Embed Size (px)
Citation preview

2 33
Pathophysiology, Clinical Recognition, and
Treatment of Congenital Heart Disease
Steven R. Neish and Jeffrey A. Towbin
Key Points
• Atrial level communications include any of the follow-ing: (1) ostium secundum atrial septal defect (ASD), (2) ostium primum atrial septal defect, (3) sinus venosus atrial septal defect, and (4) coronary sinus atrial septal defect. The common ASD among these are the ostium secundum ASD representing approximately 70% of all ASDs. With all of the ASDs, signifi cant pulmonary hyper-tension rarely occurs before the third to fourth decade.
• Fifty percent or more of ventricular septal defects (VSDs) close spontaneously in early childhood, even as late as adolescence. The VSDs with large left-to-right shunts may lead to severe pulmonary hypertension with bidirec-tional or right-to-left shunting and cyanosis in the adult.
• Patent ductus arteriosus leads to a continuous systolic diastolic murmur under the left clavicle and places the patient at risk for infection at the ductus site, and when it leads to a large left-to-right shunt, may cause severe pulmonary hypertension.
• Aortic valve stenosis in the child and early in adulthood is usually caused by a bicuspid or a unicuspid valve.
• Supravalvular aortic stenosis occurs in two settings. Williams syndrome is a dysmorphic syndrome caused by a mutation of the elastin gene or chromosome 7, often associated with hypercalcemia in infancy. The second group of patients with supravalvular aortic stenosis have a familial form without hypercalcemia, dysmorphic fea-tures, or behavioral abnormalities.
• Subvalvular aortic stenosis has three types: membranous subaortic stenosis, a fi bromuscular tunnel type, and an idiopathic hypertrophic subaortic stenosis type.
• Pulmonary stenosis may be valvular, supravalvular, or subvalvular.
• Congenital mitral stenosis in adults is uncommon. Shone syndrome describes the occurrence of multiple levels of obstruction to blood fl ow into and out of the left ventricle and in the aorta. The classic picture includes mitral ste-nosis with a parachute mitral valve (single papillary muscle), subaortic stenosis, a bicuspid aortic valve, and coarctation of the aorta.
• Tricuspid stenosis is generally acquired rather than congenital and causes include rheumatic heart disease, carcinoid syndrome, and right atrial myxoma.
• Ebstein’s anomaly consists of downward displacement of the tricuspid valve “atrializing” the infl ow tract of the right ventricle. Tricuspid insuffi ciency, right-to-left shunts at the atrial level, and supraventricular arrhyth-mias often coexist.
• Coarctation of the aorta as manifested in the adult is almost always at or just distal to the ligamentum arterio-sum and the take-off of the left subclavian artery. This often leads to hypertension in the arms and reduced pres-sures and pulses in the lower extremities sometimes with some growth retardation in the legs.
• Tetralogy of Fallot consists of a large VSD, pulmonary stenosis, either valvular or infundibular or both, right ventricular hypertrophy, and overriding of the ventricu-lar septum by a dilated aorta.
10
Left-to-Right Shunt Lesions . . . . . . . . . . . . . . . . . . . . . . . 235Congenital Valve Abnormalities . . . . . . . . . . . . . . . . . . . 247Complex Congenital Heart Disease. . . . . . . . . . . . . . . . . 256Great Vein Malpositions . . . . . . . . . . . . . . . . . . . . . . . . . . 268
Coronary Artery Anomalies . . . . . . . . . . . . . . . . . . . . . . . 269Aneurysms of the Sinus of Valsalva. . . . . . . . . . . . . . . . . 269Pregnancy and Congenital Heart Disease. . . . . . . . . . . . 270
CAR010.indd 233CAR010.indd 233 11/24/2006 10:40:45 AM11/24/2006 10:40:45 AM

2 3 4 c h a p t e r 10
• Transposition of the great arteries in the adult exists as D-transposition, L-transposition, and double-outlet RV.
• Truncus arteriosus exists as three types. Type I has a common trunk, but this gives rise to separate ascending aorta and pulmonary trunk. In type II, the truncus extends up to the right and left pulmonary artery bifurca-tions. In type III, there is no separate pulmonary trunk.
In 1897, Maude Abbott wrote her fi rst paper on heart murmurs. The following year she was made curator of the McGill University medical museum in Montreal. She began a formal study of congenital heart disease by studying patho-logic specimens, encouraged by Sir William Osler. In 1908, Osler included Abbott’s chapter on congenital heart disease in his text called Modern Medicine.1 That chapter was the defi nitive statement on congenital heart disease in the early 20th century. In 1936, Abbott published her classic text, Atlas of Congenital Cardiac Disease,2 in which she described hundreds of pathologic specimens and provided the frame-work that allowed congenital heart disease to move past being a curiosity.
In 1930, early in her career, Helen Taussig was appointed to head the cardiac clinic at the Harriet Lane home at Johns Hopkins Hospital in Baltimore. In a revolutionary way, she began to understand the patterns that differentiated children born with various forms of congenital heart disease. She recognized that the degree of pulmonary blood fl ow pro-foundly affected the long-term course of many children with more severe congenital heart disease. She began to look for a way to apply this observation. In 1941, Alfred Blalock moved from Vanderbilt University to Johns Hopkins Hospi-tal and, shortly after Blalock’s move, Taussig approached Blalock and proposed that he attempt to attach the subcla-vian artery to the pulmonary artery to augment pulmonary blood fl ow in children with critically restricted pulmonary blood fl ow. In 1944, the fi rst operation was performed that came to be known as the Blalock-Taussig operation. The fi rst three applications of this “B-T shunt” were described in 1945 and the door to real therapy for children with congenital heart disease was opened.3
Successful open-heart surgery to correct septal defects in humans was ushered in by the pioneering work of C. Walton Lillehei,4 who closed a ventricular septal defect in a 4-year-old boy, using the boy’s father for cross-circulation in 1954. In the early days of open-heart surgery, the results of surgery frequently were uncertain. Also, there were many adults with congenital heart disease who had grown up before the development of heart surgery. Congenital heart disease clinics were fi lled with patients with unoperated congenital heart disease, some of whom had survived into adulthood. Today, most children with signifi cant congenital heart disease have palliative or corrective surgery in infancy or childhood. Some of these operations are “reparative” while others are best described as complex palliations. Today, the cardiologist with an interest in congenital heart disease in adults sees a combination of patients whose course is often characterized by when and where they were born.5 There are still patients with Eisenmenger’s syndrome (pulmonary hypertension with severe cyanosis due to unrepaired con-genital heart disease associated with chronic shunts), surviv-ing into adulthood, who would have avoided that tragic
complication through reparative surgery if they had been born a decade later.6 Others have been surgically corrected but may have sequelae related to either their native disease or some complication of therapy. Some remain with signifi -cant cardiovascular disease because their defect was too complex to repair. And fi nally, there are patients with condi-tions that escape detection during routine physical examina-tion, such as atrial septal defect and bicuspid aortic valve.
It has become increasingly apparent that patients who have undergone surgical correction of congenital heart disease frequently have complications later in adulthood. The ligation and division of a patent ductus arteriosus perhaps comes closest to a complete cure. Even patients with closed atrial septal defects occasionally are plagued in later life by supraventricular tachyarrhythmias, especially atrial fi brilla-tion and atrial fl utter. Therefore, most patients seen by car-diologists interested in adult congenital heart disease are those with previously “corrected” congenital heart disease who have sequelae later in life. However, a signifi cant minor-ity consists of those with hitherto undiagnosed congenital heart disease, inoperative heart disease, or congenital heart disease for which the patient has refused surgery. Signifi cant advances in therapy, including surgery, pacemakers, better treatment modalities for heart failure, and improved man-agement of hyperviscosity syndromes, have increased the life expectancy of such patients.
In earlier eras of congenital heart disease care, the physi-cian who cared for adults could ignore some of the more common defects, such as hypoplastic left heart syndrome (HLHS), as children with HLHS and some of the other complex defects never survived to adulthood. Some defects, such as bicuspid aortic valve or ventricular septal defect are much more common, and survival into adulthood has been the rule for decades. This chapter concentrates on both the most common congenital heart defects, as well as some of the rarer defects that have the potential to present on a regular basis to adult cardiology clinics.
The incidence of congenital heart disease varies with the population studied and the age of the patients in the study. In general, though, the order of frequency for the 10 most common congenital heart defects is as follows:
1. Ventricular septal defect (VSD) 2. Atrial septal defect (ASD) 3. Aortic stenosis (AS) 4. Pulmonic stenosis (PS) 5. Coarctation of the aorta (COA) 6. Patent ductus arteriosus (PDA) 7. Tetralogy of Fallot (TOF) 8. Transposition of the great arteries (TGA) 9. Atrioventricular septal defect (AVSD)10. Hypoplastic left heart syndrome (HLHS)
Congenital anomalies of the aortic valve actually are more common than is represented by the above list, but many, if not most, patients with a functionally bicuspid aortic valve are not diagnosed until adulthood.7
The cardiologist treating adults should still be able to diagnose congenital heart disease, or at least to narrow down the diagnostic possibilities by both clinical and laboratory methods.8 This is equally true in the long-term follow-up of such patients, especially if they have undergone surgery,
CAR010.indd 234CAR010.indd 234 11/24/2006 10:40:45 AM11/24/2006 10:40:45 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 3 5
because the natural history of the disease will have been altered by the operation itself. This demands thorough knowledge of the operative techniques used at the time of surgical intervention—many of which have changed consid-erably in recent years.8
Left-to-Right Shunt Lesions
The most frequent physiologic abnormality caused by con-genital heart disease is left-to-right shunting, resulting in pulmonary overcirculation.9 The term left-to-right shunt refers to blood in the systemic circulation (i.e., pulmonary veins, left atrium, left ventricle, or aorta) shunting into the circulation somewhere after the blood leaves the systemic capillary bed and before it reaches the pulmonary capillary bed. Abnormal communications can exist at atrial or ven-tricular levels, the shunting can be atrioventricular or aorto-pulmonary, or the pulmonary veins can connect somewhere other than the left atrium. Such communications result in shunting of blood, the direction of fl ow being determined by the pressure gradient or the difference in resistance between pulmonic and systemic circulation. Typically, the dominant direction of fl ow at these abnormal connections is from the systemic circulation into the pulmonary circulation (i.e., left to right) because the impedance of the pulmonary vascula-ture is much lower than the impedance in the systemic circulation.
A unique physiologic situation exists if the defect results in formation of a common mixing chamber. For example, a subset of complete atrioventricular (AV) canal defects con-sists of complete absence of the atrial septum (common atrium). In total anomalous pulmonary venous connection (TAPVC), the right atrium (RA) serves as the common mixing chamber. In both common atrium and TAPVC, the systemic venous return and the pulmonary venous return mix com-pletely at the atrial level, before entering the ventricles. A functionally single or common ventricle in which the other ventricle is rudimentary would constitute a mixing chamber. Finally, the septum between the aorta and the pulmonary artery and the outlet portion of the ventricles may be absent (i.e., truncus arteriosus). In all four of these situations, there will always be some degree of arterial desaturation, regard-less of the pulmonary vascular resistance (PVR).
Atrial Septal Defect
Atrial level communications may include any of the following9–11:
1. Ostium secundum atrial septal defect, representing non-closure of the foramen ovale
2. Ostium primum atrial septal defect, which is a subset of AV septal defect or an endocardial cushion defect
3. Sinus venosus atrial septal defect, resulting from failure of the proximal portion of the sinus venosus to be incor-porated into the RA
4. Coronary sinus atrial septal defect, in which there is a communication between the coronary sinus and the left atrium (LA), resulting in fl ow from the LA to the RA via the orifi ce of the coronary sinus.5
In practice, the overwhelming majority of atrial septal defects (ASDs) are ostium secundum ASDs, representing about 70% of all atrial communications. Ostium primum ASD is second in prevalence. Sinus venosus ASD is uncom-mon, and coronary sinus ASD is extremely rare. Patients with uncomplicated atrial communications frequently arrive at adulthood undiagnosed (Table 10.1).
Atrial Septal Defect, Ostium Secundum Type
Atrial septal defect, ostium secundum type, is the most common newly diagnosed congenital heart disease in the adult, possibly matched only by the bicuspid aortic valve.7,9,10,12,13 It is helpful here to review the circulatory phys-iology in the fetus in order to describe how ASDs develop and physiologically present during life. During fetal life, the pressures in the pulmonary artery and the aorta are equal; the fetal right ventricle (RV), being adapted for pressure, has the same compliance as that of the left ventricle (LV). Blood returning from the placenta (oxygenated blood) fl ows via the umbilical vein through the ductus venosus and preferen-tially shunts across the foramen ovale. Vestigial preferential right-to-left streaming from the inferior vena cava can be demonstrated in adults with atrial septal defects by echocar-diography or by indicator dilution techniques; the clinical counterpart is paradoxical embolization. At birth, with the infant’s fi rst breath, there is an immediate drop in PVR, which gradually decreases to normal in the fi rst few months of life. At the same time, the RV undergoes regression of myocardial hypertrophy, gradually changing from its cylin-drical confi guration and thick walls to that characteristic of the adult, in which the cavity is more crescentic and the wall thinner than that of the LV. Therefore, in the fetus, there is virtually no left-to-right shunting across the defect. If the defect persists, as the PVR falls and the compliance of the RV increases, left-to-right shunting results and pulmonary fl ow may be two to fi ve times the systemic fl ow. With time, both the RA and the RV enlarge.
Signifi cant pulmonary hypertension seldom occurs before the third or fourth decade.12–14 The mechanism by which pulmonary hypertension develops is not well under-stood. It may rarely start in childhood. Although high fl ow is implicated, it takes many decades for pulmonary hyper-tension to develop, and not all patients develop pulmonary hypertension.15 Progressive right ventricular enlargement and hypertrophy may lead to decreased compliance of the RV
TABLE 10.1. Paradigm to left-to-right shunts at the atrial level
Electrocardiogram, Cyanosis frontal plane axis
Atrial septal defect, 0 Normal secundum typeAtrioventricular canal Ostium primum 0 Left Common atrium + Left Complete + LeftAnomalous pulmonary venous connectionPartial 0 NormalComplete + Normal
CAR010.indd 235CAR010.indd 235 11/24/2006 10:40:45 AM11/24/2006 10:40:45 AM
2 3 6 c h a p t e r 10
compared with that of the LV, and therefore, the exclusive left-to-right shunt also yields some right-to-left shunting. Right-to-left shunting is not a direct effect of the relative pressures of the pulmonary artery and of the aorta, but rather of the compliance of the two ventricles. If left ventricular compliance also decreases, the atrial pressures rise, and classic signs of congestive heart failure (CHF) may be present. The pulmonary artery pressure in atrial septal defects with shunt reversal is almost always signifi cantly lower than the systemic pressure. This contrasts with ventricular septal and aortopulmonary defects, in which shunt reversal and pres-sure equalization go hand in hand.
Approximately 10% of patients with atrial septal defects have one or more anomalously connected pulmonary veins. Mitral insuffi ciency may coexist in 10% to 20% of these patients.16 The mitral insuffi ciency may be due to prolapse of the posterior leafl et of the mitral valve associated with secundum ASD and signifi cant right ventricular enlarge-ment.16 An interesting syndrome has been described (Lutem-bacher’s syndrome) in which an atrial septal defect coexists with mitral stenosis;17 in this disorder, patients remain rela-tively asymptomatic until pulmonary hypertension develops because the atrial septal defect decompresses the LA, and therefore the left atrial pressure is not elevated despite sig-nifi cant mitral valve obstruction.17–19 Patients with ostium secundum-type ASDs are seldom symptomatic until they begin to experience pulmonary hypertension, usually after the fourth decade,20 if it occurs. It may not occur, however. Unlike the other atrial communications, there is a 2 : 1 female to male preponderance in cases of secondary ASD and this defect is sometimes familial.21
There is usually a soft systolic fl ow murmur due to mark-edly increased pulmonary blood fl ow. The clinically diagnos-tic feature, however, is a wide splitting of the second heart sound (S2), which does not noticeably change with the respi-ratory cycle. This wide, fi xed splitting of the S2 is variably attributed to a variety of theoretical physiologic phenomena. Most likely, it is caused by increased capacitance of the pul-monary arterial circulation and corresponding decreased impedance. This increased capacitance leads to a prolonged “hangout interval” at the end of systole. Respiration has a minimal effect on capacitance in this setting, so the splitting of S2 is wide and fi xed. The electrocardiogram (ECG) almost always shows an rSR’ confi guration in lead V1. The R’ is thought to be caused by late activation of the crista supra-ventricularis. This may explain why, after successful closure of the atrial septal defect, wide splitting of the second heart sound often persists. The development of pulmonary hyper-tension accentuates the pulmonary component of the second heart sound, P2, and ultimately cyanosis appears, at least with exercise if not at rest.
Laboratory Findings
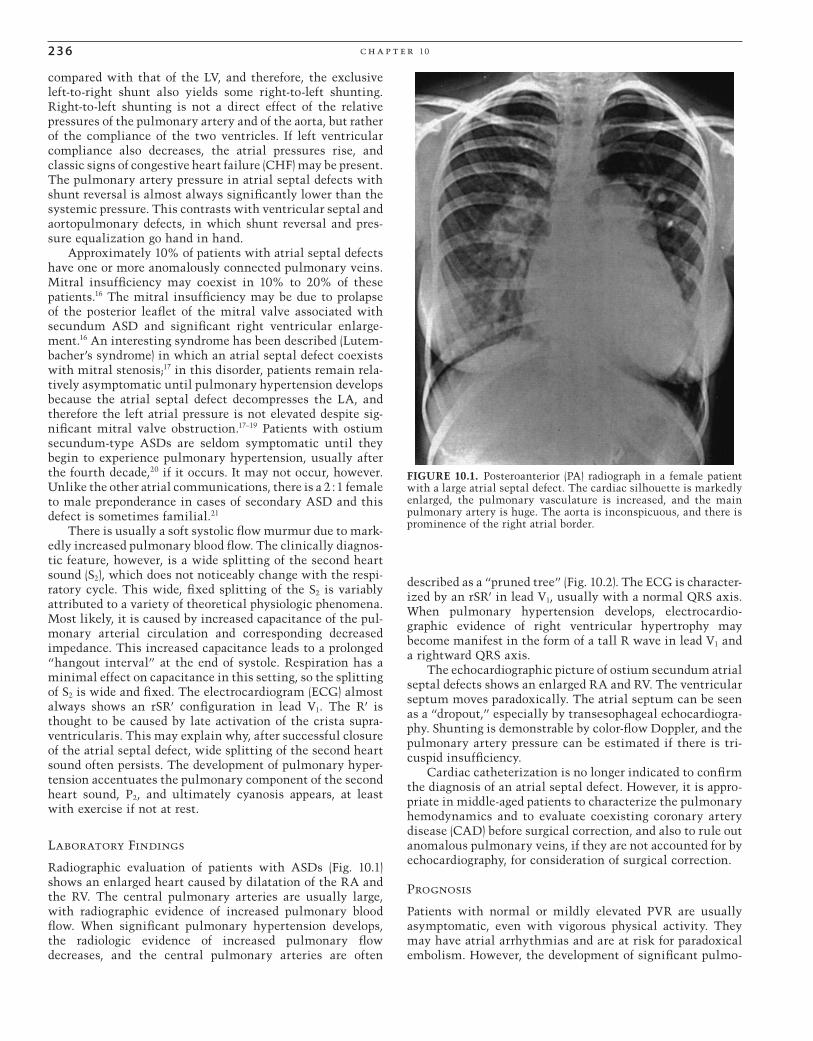
Radiographic evaluation of patients with ASDs (Fig. 10.1) shows an enlarged heart caused by dilatation of the RA and the RV. The central pulmonary arteries are usually large, with radiographic evidence of increased pulmonary blood fl ow. When signifi cant pulmonary hypertension develops, the radiologic evidence of increased pulmonary fl ow decreases, and the central pulmonary arteries are often
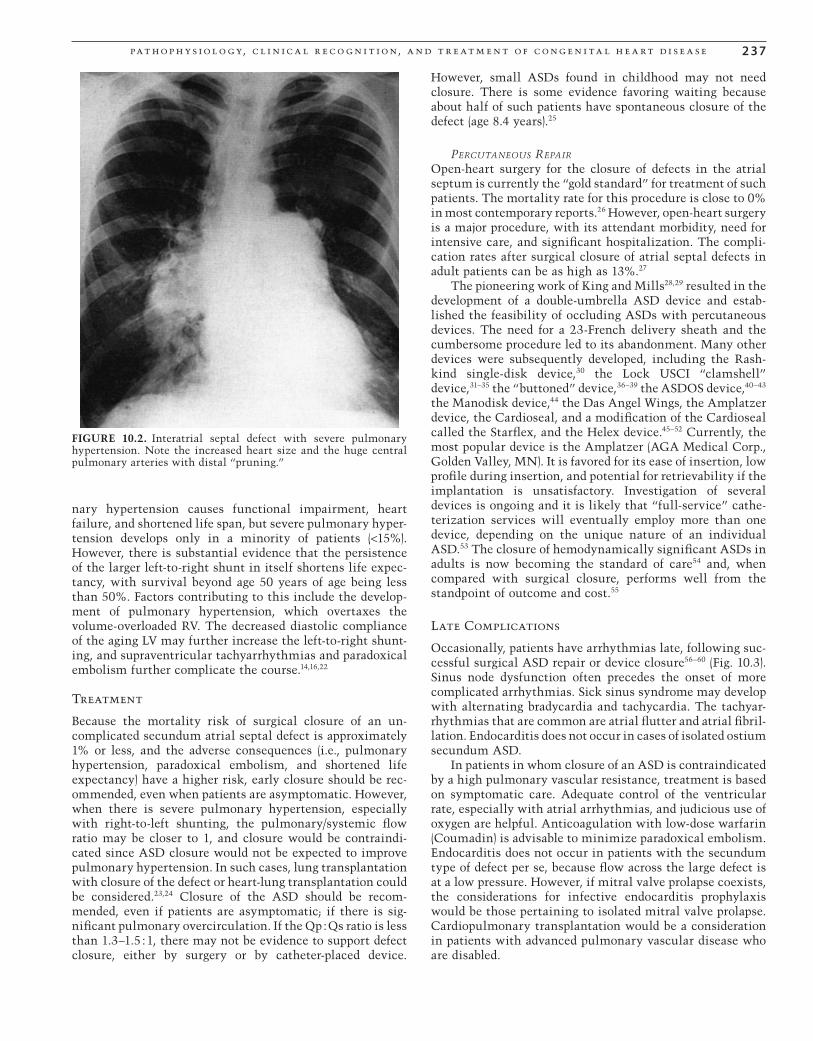
described as a “pruned tree” (Fig. 10.2). The ECG is character-ized by an rSR’ in lead V1, usually with a normal QRS axis. When pulmonary hypertension develops, electrocardio-graphic evidence of right ventricular hypertrophy may become manifest in the form of a tall R wave in lead V1 and a rightward QRS axis.
The echocardiographic picture of ostium secundum atrial septal defects shows an enlarged RA and RV. The ventricular septum moves paradoxically. The atrial septum can be seen as a “dropout,” especially by transesophageal echocardiogra-phy. Shunting is demonstrable by color-fl ow Doppler, and the pulmonary artery pressure can be estimated if there is tri-cuspid insuffi ciency.
Cardiac catheterization is no longer indicated to confi rm the diagnosis of an atrial septal defect. However, it is appro-priate in middle-aged patients to characterize the pulmonary hemodynamics and to evaluate coexisting coronary artery disease (CAD) before surgical correction, and also to rule out anomalous pulmonary veins, if they are not accounted for by echocardiography, for consideration of surgical correction.
Prognosis
Patients with normal or mildly elevated PVR are usually asymptomatic, even with vigorous physical activity. They may have atrial arrhythmias and are at risk for paradoxical embolism. However, the development of signifi cant pulmo-
FIGURE 10.1. Posteroanterior (PA) radiograph in a female patient with a large atrial septal defect. The cardiac silhouette is markedly enlarged, the pulmonary vasculature is increased, and the main pulmonary artery is huge. The aorta is inconspicuous, and there is prominence of the right atrial border.
CAR010.indd 236CAR010.indd 236 11/24/2006 10:40:45 AM11/24/2006 10:40:45 AM
pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 37
nary hypertension causes functional impairment, heart failure, and shortened life span, but severe pulmonary hyper-tension develops only in a minority of patients (<15%). However, there is substantial evidence that the persistence of the larger left-to-right shunt in itself shortens life expec-tancy, with survival beyond age 50 years of age being less than 50%. Factors contributing to this include the develop-ment of pulmonary hypertension, which overtaxes the volume-overloaded RV. The decreased diastolic compliance of the aging LV may further increase the left-to-right shunt-ing, and supraventricular tachyarrhythmias and paradoxical embolism further complicate the course.14,16,22
Treatment
Because the mortality risk of surgical closure of an un -complicated secundum atrial septal defect is approximately 1% or less, and the adverse consequences (i.e., pulmonary hypertension, paradoxical embolism, and shortened life expectancy) have a higher risk, early closure should be rec-ommended, even when patients are asymptomatic. However, when there is severe pulmonary hypertension, especially with right-to-left shunting, the pulmonary/systemic fl ow ratio may be closer to 1, and closure would be contraindi-cated since ASD closure would not be expected to improve pulmonary hypertension. In such cases, lung transplantation with closure of the defect or heart-lung transplantation could be considered.23,24 Closure of the ASD should be recom-mended, even if patients are asymptomatic; if there is sig-nifi cant pulmonary overcirculation. If the Qp : Qs ratio is less than 1.3–1.5 : 1, there may not be evidence to support defect closure, either by surgery or by catheter-placed device.
However, small ASDs found in childhood may not need closure. There is some evidence favoring waiting because about half of such patients have spontaneous closure of the defect (age 8.4 years).25
PERCUTANEOUS REPAIR
Open-heart surgery for the closure of defects in the atrial septum is currently the “gold standard” for treatment of such patients. The mortality rate for this procedure is close to 0% in most contemporary reports.26 However, open-heart surgery is a major procedure, with its attendant morbidity, need for intensive care, and signifi cant hospitalization. The compli-cation rates after surgical closure of atrial septal defects in adult patients can be as high as 13%.27
The pioneering work of King and Mills28,29 resulted in the development of a double-umbrella ASD device and estab-lished the feasibility of occluding ASDs with percutaneous devices. The need for a 23-French delivery sheath and the cumbersome procedure led to its abandonment. Many other devices were subsequently developed, including the Rash-kind single-disk device,30 the Lock USCI “clamshell” device,31–35 the “buttoned” device,36–39 the ASDOS device,40–43 the Manodisk device,44 the Das Angel Wings, the Amplatzer device, the Cardioseal, and a modifi cation of the Cardioseal called the Starfl ex, and the Helex device.45–52 Currently, the most popular device is the Amplatzer (AGA Medical Corp., Golden Valley, MN). It is favored for its ease of insertion, low profi le during insertion, and potential for retrievability if the implantation is unsatisfactory. Investigation of several devices is ongoing and it is likely that “full-service” cathe-terization services will eventually employ more than one device, depending on the unique nature of an individual ASD.53 The closure of hemodynamically signifi cant ASDs in adults is now becoming the standard of care54 and, when compared with surgical closure, performs well from the standpoint of outcome and cost.55
Late Complications
Occasionally, patients have arrhythmias late, following suc-cessful surgical ASD repair or device closure56–60 (Fig. 10.3). Sinus node dysfunction often precedes the onset of more complicated arrhythmias. Sick sinus syndrome may develop with alternating bradycardia and tachycardia. The tachyar-rhythmias that are common are atrial fl utter and atrial fi bril-lation. Endocarditis does not occur in cases of isolated ostium secundum ASD.
In patients in whom closure of an ASD is contraindicated by a high pulmonary vascular resistance, treatment is based on symptomatic care. Adequate control of the ventricular rate, especially with atrial arrhythmias, and judicious use of oxygen are helpful. Anticoagulation with low-dose warfarin (Coumadin) is advisable to minimize paradoxical embolism. Endocarditis does not occur in patients with the secundum type of defect per se, because fl ow across the large defect is at a low pressure. However, if mitral valve prolapse coexists, the considerations for infective endocarditis prophylaxis would be those pertaining to isolated mitral valve prolapse. Cardiopulmonary transplantation would be a consideration in patients with advanced pulmonary vascular disease who are disabled.
FIGURE 10.2. Interatrial septal defect with severe pulmonary hypertension. Note the increased heart size and the huge central pulmonary arteries with distal “pruning.”
CAR010.indd 237CAR010.indd 237 11/24/2006 10:40:46 AM11/24/2006 10:40:46 AM

