Embed Size (px)
DESCRIPTION
NTU Lecnotes
Citation preview

Cryptic splice sites and split genesYuri Kapustin1,*, Elcie Chan2, Rupa Sarkar2, Frederick Wong2, Igor Vorechovsky3,
Robert M. Winston2, Tatiana Tatusova1 and Nick J. Dibb2,*
1National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health,Bethesda, MD 20814, USA, 2Institute of Reproductive and Developmental Biology, Imperial College London,Du Cane Road, London W12 ONN and 3Division of Human Genetics, University of Southampton MedicalSchool, Southampton SO16 6YD, UK
Received October 6, 2010; Revised March 17, 2011; Accepted March 22, 2011
ABSTRACT
We describe a new program called cryptic splicefinder (CSF) that can reliably identify cryptic splicesites (css), so providing a useful tool to help inves-tigate splicing mutations in genetic disease. Wereport that many css are not entirely dormant andare often already active at low levels in normalgenes prior to their enhancement in genetic disease.We also report a fascinating correlation between thepositions of css and introns, whereby css within theexons of one species frequently match the exactposition of introns in equivalent genes from anotherspecies. These results strongly indicate that manyintrons were inserted into css during evolution andthey also imply that the splicing information that liesoutside some introns can be independently recogn-ized by the splicing machinery and was in place priorto intron insertion. This indicates that non-intronicsplicing information had a key role in shaping thesplit structure of eukaryote genes.
INTRODUCTION
Eukaryotic genomes contain large numbers of splice sites,known as cryptic splice sites (css), which are generally heldto be disadvantageous sites that are dormant or used onlyat low levels unless activated by mutation of nearby au-thentic or advantageous splice sites (1,2). Once activated,css may be used extremely efficiently, resulting in a widerange of genetic disease (3–5). It is generally accepted thatcss are suppressed by nearby stronger splice sites and thatsplice site selection can be viewed as a competitionbetween the various potential splice sites in a pre-mRNAfor the splicing machinery (1,2).
For genes with many introns it is suspected that up to50% of mutations that cause disease do so by affectingsplicing, either through the activation of css, exon
skipping or disruption of alternative splicing (4–7). Cssare found in exons as well as introns and their recognitionby the splicing machinery is similar to splice site recogni-tion in general and is dependent upon information both atthe splice site and outside this region at enhancer andsilencer sequences (8–10).It is important to be able to predict the positions of css
that might be activated in genetic disease and a number ofDNA-sequence scanning programs have been developedfor this purpose. Such programs are often highly inform-ative but are handicapped by the complex nature of thenucleotide information that is required to define a splicesite (4,8,11,12).Our previous work indicates a connection between css
and introns. We identified a small number of css in theexon regions of actin genes by experiment and discoveredthat eight out of nine of these exonic css sites exactlymatch the positions of introns in actin genes from otherspecies, which led us to conclude that these particularactin introns were inserted into css during evolution(13,14). This finding may help to explain why and howeukaryotes acquired introns; however, it is important toestablish if our results for the actin gene family are gener-ally applicable.We have been unable to identify a DNA-scanning
program that reveals the same strong correlation betweenpredicted actin css and intron positions that we observedthrough experiments (13). However, this is probablybecause DNA-scanning programs were not designed spe-cifically for this purpose and because of the difficultiessuch programs face in distinguishing between css andfalse-positive non-functional splice sites (12). Here, we de-scribe a program called cryptic splice finder (CSF) that canreliably identify css by EST-to-genomic alignment. It doesthis by identifying transcripts that have been generatedthrough the low level use of css by normal genes. Unlikethe scanning programs, CSF cannot predict the positionof splice sites that are created de novo by gene mutation.However, this program provides a useful complementary
*To whom correspondence should be addressed. Tel: +212 813 8774; Fax: +212 826 8280; Email: [email protected] may also be addressed to Nick J. Dibb. Tel: +020 7594 2103; Fax: +020 759 42154; Email: [email protected]
Nucleic Acids Research, 2011, 1–8doi:10.1093/nar/gkr203
� The Author(s) 2011. Published by Oxford University Press.This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.5), which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Nucleic Acids Research Advance Access published April 5, 2011 by guest on January 13, 2013
http://nar.oxfordjournals.org/D
ownloaded from

resource for studies of genetic disease and it also enabledus to establish that there is a strong and general correl-ation between the positioning of css and of introns. Theevolutionary implications of this finding are discussed.
MATERIALS AND METHODS
CSF
CSF (http://www.ncbi.nlm.nih.gov/IEB/Research/csf)predicts css based on spliced alignment of ESTs. EachEST is aligned against the genome independently fromother ESTs. In order to be considered by CSF, a gap inthe alignment must be flanked by a minimum number (25by default) of matching residues. CSF searches for ESTalignments that form the patterns illustrated in Figure 1A.It can be seen that the majority of ESTs must share acommon gap or deletion and in addition must includeminor transcripts that share only one of the commondeletion endpoints. CSF defines the common deletionendpoints as authentic splice sites and the deletion endpoint of the minor transcript(s) as cryptic or alternativesplice sites (arrowed). For constitutively spliced genes,these minor deletion endpoints occur very infrequentlyand are therefore candidate css. For alternatively splicedgenes, CSF identifies both css and more frequentlyoccurring alternative splice sites (see ‘Results’ section).As illustrated in Figure 1A, css can be 50 or 30 andupstream or downstream of the alternative authenticsplice site. In the majority of cases the authentic splice
sites defined by CSF are the same as the splice sitesdefined by reference sequences (i.e. NM_000518.4) andso represent commonly used splice sites (see below forexceptions).
In more detail, coordinates of the splice sites fromadjacent exon–intron–exon sequences are pooled intofour-tuples which are then loaded into a relationaldatabase alongside the data linking them to their align-ments. The database runs a query to detect tuple pairssatisfying the css condition: the overlapping intronsmatch at one end and mismatch at the other, with a mis-matching intron end from one tuple residing within theexon from the other tuple. For each tuple pair returnedby the query, the splice sites of the intron that has moresupporting ESTs are declared authentic and the remainingsite is declared cryptic. Splice site coordinates are mappedagainst the NCBI36/hg18 human genome assembly.
As illustrated (Figure 1A), a css detected by CSF is fur-ther classified as to whether it is a 50 or 30 css and whetherit is located in an exon or intron. The number of tran-scripts supporting a splice (either authentic or css), isprinted and the complete list of such ESTs is linkedunder the count. Where applicable, the count is followedby a number in parentheses. For exonic css, parenthesesalways appear on the left-hand (authentic) side of the CSFreport and on the right-hand side (cryptic) for intronic css.The numbers in parentheses show the transcripts thatformally satisfy the css conditions (see Notes about CSFsection).
Notes about CSF. CSF only lists authentic 50- and30-splice sites when one of the two authentic splice sitesis a more common alternative for another nearby splicesite that is listed under the cryptic (alternative) column.This means that CSF only lists a subset of the totalnumber of authentic splice sites for any one gene.
CSF provides a very good way of screening largenumbers of transcripts for the minority that are likely tohave been generated by use of a css or nearby alternativesplice site. However, candidate transcripts do need to bechecked, by for example, using the Splign alignment option(15). CSF classifies a minority of less well-supported dele-tions as authentic splices because these happen to meetthe CSF conditions. These sites can be easily recognizedfrom the CSF output because they have lower levels ofsupport than other authentic splice sites and do notmatch reference sequence splice sites. Caution needs tobe taken with the interpretation of these particular‘authentic’ splice sites and also with the interpretationof css that are paired with authentic splice sites thathave a low-parenthesis score. See Supplementary Datafor illustrative examples.
Searching CSF. Two methods of searching for css areprovided (Supplementary Figure S1). In the first method,a landmark EST accession is submitted and CSF returns alist of css within the genomic range of the entered EST.In the second method, an arbitrary genomic interval canbe specified and CSF will return a hierarchic list of css forthat interval. Entire chromosomes can also be enteredsuch as NC_000001 (human chromosome 1) for which
B
(i)
(ii)
’3’5
3’ss 5’ss 3’ss5’ss
5’css
3’ss
A
5’ss
5’ss
3’ss
3’ss
(i)
5’ss
5’ss
3’ss
3’ss
(iv)
(ii)
(iii)
(5204752)
(5204605)(5204736)
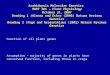
Figure 1. (A) CSF searches for transcript alignments that form one offour patterns (i–iv). All of these patterns contain a group of majortranscripts that share a common deletion and a minor transcript thatshares only one of the deletion endpoints. CSF defines the commondeletion endpoints as authentic splice sites and the less commondeletion endpoint of the minor transcript(s) as cryptic or alternativesplice sites (arrowed). (B) Schematic of the HBB gene for humanb-globin, which contains two introns that are constitutively splicedfrom pre-mRNA. As illustrated the vast majority of ESTs align asshown and define the three exons of this gene. The circle shows apattern of ESTs that CSF is designed to recognize and that isreported in Figure 2A. The numbers in brackets show the genome co-ordinates of the three splice sites that are identified and listed by CSF(Figure 2A).
2 Nucleic Acids Research, 2011
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from

