Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY
Prrnted in U.S.A. Vol. 258, No. 1, Issue of January 10, pp. 396-407, 1983
'13Cd Nuclear Magnetic Resonance of Cd(I1) Alkaline Phosphatases"
(Received for publication, March 18, 1982)
Peter Gettins and Joseph E. Coleman From the Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06510
'I3Cd NMR spectra of 113Cd(II)-substituted Esche- richia coli alkaline phosphatase have been recorded over a range of pH values, levels of metal site occu- pancy, and states of phosphorylation. Under all condi- tions resonances attributable to cadmium specifically bound at one or more of the three pairs of metal-binding sites (A, B, and C sites) are detected. By following changes in both the '13Cd and 31P NMR spectra of '13Cd(II)z alkaline phosphatase during and after phos- phorylation, it has been possible to assign the cadmium resonance that occurs between 140 and 170 ppm to Cd(I1) bound to the A or catalytic site of the enzyme and the resonance occurring between 51 and 76 ppm to Cd(I1) bound to B site, which from x-ray data is located 3.9 A from the A site. The kinetics of phosphorylation show that cadmium migration from the A site of one subunit to the B site of the second subunit follows and is a consequence of phosphate binding, thus precluding the migration as a sufficient explanation for half-of- the-sites reactivity. Rather, there is evidence for sub- unit-subunit interaction rendering the phosphate bind- ing sites inequivalent. Although one metal ion, at A site, is sufficient for phosphate binding and phospho- rylation, the presence of a second metal ion at B site greatly enhances the rate of phosphorylation. In the absence of phosphate, occupation of the lower affinity B and C sites produces exchange broadening of the cadmium resonances. Phosphorylation abolishes this exchange modulation. Magnesium at high concentra- tion broadens the resonances to the point of undetect- ability. The chemical shift of l13Cd(II) in both A and B sites (but not C site) is different depending on the state of the bound phosphate (whether covalently or nonco- valently bound) and gives separate resonances for each form. Care must be taken in attributing the initial distribution of cadmium or phosphate in the reconsti- tuted enzyme to that of the equilibrium species in sam- ples reconstituted from apoenzyme. Both 'I3Cd NMR and 31P NMR show that some conformational changes consequent to metal ion or phosphate binding require several days before the final equilibrium species is formed.
The definitive interpretation of much of the NMR data on the dimeric zinc metalloenzyme, alkaline phosphatase from Escherichia coli, requires a detailed knowledge of the ther-
* This work was supported by Grant AM09070 from the National Institutes of Health and Grant PCM 7682231 from the National Science Foundation. The 200 MHz NMR facility is partially sup- ported by Grants PCM 77-18941 and CHE 79-16210 from the National Science Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
modynamic and kinetic aspects of metal ion binding to the apoenzyme as reported in the preceding paper (1). Sample preparation is, however, greatly complicated by interactions between three closely spaced metal ion binding sites on each monomer. The binding affinities and, hence, the relative sta- bility constants for each site are altered by binding of metal ions to the other two sites. Binding of more than one metal ion to each monomer is, thus, cooperative and multiple con- formational equilibria, some of them slow, appear to distin- guish the apoenzyme from various forms of the metalloenzyme and its phosphorylated derivatives. This may in part account for reported variations in the number of metal ions per dimer required to restore full activity to the metal-free apoenzyme (2-8).
In our fiist report of cadmium-113 NMR studies on alkaline phosphatase (9) the addition of 2 Il3Cd(II) ions per dimer at the relatively low pH 6.5 was shown to result in the generation of a single I1"Cd resonance corresponding to all the "3Cd(II) bound to the protein. This was interpreted as showing that both metal ions are bound to identical sites, one in each monomer of a symmetrical dimer. These sites were designated as type A to correspond to the two identical sites of highest occupancy observed in the crystal structure of the cadmium enzyme (10). In contrast, the phosphoseryl derivative of the 113Cd(II)2' enzyme showed two ""Cd resonances of equal in- tensity, one close, but not identical, to the position of the original A site resonance and a second 90 ppm to higher field (11). The original interpretation was that formation of the phosphorylated enzyme produces a change in the conforma- tion of the phosphorylated subunit which is propagated to the second subunit such that both differ in environment and chemical shift from the original symmetrical pair of A sites. A re-evaluation of these llJCd NMR data together with comple- mentary and '"C data (12, 13) led to an alternative expla- nation, uiz. that as a consequence of phosphorylation the Il3Cd(II) ion migrates from the A site of one monomer to a second or B site (corresponding to the new more upfield '13Cd resonance) on the other monomer, thereby leaving one mono- mer devoid of metal ions and the other phosphorylated mono- mer containing '"Cd(I1) ions at both A and B sites. It was then suggested that such a migration might be the reason for "half-of-the. sites" reactivity, since phosphorylation may re- quire two metal ions, A and B, at each active centre (12). For such a model to be valid cadmium migration would have to precede or coincide with phosphorylation. Data to be pre- sented here show that, on the contrary, phosphorylation OC-
curs prior to migration and stimulates migration by increasing
' The abbreviations used in this manuscript are: Me(II)*AP, Me(II)4AP, or Me(II)2Mg(II)2AP refer to alkaline phosphatase con- taining metal ions at A sites alone, the same metal at A and B sites or Me(I1) at A sites and Mg(I1) at B sites. Me(I1) = Zn(II), or Cd(1I). E-P is the phosphoryl enzyme while E . P refers to the noncovalent complex with inorganic phosphate.
396

'I3Cd NMR of Alkaline Phosphatase 397
the affinity of the vacant B site for cadmium. The B site is probably the site shown by X-ray crystallography to be within 3.9 of the A site.2
Our initial work indicated that as the pH of the cadmium enzyme is raised much above 6.5 it becomes difficult or impossible to observe NMR resonances from bound "'Cd. Hence, all previous "'Cd NMR on alkaline phosphatase has been reported at pH 6.5 (11, 13, 14). It is now apparent that failure to observe resonances at higher pH values has been due to either broad resonances, splitting of resonances into pairs or clusters, or to the presence of inhomogeneous site occupancy at cadmium:enzyme stoichiometries of less than 6 per dimer.
The ""Cd NMR spectra of "'Cd(II)-substituted alkaline phosphatase containing 2, 4, and 6 metal ions have now been observed from pH 6 to 10, the range over which the major phosphoenzyme species present at equilibrium shifts from the covalent phosphoseryl form (E-P) to the noncovalent complex (E. P). This reveals much about metal ion site occupancy, the dynamics of phosphorylation, and the 'I%d chemical shifts characteristic of the various forms of the enzyme.