2 3 8 c h a p t e r 10
Ostium Primum Atrial Septal Defect
Ostium primum ASD is an endocardial cushion defect in which the septum primum (i.e., the lower portion of the atrial septum) fails to develop, as does the atrioventricular septum. In this situation, the anterior leafl et of the mitral valve is attached to the ventricular septum in a somewhat lower position than normal and is therefore at the same level as the tricuspid valve. The anterior leafl et is cleft and, because of its lower position, has characteristic angiographic61 and echocardiographic20 features. The mitral valve cleft is associ-ated with various degrees of mitral regurgitation. Typically, the hemodynamic disturbance of an ostium primum ASD is more severe than an ostium secundum ASD. This is particu-larly true if there is signifi cant mitral valve insuffi ciency. It is rare for a patient with an ostium primum ASD to reach adulthood undiagnosed.
The clinical diagnosis of an ostium primum ASD has many of the features of those associated with a secundum-type ASD (i.e., hyperactive RV; wide, fi xed splitting of the second heart sound; and a pulmonary fl ow murmur). However, there is usually an additional murmur of mitral insuffi ciency. This does not necessarily radiate to the axilla, because the regurgitant jet is directed more medially than toward the free wall of the LA. If the mitral regurgitation is severe or if pulmonary hypertension develops, decreased exercise tolerance and exertional dyspnea can be expected. Examination fi nding may also include a left ventricular fi lling sound [the third heart sound (S3) of mitral insuffi -ciency] and a fourth heart sound (S4). The chest x-ray result
is very similar to that of a secundum-type ASD unless there is severe mitral insuffi ciency, in which case there is evidence of left atrial enlargement as well. The ECG has the charac-teristic rSR’ in lead V1, but in addition, the frontal plane QRS axis is always leftward, and there is often fi rst-degree heart block (Fig. 10.4). With pulmonary hypertension, the chest leads show right ventricular hypertrophy (Fig. 10.5).
Until and unless pulmonary hypertension develops, the atrial shunting is left to right, and there is no cyanosis. When the mitral insuffi ciency is severe, there may not be a signifi -cant elevation of atrial pressures, because the RA has high capacitance and the RV is adapted for volume overload and accepts the increased left-to-right shunt. However, with a decrease in compliance of a failing RV due to chronic volume overload or a hypertrophied RV because of pulmonary hyper-tension, atrial pressures rise, and the pulmonary venous pressure can be estimated from inspection of the neck veins.
The natural history of ostium primum ASDs differs signifi cantly from that of a secundum-type ASD. Ostium primum defects are susceptible to infective endocarditis of the cleft mitral valve. If the mitral regurgitation is severe, patients may have dyspnea and left ventricular dysfunction, and if the right ventricular compliance is compromised, there are signs of both right ventricular and left ventricular failure.
Patients with complete AV canal seldom survive to late adulthood without cardiac surgery. The ECG shows left axis deviation, right ventricular hypertrophy, large P waves, and various degrees of AV block (Fig. 10.6).
I II
III
aVR aVL aVF
V1V2
V3
V4 V5 V6
FIGURE 10.3. Electrocardiogram of a patient with an ostium primum defect, severe pulmo-nary hypertension, and mitral insuffi ciency through a cleft anterior leafl et. Note the left-axis deviation, right ventricular hypertrophy, and atrial fl utter.
CAR010.indd 238CAR010.indd 238 11/24/2006 10:40:46 AM11/24/2006 10:40:46 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 3 9
1 aVRV1
V2
V3 V6
V5
V4
aVL
aVF
2
3
FIGURE 10.4. Electrocardiogram of a patient with an ostium primum. Note the left-axis deviation, incomplete right bundle branch block in V2, and fi rst- and second-degree atrioventricular block.
I
II
III
aVR aVL aVF
V1
V2
V3
V4
V5
V6
FIGURE 10.5. Electrocardiogram of a patient with an atrial septal defect of secundum type with severe pulmonary hypertension. Note the right-axis deviation and right ventricular hypertrophy. This
patient has slow fl utter with varying atrioventricular conduction, which resembles “paroxysmal atrial tachycardia with block” and may be mistaken for digitalis intoxication.
Laboratory Studies
The echocardiogram classically demonstrates a low-lying ASD with no continuity between the anterior leafl et of the mitral valve and the aortic valve, and a cleft in the anterior leafl et of the mitral valve.20 The mitral and tricuspid valves
are at the same level. There is mitral insuffi ciency of varying degree, an enlarged RV, paradoxical septal motion, and enlarged atria. Angiographically, the left ventriculogram reveals a “goose-neck” deformity (Fig. 10.7), related to the unusual attachment of the anterior leafl et of the mitral valve, and some degree of mitral insuffi ciency.61
CAR010.indd 239CAR010.indd 239 11/24/2006 10:40:46 AM11/24/2006 10:40:46 AM

2 4 0 c h a p t e r 10
1
2
3
aVR
aVL
aVF
V1
V2
V3
V4
V5
V6
FIGURE 10.6. Electrocardiogram of a patient with an ostium primum. Note the left-axis deviation, incomplete right bundle branch block in V2, and fi rst- and second-degree atrioventricular block.
FIGURE 10.7. (A,B) Left ventriculogram of a 21-year-old woman after surgery for closure of an ostium primum defect. Note the usual “bite” out of the inferomedial portion of the left ventricle, giving rise to the so-called goose-neck deformity. This is due to the lower
detachment of the anterior leafl et of the mitral valve to the ven-tricular septum so it is at the same level as the tricuspid valve, whereas normally it is attached more superiorly.
Prognosis
The large left-to-right shunt may eventually lead to pulmo-nary hypertension. The cleft mitral leafl ets result in increas-ing mitral regurgitation and left ventricular dysfunction and is susceptible to infective endocarditis. Finally, AV block may develop as part of the natural history of endocardial cushion defects.
Treatment
The defi nitive treatment for an ostium primum defect is early surgical closure.62 The condition is not suitable for
transcatheter closure, in contrast to the secundum type of septal defect, because the lower border of the defect is in direct continuity with the anterior leafl et of the mitral valve. Precautions during surgical closure include not encroaching on or injuring the AV node and the bundle of His. Therefore, patching rather than direct suturing is required. The treat-ment of the cleft mitral valve depends on the degree of mitral regurgitation. However, valvuloplasty is often feasible in skillful hands. If closure of the cleft in the anterior leafl et of the mitral valve is overly aggressive or if there is insuffi cient native mitral valve tissue, mitral stenosis may occur. Should the patient show evidence of advanced second- or third-
CAR010.indd 240CAR010.indd 240 11/24/2006 10:40:48 AM11/24/2006 10:40:48 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 41
degree AV block, biventricular or dual chamber (DDD) pacing would be advisable but only after the ASD has been closed, because the electrodes are thrombogenic and paradoxical embolization may occur.
Prognosis
The prognosis for ostium primum atrial septal defects that have undergone surgical closure depends on the degree of pulmonary vascular disease, complications associated with mitral valve prostheses, and left ventricular function. If the mitral insuffi ciency has been relatively severe and long-standing, even mitral valve replacement may provide only a short-term reprieve from eventual failure of the LV.
Common Atrium
Common atrium is a variant of AV canal defect. It is char-acterized by a total absence of the atrial septum and a cleft in the anterior leafl et of the mitral valve. The clinical profi le differs from classic AV canal in that these patients invari-ably have right-to-left shunting at the atrial level. Frequently, patients with a common atrium have some cyanosis, if not at rest at least with exercise, because the common atrium acts as an incomplete mixing chamber. The ECG is essen-tially the same as with an ostium primum atrial septal defect. The echocardiogram differs in that there is complete absence of the atrial septum. Because of the pulmonary overcirculation, these patients are symptomatic at an earlier age than other atrial septal defects, and surgical creation of an atrial septum is usually performed in the fi rst year of life.
Sinus Venosus Atrial Septal Defect
Sinus venosus ASD is an ASD located posteriorly subjacent to the superior vena cava. It is almost always associated with the anomalous insertion of at least one pulmonary vein adja-cent to the superior vena cava. Clinically, it is indistinguish-able from a secundum type of atrial septal defect. On echocardiography, the defect is seen posteriorly rather than in the area of the foramen ovale, and an anomalous pulmo-nary vein can be identifi ed. Successful surgical closure of this defect is more diffi cult because of the presence of an anomalous pulmonary vein adjacent to the superior vena cava, and some degree of dehiscence of the patch near that area is common. As long as the residual left-to-right shunt is small, it should not cause diffi culty.63,64
Coronary Sinus Atrial Septal Defect
A congenital defect may occur between the coronary sinus and the LA, which results in a left-to-right shunt that physi-ologically resembles an atrial septal defect.65 The cause of a coronary sinus ASD is unroofi ng of the coronary sinus into the left atrium. This entity can be suspected when echocar-diography fails to identify an ASD in the presence of clinical and radiographic evidence of a left-to-right shunt. Cardiac catheterization yields an oximetry series that is consistent with a shunt at the atrial level. A useful maneuver is to introduce the catheter into the coronary sinus, which is not
diffi cult in the hands of an experienced operator. The coro-nary sinus yields a rather high oxygen saturation, in contrast to the normally very low saturation. It may be diffi cult to distinguish between a coronary sinus ASD and partial anom-alous pulmonary venous connection to the coronary sinus. However, the coronary sinus ASD is more proximal, and sampling of the more distal blood in the coronary sinus should yield a low saturation, whereas with an anomalous pulmonary venous connection to the coronary sinus, there is a higher saturation throughout the coronary sinus. This entity is of some clinical importance because the coronary sinus ostium can be rather large, and there have been rare instances in which the coronary sinus ostium has been mis-takenly closed for an atrial septal defect.
Partial Anomalous Pulmonary Venous Connection
The connection of one or more pulmonary veins to a struc-ture other than the LA should be considered in patients with clinical or radiographic evidence of left-to-right shunting at the atrial level.66 Partial anomalous pulmonary venous con-nection may occur with no atrial communication.67 It may coexist in approximately 10% of secundum-type ASDs. The insertion may be directly into the superior vena cava; into a vertical vein that goes into the innominate vein and then into the superior vena cava; into the RA directly; to the coro-nary sinus68; or, rarely, into the inferior vena cava (scimitar syndrome)69 (Fig. 10.8). If the cardiologist is alert to the pos-sibility, the echocardiographer who is warned is usually able
FIGURE 10.8. Posteroanterior radiograph of patient with partial anomalous venous return to the inferior vena cava (scimitar syn-drome). The heart is normal in size, as is the pulmonary vascula-ture. The anomalous drainage through a large scimitar vein (arrows) is well demonstrated.
CAR010.indd 241CAR010.indd 241 11/24/2006 10:40:50 AM11/24/2006 10:40:50 AM

2 4 2 c h a p t e r 10
to identify these. They may also be identifi ed through cardiac catheterization techniques using selective pulmonary arte-riography (including the levophase) or by means of selective indicator dilution techniques. It is important to be alert to these veins because the closure of an ostium secundum-type ASD alone only partially decreases the left-to-right shunt if several anomalous pulmonary veins coexist.
Ventricular Septal Defects
Isolated ventricular septal defects (VSDs) are infrequently seen in adults.70 Fifty percent or more of VSDs close sponta-neously in early childhood, even as late as adolescence.71,72 If the VSD is small, these defects are associated with little or no hemodynamic disturbance of the LV and result in only a small left-to-right shunt and no pulmonary hypertension. Large defects are associated with equalization of pressure in the two ventricles and therefore in the pulmonary artery. In a large VSD, the direction and the degree of shunting are determined by the relative resistances of the pulmonary and systemic circuits. The right-to-left and the left-to-right shunts are more or less balanced if the resistances in the systemic and pulmonary circulations are equal. This is the classic Eisenmenger complex. Large defects with a low to mildly elevated PVR have large left-to-right shunts with severe volume overload of the LV. These are almost invari-ably discovered by a pediatric cardiologist, and the defect is closed.72 Therefore, in adults, the common forms of congeni-tal ventricular septal defect are either large ones of the Eisen-menger physiology or those in association with pulmonary stenosis, in which the pulmonary circulation is “protected” from the development of pulmonary vascular disease. Occa-sionally, one may see patients who have a moderate-size ventricular septal defect, a large left-to-right shunt, and an enlarged LV and are symptomatic who present as adults (often coming from areas of the world without sophisticated medical care). Unless there are separate, serious comorbid factors, and pulmonary vascular resistance is not severely elevated, the defect should be closed.
The most common form of congenital isolated ventricu-lar septal defect is of the so-called perimembranous type, which is posterior and inferior to the crista supraventricu-laris, involving what would be the membranous septum and some of the adjacent muscular septum.73 This is situated just under the septal leafl et of the tricuspid valve and is sub-tended by the aortic valve. The bundle of His courses along the posterior rim of this defect and therefore is not affected, but it is vulnerable during surgical closure of the defect. Single or multiple muscular septal defects may also occur as isolated congenital lesions.73 In early infancy, up to 50% of isolated VSDs are in the trabecular septum. Later, muscular VSDs are present in about 10% of the cases of VSD. They are generally multiple and small, so that even in the presence of large shunts, there is seldom signifi cant elevation of the right ventricular or pulmonary artery pressure. Although many of these defects close spontaneously, they may persist and are diffi cult to close completely at the time of surgery because of heavy trabeculation on the right ventricular aspect of the ventricular septum. The seemingly logical left ventricular approach to closure of the lesion would seriously compro-mise the contractility of the LV.
The predominant type of congenital isolated VSD seen in adults is that termed the Eisenmenger complex (i.e., a large VSD with severe pulmonary vascular obstruction and bidi-rectional shunting).74
Small Ventricular Septal Defect (Maladie de Roger)
Patients with small VSDs are totally asymptomatic. The volume overload of the LV is minimal to mild. The heart size remains normal, and pulmonary hypertension does not develop on the basis of this defect alone. The sole risk is that of infective endocarditis.
Rarely, a moderate-size defect may be converted to a smaller defect by adhesion of the septal leafl et of the tricus-pid valve, aneurysm formation of the membranous septum, or prolapse of an aortic cusp. This prolapsed cusp may in time become adherent to the VSD, partly occluding it func-tionally. The patient may then present with primarily aortic regurgitation and a small VSD.
The diagnosis of a small VSD is made clinically by the characteristic holosystolic murmur, starting with the fi rst heart sound. This holosystolic murmur should not be mis-taken for mitral insuffi ciency. It is loudest at the left sternal border but does not radiate to the neck. In mitral regurgita-tion caused by a redundant anterior leafl et, the murmur is transmitted to the axilla, whereas if it is associated with a redundant posterior leafl et, it radiates medially and often is transmitted to the neck. The chest x-ray study and ECG are usually normal. The diagnosis is best confi rmed echocardio-graphically with the color-fl ow Doppler technique.75,76 It is also readily visualized on the left ventriculogram (Fig. 10.9). If this is truly a small VSD and not one that has been made small by the prolapse of an aortic cusp, surgery is not indi-cated unless there are other unrelated cardiac defects that require surgical correction. The prognosis is good, and the chief precaution is the need for prophylaxis against infective endocarditis. If a “small” VSD is associated with signifi cant aortic regurgitation due to a prolapsed cusp, the major con-sideration is that of the aortic regurgitation.
Large Ventricular Septal Defect
The rare patient who survives into adulthood with a large VSD and a large left-to-right shunt but without severely ele-vated pulmonary vascular resistance may exhibit symptoms similar to those of patients with signifi cant mitral insuffi -ciency, both representing examples of left ventricular volume overload. The chief manifestation is exertional dyspnea as a consequence of elevated left ventricular fi lling pressure causing elevated pulmonary venous pressure. Radiographic studies show cardiomegaly, primarily of the LV, and enlarged central pulmonary arteries with radiologic evidence of increased pulmonary fl ow. The ECG may show tall voltages in the chest leads associated with left ventricular volume overload. Echocardiography is essential not only to confi rm the large left-to-right shunt but also to determine the type and the location.77 Cardiac catheterization is helpful in defi n-ing the pulmonary hemodynamics for surgical risk assess-ment and for ultimate prognosis. Left ventricular angiography
CAR010.indd 242CAR010.indd 242 11/24/2006 10:40:50 AM11/24/2006 10:40:50 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 4 3
in several views is very helpful. Some of these patients may unexpectedly experience a systolic gradient between the RV and the pulmonary artery (Gasul’s phenomenon) without having true pulmonary stenosis.78
Surgical closure of VSDs is associated with conduction abnormalities in as many as 15% of cases, consisting of right
bundle branch block and left-axis deviation79,80 (Fig. 10.10). Rarely in the current era, damage to the conduction system can result in complete AV block, necessitating placement of a pacemaker. There are no extensive data on the late occur-rence of complete AV block, but it appears to be uncommon.79 These conduction abnormalities are related to the close
FIGURE 10.9. (A,B) Left ventriculogram of a small ventricular septal defect, best seen in the lateral view (B). Note the normal size of the left ventricle.
Lead 1
Lead 2
Lead 3
Trial rate
6 Sec.
6 Sec.
6 Sec.
V1
V2
V3
V4
V5
V6
aVR aVL aVF
Entricular rateHythmWavesEmarks
P-R IntervalQRS IntervalQ-T IntervalT Waves
Patient positionElectrical axisS-T Segment
FIGURE 10.10. Electrocardiogram of a patient who underwent closure of a ventricular septal defect. Note the left-axis deviation and right bundle branch block, which are commonly seen after ventricular septal defect closure whether as an isolated defect or as
part of another entity, such as tetralogy of Fallot. This bifascicular block may uncommonly progress to complete atrioventricular block.
CAR010.indd 243CAR010.indd 243 11/24/2006 10:40:50 AM11/24/2006 10:40:50 AM

2 4 4 c h a p t e r 10
proximity of the bundle of His to the posterior wall of the VSD. Currently, transcatheter device closure of certain VSDs, particularly certain muscular VSDs and perimembranous defects, are being used effectively.81–85
Eisenmenger’s Complex
Eisenmenger’s complex is by far the most common presen-tation of a large VSD in adults.74 These patients exhibit equalized pressures and resistances in the pulmonary and systemic circuits from early childhood onward (Fig. 10.10). At rest, the patients have a bidirectional shunt. With physi-cal exertion and a decrease in systemic vascular resistance (SVR), the right-to-left shunting increases, resulting in increased arterial desaturation and fatigue. There is an inability to increase pulmonary blood fl ow with exercise, so the magnitude of right-to-left shunt increases as systemic oxygen delivery demands increase with exercise and sys-temic blood fl ow increases. Therefore, these patients are self-restricting and seldom need to have physical restric-tions arbitrarily imposed. The longevity of such patients is variable. Causes of death include sudden cardiac death, infective endocarditis, brain abscess, massive hemoptysis, and hyperviscosity. By x-ray study, the heart size may be normal, the central pulmonary arteries are markedly dilated, and there is no evidence of increased pulmonary fl ow (Fig. 10.11).
Echocardiography characteristically demonstrates the location of a large VSD and bidirectional shunting.86 It can also differentiate between an isolated VSD with normally related great vessels from one associated with congenitally corrected transposition with double-outlet RV.
Cardiac catheterization characterizes the hemodynamics (i.e., a large VSD through which the venous catheter can be manipulated into both the aorta and the pulmonary artery), severe pulmonary hypertension with markedly elevated PVR, and bidirectional shunting.87
Continued survival of such patients requires careful follow-up and management.88 The erythrocytosis is a conse-quence of arterial desaturation, and symptoms of hypervis-cosity are peculiar to each patient. Most patients have correlating symptoms, such as visual disturbances, light-headedness, or a “funny feeling,” at which time it may be appropriate to phlebotomize them, simultaneously replacing the blood loss with intravenous saline. It is seldom necessary to bring the hemoglobin to much below 20 g. The use of phlebotomy is controversial; it is variably applied at different adult congenital heart centers. The patients’ symptoms appear to be a better guide than arbitrary numeric guides. A word of caution is necessary here, however. Repeated phlebotomies without iron supplementation result in an iron-defi ciency anemia that can worsen symptoms of hypervisco-sity due to the effects of iron defi ciency on red cell function. In iron defi ciency, the red blood cells are small and more rigid, which increases viscosity. Therefore, iron supplementation may be necessary. Rapid erythrocyte turn-over may also lead to folate defi ciency, which may require folic acid supplementation. These patients should also be cautioned to maintain adequate fl uid intake during hot weather and during exertion.
FIGURE 10.11. (A–E) Eisenmenger’s complex (ventricular septal defect with severe pulmonary hypertension and equal resistances in the pulmonary and systemic circuits). Patient at age 2 (A), at age 10 (B), and at age 20 (E). The left ventriculogram (C and D) shows the normally related great arteries and markedly dilated pulmonary trunk and left pulmonary artery. Commonly, adults with Eisen-menger’s complex have this physiology from early life.
Patients with Eisenmenger’s complex do not progress to CHF unless other factors supervene, such as ventricular dysfunction. The RV is adapted to pressure work, and there is no volume overload. However, injudicious and excessive
CAR010.indd 244CAR010.indd 244 11/24/2006 10:40:51 AM11/24/2006 10:40:51 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 4 5
phlebotomies may render the patient relatively anemic, espe-cially if associated with iron defi ciency, and CHF may ensue. In some patients, moderate pulmonary insuffi ciency may develop, creating a volume overload situation that can also lead to CHF. Treatment of CHF in Eisenmenger’s syndrome is controversial. Historically, digoxin and cautious diuresis were employed. There is some evidence to support judicious use of afterload reduction, but this therapy has the risk of increasing the right-to-left shunt and aggravating the cyano-sis. If the patient has an iron-defi ciency anemia, often owing to repeated phlebotomies, cautious transfusion of packed red blood cells is sometimes helpful, as is iron replacement. Current treatment regimens include bosentan, sildenafi l, and iloprost, and outcomes appear to be improved.88
Many patients with Eisenmenger’s complex manage to do reasonably well, holding down full-time jobs and perform-ing many normal activities, although they are not capable of performing strenuous exertion. Ultimately, increasing pul-monary vascular disease with increasing right-to-left shunt-ing and increasing erythrocytosis make existence diffi cult, and such patients should be considered for heart-lung transplantation.
Patent Ductus Arteriosus
The ductus arteriosus is the main fetal path through which oxygenated umbilical cord blood perfuses distal to the aortic arch of the fetus. With delivery and the baby’s fi rst breath, there is an immediate drop in PVR, resulting in a reversal of the shunt through the ductus arteriosus. Usually, the ductus arterious constricts and is functionally closed by 18 hours after birth. Patency may persist for several weeks without any consequences. The pathophysiologic consequences of persistent patency of the ductus arteriosus depend on the size of the ductus and, to a lesser extent, on the length.89 There is a continuous left-to-right shunt during systole and diastole through the ductus as long as the PVR is lower than the systemic resistance. Predisposing factors for a patent ductus arteriosus (PDA) are maternal rubella in the fi rst trimester of pregnancy, prematurity, and high altitude.
Restrictive Patent Ductus Arteriosus
Most patients with restrictive PDA are asymptomatic because the left-to-right shunt is generally mild to moderate. The pathophysiology is similar to that of aortic regurgitation, and there is a rapid run-off with some degree of left ventricular volume overload. The classic physical fi nding is the machin-ery murmur of Gibson,90 heard best in the left subclavicular region and continuous throughout the whole cardiac cycle. This is not to be confused with long murmurs in systole and diastole, such as may occur in combined aortic stenosis and regurgitation, in which the directional shift in fl ow is marked by a short hiatus between the systolic and the diastolic com-ponents. The murmur is identical to that heard in an AV fi stula, which it in fact resembles physiologically. The radio-graphic study in patients with a small PDA shows a normal heart size with normal pulmonary vasculature. There is a tendency, however, for the aortic arch to be somewhat wider than usual for the patient’s age. With a moderate left-to-right shunt, the heart size may increase somewhat over time, and
there may be a suggestion of increased pulmonary blood fl ow.
The echocardiogram shows evidence of fl ow into the left pulmonary artery because the ductus is usually between the distal aortic arch and the proximal left pulmonary artery91 shortly after bifurcation. During cardiac catheterization, it is usually possible to manipulate the venous catheter from the proximal left pulmonary artery through the ductus into the descending aorta. The pulmonary artery pressure should be measured simultaneously with the systemic arterial pressure.
PROGNOSIS
The prognosis for PDAs with small-to-moderate left-to-right shunt is excellent, the chief risk being endocarditis. Over time, though, the likelihood of endocarditis is signifi cant. Cardiac catheterization can help to determine the status of the pulmonary vasculature.
Because the PDA is extracardiac, its ligation and division do not require open-heart surgery, making the surgical risk virtually the risk of anesthesia only. At present, however, there are transcatheter techniques for closure of a patent ductus, and these are quite suitable for most patients with a PDA who have persistent patency after the fi rst few months of life.92
Large Patent Ductus
With normal or even moderately increased PVR, patients with large PDAs have a large continuous left-to-right shunt (Fig. 10.12). They are only rarely seen as adults because the
FIGURE 10.12. Posteroanterior radiograph of patient with a large patent ductus arteriosus. There is moderate cardiomegaly, with increased pulmonary vasculature and marked enlargement of the main pulmonary artery segment (arrow in prominence of the aorta).
CAR010.indd 245CAR010.indd 245 11/24/2006 10:40:52 AM11/24/2006 10:40:52 AM

