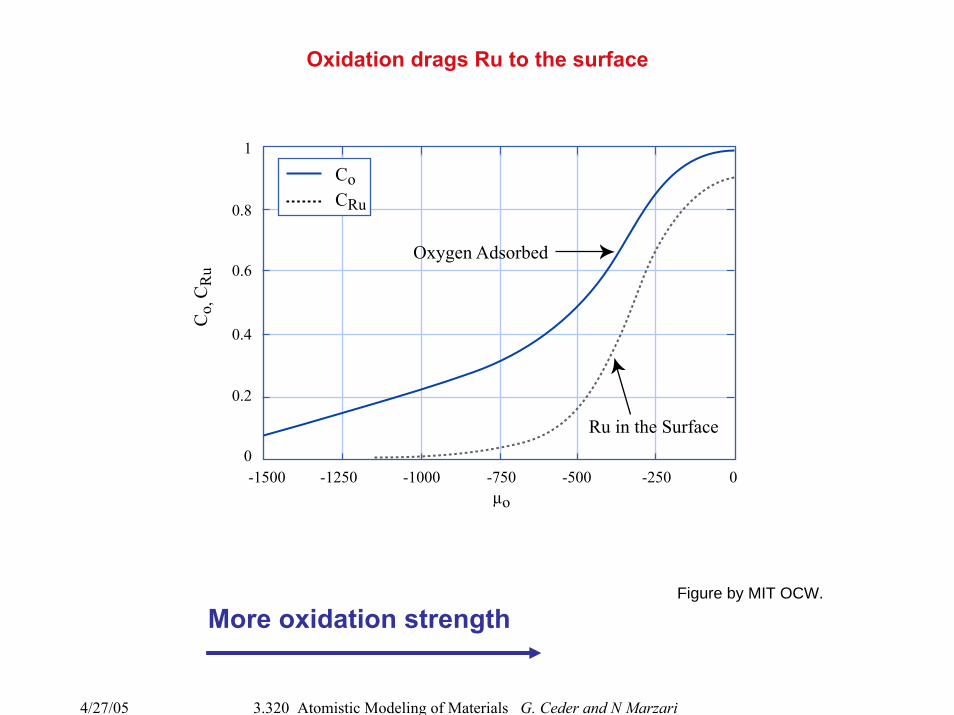
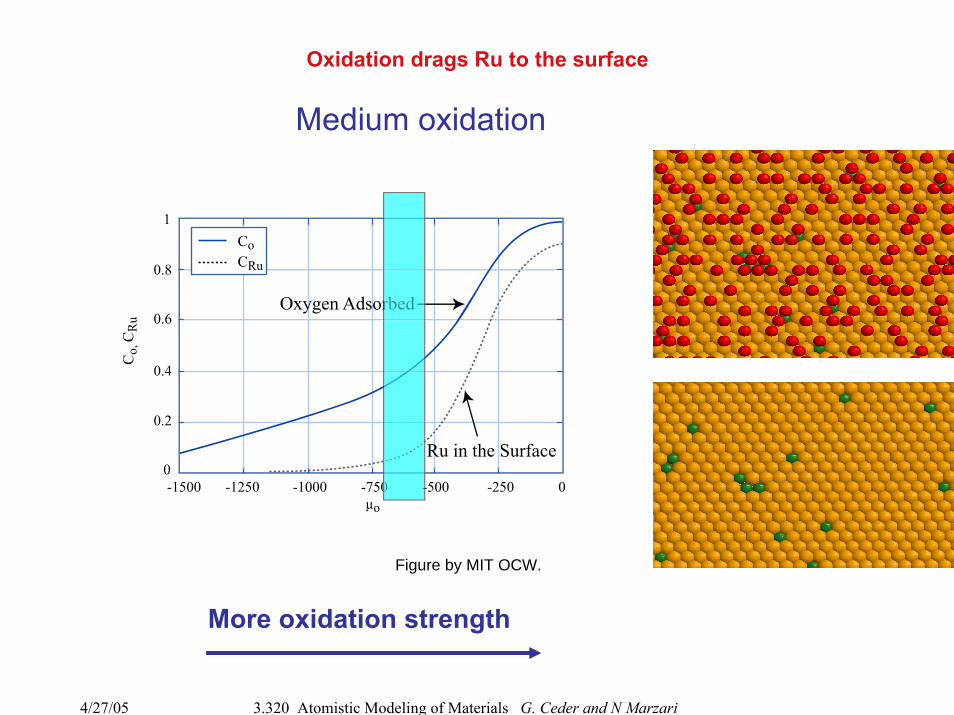
Oxidation drags Ru to the surface
More oxidation strength
1
0.8
0.6
0.4
0.2
0-1500 -1250 -1000 -750 -500 -250 0
Ru in the Surface
Oxygen Adsorbed
µo
CoCRu
Co,
CR
u
Figure by MIT OCW.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
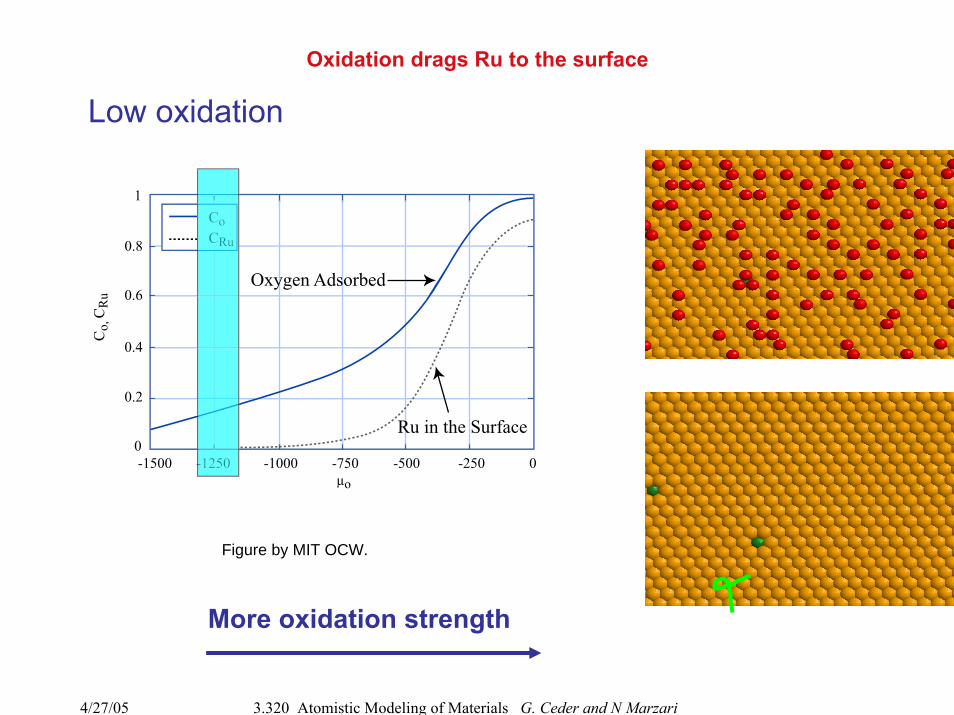
Oxidation drags Ru to the surface
More oxidation strength
Low oxidation
Figure by MIT OCW.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Oxidation drags Ru to the surface
More oxidation strength