MATERIALS AND METHODS
Enzyme Preparation-Alkaline phosphatase was isolated from E. coli (strain CW3747) by osmotic shock and purified by heat treatment, ammonium sulfate precipitation, and gel chromatography using Seph- adex G-100 superfine (2). Apoenzyme was prepared by dialysis of sample (IO mg/ml) against 2 liters of 2 M (NHa)zSOd (enzyme grade, Schwartz/Mann), pH 9.0, 4 "C for 24 h. Two changes of dialysate were made. Ammonium sulfate was removed by dialysis against 2 liters of 0.01 M Tris-HC1,O.Ol M sodium acetate, 0.1 M NaCl at pH 6.5 or pH 9 with two changes of dialysate. Enzyme concentrations were determined spectrophotometrically at 278 nm using E'?::= 0.72 (15) and a molecular weight for the dimer of 94,000 calculated from the amino acid sequence (16). Enzyme activity was measured by hydrol- ysis of p-nitrophenyl phosphate (Sigma) in 1 M Tris-HC1, pH 8 at 22 "C (17). The native enzyme had a specific activity of 2500 f 500 pmol of substrate hydrolyzed/h/mg of protein. Buffer solutions con- tained 0.01 M Tris-HC1, 0.01 M acetate 0.1 M NaCl and were made metal-free as described previously (9). Concentration of enzyme sam- ples was carried out at 4 "C in a metal-free Amicon ultrafiltration cell using a PM-30 membrane. The desired metal ion compositions were achieved by addition of stoichiometric amounts of stock solutions of 112Cd(CH3C00)2 or Ii3Cd(CH3COO),, the latter prepared from the 96% isotopically enriched oxide (Oak Ridge National Laboratory). Metal ion concentrations of reconstituted metalloenzyme were checked by atomic absorption spectroscopy as described in the pre- vious paper (1).
NMR Studies--"'CCd NMR spectra were recorded both at 19.96 MHz, using an extensively modified Bruker HFX 90 spectrometer (18). and at 44.37 MHz on a Bruker CXP-200 spectrometer equipped with a broad-band tunable probe. Spectra were recorded at 293 K with sweep widths of 5000 Hz and 10,000 Hz at the lower and higher frequencies, respectively. Samples of 1-2 ml were used in 10-mm NMR tubes fitted with vortex plugs and a co-axial capillary containing D20 for the external field-frequency lock. Proton decoupling was not employed because of the negative NOE of "'Cd with a long correlation time. Chemical shifts are reported on the 6 scale relative to 0.1 M
' Analysis of the electron density map obtained from the Cd(II)2 phosphoryl-enzyme using anomalous dispersion shows that the metal binding site originally designated A in the crystal structure (10) consists of two separate cadmium sites 3.9 A apart (A and Az) (J. M. Sowadski, M. Handschumacher, K. Murthy, and €3. W. Wyckoff, (1982) Nature (Lond.), submitted for publication). These probably correspond to the spectroscopic A and B sites. The site originally labeled B in the crystal structure appears to consist exclusively of carboxylate ligands and probably corresponds to the site which gives rise to the ""Cd resonance at 2 ppm and which has been designated C in the spectroscopic work. The upfield chemical shift of C site ""Cd(I1) also suggests carboxylate ligands to the metal. The new site Az, much closer to A and close to the phosphoseryl residue, is likely to correspond to the spectroscopic B site; the site that is stabilized by the formation of the phosphoenzyme.
100 - /*\
/ \ / \
(I) z iLI
t
9 50
-
3
: / / / / ( ~~~~~ \ \ I i - 4 U
/ 0
0 I I , x 10 20 3.0 4-0
'"CdlAP DIMER
FIG. 1. Variation in amplitude of the 113Cd resonance (169 ppm) of Cd(I1)-substituted alkaline phosphatase (3.0 m, pH 6.5) as a function of 'I3Cd(II) stoichiometry. 0 represents the integrated area of the resonance. Spectra were recorded at 19.96 MHz and are the average of 46,000 transients.
( (1 )
J 113CdlI112
J I 1 1
200 150 100 50 0 w m
FIG. 2. '13Cd NMR spectra (19.96 MHz) of unphosphorylated cadmium alkaline phosphatase species. a, "3Cd(II)2AP (2.50 mM) at pH 6.30 (120,000 scans); b, i1JCd(II)4AP (2.72 mM) at pH 6.00 (60,000 scans); c, ""Cd(1I)bAP (2.69 mM) at pH 6.05 (64,000 scans). Note that vertical scales are not identical. For situations where S/N limits the resolution of NMR spectra (e.g. Il3Cd NMR at millimolar concentrations as used here) the ability to detect resonances may become difficult or impossible if an additional broadening occurs. The broadening of the A site ""Cd(I1) resonance in Cd(II),AP ( 6 ) is a good example and we have drawn in the approximate lineshape of this resonance at 170 ppm.

398 1'3Cd NMR of Alkaline Phosphatase
FIG. 3. Time course of phospho- rylation of 113Cd(II)z alkaline phos- phatase (2.47 m ~ , pH 6.5) followed by 31P NMR (80.9 MHz) and 'I3Cd NMR (44.37 MHz). At t = 0, 2 equiva- lents of inorganic phosphate were added and 31P and ""Cd spectra recorded at the times indicated. Each "'P spectrum is the average of 1500 scans (30' pulse angle, 3 s repeat time). The ""Cd spectra are the average of 60,000 scans. The extreme upfield cadmium resonance is an exter- nal I I : % Cd(acetate)z standard. Times are given for the midpoint of each accumu- lation.
31 P
ESP E-F t = l h \ /
4 h n E-< E-F
17 h - Uh E- F I
I I I I
30 20 10 0 ppm
'13Cd A
t = o
14h ,
A B
I I 1 1 1
200 100 0 PPm
Cd(ClO&. A line broadening of 40 Hz was employed. Pulse widths of -30" were used unless stated otherwise. pH values and the number of transients accumulated are as indicated. Conditions of the '"P NMR data collection are described in the following paper.
RESULTS
"'Cd NMR of 'l%'d(II,J Alkaline Phosphatases-Titration of ""Cd(I1) into apoalkaline phosphatase at pH 6.5 results in the appearance of a single ""Cd resonance at 169 pprn with a line width of 35 Hz (Figs. 1 and 2A). It increases linearly in area up to a cadmium:enzyme ratio of 21. Additional Cd(I1) results in an approximately linear decrease in resonance area until a ratio of 4:l is reached, at which point the original resonance has disappeared completely (Fig. 1). Collection of more transients for the 1':3Cd(II)4AP sample reveals that the A site resonance has been replaced by a broad resonance with a line width of 150 Hz located at slightly lower field than the original resonance of ""Cd(II),AP. This signal is visualized somewhat better at pH 6.0 and occurs a t -170 ppm. The area of this resonance accounts for no more than 50% of the four equivalents of cadmium present and may, therefore, represent only the A site Cd(I1) of both subunits, while resonance from B site Cd(I1) is invisible because of even greater broadening. Since no broadening of the original narrow resonance at 169 ppm (characteristic of the ""Cd(II)2AP species) occurs as it diminishes in intensity during the titration (Fig. l), the two species giving rise to the sharp and broad type resonances
must interconvert relatively slowly, especially since the chem- ical shift difference between them is only 1 ppm. At a cad- mium:enzyme stoichiometry of 6:l there are two resonances of approximate relative areas 1:2 a t 158 pprn and 22 ppm, respectively. The 158 pprn resonance is probably from A site bound cadmium, while the upfield resonance is at approxi- mately the chemical shift expected for free ":'Cd(II) in this buffer system (Fig. 2C).
Phosphorylation of "'Cd(II)&kaline Phosphatase- Phosphorylation of the "'CCd(II),AP at pH 6.5 induces a slow rearrangement of cadmium from the A site of one subunit to the B site of the other subunit (12). At enzyme concentrations of 2 mM it is possible to follow the time course of formation of the phosphoseryl residue by :"P NMR and the ""Cd(I1) mi- gration by Il3Cd NMR as illustrated in Fig. 3.