2 4 6 c h a p t e r 10
volume overload of the LV and the resultant symptoms betray the diagnosis in childhood, at which time the defect is cor-rected. A large PDA seen in adulthood is almost always one with a PVR slightly less than, equal to, or even greater than the SVR, with either a small bidirectional shunt or an exclu-sively right-to-left shunt. In many respects, this resembles the Eisenmenger complex associated with a ventricular septal defect (Fig. 10.13).93 The patients experience signifi -cant exercise limitations because peripheral vasodilation serves to increase right-to-left shunting and further arterial desaturation. However, unlike the situation in the Eisen-menger complex with a ventricular septal defect, the natural history of a high-pressure, high-resistance PDA may lead to right-sided CHF if the pulmonary artery dilation results in signifi cant pulmonary insuffi ciency. The resulting volume overload of the RV with an intact ventricular septum can lead to decompensation.
The clinical history is that of exercise limitation and sometimes angina-like symptoms with exertion, associated with defi nite cyanosis. Classically, the cyanosis should involve the left hand and the feet and not so much the right hand, although this is occasionally seen (Fig. 10.14). Far more common, however, is cyanosis and clubbing of all four extremities, possibly because of reversal of fl ow in the ascending aorta during early diastole. Sometimes there is hoarseness caused by compression of the recurrent laryngeal nerve, especially if the PDA becomes aneurysmal. Hemop-tysis may occur, presumably because of the pulmonary hypertension.94
LABORATORY STUDIES
The chest x-ray evaluation is characterized by a prominent aortic arch and large central pulmonary arteries. If the PVR is high, the peripheral pulmonary vasculature appears “pruned,” as in the Eisenmenger complex. The heart is usually of normal size. Examination of the heart may dis-close only a systolic murmur or no murmur, prominent pal-pable pulsation of the main pulmonary artery, and a loud P2. Radiographically, it is possible to see the PDA as a slightly oblique shadow between the distal portion of the aortic arch and the pulmonary artery. This is especially evident when there is calcifi cation of the ductus (Fig. 10.13).
Echocardiographically, a high-pressure PDA is best seen by the transesophageal technique. Cardiac catheterization is used to defi ne the pulmonary hemodynamics, and the PDA can be visualized angiographically for the surgeon.
PROGNOSIS
In a large PDA in which there is still an appreciable left-to-right shunt and in which the pulmonary/systemic fl ow ratio is greater than 1.5 : 1, the ductus can still be divided and ligated with the anticipation that the pulmonary artery pres-sure will fall. However, the PVR will remain somewhat ele-vated. When pulmonary fl ow and systemic fl ow are equal, as are their respective resistances, closure of the PDA is contra-indicated, and the only treatment would be heart-lung transplantation.24,95
Endocardial Cushion Defects
The crux of the heart refers to the area where the atrial septum, the ventricular septum, the mitral valve, and the tricuspid valve all come together. The structures that make up this part of the heart are formed by endocardial cushion tissue during development as the common atrioventricular canal is divided. Defects in this part of the heart have been
FIGURE 10.13. Posteroanterior radiograph of patient with patent ductus arteriosus and severe pulmonary vascular disease, as indi-cated by calcifi ed plaque in the pulmonary arteries (curved white arrow). The ductus arteriosus itself is also calcifi ed (black arrow). Pulmonary artery segment and aortic knob are markedly enlarged.
FIGURE 10.14. Differential clubbing in a 23-year-old patient with reversing ductus arteriosus. Note the marked clubbing of the toes and of the fi ngers of the left hand, with normal nails and fi ngers on the right hand. Although classic, this is less common than cyanosis and clubbing of all fi ngers and toes in reversing ductus arteriosus.
CAR010.indd 246CAR010.indd 246 11/24/2006 10:40:52 AM11/24/2006 10:40:52 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 47
variably referred to as endocardial cushion defects, atrioven-tricular canal defects, and atrioventricular septal defects. Each nomenclature has a group of passionate defendants. The severity of disease caused by abnormal development and division of the atrioventricular canal ranges from minor abnormalities of the mitral valve such as a cleft in the ante-rior leafl et to much more severe disturbances such as a complete atrioventricular canal defect or complete atrioven-tricular septal defect. The anterior leafl et of the mitral valve is almost invariably cleft in this group of defects. Defi ciency of the primum atrial septum at the crux is referred to as an ostium primum ASD. Alternatively, this abnormality has been referred to as a partial atrioventricular canal defect. The combination of an ostium primum ASD and a pressure-restrictive VSD is conventionally referred to as a transitional atrioventricular canal defect. A large ASD and a large inlet VSD is referred to as a complete AV canal (septal) defect. Typically, there is a large common AV valve that sits over both ventricles. This common AV valve has four to six leaf-lets, including an anterior bridging leafl et and posterior bridging leafl et that “bridge” the ventricular septum. Defects of the AV septum make up the most common type of defects seen in Down syndrome.
Clinical Presentation
The clinical presentation of ostium primum ASD and its close relative, common atrium, was discussed earlier.
Complete AV canal defect in adults may present in asso-ciation with either pulmonary stenosis or pulmonary hyper-tension. In the latter case, it has many of the characteristics of the Eisenmenger syndrome. These patients have a high incidence of cyanosis. The heart is enlarged, and there is a loud P2. There is an absent P2 in cases with associated pul-monary stenosis, and a loud ejection systolic murmur is heard if pulmonary stenosis is present. Evidence of AV valve regurgitation may be observed by evaluation of deep jugular pulses.
Laboratory Studies
The chest x-ray shows an enlarged heart with a large pulmo-nary arterial tree and, if there is no pulmonary stenosis, evidence of increased pulmonary blood fl ow. Both ventricles and both atria are enlarged. The ECG may be similar to that of an ostium primum ASD (i.e., rSR’ in the anterior chest leads, left-axis deviation, and often fi rst-degree AV block). Echocardiography visualizes the low-lying ASD, a corre-sponding VSD, and the characteristic straddling AV valve.20 If there is pulmonary stenosis, this can be seen with color-fl ow Doppler techniques, and the degree of stenosis can be estimated. Measurements should be made of the pulmonary hemodynamics with quantifi cation of the left-to-right and the right-to-left shunts and the respective resistances. Because of the cyanosis, there is secondary erythrocytosis, which must be carefully managed as discussed earlier.
Prognosis
Long-term survival with a complete atrioventricular septal defect, without surgery, is uncommon, and such patients rarely survive into adulthood.
Treatment
Treatment is surgical unless there is irreversible pulmonary vascular disease. If there is severe pulmonary vascular disease, heart-lung transplantation is the only option. In earlier eras, patients with Down syndrome were often denied surgical therapy. More recently, though, this has not been a consideration, and children with Down syndrome routinely do well following surgery. Over the long term, postoperative morbidity is related to the adequacy of the mitral valve func-tion. Repair of the cleft mitral valve in endocardial cushion defects is challenging and patients are almost always left with some degree of mitral insuffi ciency. This can be severe enough to require a second operation to repair or replace the mitral valve. Additionally, if the atrioventricular tissue is inadequate or the cleft in the anterior leafl et is repaired overaggressively, there can be concomitant mitral stenosis following repair.
Congenital Valve Abnormalities
Aortic Stenosis
Congenital obstruction of the outfl ow tract of the left ven-tricle in the adult is overwhelmingly due to valvar disease and commonly results from the development of aortic steno-sis because of a congenital bicuspid or unicuspid valve.96,97 The incidence of aortic valve disease is four to fi ve times higher in males than in females.98 Idiopathic hypertrophic subaortic stenosis and asymmetric septal hypertrophy, now known as hypertrophic cardiomyopathy (HCM), are different disorders from valvular aortic stenosis and are discussed separately.
Aortic Valve Stenosis
In nonelderly adults, aortic stenosis typically develops because of a bicuspid (or unicuspid) aortic valve.99,100 This is sometimes seen in association with coarctation of the aorta.101 In general, the valve causes little or no hemody-namic disturbance for the fi rst several decades of life and, in fact, may never cause any hemodynamic disturbance at all. Progressive aortic regurgitation may develop, especially in young adulthood. It appears that most valves with this abnor-mality develop progressive thickening and fi brosis after the third or fourth decade of life. Over time, increasing calcifi ca-tion and progressively severe aortic stenosis (AS) develops. The presence of mild AS may be associated with progres-sively severe aortic regurgitation when inadequate coapta-tion outweighs inadequate opening of the valve.101 Bacterial endocarditis may also transform the pathophysiologic dis-turbance from predominant stenosis to predominant regurgitation.
The course of the disease, once stenosis begins, tends to be relentless but has a highly variable timetable. In general, the progression to severe AS may take decades, although on rare occasion may be rapid. After signifi cant AS develops, the natural history is similar to that of acquired AS. The classic ominous symptoms are syncope, angina, and CHF. Sudden death is a threat even when mild symptoms are noted.98,102–104 However, evaluation for surgery should be undertaken if
CAR010.indd 247CAR010.indd 247 11/24/2006 10:40:52 AM11/24/2006 10:40:52 AM

2 4 8 c h a p t e r 10
there has been any subjective change in exercise tolerance or in the patient’s general sense of well-being. Most patients with predominant AS have well-preserved left ventricular function if exertional syncope is the primary manifestation. Although angina may occur in severe AS without concomi-tant CAD, CAD is not uncommon in the presence of AS and should always be evaluated when surgery on the aortic valve is contemplated.105–107 The appearance of overt CHF is usually of ominous prognostic consequences, although some patients may recover virtually normal left ventricular function after successful surgery. Approximately 5% of patients with a bicuspid aortic valve (BAV) have associated cystic medial disease of the aorta, which can become the basis of an aortic dissection. Coarctation of the aorta is sometimes associated with a BAV (it is far more common for coarctation of the aorta to be associated with a BAV, rather than the reverse), which has its own inherent natural history and complica-tions.108 Before the aortic valve has become heavily calcifi ed, the presence of a BAV has classic physical examination fi nd-ings. The most characteristic feature of a BAV is a loud sys-tolic ejection click, which coincides with the doming of the aortic valve toward the end of isovolumetric contraction of the LV.109 In a younger age group, this may be the only physi-cal examination fi nding, although commonly there is an aortic systolic murmur that starts after the ejection sound. The ejection click is usually somewhat pronounced, and it is likely that the doming of the aortic valve prolongs the phase of isovolumetric contraction.109,110 The aortic systolic murmur, if present, starts with the ejection click and may be of variable intensity.98,111
The second heart sound characteristically preserves both components (A2 and P2) until signifi cant AS develops, when the second sound becomes single. There may or may not be
an early diastolic blowing murmur of aortic regurgitation. The peripheral pulse contour characteristically shows an anacrotic notch. In general, the more severe the stenosis, the more delayed the upstroke and the lower the anacrotic notch on the upstroke.98,112 Classically, the heart is not enlarged.113 If the aortic valve is heavily calcifi ed, it can be visualized, especially in the lateral view of the chest fi lm, and can be readily seen on fl uoroscopy. A characteristic associated fi nding is dilation of the ascending aorta (Fig. 10.15).114 This dilation is asymmetric, anterior, and to the right and is believed to be due to the direction of the abnormal fl ow pattern across the bicuspid aortic valve. This “poststenotic dilatation” can be observed even when there is no systolic gradient across the aortic valve. The ECG may be unremark-able throughout the course of the disease up to the time of surgery, although it may demonstrate a characteristic “strain” pattern of left ventricular hypertrophy in some patients.
DIAGNOSIS
The clinical diagnosis of aortic valve stenosis is made by auscultating the characteristic ejection systolic murmur, which is harsh, heard in the upper right sternal border and suprasternal notch, and radiates to the neck.98 This murmur may be mimicked by mitral insuffi ciency associated with an “overshooting” of the posterior mitral valve leafl et directing the regurgitant jet anteriorly and medially, giving rise to a murmur best heard along the left sternal border and some-times heard in the neck.115–117 Valvar pulmonary stenosis may also be heard in the neck. An aortic ejection sound strongly suggests the presence of a BAV. With that, many or most such aortic valves become more stenotic.7,118–121 The ejection sound may gradually disappear, and its absence is associated with increasing fi brosis and calcifi cation of the valve leafl ets.122,123
FIGURE 10.15. Aortic stenosis on a congenitally bicuspid aortic valve. Note the eccentric poststenotic dilatation of the ascending aorta, with a normal-sized aortic ring. The ventriculogram illus-trates the contraction of the severely hypertrophied left ventricle to a very small end-systolic volume.
CAR010.indd 248CAR010.indd 248 11/24/2006 10:40:53 AM11/24/2006 10:40:53 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 4 9
The fi rst heart sound is characteristically soft, and the A2 component of S2 gradually diminishes as the stenosis becomes progressively more severe. A soft diastolic murmur of aortic insuffi ciency is not uncommon as the cusps become more rigid. As the stenosis becomes more severe, an S4 becomes prominent and may sometimes be mistaken for S1. The peripheral pulses show an anacrotic notch and a somewhat delayed upstroke. In general, the lower the notch, the more severe the stenosis. The heart is usually not grossly enlarged unless there is a signifi cant degree of aortic insuffi ciency.
Radiographically, the heart is typically of normal size unless there is signifi cant concomitant regurgitation, possi-bly with some calcifi cation of the aortic valve and “postste-notic” dilatation of the ascending aorta, as previously described (Fig. 10.16). The ECG, even in cases of severe aortic stenosis, may range from normal to that characteristic of left ventricular hypertrophy with a “strain” pattern (Fig. 10.17) and may not be helpful in the clinical evaluation of severity. The echocardiogram shows a thickened aortic valve, which becomes increasingly immobile as it becomes more calcifi ed. The LV exhibits concentric hypertrophy, and the systolic gradient across the aortic valve can be estimated by the velocity of the systolic jet.124,125 Similarly, aortic insuffi ciency can be visualized.
On cardiac catheterization, the LV can be entered either retrograde across the stenotic aortic valve or by transseptal puncture from the femoral venous approach to the LA and then the LV. Angiography shows the characteristic postste-notic dilatation of the ascending aorta, which does not involve the aortic ring or sinuses. Simultaneous pressures
across the aortic valve can be measured, and with simultane-ous measurement of cardiac output, the valve area can be calculated from the Gorlin formula,126,127 assuming inconse-quential aortic regurgitation. If the patient has CHF, the clinical picture must be modifi ed to include left ventricular dilation, decreased contractility, and a ventricular gallop rhythm.
TREATMENT
There is no absolute aortic valve area that clearly dictates intervention. In general, the patient who is normally active, who has no symptoms, and in whom the clinical signs and the echocardiogram do not suggest severe aortic stenosis can be safely watched. Even when the evidence points to signifi -cant aortic stenosis, the total absence of symptoms should outweigh the objective measurements of valve orifi ce size in continuing careful observation and follow-up.98 However, with even mild changes in status that can be ascribed to the heart, together with the physical signs of signifi cant aortic stenosis that are confi rmed by echocardiography and cathe-terization, surgical replacement of the aortic valve should be seriously considered. These changes may involve merely a subtle change in well-being or a slight decrease in exercise tolerance.128 It is advisable to proceed with cardiac catheter-ization when it is felt that surgery is imminent, both to
FIGURE 10.16. Posteroanterior radiograph of a patient with con-genital aortic stenosis. The heart is slightly enlarged and has a left ventricular confi guration. The pulmonary vasculature is normal. There is striking prominence of the ascending aorta (arrow) as a result of poststenotic dilatation.
59-year-old man 12364016No aortic systolic gradient
67-year-old man 12364016Severe aortic stenosis, symptomatic
FIGURE 10.17. Electrocardiograms of a patient who developed severe symptomatic aortic stenosis within less than 8 years. Tight stenosis and angina were corrected by valve replacement. Note the absence of signifi cant electrocardiographic changes during this interval.
CAR010.indd 249CAR010.indd 249 11/24/2006 10:40:53 AM11/24/2006 10:40:53 AM

2 5 0 c h a p t e r 10
confi rm the clinical and echocardiographic evidence and to visualize the coronary arteries. It is not wise to wait until major symptoms arise, because the incidence of sudden death rises dramatically.129 In patients with AS and angina, there is a signifi cant incidence of coexisting CAD.105,106 Treatment involves surgical replacement of the calcifi ed valve130,131 by a prosthetic valve, and coronary artery bypass grafting, if indicated. Even patients older than 80 years of age may benefi t from valve replacement; they have an opera-tive mortality of 9.4% and a good outcome in 81% of cases described. Porcine bioprosthesis is especially suitable in the elderly, in whom the longevity of such prostheses is better than when they are implanted in younger patients. Balloon valvuloplasty has been useful in children but disappointing (but improving) in adults, for whom the benefi ts are of limited duration, especially with a stiff, calcifi ed valve.132–135 More recently, innovations have occurred including percuta-neous valve repair and replacement approaches, which appear to have promise.135,136
Long-term follow-up on a large cohort of 462 patients with congenital aortic stenosis (mostly children) showed that with gradients of less than 50 mm Hg, medical follow-up is appropriate, whereas with gradients of more than 80 mm Hg, intervention is clearly indicated.130
Supravalvar Aortic Stenosis
Supravalvar aortic stenosis typically occurs in two set-tings.137,138 Williams syndrome is a dysmorphic syndrome caused by a mutation of the elastin gene on chromosome 7,139 most typically a deletion. Often, there is hypercalcemia in infancy. Affected people have characteristic facies and char-acteristic personality traits and limits in learning. As infants, there is often pulmonary artery branch stenosis, which regresses with time. At the time of diagnosis in infancy, there may be no real supravalvar aortic obstruction, but over time the aortic obstruction tends to be progressive. There is also another group of patients with supravalvar aortic steno-sis that is familial. These individuals appear to have autoso-mal dominant transmission and do not have the fi ndings of dysmorphic features, hypercalcemia in infancy, or behav-ioral abnormalities. On physical examination, there is usually no ejection click, but there is a harsh ejection murmur at the right upper sternal border that transmits prominently into the carotid arteries. Additionally, there is a peculiar physical examination fi nding, caused by the Coanda effect. The Coanda effect causes the jet of blood passing through the supravalvar narrowing to track along the greater curve of the aorta and into the innominate artery and subsequently into the right subclavian artery. This creates a higher systolic blood pressure in the right arm than in the left arm or the legs. While the pathophysiology seems similar to aortic valve stenosis, there are some important differences. The coronary artery origins are proximal to the obstruction. Since coro-nary fl ow is most prominent in diastole, coronary blood fl ow may be compromised in severe obstruction because blood fl ow back from the aorta into the coronary arteries is com-promised. Also, ostial stenosis is often present, causing even more coronary compromise. As the obstruction tends to be progressive, treatment typically consists of surgical repair once there is more than mild stenosis.
Subaortic Stenosis
Subvalvar aortic stenosis has three types: membranous sub-aortic stenosis,98,140 a fi bromuscular tunnel type,98,141 and an idiopathic hypertrophic subaortic stenosis type. Discrete subaortic stenosis may be familial.98,142
Membranous subaortic stenosis or subaortic rings are uncommon in adults. An aortic systolic murmur and often an aortic diastolic murmur are present, the latter resulting from aortic valvar regurgitation. This is postulated to be related to damage to the aortic valve caused by the jet of blood fl owing through the obstructive membrane.143 In patients who have a predominantly muscular ring, a charac-teristic Brockenbrough’s sign may be observed after prema-ture beats (i.e., a fall in the aortic systolic pressure and an increase in the ventricular pressure after an extrasystole). Treatment of this condition involves resection of the ring. At surgery, the resection may be incomplete, to avoid damage to the conduction system or the anterior leafl et of the mitral valve.143,144
Idiopathic hypertrophic subaortic stenosis is an older term for one of the fi ndings in some patients with hypertro-phic cardiomyopathy. This disorder is not discussed in this chapter.
Pulmonary Stenosis
Pulmonary stenosis (PS) can be valvar, supravalvar, or sub-valvar. Although pulmonary stenosis may occur as an iso-lated anomaly, it is often associated with an ASD, VSD, or transposition of the great arteries (TGA).
Pulmonary Valve Stenosis
Pulmonary valvar stenosis typically is caused by commis-sural fusion and is associated with a dome-shaped valve. Alternatively the valve may be dysplastic.145,146 The conse-quent obstruction to right ventricular outfl ow results in right ventricular hypertrophy and, when severe, an increase in the right ventricular fi lling pressure. Patients with iso-lated mild PS usually do well,147–149 whereas those with mod-erate (peak-to-peak systolic gradient of 50 to 80 mm Hg) or severe stenosis (systolic gradient >80 mm Hg) should be con-sidered for intervention.150,151
Multiple peripheral PSs are rare in the adult (Fig. 10.18). These are almost always associated with one of several clini-cal syndromes, including the following:
1. Williams syndrome137–139
2. Noonan syndrome152,153
3. Congenital rubella syndrome154
In patients who have an associated patent foramen ovale, right-to-left shunting at rest may occur during atrial systole (during which the right atrial pressure exceeds that of the LA) or during exercise or tachycardia, when the pressure in the RA may exceed that of the LA throughout the cardiac cycle, resulting in arterial desaturation. These elevated right atrial pressures result from the decreased compliance of the hypertrophied RV. The Valsalva maneuver at rest may cause a transient increase in the right atrial pressure relative to that of the LA during its release.
CAR010.indd 250CAR010.indd 250 11/24/2006 10:40:53 AM11/24/2006 10:40:53 AM
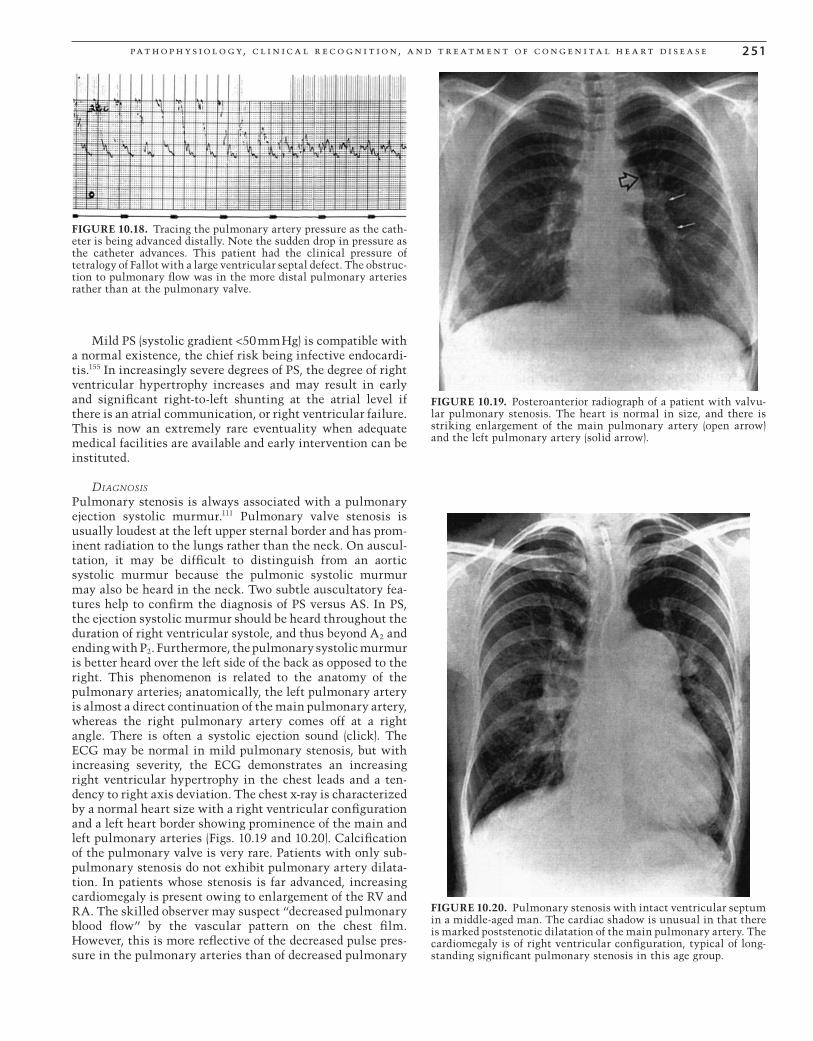
pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 51
Mild PS (systolic gradient <50 mm Hg) is compatible with a normal existence, the chief risk being infective endocardi-tis.155 In increasingly severe degrees of PS, the degree of right ventricular hypertrophy increases and may result in early and signifi cant right-to-left shunting at the atrial level if there is an atrial communication, or right ventricular failure. This is now an extremely rare eventuality when adequate medical facilities are available and early intervention can be instituted.
DIAGNOSIS
Pulmonary stenosis is always associated with a pulmonary ejection systolic murmur.111 Pulmonary valve stenosis is usually loudest at the left upper sternal border and has prom-inent radiation to the lungs rather than the neck. On auscul-tation, it may be diffi cult to distinguish from an aortic systolic murmur because the pulmonic systolic murmur may also be heard in the neck. Two subtle auscultatory fea-tures help to confi rm the diagnosis of PS versus AS. In PS, the ejection systolic murmur should be heard throughout the duration of right ventricular systole, and thus beyond A2 and ending with P2. Furthermore, the pulmonary systolic murmur is better heard over the left side of the back as opposed to the right. This phenomenon is related to the anatomy of the pulmonary arteries; anatomically, the left pulmonary artery is almost a direct continuation of the main pulmonary artery, whereas the right pulmonary artery comes off at a right angle. There is often a systolic ejection sound (click). The ECG may be normal in mild pulmonary stenosis, but with increasing severity, the ECG demonstrates an increasing right ventricular hypertrophy in the chest leads and a ten-dency to right axis deviation. The chest x-ray is characterized by a normal heart size with a right ventricular confi guration and a left heart border showing prominence of the main and left pulmonary arteries (Figs. 10.19 and 10.20). Calcifi cation of the pulmonary valve is very rare. Patients with only sub-pulmonary stenosis do not exhibit pulmonary artery dilata-tion. In patients whose stenosis is far advanced, increasing cardiomegaly is present owing to enlargement of the RV and RA. The skilled observer may suspect “decreased pulmonary blood fl ow” by the vascular pattern on the chest fi lm. However, this is more refl ective of the decreased pulse pres-sure in the pulmonary arteries than of decreased pulmonary
FIGURE 10.18. Tracing the pulmonary artery pressure as the cath-eter is being advanced distally. Note the sudden drop in pressure as the catheter advances. This patient had the clinical pressure of tetralogy of Fallot with a large ventricular septal defect. The obstruc-tion to pulmonary fl ow was in the more distal pulmonary arteries rather than at the pulmonary valve.
FIGURE 10.19. Posteroanterior radiograph of a patient with valvu-lar pulmonary stenosis. The heart is normal in size, and there is striking enlargement of the main pulmonary artery (open arrow) and the left pulmonary artery (solid arrow).
FIGURE 10.20. Pulmonary stenosis with intact ventricular septum in a middle-aged man. The cardiac shadow is unusual in that there is marked poststenotic dilatation of the main pulmonary artery. The cardiomegaly is of right ventricular confi guration, typical of long-standing signifi cant pulmonary stenosis in this age group.
CAR010.indd 251CAR010.indd 251 11/24/2006 10:40:53 AM11/24/2006 10:40:53 AM