Medium oxidation
Figure by MIT OCW.

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Oxidation drags Ru to the surface
More oxidation strength
High oxidation
Figure by MIT OCW.
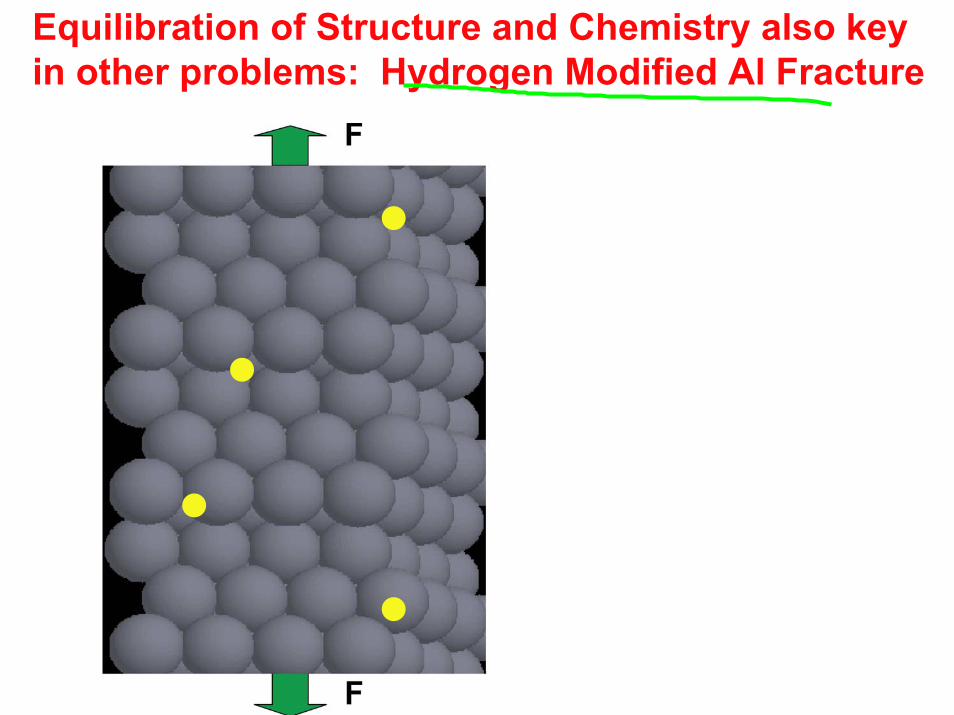
Equilibration of Structure and Chemistry also key in other problems: Hydrogen Modified Al Fracture

For slow separation impurities can flow in
Free energy of separation (and force displacement relation) depend on amount and arrangement of impurity
Need to equilibrate both amount and arrangement of H on Al(111) for each separation