CSF currently lists 3232 css. Links are provided in orderto view the sequence alignments of individual css. CSF canpredict css for Homo sapiens, Bos taurus, Mus musculus,Danio rerio and Arabidopsis thaliana and we intend toexpand this range as further transcript data becomesavailable
Statistical analysis. There are 91 known different intronpositions within the coding region of the actin gene family,these have been identified by sequencing actin genesfrom over 160 different species (16–18). The positions ofall of these introns together with the 14 css that we haveidentified are plotted in Supplementary Figure S2A. Actingenes usually have 375 codons and therefore three timesthis number of possible positions for introns or css and sothe probability of a single css exactly matching an intronposition by chance is 91/(3� 375). The exact Fisher’s testgives the P-value of 1.6� 10�10 for 11 or more matchesout of 14 occurring by chance. Similarly, we identified 135css in the coding region of 51 different genes of the ribo-somal protein gene database. The 51 genes are 190 codonsin size on average and have a total of 957 introns (from22 species). The probability of a css exactly matching anintron by chance is therefore 957/(190� 3� 51)=0.033.The binomial probability of 33 or more matches out of135 occurring by chance is P=2.2� 10�15.
RT–PCR. Total RNA was extracted using Trizol and thecDNA first strand was synthesized from 0.1 mg of RNAusing superscript III (Invitrogen) and random hexamers.PCR products were generated with Taq polymerase (NBI)for 25 or 35 cycles and separated on 5% native polyacryl-amide gels. PCR bands were excised and cloned intopGEM-T Easy vectors (Promega) for sequencing bycolony PCR followed by ABI Prism Big Dye Terminatorcycle sequencing (Applied Biosystems).
RESULTS
Principle of css detection by CSF
Css are often used highly efficiently in genetic disease fol-lowing the mutation of nearby more competitive splicesites. We, therefore, reasoned that css might be used at alow but detectable frequency by normal genes. Figure 1Ashows the patterns of EST alignments that CSF is designedto identify. It can be seen that CSF identifies groups ofESTs or transcripts that share a common deletion, whenaligned to the genome, together with a minor transcript(s)that shares just one of the common deletion endpoints.CSF defines the common deletion endpoints as authenticsplice sites and the unusual deletion endpoint of the minortranscript as a cryptic or alternative splice site (see‘Materials and Methods’ section for further details).Figure 1B illustrates how ESTs align to the human HBBgene for b-globin, which has two introns. The circleidentifies a pattern of alignments that is recognized byCSF because it includes a minor transcript that has anunusual deletion endpoint (position 5 204 752) that is apredicted css (Figures 1B and 2A). The reason why suchcss predictions turn out to be accurate (see below) is
because of the restriction that the predicted css is pairedwith a commonly used splice site. This distinguishesmRNA deletions that are generated by low-level aberrantsplicing from deletions that are generated by non-splicingmechanisms such as errors during transcription or duringthe generation of the EST.Figure 2A shows the CSF output for HBB, which is one
of the first genes in which css were identified (19). CSFidentifies a single EST called BU198526 as having beengenerated by aberrant splicing. BU198526 aligns to HBBas illustrated in Figure 1B(ii), this alignment can be viewedin detail by a link to Splign (Figure 2). As explainedabove, the reason why BU198526 was identified by CSFis because it forms a pattern alignment with other ESTs asillustrated in Figure 1A(ii). The CSF output (Figure 2A)shows that BU198526 shares a deletion endpoint atposition 52 044 605 that is in common with 703 otherESTs (link provided) but differs in having an unusual 50
deletion endpoint at position 5 204 752, which is the pre-dicted css. A comparison with the known css of HBBconfirms that 5 204 752 is indeed a css (SupplementaryTable S1). We show below that css predictions by CSFare very reliable even if supported by just a single EST,as in this case. However, HBB has 11 known css(Supplementary Table S1), which illustrates that CSF islimited in its predictions, most probably by the amount ofavailable transcript data.For alternatively spliced genes, CSF identifies both css
and also nearby alternative splice sites that act to shortenor lengthen exons. This is because the two types of splicesites are rather similar. For example, the css identified inHBB (Figure 2A) might be considered an alternative splicesite if used at a greater frequency than 1 in 703.WT1 is an example of gene that encodes at least three
alternatively spliced mRNAs and for which there are only80 ESTs. CSF identifies two css (Figure 2B), however, theCSF output also shows that the predicted css at32 370 103, for example, is supported by five ESTsincluding the reference sequence NM_000378.3 and thatthe ‘authentic’ splice site nine bases away at 32 370 094 issupported by only eight transcripts. The similar usage ofthe authentic and css shows that these are really alterna-tive 50 ss. However, it is useful to have this type of infor-mation because alternative splice sites are also implicatedin genetic disease and in this particular case disruption ofthe splice site at position 32 370 094 gives rise to Frasiersyndrome, possibly due to the increased use of splice site32 370 103 (20,21).To test CSF we analyzed a database called DBASS
(database of aberrant splice sites), which lists 340 humangenes that have one or more css that are activated ingenetic disease (3). There are 814 different css listed inDBASS and CSF predicts 609 css from the same set of340 genes. Fifty-eight percent of these predictions are sup-ported by only single ESTs (Supplementary Table S1).Before comparing the css identified by CSF with thoseof DBASS, we first asked whether the css predictionsthat are supported by just single ESTs were likely tohave been generated by aberrant splicing. We reasonedthat because CSF identifies deletion endpoints irrespectiveof their sequence, then if these rare deletion endpoints
Nucleic Acids Research, 2011 3
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from
were generated by splicing they should look like splicesites. The predictions were divided into 50 and 30 css andTable 1 shows that the predicted css have a very goodmatch to the expected 50- or 30-splice site consensus se-quence, which is a strong proof that the vast majority ofeven the rarest deletions identified by CSF were generatedby aberrant splicing.Of the two sets of DBASS and CSF css, only 46 are in
common (Supplementary Tables S1 and S2). However,there are only 61 cases where the DBASS and CSF cssare located within the same exon or intron(Supplementary Table S1, column 5), giving a matchrate of 46/61 or 75%. Thirteen of the 15 DBASS cssthat did not match were either created de novo bymutation and would, therefore, not be expected to beidentified by CSF or were of opposite type, for example,a 50 css in DBASS and a 30 css in CSF and would, there-fore, be unlikely to match (Supplementary Table S1). Onlytwo out of 48 css predictions that could be meaningfullycompared with DBASS did not exactly match(Supplementary Table S1). This result together with theclear consensus sequence results shown in Table 1 indi-cates that CSF predictions are highly reliable and canonly generate a very low level of false positives evenwhen supported by single ESTs. The small number of 46css in common between the CSF database and DBASS(Supplementary Table S1) presumably indicates thatboth DBASS and CSF have identified relatively few ofall possible css within this set of 340 genes.We confirmed that css predicted by CSF could also be
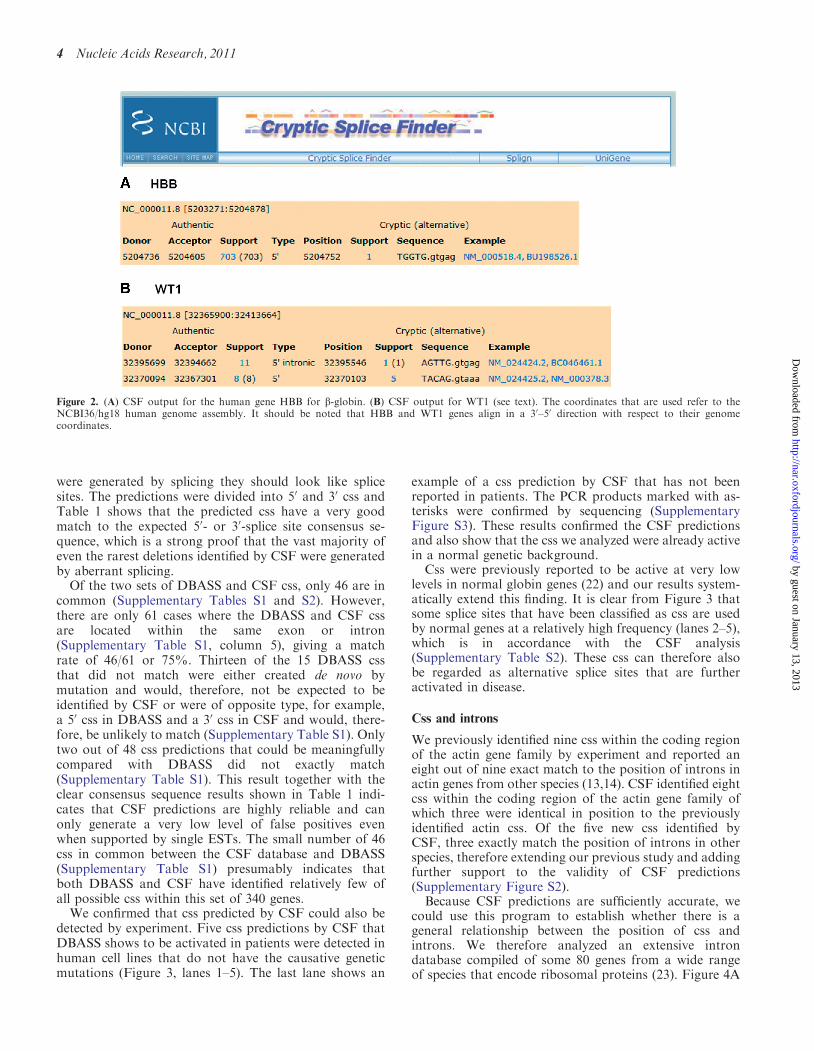
detected by experiment. Five css predictions by CSF thatDBASS shows to be activated in patients were detected inhuman cell lines that do not have the causative geneticmutations (Figure 3, lanes 1–5). The last lane shows an
example of a css prediction by CSF that has not beenreported in patients. The PCR products marked with as-terisks were confirmed by sequencing (SupplementaryFigure S3). These results confirmed the CSF predictionsand also show that the css we analyzed were already activein a normal genetic background.
Css were previously reported to be active at very lowlevels in normal globin genes (22) and our results system-atically extend this finding. It is clear from Figure 3 thatsome splice sites that have been classified as css are usedby normal genes at a relatively high frequency (lanes 2–5),which is in accordance with the CSF analysis(Supplementary Table S2). These css can therefore alsobe regarded as alternative splice sites that are furtheractivated in disease.
Css and introns
We previously identified nine css within the coding regionof the actin gene family by experiment and reported aneight out of nine exact match to the position of introns inactin genes from other species (13,14). CSF identified eightcss within the coding region of the actin gene family ofwhich three were identical in position to the previouslyidentified actin css. Of the five new css identified byCSF, three exactly match the position of introns in otherspecies, therefore extending our previous study and addingfurther support to the validity of CSF predictions(Supplementary Figure S2).
Because CSF predictions are sufficiently accurate, wecould use this program to establish whether there is ageneral relationship between the position of css andintrons. We therefore analyzed an extensive introndatabase compiled of some 80 genes from a wide rangeof species that encode ribosomal proteins (23). Figure 4A
Figure 2. (A) CSF output for the human gene HBB for b-globin. (B) CSF output for WT1 (see text). The coordinates that are used refer to theNCBI36/hg18 human genome assembly. It should be noted that HBB and WT1 genes align in a 30–50 direction with respect to their genomecoordinates.
4 Nucleic Acids Research, 2011
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from
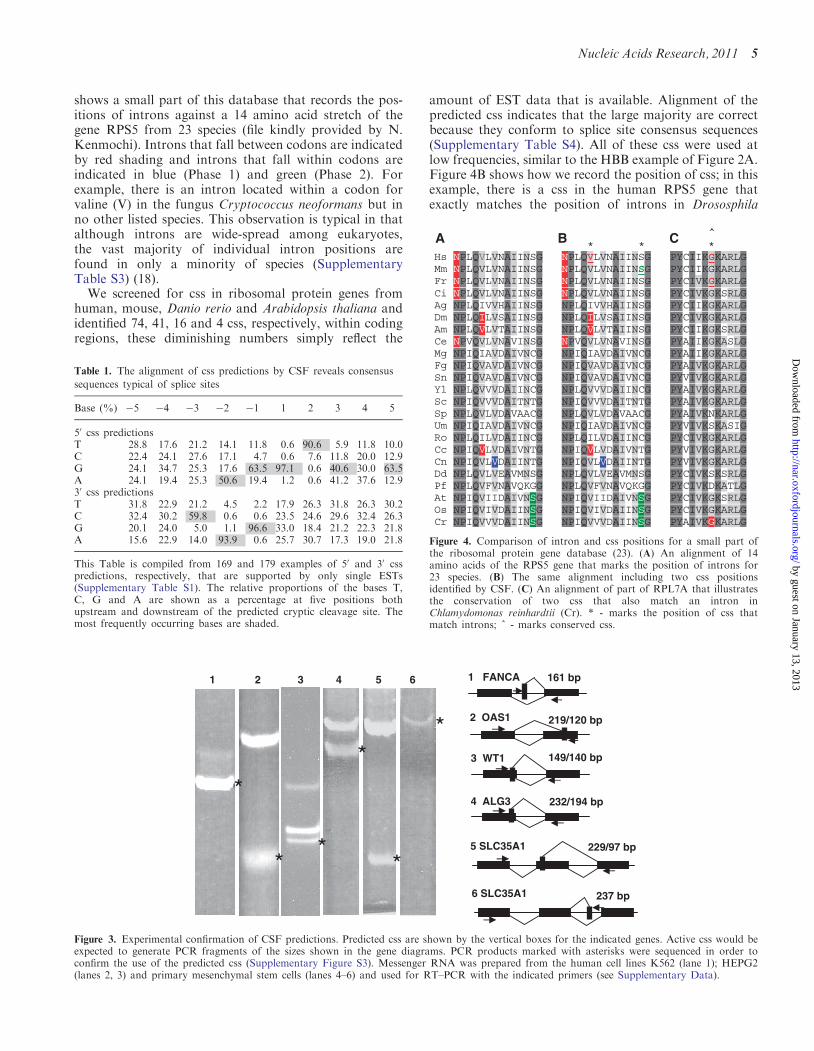
shows a small part of this database that records the pos-itions of introns against a 14 amino acid stretch of thegene RPS5 from 23 species (file kindly provided by N.Kenmochi). Introns that fall between codons are indicatedby red shading and introns that fall within codons areindicated in blue (Phase 1) and green (Phase 2). Forexample, there is an intron located within a codon forvaline (V) in the fungus Cryptococcus neoformans but inno other listed species. This observation is typical in thatalthough introns are wide-spread among eukaryotes,the vast majority of individual intron positions arefound in only a minority of species (SupplementaryTable S3) (18).
We screened for css in ribosomal protein genes fromhuman, mouse, Danio rerio and Arabidopsis thaliana andidentified 74, 41, 16 and 4 css, respectively, within codingregions, these diminishing numbers simply reflect the
amount of EST data that is available. Alignment of thepredicted css indicates that the large majority are correctbecause they conform to splice site consensus sequences(Supplementary Table S4). All of these css were used atlow frequencies, similar to the HBB example of Figure 2A.Figure 4B shows how we record the position of css; in thisexample, there is a css in the human RPS5 gene thatexactly matches the position of introns in Drososphila
5 SLC35A1 229/97 bp
4 ALG3 232/194 bp
3 WT1 149/140 bp
2 OAS1 219/120 bp
161 bp1 FANCA321 4 5 6
6 SLC35A1 237 bp
Figure 3. Experimental confirmation of CSF predictions. Predicted css are shown by the vertical boxes for the indicated genes. Active css would beexpected to generate PCR fragments of the sizes shown in the gene diagrams. PCR products marked with asterisks were sequenced in order toconfirm the use of the predicted css (Supplementary Figure S3). Messenger RNA was prepared from the human cell lines K562 (lane 1); HEPG2(lanes 2, 3) and primary mesenchymal stem cells (lanes 4–6) and used for RT–PCR with the indicated primers (see Supplementary Data).
^ * * *
Hs NPLQVLVNAIINSG NPLQVLVNAIINSG PYCIIKGKARLGMm NPLQVLVNAIINSG NPLQVLVNAIINSG PYCIIKGKARLG Fr NPLQVLVNAIINSG NPLQVLVNAIINSG PYCIVKGKARLGCi NPLQVLVNAIINSG NPLQVLVNAIINSG PYCIVKGKSRLGAg NPLQIVVHAIINSG NPLQIVVHAIINSG PYCIIKGKARLG Dm NPLQILVSAIINSG NPLQILVSAIINSG PYCIVKGKARLG Am NPLQVLVTAIINSG NPLQVLVTAIINSG PYCIIKGKSRLG Ce NPVQVLVNAVINSG NPVQVLVNAVINSG PYAIIKGKASLG Mg NPIQIAVDAIVNCG NPIQIAVDAIVNCG PYAIIKGKARLG Fg NPIQVAVDAIVNCG NPIQVAVDAIVNCG PYAIVKGKARLG Sn NPIQVAVDAIVNCG NPIQVAVDAIVNCG PYVIVKGKARLG Yl NPLQVVVDAIINCG NPLQVVVDAIINCG PYAIVKGKARLG Sc NPIQVVVDAITNTG NPIQVVVDAITNTG PYAIVKGKARLG Sp NPLQVLVDAVAACG NPLQVLVDAVAACG PYAIVKNKARLG Um NPIQIAVDAIVNCG NPIQIAVDAIVNCG PYVIVKSKASIG Ro NPLQILVDAIINCG NPLQILVDAIINCG PYCIVKGKARLG Cc NPIQVLVDAIVNTG NPIQVLVDAIVNTG PYVIVKGKARLG Cn NPIQVLVDAIINTG NPIQVLVDAIINTG PYVIVKGKARLG Dd NPLQVLVEAVMNSG NPLQVLVEAVMNSG PYCIVKSKSRLG Pf NPLQVFVNAVQKGG NPLQVFVNAVQKGG PYCIVKDKATLG At NPIQVIIDAIVNSG NPIQVIIDAIVNSG PYCIVKGKSRLG Os NPIQVIVDAIINSG NPIQVIVDAIINSG PYCIVKGKARLG Cr NPIQVVVDAIINSG NPIQVVVDAIINSG PYAIVKGKARLG
A B C
Figure 4. Comparison of intron and css positions for a small part ofthe ribosomal protein gene database (23). (A) An alignment of 14amino acids of the RPS5 gene that marks the position of introns for23 species. (B) The same alignment including two css positionsidentified by CSF. (C) An alignment of part of RPL7A that illustratesthe conservation of two css that also match an intron inChlamydomonas reinhardtii (Cr). * - marks the position of css thatmatch introns; ^ - marks conserved css.
Table 1. The alignment of css predictions by CSF reveals consensus
sequences typical of splice sites
Base (%) �5 �4 �3 �2 �1 1 2 3 4 5
50 css predictionsT 28.8 17.6 21.2 14.1 11.8 0.6 90.6 5.9 11.8 10.0C 22.4 24.1 27.6 17.1 4.7 0.6 7.6 11.8 20.0 12.9G 24.1 34.7 25.3 17.6 63.5 97.1 0.6 40.6 30.0 63.5A 24.1 19.4 25.3 50.6 19.4 1.2 0.6 41.2 37.6 12.930 css predictionsT 31.8 22.9 21.2 4.5 2.2 17.9 26.3 31.8 26.3 30.2C 32.4 30.2 59.8 0.6 0.6 23.5 24.6 29.6 32.4 26.3G 20.1 24.0 5.0 1.1 96.6 33.0 18.4 21.2 22.3 21.8A 15.6 22.9 14.0 93.9 0.6 25.7 30.7 17.3 19.0 21.8
This Table is compiled from 169 and 179 examples of 50 and 30 csspredictions, respectively, that are supported by only single ESTs(Supplementary Table S1). The relative proportions of the bases T,C, G and A are shown as a percentage at five positions bothupstream and downstream of the predicted cryptic cleavage site. Themost frequently occurring bases are shaded.
Nucleic Acids Research, 2011 5
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from

(Dm) the honeybee (Am) and a fungus (Cc) and a nearbycss in the mouse (Mm) that exactly matches the position ofintrons in three plant species (At, Os, Cr). We haveidentified 135 css to date and of these at least five areconserved between species (Figure 4C andSupplementary Figure S4). Thirty-three out of 135 css ofthe ribosomal gene family exactly match the position ofintrons in other species (Supplementary Figure S4). Thisproportion is smaller than the 11/14 match observed forthe actin gene family (Supplementary Figure S2), but isstill highly significant (P=2.2� 10�15, see ‘Materials andMethods’ section).
DISCUSSION
The CSF program is designed to identify transcripts thatare generated through the low level use of css by normalgenes. In addition, CSF also identifies a subset of alterna-tive splice sites that are similar to css, but are used at agreater frequency. Both types of splice sites are reported tobe activated in genetic disease as a result of mutations thatdisrupt the function of nearby competitive splice sites(4,5). Our analyses show that css predictions by CSF arevery reliable and so we would expect, for example, thatmany more of the predicted css and alternative splice sitesof Supplementary Table S1 will be discovered to beassociated with genetic disease. The identification of cssby CSF is limited by the amount of available transcriptdata but this will improve as further transcript sequencesbecome available, particularly with the advent of mRNAdeep sequencing (24).CSF was designed primarily to identify css in highly
conserved gene families such as actin in order toadvance our understanding of intron origin. We foundthat about 25% of the css within the coding sequence ofthe large family of genes that encode ribosomal proteinsexactly match the position of introns that are present inequivalent genes from other species (Figure 4 andSupplementary Figure S4). This compares to a 78%match between actin css and introns, however, there isfar more phylogenetic data available for the actin genefamily and so relatively more introns have been dis-covered. Consequently, the css that are recorded inSupplementary Figure S4 predict the positions of as yetundiscovered introns in the ribosomal gene family.The well-known and valuable early and late models of
intron origin both assume that the splicing machineryevolved for the purpose of removing introns that wereeither present in the most ancestral genomes or wereinserted after the separation of the prokaryotes (25–27).At the time the models were proposed, non-intronicsplicing information was not generally thought to be ofmajor relevance and so is not an important feature ofeither model (28).However, it is now established that exon junction se-
quences are older and better conserved than most if notall introns and were sites for intron insertions during evo-lution (18,29–33). Indeed, a number of intron propertiessuch as their phasing with respect to the coding sequence(29–30,34) and their location with respect to protein
structure have now been largely attributed to theflanking exon junction sequence (35,36).
Consequently, our finding that css often match theposition of exon junction sequences in gene homologues,strongly indicates that css were targeted by intron inser-tions during evolution. Our data also indicates that theinformation that lies outside some introns is not onlyconserved with homologs that lack such introns but isalso capable of being independently recognized by thesplicing machinery and can define the position of the‘missing’ introns (13). This is a striking observationbecause although the splicing information that flanksintrons contributes to their recognition, there is no mech-anistic reason for this information to be autonomousrather than auxiliary in nature for the purpose of intronremoval (37). It is unlikely that a css is recognized fromthe immediate splice-site information alone (8–10), sug-gesting that other information such as splicing enhancersequences are also conserved between some gene homo-logues independently of intron presence.
All of the available evidence indicates that the splicinginformation that flanks introns was largely in place priorto their insertion (18,29–33). The key question we havestarted to address here is whether such informationmight have been functional. Our data so far does notprove but it certainly supports the suggestion that infor-mation of this nature could have been used by the splicingmachinery for splicing purposes prior to the arrival ofintrons, which is a rather different concept to the earlyand late models of intron origin (28).
The splicing information that flanks introns is perhapssimilar to the splicing information that enables the mRNAof intronless genes to interact with components of thesplicing machinery for the purpose of mRNA biogenesis,nonsense mediated decay or alternative splicing (38–43).For genes that have introns there are still many reports ofintron-independent splicing between exonic splice sites(44–53). If intron-independent splicing is an ancestralmechanism then it may be far more prevalent acrossspecies than is currently realized.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are grateful to Naoya Kenmochi for his help andwould also like to thank Anil Chandrashekran, AndyNewman, Peter Rogan, Alison Russell, Malcolm ParkerTerrie Sadusky and Arlin Stoltzfus.
FUNDING
Reserch grants from Biotechnology and BiosciencesResearch Council and Atazoa. Funding for open accesscharge: Genesis Trust.
Conflict of interest statement. None declared.
6 Nucleic Acids Research, 2011
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from