2 52 c h a p t e r 10
fl ow because it is the same as the cardiac output. The echo-cardiogram demonstrates a dome-shaped stenotic pulmonary valve, the severity of which can be gauged by Doppler deter-mination of the pressure gradient and assessment of coexist-ing infundibular stenosis. Pulse oximetry can be useful to detect an associated right-to-left atrial level shunt.
When stenosis is mild, the ECG is usually normal. With moderate to more severe PS (systolic gradient >50 mm Hg), right ventricular hypertrophy is seen in the chest leads, and “P pulmonale” will be increasingly evident (Fig. 10.21).
Cardiac catheterization for direct measurement of the systolic gradient across the pulmonic valve, combined with simultaneous cardiac output measurements, permits calcu-lation of valve orifi ce size by the Gorlin formula.126 The catheter may cross a probe-patent foramen ovale, and, if so, a comparison of the oxygen saturation of the pulmonary vein and that of the systemic artery indicates whether a signifi -cant right-to-left shunt is present. Angiography in the RV visualizes the valve and right ventricular outfl ow tract stenosis.
PROGNOSIS
In the modern era, the prognosis is usually good because the striking murmur demands cardiac evaluation and, when indicated, intervention. However, such patients are always at risk for infective endocarditis.
TREATMENT
Treatment of isolated pulmonary valvar stenosis formerly entailed surgical valvuloplasty, but increasingly, the treat-ment of choice is percutaneous transvenous balloon valvu-loplasty.156–159 This is the noninvasive analogue of the older Brock procedure (pulmonary valvotomy)160 but with more adequate dilatation of the valve. Treatment with balloon valvuloplasty is highly successful and can be repeated if the dilatation is inadequate or restenosis occurs.161–163 If there is a coexisting probe-patent foramen ovale, consideration should be given to its closure because late paradoxical embolism is possible, but during balloon dilation, it may be protective.164 Several devices are now able to close atrial communications by transcatheter techniques.28–51,165,166
Long-term follow-up of patients with PS shows that the probability of survival is similar to that of the general popu-lation after 25 years (95.7%) whether medically or, when indicated, surgically/catheter treated. Survival is somewhat shorter when associated cardiomegaly is present.145,167
Subpulmonary Stenosis
Subpulmonary stenosis is relatively uncommon as an iso-lated lesion but is the more common type of pulmonary ste-nosis associated with a ventricular septal defect (tetralogy of Fallot).168 Pulmonary stenosis can be either membranous or infundibular and may be clinically indistinguishable from pulmonary valvar stenosis. However, pulmonary valvar ste-nosis is associated with poststenotic dilatation of the main pulmonary artery, which is readily seen on chest x-ray, and the echocardiogram is defi nitive. When the condition is severe, the treatment is surgical, with either resection of the obstruction or roofi ng of the outfl ow tract by patching.169,170 On rare occasion, the “stenosis” may be caused by a tricuspid valve aneurysm during systole.171
Mitral Stenosis
Congenital mitral stenosis (MS) in adults is uncommon. Shone syndrome is the term used to describe the occurrence of multiple levels of obstruction to blood fl ow at levels of fl ow into and out of the left ventricle and in the aorta.172,173 The classic picture includes MS with a parachute mitral valve (a single papillary muscle), subaortic stenosis, a bicus-pid aortic valve, and coarctation of the aorta. These patients typically require intervention in infancy and often repair will be staged with different lesions requiring intervention at different ages. In adulthood, the clinical picture may resemble that of rheumatic mitral stenosis, in that there is a long and initially asymptomatic history followed by gradu-ally increasing exertional dyspnea and fatigability. Atrial fi brillation may develop secondary to enlargement of the LA as a consequence of the stenosis.
Cardiac examination does not identify the opening snap characteristic of rheumatic mitral stenosis, and a dia-stolic rumble may or may not be present. However, there may be signs of pulmonary hypertension associated with a loud P2 and an active RV. The defi nitive examination is echocar-diography, both transthoracic and transesophageal. However, hemodynamic studies at cardiac catheterization are impor-tant for the characterization of pulmonary hemodynamics and for the search for other features of the Shone syndrome.
Treatment consists of excision of the mitral valve with valve replacement, and removal of the supravalvar ring if this is signifi cantly stenotic.
Tricuspid Stenosis
Congenital tricuspid stenosis is extremely rare as an isolated lesion.174,175 Most cases of adult tricuspid stenosis are acquired, such as those associated with rheumatic fever, carcinoid syn-drome, or right atrial myxoma.
Echocardiography provides a defi nitive diagnosis by dem-onstrating a stenotic tricuspid valve with an enlarged RA.
FIGURE 10.21. Electrocardiogram of a patient with severe pulmo-nary stenosis. Note the right-axis deviation and severe right ven-tricular hypertrophy.
CAR010.indd 252CAR010.indd 252 11/24/2006 10:40:55 AM11/24/2006 10:40:55 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 53
Cardiac catheterization is usually unnecessary unless the coexistence of other anomalies cannot be entirely excluded by echocardiography.
Atrioventricular Valvar Regurgitation
Atrioventricular (AV) valvar regurgitation, long held to always be pathologic, is now seen as a phenomenon that may at times be “normal” under certain conditions when it is minimal. This has become clear with newer, highly sensitive echocardiographic techniques. Near-instant closure of the AV valves is dependent on normal sinus rhythm, an optimal PR interval, and normal ventricular function. The swiftness of AV valve closure is greatest when there is sinus rhythm with a normal PR interval, resulting in a ventricular contrac-tion that generates the steepest ventricular pressure rise as a consequence of the presystolic atrial “boost.” Therefore, in atrial fi brillation, mild valve regurgitation may occur in the presence of a normal valve. Similarly, in marked fi rst-degree AV block, mild regurgitation may occur because atrial systole is not timely. In AV dissociation or third-degree heart block, mild AV valve regurgitation may be intermittent, depending on the timing of atrial contraction in relation to ventricular systole. Normally, one may see mild tricuspid regurgitation, even with sinus rhythm. Thus, pathologic valvar regurgita-tion entails more than trivial regurgitation in the presence of sinus rhythm and a normal PR interval.
Mitral Valve Regurgitation
Isolated congenital mitral regurgitation is rare. Rarely, an isolated cleft in the anterior leafl et of the mitral valve can lead to mitral regurgitation. A cleft in the anterior leafl et of the mitral valve usually is in the spectrum of AV canal or endocardial cushion defects.
Aortic Valve Regurgitation
Congenital aortic valve insuffi ciency is extremely uncom-mon. There have been rare reports of absence of the aortic valve. These patients have severe aortic valve insuffi ciency with severe consequences on cardiac output and ventricular function. Patients born with an absent aortic valve require surgical valve replacement, most typically with a homograft valve, early in the neonatal period.
Ebstein’s Anomaly
Ebstein’s anomaly consists of downward displacement of the tricuspid valve so that the septal and posterior leafl ets are adherent to the right ventricular wall, thus to a greater or lesser extent “atrializing” the infl ow tract of the RV.176,177 The tricuspid valve is deformed and may assume a cribriform appearance, and it is invariably associated with tricuspid regurgitation. Some evidence has linked this anomaly to maternal use of lithium.178
The physical fi ndings show a characteristic “quadruple cadence” consisting of S1, one or multiple clicks (“sail sound”) produced by the upward motion of the billowing anterior leafl et, and wide splitting of S2 due to right bundle branch block.
In mild cases of Ebstein’s anomaly, there is little or no functional disturbance, and the condition is compatible with a normal life expectancy. When the condition is severe, the entire infl ow tract is functionally atrialized, and there is little pump function of the RV. There is an associated atrial communication, with a patent foramen ovale or an ASD in 50% of patients. If right-to-left shunting occurs, cyanosis and fatigability ensue. The large “atrium” also predisposes to atrial fi brillation. The chest x-ray is characteristic and has been described as a “pumpkin” heart (Fig. 10.22).
The diagnosis is made by echocardiography to delineate the tricuspid valve, assess right ventricular function, and look for right-to-left shunting.177,179
The ECG may show one of two patterns: Wolff-Parkinson-White pattern180 or an unusual right bundle branch block with a “splintered” QRS complex in V1 or V2 (Fig. 10.23). These patients may have an accessory AV pathway that predisposes to supraventricular tachyarrhythmias (SVTs).
Treatment
No treatment is needed when the condition is mild.181 If there is considerable atrialization of the RV and some function in the remaining RV, tricuspid valve replacement or reconstruc-tion with closure of the ASD may be indicated.182 Electro-physiologic studies are appropriate so that an accessory pathway can be interrupted at operation. When arrhythmias are signifi cant, they require treatment. These include atrial ectopic tachycardia, atrial fl utter and fi brillation, atrial reentry tachycardia, and ventricular tachycardia (VT).183
FIGURE 10.22. Posteroanterior radiograph of a patient with Ebstein’s malformation of the tricuspid valve with right-to-left shunt. The cardiac silhouette is huge. There is prominence of the right atrial border. The pulmonary artery segment and aorta are inconspicuous. The pulmonary vasculature is frankly decreased.
CAR010.indd 253CAR010.indd 253 11/24/2006 10:40:55 AM11/24/2006 10:40:55 AM

2 5 4 c h a p t e r 10
Therapy may include radiofrequency ablation.183 Severe cases of Ebstein’s anomaly, however, do not do well over the long term. In some cases, cardiac transplantation is required.184
Coarctation of the Aorta
By far, the most common congenital aortic obstruction in adults is coarctation of the aorta (COA, which in adults is almost always at or just distal to the ligamentum arteriosum and the takeoff of the left subclavian artery. This anomaly is seen in adults who have hypertension in the arms and in whom there is a decrease in the pulse pressure of the femoral arteries. It is twice as common in men and is sometimes seen in patients with Turner syndrome.185–187 Sometimes there is a discrepancy between the blood pressure in the two arms. This may occur because the coarctation is proximal to the left subclavian artery stenosis or because there is an anoma-lous origin of the right subclavian artery distal to the coarc-tation or because the origin of the left subclavian artery is involved in the coarctation and is stenotic.
Physiologically, there is hypertension of the arterial system proximal to the coarctation and normal blood pres-sure distal to the COA. Outside of infancy, the decreased or absent pulses in the distal arteries are due not to low fl ow but to a narrow pulse pressure. Invariably, in untreated adults, the mean pressure in the distal arterial tree is normal. Flow to the distal aorta is by a number of collaterals via arte-rial anastomoses involving the internal mammary, intercos-tal, and superior and inferior epigastric arteries. It has been well demonstrated by Shepherd188 that fl ow distal to the coarctation is normal at rest and in exercise. This is often not appreciated if it is assumed that impalpable pulses are associated with low fl ow. The difference in the systolic pres-sure between the upper and the lower halves of the body is determined by the degree of coarctation and by the caliber and the number of the collateral vessels. Hypertension results both from mechanical obstruction, and damping of pulsatile fl ow distal to the coarctation may affect the renin-angioten-sin system via the juxtaglomerular apparatus leading to a high renin state.
Mild COA initially may not be associated with hyperten-sion. In general, though, the severity of obstruction is pro-gressive and the natural history of unoperated COA in the
presurgical era was ominous with only 10% of patients sur-viving to age 50 years. Coarctation of the aorta is the most common congenital cardiovascular disease in Turner syndrome.187,189
Diagnosis
There are seldom symptoms from hypertension alone. Because blood fl ow distal to the coarctation is normal, lower body development is normal, and claudication is not neces-sarily a part of the clinical picture. In the adult, no symptoms occur until one or more complications develop.190 The diag-nosis is made clinically by the faint or absent pulses in the femoral arteries in the presence of hypertension in the arms. In some instances, the femoral artery pulsations are good, but simultaneous pressure measurements in the arms and legs reveal the systolic pressure in the lower extremities to be somewhat lower than in the arms. A bicuspid aortic valve is present in more than half the cases.190,191 Sometimes, the intercostal collaterals are palpable, and murmurs are audible. The chest x-ray study often shows a notch at the site of the coarctation just distal to the aortic arch, the so-called “3” sign (Fig. 10.24), caused by dilatation of the proximal aorta because of the hypertension and poststenotic dilatation distal to the coarctation. There is frequently notching of the pos-teroinferior border of the ribs after the second rib (Fig. 10.25). These are not directly due to increased fl ow per se in the intercostal arteries but rather to the tortuosity and resulting “knuckling” of the arteries from increased fl ow, the notches corresponding to the knuckling. Mild cases of COA are sometimes found incidentally in the evaluation of a bicuspid aortic valve192 (Fig. 10.26).
Echocardiography is useful to assess the anatomy and function of the aortic valve and the degree of left ventricular hypertrophy and to follow the diameter of the ascending aorta, especially after surgery. Transesophageal echocardiog-raphy may or may not visualize the coarctation optimally.
FIGURE 10.23. Electrocardiogram of a 24-year-old man with Ebstein’s anomaly. Note the right bundle branch block pattern, with “splintering” of the QRS complex in the V2 (i.e., R’SS’S).
FIGURE 10.24. Coarctation of the aorta in a 24-year-old man. Note the “3” sign just beyond the aortic arch, where the indentation (arrow) is the site of the coarctation with a dilated aortic arch proxi-mal to the coarctation because of the hypertension.
CAR010.indd 254CAR010.indd 254 11/24/2006 10:40:55 AM11/24/2006 10:40:55 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 5 5
Magnetic resonance imaging (MRI) has emerged as the most valuable tool to completely outline the anatomy of the aortic arch in coarctation.193,194 It can provide important informa-tion that is helpful to direct surgical or interventional cath-eterization procedures.195,196
Prognosis
The frequently associated congenital bicuspid aortic valve discussed elsewhere infl uences prognosis separately if it pro-gresses to signifi cant aortic stenosis or refl ux.96–98 There is
an increased incidence of cystic medial changes in the aorta,197 which may predispose to aortic dissection, the ini-tiation of which may be just above the aortic valve or in the aortic arch.198,199 Aortic rupture may occur as an infrequent complication of dissection. Infective endocarditis may occur at the site of coarctation or, more commonly, on the bicuspid aortic valve. The presence of arterial hypertension from birth predisposes to premature cerebrovascular disease and CAD if the hypertension persists into adulthood.186,197 Cerebral hemorrhage may occur in 20% of cases as a result of a fre-quently associated berry aneurysm of the circle of Willis.200 In women, pregnancy increases the risk for aortic rupture, ventricular dysfunction, and increased hypertension.201,202 Most patients who survive childhood reach adulthood without having symptoms but hypertension, until one of the complications supervenes.
Treatment
Treatment in adults should be undertaken as soon as the diagnosis is made. Balloon dilatation is often performed in the pediatric age group, but recurrence is common in native coarctation and has limited enthusiasm.203,204 Currently, stents have been used with better outcomes in adolescents and adults.204 In adults, the most common form of coarcta-tion is a discrete constriction, which is best resected with end-to-end anastomosis.205,206 Sometimes signifi cant resteno-sis occurs, necessitating reintervention. Long tubular coarc-tation is rare in adults. Cross-clamping of the aorta during surgery does not usually cause ischemia of the kidneys or spinal cord, because most of the distal aortic fl ow is via col-laterals. Overall, most patients have been treated with surgery over the years. There appears to be a place for balloon angioplasty with stent placement as an alternative to surgery is some patients. After successful correction in the adult, hypertension often persists but is more readily controlled medically, especially with repair in later adulthood. This persistent hypertension appears to be related to barometer resetting and activation of the renin-angiotensin system. Patients must be followed clinically throughout their lives because of the potential complications associated with
FIGURE 10.25. Posteroanterior radiograph of a patient with coarc-tation of the aorta. The heart is not enlarged, and the pulmonary vasculature is normal. The aortic arch is inconspicuous owing to tubular hypoplasia of the distal segment. Rib notching (arrows) is demonstrated.
FIGURE 10.26. (A,B) Mild coarctation found incidentally during evaluation of aortic stenosis on a bicuspid aortic valve. Note the characteristic poststenotic dilatation.
CAR010.indd 255CAR010.indd 255 11/24/2006 10:40:57 AM11/24/2006 10:40:57 AM

2 5 6 c h a p t e r 10
hypertension and the frequently associated bicuspid aortic valve.
Complex Congenital Heart Disease
Complex congenital heart disease is a group of diseases in which more than one cardiac abnormality is present, which may or may not be associated with cyanosis.
Without Cyanosis
Congenital heart diseases without cyanosis have been alluded to in separate categories. Their clinical presentation is a composite of their individual pathophysiology. Such combi-nations include atrial septal defects with one or more anoma-lous pulmonary veins,66–69 Shone’s syndrome,172 Williams syndrome (supravalvar aortic stenosis) and peripheral pulmo-nary artery stenoses (with characteristic facies), aortic insuf-fi ciency and mitral insuffi ciency with Marfan syndrome,207 coarctation of the aorta or ventricular septal defect with Turner syndrome,187 pulmonic stenosis or AV canal defect in Noonan syndrome,153 and various degrees of AV block with endocardial cushion defects, from fi rst- to second- to third-degree AV block.
With Cyanosis
These conditions involve TGA. Three categories of great vessel transposition can be seen in the adult: D-transposition, L-transposition, and double-outlet RV.
D-Transposition (Complete Transposition) of the Great Arteries (D-TGA)
This form of transposition of the great arteries involves vir-tually a direct switch between the two great arteries so that the aorta arises anteriorly from the RV and the pulmonary artery arises posteriorly from the LV208 (Figs. 10.27, 10.28, 10.29). It is evident that without intercommunication, these would be two closed circuits that are incompatible with life. Therefore, there must be a shunt at the atrial level (most common) or at the ventricular level, or else a large PDA. In patients with an ASD or VSD, pulmonary stenosis may be present, which “protects” the pulmonary vasculature.209 Uncorrected D-TGA is compatible with survival into adult-hood, but patients are invariably cyanotic. Because the dis-ability is obvious in childhood, these patients now undergo surgery before they reach adulthood. Echocardiography208,210 is the diagnostic modality of choice. Cardiac catheterization is essential to characterize the pulmonary vasculature for any consideration of surgery. Surgical treatment for this con-dition has undergone considerable evolution, from “atrial switching” (actually rerouting of venous blood to the appro-priate ventricle) to present-day methods, including arterial switching.208,211
Tetralogy of Fallot
Tetralogy of Fallot (TOF) consists of a large ventricular septal defect in the usual position, together with pulmonary steno-
sis, either valvar or infundibular, or both.212 The large VSD results in equal pressure in the two ventricles. The two other features constituting the “tetralogy” are right ventricular hypertrophy and overriding of the ventricular septum by the dilated aorta. This malformation is compatible with surviv-ing into adulthood.213,214 The more severe the PS, the greater the degree of right-to-left shunting at the ventricular level (Fig. 10.30). The more complete the arterial desaturation, the greater the disability. Such patients not only are prone to the usual complications of cyanotic heart disease (e.g., endocar-ditis, brain abscess, and complications of marked erythrocy-tosis) but may also experience sudden death, which has been attributed to spasm of the infundibulum.215 Lesser degrees of PS result in a lesser degree of right-to-left shunting, with a modestly decreased exercise capacity but with the ability to perform most normal activities. In some patients, the degree of PS is such that the patient is not visibly cyanotic at rest but only with exercise; such patients are said to have tardive cyanosis.216
The usual clinical fi ndings in the adult are cyanosis and clubbing of the digits and a loud, harsh pulmonary ejection murmur with spilling over A2 to a very soft P2 of the second heart sound, or P2 may even be absent. This murmur is often well heard in the neck, and such radiation should not mislead one into thinking that this is of aortic origin. Frequently, there is a systolic ejection sound that arises in the dilated ascending aorta. A faint blowing diastolic murmur of pul-monary insuffi ciency is sometimes heard either starting with P2 or, when P2 is inaudible, starting after a gap from A2.
FIGURE 10.27. Posteroanterior radiograph of a patient with trans-position of the great vessels, ventricular septal defect, and pulmo-nary stenosis. The heart size is at the upper limits of normal, and the pulmonary vasculature is slightly accentuated. The main pul-monary artery segment is absent.
CAR010.indd 256CAR010.indd 256 11/24/2006 10:40:57 AM11/24/2006 10:40:57 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 57
RPALPA
MPA
RV
Complete transposition, VSD, hypoplasia LPAA B
DC
AO PA
Inf
RV
FIGURE 10.28. (A–D) D-transposition in a 28-year-old man. (D) The completely switched position of the pulmonary artery (PA) and the aorta (AO) is evident, with the aortic valve anterior to the pul-monary trunk and the pulmonary valve. LPA, left PA; MPA, main PA; RPA, right PA; RV, right ventricle.
FIGURE 10.29. (A,B) Double-outlet right ventricle in a 25-year-old man with a supracristal ventricular septal defect with signifi cant left-to-right shunting. Note the kyphoscoliosis, which is not uncommon in cyanotic congenital heart disease.
CAR010.indd 257CAR010.indd 257 11/24/2006 10:40:58 AM11/24/2006 10:40:58 AM

