Embed Size (px)
Citation preview

1
Epstein-Barr virus (EBV)-induced resistance to drugs that 1
activate the mitotic spindle assembly checkpoint in Burkitt’s 2
lymphoma cells 3
4
Maria Leao1, Emma Anderton, Mark Wade
2, Kiran Meekings and 5
Martin J Allday* 6
Department of Virology, Faculty of Medicine, Imperial College London, Norfolk Place, 7
London W2 1PG. UK. 8
Present address: 1Ludwig Institute for Cancer Research 9
Cell and Molecular Biology 10
Courtauld Building 11
91 Riding House Street 12
London W1 W7BS. 13
2Gene Expression Laboratory 14
The Salk Institute for Biological Studies 15
La Jolla 16
CA 92037 USA. 17
*Corresponding Author. Mailing Address: Department of Virology, Faculty of 18
Medicine, Imperial College London, Norfolk Place, London, W2 1PG, UK. 19
Phone: 44 (0) 2075943836. Fax: 44 (0) 2075943973. E.mail: [email protected] 20
Running title: EBV-induced resistance to microtubule disrupting drugs 21
Keywords: EBV, mitosis, spindle checkpoint, nocodazole, apoptosis. 22
Abstract: 239 23
24
ACCEPTED
Copyright © 2006, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved.J. Virol. doi:10.1128/JVI.01096-06 JVI Accepts, published online ahead of print on 11 October 2006
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

2
Epstein-Barr virus (EBV) is associated with a number of human cancers and latent 1
EBV gene expression has been reported to interfere with cell cycle checkpoints and 2
cell death pathways. Here we show that latent EBV can compromise the mitotic 3
spindle assembly checkpoint and rescue Burkitt’s lymphoma (BL)-derived cells 4
from caspase-dependent cell death initiated in aberrant mitosis. This leads to 5
unscheduled mitotic progression resulting in polyploidy, multi- and/or micro-6
nucleation. The EBV latent genes responsible for this phenotype are expressed 7
from the P3HR1 strain of virus and several viruses with similar genomic deletions 8
that remove the EBNA2 gene. Although EBNA2 and the latent membrane proteins 9
(LMPs) are not expressed, the EBNA3 proteins are present in these BL cells. 10
Survival of the EBV-positive cells is not consistently associated with EBV lytic gene 11
expression, nor the genes that are expressed in EBV latency I BL cells (ie EBNA1, 12
EBERs and BARTs), but correlates with reduced expression of the cellular pro-13
apoptotic BH3-only protein Bim. These data suggest that a subset of latent EBV 14
gene products may increase the likelihood of damaged DNA being inherited 15
because of the impaired checkpoint and enhanced survival capacity. This could 16
lead to greater genetic diversity in progeny cells and contribute to tumorigenesis. 17
Furthermore, since it appears that this restricted latent EBV expression interferes 18
with the responses of Burkitt’s lymphoma-derived cells to cytotoxic drugs, the 19
results of this study may have important therapeutic implications in the treatment 20
of some BL. 21
22
23
24
25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