REFERENCES
1. Padgett,R.A., Grabowski,P.J., Konarska,M.M., Seiler,S. andSharp,P.A. (1986) Splicing of messenger RNA precursors.Annu. Rev. Biochem., 55, 1119–1150.
2. Green,M.R. (1986) Pre-mRNA splicing. Annu. Rev. Genet., 20,671–708.
3. Buratti,E., Chivers,M., Hwang,G. and Vorechovsky,I. (2011)DBASS3 and DBASS5: databases of aberrant 30- and 50-splicesites. Nucleic Acids Res., 39, D86–D91.
4. Buratti,E., Chivers,M., Kralovicova,J., Romano,M., Baralle,M.,Krainer,A.R. and Vorechovsky,I. (2007) Aberrant 50 splice sites inhuman disease genes: mutation pattern, nucleotide structure andcomparison of computational tools that predict their utilization.Nucleic Acids Res., 35, 4250–4263.
5. Wang,G.S. and Cooper,T.A. (2007) Splicing in disease: disruptionof the splicing code and the decoding machinery.Nat. Rev. Genet., 8, 749–761.
6. Hastings,M.L., Resta,N., Traum,D., Stella,A., Guanti,G. andKrainer,A.R. (2005) An LKB1 AT-AC intron mutation causesPeutz-Jeghers syndrome via splicing at noncanonical cryptic splicesites. Nat. Struct. Mol. Biol., 12, 54–59.
7. Lopez-Bigas,N., Audit,B., Ouzounis,C., Parra,G. and Guigo,R.(2005) Are splicing mutations the most frequent cause ofhereditary disease? FEBS Lett., 579, 1900–1903.
8. Wimmer,K., Roca,X., Beiglbock,H., Callens,T., Etzler,J.,Rao,A.R., Krainer,A.R., Fonatsch,C. and Messiaen,L. (2007)Extensive in silico analysis of NF1 splicing defects uncoversdeterminants for splicing outcome upon 50 splice-site disruption.Hum. Mutat., 28, 599–612.
9. Kralovicova,J. and Vorechovsky,I. (2007) Global control ofaberrant splice-site activation by auxiliary splicing sequences:evidence for a gradient in exon and intron definition.Nucleic Acids Res., 35, 6399–6413.
10. Russo,A., Siciliano,G., Catillo,M., Giangrande,C., Amoresano,A.,Pucci,P., Pietropaolo,C. and Russo,G. (2010) hnRNP H1 andintronic G runs in the splicing control of the human rpL3 gene.Biochim. Biophys. Acta., 1799, 419–428.
11. Divina,P., Kvitkovicova,A., Buratti,E. and Vorechovsky,I. (2009)Ab initio prediction of mutation-induced cryptic splice-siteactivation and exon skipping. Eur. J. Hum. Genet., 17, 759–765.
12. Betz,B., Theiss,S., Aktas,M., Konermann,C., Goecke,T.O.,Moslein,G., Schaal,H. and Royer-Pokora,B. (2009) Comparativein silico analyses and experimental validation of novel splice siteand missense mutations in the genes MLH1 and MSH2.J. Cancer Res. Clin. Oncol., 136, 123–134.
13. Sadusky,T., Newman,A.J. and Dibb,N.J. (2004) Exon junctionsequences as cryptic splice sites: implications for intron origin.Curr. Biol., 14, 505–509.
14. Stoltzfus,A. (2004) Molecular evolution: introns fall into place.Curr. Biol., 14, R351–352.
15. Kapustin,Y., Souvorov,A., Tatusova,T. and Lipman,D. (2008)Splign: algorithms for computing spliced alignments withidentification of paralogs. Biol. Direct., 3, 20.
16. Bhattacharya,D. and Weber,K. (1997) The actin gene of theglaucocystophyte Cyanophora paradoxa: analysis of the codingregion and introns, and an actin phylogeny of eukaryotes.Curr. Genet., 31, 439–446.
17. Flakowski,J., Bolivar,I., Fahrni,J. and Pawlowski,J. (2006) Tempoand mode of spliceosomal intron evolution in actin offoraminifera. J. Mol. Evol., 63, 30–41.
18. Qiu,W.G., Schisler,N. and Stoltzfus,A. (2004) The evolutionarygain of spliceosomal introns: sequence and phase preferences.Mol. Biol. Evol., 21, 1252–1263.
19. Treisman,R., Orkin,S.H. and Maniatis,T. (1983) Specifictranscription and RNA splicing defects in five clonedbeta-thalassaemia genes. Nature, 302, 591–596.
20. Barbaux,S., Niaudet,P., Gubler,M.C., Grunfeld,J.P., Jaubert,F.,Kuttenn,F., Fekete,C.N., Souleyreau-Therville,N., Thibaud,E.,Fellous,M. et al. (1997) Donor splice-site mutations in WT1 areresponsible for Frasier syndrome. Nat. Genet., 17, 467–470.
21. Niaudet,P. and Gubler,M.C. (2006) WT1 and glomerular diseases.Pediatr. Nephrol., 21, 1653–1660.
22. Haj Khelil,A., Deguillien,M., Moriniere,M., Ben Chibani,J. andBaklouti,F. (2008) Cryptic splicing sites are differentially utilizedin vivo. FEBS J., 275, 1150–1162.
23. Yoshihama,M., Nguyen,H.D. and Kenmochi,N. (2007) Introndynamics in ribosomal protein genes. PLoS ONE, 2, e141.
24. Pickrell,J.K., Pai,A.A., Gilad,Y. and Pritchard,J.K. (2010) Noisysplicing drives mRNA isoform diversity in human cells.PLoS Genet., 6, e1001236.
25. Cavalier-Smith,T. (1985) Selfish DNA and the origin of introns.Nature, 315, 283–284.
26. Gilbert,W. (1978) Why genes in pieces? Nature, 271, 501.27. Roy,S.W. and Gilbert,W. (2006) The evolution of spliceosomal
introns: patterns, puzzles and progress. Nat. Rev. Genet., 7,211–221.
28. Dibb,N.J. (1993) Why do genes have introns? FEBS Lett., 325,135–139.
29. Nguyen,H.D., Yoshihama,M. and Kenmochi,N. (2006) Phasedistribution of spliceosomal introns: implications for intron origin.BMC Evol. Biol., 6, 69.
30. Ruvinsky,A., Eskesen,S.T., Eskesen,F.N. and Hurst,L.D. (2005)Can codon usage bias explain intron phase distributions and exonsymmetry? J. Mol. Evol., 60, 99–104.
31. Bhattacharya,D., Simon,D., Huang,J., Cannone,J.J. andGutell,R.R. (2003) The exon context and distribution ofEuascomycetes rRNA spliceosomal introns. BMC Evol. Biol., 3,7.
32. Dibb,N.J. and Newman,A.J. (1989) Evidence that introns arose atproto-splice sites. EMBO J., 8, 2015–2021.
33. Sverdlov,A.V., Rogozin,I.B., Babenko,V.N. and Koonin,E.V.(2004) Reconstruction of ancestral protosplice sites. Curr. Biol.,14, 1505–1508.
34. Long,M., de Souza,S.J., Rosenberg,C. and Gilbert,W. (1998)Relationship between ‘‘proto-splice sites’’ and intron phases:evidence from dicodon analysis. Proc. Natl Acad. Sci. USA, 95,219–223.
35. De Kee,D.W., Gopalan,V. and Stoltzfus,A. (2007) Asequence-based model accounts largely for the relationship ofintron positions to protein structural features. Mol. Biol. Evol.,24, 2158–2168.
36. Whamond,G.S. and Thornton,J.M. (2006) An analysis of intronpositions in relation to nucleotides, amino acids, and proteinsecondary structure. J. Mol. Biol., 359, 238–247.
37. Burge,C.B., Tuschl,T. and Sharp,P.A. (eds), (1999) Splicing ofPrecursors to mRNAs by the Spliceosomes, 2nd edn. Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
38. Brogna,S. and Wen,J. (2009) Nonsense-mediated mRNA decay(NMD) mechanisms. Nat. Struct. Mol. Biol., 16, 107–113.
39. Guang,S., Felthauser,A.M. and Mertz,J.E. (2005) Binding ofhnRNP L to the pre-mRNA processing enhancer of the herpessimplex virus thymidine kinase gene enhances bothpolyadenylation and nucleocytoplasmic export of intronlessmRNAs. Mol. Cell. Biol., 25, 6303–6313.
40. Guang,S. and Mertz,J.E. (2005) Pre-mRNA processing enhancer(PPE) elements from intronless genes play additional roles inmRNA biogenesis than do ones from intron-containing genes.Nucleic Acids Res., 33, 2215–2226.
41. Juneau,K., Nislow,C. and Davis,R.W. (2009) Alternative splicingof PTC7 in Saccharomyces cerevisiae determines proteinlocalization. Genetics, 183, 185–194.
42. Pozzoli,U., Riva,L., Menozzi,G., Cagliani,R., Comi,G.P.,Bresolin,N., Giorda,R. and Sironi,M. (2004) Over-representationof exonic splicing enhancers in human intronless genes suggestsmultiple functions in mRNA processing. Biochem. Biophys. Res.Commun., 322, 470–476.
43. Brody,Y., Neufeld,N., Bieberstein,N., Causse,S.Z., Bohnlein,E.M.,Neugebauer,K.M., Darzacq,X. and Shav-Tal,Y. (2011) TheIn Vivo Kinetics of RNA Polymerase II Elongation duringCo-Transcriptional Splicing. PLoS Biol., 9, e1000573.
44. Ng,B., Yang,F., Huston,D.P., Yan,Y., Yang,Y., Xiong,Z.,Peterson,L.E., Wang,H. and Yang,X.F. (2004) Increasednoncanonical splicing of autoantigen transcripts providesthe structural basis for expression of untolerized epitopes.J. Allergy Clin. Immunol., 114, 1463–1470.
Nucleic Acids Research, 2011 7
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from

45. Rovescalli,A.C., Cinquanta,M., Ferrante,J., Kozak,C.A. andNirenberg,M. (2000) The mouse Nkx-1.2 homeobox gene:alternative RNA splicing at canonical and noncanonical splicesites. Proc. Natl Acad. Sci. USA, 97, 1982–1987.
46. Baumbusch,L.O., Myhre,S., Langerod,A., Bergamaschi,A.,Geisler,S.B., Lonning,P.E., Deppert,W., Dornreiter,I. andBorresen-Dale,A.L. (2006) Expression of full-length p53 and itsisoform Deltap53 in breast carcinomas in relation to mutationstatus and clinical parameters. Mol. Cancer, 5, 47.
47. Cagliani,R., Bardoni,A., Sironi,M., Fortunato,F., Prelle,A.,Felisari,G., Bonaglia,M.C., D’Angelo,M.G., Moggio,M., Bresolin,N.et al. (2003) Two dystrophin proteins and transcripts in a milddystrophinopathic patient. Neuromuscul. Disord., 13, 13–16.
48. Chikaev,N.A., Bykova,E.A., Najakshin,A.M., Mechetina,L.V.,Volkova,O.Y., Peklo,M.M., Shevelev,A.Y., Vlasik,T.N.,Roesch,A., Vogt,T. et al. (2005) Cloning and characterization ofthe human FCRL2 gene. Genomics, 85, 264–272.
49. Cocquet,J., Chong,A., Zhang,G. and Veitia,R.A. (2006) Reversetranscriptase template switching and false alternative transcripts.Genomics, 88, 127–131.
50. Cox,P.R., Siddique,T. and Zoghbi,H.Y. (2001) Genomicorganization of Tropomodulins 2 and 4 and unusual intergenicand intraexonic splicing of YL-1 and Tropomodulin 4.BMC Genomics, 2, 7.
51. Galante,P.A., Sakabe,N.J., Kirschbaum-Slager,N. and deSouza,S.J. (2004) Detection and evaluation of intron retentionevents in the human transcriptome. RNA, 10, 757–765.
52. Lukas,J., Gao,D.Q., Keshmeshian,M., Wen,W.H., Tsao-Wei,D.,Rosenberg,S. and Press,M.F. (2001) Alternative and aberrantmessenger RNA splicing of the mdm2 oncogene in invasive breastcancer. Cancer Res., 61, 3212–3219.
53. Aebi,M., Hornig,H., Padgett,R.A., Reiser,J. and Weissmann,C.(1986) Sequence requirements for splicing of higher eukaryoticnuclear pre-mRNA. Cell, 47, 555–565.
8 Nucleic Acids Research, 2011
by guest on January 13, 2013http://nar.oxfordjournals.org/
Dow
nloaded from

Dispatch R367
DNA replication: Building the perfect switchJohn F.X. Diffley
A sophisticated molecular switch ensures thatreplication origins are activated just once in each cellcycle. Recent work reveals how the proteolysis of a keyreplication inhibitor, geminin, by the anaphasepromoting complex/cyclosome is an importantcomponent of this switch.
Address: ICRF Clare Hall Laboratories, South Mimms, EN6 3LD, UK.E-mail: [email protected]
Current Biology 2001, 11:R367–R370
0960-9822/01/$ – see front matter © 2001 Elsevier Science Ltd. All rights reserved.
The sequencing of the human genome has been hailed asone of humankind’s great achievements, in part, becauseof the sheer magnitude of the endeavour. The accuratesequence of the three billion or so nucleotides of the humangenome has involved many scientists and has taken yearsto assemble. Yet, every proliferating human cell is facedwith the prospect of having to copy accurately and pre-cisely this same information in the space of only a fewhours during the cell cycle. Either incomplete replicationor over-replication would cause cell death, or worse, wouldgenerate the kinds of genetic alterations associated withdiseases like cancer.
To accomplish this feat in the allotted time, eukaryoticcells have developed a ‘divide and conquer’ strategy.Unlike their prokaryotic counterparts, eukaryotic genomesare replicated from multiple replication origins distributedalong their chromosomes. In human somatic cells, replica-tion occurs from 10,000–100,000 such replication origins;thus, each replication origin is only responsible for thereplication of a relatively small portion of the genome.
This strategy can allow rapid replication of large genomesbut brings with it a serious bookkeeping problem. Howcan the cell keep track of all of these origins, ensuring thateach one fires efficiently during S phase while also ensur-ing that no origin fires more than once? To cope with this,eukaryotic cells have evolved a remarkable molecular switchwhich, when turned on, promotes just a single initiationevent from each origin. Two recent studies [1,2] of DNAreplication in Xenopus show in greater detail the workingsof this switch.
At its heart is the tightly regulated assembly of pre-replicative complexes (pre-RCs) at replication origins in areaction known as ‘licensing’ (Figure 1). Pre-RCs assem-ble in a stepwise manner: the origin recognition complex(ORC), a sequence-specific DNA binding protein, binds
first and remains bound to origins during most or all of thecell cycle. Cdc6 then enters the complex, and cooperateswith ORC to load six different but related polypeptidesknown as the Mcm2-7 complex [3]. Recent work in fissionyeast and Xenopus has identified another pre-RC compo-nent, Cdt1, which enters the pre-RC independently ofCdc6 and is also required to load the Mcm2-7 complex[4,5]. The Mcm2-7 complex then plays a crucial roleduring initiation and during the ensuing elongation phaseof DNA replication, perhaps acting as a replicative DNAhelicase [6].
Pre-RCs can only assemble at origins during a short periodof the cell cycle between the end of mitosis and a pointlate in G1 phase (Figure 1). This temporal separation ofpre-RC assembly and origin activation is a key feature ofthe switch because it ensures that new pre-RCs cannotassemble on origins which have fired and, thus, origins canfire just once in each cell cycle [7]. Understanding howlicensing is prevented after S phase begins, therefore, hasbeen the focus of much research in the field.
Cyclin-dependent kinases (Cdks) are central to this regula-tion. Cdks are essential for triggering the initiation of DNAreplication from origins that contain pre-RCs. At the sametime, Cdks appear to play a direct role in preventing theassembly of new pre-RCs [7]. Because Cdk activityremains high from the onset of S phase until the end of thefollowing mitosis, re-licensing of origins cannot occur untilthe beginning of the next cell cycle. Although the pictureis far from complete, it appears that Cdks prevent pre-RCassembly in multiple, redundant ways. In budding yeast,for example, Cdks target Cdc6 for SCF-dependent, ubiq-uitin-mediated degradation [8–10] and trigger the export ofthe Mcm2-7 complex from the nucleus [11,12]. It is likelythat Cdks also act in other ways to prevent re-replication.
Recent work in Xenopus [1,2,13,14] has revealed thatanother key cell cycle regulator, the anaphase promotingcomplex/cyclosome (APC/C), plays an important role inconstraining licensing to the Cdk cycle. The APC/C is anE3 ubiquitin ligase whose activity is regulated by Cdks:it is activated in mitosis by Cdks associated with mitoticcyclins and inactivated in late G1 phase by Cdks associ-ated with G1 cyclins [13]. In a screen for novel APC/Csubstrates, McGarry and Kirschner identified a proteinthey called ‘geminin’ [14]. Consistent with it being anAPC/C substrate, geminin is degraded in mitosis anddegradation requires a cyclin-like destruction box nearits amino-terminus. By using a destruction box mutantthat cannot be degraded, these authors showed that