2 5 8 c h a p t e r 10
The chest x-ray (Fig. 10.31) is characterized by a some-what globular heart of normal size, and on the lateral view a prominent RV can be seen “hugging” the sternum. There may be a right-sided aortic arch, which deviates the esopha-gus and the sternum slightly to the left. If the pulmonary stenosis is purely valvar and tricuspid, as occurs in about 20% of these cases, poststenotic dilation of the pulmonary trunk and left pulmonary artery occurs.217 With the much more common infundibular stenosis, the left heart border is concave because the pulmonary trunk is relatively small. This stenosis usually creates a “third chamber” just subja-cent to the pulmonary valve.206,207 The ECG shows RV hyper-trophy (RVH), often with a right bundle branch block pattern. Echocardiography shows the large VSD, the overriding large aorta, the pulmonary valvar or infundibular stenosis with the systolic gradient by Doppler, and RVH. Cardiac catheter-ization can quantify the pulmonary artery pressure and the PVR and can measure the systolic gradient between the pul-monary artery and the RV. By careful catheter withdrawal, it is possible to quantify the systolic gradient across the valve and also across the infundibulum, if both coexist. Right ventriculography can indicate the size and the location of the ventricular septal defect, and biplanar angiography demon-strates the extent of the infundibular stenosis, if present (Fig. 10.32). Sometimes, unusual moderator bands can mimic classic infundibular stenosis.
PROGNOSIS
Patients who reach adulthood with uncorrected tetralogy of Fallot used to be common, but now the vast majority of such patients are diagnosed in childhood and undergo surgery.169,170 Patients who have had a surgically placed Potts shunt or a Waterston anastomosis may experience signifi cant rise in PVR,169,170 unlike those who have undergone the single Blalock-Taussig anastomosis,218 in which pulmonary vascu-lar changes are exceedingly rare. However, a double Blalock-Taussig anastomosis may result in volume overload of the LV (Fig. 10.33).
FIGURE 10.30. This 22-year-old man had origin of both great vessels from the right ventricle with the ventricular septal defect under the pulmonary valve. (A) In this chest fi lm note the unusual left-sided heart border caused by the markedly dilated pulmonary artery (PA). (B) This ventriculogram shows both great vessels arising from the right ven-tricle. This patient had an Eisenmenger physiology. AO, aorta.
FIGURE 10.31. Posteroanterior radiograph of a patient with tetral-ogy of Fallot. The heart is normal size and boot shaped in confi gura-tion. The pulmonary vasculature is decreased. The esophagus is displaced toward the left by a right-sided aortic arch.
TREATMENT
The fi rst palliative operation for cyanotic congenital heart disease with pulmonic stenosis was the Blalock-Taussig anastomosis (B-T shunt), anastomosing the subclavian artery to the pulmonary artery, resulting in signifi cant functional improvement, with many patients reaching adulthood.218 Surgical correction now consists of closure of the VSD and relief of the PS.169,170 If the PS is valvar, the entire procedure can be performed transatrially for both the closure of the VSD and the pulmonary valvotomy. In patients in whom the
CAR010.indd 258CAR010.indd 258 11/24/2006 10:40:58 AM11/24/2006 10:40:58 AM
pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 5 9
PS is largely infundibular, it may be necessary to expand and roof the outfl ow tract and the pulmonary trunk by incision and patching. In general, the postoperative results of such surgery have been extremely gratifying, with restoration of full functional capacity.169,170,219,220 However, late sequelae may include recurrent supraventricular tachyarrhythmias related to the atriotomy, ventricular tachycardias related to ventriculotomy, and mild right-sided CHF resulting from ventriculotomy and pulmonary regurgitation.169,170,219,220 Such CHF is uncommon, but when it exists, it occurs mostly in patients who have undergone ventriculotomy, either to close the VSD or to open up the infundibular stenosis rather than using the transatrial approach.
Many patients with TOF have a mild degree of aortic insuffi ciency, which is related more to the dilatation of aortic root than to intrinsic valvar disease. This is almost never of clinical signifi cance. Over the long term, many problems remain in patients who have undergone “total correction”: impaired right heart function, decreased exercise intoler-ance, and especially rhythm disorders.169,170,219–222
Pulmonary Atresia with Ventricular Septal Defect (Pseudotruncus Arteriosus)
Pulmonary atresia may be associated with a large VSD and is sometimes considered the extreme version of the tetralogy of Fallot.168 A more uncommon situation exists when the pulmonary atresia is seen with an intact ventricular septum (PA-IVS) and a small RV that is drained by large coronary sinusoids. The latter is almost never seen by cardiologists treating adults.223
Pulmonary atresia associated with a VSD (PS-VSD) is more usefully thought of clinically as an entity apart from the tetralogy of Fallot.168 The clinical manifestations and the surgical considerations are quite different. The pulmonary circulation is derived entirely from large anomalous arteries that arise from the descending aorta, often called bronchial arteries224 (Fig. 10.34). At the junction of these arteries with the pulmonary arteries, it is common to observe signifi cant stenosis, so that the distal pulmonary arteries may be of relatively normal pressure. Pulmonary atresia can range
PA
Inf
PA
Inf PA
Inf
PA
Inf
PA
Inf
PA
InfP.V.
VSD
LVRVRV
Im.V.
VSD
RV
LV
AO
P.V.
RV RV
FIGURE 10.32. Right ventriculogram of a 40-year-old patient with tetralogy of Fallot. In the upper panels are anteroposterior views showing the normal relationship of the pulmonary trunk and the ascending aorta (AO). The lower panels show an infundibular
chamber just under the pulmonary valve (Pulm. V.) and also show contrast from the right ventricle (RV) passing through a high ven-tricular septal defect (VSD) into the left ventricle (LV). The ventricu-lar septum can be clearly seen.
CAR010.indd 259CAR010.indd 259 11/24/2006 10:40:58 AM11/24/2006 10:40:58 AM
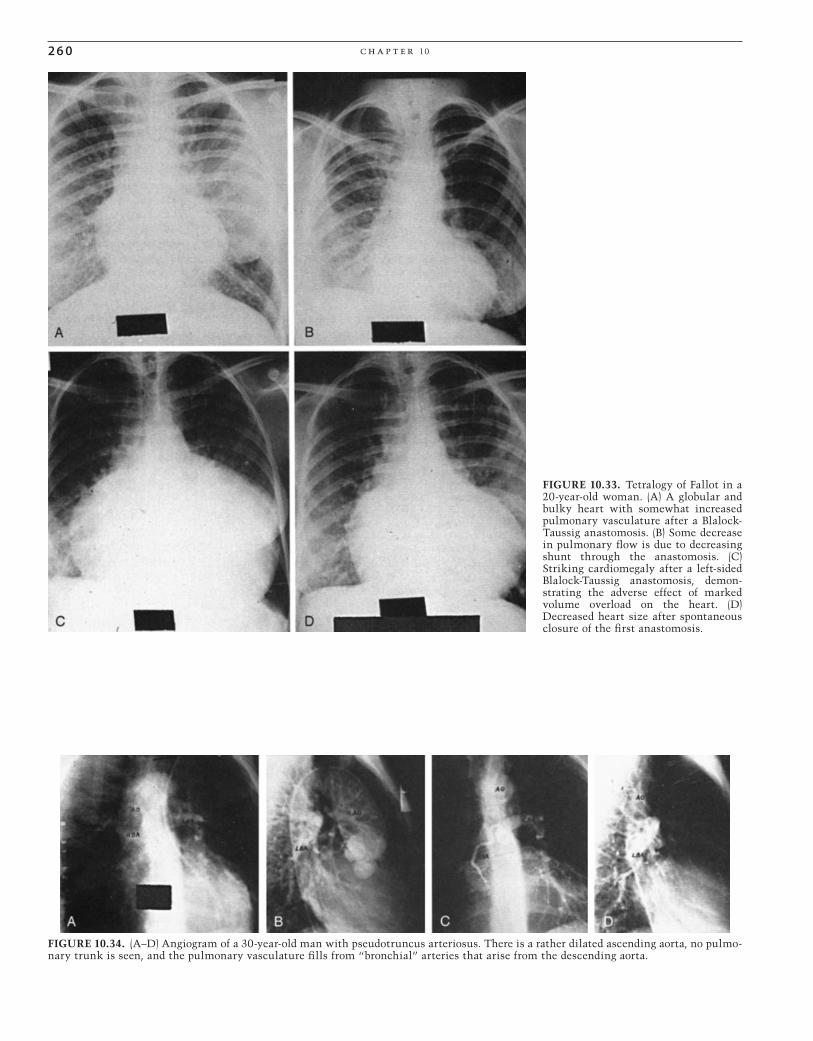
2 6 0 c h a p t e r 10
FIGURE 10.33. Tetralogy of Fallot in a 20-year-old woman. (A) A globular and bulky heart with somewhat increased pulmonary vasculature after a Blalock-Taussig anastomosis. (B) Some decrease in pulmonary fl ow is due to decreasing shunt through the anastomosis. (C) Striking cardiomegaly after a left-sided Blalock-Taussig anastomosis, demon-strating the adverse effect of marked volume overload on the heart. (D) Decreased heart size after spontaneous closure of the fi rst anastomosis.
FIGURE 10.34. (A–D) Angiogram of a 30-year-old man with pseudotruncus arteriosus. There is a rather dilated ascending aorta, no pulmo-nary trunk is seen, and the pulmonary vasculature fi lls from “bronchial” arteries that arise from the descending aorta.
CAR010.indd 260CAR010.indd 260 11/24/2006 10:40:59 AM11/24/2006 10:40:59 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 61
from mere atresia of the pulmonary valve, which is uncom-mon, to atresia of the pulmonary trunk and often atresia of the proximal left and right pulmonary arteries. These patients are markedly cyanotic. A characteristic feature on clinical examination is the presence of a continuous murmur throughout the chest, arising from the stenoses of the bron-chopulmonary anastomoses. The chest x-ray demonstrates a large ascending aorta and aortic arch, and absence of the pulmonary trunk and the left and right pulmonary arteries (Fig. 10.35). There are bronchial arteries arising from the descending aorta. Echocardiography can identify the absence of a pulmonary artery in the presence of a large ventricular septal defect. Cardiac catheterization is essential for the measurement of pressures in the distal pulmonary arteries. This is a very diffi cult procedure, which involves cannulat-ing the bronchial arteries arising from the descending aorta and threading the catheter into the distal arteries beyond the stenoses. All the arteries arising from the descending aorta that are supplying the pulmonary vasculature should be thus cannulated.
Arteriography of each of these vessels is also important to defi ne the anatomy. If the anatomy is suitable and the PVR acceptable, some of these patients can be considered for staged open-heart surgery to establish continuity between the RV and the pulmonary arteries.223 This can be performed only in highly selected individuals and carries a signifi cant surgical risk.
Hypoplastic Left Heart Syndrome
Hypoplastic left heart syndrome is an unusual lesion in adult congenital cardiology clinics. Until the 1980s, this complex was almost universally fatal in the fi rst year of life.225 In 1982, Norwood reported his initial experience with a complex palliation that has subsequently become known as the Norwood procedure.226 Classic hypoplastic left heart syn-drome (HLHS) consists of mitral atresia or severe mitral ste-nosis, aortic atresia or severe aortic stenosis, left ventricular hypoplasia which is usually severe, and hypoplasia of the
ascending aorta. Over the past two decades, the Norwood procedure has become standard therapy in most centers, and in the future a cadre of these patients will survive into adulthood.227–229
Double-Outlet Right Ventricle
A double-outlet RV (DORV) is rarely seen in the adult. Both the aorta and the pulmonary artery arise from the RV.230 The relationship of the great arteries is more or less usual. There is an obligatory VSD.230 When the VSD is under the aortic valve (Taussig-Bing anomaly), cyanosis is minimal or mild, with left ventricular blood fl owing preferentially into the aorta and pulmonary fl ow coming chiefl y from the RV. Right ventricular blood goes to both great vessels but primarily to the pulmonary artery. When the VSD is subjacent to the pulmonary valve, the situation physiologically resembles that of D-transposition of the great vessels with ventricular septal defect. Diagnostic confi rmation is by echocardiogra-phy230 and by cardiac catheterization and angiography230 for consideration of surgical correction.231,232
Tricuspid Atresia
Tricuspid atresia233 is unusual but is compatible with sur-vival to adulthood, providing that there is a coexisting VSD.233,234 This anomaly never occurs alone because live birth obligates patency of the foramen ovale or, more com-monly, a secundum type of ASD. In patients who survive to adulthood, coexisting PS or subpulmonary stenosis is virtu-ally always present. About one fourth of the patients also will have transposition of the great arteries, with the aorta arising from a hypoplastic right ventricle. Patients with tri-cuspid atresia are invariably cyanotic because there is mixing at both the atrial and at the ventricular level. The degree of disability largely depends on the degree of arterial desatura-tion. This, in turn, depends on the magnitude of pulmonary blood fl ow. Patients with a large VSD and mild PS will have the most pulmonary blood fl ow and be the least cyanotic
FIGURE 10.35. (A,B) Pseudotruncus arteriosus in a 24-year-old man. Note the large ascending aorta and aortic arch and marked concavity of the left heart border owing to the absence of the central pulmonary arteries.
CAR010.indd 261CAR010.indd 261 11/24/2006 10:40:59 AM11/24/2006 10:40:59 AM

2 6 2 c h a p t e r 10
initially. Over time, though, there will be high pulmonary blood fl ow and pulmonary vascular disease will develop. If pulmonary vascular disease develops, standard single ven-tricle palliations such as the Fontan procedure will be impossible.235
Clinically, there is always marked cyanosis with club-bing. The characteristic murmur of pulmonary stenosis can be heard. There is some degree of cardiomegaly. On chest x-ray study, the enlarged heart is somewhat bottle shaped, with normal or somewhat decreased pulmonary vasculature (Fig. 10.36). There may be associated skeletal anomalies, such as pectus excavatum and kyphoscoliosis.
The ECG shows left axis deviation in patients with nor-mally related great arteries and normal axis in patients with
D-TGA. The P waves are often large and bizarre (Figs. 10.37 and 10.38). Atrial arrhythmias are common, especially atrial fi brillation and atrial fl utter (Fig. 10.39). In some patients, preexcitation or Wolff-Parkinson-White (WPW) syndrome is notable on the ECG.236
The echocardiogram demonstrates an absence of the infl ow portion of the right ventricle. There is an ASD with obligate right-to-left shunting, a functionally single ventricle communicating with a rudimentary ventricle via a VSD, and often PS. There may be transposition of the great arteries.
Cardiac catheterization provides important information if a Fontan procedure is contemplated.235 The PVR must be carefully quantifi ed and adequacy of ventricular function determined. These can be accurately obtained only with direct measurements of pressures in the pulmonary artery and the two atria and measurement of simultaneous fl ow by the Fick method. Measurement of pulmonary artery pressure is needed to calculate the PVR. Because even a mild elevation of PVR greatly decreased the success of the Fontan proce-dure,237 direct measurement is vital.
FIGURE 10.36. Tricuspid atresia in a 16-year-old girl. Note that there is unusual cardiomegaly due to the greatly dilated “right atrium,” which is actually composed of the true right atrium and the thin-walled, atrialized right ventricle.
FIGURE 10.37. A 34-year-old man with tricuspid atresia and nor-mally related great vessels. Note the left-axis deviation and the unusual P waves.
I
II
III
RHY THM STRIP: II25 mm/sec; I cm/mV
LOC 50000-0006
aVF
aVL
aVR V1
V2
V3 V6
V5
V4
15569
FIGURE 10.38. A 25-year-old man with tricuspid atresia. Note the marked right-axis deviation in the frontal plane. This patient had L-transposition of the great arteries.
CAR010.indd 262CAR010.indd 262 11/24/2006 10:40:59 AM11/24/2006 10:40:59 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 6 3
TREATMENT OF TRICUSPID ATRESIA WITH PULMONARY STENOSIS
Earlier treatment modalities were directed toward increasing pulmonary blood fl ow by creating systemic artery to pulmo-nary artery shunts.238–242 The earliest treatment was the Blalock-Taussig anastomosis, originally devised for tetralogy of Fallot.218 In the Blalock-Taussig operation, a signifi cant portion of the partially oxygenated arterial blood is recycled into the pulmonary vasculature for more complete oxygen-ation. The Glenn procedure (anastomosis of the superior vena cava to the right pulmonary artery) is another palliative modality.243 In the Glenn procedure, no true shunt is involved, because the operation allows the superior vena cava to bypass the right heart and empty directly into the pulmonary artery. However, blood from the inferior vena cava continues to shunt from right to left, so that although there is some clini-cal improvement, the increasing desaturation of the inferior vena cava blood associated with work by the lower extremi-ties, such as walking and running, results in shunting of increasingly desaturated blood and therefore a considerable limitation of exercise capacity.
In 1971, Fontan and colleagues237 devised an ingenious procedure to separate the pulmonary and systemic circula-tions. The original procedure entailed closing the ASD, closing the stenotic pulmonary valve, and anastomosing the RA to the pulmonary artery by means of a conduit. A modi-fi cation in which the communication between the RA and the pulmonary artery is achieved by anastomosing the right atrial appendage to the pulmonary artery avoids the use of prosthetic material.244 In this arrangement, fl ow proceeds from the venous system to the LA entirely by the pressure gradient between the RA and the LA. Two obligatory condi-
tions must be met for this to succeed: a virtually passive pulmonary vascular bed (i.e., an unequivocally normal PVR) and a completely normal left ventricular fi lling pressure.245 When these conditions are rigidly met, this operation is sur-prisingly successful and the patients are able to live relatively normal lives with normally saturated arterial blood. However, the cardiac output response to exercise is somewhat limited and, therefore, so is the maximal exercise capacity.245 The long-term results are uncertain but it appears that a signifi -cant portion of this increasingly large patient population will require cardiac transplantation.246–249 In adulthood, palliative shunts should be performed only when the physiologic condi-tions preclude a Fontan procedure and the degree of cyanosis is severe.
Truncus Arteriosus
Truncus arteriosus is an incomplete septation of the ascend-ing aorta and the pulmonary trunk.250,251 The semilunar valve is a single truncus valve, which in most cases consists of three cusps but may be either quadricuspid or bicuspid. It is commonly insuffi cient, sometimes markedly so, and strad-dles a VSD.250 Truncus defects are divided into three types.250 Type I has a common trunk, but this gives rise distally to recognizably separate ascending aorta and pulmonary trunk (Fig. 10.40). In type II, the truncus extends up to the right and left pulmonary artery bifurcations, there being no sepa-rate pulmonary trunk (Fig. 10.41). In type III, the left and right pulmonary arteries arise from either side of the truncus. In adults, the most common type is a type I truncus.252 Such patients have a regurgitant truncal valve, a common mixing chamber, and Eisenmenger’s physiology. In the very young
V.P. # 844 379Tricuspid atresia
17
I
II
III
aVR aVL aVF
V1
V2.5
V3.5
V4
V5
V6
V4RFIGURE 10.39. Electrocardiogram of a young woman with tricuspid atresia and chronic atrial fl utter.
CAR010.indd 263CAR010.indd 263 11/24/2006 10:41:02 AM11/24/2006 10:41:02 AM
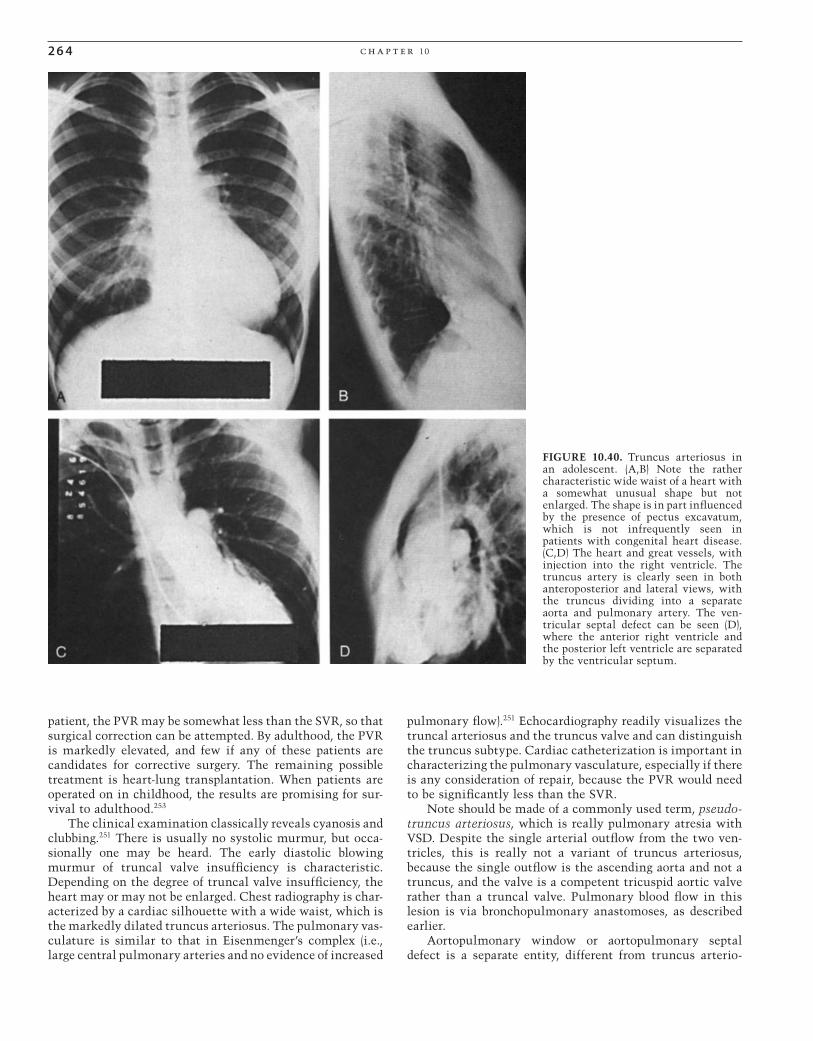
2 6 4 c h a p t e r 10
patient, the PVR may be somewhat less than the SVR, so that surgical correction can be attempted. By adulthood, the PVR is markedly elevated, and few if any of these patients are candidates for corrective surgery. The remaining possible treatment is heart-lung transplantation. When patients are operated on in childhood, the results are promising for sur-vival to adulthood.253
The clinical examination classically reveals cyanosis and clubbing.251 There is usually no systolic murmur, but occa-sionally one may be heard. The early diastolic blowing murmur of truncal valve insuffi ciency is characteristic. Depending on the degree of truncal valve insuffi ciency, the heart may or may not be enlarged. Chest radiography is char-acterized by a cardiac silhouette with a wide waist, which is the markedly dilated truncus arteriosus. The pulmonary vas-culature is similar to that in Eisenmenger’s complex (i.e., large central pulmonary arteries and no evidence of increased
pulmonary fl ow).251 Echocardiography readily visualizes the truncal arteriosus and the truncus valve and can distinguish the truncus subtype. Cardiac catheterization is important in characterizing the pulmonary vasculature, especially if there is any consideration of repair, because the PVR would need to be signifi cantly less than the SVR.
Note should be made of a commonly used term, pseudo-truncus arteriosus, which is really pulmonary atresia with VSD. Despite the single arterial outfl ow from the two ven-tricles, this is really not a variant of truncus arteriosus, because the single outfl ow is the ascending aorta and not a truncus, and the valve is a competent tricuspid aortic valve rather than a truncal valve. Pulmonary blood fl ow in this lesion is via bronchopulmonary anastomoses, as described earlier.
Aortopulmonary window or aortopulmonary septal defect is a separate entity, different from truncus arterio-
FIGURE 10.40. Truncus arteriosus in an adolescent. (A,B) Note the rather characteristic wide waist of a heart with a somewhat unusual shape but not enlarged. The shape is in part infl uenced by the presence of pectus excavatum, which is not infrequently seen in patients with congenital heart disease. (C,D) The heart and great vessels, with injection into the right ventricle. The truncus artery is clearly seen in both anteroposterior and lateral views, with the truncus dividing into a separate aorta and pulmonary artery. The ven-tricular septal defect can be seen (D), where the anterior right ventricle and the posterior left ventricle are separated by the ventricular septum.
CAR010.indd 264CAR010.indd 264 11/24/2006 10:41:03 AM11/24/2006 10:41:03 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 6 5
sus.254 In this condition, there is a “window” in the septum between the aorta and the pulmonary trunk. The aorta and the pulmonary trunk have separate semilunar valves, and the window does not go down to and involve the semilunar valves. If the window is large, the adult patient presents with Eisenmenger’s syndrome (Fig. 10.42). Smaller windows may protect the pulmonary vasculature and allow closure of the defect.
Congenitally Corrected Transposition
Numerous attempts at standardizing the nomenclature of this anomaly have been attempted but none has ever been completely satisfactory. These include L-transposition of the great arteries (L-TGA), corrected transposition (CC-TGA), congenitally corrected transposition, and ventricular inver-sion. The most precise description is atrioventricular discor-dance or ventricular inversion. This “inversion” involves both the AV valves and the semilunar valves, so that the venous ventricle is the anatomic LV, the arterial ventricle is the anatomic RV, and the crista supraventricularis is on the left, as is the tricuspid valve, whereas the mitral valve is the right AV valve. The AV conduction system goes down the left side of the heart. The atria are not involved, and therefore the venous return to both atria and the coronary sinus is in the normal position. However, the aortic root and the pul-monary trunk do not have an anteroposterior relationship but rather a side-by-side relationship, so that the aortic root forms the left heart border and the pulmonary trunk arises medially. Were there no other abnormalities, this would be a functionally “normal” heart and therefore would not belong
under the rubric of cyanotic congenital heart disease.255 However, such normal hearts are uncommon, and in such cases, two common features in the natural history make it unlikely that these patients will have a normal heart for the rest of their natural lives. These patients are prone to develop AV block, which may progress to third-degree block with a narrow QRS.255,256 Although they may have Adams-Stokes attacks, this is a less common manifestation, but exercise tolerance may become limited because of the inadequate chronotropic response to exercise. Furthermore, the left-sided AV valve (i.e., the tricuspid valve) is prone to dysplasia or to Ebstein’s anomaly.254 If the patient survives into adult-hood, this Ebstein’s anomaly is not apt to be severe, but the valve becomes insuffi cient because of systemic pressure in the arterial ventricle. This may therefore necessitate subse-quent valve replacement.255,256
In the vast majority of patients who reach adulthood, there is communication at the ventricular level associated with pulmonary stenosis. A ventricular septal defect or sometimes even a functionally common ventricle may be present. Such patients are always cyanotic but have a pulmo-nary vasculature that is “protected” against the development of pulmonary hypertension. The physical examination fi nd-ings are those of severe PS, together with cyanosis and club-bing. The chest x-ray shows a normal heart size, very often with a straight left heart border, this being the ascending aorta (Figs. 10.43 and 10.44). The heart may have an unusual contour if the inverted RV is prominent (Fig. 10.45). The ECG
FIGURE 10.41. Posteroanterior radiograph of a patient with truncus arteriosus type II. The heart is markedly enlarged, and no main pulmonary artery segment is demonstrated. The right and left pul-monary arteries are prominent, as is the aorta. The left pulmonary artery is in an abnormally high position (arrow).
FIGURE 10.42. Posteroanterior radiograph of a patient with ventricular septal defect and severe pulmonary vascular disease (Eisenmenger’s syndrome). The heart is moderately enlarged, and the central pulmonary artery is distinctly enlarged, whereas the peripheral pulmonary vasculature is normal. The main pulmonary artery is huge (arrow) as a result of long-standing pulmonary hypertension.
CAR010.indd 265CAR010.indd 265 11/24/2006 10:41:04 AM11/24/2006 10:41:04 AM