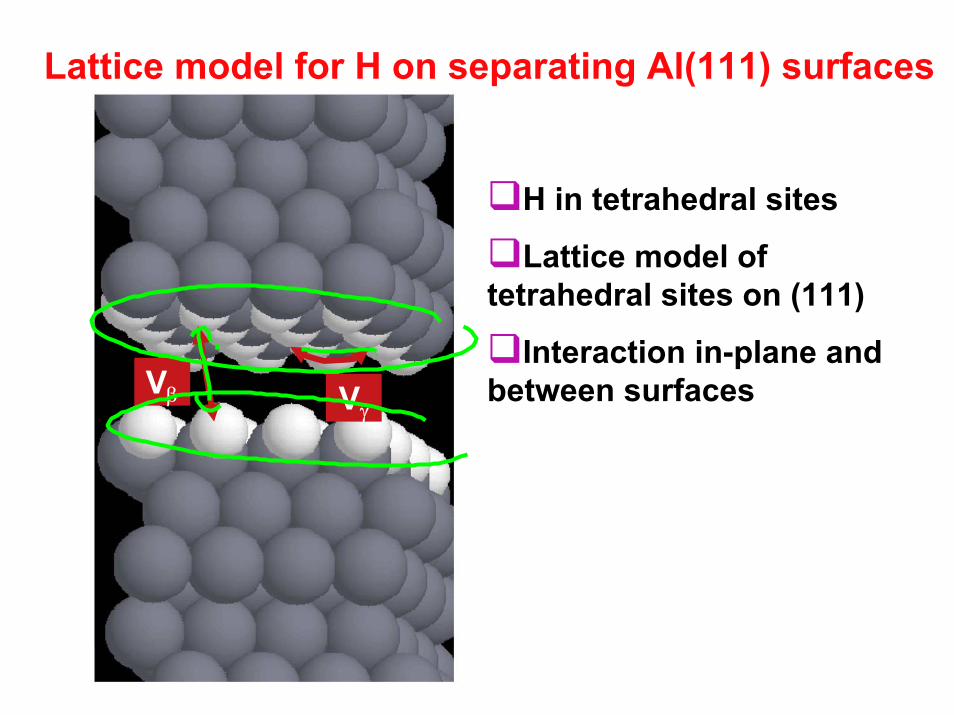
Lattice model for H on separating Al(111) surfaces
Vβ Vγ
H in tetrahedral sites
Lattice model of tetrahedral sites on (111)
Interaction in-plane and between surfaces

ProcedureCalculate energy of different H configurations on surface at different plane separations.
Cluster expand H configuration energy at each plane separation
Monte Carlo simulation at each plane separation
Take derivatives of free energy (to get force)
Construct grand potentials and construct equilibrium “trajectory”