The "'Cd spectrum prior to addition of phosphate shows the expected single sharp resonance from two identical A site "'Cd(I1) ions (Fig. 3, t = 0). Seventy-two hours after the addition of phosphate this single resonance is replaced by two resonances of equal intensity at 141 and 55 pprn (Fig. 3, t = 72 h). The two resonances are characteristic of ""Cd in A and B sites. In contrast, the "'Cd spectrum taken at the intermediate time of 14 h after the phosphate addition shows three closely spaced '%d resonances near the chemical shift of the original A site lI3Cd(II), two of approximately equal intensity at 163 pprn and 152 ppm and the third, somewhat smaller resonance at 141 ppm (Fig. 3, t = 14 h).

1'3Cd NMR of Alkaline Phosphatase
The "p spectra from the same sample can be recorded at more frequent intervals because of the greater sensitivity of l3IP NMR. At equilibrium Cd(1I)tAP at pH 6.5 forms 1 mol of phosphoseryl intermediate (E-P) per mol of dimer and no E - P is present a t this pH (11). At t = l h, however, the bound phosphate is predominantly present as the noncovalent com- plex (E. P) as shown by its characteristic '"E' resonance at 12.79 ppm (19) (Fig. 3, t = 1 h). The E.P resonance slowly disappears to be replaced by the resonance at 8.30 pprn characteristic of the E-P species (19). By t = 4 h, 0.6 equiva- lents of phosphate are bound and by t = 17 h the maximum formation of 1 equivalent of E-P has been completed (Fig. 3).
The '3'p spectrum a t 17 h was obtained immediately after completion of the intermediate ""Cd spectrum shown in Fig. 3 a t t = 14 h. Hence, phosphorylation has occurred before significant cadmium rearrangement has taken place. The E-P resonance a t this stage consists of two overlapping resonances of slightly different chemical shift, One is the chemical shift observed for E-P a t final equilibrium (72 h) and the other, slightly downfield, is the chemical shift first observed for E-P at 1-4 h (Fig. 3, t = 1, 2, and 4 h). This suggests that two different conformations of E-P exist early in the phosphoryl- ation. The three '13Cd resonances in Fig. 3 (t = 14 h) thus all appear to arise from A site ""Cd(II), the most downfield from unphosphorylated active centers, while the two upfield arise from active centers containing the two conformers of E-P. Over the next 48 h, the ""Cd(11) rearranges and gives rise to two ""Cd resonances of equal intensity, one at the most upfield position (141 ppm) of the A site group and a new one far upfield at 55 pprn which arises from the B site ""Cd(I1). The total amount of E-P has not changed during this rear- rangement, but the E-P resonance has become homogeneous (Fig. 3, t = 66 h) and the system has reached equilibrium.
These results, together with other experiments to be shown below, allow a confident assignment of the 141 ppm and 55 ppm resonances to ""Cd(I1) bound to A and B sites, respec- tively. This assignment simplifies description and understand- ing of the remainder of the results to be presented and will be assumed prior to presentation of the supporting evidence to be shown below.
p H Titration of "'Cd(I& Alkaline Phosphatase-The rearrangement of the Cd(I1) ions induced by phosphorylation of alkaline phosphatase at pH 6.5 can be mimicked by raising the pH of an unphosphorylated sample of "'Cd(II).LAP (Fig. 4). In the presence of 0.1 M NaCl and at pH values as low as 5.4, a single A site resonance of intensity 2 appears a t 168 ppm. As the pH is raised to 7.0 this resonance titrates upfield to 164 ppm with an associated loss in intensity. These spectra are not accumulated under fully relaxed conditions, nor have we attempted to scale resonances according to individual TI values. The latter appear to be between 5 and 8 s based on previous data (12) and our unpublished results. Despite pos- sible changes in TI, it is likely that the reduction in intensity of the sharp 164 ppm resonance in the spectrum at pH 7.0 is accounted for by the appearance of a new, barely discernible resonance at 144 ppm. By pH 7.5 the original resonance has been reduced to -20% of its initial area, while two new narrow resonances of equal intensity have appeared at 144 ppm and 52 ppm with line widths of 33 and 40 Hz, respectively. Above pH 7.8 the equilibrium favors the rearranged species almost exclusively and the 162 ppm resonance is no longer seen. Further increase in pH results in broadening of the downfield resonance and by pH 9.5 an apparent loss of overall signal intensity appears to result from Cd(I1) dissociation from the unphosphorylated enzyme.
pH Titration of ""Cd(I& Alkaline Phosphatase-At pH 6.0 the addition of four equivalents of li."Cd(1I) to apoalkaline
A PH
5.4
,.i 7.0
n
200 150 100 50 0 PPm -
FIG. 4. pH titration of "'jCd(I1)z alkaline phosphatase (2.50 mM) followed by 'I3Cd NMR (19.96 MHz). Spectra are the average of 120,000 scans. The designations A and B correspond to the metal binding site responsible for the resonance.
phosphatase gives rise to a cadmium NMR spectrum consist- ing of one very broad resonance at 170 ppm, assigned above to A site ll3Cd(11) broadened by metal ion exchange a t B (Fig. 2b). As the pH is raised to 7.5 the resonance moves upfield to 142 ppm (Fig. 5 ) . Above this pH a new, initially very broad resonance with vlrJ E 250 Hz appears at 52 ppm, without

400 '13Cd NMR of Alkaline Phosphatase
200 150 100 50 0 ppm
I I 1 I I
200 150 100 50 0 ppm
FIG. 5. pH titration of 113Cd(II)4 alkaline phosphatase (2.72 m) followed by 'I3Cd NMR (19.96 MHz). Spectra are the average of 60,000 scans. Resonances A and B are as in Fig. 4.
resulting in any diminution in intensity of the low field 142 ppm resonance. Finally, between pH 8 and 9 both resonances of "''Cd(II),AP narrow, become similar in intensity and with chemical shifts of 143 ppm and 52 ppm.
Thus the Cd(I1) binding sites present in "3Cd(II)4 and "'Cd(II), alkaline phosphatases show similar NMR properties as a function of pH. At acid pH both start with a single low field resonance (169-170 ppm), which is replaced by two resonances at higher pH. The major differences occur in the relative intensities and line widths of the corresponding reso- nances. Whereas the initial resonance of "'Cd(II),AP at low pH is narrow and accounts for all "'Cd(I1) ions, the low field resonance of the '1"Cd(II)4 enzyme is much broader and rep- resents only -50% of the "3Cd(II). The latter probably rep- resents only the A sites on both monomers, while the reso- nance from "'Cd(I1) at the B sites remains too broad to be detected until the pH exceeds 7.5. Consistent with this is the appearance of more total signal intensity when resonances corresponding to both A and B sites are seen at high pH. At no time, however, are the resonances of "'Cd(11)4AP as narrow as those from "3Cd(II)2AP (95-110 Hz uersus 40-45 Hz, re- spectively).