R368 Current Biology Vol 11 No 9
geminin could inhibit DNA replication and that thisinhibition of replication correlated with an inhibition ofMcm loading.
Two groups have recently identified Cdt1 as the target oflicensing inhibition by geminin [1,2]. Wolfschlegel et al.[1] found human Cdt1 as a protein that is tightly associatedwith geminin in co-immunoprecipitation experimentsfrom human cell extracts. Using cell-free replication extractsfrom Xenopus eggs, they showed that the inhibition ofDNA replication by geminin could be overcome by addi-tion of excess Cdt1 suggesting that geminin may act byinhibiting Cdt1.
Biochemical approaches from Blow and colleagues [15]had identified two protein fractions required for licensing,termed RLF-B and RLF-M. RLF-M was previously shownto comprise a heterohexameric complex of the Mcm2-7proteins [15]. In their recent work, Tada et al. [2] show thatgeminin acts by inhibiting RLF-B. They used a geminin-affinity chromatography to purify RLF-B and showed thatRLF-B appears to be identical to Cdt1. Elution of Cdt1from the geminin affinity column required 4M urea, attest-ing to the tight interaction between these two proteins.
Although the regulated appearance and disappearance ofgeminin could be sufficient to explain how replicationoccurs only once per cell cycle, it is likely that there will bemore to the story. McGarry and Kirschner [14] showedthat geminin-depleted Xenopus extracts, while supportingefficient DNA replication, did not re-replicate their DNA.This demonstrates that there must be something in theseextracts besides geminin which can block re-initiation.
A clue to the nature of this inhibitor may come from theexperiments of Tada et al. [2] who showed that the partiallicensing activity in geminin-depleted metaphase extracts
could be significantly enhanced by treatment of extractswith chemical inhibitors of Cdks. This suggests that, evenin the absence of geminin, Cdks are able to inhibit licens-ing to some extent. This is consistent with previous workshowing that Cdk2 can prevent licensing in Xenopus eggextracts [16].
How does this inhibition work? Experiments in humancells and Xenopus [17–19] have shown that phosphorylationof Cdc6, presumably by cyclin A-associated kinase causesits export from nuclei. This may be important for blockinglicensing during S and G2 phases, however, it cannotexplain the results of Tada et al. [2] since licensing inmetaphase extracts occurs in the absence of a nuclearenvelope. Therefore, there must be some additional andas yet unidentified way in which Cdks can prevent licens-ing. Thus, in Xenopus, as in yeast, Cdks block licensing inmultiple, redundant ways. First, they inactivate theAPC/C during G1, allowing the accumulation of the licens-ing inhibitor, geminin. Second, they cause the export ofCdc6 from the nucleus, and third, they act on at least oneadditional, unidentified target. This may be important inmaking absolutely sure that re-initiation never occurs andillustrates that such redundancy may be a general featureof the eukaryotic cell cycle.
This points to another emerging trend: Cdks prevent re-replication by different means in different organisms(Figure 2). In budding yeast and fission yeast, Cdc6 —Cdc18 in fission yeast — protein levels are regulated;Cdc6 transcription is limited to late mitosis/early G1 phaseand the Cdc6 protein is targeted for ubiquitin-mediateddegradation when Cdks are activated in late G1 phase[8–10,20–23]. In Xenopus, Cdc6 remains stable during thecell cycle but, instead, Cdk phosphorylation triggers itsexport from the nucleus [19]. As in Xenopus, the nuclearlocalisation of Cdc6 is also regulated by Cdks in human
Figure 1
Regulation of licensing in eukaryotic cells. Atthe end of mitosis, the anaphase promotingcomplex/cyclosome (APC/C) is activated byCdks. Active APC/C then contributes to theinactivation of Cdks by targeting the essentialcyclin subunits for ubiquitin mediateddegradation. In this state — CDKs off, APC/Con — cells are competent to assemble pre-RCs at their origins. However, the presence ofactive APC/C prevents accumulation ofnecessary S phase promoting factors like Sphase cyclins and the Cdc7 regulatorysubunit, Dbf4. At the end of G1 phase, aswitch is thrown which converts cells to a verydifferent state in which origin firing ispromoted while licensing is prevented — Cdkson, APC/C off. Both the activation of origin
firing and the prevention of licensing requiresthe activation of Cdks, which in turn requires
Cdk-dependent inactivation of the APC/C.Further details are provided in the text.
Current Biology
MCM2-7
Cdks OFFAPC/C ONLicensing allowedOrigin firing prevented
Cdks ONAPC/C OFFLicensing preventedOrigin firing allowed
Pre-RC
G1 Phase S, G2 Phase
Cdc6Cdt1
ORC
ORC
ORC

[17,18] and, in addition, Cdc6 is targeted for degradationby the APC/C [24,25].
Cdt1 also appears to be regulated differently in differentorganisms. Both transcription and proteolysis of Cdt1 arevery similar to that of Cdc6 in fission yeast [5]. However,in Xenopus, Cdt1 is regulated by geminin which, in turn istargeted for degradation by the APC/C [1,2,12]. In humancells, Cdt1 appears to be regulated by both geminin and,perhaps, cell cycle-regulated proteolysis [1,14]. If a geminin-like licensing inhibitor exists in budding yeast, it is notregulated solely by the APC/C since Cdk inactivationbypasses any requirement for the APC/C in licensing [26].In a different way, the APC/C may have some role in pre-venting re-replication in budding yeast, for example, bytargeting S phase promoting factors like Clb5 and theCdc7 regulatory subunit Dbf4 for degradation [27–30].
In conclusion, a single round of DNA replication per cellcycle is achieved by an intricate molecular mechanismwhich ensures that the ‘loading’ and ‘firing’ of replicationorigins cannot occur in a cell at the same time. The cou-pling of licensing to the Cdk cycle may be a universalfeature of this switch. How this is ultimately achievedmay differ in different organisms. Finally, this shortreview has only addressed the ‘negative’ side of theswitch — how licensing is prevented after S phase begins.The other side of the switch — the mechanism by whichCdks activate replication origins is currently far less wellunderstood and likely to be an area of intense interest inthe future.
References1. Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A:
Inhibition of eukaryotic DNA replication by geminin binding toCdt1. Science 2000, 290:2309-2312.
2. Tada S, Li A, Maiorano D, Mechali M, Blow JJ: Repression of originassembly in metaphase depends on inhibition of RLF-B/Cdt1 bygeminin. Nat Cell Biol 2001, 3:107-113.
3. Takisawa H, Mimura S, Kubota Y: Eukaryotic DNA replication: frompre-replication complex to initiation complex. Curr Opin Cell Biol2000, 12:690-696.
4. Maiorano D, Moreau J, Mechali M: XCDT1 is required for theassembly of pre-replicative complexes in Xenopus laevis. Nature2000, 404:622-625.
5. Nishitani H, Lygerou Z, Nishimoto T, Nurse P: The Cdt1 protein isrequired to license DNA for replication in fission yeast. Nature2000, 404:625-628.
6. Labib K, Diffley JFX: Is the MCM2-7 complex the eukaryotic DNAreplication fork helicase? Curr Opin Genet Dev 2001, 11: 64-70.
7. Diffley JFX: Once and only once upon a time: Specifying andregulating origins of DNA replication in eukaryotic cells. GenesDev 1996, 10:2819-2830.
8. Elsasser S, Chi Y, Yang P, Campbell JL: Phosphorylation controlstiming of Cdc6p destruction: a biochemical analysis. Mol Biol Cell1999, 10:3263-3277.
9. Drury LS, Perkins G, Diffley JFX: The Cyclin dependent kinaseCdc28p regulates distinct modes of Cdc6p proteolysis during thebudding yeast Cell cycle. Curr Biol 2000, 10:231-240.
10. Calzada A, Sanchez M, Sanchez E, Bueno A: The stability of theCdc6 protein is regulated by cyclin-dependent kinase/cyclin Bcomplexes in Saccharomyces cerevisiae. J Biol Chem 2000,275:9734-9741.
11. Labib K, Diffley JFX, Kearsey SE: G1-phase and B-type cyclinsexclude the DNA-replication factor Mcm4 from the nucleus.Nat Cell Biol 1999, 1:415-422.
12. Nguyen VQ, Co C, Irie K, Li JJ: Clb/Cdc28 kinases promote nuclearexport of the replication initiator proteins Mcm2-7. Curr Biol 2000,10:195-205.
13. Zachariae W, Nasmyth K: Whose end is destruction: cell divisionand the anaphase-promoting complex. Genes Dev 1999,13:2039-2058.
14. McGarry TJ, Kirschner MW: Geminin, an inhibitor of DNAreplication, is degraded during mitosis. Cell 1998, 93:1043-1053.
Dispatch R369
Figure 2
Different ways of licensing inhibition indifferent organisms. Details are provided in thetext.
Cdk
SCF dependentCdc6 proteolysis
MCM2-7 nuclear export
?
Cdk
SCF dependentCdc6 (Cdc18) proteolysis
Cdt1 proteolysis?
Current Biology
Cdk
APC/C inactivation
Cdc6 nuclear export
?
Geminin accumulation,Cdt1 inhibition
Budding yeast Fission yeast
Xenopus, humans

15. Chong, JPJ, Mahbubani HM, Khoo C-Y, Blow JJ: Purification of anMCM-containing complex as a component of the DNA replicationlicensing system. Nature 1995, 375:418-421.
16. Hua XH, Yan H, Newport J: A role for Cdk2 kinase in negativelyregulating DNA replication during S phase of the cell cycle. J CellBiol 1997, 137:183-192.
17. Peterson BO, Lukas J, Sorenson CS, Bartek J, Helin K:Phosphorylation of mammalian CDC6 by Cyclin A/CDK2 regulatesits subcellular localization. EMBO J 1999, 18:396-410.
18. Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M,Parvin JD, Dutta A: Human CDC6/Cdc18 associates with Orc1 andcyclin-cdk and is selectively eliminated from the nucleus at theonset of S phase. Mol Cell Biol 1998, 18:2758-2767.
19. Pelizon C, Madine MA, Romanowski P, Laskey RA:Unphosphorylatable mutants of Cdc6 disrupt its nuclear exportbut still support DNA replication once per cell cycle. Genes Dev2000, 14:2526-2533.
20. Jallepalli PV, Tien D, Kelly TJ: sud1(+) targets cyclin-dependentkinase-phosphorylated Cdc18 and Rum1 proteins for degradationand stops unwanted diploidization in fission yeast. Proc Natl AcadSci USA 1998, 95:8159-8164.
21. Baum B, Nishitani H, Yanow S, Nurse P: Cdc18 transcription andproteolysis couple S phase to passage through mitosis. EMBO J1998, 17:5689-5698.
22. Wolf DA, McKeon F, Jackson PK: F-box/WD-repeat proteins pop1pand Sud1p/Pop2p form complexes that bind and direct theproteolysis of cdc18p. Curr Biol 1999, 9:373-376.
23. Kominami K, Toda T: Fission yeast WD-repeat protein pop1regulates genome ploidy through ubiquitin-proteasome-mediateddegradation of the CDK inhibitor Rum1 and the S-phase initiatorCdc18. Genes Dev 1997, 11:1548-1560.
24. Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M,Denchi EL, Gieffers C, Matteucci C, Peters JM, Helin K: Cell cycle-and cell growth-regulated proteolysis of mammalian CDC6 isdependent on APC-CDH1. Genes Dev 2000, 14:2330-2343.
25. Mendez J, Stillman B: Chromatin association of human originrecognition complex, cdc6, and minichromosome maintenanceproteins during the cell cycle: assembly of prereplicationcomplexes in late mitosis. Mol Cell Biol 2000, 20:8602-8612.
26. Noton EA, Diffley JFX: CDK inactivation is the only essentialfunction of the APC/C and the mitotic exit network proteins fororigin resetting during mitosis. Mol Cell 2000, 5:85-95.
27. Shirayama M, Toth A, Galova M, Nasmyth K: APC(Cdc20) promotesexit from mitosis by destroying the anaphase inhibitor Pds1 andcyclin Clb5. Nature 1999, 402:203-207.
28. Cheng L, Collyer T, Hardy CF: Cell cycle regulation of DNAreplication initiator factor Dbf4p. Mol Cell Biol 1999,19:4270-4278.
29. Oshiro G, Owens JC, Shellman Y, Sclafani RA, Li JJ: Cell cyclecontrol of Cdc7p kinase activity through regulation of Dbf4pstability. Mol Cell Biol 1999, 19:4888-4896.
30. Godinho Ferreira M, Santocanale C, Drury LS, Diffley JFX: Dbf4p, anessential S phase promoting factor, is targeted for degradation bythe anaphase promoting complex. Mol Cell Biol 2000, 20:242-248.
R370 Current Biology Vol 11 No 9

The role of endogenous and exogenous DNAdamage and mutagenesisErrol C Friedberg�, Lisa D McDaniel and Roger A Schultz
The field of DNA damage responsiveness in general, and the
consequences of endogenous and exogenous base damage
in DNA, in particular, has made new and exciting contributions
to our increasing understanding of the initiation and
progression of neoplasia in humans. This article presents
some of the highlights in this area of investigation, with a
particular emphasis on DNA repair, the tolerance of DNA damage
and its contribution to mutagenesis, and DNA damage
checkpoint regulation.
AddressesLaboratory of Molecular Pathology, Department of Pathology,University of Texas Southwestern Medical Center, Dallas TX,
75390-9072, USA�e-mail: [email protected]
Current Opinion in Genetics & Development 2004, 14:5–10
This review comes from a themed issue on
Oncogenes and cell proliferation
Edited by Zena Werb and Gerard Evan
0959-437X/$ – see front matter
� 2003 Elsevier Ltd. All rights reserved.
DOI 10.1016/j.gde.2003.11.001
AbbreviationsAAF acetylaminofluorene
APOBEC3G apolipoprotein B mRNA editing enzyme catalytic
polypeptide-like 3G
ATM ataxia telangiectasia mutated
ATR ataxia telangiectasia and Rad3-related
ATRIP ATR-interacting protein
CSA Cockayne syndrome group A gene
CSN COP9 signalosome
FA Fanconi anemiaMAC mutagenesis in aging colonies
MMC mitomycin C
MMS methylmethane sulfonate
NER nucleotide excision repair
NHEJ non-homologous end joining
ORF open reading frame
RNAi RNA interference
RPA replication protein A
XP xeroderma pigmentosum
IntroductionThe role of both endogenous (spontaneous) and exogen-
ous (environmental) DNA damage in the initiation and
progression of neoplasms is unassailable. Genes that
participate in various mechanisms that protect cells from
the generation of mutations in somatic cells — except in
selected physiological situations, such as the generation
of mutations to promote variability in immunoglobulin
genes — are now appropriately designated as tumor
suppressor or gatekeeper genes [1].
Cells have evolved multiple, and often apparently redun-
dant, biological responses to DNA damage that are con-
veniently classified as either ‘DNA repair’ or ‘DNA
damage tolerance’ [2] (Figure 1). As the name implies,
DNA repair embraces mechanisms for the enzyme-
catalyzed reversal of damage, the excision of base damage
(including inappropriate bases such as uracil), as well as
nucleotides that are incorrectly incorporated during DNA
replication (Figure 1). In addition to base damage, our
understanding of DNA repair now embraces the restora-
tion of both single- and double-strand breaks in the
genome [3,4]. The tolerance of DNA damage involves
several distinct cellular responses, by which the poten-
tially lethal effects of arrested DNA replication by
damaged bases are mitigated (Figure 1).
Both the efficiency and kinetics of DNA repair and DNA
damage tolerance are influenced by regulatory responses.
With the advent of microarray technologies, we are just
beginning to appreciate the magnitude, variation and
significance of the extensive transcriptional responses
to various types of genomic insult (Figure 1). Addition-
ally, the role of DNA damage in the activation of cell
cycle checkpoints is now a burgeoning field, involving
multiple complex pathways that transduce signals from
sites of DNA damage and altered DNA replication to
repair and damage tolerance effector pathways (Figure 1).
In this review, we highlight some recent advances in
selected aspects of the plethora of biological responses
to DNA damage, with a particular emphasis on mechan-
isms of mutagenesis from endogenous and exogenous
damage.
New insights into DNA repair by the reversalof base damageSeveral forms of base damage to DNA are repaired by
biochemical reactions that directly reverse the damage,
restoring affected bases to their native chemistry and
conformation. Among the several genes that are required
for the maintenance of genomic integrity in the face of
alkylation damage to DNA in Escherichia coli is one called
alkB, the specific function of which was unknown until
very recently. In a model example of the utility of
bioinformatics, Eugene Koonin and his colleagues [5]
have identified a domain in the translated alkB sequence
that is suggestive of an a-ketoglutarate- and Fe(II)-
dependent dioxygenase. Two groups [6��,7��] have
www.sciencedirect.com Current Opinion in Genetics & Development 2004, 14:5–10

now independently shown that purified AlkB protein
from E. coli repairs the cytotoxic lesions 1-methyladenine
and 3-methylcytosine, by reversing such damage via a
deoxygenase reaction that requires oxygen, a-ketogluta-
rate and Fe(II). The bacterial alkB gene is conserved in
eukaryotes, including human cells — in which there are
two structural homologs, hABH2 and hABH3 — where it,
presumably, subserves the same function.
More recent studies [8] have demonstrated that the E. colialkB and human hABH3 gene products also repair these
lesions in RNA, extending the range of the repair of
biological macromolecules to this class of polynucleo-
tides. The repair of proteins [9] and of deoxyribonucleo-
side triphosphate precursors of DNA [10] has been
documented previously.
New complexities in nucleotide excisionrepairNucleotide excision repair (NER) is a major form of repair
for DNA base damage that results in distortions of DNA
structure that (among other effects) interfere with normal
base pairing. As such, it is suited to many forms of
exogenous base damage, such as cyclobutane pyrimidine
dimers induced by exposure to sunlight, a potent evolu-
tionary driving force. NER encompasses the repair of
both transcriptionally silent and transcriptionally active
regions of the genome, by somewhat distinct mechanisms
[11]. These distinctions primarily center around the
recognition of base damage. In particular, it is believed
that damage recognition in transcriptionally active DNA
is effected by arrest of the transcriptional machinery. One
of the gene products required for transcription-coupled
NER is the CSA protein, the product of the Cockayne
syndrome group A gene (CSA) [11].
A recent study [12��] has identified human CSA protein in
a multiprotein complex that includes the COP9 signalo-
some (CSN), a regulator of cullin-based ubiquitin ligases.
This study also identified a second multiprotein complex,
containing the COP signalosome, except that instead of
CSA protein, this complex contains another protein that is
required for NER, DDB2, a 48 kDa protein, which,
together with DDB1 protein, comprises a heterodimer
that is involved in DNA damage recognition during
transcriptionally independent NER [12��]. Mutations in
the DDB2 gene have been identified in several indivi-
duals with the NER-defective and skin-cancer-prone
hereditary disease xeroderma pigmentosum (XP),
belonging to genetic complementation group E [13,14].
The authors suggest that, following exposure to UV
radiation, the DDB2 complex binds to chromatin and
the COP signalosome dissociates and activates ubiquitin
ligase E3 activity. By contrast, when the transcriptional
Figure 1
Current Opinion in Genetics & Development
COP signalosomeUbiquitin ligase
Global NER activation
Transcription-coupledNER inactivation
Cadmium inactivation
DNA damagetolerance
DNA damagerepair
Check point
Signaltransduction
Transcriptionalactivation
Cell cyclearrest
DNA damagebypass
polymerases
DNA damage
MAC
Novel genediscovery
alkB damagereversal
hMLH1≠
hMLH1+hMSH6
Artemis Lig4
RPAssDNA
BRCA2/FANCD1/FANCB
Nucleotide excision repair
Base excision repair
Mismatch repair
Single-strand break repair
Double-strand break repair
APOBEC3G
Cellular responses to DNA damage. The response to DNA damage (yellow box) results in either tolerance or damage repair (blue boxes), with
damage repair represented by a variety of specific damage repair pathways. These events are modulated and facilitated by cell cycle checkpoint
mechanisms that arrest cell cycle progression at various points (orange box). The recent insights into the complexities of these events, as summarized
in this review, are shown here to depict their newly discovered role(s) in these processes (green boxes).
6 Oncogenes and cell proliferation
Current Opinion in Genetics & Development 2004, 14:5–10 www.sciencedirect.com