3
INTRODUCTION 1
Epstein-Barr virus (EBV) is a �amma-herpesvirus that produces an asymptomatic 2
infection in the majority of the human population. However, EBV is also associated 3
with a number of human tumours of B cell, T cell and epithelial origin. These include 4
Burkitt’s lymphoma (BL), some types of Hodgkin’s lymphoma, immunoblastic B-5
lymphoma in the immunosuppressed, nasopharyngeal carcinoma and gastric carcinoma. 6
Although the precise contribution EBV makes to the development of these diseases is 7
not yet known, it has been suggested that interference with cell cycle checkpoints and 8
cell death pathways by EBV may play an important role in B-lymphomagenesis 9
[reviewed in (20)]. 10
In vitro, EBV has the ability to infect and transform primary B cells into continuously 11
proliferating lymphoblastoid cell lines (LCLs) that have a pattern of viral gene 12
expression known as latency III. This is characterised by the expression of nine viral 13
proteins: six Epstein-Barr nuclear antigens [(EBNAs) 1, 2, 3A, 3B, 3C and -LP] and 14
three latent membrane proteins [(LMPs) 1, 2A and 2B]. Additional RNA species [the 15
EBV-encoded RNAs (EBERs) and the BamH1A Rightward Transcripts (BARTs)] are 16
also expressed during latency III, but their significance is not fully understood (3). It has 17
been reported from studies with recombinant EBV that only six of the latency III 18
proteins (EBNAs 1, 2, 3A, 3C, -LP and LMP1) are essential for efficient transformation 19
of B cells into LCLs (3). 20
Current data on the persistence of EBV in humans are consistent with the viral genome 21
residing long-term in a resting memory B cell population. However, to establish 22
persistence EBV is thought to infect naïve B cells and drive these to proliferate as 23
activated LCL-like B-blasts. This expansion of an infected B-blast population in vivo is 24
accompanied by their differentiation into centroblasts and centrocytes and finally resting 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

4
memory B cells. The precise series of events that the EBV-positive B cells undergo to 1
reach the memory compartment is not yet known, however, it appears to involve 2
regulated shut-down of latent EBV gene expression from latency type III in the B-3
blasts, via a state called latency II (EBNA1, LMP1 & 2 and the RNAs) until in 4
quiescent memory B cells no EBV proteins can be detected in a state termed latency 0 5
(30). 6
Generally, oncogenic viruses are able to disrupt cell cycle checkpoints that are induced 7
by genotoxic stress and by other forms of cellular damage such as microtubule 8
disruption [reviewed in (20)]. Although the tumour suppressor p53 is one of the major 9
targets of oncogenic viruses, several studies have shown that EBV does not specifically 10
target p53 during the transformation of normal B cells into LCLs (1, 2). Nevertheless, 11
recent studies demonstrated that although not able to directly target p53, EBV may still 12
interfere with cell cycle checkpoints at both G1/S and G2/M phases (15, 21, 31). 13
Following genotoxic stress induced by cisplatin, EBV suppresses a checkpoint 14
downstream of p53 by preventing the inactivation of cdk2 by p21WAF1/CIP1
and this 15
appears to involve the modulation of p21WAF1/CIP1
stability (21). In addition to 16
deregulating a G1 checkpoint, EBV has also been shown to interfere with checkpoints 17
acting at the G2/M transition. Treatment of EBV-negative Burkitt’s lymphoma-derived 18
cell lines with cisplatin, etoposide or doxorubicin activates a p53- and p73-independent 19
checkpoint leading to G2/M arrest and default apoptosis. In contrast, similar Burkitt’s 20
lymphoma-derived cell lines infected with the B95.8 strain of EBV (or a deletion 21
mutant virus – P3HR1) fail to arrest or to undergo apoptosis after treatment with these 22
genotoxins (31). It has also been reported that latent EBV can disrupt the regulation of a 23
checkpoint activated in G2 by a histone deacetylase inhibitor azelaic bishydroxamine 24
and this appears to be associated with expression of the EBNA3 family of proteins (15, 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