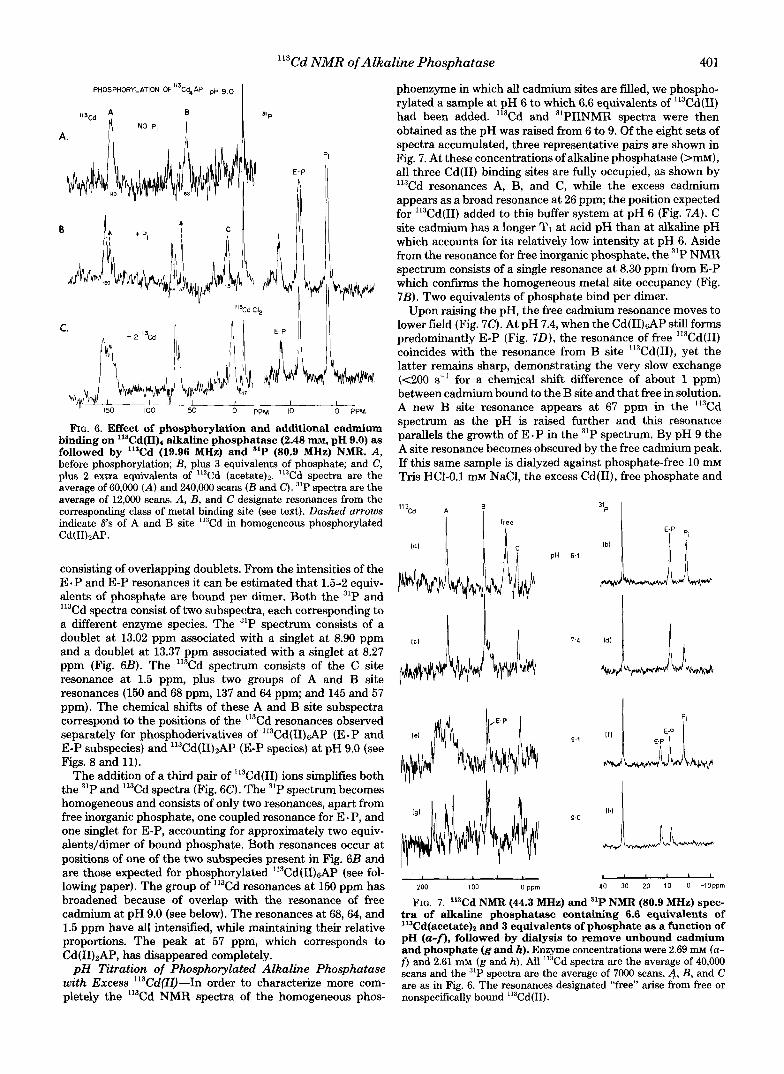
Phosphorylation of "'Cd(Iq4 Alkaline Phosphatase at p H 9.0-The 'I3Cd spectrum of "'Cd(II),AP at pH 9.0 is repeated in Fig. 6 A and compared with the spectrum of the same sample after the addition of excess phosphate (Fig. 6B). There are substantial changes in the pattern of ""Cd resonances accompanying formation of the phosphoenzyme derivatives (both E - P and E-P are formed at this pH) (Fig. 6B). The most striking change is the appearance of a new resonance (C site) at 1.5 ppm. At the same time the original two I1%d resonances have each split into 3 peaks. These resonances are much sharper than those of the unphosphorylated enzyme, while the total integrated intensity remains approximately the same before and after phosphorylation.
The 31P NMR spectrum of phosphorylated "'Cd(II), shows it to be heterogeneous (Fig. 6 B ) . The "P resonances of E-P for both zinc and cadmium-containing alkaline phosphatases occur in the region 7.9-8.9 ppm, whereas the E e P resonances of cadmium alkaline phosphatase occur between 12 and 13.4 ppm and show a 33 Hz "P-0-"'Cd coupling (see following paper). There is, therefore, little uncertainty in assigning the two resonances at 8.90 and 8.27 ppm to two forms of E-P and the asymmetric peak at 13.27 ppm to two forms of E - P
'%d NMR of Alkaline Phosphatase 401
PHOSPHORYLATION OF113Cd4AP p~ 9.0
C.
"P
Il3Cd CI, /I II
FIG. 6. Effect of phosphorylation and additional cadmium binding on 11SCd(II)4 alkaline phosphatase (2.48 nm, pH 9.0) as followed by '13Cd (19.96 MHz) and 31P (80.9 MHz) NMR. A, before phosphorylation; B, plus 3 equivalents of phosphate; and C, plus 2 extra equivalents of 'I3Cd (acetate)*. 'I3Cd spectra are the average of 60,000 (A) and 240,000 scans ( B and C ) . 31P spectra are the average of 12,000 scans. A, B, and C designate resonances from the corresponding class of metal binding site (see text). Dashed arrows indicate 6's of A and B site 'I3Cd in homogeneous phosphorylated Cd(1I)tAP.
consisting of overlapping doublets. From the intensities of the E. P and E-P resonances it can be estimated that 1.5-2 equiv- alents of phosphate are bound per dimer. Both the 31P and
Cd spectra consist of two subspectra, each corresponding to a different enzyme species. The 31P spectrum consists of a doublet at 13.02 ppm associated with a singlet at 8.90 ppm and a doublet at 13.37 ppm associated with a singlet at 8.27 ppm (Fig. 6B). The '13Cd spectrum consists of the C site resonance at 1.5 ppm, plus two groups of A and B site resonances (150 and 68 ppm, 137 and 64 ppm; and 145 and 57 ppm). The chemical shifts of these A and B site subspectra correspond to the positions of the 'I3Cd resonances observed separately for phosphoderivatives of '13Cd(II)&P (E-P and E-P subspecies) and 113Cd(II)2AP (E-P species) at pH 9.0 (see Figs. 8 and 11).
The addition of a third pair of "3Cd(II) ions simplifies both the 31P and 'I3Cd spectra (Fig. 6C). The 31P spectrum becomes homogeneous and consists of only two resonances, apart from free inorganic phosphate, one coupled resonance for E - P, and one singlet for E-P, accounting for approximately two equiv- alents/dimer of bound phosphate. Both resonances occur at positions of one of the two subspecies present in Fig. 6B and are those expected for phosphorylated Il3Cd(II)sAP (see fol- lowing paper). The group of '13Cd resonances at 150 ppm has broadened because of overlap with the resonance of free cadmium at pH 9.0 (see below). The resonances at 68,64, and 1.5 ppm have all intensified, while maintaining their relative proportions. The peak at 57 ppm, which corresponds to Cd(II)2AP, has disappeared completely.
pH Titration of Phosphorylated Alkaline Phosphatase with Excess 113Cd(ll)-In order to characterize more com- pletely the '13Cd NMR spectra of the homogeneous phos-
113
phoenzyme in which all cadmium sites are filled, we phospho- rylated a sample at pH 6 to which 6.6 equivalents of lI3Cd(II) had been added. l13Cd and 31PIINMR spectra were then obtained as the pH was raised from 6 to 9. Of the eight sets of spectra accumulated, three representative pairs are shown in Fig. 7. At these concentrations of alkaline phosphatase (>mM), all three Cd(I1) binding sites are fully occupied, as shown by '13Cd resonances A, B, and C, while the excess cadmium appears as a broad resonance at 26 ppm; the position expected for '13Cd(II) added to this buffer system at pH 6 (Fig. 7A). C site cadmium has a longer TI at acid pH than at alkaline pH which accounts for its relatively low intensity at pH 6. Aside from the resonance for free inorganic phosphate, the 31P NMR spectrum consists of a single resonance at 8.30 ppm from E-P which confirms the homogeneous metal site occupancy (Fig. 7B). Two equivalents of phosphate bind per dimer.