A. Van der Ven, G. Ceder, Acta Materialia 52, (2004) 1223-1235.
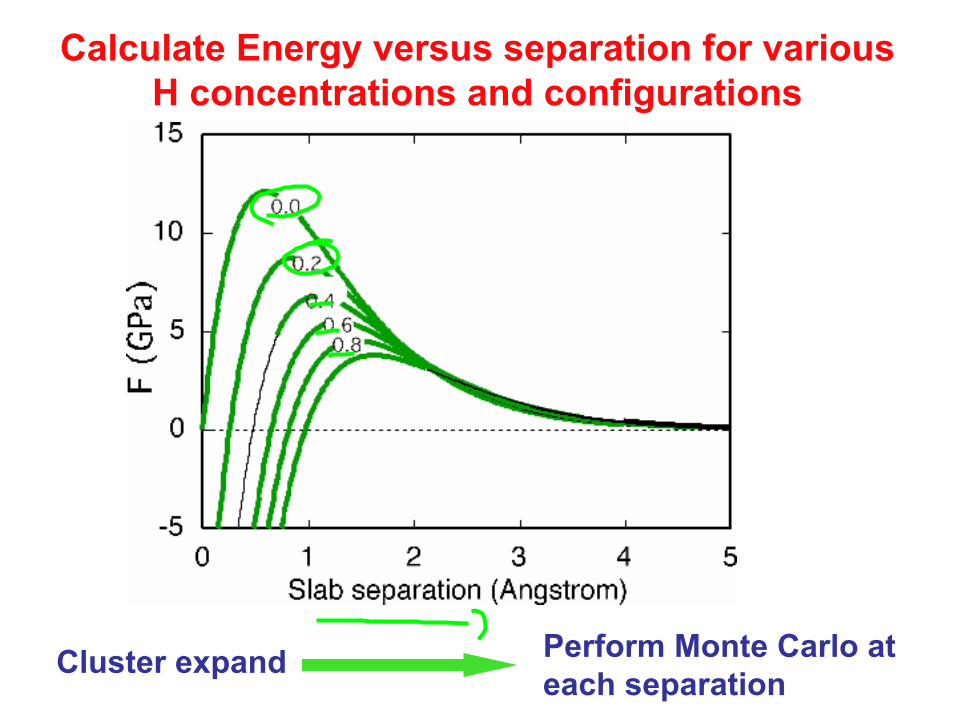
Calculate Energy versus separation for various H concentrations and configurations
Perform Monte Carlo at each separation
Cluster expand
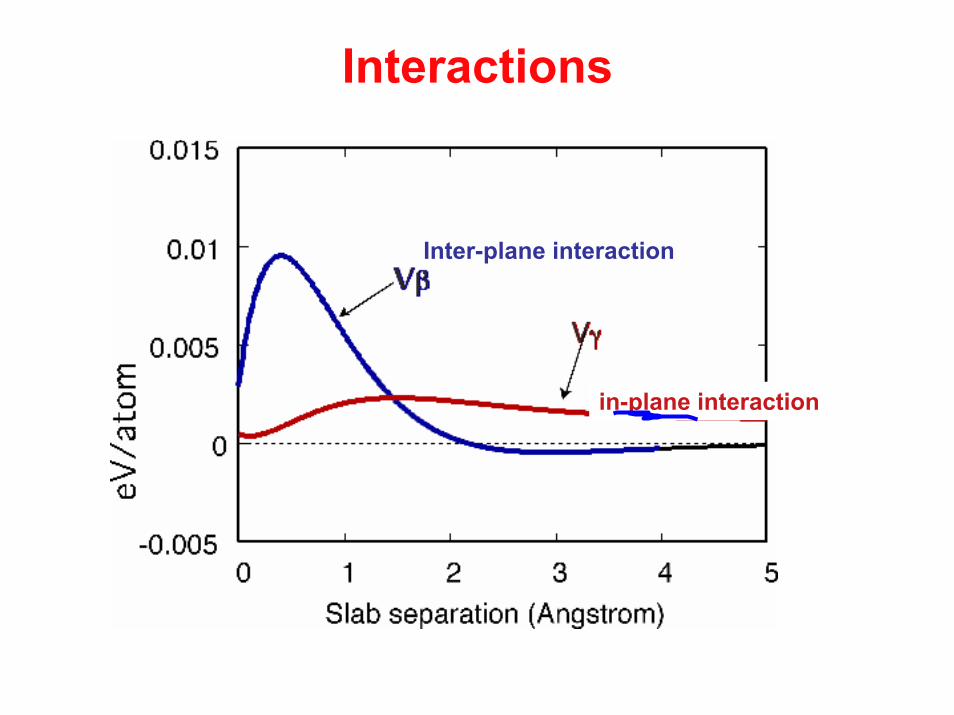
Interactions
Inter-plane interaction
in-plane interaction
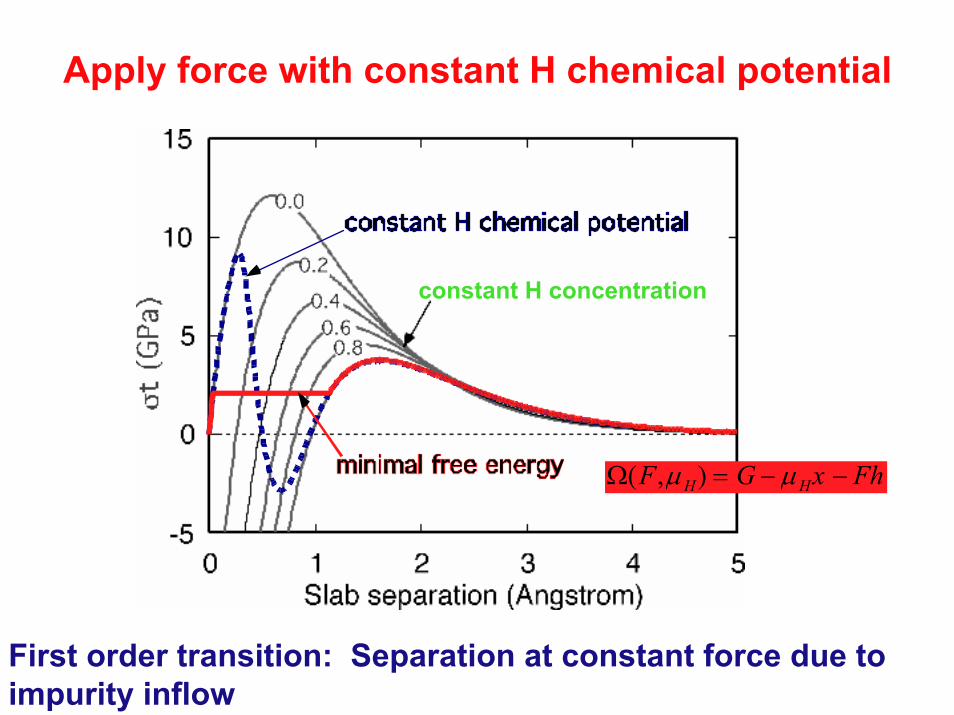
Apply force with constant H chemical potential
constant H concentration
First order transition: Separation at constant force due to impurity inflow
Ω(F,µH) = G − µHx − Fh
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
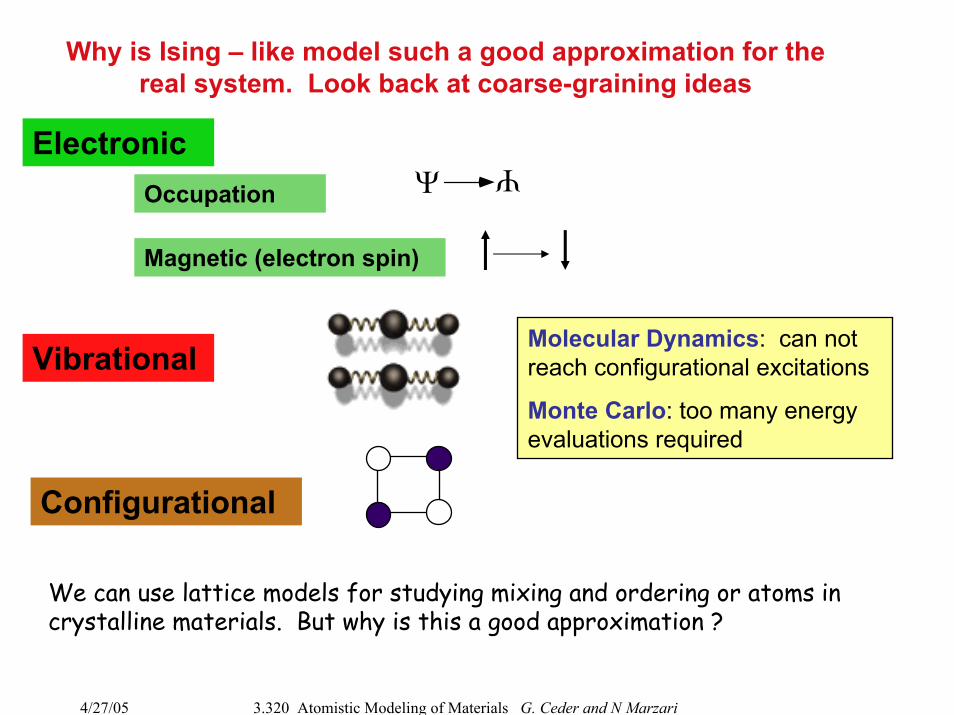
Why is Ising – like model such a good approximation for the real system. Look back at coarse-graining ideas
ElectronicΨ
Ψ
Occupation
Magnetic (electron spin)
Molecular Dynamics: can not reach configurational excitations
Monte Carlo: too many energy evaluations required
Vibrational
Configurational
We can use lattice models for studying mixing and ordering or atoms in crystalline materials. But why is this a good approximation ?

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Coarse-graining: The concept
Can we integrate partition function over fast degrees of freedom to obtain an effective Hamiltonian for the slower degrees of freedom ?
e.g. for an alloy: Can we find an effective free energy function for the substitutional arrangement of an alloy that includes the entropic effect of vibrations and electronic excitations ?