2 6 6 c h a p t e r 10
FIGURE 10.43. Congenitally corrected transposition in a 35-year-old man with ventricular septal defect and no pulmonary stenosis. Note the straight left-sided heart border, which is formed by the L-transposed ascending aorta and the inverted right ventricle.
FIGURE 10.44. (A–D) An 18-year-old woman with congenitally corrected transposition of the great vessels and a common ventricle. (A,B) The anteropos-terior views show that the ascending aorta forms the left-sided heart border and the pulmonary trunk arises more medially. (C,D) The lateral views show that the semilunar valves are more side-to-side than anteroposterior to each other.
shows evidence of right ventricular hypertrophy and fi rst-, second-, or third-degree AV block (Figs. 10.46 and 10.47).
The echocardiogram is characteristic.255 The inversion of the ventricles can be identifi ed by the presence of the crista supraventricularis in the arterial ventricle, the clearly switched AV valves, and the unusual position of the two semilunar valves. Because the aorta and the tricuspid valve are now on the same side, the aortic valve and the left-sided (tricuspid) AV valve are not in continuity, as would be the relationship between the aortic and the mitral valves in the normal heart. Ebstein’s anomaly of the left-sided AV valve can also be visualized if present. The VSD and PS can be further identifi ed. For purposes of possible surgical treat-ment, one needs to know whether the ventricular communi-cation is a functionally common ventricle or a large VSD.
Cardiac catheterization is important for characterizing the hemodynamics, including ventricular function, the degree of pulmonary stenosis, and the PVR. These data are especially important in patients who have a common ven-tricle, with the possibility of a modifi ed Fontan procedure as a therapeutic option. Angiography can quantify the degree of left AV valve insuffi ciency and the respective positions of the two great arteries. The pulmonary stenosis can be confi rmed as being either valvar or subvalvar. Angiography also helps
CAR010.indd 266CAR010.indd 266 11/24/2006 10:41:04 AM11/24/2006 10:41:04 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 67
to detail the anatomy of the coronary arteries, which undergo a curious “rotation.”
Patients who start out in life with a “normal” heart usually have trouble eventually with the supervention of various degrees of heart block and with insuffi ciency of the left-sided AV valve. The prognosis for patients who have VSD with PS has improved considerably with modern surgical therapy. However, these patients are still susceptible to infec-tive endocarditis. Even with correction of the left-sided AV valve insuffi ciency, the anatomic RV may not perform opti-mally over the years.257,258
TREATMENT
The treatment of this lesion is surgical.255,258 Because the clinical profi le of such patients closely resembles that of tetralogy of Fallot, many of them have undergone a Blalock-Taussig shunt anastomosis with marked improvement in functional capacity and arrive at adulthood only mildly cya-notic.259 Although they may not be functional class I, they may still be quite functional but receptive to future surgical procedures.260,261
For patients who have an ventricular septal defect and pulmonary stenosis, closure of the VSD and pulmonary val-votomy would seem reasonable. The approach to the defect from the right side in this lesion, however, is technically
FIGURE 10.45. Posteroanterior radiograph of a patient with cor-rected transposition, ventricular septal defect, and pulmonary ste-nosis. The heart is slightly enlarged, and the pulmonary vasculature is within normal limits. There is a prominent bulge at the left heart contour (arrows), representing the inverted right ventricle. The pul-monary segment is absent because the main pulmonary artery lies centrally within the mediastinum and is no longer border forming.
38EC 38EC
I
II
III
V1
V1
V2
V3
V2 V5
V6V3
V1
V4
aVF
aVL
aVR
3 0.5
.5
1.0
0.5
.5
1.0 1.0
1.0
1.0
1.0
M
F
Au
R
FIGURE 10.46. Electrocardiogram of a 43-year-old patient with congenitally corrected transposition with pulmonary stenosis and ventricular septal defect. Note the absence of Q wave in the lateral chest leads. This is because of the ventricular inversion, with the septum depolarizing from right to left rather than from left to right.
FIGURE 10.47. Electrocardiogram of a 54-year-old woman with a third-degree heart block. She did well over the years, but eventually, the inadequate chronotropic response to exercise necessitated a pacemaker.
CAR010.indd 267CAR010.indd 267 11/24/2006 10:41:04 AM11/24/2006 10:41:04 AM

2 6 8 c h a p t e r 10
diffi cult. Patients with a common ventricle and pulmonary stenosis may be eligible for a modifi ed Fontan proce-dure,235,237,244 provided that the PVR and ventricular fi lling pressure are normal. This can be accomplished by converting the heart into one with tricuspid atresia by closing the tri-cuspid valve and placing a conduit between the RA and the pulmonary artery.
Great Vein Malpositions
Partial Anomalous Pulmonary Venous Return (Connection)
Partial anomalous pulmonary venous return (PAPVR) is physiologically similar to left-to-right shunting at the atrial level.262 There may be one, two, or three pulmonary veins that are anomalously connected, and they may drain together or separately into the innominate–superior vena cava system, coronary sinus, RA, or inferior vena cava. These constitute pure left-to-right shunts. When all four pulmonary veins connect anomalously (i.e., total anomalous pulmonary venous connection, TAPVC) and drain eventually into the RA, there is an obligatory ASD, the RA becoming a mixing chamber, with right-to-left shunting at the atrial level.
Total Anomalous Pulmonary Venous Connection
Total anomalous pulmonary venous connection is uncom-mon in the adult.262 The most common form is type I, in which the pulmonary veins drain via a large ascending verti-cal vein into the left innominate vein and then to the supe-rior vena cava. Less common is type II, in which the veins drain into a left superior vena cava and then into the coro-nary sinus. In type III, the pulmonary veins join to form a long anomalous vein into the inferior vena cava or even into the portal vein. Type III is not seen in adults and, if not cor-rected during the fi rst few weeks of life, is usually fatal. One may also see a combination of connections, with the veins draining separately but ultimately into the RA.262
In all situations, the oxygen saturation in all four cham-bers should in theory be identical, but in fact, this is not necessarily so. Drainage into the inferior vena caval system causes the relatively more oxygenated blood to stream pref-erentially through the ASD so that the left heart chambers are somewhat more saturated than the right heart chambers. When the connections are by the superior vena caval system, there is apt to be more nearly equal saturation in all four chambers. If all four veins drain to the coronary sinus, the right ventricle and the pulmonary artery may have a higher oxygen content than the left side of the heart.
Clinically, patients may exhibit cyanosis and clubbing, but the cardiac fi ndings are similar to those of the secundum type of ASD, with a wide, fi xed splitting of the second heart sound and a hyperactive LV. The ECG is also similar, with incomplete right bundle branch block with rSR´ in lead V1 and a normal frontal plane QRS axis. The chest x-ray is most striking if all four pulmonary veins drain into a venous sinus, then into a common vertical vein and into the innomi-nate vein, and then to the superior vena cava. The marked dilatation of this system gives rise to the so-called “snowman”
FIGURE 10.48. Posteroanterior radiograph of a patient with total anomalous venous return to the left superior vena cava. The heart is mildly enlarged, the pulmonary vasculature is increased, and the dilated left and right portions of the superior vena cava are clearly demonstrated (arrows); snowman heart.
or “fi gure-eight” confi guration of the heart and great vessels (Fig. 10.48). The echocardiogram can identify the drainage site of most or all of the pulmonary veins if the echocardiog-rapher is compulsive in identifying all pulmonary veins or alerted beforehand to the diagnosis. Sometimes a transesoph-ageal echocardiogram may be required. Cardiac catheteriza-tion is helpful in obtaining the oxygen saturations in all cardiac chambers and in the pulmonary artery and aorta to give one a clue as to the likely connections. The anomalous connections can be individually cannulated, and angiograms of these anomalous veins and the exact anatomy are extremely helpful to the surgeon, especially if they drain to different sites (Fig. 10.49). It may be necessary to explore the superior vena cava and innominate vein, the coronary sinus, and the inferior vena cava. Selective indicator dilution curves in the four pulmonary arteries or selective angiography of these vessels can help identify the connections if the anoma-lous veins are not draining together. Today, computed tomog-raphy (CT) and MRI are increasingly used to identify the pulmonary venous anatomy.263
Treatment
The treatment is surgical. The type of connection that is most readily amenable to surgery is anomalous pulmonary venous connection of the snowman type, by a side-to-side anastomosis of the common venous sinus to the LA and liga-tion of the vertical vein.
CAR010.indd 268CAR010.indd 268 11/24/2006 10:41:06 AM11/24/2006 10:41:06 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 2 6 9
Coronary Artery Anomalies
Coronary artery anomalies can be divided into four types.265–267 The fi rst is anomalous origin of the right coro-nary artery from the left coronary artery, or a branch of the left coronary artery from the right coronary artery, or if the left anterior descending coronary artery and the circumfl ex artery have separate ostia. These patients are asymptomatic and do well unless they acquire coronary atherosclerosis.
A second type is an ectopic course of the left coronary artery, so that instead of coursing anterior to the pulmonary artery, it goes between the aorta and the pulmonary artery.266 Sudden deaths have been reported with this anomaly. It is likely that the pathophysiology is not compression of the coronary artery between the aorta and the pulmonary artery, as has been suggested, because the pulmonary artery is essentially a low-pressure vessel. Rather, the course of the coronary artery shortly after its origin from the aorta is at a somewhat acute angle, which, during vigorous exercise, may become more acute, with resultant myocardial ischemia. The other anomaly in this group is myocardial bridging, in which the epicardial coronary artery dives under the myo-cardium and then reemerges. During systole, the vessel undergoes compression. Most such cases are asymptomatic, but an occasional patient has angina and a positive stress test result that is relieved when the bridging is unroofed.268,269
A third anomaly involves fi stulas between the coronary arteries and the ventricular cavity.270 A coronary artery may empty distally directly into a cardiac chamber. This more commonly involves the right coronary artery, which drains into the RV. More often, the coronary artery drains into a telangiectatic structure, which then empties into the RV. Clinically, such patients are usually asymptomatic, but the physical examination may reveal a soft, continuous murmur over the precordium, more commonly near the left sternal border if the right coronary artery is involved. If the shunt is large, there may be a “steal” phenomenon, which may rarely cause angina. The treatment may be either surgical or selective embolization of the distal telangiectatic lesion. Angiographically, this lesion is seen at the terminus of a
greatly enlarged coronary artery as a “puddle” from which contrast enters the RV. Fistulas of small coronary arterial branches are sometimes seen in routine angiograms and are of no hemodynamic signifi cance.
If coronary artery fi stulas are asymptomatic, it seems that they can be safely watched. Although the details of the lifelong mortality risk of such an anomaly are not entirely known, the evidence that exists suggests that the mortality risk of such fi stulas is extremely small and would not neces-sarily warrant surgical intervention.271
Finally, there may be an anomalous origin of a coronary artery from a pulmonary artery.272 In this situation, the effect is that of a large coronary artery steal because the pulmonary artery is under low pressure. Therefore, anomalous origin of the left coronary artery from the pulmonary artery is seen less commonly in adults because it is usually symptomatic or fatal in childhood unless it is treated. Formerly, the recom-mended treatment was ligation of the anomalous artery, but current treatment involves both ligation of the anomalous coronary artery at its takeoff from the pulmonary artery and bypass grafting. In the adult, anomalous origin of the right coronary artery from the pulmonary artery is somewhat more common and is usually asymptomatic, although it has been implicated in sudden death.
Aneurysms of the Sinus of Valsalva
Congenital aneurysm of the sinus of Valsalva is caused by a weakness at the junction of the aortic media and annular fi brosis of the aortic valve.273 By far the most commonly involved sinus is the right aortic sinus. The aneurysm usually protrudes into the outfl ow tract of the RV or, less commonly, into the RA. Aneurysm of the posterior sinus is much less common, and that of the left coronary sinus is extremely rare. An aneurysm of the sinus of Valsalva is asymptomatic until it ruptures, at which time a sudden large left-to-right shunt develops into the RV or RA, with dyspnea due to pulmonary congestion and even pulmonary edema.274 The physical fi ndings are those of an arteriovenous
FIGURE 10.49. This 35-year-old patient had total anomalous pulmonary venous connection via a large vertical vein to the superior vena cava. (A) Chest x-ray shows the characteristic “snowman” or fi gure-eight confi guration. (B) Angio-gram shows the large horizontal vein, being the confl uence of the pulmonary veins, which drains by a vertical vein into the innominate vein and thence into a greatly dilated superior vena cava.
CAR010.indd 269CAR010.indd 269 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

270 c h a p t e r 10
fi stula (i.e., a loud, continuous murmur over the precor-dium). The diagnosis is made clinically by the history and physical fi ndings and is corroborated by echocardiography, which can locate the fi stula and visualize the shunt. This can also be accomplished by aortic root angiography. The treatment is surgical.
The sinus of Valsalva aneurysm itself is a potential site for infective endocarditis and occasionally is the cause of rupture.
Pregnancy and Congenital Heart Disease
During pregnancy, there is an increase in intravascular volume starting in the second trimester, reaching a peak at 32 weeks, and declining slightly thereafter until term.201,220,275 There is a corresponding rise in cardiac output owing to the increased volume and the vascularity of the gravid uterus.276,277 During the fi rst stage of labor, the hemodynamic alterations are intermittent and transient. The pulmonary artery wedge pressure may rise with each uterine contraction and fall promptly with relaxation, the rise in pulmonary wedge pres-sure being due to a sudden increase in intravascular volume by up to 500 mm Hg, caused by the contracting uterus. During the second stage of labor, however, vigorous bearing down constitutes a series of giant, prolonged Valsalva maneu-vers. During the third stage, there is a loss of blood of about 500 mL and a transient rise in cardiac output without a sig-nifi cant change in arterial blood pressure, which translates into a lower SVR.277–279
Patients with noncyanotic heart disease with left-to-right shunting and normal to moderately elevated PVRs usually tolerate pregnancy well.280,281 Patients with ASD, PDAs, small-to-moderate VSDs, and mild valvar stenosis or insuf-fi ciency can also be safely carried through pregnancy. Coarc-tation of the aorta carries a higher risk during pregnancy because of possible aortic dissection, aortic rupture, or sub-arachnoid hemorrhage from a berry aneurysm, which is sometimes associated with coarctation of the aorta.282 In all of these situations, salt restriction to prevent a dispropor-tionate rise in the intravascular volume is of great impor-tance. It is especially important in coarctation of the aorta, in which management of the intravascular volume should coincide with control of hypertension, if present. Salt restric-tion may also help to control hypertension. However, if drug therapy becomes necessary, the inevitable question of fetal toxicity arises. Verapamil has been used during pregnancy to treat fetal tachycardias without documented fetal toxicity. However, nifedipine has been associated with a high rate of cesarean section and low-birth-weight infants. The beta-blockers propranolol and atenolol have been associated with low birth weight, tachycardia, and hypoglycemia. Metoprolol has been well tolerated, as has labetalol. There have been rare reports of adverse effects on the fetus with angiotensin-con-verting enzyme (ACE) inhibitors. Diuretics have not been shown to be directly deleterious to the fetus, although they may decrease placental perfusion. The combination of thia-zide diuretics and methyldopa has apparently been shown to be safe and effective. There have been reports, however, that thiazides, which cross the placental barrier, are associated with neonatal thrombocytopenia and hyponatremia. Thus,
drugs should be reserved for pregnant women who have clear-cut hypertension that cannot be controlled with salt restric-tion alone.281
In patients who have mechanical cardiac prostheses280 or who might have thromboembolic phenomena caused by secondary erythrocytosis in cyanotic heart disease, use of warfarin should be avoided because of its substantial terato-genicity and fetal wastage. Heparin does not cross the pla-cental barrier because of its molecular size and is the drug of choice for anticoagulation.283 In the presence of erythro-cytosis, self-administration of subcutaneous heparin is prac-tical.284 For mechanical prostheses, however, more careful monitoring of the activated partial thromboplastin time is necessary, and intravenous administration of heparin via a heparin lock may be necessary. It is precisely these consid-erations that may make it advisable for women of childbear-ing age who have severe congenital valve disease necessitating valve replacement to opt for a bioprosthesis, if they wish to bear children, with the full understanding that a second operation will in all likelihood be necessary some years later, after childbearing has been completed.
For cardiac arrhythmias, digoxin has a long history of safety in pregnancy. Although by itself, it may not be espe-cially effective, in combination with other drugs, it can be useful.285 In general, quinidine has had a reasonable record of safety, although rarely it causes damage to the eighth cranial nerve of the fetus. Disopyramide, verapamil, meto-prolol, and labetalol have a reasonable safety record.286,287 The conduct of labor in patients with serious valve disease and cyanotic heart disease optimally should include Swan-Ganz catheterization for monitoring of the pulmonary artery wedge and right atrial pressures, constant blood pressure monitoring, and oximetry.288 Vaginal delivery, with shorten-ing of the second stage of labor by forceps delivery, if neces-sary, is feasible in most patients. However, intractable and serious elevation of the pulmonary artery wedge pressure, signifi cant fall in arterial oxygen saturation, or intractable hypertension or hypotension or fetal distress should raise a serious consideration of cesarean section.
The fi rst stage of labor usually does not cause serious hemodynamic disturbances, except transiently during con-tractions. However, the second stage should be shortened by forceps if safely feasible, and anesthesia, which minimizes vasodilatation, should be given. The third stage may be dan-gerous in cyanotic patients, in whom the fall in SVR after delivery, combined with blood loss, may suddenly increase right-to-left shunting. The fall in blood pressure and arterial desaturation may cause progressive deterioration, with a fatal outcome. Therefore, every effort should be made to replenish lost intravascular volume and to maintain blood pressure and adequate oxygenation. Such patients should have been typed and cross-matched, with blood available and arterial vasoconstrictors on hand to maintain the blood pres-sure and thus increase the SVR if necessary if sudden hypo-tension occurs. Cesarean section itself is not without risk. There is the risk posed by general anesthesia, the greater blood loss, and the supine hypotension phenomenon of preg-nancy (postulated to be due to compression of the inferior vena cava by the gravid uterus). Therefore, cesarean section is an option only when it appears that vaginal delivery is posing an unacceptable risk.277,289
CAR010.indd 270CAR010.indd 270 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 271
Eisenmenger’s syndrome carries a high risk, as high as 38%, and causes considerable fetal wastage.281
Intrapartum open-heart surgery is almost never neces-sary with careful medical management and is associated with a high incidence of fetal wastage. Patients with tight aortic or pulmonic valve stenosis may be considered for balloon dilatation, preferably well after the fi rst trimester but before the intravascular volume has peaked.290
In general, with careful medical management, most women with congenital heart disease can carry a pregnancy safely to term, with the risk being somewhat higher in cya-notic congenital heart disease. The risk is not only to the mother but also to the fetus, because fetal wastage is higher in cyanotic conditions. The causes are not entirely clear but are probably multifactorial because fetal hemoglobin satu-rates at relatively low oxygen tensions. The placental circula-tion is probably adversely affected by low oxygen tension and erythrocytosis.291 Women with repaired cyanotic heart disease, such as transposition of the great arteries, may suc-cessfully complete pregnancy292; however, maternal and fetal complications, as well as genetic risks do play a role.
References
1. Kelen S. Maude Abbott: a biography. Can J Cardiol 2000;16:893–898.
2. Waugh D. Maudie: The life and times of McGill’s Maude Abbott. Mod Pathol 1992;5:597–599.
3. Taussig HB, King JT, Bauersfeld R, Padvamati-Iyer S. Results of operation for pulmonary stenosis and atresia; (report of 1000 cases). Trans Assoc Am Physicians 1951;65:67–73.
4. Lillehei CW, Cohen M, Warden HE, et al. The results of direct vision closure of ventricular septal defects in 8 patients by means of controlled cross circulation. Surg Gynecol Obstet 1955;101:446.
5. Shirodaria CC, Gwilt DJ, Gatzoulis MA. Joint outpatient clinics for the adult with congenital heart disease at the district general hospital: an alternative model of care. Int J Cardiol 2005;103:47–50.
6. Berman EB, Barst RJ. Eisenmenger’s syndrome: current man-agement. Prog Cardiovasc Dis 2002;45:129–138.
7. Braverman AC, Guven H, Beardslee MA, Makan M, Kates AM, Moon MR. The bicuspid aortic valve. Curr Probl Cardiol 2005;30:470–522.
8. Engelfriet P, Tijssen J, Kaemmerer H, et al. Adherence to guide-lines in the clinical care for adults with congenital heart disease: The Euro Heart Survey on adult congenital heart disease. Eur Heart J 2006;27:737–745.
9. Qu JZ. Congenital heart diseases with right-to-left shunts. Int Anesthesiol Clin 2004;42:59–72.
10. Bedford DE. The anatomical types of atrial septal defect: their incidence and clinical diagnosis. Am J Cardiol 1960;6:568, 1960.
11. Vick GW III. Defects of the atrial septum including atrioven-tricular septal defects. In: Garson A Jr, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiol-ogy, 2nd ed. Philadelphia: Williams & Wilkins, 1998:1141–1180.
12. Craig RJ, Selzer A. Natural history and prognosis of atrial septal defect. Circulation 1968;37:805.
13. Mattila S, Merikallio E, Talia T. ASD in patients over 40 years of age. Scand J Thorac Cardiovasc Surg 1979;13:21.
14. Steele PM, Foster V, Cohen M, et al. Isolated atrioseptal defect with pulmonary vascular obstructive disease: long-term follow-
up and prediction of outcome after surgical correction. Circula-tion 1987;76:1037.
15. Sutton MGS, Tajaik A, McGoon DC. Atrial septal defects in patients ages 60 years or older. Circulation 1982;64:402.
16. Leachman RD, Cokkinos DV, Cooley DA. Association of ostium secundum atrial septal defects with mitral valve pro-lapse. Am J Cardiol 1976;38:167.
17. Lutembacher R. De la stenose mitrale avec communication interauriculaire. Arch Mal Coeur 1916;9:237.
18. McGinn S, White PD. Interauricular septal defect associated with mitral stenosis. Am Heart J 1933;9:1.
19. Goldfarb B, Wang Y. Mitral stenosis and left-to-right shunt at the atrial level: a broad concept of the Loutembacher syndrome. Am J Cardiol 1966;17:3, 1966.
20. Smallhorn JF, Tommasini G, Anderson RH. Assessment of atrioventricular septal defects by two-dimensional echocar-diography. Br Heart J 1982;47:109.
21. Caputo S, Capozzi G, Russo MG, et al. Familial recurrence of congenital heart disease in patients with ostium secundum atrial septal defect. Eur Heart J 2005;26:2179–2184.
22. Wu JC, Child JS. Common congenital heart disorders in adults. Curr Probl Cardiol 2004;29:641–700.
23. Curran WD, Akindipe O, Staples ED, Baz MA. Lung transplan-tation for primary pulmonary hypertension and Eisenmenger’s syndrome. J Cardiovasc Nurs 2005;20:124–132.
24. Waddell TK, Bennett L, Kennedy R, Todd TR, Keshavjee SH. Heart-lung or lung transplantation for Eisenmenger syndrome. J Heart Lung Transplant 2002;21:731–737.
25. Brassard M, Fouron JC, van Doesburg NH, et al. Outcome of children with atrial septal defect considered too small for sur-gical closure. Am J Cardiol 1999;83:1552.
26. Hopkins RA, Bert AA, Buchholz B, Guarino K, Meyers M. Surgical patch closure of atrial septal defects. Ann Thorac Surg 2004;77:2144–2149.
27. Horvath KA, Burke RP, Collins JJ Jr, Cohn LH. Surgical treat-ment of adult atrial septal defect: early and long-term results. J Am Coll Cardiol 1992;20:1156–1159.
28. King TD, Mills NL. Nonoperative closure of atrial septal defects. Surgery 1974;3:383–388.
29. King TD, Mills NL. Secundum atrial septal defects: nonopera-tive closure during cardiac catheterization. JAMA 1976;235:2506–2509.
30. Rashkind WJ. Interventional cardiac catheterization in con-genital heart disease. Int J Cardiol 1985;7:1–11.
31. Lock JE, Cockerham JT, Keane JF, et al. Transcatheter umbrella closure of congenital heart defects. Circulation 1987;75:593–599.
32. Rome JJ, Keane JF, Perry SB, et al. Double umbrella closure of atrial septal defects: initial clinical applications. Circulation 1990;82:751–758.
33. Perry SB, van der Velde ME, Bridges ND, et al. Transcatheter closure of atrial and ventricular septal defects. Herz 1993;18:135–142.
34. O’Laughlin MP, Bricker JT, Mullins CE, et al. Transcatheter closure of residual atrial septal defect following cardiac trans-plantation. Cathet Cardiovasc Diagn 1993;28:162–163.
35. Ruiz CE, Gamra H, Mahrer P, et al. Percutaneous closure of a secundum atrial septal defect and double balloon valvotomies of a severe mitral and aortic valve stenosis in a patient with Lutembacher’s syndrome and severe pulmonary hypertension. Cathet Cardiovasc Diag 1992;25:309–312.
36. Sideris EB, Sideris SE, Thampoulos BD, et al. Transvenous atrial septal defect occlusion by the buttoned device. Am J Cardiol 1990;66:1524–1526.
37. Rao PS, Sideris EB, Chopra PS. Catheter closure of atrial septal defects: successful use in a 3.6 kg infant. Am Heart J 1991;121:1826–1829.
CAR010.indd 271CAR010.indd 271 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