Upon raising the pH, the free cadmium resonance moves to lower field (Fig. 7C). At pH 7.4, when the Cd(I1)sAP still forms predominantly E-P (Fig. 70), the resonance of free l13Cd(II) coincides with the resonance from B site l13Cd(II), yet the latter remains sharp, demonstrating the very slow exchange (e200 s-' for a chemical shift difference of about 1 ppm) between cadmium bound to the B site and that free in solution. A new B site resonance appears at 67 ppm in the '13Cd spectrum as the pH is raised further and this resonance parallels the growth of E - P in the 31P spectrum. By pH 9 the A site resonance becomes obscured by the free cadmium peak. If this same sample is dialyzed against phosphate-free 10 mM Tris HC1-0.1 mM NaC1, the excess Cd(II), free phosphate and
I hl
B " " L 40 30 20 10 0 -1Opprn
FIG. 7. '13Cd NMR (44.3 MHz) and "P NMR (80.9 MHz) spec- tra of alkaline phosphatase containing 6.6 equivalents of %d(acetate)z and 3 equivalents of phosphate as a function of pH (a-f), followed by dialysis to remove unbound cadmium and phosphate (g and h). Enzyme concentrations were 2.69 mM (a- f ) and 2.61 mM (g and h). All '13Cd spectra are the average of 40,000 scans and the 31P spectra are the average of 7000 scans. A , B, and C are as in Fig. 6. The resonances designated "free" arise from free or nonspecifically bound "3Cd(II).

402 1'3Cd NMR of Alkaline Phosphatase
a portion of the enzyme-bound phosphate is removed (Fig. 7H). This leaves B and C site "'Cd resonances unaffected, but resolves the broad resonance around 150 ppm into two sharp resonances at 153 and 137 ppm (Figs. 7G and 8A). Two additional ""'Cd resonances have appeared at 180 and 174 ppm and appear to arise from A sites of alkaline phosphatase dimers from which the phosphate has dissociated. These two resonances can, indeed, be made to shift to the positions of the normal phosphoenzyme resonances by readdition of in- organic phosphate, leaving only three major resonances cor- responding to the E-P species and two minor resonances corresponding to A and B site ""Cd(I1) in the E - P species (Fig. 8B). From the proportions of E.P and E-P present, as indicated by the '"P NMR spectrum (Fig. 8, A and B), it is clear that subunits of Cd(1I)GAP carrying E . P have 'I3Cd resonances from A and B sites at 137 and 65 ppm, respectively, whereas subunits carrying E-P have '13Cd resonances from A and B sites at 153 and 70 ppm, respectively. The chemical shifts of these and other cadmium-containing alkaline phos- phatase species are summarized in Table I.
Effect of Mg(II) on Enzyme-bound ""Cd NMR Reso- nances-As was outlined in the preceding paper (l) , Mg(I1) at a concentration of 10 mM appears to complete effectively for one pair of Cd(I1) binding sites. Since Mg(I1) binding has been shown to require the prior binding of Zn(I1) (20), the competition is most probably at the B site. We t.herefore
examined the phosphorylated derivative of '13Cd(II)2AP in the presence of 10 mM Mg(I1) for which one might expect competition at the B site to drive both cadmium ions into the A sites. As expected, the initial ""Cd spectrum of 113Cd(II)2AP- P in the presence of 2 mM Mg(I1) (not enough to saturate the binding sites (1)) has two sharp resonances at 158 and 76 ppm from the A and B site cadmium ions at pH 6.5 (Fig. 9). In the absence of two equivalents of magnesium the spectrum is qualitatively the same, but has resonances at 141 ppm and 56 ppm, suggesting that even undersaturating Mg(I1) has some effect.
Addition of excess Mg(I1) to a total concentration of 10 mM (sufficient to compete for B site and accelerate phosphoryla- tion (1)) completely abolishes the two cadmium resonances (Fig. 9B). No significant change is observed in the "P spec- trum, which shows exclusively E-P at pH 6.5. This must be due to conformational modulation caused by exchange of Mg(I1) between free and bound forms, a modulation trans- mitted to the "'Cd(11) sites and which results in exchange broadening of the latter's resonance to the point of undetect- ability. While not proved by these results, exchanging Mg(1I) at the B site with lI3Cd(II) in the A sites 3.9 8, away appears to be a likely means of producing such modulation. If two additional ""Cd(I1) ions are added, the ""Cd resonances typ- ical of phosphorylated "'Cd(II)4AP appear (Fig. 9C). This would appear to reflect successful competition by cadmium
E*P E-P
~
200 150 100 50 0 PPm 15 10 0 PPm ~~~ ~ ~~~
FIG. 8. Re-establishment of homogeneity in dialysed, phosphorylated "3Cd(II)&P by addition of excess phosphate. a, "'Cd NMR spectra (44.3 MHz) (left) and "P NMR spectra (80.9 MHz) (right) of phosphorylated ''3Cd(11)66 alkaline phosphatase (2.61 mM) after dialysis to remove unbound cadmium and phosphate. The spectra are those shown in Fig. 7, g and h, respectively. b, sample a plus 2 equivalents of inorganic phosphate, "'Cd spectrum (19.96 MHz) (160,000 scans) and "P spectrum (12,OOO scans).

1'3Cd NMR of Alkaline Phosphatase 403
TABLE I "'Cd chemical shifts of A, B, and C site-bound "'Cd(II) in
cadmium alkalinephosphatase species From present work, except as noted.
Enzyme Chemical shifts
A E C
Cd(I1)zAP Cd(1I)zAP Cd(1I)ZAP E-P 6.5" 163, 152
ppm 6.5 169 (Ref. 12) 8.3 144 52
6.5' 141 55 9.0 144 57
50 6.0 170 ND' 8.7 142 53
Cd(II),AP E.P 9.0 168 Cd(I1)dAP
Cd(1I)tiAP E-P 8.5 153 70 2 Cd(1I)tiAP E.P 8.5 137 65 2 Cd(II),Mg(II),AP E-P 6.5 158 Cd(II),Mg(II)ZAP E-P 6.5 159 76
76 (Ref. 12)
After phosphorylation, but before cadmium migration After migration. ' ND, not detectable.
addition of Cd(I1) isotopes in pairs does not result in unique site occupancy, apparently because of rapid intersite exchange associated with phosphorylation. Two of the orders of addition experiments, however, reveal significant dynamic aspects of Cd(I1) binding to the enzyme and are reported here.
Three experiments are described, all conducted at pH 6.5, since the initial operational definition of A site is that it is the site fiist occupied by Cd(I1) at this pH. To a dilute (0.27 mM) solution of apoalkaline phosphatase at pH 6.5 and 4 "C were added 2 equivalents of "'Cd(I1). Two equivalents of inorganic phosphate were then added followed immediately by 4 equiv- alents of "'Cd(I1) and the sample was left for 20 min to ensure complete phosphorylation before concentrating in an ultrafil- tration unit to the final NMR concentration. The "'Cd NMR spectrum of this sample is the average over the fiist 13 h after addition of L1lCd(II) and shows two resonances of approxi- mately equal area at positions corresponding to A and B site
I I I I I
200 150 100 50 o ppm
FIG. 9. The effect of Mg(I1) on the '13Cd resonances (44.37 MHz) of phosphorylated '13Cd(II) alkaline phosphatase. a, "'Cd(II),sAP, (1.2 mM) plus 2.4 mM Mg(II) and 2.4 mM inorganic phosphate (20,000 scans); b as a but 10 mM Mg(I1) (14,000 scans); c as b plus 2 equivalents of ""Cd (acetate), (20,000 scans).