machinery is arrested by UV radiation induced base
damage, the CSA complex recruits the COP signalosome
and inactivates the ubiquitin ligase [12��].
Something new in mismatch repairOne of the central debates about neoplastic transforma-
tion concerns the strong mutator phenotype of neoplasms,
which cannot be accounted for by the multiplicative
sum of the spontaneous mutation frequency in individual
genes. An in-depth coverage of this topic is outside the
scope of this review; however, among the possible ex-
planations for this strong mutator phenotype is the
notion that genes involved in mismatch repair of DNA
become inactivated by one or another genetic and/or
epigenetic mechanisms [15,16], and that the ensuing
mutator phenotype increases the probability (by random
or possibly non-random mechanisms) of inactivation of
other DNA repair genes. In support of this notion, a
recent study examined clones of cells with either an
inactive or active hMSH6 gene [17] in which expression
of hMLH1 was silenced by promoter hypermethylation.
The additional inactivation of this gene in cells mutant
for hMSH6 resulted in a higher mutation rate and a
different mutational spectrum than cells that are wild-
type for hMSH6 [17].
Most mutations that are associated with defective mis-
match repair are the result of inactivation of genes for this
DNA repair process. A recent study, however, has
demonstrated that some exogenous mutagens can inac-
tivate mismatch repair proteins directly [18�]. Specific-
ally, chronic exposure of yeast to cadmium inactivates
mismatch repair in vivo and this effect can be reproduced
in vitro. This is the first clear demonstration of an exo-
genous agent promoting genomic instability through
direct effects on ‘guardians of the genome’, rather than
on the genome itself, and begs more extensive examina-
tion of this mechanism of environmentally induced geno-
mic instability.
What’s new in strand-break repair?Our understanding of the details of DNA-strand-break
repair, particularly that in mammalian cells, has, until
recently, lagged behind that of the repair of base damage:
however, the past 4–5 years have witnessed impressive
progress in this area. We now know that double-strand
breaks can be repaired by either homologous recombina-
tion by a variety of different mechanisms, or by the direct
fusion of broken ends (non-homologous end joining
[NHEJ]), an area that has significant overlap with
V(D)J recombination in the immune system. A newly
discovered gene called Artemis has been shown to be
involved in V(D)J recombination in the immune system
[19]. More recently, it was demonstrated that the Artemis
protein is a single-strand-specific exonuclease. It also
complexes with the DNA-dependent protein kinase
(DNA-PKCS), an integral component of the NHEJ
machinery [20��]. Complex formation is followed by phos-
phorylation of Artemis by DNA-PKCS,, an event that
converts it to an endonuclease that can open DNA hairpins
that are generated during V(D)J recombination [20��].
Defects in NHEJ in the immune system accelerate the
formation of lymphomas in mice. However, this process
of strand-break rejoining can also suppress tumors in cells
that do not undergo V(D)J recombination. A recent study
has demonstrated that haploinsufficiency for the Lig4gene (which encodes DNA ligase IV) results in the
development of non-lymphomatous tumors in a cancer-
prone mouse strain [21]. Hence, even a modest reduction
in NHEJ activity promotes tumorigenesis in mice.
DNA damage tolerance: the role(s) oferror-prone DNA polymerasesAnother area of significant progress has emerged from the
discovery of a large repertoire of DNA polymerases
(especially in mammalian cells), endowed with the ability
to bypass many types of spontaneous and exogenously
generated forms of base damage, often (but not always)
leading to mutations [22]. In E. coli, one of these poly-
merases, called Pol IV and encoded by the dinB gene, has
been implicated in spontaneous mutagenesis [23�]. Spon-
taneous mutagenesis can occur in rapidly growing and in
stationary phase E. coli by different processes. There has
been conflicting data as to the requirement for Pol IV for
the latter process. A recent study indicates that this
confusion apparently originates from the type of mutant
strain used. The dinB gene is part of a four-gene operon
with three downstream genes of unknown function.
Hence, some mutations in the dinB gene can result in
polar effects. Non-polar mutants do not result in sponta-
neous mutations in rapidly growing cells [23�].
It is believed that when replication is blocked by DNA
damage in a strand (either leading or lagging), polymerase
switching transpires, enabling the bypass polymerase(s)
to transiently occupy the primer template for replicative
bypass and then to reoccupy this site when translesion
synthesis is completed. If the replicative machinery is
physically displaced from the primer-template during this
process, replication of both DNA strands might be
expected to arrest. An in vivo system was established
in E. coli to address this question [24��]. It was observed
that an acetylaminofluorene (AAF) lesion in the leading
strand did not affect the kinetics of lagging strand repli-
cation and vice versa. Hence, whatever the nature of the
polymerase switching events during replicative bypass of
base damage (translesion DNA synthesis), the replicative
machinery does not appear to be completely disengaged
from the arrested fork.
Tuberculosis is a pulmonary infection that is sometimes
prone to lethal antibiotic resistance. A recent study has
demonstrated that when Mycobacterium tuberculosis is
The role of endogenous and exogenous DNA damage and mutagenesis Friedberg, McDaniel and Schultz 7
www.sciencedirect.com Current Opinion in Genetics & Development 2004, 14:5–10

exposed to various types of DNA base damage, a gene
called dnaE2 (believed to encode a novel DNA polymer-
ase) is upregulated and results in an increased mutation
frequency in the bacterium. It is suggested that mutations
associated with spontaneous DNA damage might form
the basis of the antibiotic resistance that is manifested by
this organism [25�].
Other aspects of spontaneous mutagenesisIt is well established that, in laboratory-derived strains of
E. coli, mutagenesis can be promoted by stress conditions.
However, the general evolutionary significance of this
phenomenon has been questioned because laboratory
strains are not representative of strains in the wild,
growing in different natural ecological niches. A recent
study collected nearly 800 E. coli isolates from around the
world and examined mutagenesis in aging colonies
(MAC) by exposure to starvation after a period of expo-
nential growth [26�]. Most natural isolates exhibited
increased MAC. Although the nature of the mutagenesis
was characteristic for each strain — the particular ecolog-
ical niche from which the strain was isolated being a major
determinant of the mutator phenotype — the study
supports the notion that adaptive mutagenesis associated
with stress-induced mutations is a general evolutionary
strategy in E. coli.
The high spontaneous mutation of HIV is a principal
scourge of the disease AIDS, but the primary mech-
anism(s) of this mutagenesis remains to be established.
No less than three recent studies [27�–29�] have demon-
strated that APOBEC3G (apolipoprotein B mRNA editing
enzyme catalytic polypeptide-like 3G), an endogenous
inhibitor of HIV-1 replication, is a cytidine deaminase that
generates G!A mutations in the viral DNA, presumably
by converting cytidine to uracil in the viral DNA minus
strand, thereby promoting the formation of U:A base pairs.
Whereas this hypermutation is considered to have evolved
as a viral defense mechanism leading to inactivation of
the virus, the accumulation of APOBEC3G-induced non-
lethal mutations could potentially promote variation in
primate lentiviral populations, including HIV.
Checkpoint control and initiating signals forresponses to DNA damageATR (ataxia telangiectasia and Rad3-related) and ATM
(ataxia telangiectasia mutated) are kinases that play cen-
tral roles in responses to various types of DNA damage,
notably that produced by ionizing radiation [30,31]. ATR
is known to phosphorylate substrates such as Brca1, Chk1
protein, p53 and Rad17. These phosphorylated sub-
strates, in turn, mediate inhibition of DNA replication
and progression through the cell cycle and promote DNA
repair and other effector responses [30,31]. Many DNA-
damaging agents can elicit the ATR-mediated DNA
damage response, therefore an issue of considerable
interest is whether different sensors function for different
types of damage, or whether all are processed to a com-
mon intermediate. A recent study has demonstrated that
RPA (replication protein A; a single-stranded DNA bind-
ing protein) stimulates the binding of the ATR–ATRIP
(ATR-interacting protein) complex to single-stranded
DNA, stimulating the phosphorylation of Rad17 protein
that is bound to DNA [32��]. These studies suggest that
single-stranded DNA coated with RPA is the key
denominator of varying types of DNA damage that
recruits ATR–ATRIP and initiates the DNA-damage-
signaling cascade.
It has also been proposed that the process of retroviral
DNA integration is ‘sensed’ as DNA damage by host
cells. In an independent study, it was demonstrated that
the ATR kinase (but not the ATM kinase) is required for
the successful integration of retroviral RNA [33�].
New genes for biological responses to DNAdamage?The genomics era has facilitated the identification of new
genes that are involved in biological responses to DNA
damage. In addition to expression-array studies, other
techniques have been employed in attempts to gain such
insights. One recent study identified six novel genes that
are involved in biological responses to UV radiation or
methylmethane sulfonate (MMS) by screening a collec-
tion of >2800 yeast-deletion mutants. In each mutant, a
single ORF was replaced by a cassette, containing two
unique sequence tags, thus allowing for their identifica-
tion by hybridization to a high-density oligonucleotide
array [34�].
Another study used RNAi to uncover genes in the nema-
tode worm Caenorhabditis elegans. A total of 61 genes were
found to affect genomic stability in somatic cells and
spontaneous mutagenesis in the germ line [35�]. Many of
the genes uncovered are novel ORFs with no known
function.
Fanconi anemia and BRCAFanconi anemia (FA) is a clinically heterogeneous human
disorder that is associated with genomic instability, cancer
predisposition and cellular sensitivity to certain DNA
damaging agents, including mitomycin C (MMC). The
disease is genetically complex, represented by eight
genetic complementation groups. Genes representing
six of the FA complementation groups had been pre-
viously cloned and elucidation of the function of the
products of these genes over the last decade has provided
insight into a previously unrecognized regulatory pathway
for DNA-damage response. Five of these (groups FA-A,
-C, -E, -F and -G) were shown to participate in the
formation of a complex that directs the monoubiquitina-
tion of the product of the gene mutated in a sixth group
(FA-D2). Moreover, it was recognized that this mono-
ubiquitination leads to the targeting of the FANCD2
8 Oncogenes and cell proliferation
Current Opinion in Genetics & Development 2004, 14:5–10 www.sciencedirect.com

protein to BRCA1 nuclear foci and subsequent signaling
through BRCA2 and RAD51 for the repair of DNA
damage. RAD51 is a crucial component of homologous
recombination and this, therefore, suggests that the FA
complex mediates error-free homologous recombination
during S-phase arrest. These results are consistent with
studies that have reported an increased mutation fre-
quency in FA cells, relative to normal controls and, more
specifically, an increase in the occurrence of deletion
mutations. What remained a mystery here was which
genes were mutated in the other FA groups (FA-B and
-D1) and how the protein products of these two uniden-
tified genes participated in the same DNA-damage-
signaling pathway.
A recent report by Howlett et al. [36��] demonstrates
biallelic mutations in the BRCA2 gene in both of these
complementation groups. The findings illustrate that all
eight of the FA gene products are involved in a single
pathway that mediates DNA repair in response to DNA
damage caused by agents such as MMC and diepoxybu-
tane, which are both known to produce crosslinks in
DNA. The MMC sensitivity of FA-D1 fibroblast was
complemented, following expression of the wild-type
BRCA2 protein. The fact that two distinct complementa-
tion groups are defined by mutations in a single gene
suggests interallelic complementation or dominant activ-
ity for certain mutant alleles.
ConclusionsThe multiple mechanisms by which both natural (spon-
taneously-derived) and exogenous (environmentally-
derived) mutations arise in cells and, hence, can trigger
neoplasia are becoming clearer. Mutagenesis is a univer-
sal fact of life: the genetic diversity it generates in germ
cells is essential for Darwinian evolution. From an evolu-
tionary point of view, there is no intrinsic reason to
suppress mutations in somatic cells, provided that they
do not result in deleterious phenotypes before reproduc-
tive age. However, diseases such as XP, in which a
hereditary defect in a specific DNA repair modality
predisposes the individual to lethal multiple skin cancers
well before the onset of puberty, provide dramatic evi-
dence that preventing an excessive mutational burden in
somatic cells is as essential to life as the generation of a
threshold of mutations in the germ line. Biological evolu-
tion has apparently achieved a delicate balance in which
mutations are tolerated at certain levels in both germ line
and somatic cells. The preponderance of the evidence
indicates that mutations are stochastic events in genes.
Hence, should they transpire in genes that are crucial for
normal cellular proliferation, the price might be neopla-
sia. As there is no a priori reason to select against neoplasia
in organisms past their reproductive life, there is no
reason to expect selection for anti-mutagenic mechanisms
that specifically favor oncogenes and tumor suppressor
genes. Nor is there evidence of such mechanisms.
Meanwhile, our understanding of the multiple ways in
which cells are subjected to genomic insult and the
diverse responses to such damage continues to expand.
Such responses appear, primarily, to be designed to avoid
cell death, not mutations. Indeed, in some cases, muta-
tions are imposed on cells as a strategy to avoid death.
AcknowledgementsWe apologize to the many authors of outstanding papers that were notincluded here due to space limitations and the general scope ofthis issue.
References and recommended readingPapers of particular interest, published within the annual period ofreview, have been highlighted as:
� of special interest��of outstanding interest
1. Levine AJ: p53, the cellular gatekeeper for growth and division.Cell 1997, 88:323-331.
2. Sutton MD, Smith BT, Godoy VG, Walker GC: The SOS response:recent insights into umuDC-dependent mutagenesis and DNAdamage tolerance. Annu Rev Genet 2000, 34:479-497.
3. Lisby M, Mortensen UH, Rothstein R: Colocalization of multipleDNA double-strand breaks at a single Rad52 repair centre.Nat Cell Biol 2003, 5:572-577.
4. Taylor RM, Thistlethwaite A, Caldecott KW: Central role for theXRCC1 BRCT I domain in mammalian DNA single-strand breakrepair. Mol Cell Biol 2002, 22:2556-2563.
5. Aravind L, Koonin EV: The DNA-repair protein AlkB, EGL-9,and leprecan define new families of 2-oxoglutarate- andiron-dependent dioxygenases. Genome Biol 2001,2:research0007.1-0007.8.
6.��
Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B:Oxidative demethylation by Escherichia coli AlkB directlyreverts DNA base damage. Nature 2002, 419:174-178.
See annotation [7��].
7.��
Falnes PO, Johansen RF, Seeberg E: AlkB-mediated oxidativedemethylation reverses DNA damage in Escherichia coli.Nature 2002, 419:178-182.
These two papers [6��,7��] were published together and are the first directdemonstration of the repair of 1-methyladenine and 3-methylcytosine byalkB protein by direct reversal of the lesion.
8. Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M,Sundheim O, Bjoras M, Slupphaug G, Seeberg E et al.: Human andbacterial oxidative demethylases repair alkylation damage inboth RNA and DNA. Nature 2003, 421:859-863.
9. Schubert HL, Blumenthal RM, Cheng XD: Many paths tomethyltransfer: a chronicle of convergence. Trends Biochem Sci2003, 28:329-335.
10. Hayakawa H, Taketomi A, Sakumi K, Kuwano M, Sekiguchi M:Generation and elimination of 8-oxo-7,8-dihydro-2(-deoxyguanosine 5(-triphosphate, a mutagenic substratefor DNA synthesis, in human cells. Biochemistry 1995,34:89-95.
11. Friedberg EC, Walker GC, Siede W: DNA Repair and Mutagenesis.Washington DC: ASM Press; 1995.
12.��
Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M,Drapkin R, Kisselev AF, Tanaka K, Nakatani Y: The ubiquitin ligaseactivity in the DDB2 and CSA complexes is differentiallyregulated by the COP9 signalosome in response to DNAdamage. Cell 2003, 113:357-367.
This study yields new and provocative information concerning the pos-sible role of the COP9 signalosome in nucleotide excision repair. TheCOP9 signalosome (CSN), a regulator of cullin-based ubiquitin ligases ispart of two different complexes — one containing DDB and anothercontaining CSA. Differential regulation of the CSN ubiquitin ligase activityof the DDB2 and CSA complexes in response to UV irradiation isdemonstrated.
The role of endogenous and exogenous DNA damage and mutagenesis Friedberg, McDaniel and Schultz 9
www.sciencedirect.com Current Opinion in Genetics & Development 2004, 14:5–10