27 2 c h a p t e r 10
38. Rao PS, Wilson AD, Chopra PS. Transcatheter closure of atrial septal defect by “buttoned” devices. Am J Cardiol 1992;69:1056–1061.
39. Zamora R, Lax D, Donnerstein RL, Lloyd TR. Transcatheter closure of residual atrial septal defect following implantation of buttoned device. Cathet Cardiovasc Diagn 1995;36:242–246.
40. Babic UU, Grujicic S, Djurisic A, Vucinic M. Transcatheter closure of atrial septal defects. Lancet 1990;336:566–567.
41. Sievert H, Babic UU, Ensslen R, et al. Transcatheter closure of atrial septal defects. Lancet 1990;336:566–567.
42. Hausdorf G, Schneider M, Franzbach B, et al. Transcatheter closure of large atrial septal defects with the Babic system. Cathet Cardiovasc Diagn 1995;36:232–240.
43. Sievert H, Dirks J, Rux S, et al. ASD and PFO closure in adults with the second generation ASDOS device. J Am Coll Cardiol 1997;29:143A.
44. Pavcnik D, Wright KC, Wallace S. Monodisk: device for percu-taneous transcatheter closure of cardiac septal defects. Cardio-vasc Interventent Radiol 1993;16:308–312.
45. Das GS, Voss G, Jarvis G, et al. Experimental atrial septal defect closure with a new, transcatheter, self-centering device. Circulation 1993;88:1754–1764.
46. Sharafudin MJ, Gu X, Titus JL, Amplatz K. Secundum-ASD closure with a new self-expanding prosthesis in swine. Circula-tion 1996;94:1–57.
47. Boutin C, Musewe NN, Smallhorn JF, et al. Echocardiographic follow-up of atrial septal defect after catheter closure by double-umbrella device. Circulation 1993;88:621–627.
48. Celiker A, Ozkutlu S, Karagoz T, Ayabakan C, Bilgic A. Trans-catheter closure of interatrial communications with Amplatzer device: results, unfulfi lled attempts and special considerations in children and adolescents. Anadolu Kardiyol Derg 2005;5:159–164.
49. Purcell IF, Brecker SJ, Ward DE. Closure of defects of the atrial septum in adults using the Amplatzer device: 100 consecutive patients in a single center. Clin Cardiol 2004;27:509–513.
50. Butera G, Carminati M, Chessa M, et al. CardioSEAL/STAR-fl ex versus Amplatzer devices for percutaneous closure of small to moderate (up to 18 mm) atrial septal defects. Am Heart J 2004;148:507–510.
51. Kay JD, O’Laughlin MP, Ito K, Wang A, Bashore TM, Harrison JK. Five-year clinical echocardiographic evaluation of the Das Angel Wings atrial septal occluder. Am Heart J 2004;147:361–368.
52. Pedra CA, Pedra SF, Esteves CA, et al. Initial experience in Brazil with the Helex septal occluder for percutaneous occlu-sion of atrial septal defects. Arq Bras Cardiol 2003;81:435–452.
53. Schrader R. Catheter closure of secundum ASD using “other” devices. J Intervent Cardiol 2003;16:409–412.
54. Mills NL, King TD. Late follow-up of nonoperative closure of secundum atrial septal defects using the King-Mills double-umbrella device. Am J Cardiol 2003;92:353–35.
55. Vida VL, Barnoya J, O’Connell M, Leon-Wyss J, Larazabal LA, Castaneda AR. Surgical versus percutaneous occlusion of ostium secundum atrial septal defects: results and cost-effective considerations in a low-income country. J Am Coll Cardiol 2006;47:326–331.
56. Celiker A, Ozkutlu S, Karakurt C, Karagoz T. Cardiac dys-rhythmias after transcatheter closure of ASD with Amplatzer device. Turk J Pediatr 2005;47:323–326.
57. Suda K, Raboisson MJ, Piette E, Dahdah NS, Miro J. Reversible atrioventricular block associated with closure of atrial septal defects using the Amplatzer device. J Am Coll Cardiol 2005;45:1306.
58. Silversides CK, Siu SC, McLaughlin PR, et al. Symptomatic atrial arrhythmias and transcatheter closure of atrial septal defects in adult patients. Heart 2004;90:1194–1198.
59. Roos-Hesselink JW, Meijboom FJ, Spitaels SE, et al. Excellent survival and low incidence of arrhythmias, stroke and heart failure long-term after surgical ASD closure at young age. A prospective follow-up study of 21–33 years. Eur Heart J 2003;24:190–197.
60. Bink-Boelkens MT, Velvis H, van der Heide JJ, Eygelaar A, Hardjowijono RA. Dysrhythmias after atrial surgery in chil-dren. Am Heart J 1983;106:125–130.
61. Girod D, Raghib G, Wang Y, Amplatz K. Angiographic charac-teristics of persistent atrioventricular canal. Radiology 1965;85:442.
62. McGrath LB, Gonzalez-Lavin L. Actuarial survival, freedom from reoperation, and other events after repair of atrioventricu-lar septal defects. Circulation 1993;88:621–627.
63. Kyger ER, Frazier OH, Cooley DA, et al. Sinus venosus atrial septal defect: early and late results following closure in 109 patients. Am Thorac Surg 1978;25:44–50.
64. Trusler GA, Kazenleson G, Freedom RM, et al. Late results following repair of partial anomalous pulmonary venous con-nection with sinus venosus atrial septal defect. J Thorac Car-diovasc Surg 1980;79:776–781.
65. Craig JD. Communications between the left atrium and the coronary sinus. Thesis, University of Minnesota, 1952.
66. McGaughey MD, Trail TA, Brinker JA. Partial left anomalous pulmonary venous return: a diagnostic dilemma. Cathet Car-diovasc Diagn 1986;12:110–115.
67. Stewart JR, Schaff HV, Fortunin NJ, Brawley RK. Partial anom-alous pulmonary venous return with intact atrial septum: report of four cases. Thorax 1983;38:859–862.
68. Ward KE, Mullins CE. Anomalous pulmonary venous connections, pulmonary vein stenosis, and atresia of the common pulmonary vein. In: Garson A Jr, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardi-ology, 2nd ed. Philadelphia: Williams & Wilkins, 1998:1431–1462.
69. Zagol B, Book S, Krasuski RA. Late “adult form” scimitar syn-drome presenting with “infant form” complications. J Invasive Cardiol 2006;18:E82–E85.
70. Patrianakos AP, Parthenakis FI, Chrysostomakis SI, Vardas PE. Ventricular special defect in the elderly: An uncommon clini-cal entity. Hellenic J Cardiol 2005;46:158–160.
71. Onat T, Ahunbay G, Batmaz G, Celebi A. The natural course of isolated ventricular septal defect during adolescence. Pediatr Cardiol 1998;19:230–234.
72. Mehta AV, Goenka S, Chidambaram B, Hamati F. Natural history of isolated ventricular septal defect in the fi rst fi ve years of life. Tenn Med 2000;93:136–138.
73. van Praagh R, Geva T, Kreutzer J. Ventricular septal defects: how should we describe, name and classify them? J Am Coll Cardiol 1989;14:1298.
74. Eisenmenger V. Ursprung de Aorta aus beiden Ventrikeln beim Defect der Septum ventriculoren. Wein Klin Wochenschr 1898;11:26, 1898.
75. Helmcke F, Souza A, Nanda NC, et al. Two-dimensional color Doppler assessment of ventricular septal defect of congenital origin. Am J Cardiol 1989;63:1112.
76. Sharif DS, Huhta JC, Marantz P, et al. Two-dimensional echo-cardiographic determination of ventricular septal defect size: correlation of autopsy. Am Heart J 1989;117:133.
77. Kurokawa S, Takahashi M, Kato HY, et al. Noninvasive evalu-ation of the ratio of pulmonary to systemic fl ow in ventricular septal defect by means of Doppler two-dimensional echocar-diography. Am Heart J 1988;116:1033.
CAR010.indd 272CAR010.indd 272 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 27 3
78. Gasul BM, Dillon RF, Vrla V, Hait G. Ventricular septal defects: their natural transformation into those with infundibular ste-nosis or into the cyanotic or noncyanotic type of tetralogy of Fallot. JAMA 1957;164:847, 1957.
79. Roos-Hesselink JW, Meijboom FJ, Spitaels SE, et al. Outcome of patients after surgical closure of ventricular septal defect at young age: longitudinal follow-up of 22–34 years. Eur Heart J 2004;25:1057–1062.
80. Godman MJ, Roberts NK, Izukawa T. Late post operative con-duction disturbances after repair of ventricular septal defect in tetralogy of Fallot. Circulation 1949;49:214.
81. Chessa M, Carrozza M, Butera G, et al. The impact of interven-tional cardiology for the management of adults with congenital heart defects. Catheter Cardiovasc Intervent 2006;67:258–264.
82. Knauth AL, Lock JE, Perry SB, et al. Transcatheter device closure of congenital and postoperative residual ventricular septal defects. Circulation 2004;110:501–507.
83. Moodie DS. VSD closure device in the setting of adult con-genital heart disease. Catheter Cardiovasc Intervent 2005;64:213.
84. Pawelec-Wojtalik M, Wojtalik M, Mrowczynski W, Surmacz R. Closure of perimembranous ventricular septal defect using transcatheter technique versus surgical repair. Kardiol Pol 2005;63:595–602.
85. Bacha EA, Cao QL, Galantowicz ME, et al. Multicenter experi-ence with preventricular device closure of muscular ventricu-lar septal defects. Pediatr Cardiol 2005;26:169–175.
86. Tulloh RM. Congenital heart disease in relation to pulmonary hypertension in paediatric practice. Paediatr Respir Rev 2005;6:174–180.
87. Guillinta P, Peterson KL, Ben-Yehuda O. Cardiac catheteriza-tion techniques in pulmonary hypertension. Cardiol Clin 2004;22:401–415.
88. Agapito AF, Sousa L, Oliveira JA, Feliciano J, Cacela D, Quininha J. Eisenmenger syndrome in the adult experience with new drugs for the treatment of pulmonary hypertension. Rev Port Cardiol 2005;24:421–431.
89. Campbell M. Natural history of persistent ductus arteriosus. Br Heart J 1968;30:4.
90. Gibson GA. Diseases of the heart and aorta. London: Young J. Pentland, 1898.
91. Liao PK, Su WJ, Hung JS. Doppler echocardiographic fl ow char-acteristics of isolated patent ductus arteriosus: better delinea-tion by Doppler color-fl ow mapping. J Am Coll Cardiol 1988;12:1285.
92. Moore JW, Levi DS, Moore SD, Schneider DJ, Berdjis E. Inter-ventional treatment of patent ductus arteriosus in 2004. Cath-eter Cardiovasc Intervent 2005;64:91–101.
93. McManus BM, Hahn PF, Smith JA, et al. Eisenmenger type patent ductus arteriosus with prolonged survival. Am J Cardiol 1984;54:462.
94. Ng AS, Liestra RE, Smith HC, et al. Patent ductus arteriosus in patients over 50 years. J Am Coll Cardiol 1984;3:599.
95. Bessenger FB Jr, Blieden LC, Edwards JE. Hypertensive pulmo-nary vascular disease associated with patent ductus arteriosus. Circulation 1975;52:157.
96. Fenoglio JJ, McAlister HA Jr, deCastro MC, et al. Congenital bicuspid aortic valve after age 20. Am J Cardiol 1977;39:164.
97. Edwards JE. Calcifi c aortic stenosis: pathologic features. Mayo Clin Proc 1961;36:444.
98. Latson LA. Aortic stenosis: Valvar, supravalvar, and fi bromus-cular subvalvar. In: Garson A Jr, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology, 2nd ed. Philadelphia: Williams & Wilkins, 1998:1257–1276.
99. Borger MA, David TE. Management of the valve and ascending aorta in adults with bicuspid aortic valve disease. Semin Thorac Cardiovasc Surg 2005;17:143–147.
100. Roberts WC. The congenitally bicuspid aortic valve: a study of 85 autopsy cases. Am J Cardiol 1970;26:72.
101. Baxter BT. Heritable diseases of the blood vessels. Cardiovasc Pathol 2005;14:185–188
102. Peckham GB, Keith JD, Evans JR. Congenital aortic stenosis: some observations in the natural history and clinical assess-ment. Can Med Assoc J 1964;91:639.
103. Johnson AM. Aortic stenosis, sudden death and left ventricular baroreceptors [editorial]. Br Heart J 1971;33:1.
104. Graham TP Jr, Driscoll DJ, Gersony WM, Newburger JW, Rocchini A, Towbin JA. Task Force 2: Congenital heart disease. J Am Coll Cardiol 2005;45:1326–1333.
105. Paquay PA, Anderson G, Diefenthal H, et al. Chest pain as a predictor of coronary artery disease in patients with obstruc-tive aortic valve disease. Am J Cardiol 1976;38:863.
106. Berndt TB, Hancock EW, Shumway NE, et al. Aortic valve replacement with and without coronary artery bypass surgery. Circulation 1974;50:967.
107. Rosenhek R, Klaar U, Schemper M, et al. Mild and moderate aortic stenosis. Natural history and risk stratifi cation by echo-cardiography. Eur Heart J 2004;25:199–205.
108. Campell M. Natural history of coarctation in the aorta. Br Heart J 1970;32:633, 1970.
109. Hancock EW. The ejection sound in aortic stenosis. Am J Med 1966;40:569.
110. Glancy DL, Epstein SE. Differential diagnosis of type and severity of obstruction of left ventricular outfl ow. Prog Cardio-vasc Dis 1971;14:153,.
111. Leatham A. Systolic murmurs. Circulation 1958;17:601. 112. Wood P. Aortic stenosis. Am J Cardiol 1958;1:553. 113. Rodan BA, Chen JTT, Halber MD, et al. Chest roentgenographic
evaluation of the severity of aortic stenosis. Invest Radiol 1982;17:453.
114. Klatte EC, Tampas JP, Campbell JA, Lurie PR. The roentgeno-graphic manifestations of aortic stenosis in aortic valvular insuffi ciency. AJR Radium Ther Nucl Med 1962;88:57.
115. Merendino KA, Hessel EA. The murmur on top of the head in acquired mitral insuffi ciency. JAMA 1967;199:392.
116. Caves PK, Sutton GC, Paneth M. Nonrheumatic subvalvular mitral regurgitation: etiology and clinical aspects. Circulation 1973;47:1242.
117. Roman JA Jr, Steelman RB, DeLeon AC Jr, et al. The clinical diagnosis of acute severe mitral insuffi ciency. Am J Cardiol 1971;27:284.
118. Yap SC, Takkenberg JJ, Witsenburg M, Meijboom FJ, Roos-Hesselink JW. Aortic stenosis at young adult age. Expert Rev Cardiovasc Ther 2005;3:1087–1098.
119. Borger MA, David TW. Management of the valve and ascending aorta in adults with bicuspid aortic valve disease. Semin Thorac Cardiovasc Surg 2005;17:143–147.
120. Butany J, Collins MJ, Demellawy DE, et al. Morphological and clinical fi ndings in 247 surgically excised native aortic valves. Can J Cardiol 2005;21:747–755.
121. Fedak PW, David TW, Borger M, Verma S, Butany J, Weisel RD. Bicuspid aortic valve disease: recent insights in pathophysiol-ogy and treatment. Expert Rev Cardiovasc Ther 2005;3:295–308.
122. Stein TD, Sabbah HN, Pitha JV. Continuing disease process of calcifi c aortic stenosis: role of microthrombi and turbulent fl ow. Am J Cardiol 1977;39:159.
123. Stewart JR, Paton BC, Blount SG Jr, Swan H. Congenital aortic stenosis 10 to 20 years after valvulotomy. Arch Surg 1978;113:1248.
CAR010.indd 273CAR010.indd 273 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