for B site magnesium. Ordered Addition of Cadmium Isotopes to Apoalkaline
Phosphatase-The original objective of these experiments, in which pairs of different cadmium isotopes were added sequen- tially to apoalkaline phosphatase, was to provide an assign- ment,, A or B, of the two post-phosphorylation cadmium resonances of '13Cd(II)2AP at pH 6.5, since chemical shift alone does not provide an absolute assignment. Unfortunately,
J I 1 I I
200 150 100 50 0 ppm
FIG. 10. Ordered addition of cadmium isotopes to apoalka- line phosphatase at low pH. a, ""Cd NMR spectrum (44.3 MHz) of apoalkaline phosphatase (2.68 mM, pH 6.0) plus 2 equivalents of
by 4 equivalents of "'Cd (acetate)2. Data accumulation was started 20 min after addition of the "'Cd (acetate)? (48,000 scans requiring 13.3 h). 6, "'Cd NMR spectrum (42.4 MHz) of sample a taken 3 days after formation of the sample (62,000 scans). c, ",'Cd NMR spectrum (44.3 MHz) of apoalkaline phosphatase (2.25 mM, pH 6.52) plus 2 equivalents of ""Cd (acetate), plus 10 mM Mg(II), followed 5 min later by 2 equivalents of P, and 40 min later by 2 equivalents of "'Cd (acetate)z. The spectrum represents the first 20,000 scans (1 1 h) after reconstitution. d, ""Cd NMR spectrum (44.3 MHz) of 11"Cd(II)2
prepared by addition of 2 equivalents of ""Cd (acetate)? and 2 equiv- alents of P, to the enzyme at pH 6.4 with monitoring of P, binding by "P NMR. After 4 h, 4 equivalents of "'Cd (acetate)? were added and the Il3Cd NMR spectrum recorded. The first 24,000 scans (6.7 h ) are shown.
113 Cd (acetate)Z, plus 2 equivalents of inorganic phosphate followed
112 CdUI), alkaline phosphatase (2.10 mM) + P,. The sample was

404 '13Cd NMR of Alkaline Phosphatase
cadmium ions (Fig. 10A). The "'Cd NMR spectrum, recorded 3 days later, is nearly identical (Fig. 10B). Hence, before phosphorylation is complete, there appears to be a destabi- lized state in which facile metal exchange between A and B sites occurs even when sufficient metal is present to occupy all three pairs of binding sites. Since the phosphorylated enzyme shows a slow exchange of both A and B site cadmium (l), the scrambling observed probably occurs faster than or at least coincident with phosphorylation. With 6 equivalents of metal ion present, phosphorylation has a tI l2 of about 1 min (1).
In the other two experiments, attempts were made to pre- vent, or at least slow down, this rapid inter-site rearrangement of cadmium atoms. The fist made use of the competitive affinity of Mg(I1) ions for the second pair of Cd(I1) binding sites. After addition of two equivalents of "'Cd(I1) to apoal- kaline phosphatase, the sample was made 10 mM in Mg(I1) and then 2 equivalents each of "'Cd(I1) and phosphate were added. This does not completely inhibit rearrangement, but does trap an asymmetric distribution of 'I3Cd(II) (Fig. 1OC). The downfield resonance is much greater in intensity than the upfield. This agrees with the assignment made in Fig. 3, i.e. the low field peak arises from A site cadmium, which was the site originally containing all of the "'Cd(I1). After several days the relative peak heights approach equality (spectrum not shown).
The last experiment exploited the different rates of phos- phorylation and cadmium migration in a ""Cd(II)2AP sample. Phosphate was added to ""Cd(II)2AP and allowed to phos- phorylate for 4 h, at which time migration of cadmium from
FIG. 11. Time course of the slow phosphorylation of "3Cd(II)~ alka- line phosphatase (2.76 mM) contain- ing 2 equivalents of Pi at pH 9.0. The spectra on the right are "P NMR (80.9 MHz) and on the left are "'Cd NMR (44.3 MHz; a) and (19.96 MHz; b and c ) . Phosphate was added to ":'Cd(II)2AP, pH 9 at t = 0. For the ""Cd spectra, 37,000 ( a ) , 116,000 ( b ) , and ,162,000 ( 4 scans were recorded. For the '"P spectra, 18,000 (a), 12,000 (b) , and 6,000 (c ) scans were recorded.
A to B site is far from complete (Fig. 3). Four equivalents of 112Cd(II) were then added and the ""Cd NMR spectrum followed with time. The initial 24,000 scans again show an asymmetric distribution of "'Cd between high and low field resonance positions, with the low field, A site resonance pre- dominating (Fig. 1OD).
Slow Conformation Changes in Alkaline Phosphatase-It has been generally assumed in metal ion titration studies of alkaline phosphatase that equilibrium is achieved relatively rapidly after the addition of metal ions. Several experiments have, however, shown a time dependence associated with the generation of specific NMR signals of "3Cd(II) alkaline phos- phatase. These demonstrate that conformational transitions must occur between the apoenzyme and metalloenzyme and its various phosphoderivatives, which suggest that significant kinetic barriers exist to the establishment of the final equilib- rium species. Two experiments indicating such shifts are illustrated here.
Slow Equilibria Associated with Phosphorylation of Cd(I& Alkaline Phosphatase-In the following paper (21), we show that the equilibrium percentage of E - P increases along a pH profile related to the activity profile and typical of each metal ion stoichiometry. For the Cd(11h enzyme, where the E-P s E . P equilibrium is shifted far to alkaline pH, the phosphoenzyme, if formed at pH 6 and titrated to pH 9, still has a E-P:E.P ratio of -85:15, even at pH 9.0. In contrast, if 113Cd(II)2AP formed at pH 6.5 is fist taken to pH 9 (where migration of half the Cd(I1) ions from the A site to the B site of the opposite monomer has occurred) and is then phospho- rylated, the phosphoenzyme formed in the first hour is 85-
31P E P
t = 0 days
e' I
2 days
A 10 days
144
I I I I ' I I
200 150 100 50 ppm 15 10 5 0 ppm

''"Cd NMR of Alkaline Phosphatase 405
90% E.P as shown by the 31P NMR signal at 13.38 ppm (Fig. 1lA).
The '13Cd spectrum of this sample (E. P complex) consists of two resonances at 168 ppm (A) and 50 ppm (B') (Fig. 1 W . This is not the final equilibrium species, however. Over the next several days there is a gradual change in both the "P and "'Cd spectra of the enzyme; a change which finally results in a complete reversal of the E - P:E-P ratio. After 2 days; the E.P:E-P ratio has fallen from 9O:lO to 5050 as shown by the
NMR and the ""Cd spectrum shows two sets of ""Cd resonances with half of the original resonance intensity in each set (Fig. 11B). One set occurs at the original chemical shifts of 168 ppm and 50 ppm (A and B') and the new set at 144 and 57 ppm (A and B). Ten days after addition of phosphate the E. P:E-P ratio has fallen further to 10:90 and the "'Cd spectrum has converted almost exclusively to the species with signals a t 144 and 57 pprn (Fig. 1lC).
Slow Equilibria Associated with Cd(I4 Binding at Alka- line pH-The '13Cd NMR spectra of the alkaline forms of Cd(II)2AP, Cd(II),AP, and Cd(I1)sAP have been presented in Figs. 4-7. All of these samples were formed by titrating metal into the apoenzyme at pH 6.5 or below followed by titration to alkaline pH. If cadmium is added to apoenzyme at pH 9, however, a significant difference in behavior is observed. In the first 24 h after addition of 2 equivalents of ""Cd(II), no "'Cd resonances are observed. After 72 h incubation the typical alkaline species forms with the "'Cd(I1) ions equally distributed between A and B sites, narrow resonances a t 145 ppm and 51 ppm, respectively. Thus, there appear to be some rather slow conformational changes involved before the final stable metalloenzyme is formed at alkaline pH.