13. Dualan R, Brody T, Keeney S, Nichols AF, Admon A, Linn S:Chromosomal localization and cDNA cloning of the genes(DDB1 and DDB2) for the p127 and p48 subunits of a humandamage-specific DNA binding protein. Genomics 1995,29:62-69.
14. Rapic-Otrin V, Navazza V, Nardo T, Botta E, McLenigan M, Bisi DC,Levine AS, Stefanini M: True XP group E patients have adefective UV-damaged DNA binding protein complex andmutations in DDB2 which reveal the functional domains of itsp48 product. Hum Mol Genet 2003, 12:1507-1522.
15. Shibata D, Aaltonen LA: Genetic predisposition and somaticdiversification in tumor development and progression.Adv Cancer Res 2001, 80:83-114.
16. Kolodner R: Biochemistry and genetics of eukaryotic mismatchrepair. Genes Dev 1996, 10:1433-1442.
17. Baranovskaya S, Soto JL, Perucho M, Malkhosyan SR: Functionalsignificance of concomitant inactivation of hMLH1 and hMSH6in tumor cells of the microsatellite mutator phenotype.Proc Natl Acad Sci USA 2001, 98:15107-15112.
18.�
Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA,Resnick MA, Gordenin DA: Cadmium is a mutagen that acts byinhibiting mismatch repair. Nat Genet 2003, 34:326-329.
A fascinating study on the apparent direct effect of cadmium on mismatchrepair. The results illustrate that exogenous agent can directly interferewith DNA-repair pathways and enhance genomic instability.
19. Moshous D, Callebaut I, de Chasseval R, Corneo B,Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y,Philippe N et al.: Artemis, a novel DNA double-strand breakrepair/V(D)J recombination protein, is mutated in humansevere combined immune deficiency. Cell 2001, 105:177-186.
20.��
Ma Y, Pannicke U, Schwarz K, Lieber MR: Hairpin opening andoverhang processing by an Artemis/DNA-dependent proteinkinase complex in nonhomologous end joining and V(D)Jrecombination. Cell 2002, 108:781-794.
This study shows that Artemis protein is a single-stranded DNA-specificexonuclease and is phosphorylated by DNA-PKcs. Artemis protein isshown to possess single-strand-specific 50!30 exo-nuclease activity.After phosphorylation by DNA-PKcs, Artemis gains endonucleolytic activ-ity on 50 and 30 overhangs, as well as hairpins. In addition, the complexcan open hairpins generated by the RAG complex.
21. Sharpless NE, Ferguson DO, O’Hagan RC, Castrillon DH, Lee C,Farazi PA, Alson S, Fleming J, Morton CC, Frank K et al.: Impairednonhomologous end-joining provokes soft tissue sarcomasharboring chromosomal translocations, amplifications, anddeletions. Mol Cell 2001, 8:1187-1196.
22. Friedberg EC, Wagner R, Radman M: Specialized DNApolymerases, cellular survival, and the genesis of mutations.Science 2002, 296:1627-1630.
23.�
McKenzie GJ, Magner DB, Lee PL, Rosenberg SM: The dinBoperon and spontaneous mutation in Escherichia coli.J Bacteriol 2003, 185:3972-3977.
A study showing that the dinB gene is part of a polycistronic operon.Some mutations in dinB affect down-stream genes, whereas others donot. These results account for conflicting data regarding the role of Pol IVin spontaneous mutagenesis in E. coli.
24.��
Pages V, Fuchs RP: Uncoupling of leading- and lagging-strandDNA replication during lesion bypass in vivo. Science 2003,300:1300-1303.
This study examines the kinetics of leading and lagging strand DNAsynthesis in vivo, indicating that the synthesis of the two strands isuncoupled when a blocking lesion is on either strand. The replicativemachinery does not appear to disengage from the arrested fork while thebypass polymerase performs translesion DNA synthesis.
25.�
Boshoff HI, Reed MB, Barry CE III, Mizrahi V: DnaE2 polymerasecontributes to in vivo survival and the emergence of drugresistance in Mycobacterium tuberculosis. Cell 2003,113:183-193.
An interesting study, suggesting a possible mechanism of antibioticresistance during infection with M. tuberculosis. This appears to resultfrom the upregulation of dnaE2, a gene with homology to the majorreplicative polymerase, in response to DNA damage.
26.�
Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M,Taddei F, Matic I: Stress-induced mutagenesis in bacteria.Science 2003, 300:1404-1409.
This study supports the notion that adaptive mutagenesis associated withstress-induced mutations is a general evolutionary strategy in E. coli.
27.�
Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK,Watt IN, Neuberger MS, Malim MH: DNA deamination mediatesinnate immunity to retroviral infection. Cell 2003, 113:803-809.
See annotation [29�].
28.�
Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L:The cytidine deaminase CEM15 induces hypermutation innewly synthesized HIV-1 DNA. Nature 2003, 424:94-98.
See annotation [29�].
29.�
Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D:Broad antiretroviral defence by human APOBEC3G throughlethal editing of nascent reverse transcripts. Nature 2003,424:99-103.
These three studies [27�–29�] suggest a role of the deamination ofcytosine in the generation of mutations in lentiviruses. The gene productof APOBEC3G is a cytidine deaminase that generates G!A mutations inviral DNA. This gene might have evolved as a viral defense, however, itmay contribute to viral diversity.
30. Zhou BB, Elledge SJ: The DNA damage response: puttingcheckpoints inperspective. Nature 2000, 408:433-439.
31. Abraham RT: Cell cycle checkpoint signaling through the ATMand ATR kinases. Genes Dev 2001, 15:2177-2196.
32.��
Zou L, Elledge SJ: Sensing DNA damage through ATRIPrecognition of RPA-ssDNA complexes. Science 2003,300:1542-1548.
This study proposes that RPA, bound to single-stranded DNA, initiates asignal transduction cascade that effects checkpoint regulation by theATR–ATRIP recruitment to the site by RPA.
33.�
Daniel R, Kao G, Taganov K, Greger JG, Favorova O, Merkel G,Yen TJ, Katz RA, Skalka AM: Evidence that the retroviral DNAintegration process triggers an ATR-dependent DNA damageresponse. Proc Natl Acad Sci USA 2003, 100:4778-4783.
Retroviral integration can, apparently, activate the checkpoint response.ATR kinase, but not ATM kinase, is required for completion of retroviralintegration into the genome.
34.�
Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE:Previously uncharacterized genes in the UV- and MMS-inducedDNA damage response in yeast. Proc Natl Acad Sci USA 2002,99:10605-10610.
This study identified novel genes involved in biological responses to DNAdamage in yeast. Screening of 2,827 yeast gene deletion strains identifiedsix novel genes that were not previously known to be involved in DNAdamage. Deletion of these genes results in UV and MMS sensitivity.
35.�
Pothof J, van Haaften G, Thijssen K, Kamath RS, Fraser AG,Ahringer J, Plasterk RH, Tijsterman M: Identification of genesthat protect the C. elegans genome against mutations bygenome-wide RNAi. Genes Dev 2003, 17:443-448.
This study identified multiple genes in C. elegans that are required forgenomic stability, using an RNAi strategy. 61 genes were identified, manyof which were novel.
36.��
Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q,De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G et al.:Biallelic inactivation of BRCA2 in Fanconi anemia.Science 2002, 297:606-609.
Most of the genes responsible for the eight FA complementation groupshave been identified and belong to the same protein complex that directthe monoubiquitination of FA-D2. The resulting protein interacts withBRCA1, with subsequent signaling through BRCA2 and RAD51. Thisstudy identified mutations in the BRCA2 gene in FANCD1 and FANCBpatients.
10 Oncogenes and cell proliferation
Current Opinion in Genetics & Development 2004, 14:5–10 www.sciencedirect.com

Mechanisms of Recombination
19. Mechanisms of Recombination
Key Concepts
Recombination occurs at regions of homology between chromosomes through the breakage and reunion of DNA molecules.
Models for recombination, such as the Holliday model, involve the creation of a heteroduplex branch, or cross bridge, that can migrate and the subsequent splicing of the intermediate structure to yield different types of recombinant DNA molecules.
Recombination models can be applied to explain genetic crosses.
Many of the enzymes participating in recombination in bacteria have been identified.
Introduction
Throughout our analysis of linkage, we studied the recombination of genes by crossing-over. In this chapter, we consider molecular mechanisms for generating recombination by crossing-over. Figure 19-1 depicts a basic crossover event, in which two homologous molecules are aligned and subsequently undergo recombination. When Benzer's work and that of others revealed that recombination occurs within genes, it became evident that recombination had to be very precise, because even single-base-pair errors could disrupt the integrity of the gene. How can recognition of homologous chromosomes and recombination events be so precise? The answer lies in the power of base-pair complementarity. We shall see how base-pair complementarity and the formation of heteroduplex regions between complementary regions of homologous chromosomes lead to the recombination events that we have been studying.
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Figure 19-1. The molecular event of recombination may be schematically represented by two double-stranded molecules breaking and rejoining. Breakage and reunion of DNA molecules
The experiments discussed in Chapter 5 provide good indirect evidence in favor of breakage and reunion. One of the first direct proofs that chromosomes (in this case, viral chromosomes) can break and rejoin came from experiments on λ phage done in 1961 by Matthew Meselson and Jean Weigle. They simultaneously infected E. coli with two strains of λ. One strain, which had the genetic markers c and mi at one end of the chromosome, was “ heavy ” because the phages were produced from cells grown in heavy isotopes of carbon (13C) and nitrogen (15N). The other strain was c+14mi+ for the markers and had “light” DNA because it was harvested from cells grown on the normal light isotopes 12C and 14N. The two DNAs (chromosomes) can be represented as shown in Figure 19-2a. The multiply infected cells were then incubated in a light medium until they lysed.
The progeny phages released from the cells were spun in a cesium chloride density gradient. A wide band was obtained, indicating that the viral DNAs ranged in density from the heavy parental value to the light parental value, with a great many intermediate densities (Figure 19-2b). Interestingly, some recombinant phages were recovered with density values very close to the heavy parental value. They were of genotype c 14 mi+, and they must have arisen through an exchange event between the two markers (Figure 19-2c). The heavy density of the chromosome would be expected because only the small tip of the chromosome carrying the mi+ allele would come from the light parental chromosome. In the reciprocal cross of heavy c+ mi+ phages to light c mi, the heavy recombinants were found to be c+ mi, as expected. These results can be explained in only one way: the recombination event must have occurred through the physical breakage and reunion of DNA. Although we have to be
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
careful about extrapolating from viral to eukaryotic chromosomes, this evidence shows that the breakage and reunion of DNA strands does occur.
Figure 19-2. Evidence for chromosome breakage and reunion in λ phages. (a) The chromosomes of the two λ strains used to multiply infect E. coli. (b) Bands produced when progeny phages are spun in a cesium chloride density gradient. The fact that intermediate densities are obtained indicates a range of chromosome compositions with partly light and partly heavy components. (c) The chromosome of the heavy c 14 mi+ progeny resulting from crossover between the two markers. The density of this crossover product confirms that the crossover entailed a physical breakage and reunion of the DNA. Chiasmata: the crossover points
In Chapter 5, we made the simple assumption that chiasmata are the actual sites of crossovers. Mapping analysis indirectly supports this idea: because an average of one crossover per meiosis produces 50 genetic map units, there should be correlation between the size of the genetic map of a chromosome and the observed mean number of chiasmata per meiosis. The correlation has been made in well-mapped organisms.
However, the harlequin chromosome-staining technique (see Chapter 8) has made it possible to test the idea directly. In 1978, C. Tease and G. H. Jones prepared harlequin chromosomes in meioses of the locust. Remember that the harlequin technique produces sister chromatids: one dark and the other light. When a crossover occurs, it can be between two dark, two light, or nonsister dark and light chromatids, as shown in Figure 8-16. This last situation is crucial because mixed (part dark and part light) crossover chromatids are produced. Tease and Jones found that the dark – light transition is right at the chiasma—proving that the chiasmata are the crossover sites and settling a question that had been unresolved since the early 1900s (Figure 19-3).
Genetic results leading to recombination models
Tetrad analysis in filamentous fungi, such as Neurospora crassa, where all four products of a single meiosis can be recovered and examined (see Chapter 6), provided
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
the impetus for the first models of intragenic recombination. These crucial findings, reviewed in the following list, were gene conversion, evidence for postmeiotic segregation of gene-conversion events, polarity, and the association of gene conversion with crossing-over.
1. Gene conversion. Departures from the predicted Mendelian 4:4 segregation ratios are detectable in some asci (0.1–1.0 percent in filamentous fungi, but as high as 4 percent in yeast). Figure 19-4 gives the most common aberrant ratios obtained. It appears as though some alleles in the cross have been “converted” into the opposite alleles (Figure 19-5). The process therefore has become known as gene conversion; it can occur only where there is heterozygosity for two different alleles of a gene. In asci with a 6:2 or 2:6 ratio, one entire chromatid of a chromosome seems to have converted. In asci with a 5:3 or 3:5 ratio, only half a chromatid seems to have converted. Here, different members of a spore pair have different genotypes. Recall that each spore pair is produced by mitosis from a single product of meiosis. The 5:3 or 3:5 ratios can be explained only by the two strands of the double helix carrying information for twoM different alleles at the conclusion of meiosis. The next mitotic division is therefore a postmeiotic segregation of alleles.
Conversion cannot be explained by mutation, because the allele that is converted always changes into the other specific allele taking part in the cross, not to some other allele known for the locus but not a part of the cross.
Figure 19-4. Rare aberrant allele ratios observed in a cross of type + × m in fungi. (Ascus genotypes are represented here.) When the Mendelian ratio of 4:4 is not obtained, some of the alleles in the cross have been converted into the opposite alleles. In some asci, it appears that the entire chromatid has been converted (6:2 or 2:6 ratios). In others, it appears that only half-chromatids have been converted (5:3, 3:5, or
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
3:1:1:3 ratios).(c) Figure 19-5. Gene conversions are inferred from the patterns of alleles observed in asci. (a) In a chromatid conversion, the allele on one chromatid seems somehow to have been converted into an allele like those on the other chromatid pair. The converted allele is shown by the symbol . One spore pair is of the opposite genotype from that expected in Mendelian segregation. (b) In a half-chromatid conversion, one spore pair (*) has nonidentical alleles. Somehow, one chromatid seems to be “half converted,” giving rise to one spore of the original genotype and one spore converted into the other allele.
Figure 19-6. Diagram of chromatids taking part in a cross in regions I and II of a gene. The arrow indicates the polarity of gene conversion in the m locus, pointing toward the end with lower conversion frequency.
Figure 19-7. A specific ascus pattern can be explained by both crossover and a chromatid conversion. In this case, a conversion of m1 → ; + is accomplished by a
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
免疫学信息网 http://www.im
One of the first plausible models to account for the preceding observations was formulated by Robin Holliday. The key features of the Holliday model are the formation of heteroduplex DNA; the creation of a cross bridge; its migration along the two heteroduplex strands, termed branch migration; the occurrence of mismatch repair; and the subsequent resolution, or splicing, of the intermediate structure to yield different types of recombinant molecules. The model is depicted in Figure 19-9.
crossover in the region between a and m– 1
Figure 19-8. Sample ascus patterns obtained from a single conversion and co-conversions. Holliday model
Enzymatic cleavage and the creation of heteroduplex DNA
Looking at Figure 19-9a, we can see that two homologous double helices are aligned, although note that they have been rotated so that the bottom strand of the first helix has the same polarity as the top strand of the second helix (5′ → 3′ in this case). Then a nuclease cleaves the two strands that have the same polarity (Figure 19-9b). The free ends leave their original complementary strands and undergo hydrogen bonding with the complementary strands in the homologous double helix (Figure 19-9c). Ligation produces the structure shown in Figure 19-9d. This partially heteroduplex double helix is a crucial intermediate in recombination, and has been termed the Holliday structure.
Branch migration
The Holliday structure creates a cross bridge, or branch, that can move, or migrate, along the heteroduplex (Figure 19-9d and e). This phenomenon of branch migration is a distinctive property of the Holliday structure. Figure 19-10 portrays a more realistic view of this structure as it might appear during branch migration.
Resolution of the Holliday structure
muneweb.com