YES
Use Monte Carlo, Molecular Dynamics, or analytical methods to integrate effect of temperature on fast degrees of freedom
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
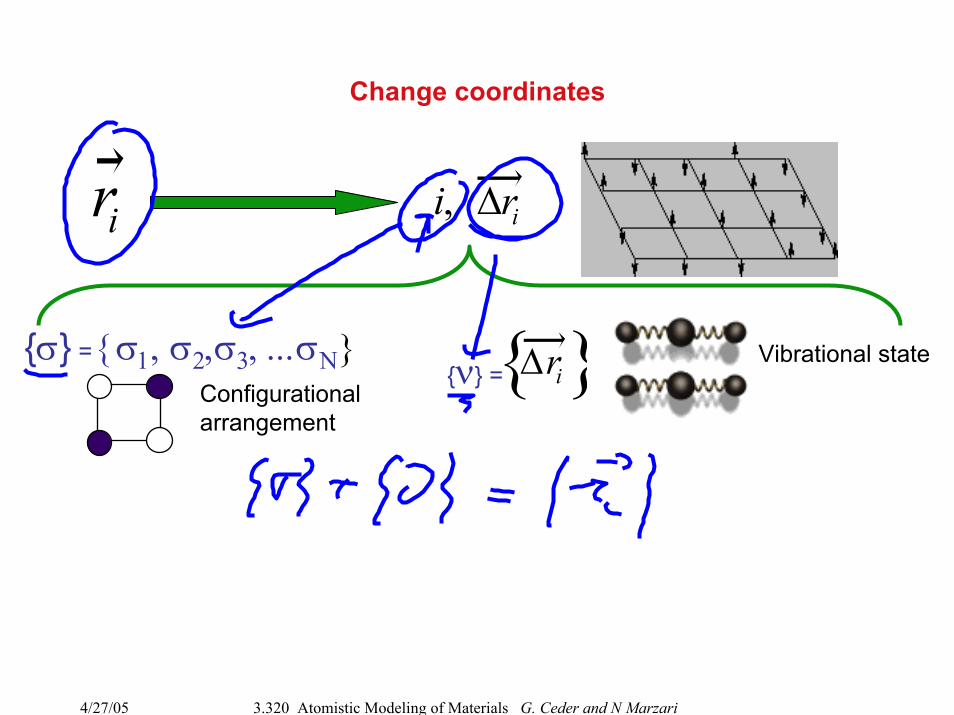
Change coordinates
ri i, ∆ri
σ = σ1, σ2,σ3, ...σΝConfigurationalarrangement
ν = Vibrational state∆ri
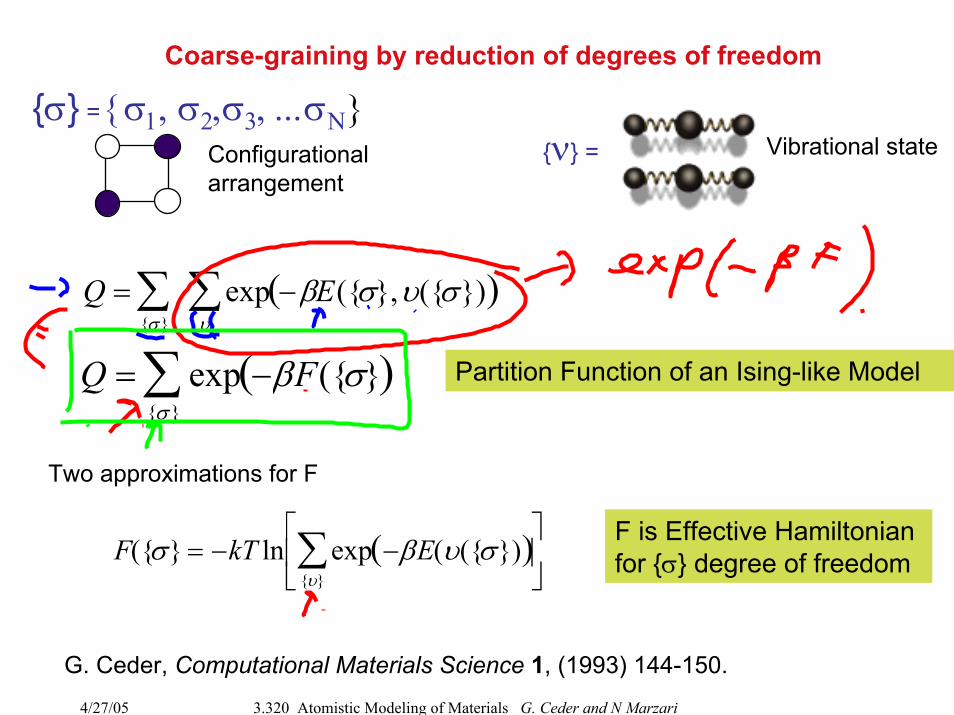
Coarse-graining by reduction of degrees of freedom
Q =σ∑ exp −βE(σ,υ(σ)( )
υ∑
Q = exp −βF(σ( )σ∑
σ = σ1, σ2,σ3, ...σΝν =Configurational
arrangementVibrational state
Partition Function of an Ising-like Model
Two approximations for F
F(σ = −kT ln exp −βE(υ(σ)( )υ∑⎡
⎣ ⎢
⎤
⎦ ⎥
F is Effective Hamiltonian for σ degree of freedom
G. Ceder, Computational Materials Science 1, (1993) 144-150.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Approximations to F(σ) determine which excitations (entropies) are included in the total free energy
F(σ = −kT ln exp −βE(υ(σ)( )υ∑⎡
⎣ ⎢
⎤
⎦ ⎥ 1. Approximate F(σ) by E(σ)
when doing Monte Carlo and free energy integration, only get configurational entropy
2. Approximate F(σ) by E(σ) -TSelectronic (σ)
when doing Monte Carlo and free energy integration, get configurational entropy and electronic
3. F(σ) = E(σ) –TSelectronic (σ) – TSvib (σ)
when doing Monte Carlo and free energy integration, get configurational entropy + electronic + vibrational

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Summary
The model on the time scale of the substitutional excitations is an Ising-like model (i.e. excitations are changes of occupation variables)
The Hamiltonian of the Ising-like model is the free energy of the faster excitations (e.g. vibrations, electronic excitations).