DISCUSSION
The substitution of Cd(I1) for Zn(1I) in zinc-containing metalloenzymes is an obvious and attractive means of opening up these systems to observation by the powerful techniques of nuclear magnetic resonance. The span of chemical shifts observed for "'Cd(I1) complexes with the potential donor groups expected to occur in proteins is -800 ppm (22). The softer thiol ligands found in metallothionein (23) are strongly deshielding with respect to nitrogen ligands (22,24,25) which are in turn deshielding with respect to oxygen ligands such as carboxyls (26-28). This large range of chemical shifts makes '13Cd a potentially sensitive probe of the structure of metal binding sites in enzymes as well as of conformational changes which may accompany binding of additional ligands. Il3Cd(II) alkaline phosphatase illustrates this sensitivity well, but also highlights the complexities associated with metal binding to multisubunit proteins with several potential metal binding sites.
One of the more interesting features of the alkaline phos- phatase molecule revealed by "."Cd NMR is the finding, perhaps not unexpected, that it is not possible to treat the protein as a rigid ligand. For example, the broadening of the A site signal on occupancy of the B site by metal ions (Figs. 1, 2, and 5) must reflect conformational flux transmitted to the A site and sensed as a modulation of the chemical shift. The frequency of such a conformational flux required to result in the intermediate chemical exchange condition depends on the chemical shift difference between the species in flux. For a chemical shift difference of 25 ppm (500 Hz a t 19.96 MHz) as estimated from the A site resonance a t 169 pprn when B site is empty and 144 ppm when B site is occupied (Fig. 4), the required rate would be between 3 X lo2 and 3 x lo4 s-'. As argued in the previous paper (l), the exchange of the second pair of Cd(I1) ions in the absence of phosphate ligand at pH 6.5 could be this rapid. The exchange of the less tightly bound
Mg(I1) ion is probably even more rapid and can modulate the
rylated species (Fig. 9). It is clear from the Ii5Cd binding studies of the preceding
paper that either raising the pH or formation of E-P induces the binding of an extra pair of cadmium ions due to an increase in the affinity of B site (1). The ""Cd NMR spectra of ""Cd(II),AP show that either of these changes causes the A site resonance (169 ppm) to split into two new resonances of half the intensity corresponding to one A site and one B site cadmium (Figs. 3 and 4). The effect of phosphorylation has been reported before (12) and been shown to arise from migration of an A site cadmium ion to the vacant B site of the adjacent monomer. Almost certainly, the same explanation is true for the split caused by raising the pH. Both are under- standable in terms of the increased B site affinity indicated by the "'Cd binding studies (1). While metal binding sites would be expected to increase their experimentally observed affinity at higher pH as the proton competition for ligand donor atoms diminishes, the relative affinity of sites would be maintained unless donors with unusually high pK, values were involved in one site and not the other. From ''3C NMR studies (13) and X-ray crystallography (10, 14) it seems that there are at least two histidine ligands to the A site metal, while the B site ligands appear to be mostly carboxyl groups but with at least one histidine ligand. Since the carboxyls are unlikely to have the high pK, values required, it seems more likely that the change in relative affinity of the two pairs of sites as the pH is raised involves some additional conforma- tional changes in the protein, which can be independently produced by phosphorylation.
From the time course of the changes in the '"P and ""Cd spectra during phosphorylation of "'Cd(II)2AP at pH 6.5 (Fig. 3), it is apparent that formation of E. P and phosphorylation of seryl 102 from this complex can occur with only the A site metal ion in place. The conformation of the active center in the E-P state is such that the potential stability of the A + B site in the presence of E-P is so great as to cause the de- population of half the A sites.
The sequence of phosphorylation of ""Cd(II)2AP over a 4- h period followed by migration of half the ""Cd(I1) from A site of the unphosphorylated monomer to the B site of the phosphorylated monomer as shown in Fig. 3 requires a ligand- induced asymmetry in the dimeric protein. This asymmetry must be introduced to explain the phosphate-induced transi- tion between the two most stable states; i.e. the initial one with Cd(I1) in two identical equally stable A sites and the final one with Cd(I1) occupying both the A and B sites of the phosphorylated monomer. No metal is present in either A or B site of the unphosphorylated monomer. One could postulate that an asymmetry is initially present in the dimeric enzyme which favors phosphorylation of only one subunit and that presently available probes are not sensitive enough to pick up the inequivalence of the monomers. These probes, however, include ""Cd NMR, which shows identical metal binding sites (Fig. 3), "C NMR of the y-"'C-labeled His residues, which shows one 13C resonance for each pair of histidines (13), and the crystal structure, which shows a molecular 2-fold axis to exist between the monomers of the dimer in the unliganded enzyme (10).
Phosphorylation of the fist active site containing the A site metal ion momentarily renders the system asymmetric by virtue of the bound phosphate, but if no ligand-induced con- formational change were propagated to the site on the other monomer, the second site should independently phosphoryl- ate and symmetry would be restored. This would remove the driving force for Cd(1I) migration and result in phosphoryla-
11"Cd resonances from the adjacent site even in the phospho-

406 "'Cd NMR of Alkaline Phosphatase
tion of both sites which does not occur in Cd(II)2AP (Fig. 3). A further example of the complications arising in the "'Cd
NMR spectra of alkaline phosphatase as a consequence of its multiple metal binding sites is the disproportionation of "'Cd(II)4AP into a mixture of Cd(II)2AP and Cd(II)6AP upon phosphorylation (Fig. 6). The broadness of the resonances for ""Cd(II),AP may actually reflect some tendency to dispro- portionation prior to phosphorylation (Fig. 5). In an earlier NMR study by "C NMR (13) the heterogeneity evident in the histidine region of ""Cd(II),AP was attributed to an insufficient difference in relative affinities of the B and C sites for Cd(I1). A simpler explanation, which would also account for the 50:50 distribution of the composite species, is dispro- portionation to Cd(1I)zAP and Cd(II),AP. Such disproportion- ation clearly indicates some subunit-subunit interactions, since it would otherwise be difficult to explain stabilization of Cd(II),AP and destabilization of Cd(II),AP on interaction with phosphate.
The disproportionation of phosphorylated Cd(II),AP also raises the question of the importance of the C sites both in stabilizing the enzyme and in affecting the properties of the active centres. There is a small but significant effect of occu- pancy of the C site upon the chemical shifts of both A and B site cadmium ions (Table I). These AS'S are small, however, compared with the changes in chemical shift accompanying the transformation from the E - P to the E-P forms of the enzyme (Figs. 8 and 11). The latter changes illustrate how sensitive the ""Cd chemical shift is to structure at the active center. Note that for the ""Cd(II)2AP, the A site resonance for the E. P form is 24 ppm downfield of that for the E-P form (Fig. l l ) , while for the ""Cd(II)6AP it is 16 ppm upfield of the E-P form (Fig. 8). This is further evidence that one active center may be influenced by metal site occupancy at the other active center of the dimer. The 11'3Cd-O-3'P coupling supports the existence of direct phosphate-cadmium coordination in E. P but not in E-P (18). The removal of the phosphate as ligand to one of the metal ions could explain the changes in chemical shift of the ""Cd(I1) on the E. P to E-P transformation.