Mechanisms of Recombination
The Holliday structure can be resolved by cutting and ligating either the two originally exchanged strands (Figure 19-9f, left) or the originally unexchanged strands (Figure 19-9f, right). The former generates a pair of duplexes that are parental, except for a stretch in the middle containing one strand from each parent. If the two parents had different alleles in this stretch, as indicated here, then the DNA will be heteroduplex. The latter resolution step generates two duplexes that are recombinant, with a stretch of heteroduplex DNA. The Holliday model also postulated that the heteroduplex DNA mismatches can be repaired by an enzymatic correction system that recognizes mismatches and excises the mismatched base from one of the two strands, filling in the excised base with the correct complementary base. The resulting molecules will carry either the wild-type or the mutant allele, depending on which allele is excised.
Figure 19-11 demonstrates one way that we can easily visualize how the Holliday structure can be converted into the recombinant structures with which we are familiar. In Figure 19-11a, we can see the structure that we arrived at in Figure 19-4e drawn out in an extended form. Compare Figures 19-9e and 19-11a until you are convinced that these two structures are indeed equivalent. If we rotate the bottom part of this structure, as shown in Figure 19-11b, we can generate the form depicted in Figure 19-11c. This last form can be converted back into two unconnected double helices by enzymatically cleaving only two strands. As indicated in 19-11c, cleavage can occur in either of two ways, each of which generates a different product (Figure 19-11d). These cleaved structures can be viewed more simply (Figure 19-11e). Repair synthesis produces the final recombinant molecules (Figure 19-11f). Note the two different types of recombinants.
Application of the Holliday model to genetic crosses
The Holliday model nicely explained the phenomena that we described previously. Gene conversion and the aberrant ratios depicted in Figure 19-4 can result as a consequence of mismatch repair, as shown in Figure 19-12 and Table 19-1. In Table 19-1, the symbols + for wild type and m for mutant are used for simplicity. When both mismatches are corrected to yield the same parental type, then 6:2 or 2:6 ratios result; when only one heteroduplex is corrected, then a 5:3 ratio results; and, when there is no correction, an aberrant 4:4 ratio is the product.
The Holliday model also accounted for polarity of gene-conversion events, because conversion takes place only within the heteroduplex DNA between the break point and the branch point at which the Holliday structure is resolved. The farther a gene locus is from the breakage point, the more likely it is to be beyond the branch point and thus not part of the heteroduplex.
It should be noted that the phenomenon of gene conversion and its association with about half the cases of crossing-over was a driving force for the formulation of the Holliday model, which entails a strand exchange that results in reciprocal crossovers
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
almost half the time. This 50 percent reciprocal crossover result is because of the resolution of the exchange point in two equally likely ways, as seen in Figure 19-9f, one of which produces crossing-over of markers outside the region of heteroduplex DNA. Coconversion is explained by the location of both sites in the region of heteroduplex DNA and by the excision of both sites in the same excision-repair act. This double excision converts both sites into the same parental type.
Meselson-Radding model
As the data from tetrad analyses accumulated, it became clear that the Holliday model could not explain everything. For instance, the two mismatches resulting from the two heteroduplexes (see Figures 19-9e and 19-12) should be manifested in the progeny from a cross, yielding aberrant 4:4 tetrads. Yet, tetrad analyses in yeast and other organisms showed that, whereas 6:2 tetrads were frequent among gene-conversion events, aberrant 4:4 tetrads were very rare. It seemed as if gene conversion and the formation of heteroduplex DNA occurred primarily in only one chromatid. The model proposed by Meselson and Radding (shown in Figure 19-13) generates the Holliday structure with one single-strand cut in only one chromosome (Figure 19-13a), in contrast with the Holliday model, in which a nick is made in one strand in each of the two homologous chromatids. This single-strand cut is followed by DNA synthesis (Figure 19-13b). After the nick, the displaced single strand invades the second duplex (Figure 19-13c), generating a loop, which is excised (Figure 19-13d). After ligation to produce a Holliday structure, followed by branch migration (Figure 19-13e), a heteroduplex is generated in each chromosome. Resolution of this intermediate (Figure 19-13f) occurs exactly as depicted in Figure 19-9 (or after rotation, as in Figure 19-11). Note the lack of symmetry in the heteroduplex DNA at resolution in Figure 19-13f (left), compared with Figures 19-9f and 19-11f. Thus, in the Meselson-Radding model (left side), the bottom chromatid duplex (in Figure 19-13) has a heteroduplex region, instead of the two chromatids having heteroduplex regions, as in the Holliday model. However, branch migration and isomerization can generate a structure that has heteroduplex regions on both duplexes (Figure 19-13, right side), which is required to explain aberrant 4:4 ratios (Table 19-1).
Double-strand break-repair model for recombination
In the Holliday and Meselson-Radding models for genetic recombination, the initiation events for recombination are single-strand nicks that result in the generation of heteroduplex DNA. However, the finding that yeast transformation is stimulated 1000-fold when a double-strand break is introduced into a circular donor plasmid provided the impetus for an additional model, the double-strand-break model shown in Figure 19-14. Originally formulated by Jack Szostak, Terry Orr-Weaver, and Rodney Rothstein, this model invokes double-strand breaks to initiate recombination. The breaks are enlarged to gaps, and the repair of the double-stranded gap results in gene conversion. The key features of this model are diagrammed in the steps in Figure 19-14: (1) a double-strand break, followed by digestion of the 5′ end of both cut sites;
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
(2) the invasion by a remaining 3′ tail of the uncut other duplex; (3) the repair synthesis of one strand; (4) the repair synthesis of the other strand, and ligation to form two Holliday junctions; (5) resolution in one of two ways, one of which generates a reciprocal crossover; and (6) mismatch repair correction to yield gene conversion.
MESSAGE
The phenomenon of gene conversion led to the development of heteroduplex models to explain the mechanism of crossing-over. Mendelian (1:1) allele ratios are normally observed in crosses because it is only rarely that a heterozygous locus is the precise point of chromosome exchange. Asci showing gene conversion at a typical heterozygous locus are relatively rare (on the order of 1 percent).
Visualization of recombination intermediates
Several of the individual steps that constitute the Holliday model have been demonstrated to occur in vivo or in vitro, such as nicking, strand displacement, branch migration, repair synthesis, and ligation. H. Potter and D. Dressler showed that DNA intermediates of the type predicted by the Holliday model can be found in recombining phages or plasmids. Figure 19-15 shows an electron micrograph of a recombinant molecule. It is formally equivalent to the central pair of DNA double helices shown in Figure 19-11, with two arms rotated to produce the central single-stranded “dia-mond,” as in Figure 19-11c, which runs between the sections of double-stranded DNA.
免疫学信息网 http://www.immuneweb.com
Mechanisms of Recombination
Figure 19-9. A prototype mechanism for genetic recombination. (a) Two homologous double helices are shown. Each pair represents a chromatid and the two pairs represent two nonsister chromatids. The helices are aligned so that the bottom strand of the first helix has the same polarity as the top strand of the second helix. (b) Two parallel or two antiparallel strands are cut. (c) The free ends become associated with the complementary strands in the homologous double helix. (d) Ligation creates partly heteroduplex double helices, the Holliday structure. (e) Migration of the branch point is by continued strand transfer by the two polynucleotide chains taking part in the
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
crossover. (f) Resolution can occur in one of two ways, which will be described in detail later. (After H. Potter and D. Dressler, Cold Spring Harbor Symposium on Quantitative Biology 43, 1979, 970. Cold Spring Harbor Laboratory, Cold Spring
Harbor, NY.)
Figure 19-10. Branch migration, the movement of the crossover point between DNA complexes. (After T. Broker, Journal of Molecular Biology 81, 1973, 1; from J. D. Watson et al., Molecular Biology of the Gene, 4th ed. Copyright © 1987 by Benjamin
免疫学信息网 http://www.immuneweb.com
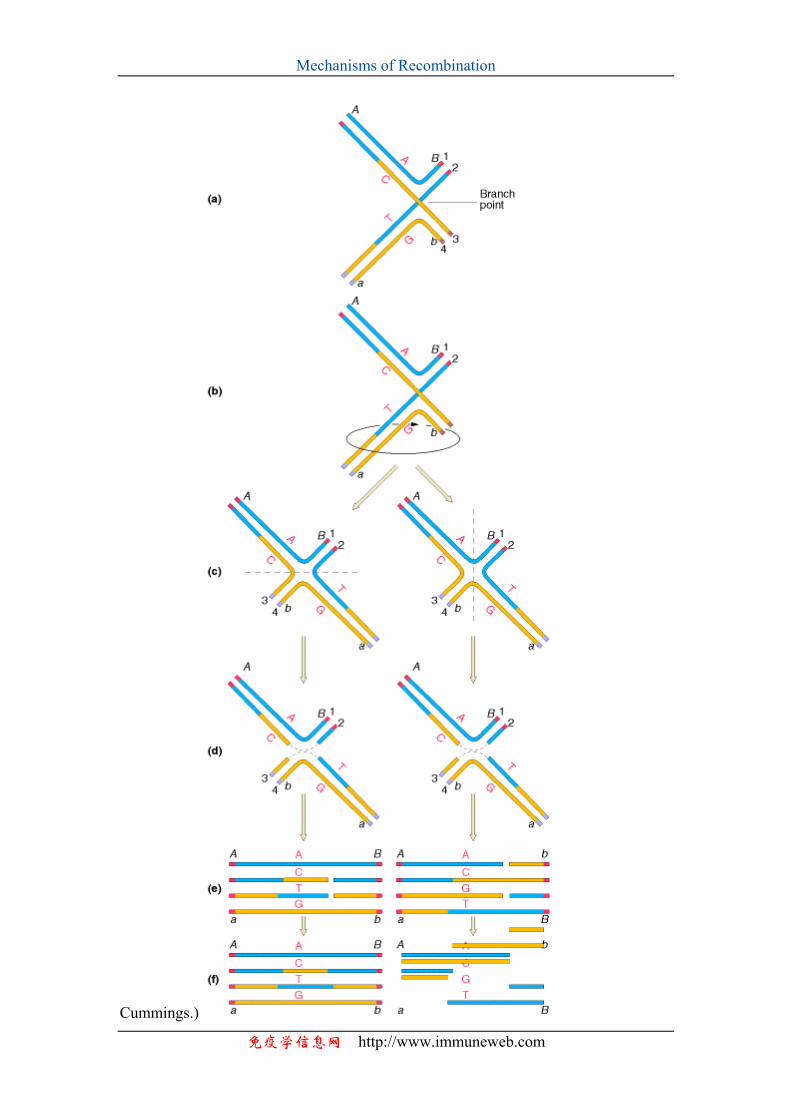
Mechanisms of Recombination
Cummings.)
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Figure 19-11. (a) The Holliday structure shown in an extended form. (b) The rotation of the structure shown in part a can yield the form depicted in part c. Resolution of the structure shown in part c can proceed in two ways, depending on the points of enzymatic cleavage, yielding the structures shown in part d. The dotted lines show which segments will rejoin to form recombinant strands for each particular cleavage scheme. The strands are shown linearly in part e and can be repaired to the forms shown in part f. (From H. Potter and D. Dressler, Cold Spring Harbor Symposium on Quantitative Biology 43, 1970, 970. Cold Spring Harbor Laboratory, Cold Spring
Harbor, NY.) Figure 19-12. When a mismatch is generated within a heteroduplex region, mismatch repair converts the mismatch into either the wild-type or the mutant sequence.
Genetic Consequences of the Meselson-Radding Model.
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Figure 19-13. The Meselson-Radding heteroduplex model. (a) A duplex is cut on one chain. (b) DNA polymerase displaces one chain. (c) The resulting single chain displaces its counterpart in the homolog. (d) This displaced chain is enzymatically digested. (e) Ligation completes the formation of a Holliday junction, which is genetically asymmetric in that only one of the two duplexes has a region of potentially heteroduplex DNA. If the junction migrates, heteroduplex DNA can arise on both duplexes. (f) Resolution of the junction occurs as in the Holliday model. (From F. W. Stahl, “The Holliday Junction on Its Thirtieth Anniversary,” Genetics 138, 1994, 241–246.)
Messelson-Radding Heteroduplex Model.
免疫学信息网 http://www.immuneweb.com
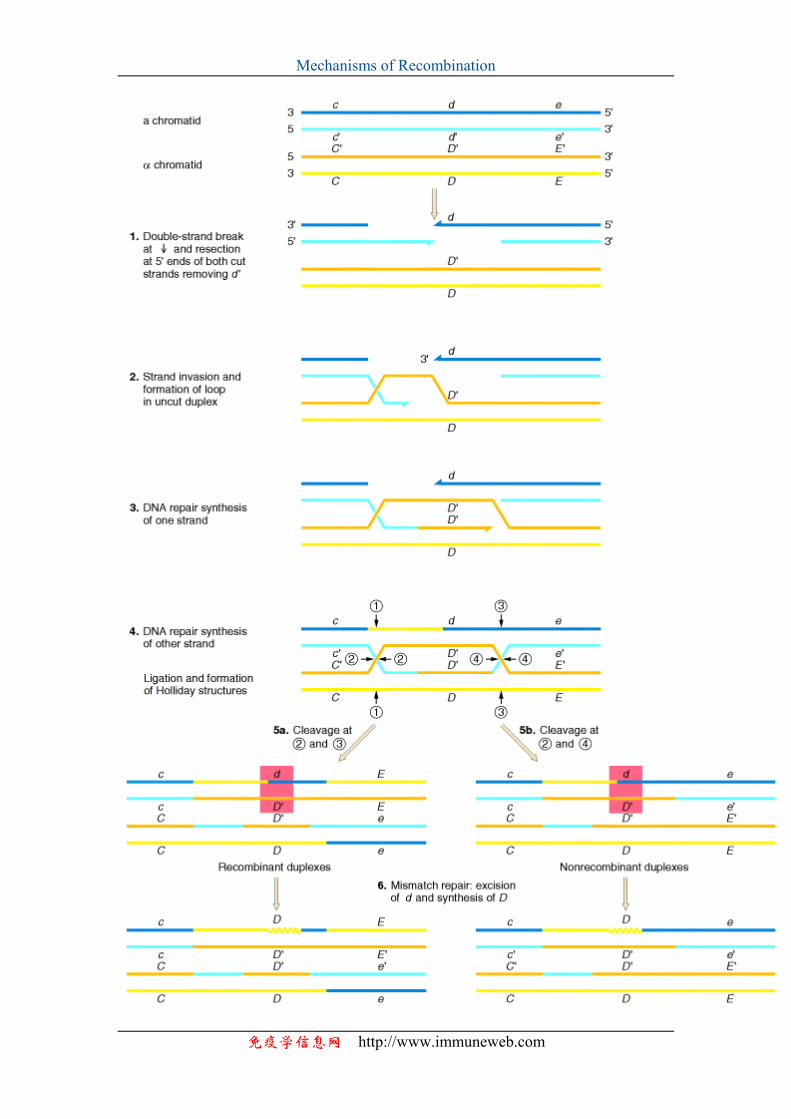
Mechanisms of Recombination
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Figure 19-14. Double-strand-break model of meiotic recombination in the yeast S. cerevisiae. The two strands of each of two chromatids (in blue and yellow) are shown. The darker and lighter colors indicate complementary strands, as do the matching primes (for example, C and C′ or c and c′ for the allele designations). A key point of this model is shown in steps 3 and 4, which picture the generation of a Holliday structure with two crossover points. Two different ways of resolving this structure lead to the recombinant or nonrecombinant duplexes seen in step 5a and b. The red patch shows the heteroduplex mismatched regions, for the alleles d and D′. Mismatch repair results in the “conversion” of d into D. (See Table 19-1 for the possible segregation patterns resulting from mismatch repair.) (From H. Lodish, D. Baltimore, A. Berk, S. L. Zipursky, P. Matsudaira, and J. Darnell, Molecular Cell Biology, 3d ed. Copyright © 1995 by Scientific American Books.) Enzymatic mechanism of recombination
By the isolation of mutants defective in some stage of recombination, much light has been shed on the enzymology of recombination. In E. coli, the products of several genes involved in general recombination—the recA, recB, recC, and recD genes—have been well characterized, as has the single-strand-DNA-binding (Ssb) protein. Mutants deficient in any of these proteins have reduced levels of recombination. In fact, three distinct recombination pathways have been identified. In addition to the major RecBCD pathway, two minor pathways, RecF and RecE, are activated in certain situations. All three pathways use the RecA protein.
Production of single-stranded DNA
An initial step in recombination is probably the nicking and unwinding of a DNA duplex by a protein complex consisting of the RecB, RecC, and RecD proteins. The complex has both helicase and nuclease activity. Figure 19-16 shows how this complex unwinds the DNA, driven by the hydrolysis of ATP as it moves and generates single strands from a duplex molecule. The nuclease activity recognizes an 8-bp sequence:
called a chi site; these chi sites appear approximately every 64 kb. As the complex unwinds the DNA, the free single-stranded DNA can be used to initiate recombination. The Ssb protein, which also takes part in DNA replication (Chapter 8), can bind to and stabilize the single strands. RecA-protein-mediated single-strand exchange
The RecA protein, which also plays a role in the induction of the SOS repair system (see Chapter 16), can bind to single strands along their length, forming a
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
nucleoprotein filament. RecA catalyzes single-strand invasion of a duplex and subsequent displacement of the corresponding strand from the duplex. The invasion and displacement take place in the presence of ATP, as shown in Figure 19-17. The displaced strand forms what is termed a D loop. Figure 19-18 depicts how this sequence can lead to Holliday junction.
Branch migration
The movement of a Holliday junction (see, for instance, Figures 19-9 and 19-10), or branch migration, increases the length of heteroduplex DNA. The RuvA and RuvB proteins catalyze branch migration, driving the reaction by the hydrolysis of ATP. The RuvA protein binds to the crossover point and then is flanked by two RuvB ATPase hexameric rings, as seen by electron microscopy. Figure 19-19 depicts a model for the action of these proteins in branch migration.
Resolution of Holliday junctions
Several enzymatic pathways have been identified that cleave across the point of strand exchange in a Holliday structure to yield two duplexes. RuvC is an endonuclease that resolves Holliday junctions by symmetric cleavage of the continuous (noncrossing) pair of DNA strands, as seen in Figure 19-20. In addition, the RecG and Rus proteins may provide alternative routes to cleavage. These reactions are summarized in Figure 19-21.
Figure 19-16. Model for the generation of single-stranded DNA by the RecBCD complex (purple), an ATP-driven helicase and nuclease. Two single-stranded loops are formed as the complex moves forward. The enzyme cleaves one of the strands
免疫学信息网 http://www.immuneweb.com
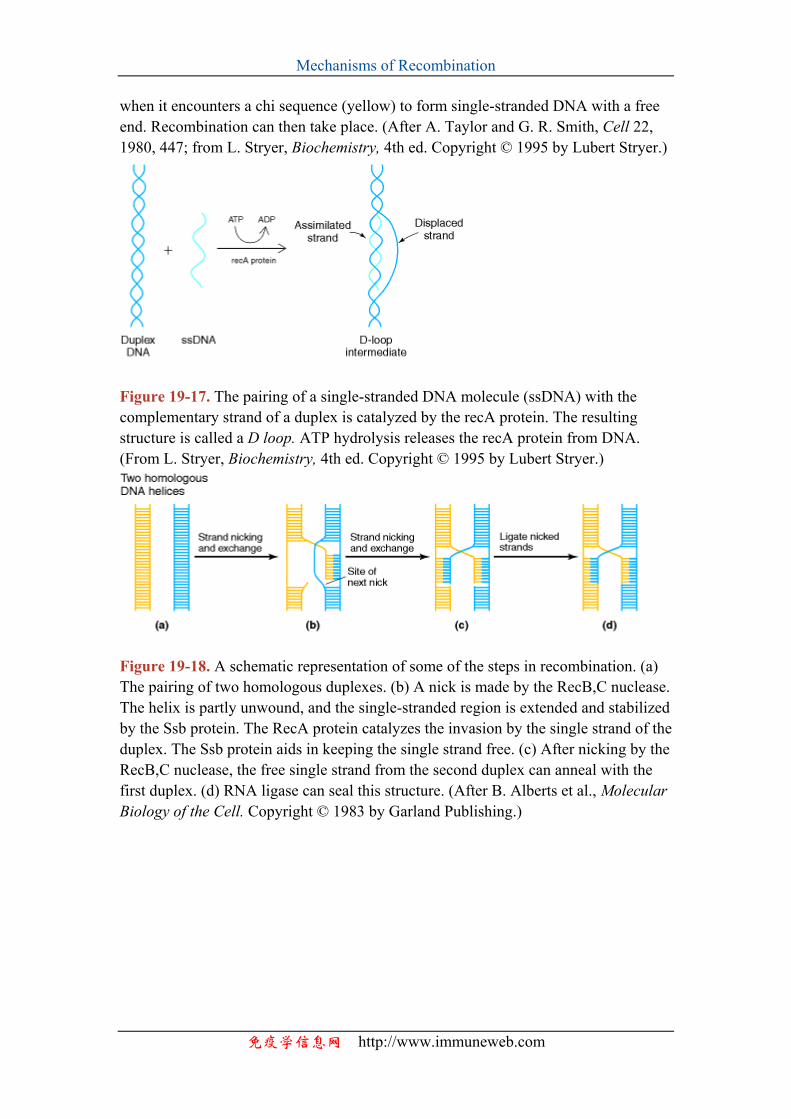
Mechanisms of Recombination
when it encounters a chi sequence (yellow) to form single-stranded DNA with a free end. Recombination can then take place. (After A. Taylor and G. R. Smith, Cell 22, 1980, 447; from L. Stryer, Biochemistry, 4th ed. Copyright © 1995 by Lubert Stryer.)
Figure 19-17. The pairing of a single-stranded DNA molecule (ssDNA) with the complementary strand of a duplex is catalyzed by the recA protein. The resulting structure is called a D loop. ATP hydrolysis releases the recA protein from DNA. (From L. Stryer, Biochemistry, 4th ed. Copyright © 1995 by Lubert Stryer.)
Figure 19-18. A schematic representation of some of the steps in recombination. (a) The pairing of two homologous duplexes. (b) A nick is made by the RecB,C nuclease. The helix is partly unwound, and the single-stranded region is extended and stabilized by the Ssb protein. The RecA protein catalyzes the invasion by the single strand of the duplex. The Ssb protein aids in keeping the single strand free. (c) After nicking by the RecB,C nuclease, the free single strand from the second duplex can anneal with the first duplex. (d) RNA ligase can seal this structure. (After B. Alberts et al., Molecular Biology of the Cell. Copyright © 1983 by Garland Publishing.)
免疫学信息网 http://www.immuneweb.com
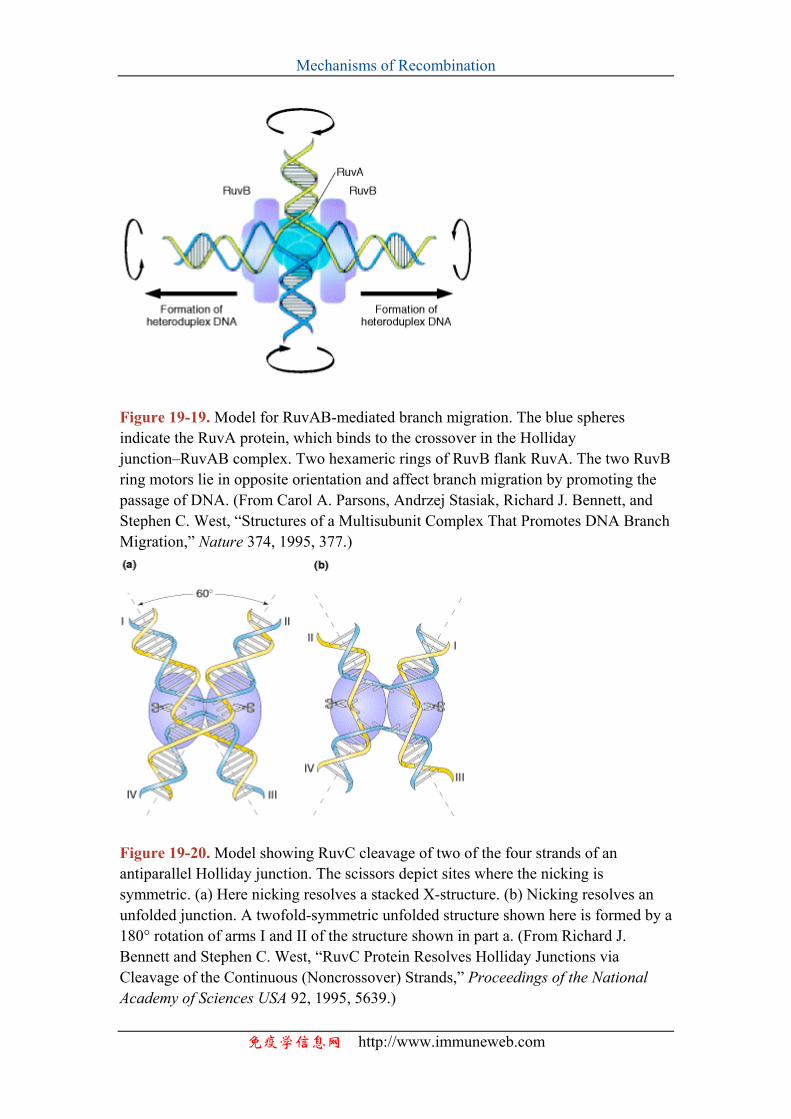
Mechanisms of Recombination
Figure 19-19. Model for RuvAB-mediated branch migration. The blue spheres indicate the RuvA protein, which binds to the crossover in the Holliday junction–RuvAB complex. Two hexameric rings of RuvB flank RuvA. The two RuvB ring motors lie in opposite orientation and affect branch migration by promoting the passage of DNA. (From Carol A. Parsons, Andrzej Stasiak, Richard J. Bennett, and Stephen C. West, “Structures of a Multisubunit Complex That Promotes DNA Branch Migration,” Nature 374, 1995, 377.)
Figure 19-20. Model showing RuvC cleavage of two of the four strands of an antiparallel Holliday junction. The scissors depict sites where the nicking is symmetric. (a) Here nicking resolves a stacked X-structure. (b) Nicking resolves an unfolded junction. A twofold-symmetric unfolded structure shown here is formed by a 180° rotation of arms I and II of the structure shown in part a. (From Richard J. Bennett and Stephen C. West, “RuvC Protein Resolves Holliday Junctions via Cleavage of the Continuous (Noncrossover) Strands,” Proceedings of the National Academy of Sciences USA 92, 1995, 5639.)
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Figure 19-21. Possible pathways for processing Holliday junction in E. coli. The center of the figure shows a Holliday junction formed by homologous pairing and strand exchange by the RecA protein. In the presence of RecA, branch migration is in the same direction as the RecA strand exchange and is promoted by RuvAB. RuvC cleavage resolves this structure (upper right). Also in the presence of RecA, RecG could drive the Holliday junction backward (upper right). However, if RecA dissociates from the junction, RecG could drive the reaction forward, with resolution coming from the Rus protein (lower left). The lower right shows the results of Rus cleavage alone, after strand exchange and branch migration by RecA. (From Gary Sharples, Sau Chan, Akeel Mahdi, Matthew Whitby, and Robert Lloyd, “Processing of Intermediates in Recombination and DNA Repair: Identification of a New Endonuclease That Specifically Cleaves Holliday Junctions,” EMBO Journal 13, 1994, 6140.) Summary
We have gained increasing knowledge of the molecular processes behind recombination, which produces new gene combinations by exchanging homologous chromosomes. Both genetic and physical evidence has led to several models of recombination that rely on common features: hetero-duplex DNA, mismatch repair, and resolution, or splicing, of the intermediate structure to yield recombinant molecules. The process of recombination itself is under genetic control, and numerous genes that affect the process have been identified.
Concept Map
Draw a concept map interrelating as many of the following terms as possible. Note that the terms are listed in no particular order.
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Chapter Integration Problem
In previous chapters, we learned about different mechanisms of generating mutations, both spontaneously and with mutagens. Describe gene conversion and how you can distinguish gene conversion from mutation. See answer
Solution
Gene conversion is a meiotic process of directed change in which one allele directs the conversion of a partner allele into its own form. Instead of ending with equal numbers of both alleles in meiosis, gene conversion results in an excess of one allele and a deficiency of the other. Models of recombination employing heteroduplex DNA offer plausible mechanisms for gene conversion. In contrast, mutation is undirected change that can occur in both meiosis and mitosis.
Solved Problems
1. In Neurospora, an ad3 double mutant consisted of two mutant sites within the ad3 gene—site 1 on the left and site 2 on the right. This mutant was crossed to wild type, with the use of parental stocks heterozygous for two closely linked flanking loci, A and B, as follows, where 1 represents wild-type sequence at the mutant positions:
Most asci were of the expected type showing regular Mendelian segregations, but there were also some unexpected types, of which several examples are represented here as I through III.
Explain the likely origin of the rare types I through III according to molecular recombination models. See answer
Solution
Ascus type I has two nonidentical sister spore pairs. Because the members of a spore
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
pair are derived from a postmeiotic mitosis (see Chapter 6), this proves that the meiotic products in these cases must have contained both 1 2 and + + information; in other words, they must have contained heteroduplex DNA. Notice also that these spore pairs are recombinant for A and B. Therefore it is likely that a crossover occurred between the A and B loci, that the heteroduplex DNA that constituted the crossover spanned both the ad3 mutant sites 1 and 2, and that there was no correction of the heteroduplex at those sites.
Type II shows a 5:3 ratio of 1 2 doubles to + +. Here again the heteroduplexes must have spanned both sites (in a noncrossover configuration), but this time there was correction of a + +/1 2 heteroduplex to 1 2/1 2, presumably by excision and repair of the + + information (co- or double conversion).
Type III reveals another noncrossover heteroduplex configuration, and this time correction occurred only at site 1; this is revealed as a 5:3 ratio for site 1, with 1 → +conversion in ascospore 5. See question
2. In fungal crosses of the following general type,
where 1 and 2 are mutant sites of a nutritional gene and M and N are flanking loci, it is possible to select rare + + prototrophic recombinants by plating on minimal medium. These prototrophs are then examined for the alleles of the flanking loci. As might be expected, the combination
is commonly encountered, but so, somewhat surprisingly, are
and
a. Explain the origin of these two genotypes in relation to molecular recombination models. b. If M + + N were more common than m + + n, what would that mean? See answer
Solution
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
a. It is likely that M + + N arose from a noncrossover heteroduplex that spanned site 2, followed by a correction of 2 → +.
Similarly, m + + n would be explained by a heteroduplex spanning site 1, and corrected 1 → +. b. Because M + + N arises from gene conversion at the right-hand site, it is likely that heteroduplex DNA is formed more commonly from the right than from the left, possibly because of a closer fixed break point. (Note: Prototrophs may also be formed by single-site correction in a heteroduplex spanning both sites, but then the inequality would require another explanation.)
Problems
1. Which of the following linear asci shows gene conversion at the arg2 locus?
See answer 3, 4, 6
2. At the light-spore locus of Ascobolus, the 1′ mutant site is in the left part of the gene and the 1″ mutant site is more to the right. When crosses are made between 1′- and 1″-bearing strains,
asci with six light and two black spores can be selected visually. They are shown to be caused by gene conversion mostly of the following type:
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
What can account for this irregularity?
3. It has been proposed that the “fixed break point” of the heteroduplex recombination model might correspond to a promoter sequence. The following data relate to this idea. In the fungus Podospora, mutants were available in adjacent spore color genes 1 through 4, and a cross was made as follows:
(The numbers 261, 136, 42, and 115 are merely names for mutant sites.) Many asci were obtained showing gene conversion, but one that was relevant to the preceding suggestion was as follows:
Interpret this ascus in relation to the promoter idea.
4. Many mutagens increase the frequency of sisterchromatid exchange. Give possible explanations for this observation.
5. Mutations in locus 46 of the Ascomycete fungus Ascobolus produce light-colored ascospores (let's call them a mutants). In the following crosses between different a mutants, asci are observed for the appearance of dark wild-type spores. In each cross, all such asci were of the genotypes indicated:
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
Interpret these results in light of the models discussed in this chapter.
See answer First, notice that gene conversion has occurred. In the first cross, a1 converted (1 : 3). In the second cross, a3 converted. In the third cross, a3 converted. Polarity obviously played a part. The results can be explained by the following map, in which hybrid DNA enters only from the left.
6. In the cross A m1 m2 B × a m3 b, the order of the mutant sites m1, m2, and m3 is unknown in relation to one another and to A/a and B/b. One nonlinear conversion ascus is obtained:
Interpret this result in light of the heteroduplex DNA theory, and derive as much information as possible about the order of the sites.
7. In the cross a1 × a2 (alleles of one locus) the following ascus is obtained:
Deduce what events may have produced this ascus (at the molecular level). See answer The ratios for a1 and a2 are both 3 : 1. There is no evidence of polarity, which indicates that gene conversion as part of recombination occurred. The best explanation is that two separate excision-repair events took place and, in both cases, the repair retained the mutant rather than the wild type.
8. G. Leblon and J.-L. Rossignol made the following observations in Ascobolus. Single-nucleotide-pair insertion or deletion mutations show gene conversions of the 6:2 or 2:6 type and only rarely of the 5:3, 3:5, or 3:1:1:3 type. Base-pair transition mutations show gene conversion of the 3:5, 5:3, or 3:1:1:3 type and only rarely of the 6:2 or 2:6 type.
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
a. In relation to the hybrid DNA model, propose an explanation for these observations. b. Leblon and Rossignol also showed that there are far fewer 6:2 than 2:6 conversions for insertions and far more 6:2 than 2:6 conversions for deletions (where the ratios are +:m). Explain these results in relation to heteroduplex DNA. (You might also think about the excision of thymine photodimers.) c. Finally, the researchers showed that, when a frameshift mutation is combined in a meiosis with a transition mutation at the same locus in a cis configuration, the asci showing joint conversion are all 6:2 or 2:6 for both sites (that is, the frameshift conversion pattern seems to have “imposed its will” on the transition site). Propose an explanation for this result. See answer a. and b. A heteroduplex that contains an unequal number of bases in the two strands has a larger distortion than does a simple mismatch. Therefore, the former would be more likely to be repaired. For such a case, both heteroduplex molecules are repaired (leading to 6 : 2 and 2 : 6) more often than one (leading to 5 : 3 or 3 : 5) or none (leading to 3 : 1 : 1 : 3). The preference in direction (that is, the addition rather than the subtraction of a base) is analogous to thymine dimer repair. In thymine dimer repair, the unpaired, bulged nucleotides are treated as correct and the strand with the thymine dimer is excised.
A mismatch more often than not escapes repair, leading to a 3 : 1 : 1 : 3 ascus.
Transition mutations would not cause as large a distortion of the helix, and each strand of the heteroduplex should have an equal chance of repair. This would lead to 4 : 4 (two repairs each in the opposite direction), 5 : 3 (one repair), 3 : 1 : 1 : 3 (no repairs or two repairs in opposite directions), and, less frequently, 6 : 2 (two repairs in the same direction). c. Because excision repair excises the strand opposite the larger buckle (that is, opposite the frameshift mutation), the cis transition mutation also is retained. The nearby genes are converted because of the length of the excision repair.
9. At the gray locus in the Ascomycete fungus Sordaria, the cross + × g1 is made. In this cross, heteroduplex DNA sometimes extends across the site of heterozygosity, and two heteroduplex DNA molecules are formed (as discussed in this chapter). However, correction of heteroduplex DNA is not 100 percent efficient. In fact, 30 percent of all heteroduplex DNA is not corrected at all, whereas 50 percent is corrected to + and 20 percent is corrected to g1. What proportion of aberrant-ratio asci will be (a) 6:2? (b) 2:6? (c) 3:1:1:3? (d) 5:3? (e) 3:5? See answer (a). 6 : 2 = 31.25 percent; (b). 2 : 6 = 5 percent; (c). 3 : 1 : 1 : 3 = 11.25 percent; (d). 5 : 3 = 37.5 percent; (e). 3 : 5 = 15 percent.
免疫学信息网 http://www.immuneweb.com