Only approximation is separation of time scales
Cluster Expansion is a practical form for the Ising-like Hamiltonian
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
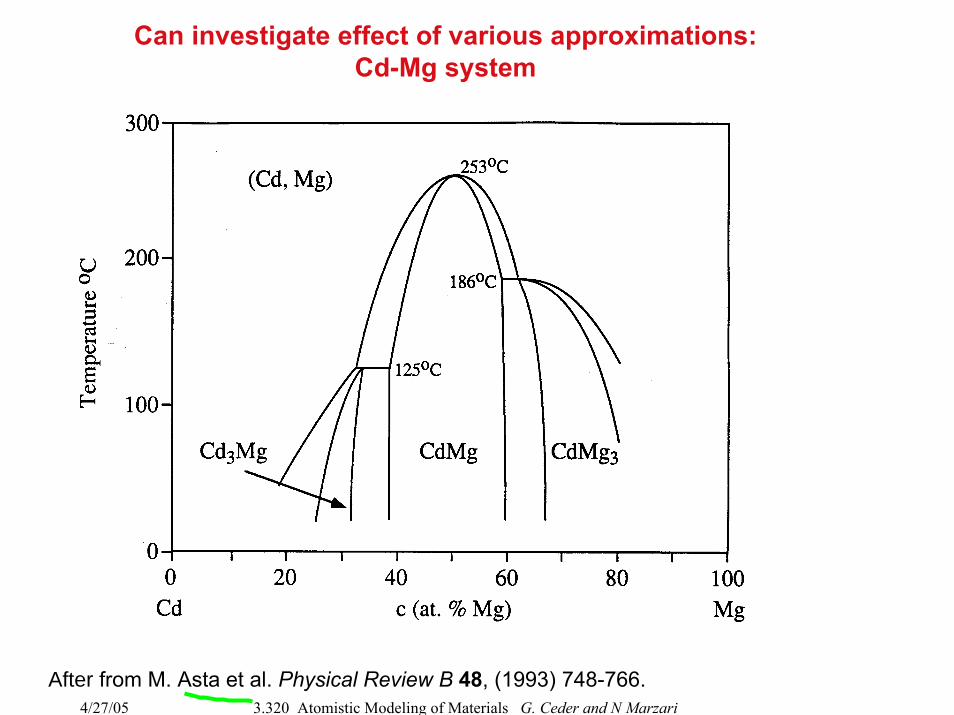
Can investigate effect of various approximations:Cd-Mg system
After from M. Asta et al. Physical Review B 48, (1993) 748-766.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
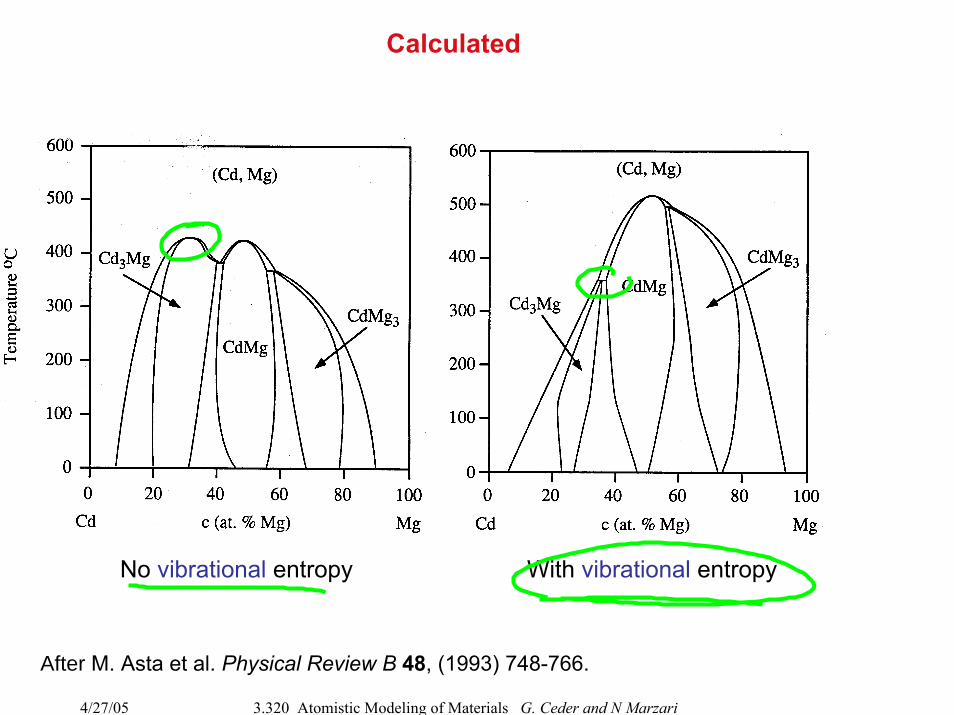
Calculated
No vibrational entropy With vibrational entropy
After M. Asta et al. Physical Review B 48, (1993) 748-766.