While both A and B site cadmium show separate resonances for the E-P and E - P forms, C site cadmium does not (Figs. 7 and 8). The ability of Mg(I1) to exchange broaden cadmium in A site seems to depend on its presence in B site (Fig. 9). When it is excluded from B site by the presence of 4 Cd(LI)/ dimer, leaving only C sites available for it to bind to, there is no longer exchange modulation of the cadmium resonances (Fig. 9). These observations all point to a somewhat unimpor- tant role for the C site metal ion. It appears to have only a small effect upon the phosphate binding centers, probably as a result of a general slight alteration of protein conformation upon binding. Despite this, one of the driving forces for disproportionation of Cd(II)4AP must be the high affinity of C site in the phosphorylated enzyme, since the latter becomes occupied in the Cd(II),;AP fraction (Fig. 6).
Formation of the phosphoenzymes by Cd(1I)zAP at pH 9 occurs within the time necessary to collect a '"P NMR spec- trum of reasonable signal to noise (-40 min) (Fig. 11). The '31P chemical shifts of the E - P and E-P species formed are the same as when the pH of phosphorylated Cd(1I)zAP is raised from 6 to 9 (see following paper), yet the E.P:E-P ratio initially seen is far from the equilibrium value and requires about 10 days before equilibrium is reached (Fig. 11). The corresponding transient splits in the ""Cd resonances merely reflect the changing proportions of E . P and E-P. The time- dependent change, therefore, does not involve a change in metal distribution or an alteration in the metal ion coordina- tion spheres, since either of these would be expected to affect the chemical shifts of the cadmium and phosphorus reso-
nances. Rather, the rate of phosphorylation of Ser 102 from E - P is very slow for Cd(II)2AP at pH 9.
For any species other than phosphorylated "3Cd(II)6AP, extreme care must be taken to ascertain the particular distri- bution of metal ions and the conformational state of the subunits. Metal ion migration, exchange broadening of reso- nances, rapid inter-site metal scrambling, and the slow attain- ment of equilibrium under some circumstances all help make the system more complex than its three pairs of metal sites, two phosphate binding sites, and two types of phosphoenzyme complex would suggest. Alkaline phosphatase does provide a good illustration of the range of phenomena likely to affect cadmium NMR spectra of proteins. None of the ""Cd NMR spectra show evidence for subunit differences when the en- zyme is fully saturated with metal ions and phosphate, though the time scale involved may be too slow to detect a more rapid averaging. Individual monomers are, however, sensitive to the metal ion distribution within the subunit as well as the adja- cent subunit of the dimer which indicates at least the potential for subunit-subunit interactions.
Phosphorylation can proceed with only A site occupied (Fig. 3), though the rate is greatly enhanced by the presence of a metal ion at the B site. The phenomenon of half-of-the- sites reactivity observed for a metal/dimer ratio of 2:l may, therefore, still be due to negative cooperativity of phosphate binding. The subsequent cadmium migration from A to B site may be merely an interesting sidelight. Lastly, the slow phos- phorylation of Ser 102 at alkaline pH for ""Cd(II)2AP may have significance in explaining the much lower turnover rate of cadmium uersus zinc enzyme. This and further discussion of the mechanism of catalysis will be covered in the following paper.
REFERENCES 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
Coleman, J. E., Nakamura, K.-I., and Chlebowski, J . F. (1982). J.
Applebury, M. L., Johnson, B. P., and Coleman, J. E. (1970) J .
Lazdunski, C., Petitclerc, C., and Lazdunski, M. (1969) Eur. J.
Csopak, H., and Szajn, H. (1973) Arch. Biochem. Biophys. 157,
Applebury, M. L., and Coleman, J. E. (1969) J. Biol. Chem. 244,
Simpson, R. T., and Vallee, B. L. (1968) Biochemistry 7, 4343-
Anderson, R. A,, Bosron, W. F., Kennedy, F. S., and Vallee, B. L.
Reynolds, J. A., and Schlesinger, M. J. (1969) Biochemistry 8,
Chlebowski, J. F., Armitage, I. M., Tusa, P. P., and Coleman, J.
Sowadski. J . M.. Foster. B. A.. and Wvckoff, H. W. (1981) J . Mol.
Biol. Chem. 257, 386-395
Biol. Chem. 245,4968-4976
Biochem. 8,510-517
374-379
709-718
4350
(1975) Proc. Natl. Acad. Sci. U. S. A. 72, 2989-2993
568-593
E. (1976) J. Biol. Chem. 251, 1207-1216 ~~
Biol. 150, 245-272 Chlebowski, J. F., Armitage, I. M., and Coleman, J. E. (1977) J .
Bioi. Chem. 252, 7053-7061 Otvos, J. D., and Armitage, I. M. (1980) Biochemistry 19, 4031-
4043 Otvos, J . D., and Armitage, I. M. (1980) Biochemistry 19, 4021-
4030 Otvos, J. D., Armitage, I. M., Chlebowski, J. F., and Coleman, J.
E. (1979) J. Biol. Chem. 254,4707-4713 Malamy, M. H., and Horecker, B. L. (1964) Biochemistry 3,1893-
1897 Bradshaw, R. A,, Cancedda, F., Ericsson, L. H., Newman, P. A.,
Piccoli, S. P., Schlesinger, M. J., Shriefer, K., and Walsh, K. A. (1981) Proc. Natl. Acad. Sei. U. S. A. 78, 3473-3477
Applebury, M. L., and Coleman, J. E. (1969) J. Biol. Chem. 244, 308-318
Armitage, I. M., Schoot-Uiterkamp, A. J. M., Chlebowski, J. F., and Coleman, J . E. (1978) J . Mag. Res. 29, 375-392
Otvos, J . D., Alger, J. R., Coleman, J. E., and Armitage, I. M.

““Cd NMR of Alkaline Phosphatase 407
(1979) J. Biol. Chern. 254, 1778-1780.
V d e e , B. C. (1977) Biochemistry 16,610-614 20. Bosron, W. F., Anderson, R. A., Falk, M. C., Kennedy, F. S., and
21. Gettins, P., and Coleman, J. E. (1982) J. Biol. Chem. 257,408-416 22. Bailey, D. B., Ellis, P. D., and Fee, J. A. (1980) Biochemistry 19,
591-596 23. Otvos, J. D., and Armitage, I. M. (1980) Proc. Nutl. Acad. Sci. U.
S. A . 77, 7094-7098 24. Armitage, I. M., Pajer, R. T., Schoot-Uiterkamp, A. J. M., Chle-
bowski, J. F., and Coleman, J. E. (1976) J . Am. Chem. SOC. 98,
5710-5712 25. Schoot-Uiterkamp, A. J. M., Armitage, I. M., and Coleman, J. E.
26. Bailey, D. B., Ellis, P. D., and Cardin, A. D. (1978) J. Am. Chem.
27. Drakenberg, T., Lindman, B., Cave, A., and Parello, J. (1978)
28. Forsen, S., Thulin, E., Drakenberg, T., Krebs, J., and Seamon, K.
(1980) J. Biol. Chem. 255, 3911-3917
SOC. 100,5236-5237
FEBS Lett. 92, 346
(1980) FEBS Lett. 117, 189-194
















![Docking interactions in protein kinase and phosphatase ...interacting protein–protein motifs for MAP kinases and tyrosine phosphatases [12,13]. Docking interactions in protein phosphatases](https://img.pdfslide.us/doc/110x75/60ee63efe2bdd8639d7712a5/docking-interactions-in-protein-kinase-and-phosphatase-interacting-proteinaprotein.jpg)