Mechanisms of Recombination
10. Noreen Murray crossed α and β, two alleles of the me-2 locus in Neurospora. Included in the cross were two markers, trp and pan, which each flank me-2 at a distance of 5 m.u. The ascospores were plated onto a medium containing tryptophan and pantothenate but no methionine. The methionine prototrophs that grew were isolated and scored for the flanking markers, yielding the results shown in the table below.
Interpret these results in light of the models presented in this chapter. Be sure to account for the asymmetries in the classes.
11. In Neurospora, the cross A x × a y is made, in which x and y are alleles of the his-1 locus and A and a are mating-type alleles. The recombinant frequency between the his-1 alleles is measured by the prototroph frequency when ascospores are plated on a medium lacking histidine; the recombinant frequency is measured as 10−5. Progeny of parental genotype are backcrossed to the parents, with the following results. All a y progeny backcrossed to the A x parent show prototroph frequencies of 10−5. When A x progeny are backcrossed to the a y parent, two prototroph frequencies are obtained: half of the crosses show 10−5, but the other half show the much higher frequency of 10−2. Propose an explanation for these results, and describe a research program to test your hypothesis. (Note: Intragenic recombination is a meiotic function that occurs in a diploid cell. Thus, even though this organism is haploid, dominance and recessiveness could have roles in this problem.)
免疫学信息网 http://www.immuneweb.com