274 c h a p t e r 10
124. Alipour MS, Shah PA. Diagnosis of aortic stenosis in the elderly: Role of echocardiography. Am J Geriatr Cardiol 2003;12:201–206.
125. Young JB, Quinones MA, Waggoner AD, Miller RR. Diagnosis and quantifi cation of aortic stenosis with pulsed Doppler echo-cardiography. Am J Cardiol 1980;45:987.
126. Gorlin R, Gorlin SG. Hydraulic formula for calculation of area of stenotic mitral valves, other cardiac valves and central cir-culatory shunts. Am Heart J 1951;41:1.
127. Burwash IG, Dickinson A, Teskey RJ, Tam JW, Chan KL. Aortic valve area discrepancy by Gorlin equation and Doppler echo-cardiography continuity equation: relationship to fl ow in patients with valvular aortic stenosis. Can J Cardiol 2000;16:985–992.
128. Cheitlin M, Gertz E, Brundage B, et al. The rate of progression of aortic stenosis in the adult. Am Heart J 1979;98:689.
129. Pellikka PA, Sarano ME, Nishimura RA, et al. Outcome of 622 adults with asymptomatic, hemodynamically signifi cant aortic stenosis during prolonged follow-up. Circulation 2005;111:3290–3295.
130. Keane JF, Driscoll DJ, Gersony WM, et al. Second natural history study of congenital heart defects: results of treatment of patients with aortic valvar stenosis. Circulation 1993;87(suppl II) I–16.
131. Baumgartner H. Aortic stenosis: Medical and surgical manage-ment. Heart 2005;91:1483–1488.
132. Sakata Y, Syed Z, Salinger MH, Feldman T. Percutaneous balloon aortic valvuloplasty: antegrade transseptal vs. conven-tional retrograde transarterial approach. Catheter Cardiovasc Intervent 2005;64:314–321.
133. Pedra CA, Sidhu R, McCrindle BW, et al. Outcomes after balloon dilation of congenital aortic stenosis in children and adolescents. Cardiol Young 2004;14:315–321.
134. Agarwal A, Kini AS, Attanti S, et al. Results of repeat balloon valvuloplasty for treatment of aortic stenosis in patients aged 59 to 104 years. Am J Cardiol 2005;95:43–47.
135. Herrmann HC. Percutaneous valve therapies. Curr Treat Options Cardiovasc Med 2005;7:477–482.
136. Tawn Z, Himberg D, Brochet E, Messika-Zeitoun D, Iung B, Vahanian A. Percutaneous valve procedures: present and future. Int J Cardiovasc Intervent 2005;7:14–20.
137. Stamm C, Friehs I, Ho SY, Moran AM, Jonas RA, del Nido PJ. Congenital supravalvar aortic stenosis: a simple lesion? Eur J Cardiothorac Surg 2001;19:195–202.
138. Kim YM, Yoo SJ, Choi JY, Kim SH, Bae EJ, Lee YT. Natural course of supravalvar aortic stenosis and peripheral pulmonary arterial stenosis in Williams’ syndrome. Cardiol Young 1999;9:37–41.
139. Brooke BS, Bayes-Genis A, Li DY. New insights into elastin and vascular disease. Trends Cardiovasc Med 2003;13;176–181.
140. Roberts WC. Valvular subvalvular and supravalvular aortic ste-nosis: morphologic features. Cardiovasc Clin 1973;5:97.
141. Greenspan AM, Morganroth J, Perloff JK. Discrete fi bromem-branous subaortic stenosis in middle age. Cardiology 1979;64:206.
142. Petsas AA, Anastassiades LC, Constantinou EC, et al. Familial discrete subaortic stenosis. Clin Cardiol 1998;21:63.
143. Morrow AG, Fort L III, Roberts WC, Braunwald E. Discrete subaortic stenosis complicated by aortic valvular regurgitation: clinical, hemodynamic and pathologic studies and the results of operative treatment. Circulation 1965;31:163.
144. Reiss RL, Peterson LM, Mason DT, et al. Congenital, fi xed subvalvular aortic stenosis: an anatomic classifi cation and cor-relations with operative results. Circulation 1971;43(suppl I):II.
145. Latson LA, Prioto LR. Pulmonary stenosis. In: Allen HD, Gut-gesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart
Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:820–844.
146. Almeda FQ, Kavinsky CJ, Pophal SG, Klein LW. Pulmonic valvular stenosis in adults: Diagnosis and treatment. Catheter Cardiovasc Intervent 2003;69:546–557.
147. Sommer RJ, Rhodes JF, Parness IA. Physiology of critical pul-monary valve obstruction in the neonate. Catheter Cardiovasc Intervent 2001;50:473–479.
148. Waller BF, Howard J, Fess S. Pathology of pulmonic valve stenosis and pure regurgitation. Clin Cardiol 1995;18:45–50.
149. Mahoney LT. Acyanotic congenital heart disease. Atrial and ventricular septal defects, atrioventricular canal, patent ductus arteriosus, pulmonic stenosis. Cardiol Clin 1993;11:603–616.
150. Block PC, Bonhoeffer P. Percutaneous approaches to valvular heart disease. Curr Cardiol Rep 2005;7:108–113.
151. Kern MJ, Bach RG. Hemodynamic rounds series II: Pulmonic balloon valvuloplasty. Cathet Cardiovasc Diagn 1998;44:227–234.
152. Noonan J, O’Connor W. Noonan syndrome: A clinical descrip-tion emphasizing the cardiac fi ndings. Acta Paediatr Jpn 1996;38:76–83.
153. Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart disease in children with Noonan syn-drome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 1999;135:703–706.
154. Nora JJ, Nora AH. Genetics and Counseling in Cardiovascular Diseases. Springfi eld, IL: Charles C. Thomas, 1978.
155. Krabill KA, Wang Y, Einzig S, Moller JH. Rest and exercise hemodynamics in pulmonary stenosis: comparison of children and adults. Am J Cardiol 1985;56:360.
156. Kan JS, White RI, Mitchell SE, Gardner TJ. Percutaneous balloon valvuloplasty: a new method for pulmonary valve ste-nosis. N Engl J Med 1982;307:540.
157. Gibbs JL. Interventional catheterization. Opening up I: The ventricular outfl ow tracts and great arteries. Heart 2000;83:111–115.
158. Garty Y, Veldtman G, Lee K, Benson L. Late outcomes after pulmonary valve balloon dilatation in neonates, infants and children. J Invasive Cardiol 2005;17:323–325.
159. Almeda FQ, Kavinsky CJ, Pophal SG, Klein LW. Pulmonic valvular stenosis in adults: Diagnosis and treatment. Catheter Cardiovasc Intervent 2003;60:546–557.
160. Brockman RC. Pulmonary valvulotomy for relief of congenital pulmonary stenosis: report of three cases. BMJ 1948;1:1121, 1948.
161. Peppine CJ, Gessner IH, Feldman RL. Percutaneous balloon valvuloplasty for a pulmonic valve stenosis in the adult. Am J Cardiol 1982;50:1442.
162. Fawzy ME, Mercer EN, Dunn B. Late results of pulmonary balloon valvuloplasty using double balloon technique. Int J Cardiol 1988;1:35.
163. Ing FF, Grifka RG, Nihill MR, Mullins CE. Repeat dilation of intravascular stents in congenital heart defects. Circulation 1985;92:893–897.
164. Shuck JW, McCormick DJ, Cohen IS, et al. Percutaneous balloon valvuloplasty of the pulmonary valve: role of right-to-left shunting through a patent foramen ovale. J Am Coll Cardiol 1984;4:132.
165. Bridges ND, Hellenbrand W, Latson L, et al. Transcatheter closure of patent foramen ovale after presumed paradoxical embolism. Circulation 1992;86:1902.
166. Zanchetta M, Rigatelli G, Pedon L, Zennaro M, Carrozza A, Onorato E. Catheter closure of perforated secundum atrial septal defect under intracardiac echocardiographic guidance using a single Amplatzer device: feasibility of a new method. J Invasive Cardiol 2005;17:262–265.
CAR010.indd 274CAR010.indd 274 11/24/2006 10:41:07 AM11/24/2006 10:41:07 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 275
167. Roos-Hesselink JW, Meijboom FJ, Spitaels SE, et al. Long-term outcome after surgery for pulmonary stenosis (a longitudinal study of 22–33 years). Eur Heart J 2006;27:482–488.
168. Neches WH, Park SC, Ettedgui JA. Tetralogy of Fallot and Tetralogy of Fallot with pulmonary atresia. In: Garson A Jr, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology, 2nd ed. Philadelphia: Williams & Wilkins, 1998:1383–1412.
169. Pokorski RJ. Long-term survival after repair of tetralogy of Fallot. J Insur Med 2000;32:89–92.
170. Murphy JG, Gersh BJ, Mair DD, et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 1993;329:593–599.
171. Cosio FG, Wang Y, Nicoloff DM. Membranous right ventricular outfl ow obstruction. Am J Cardiol 1973;32:1000.
172. Brown JW, Ruzmetov M, Vijay P, et al. Operative results and outcomes in children with Shone’s anomaly. Ann Thorac Surg 2005;79:1358–1365.
173. Hosseinpour AR, Amanullah M, Ramnarine IR, Stumper O, Barron DJ, Brawn WJ. Combined atrial arterial switch opera-tion (double switch for hearts with Shone syndrome and pul-monary hypertension). J Thorac Cardiovasc Surg 2006;131:471–473.
174. Keefe JF, Wolk MJ, Levine HJ. Isolated tricuspid valvular ste-nosis. Am J Cardiol 1970;25:252.
175. Geron M, Hirsch M, Borman J, Appelbaum A. Isolated tricuspid valvular stenosis: the pathology and merits of surgical treat-ment. J Thorac Cardiovasc Surg 1972;63:760.
176. Ebstein W. A rare case of insuffi ciency of the tricuspid valve caused by a severe malformation of the same {translated by Schiebler GS, et al}. Am J Cardiol 1968;22;867.
177. Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly-Review of a mul-tifaceted congenital cardiac condition. Swiss Med Wkly 2005;135:269–281.
178. Zalstein E, Koran G, Einerson T, Freedom RN. A case-controlled study on the association of fi rst trimester exposure to lithium in Ebstein’s anomaly. Am J Cardiol 1990;65:817.
179. Gussenhoven WJ, Spitaels SEC, Bom N, Becker AE. Echocar-diographic criteria of Ebstein’s anomaly of tricuspid valve. Br Heart J 1988;43:31.
180. Kastor JA, Goldreier BN, Josephine ME, et al. Electrophysiolo-gic characteristics of Ebstein’s anomaly of the tricuspid valve. Circulation 1975;52:987.
181. Giuliani ER, Futer V, Brandenburg RO, Mair DD. Ebstein’s anomaly: the clinical features and natural history of the tricus-pid valve. Mayo Clin Proc 1979;54:163.
182. Di Russo GB, Gaynor JW. Ebstein’s anomaly: Indications for repair and surgical technique. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 1999;2:35–50.
183. Hebe J. Ebstein’s anomaly in adults. Arrhythmias: diagnosis and therapeutic approach. Thorac Cardiovasc Surg 2000;48:214–219.
184. Speziali G, Driscoll DJ, Danielson GK, et al. Cardiac transplan-tation for end-stage congenital heart defects: the Mayo Clinic experience. Mayo Cardiothoracic Transplant Team. Mayo Clin Proc 1998;73:923–928.
185. Volkl TM, Degenhardt K, Koch A, Simm D, Dorr HG, Singer H. Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome: the Erlangen experience. Clin Cardiol 2005;28:88–92.
186. McBride KL, Marengo L, Canfi eld M, Langlois P, Fixler D, Belmont JW. Epidemiology of noncomplex left ventricular outfl ow tract obstruction malformations (aortic valve stenosis, coarctation of the aorta, hypoplastic left heart syndrome) in Texas–2001. Birth Defects Res A Clin Mol Teratol 2005;73:555–561.
187. Bondy CA. New issues in the diagnosis and management of Turner syndrome. Rev Endocr Metab Disord 2005;6:269–280.
188. Shepherd JT. Coarctation of the aorta. In Shepherd JT (ed): Physiology of the Circulation in Human Limbs in Health and Disease. Philadelphia: WB Saunders, 1963.
189. Lacro RV, Lyons-Jones K, Benirschki K. Coarctation of the aorta in Turner’s syndrome: a pathologic study of fetuses with nuchal cystic hygromas, hydros fetalis and female genitalia. Pediatrics 1988;81:445.
190. Liberthson RR, Pennington DG, Jacobs ML, Daggett WM. Coarctation of aorta: review of 234 patients and clarifi cation of management problems. Am J Cardiol 1979;43:835.
191. Reisenstein G, Levin S, Gross R. Coarctation of the aorta: review of 104 autopsy cases of the “adult type” two years of age or older. Am Heart J 1947;33:146.
192. Lewin MB, Otto CM. The bicuspid aortic valve: adverse out-comes from infancy to old age. Circulation 2005;111:832–834.
193. Ho VB, Corse WR, Hood MN, Rowedder AM. MRA of the tho-racic vessels. Semin Ultrasound CT MR 2003;24:192–216.
194. Neimatallah MA, Ho VB, Dong Q, et al. Gadolinium-enhanced 3D magnetic resonance angiography of the thoracic vessels. J Magn Reson Imaging 1999;10:758–7709.
195. Lima JA, Desai MY. Cardiovascular magnetic resonance imaging: Current and emerging applications. J Am Coll Cardiol 2004;44:1164–1171.
196. Pemberton J, Sahn DJ. Imaging of the aorta. Int J Cardiol 2004;97(suppl 1):53–60.
197. Vriend JW, Mulder BJ. Late complications in patients after repair of aortic coarctation: Implications for management. Int J Cardiol 2005;101:399–406.
198. Hirst AE Jr, Johns BJ Jr, Kine SW Jr. Dissecting aneurysm of the aorta: review of 505 cases. Medicine (Baltimore) 1958;37:217.
199. Edwards JE. Aneurysms of the thoracic aorta complicating coarctation. Circulation 1973;48:195.
200. Hodes HL, Steinfeld L, Blumenthal S. Congenital cerebral aneu-rysms and coarctation of the aorta. Arch Pediatr 1959;76:28.
201. Chalupczak P, Kolasinska-Kloch W, Jach R, Basta A. Pregnancy in patients with heart disease. Clin Exp Obstet Gynecol 2004;31:271–273.
202. Wittemore R, Hobbins JC, Engle MA. Pregnancy and its outcome in women with and without surgical treatment of congenital heart disease. Am J Cardiol 1982;50:641.
203. Tynan M, Finley JT, Fontes V, et al. Balloon angioplasty for the treatment of native coarctation results of valvuloplasty and angioplasty of congenital anomalies registry. Am J Cardiol 1990;65:790.
204. Pedra CA, Fontes VF, Esteves CA, et al. Stenting vs balloon angioplasty for discrete unoperated coarctation of the aorta in adolescents and adults. Cathet Cardiovasc Interv 2005;64:495–506.
205. Cohen M, Fuster V, Steele PM, et al. Coarctation of the aorta: long-term follow-up and prediction of outcome after surgical correction. Circulation 1989;80:840.
206. de Bono J, Freeman LJ. Aortic coarctation repair—lost and found: the role of local long-term specialized care. Int J Cardiol 2005;104:176–183.
207. Gleason TG. Heritable disorders predisposing to aortic dissec-tion. Semin Thorac Cardiovasc Surg 2005;17:274–281.
208. Wernovsky G. Transposition of the great arteries. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:1027–1084.
209. Anderson RH, Henry GW, Becker AE. Morphologic aspects of complete transposition. Cardiol Young 1991;1:41.
210. Rigby ML, Chan KY. The diagnostic evaluation of patients with complete transposition. Cardiol Young 1991;1:26.
CAR010.indd 275CAR010.indd 275 11/24/2006 10:41:08 AM11/24/2006 10:41:08 AM

276 c h a p t e r 10
211. Hornung TS, Derrick GP, Deanfi eld JE, Redington AN. Trans-position complexes in the adult: a changing perspective. Cardiol Clin 2002;20:405–420.
212. Fallot A. Contribution a l’anatomie pathologique de la maladie bleue (cyanose cardiaque). Marseille Med 1888;25:418.
213. Abraham KA, Cherian G, Rao BD, et al. Tetralogy of Fallot in adults: a report on 147 patients. Am J Med 1979;66:811.
214. Bertranou EG, Blackstone EH, Hazelrig JB, et al. Life expec-tancy without surgery in tetralogy of Fallot. Am J Cardiol 1978;42:458.
215. McGrath LB, et al. Determination of infundibular innervation in end amine receptor content in cyanotic and acyanotic myo-cardium: relation to clinical events in tetralogy of Fallot. Pediatr Cardiol 1991;12:155.
216. Baffes TG, Johnson FR, Potts WJ, Gibson S. Anatomic varia-tions in tetralogy of Fallot. Am Heart J 1953;46:647.
217. Brinton WD, Campbell M. Necropsy in some congenital diseases of the heart, mainly Fallot’s tetralogy. Br Heart J 1953;15:335.
218. Blalock A, Taussig HB. The surgical treatment of malforma-tions of the heart. JAMA 1995;128:189.
219. Park IS, Leachman RD, Cooley DA. Total correction of tetral-ogy of Fallot in adults: Surgical results and long-term follow-up. Tex Heart Inst J 1987;14:160–169.
220. Meijer JM, Pieper PG, Drenthen W, et al. Pregnancy, fertility, and recurrence risk in corrected tetralogy of Fallot. Heart 2005;91:801–805.
221. Rosenthal A. Adults with tetralogy of Fallot: repaired yes; cured, no [editorial]. N Engl J Med 1993;329:655.
222. Murphy JG, Gersh BJ, Mair DD, et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 1993;329:593.
223. Fenton KN, Pigula FA, Gandhi SK, Russo L, Duncan KF. Interim mortality in pulmonary atresia with intact ventricular septum. Ann Thorac Surg 2004;78:1994–1998.
224. Jefferson K, Simon R, Somerville J. Systemic arterial supply to the lungs and pulmonary atresia and its relation to pulmonary artery development. Br Heart J 1972;34:418.
225. Freedom RM, Black MD, Benson LN. Hypoplastic left heart syndrome. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:1011–1026.
226. Norwood WI, Kirklin JK, Sanders SP. Hypoplastic left heart syndrome: Experience with Palliative Surgery. Am J Cardiol 1989;45:87–91.
227. Tabbutt S, Dominguez TE, Ravishankar C, et al. Outcomes after the stage I reconstruction comparing the right ventricular to pulmonary artery conduit with the modifi ed Blalock-Taussig shunt. Ann Thorac Surg 2005;80:1582–1590.
228. McGuirk SP, Griselli M, Stumper O, et al. Staged surgical management of hypoplastic left heart syndrome: a single-institution 12-year experience. Heart 2006;92:364–370.
229. Connor JA, Arons RR, Figueroa M, Gebbie KM. Clinical outcomes and secondary diagnoses for infants born with hypoplastic left heart syndrome. Pediatrics 2004;114:e160–e165.
230. Hagler OJ. Double outlet right ventricle and double outlet left ventricle. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:1102–1128.
231. Kirklin JW, Pacifi co AD, Blackstone EH, et al. Current risk and protocols for surgery for double-outlet right ventricle deriva tion of an 18 year experience. J Thorac Cardiovasv Surg 1986;92:913.
232. Cetta F, Boston US, Dearani JA, Hagler DJ. Double outlet right ventricle: Opinions regarding management. Curr Treat Options Cardiovasc Med 2005;7:385–390.
233. Epstein ML. Tricuspid atresia. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:799–809.
234. Patel MM, Overy BC, Kazonis NC, Hadley-Folkes LL. Long-term survival in tricuspid atresia. J Am Coll Cardiol 1987;9:338.
235. Gewillig M. The Fontan circulation. Heart 2005;91:839–846. 236. Hager A, Zrenner B, Brodherr-Heberlein S, Steinbauer-
Rosen thal I, Schreieck J, Hess J. Congenital and surgically acquired Wolff-Parkinson-White syndrome in patients with tricuspid atresia. J Thorac Cardiovasc Surg 2005;130:48–53.
237. Fontan F, Mounicot FB, Baudet E. Correction de l’atresie tricus-pideienne: rapport de deux cas “corriges art utilization tech-nique chirugicale nouvelle.” Ann Chir Thorac Cardiovasc 1971;20:39.
238. Peries A, Al-Hay AA, Shinebourne EA. Outcome of the con-struction of a Blalock-Taussig shunt in adolescents and adults. Cardiol Young 2005;15:368–372.
239. Blanc J, Vouhe P, Bonnet D. Potts shunt in patients with pul-monary hypertension. N Engl J Med 2004;350:623.
240. Fenchel G, Steil E, Seboldt H, Quintenz R, Apitz J, Hoffmeister HE. Early and late results of the modifi ed Waterston shunt with PTFE graft for palliation of complex congenital cyanotic heart disease in neonates. Thorac Cardiovasc Surg 1991;39:268–272.
241. Hull DA, Shinebourne E, Gerlis L, Nicholson AG, Sheppard MN. Rupture of pulmonary aneurysms in association with long-standing Waterston shunts. Cardiol Young 2001;11:123–127.
242. Arciniegas E, Farooki ZQ, Hakimi M, Perry BL, Green EW. Classic shunting operations for congenital cyanotic heart defects. J Thorac Cardiovasc Surg 1982;84:88–96.
243. DeLeon SY, Idriss FS,k Ilbawi MN, et al. The role of the Glenn shunt in patients undergoing the Fontan operation. J Thorac Cardiovasc Surg 1983;85:669.
244. Molina JE, Wang Y, Lucas RV, Moller JH. The technique of the Fontan procedure with posterior right atrium pulmonary con-nection. Ann Thorac Surg 1984;39:371.
245. Benshachar G, Fuhrman BP, Wang Y, et al. Rest, exercise and hemodynamics after the Fontan procedure. Circulation 1982;65:1043.
246. Mitchell ME, Ittenbach RF, Gaynor JW, Wernovsky G, Nicolson S, Spray TL. Intermediate outcomes after the Fontan procedure in the current era. J Thorac Cardiovasc Surg 2006;131:172–180.
247. Jayakumar KA, Addonizio LJ, Kichuk-Chrisant MR, et al. Cardiac transplantation after the Fontan or Glenn procedure. J Am Coll Cardiol 2004;44:2065–2072.
248. Mitchell MB, Campbell DN, Boucek MM. Heart transplanta-tion for the failing Fontan circulation. Semin Thorac Cardio-vasc Surg Pediatr Card Surg Annu 2004;7:56–64.
249. Michielon G, Parisi F, Squitieri C, et al. Orthotopic heart trans-plantation for congenital heart disease: an alternative for high-risk Fontan candidates? Circulation 2003;108(suppl 1):II140–149.
250. Collett RW, Edwards JE. Persistent truncus arteriosus: classifi -cation according to anatomic types. Surg Clin North Am 1949;29:1245.
251. Mair DD, Edwards WD, Julsrud PR, Seward JB, Danielson GK. Truncus arteriosus. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:910–923.
CAR010.indd 276CAR010.indd 276 11/24/2006 10:41:08 AM11/24/2006 10:41:08 AM

pat hop h y s iol o g y, c l i n ic a l r e c og n i t ion, a n d t r e at m e n t of c ong e n i ta l h e a rt di s e a s e 27 7
252. Becker AE, Becker MJ, Edwards JE. Pathology of the semilunar valve and persistent truncus arteriosus. J. Thorac Cardiovasc Surg 1971;62:16.
253. Reddy VM, Hanley F. Late results of repair of truncus arterio-sus. Semin Thorac Cardiovasc Surg Pediatr Card Surg Snnu 1998;1:139–146.
254. Newfeld HN, Lester RG, Adams P Jr, et al. Aorticopulmonary septal defect. Am J Cardiol 1962;9:12.
255. Freedom RM, Dyck JD. Congenitally corrected transposition of the great arteries. In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:1085–1101.
256. Waldo AL, Pacifi co AD, Bargeron LM Jr, et al. Electro-physiologic delineation of specialized AV conduction system in patients with corrected transposition of the great vessels and ventricular septal defect. Circulation 1975;52:435.
257. Dimas AP, Moodie DS, Strba R, Gill CC. Long-term function of the morphologic right ventricle in adult patients with cor-rected transposition of the great arteries. Am Heart J 1989;118:526.
258. Mavroudis C, Backer CL. Physiologic versus anatomic repair of congenitally corrected transposition of the great arteries. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2003;6:16–26.
259. Mazzucco A, Scalia D, Faggian G, et al. Palliative surgery for single ventricle heart malformations. Int Surg 1983;68:207–210.
260. Diller GP, Dimopoulos K, Okonko D, et al. Exercise intolerance in adult congenital heart disease: Comparative severity, corre-lates and prognostic implication. Circulation 2005;112:828–835.
261. Reybrouck T, Mertens L. Physical performance and physical activity in grown-up congenital heart disease. Eur J Cardiovasc Prev Rehabil 2005;12:498–502.
262. Geva T, Van Praagh S. Anomalies of the pulmonary veins: In: Allen HD, Gutgesell HP, Clark EB, Druscoll DJ, eds. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents, 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2001:773–798.
263. Kim YH, Marom EM, Herndon JE 2nd, McAdams HP. Pulmo-nary vein diameter, cross-sectional area, and shape: CT analy-sis. Radiology 2005;235:43–49.
264. Schlesinger AE, Hernandez RJ. Magnetic resonance imaging in congenital heart disease in children. Tex Heart Inst J 1996;23:128–143.
265. Roberts WC. Major anomalies of coronary artery origin seen in adulthood. Am Heart J 1986;111:941.
266. Kragel AH, Roberts WC. Anomalous origin of either the right or left main coronary artery from the aorta. The subsequent coursing between aorta and pulmonary trunk. Analysis of 32 necropsy cases. Am J Cardiol 1988;62:77.
267. Rigatelli G, Rigatelli G. Congenital coronary artery anomalies in the adult: a new practical viewpoint. Clin Cardiol 2005;28:61–65.
268. Herrmann J, Higano ST, Lenon RJ, Rihal CS, Lerman A. Myo-cardial bridging is associated with alteration in coronary vaso-reactivity. Eur Heart J 2004;25:2134–2142.
269. Alegria JR, Herrman J, Holmes DR Jr, Lerman A, Rihal CS. Myocardial bridging. Eur Heart J 2005;26:1159–1168.
270. Levin DC, Fellows KE, Abrams HL. Hemodynamically signifi -cant primary anomalies of the coronary arteries: angiographic aspects. Circulation 1978;58:25.
271. Sherwood MC, Rockenmacher S, Colan SD, Geva T. Prognostic signifi cance of clinically silent coronary artery fi stulas. Am J Cardiol 1999;83:407.
272. Wilson CL, Dlabal PW, Holeyfi eld RW, et al. Anomalous origin of left coronary artery from pulmonary artery: case reports and review of literature concerning teenagers and adults. J Thorac Cardiovasc Surg 1977;73:887.
273. Harkness JR, Fitton TP, Barreiro CJ, et al. A 32-year experience with surgical repair of sinus of Valsalva aneurysms. J Card Surg 2005;20:198–204.
274. Mayer ED, Ruffman K, Saggau W, et al. Ruptured aneurysms of the sinus of Valsalva. Ann Thorac Surg 1986;42:81.
275. Ueland K. Maternal cardiovascular dynamics. VII: intrapartum blood volume changes. Am J Obstet Gynecol 1976;126:171.
276. Robson SC, Hunter S, Boys RJ, et al. Serial study of factors infl uencing changes in cardiac output during pregnancy. Am J Physiol 1989;256:H1060.
277. Davies GAL, Herbert WNP. Cardiac disease in pregnancy. In: Garson A Jr, Bricker JT, Fisher DJ, Neish SR, eds. The Science and Practice of Pediatric Cardiology, 2nd ed. Philadelphia: Williams & Wilkins, 1998:2915–2928.
278. Kjelbsen J. Hemodynamic investigations during labor and delivery. Acta Obstet Gynecol Scand Suppl 1979;89:20.
279. Ueland K, Hansen JM. Maternal cardiovascular dynamics. III: labor and delivery under local caudal analgesia. Am J Obstet Gynecol 1969;103:8.
280. Whittemore R, Hobbins JC, Engle MA. Pregnancy and its outcome in women with and without surgical treatment of congenital heart disease. Am J Cardiol 1982;50:641.
281. Elkayam U, Cobb T, Gleicher N. Congenital heart disease in pregnancy. In: Elkayam U, Gleicher N, eds. Cardiac Problems in Pregnancy, 2nd ed. New York: Alan R. Liss, 1990.
282. Wachtel HL, Czarnecki SW. Coarctation of the aorta and preg-nancy. Am Heart J 1966;72:251.
283. Sareli P, England MJ, Berk HR, et al. Maternal and fetal sequelae of anticoagulation during pregnancy in patients with mechani-cal heart valve prosthesis. Am J Cardiol 1989;63:1462.
284. Wang RYC, Li PK, Chow JSF, et al. Effi cacy of low-dose subcu-taneously administered heparin in the treatment of pregnant women with artifi cial heart valves. Med J Aust 1983;2:126.
285. Mitani GM, Harrison EC, Steinberg I, et al. Digitalis, glyco-sides in pregnancy. In: Elkayam U, Gleicher N, eds. Cardiac Problems in Pregnancy, 2nd ed. New York: Alan R. Liss, 1990:617–646.
286. Ellsworth AJ, Horn JR, Raisys VA, et al. Disopyramide and N-monodesalkyodisopyramide in serum and breast milk. Drug Intell Clin Pharm 1989;23:56.
287. Dicke JM. Cardiovascular drugs in pregnancy. In: Gleicher N, Elkayam U, Galbraith RM, et al., eds. Principles of Medical Therapy in Pregnancy. New York: Plenum, 1985:646.
288. Lee W, Shah PK, Amin DK, et al. Hemodynamic monitoring of cardiac patients during pregnancy. In: Elkayam U, Gleicher N, eds. Cardiac Problems in Pregnancy, 2nd ed. New York: Alan R. Liss, 1990:47.
289. Rosenberg B, Simonberg K, Peretz BA, et al. Eisenmenger’s syndrome in pregnancy: controlled segmental epidural block for cesarean section. Reg Anaesth 1984;7:131.
290. Angel JL, Chapman C, Knappeo RA, et al. Percutaneous balloon aortic valvuloplasty in pregnancy. Obstet Gynecol 1988;72:438.
291. Gleicher N, Midwall J, Hochberger B, et al. Eisenmenger’s syn-drome in pregnancy. Obstet Gynecol Surv 1979;34:721.
292. Radford DJ, Stafford G. Pregnancy and the Rastelli operation. Aust N Z J Obstet Gynaecol 2005;45:243–247.
CAR010.indd 277CAR010.indd 277 11/24/2006 10:41:08 AM11/24/2006 10:41:08 AM

CAR010.indd 278CAR010.indd 278 11/24/2006 10:41:08 AM11/24/2006 10:41:08 AM





