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
Systems as 1994
Table removed for copyright reasons.
4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
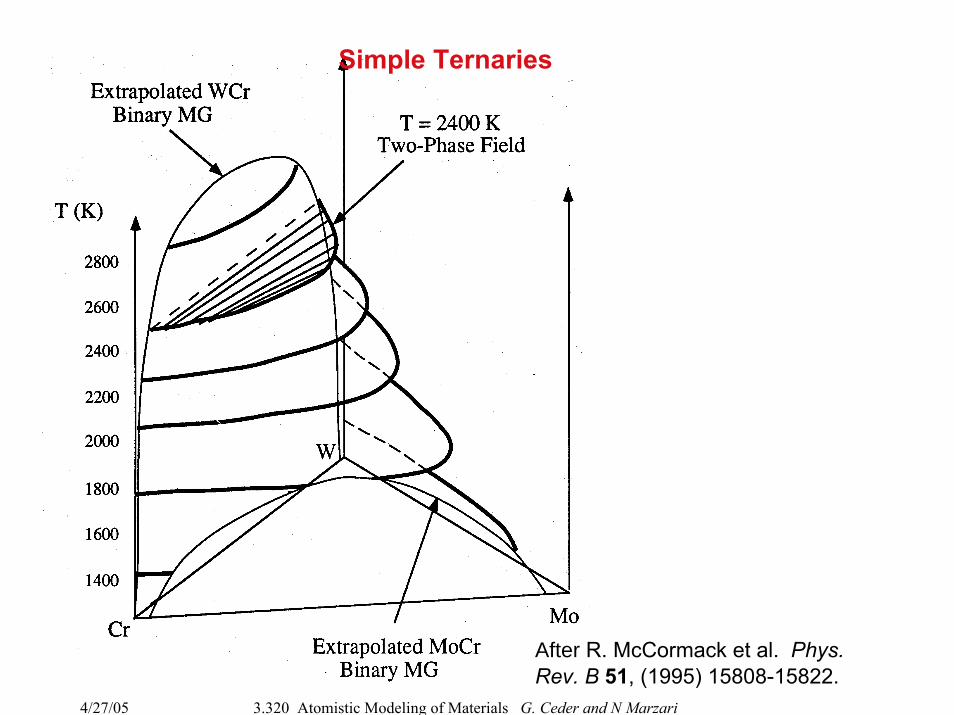
After R. McCormack et al. Phys. Rev. B 51, (1995) 15808-15822.
Simple Ternaries

4/27/05 3.320 Atomistic Modeling of Materials G. Ceder and N Marzari
References
1. D. de Fontaine, in Solid State Physics H. Ehrenreich, D. Turnbull, Eds. (Academic Press, 1994), vol. 47, pp. 33-176.2. G. Ceder, A. Van der Ven, C. Marianetti, D. Morgan, Modeling and Simulation in Materials Science and Engineering 8, (2000) 311-321.3. A. Zunger, in Statics and Dynamics of Alloy Phase Transformations P. E. A. Turchi, A. Goniss, Eds., pp. 361-419 (1994).4. A. Van de Walle, G. Ceder, J. of Phase Equilibria 23, (2002) 348-359.5. J. M. Sanchez, D. de Fontaine, Phys. Rev. B 25, (1982) 1759-1765.6. J. M. Sanchez, F. Ducastelle, D. Gratias, Physica 128A, (1984) 334-350.





