5
25). Another recent report showed that BL41 cells infected with B95.8 virus exhibited 1
significantly higher levels of micronucleus formation than EBV-negative BL41 cells. 2
This is a further indication that the latency III pattern of gene expression found in these 3
cells is associated with corrupted cell cycle checkpoints and genomic instability (10). 4
Here, we show that expression of a subset of EBV genes in Burkitt`s lymphoma-derived 5
cell lines treated with microtubule-disrupting drugs nocodazole and taxol produced 6
abnormal mitotic progression, the formation of aberrant nuclei and polyploidy. This 7
involves suppression of the mitotic spindle assembly checkpoint and evasion of 8
caspase-dependent cell death associated with mitotic slippage. 9
MATERIALS AND METHODS 10
Cell culture 11
Burkitt’s lymphoma cell lines [BL41, BL41/B95.8, Ramos, Ramos/AW, BL31, BL41-12
E2KO, BL31-E2KO, Ava-BL, Oku-BL, Sal-BL, Mutu-I (clone 179), Mutu-III and 13
Dante] were cultured in RPMI 1640 medium supplemented with 10% Serum Supreme 14
(Biowhittaker, Wycombe, UK) or 10% bovine foetal calf serum, penicillin/streptomycin 15
and glutamine at 37oC in an incubator with 10% CO2. Hygromycin B (Roche) was 16
added at a concentration of 100µg/ml to BL41-E2KO and BL31-E2KO. For routine 17
passage, cells were split 1:4 every three or four days. For experiments, all cells were re-18
suspended in fresh medium at a density of 3 x 105
cells/ml 24 hours prior to 19
manipulation. 20
Cell treatments 21
Cells were exposed to �amma-irradiation (850 rads) using a Gammacell® 1000 Elite 22
irradiator with a 137
Cs source. Nocodazole (Sigma) and taxol (Bristol-Meyers Squibb) 23
were added to the cell medium to give a final concentration of 50 ng/ml. Viable cell 24
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

6
counts were performed using a hemocytometer and based on the exclusion of trypan 1
blue dye. 2
Microscopy 3
Cells were harvested and re-suspended at 1x106
cells/ml in PBS. Approximately 8x104 4
cells were then transferred to cytospin chambers (Shandon, USA), spun onto glass 5
slides at 500xg for 1 minute and air dried before fixing in methanol:acetone (1:1) for 20 6
minutes at -20°C. After air-drying, the slides were stored at -20°C until staining. For 7
DAPI staining, slides were re-hydrated in 1xPBS for 10 minutes and excess PBS was 8
removed from the slide. Cells were covered with one drop of DAPI solution (0.001% in 9
PBS/0.6% NP40) and incubated at room temperature in the dark for 10 minutes. Slides 10
were thoroughly washed in 1xPBS, mounted in Citifluor (London, UK) and stored at 11
4°C. DAPI-stained images were captured using an Axiophot fluorescence microscope 12
(Carl Zeiss, Germany) with 40X and 63X objective lenses and Axio Vision 4 software. 13
Flow cytometry 14
All flow cytometry was performed using a Beckton-Dickinson FACSort flow cytometer 15
and analysed with CellQuest software. Doublets (two cells registering as a single event) 16
were electronically gated out of all analyses. For propidium iodide (PI) staining, 2x106
17
cells were pelleted at 700xg in a Beckman-Coulter Allegra 6R centrifuge, washed twice 18
in PBS and transferred to 1.5 ml Eppendorf tubes for fixation in 70% ethanol for at least 19
1 hour. After washing away excess ethanol, cells were re-suspended in 1 ml of 20
propidium iodide solution (18µg/ml PI with 8µg/ml RNaseA in PBS) and incubated at 21
4°C for at least one hour before flow cytometric analysis. 22
Western Immunoblot analysis 23
Briefly, protein extracts were resolved by SDS-PAGE and transferred to Protran 24
nitrocellulose membranes (Schleicher & Schuell Bioscience) and immunoblots 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

7
performed as described previously (31) using ECL chemi-luminescence (Amersham 1
Biosciences) for visualisation. Primary antibodies (unless otherwise stated used as 2
directed by the manufacturer) were: rabbit anti-PARP polyclonal (Roche); mouse anti-γ-3
tubulin monoclonal (GTU-88, Sigma); mouse anti-BZLF1 monoclonal [BZ1, (34)]; 4
mouse anti-BHRF1 monoclonal [also known as EA-R-p17; Bcl2 homologue (5B11, 5
Chemicon)] and rabbit anti-Bim polyclonal (Stressgen). The human serum EE was from 6
a patient with chronic infectious mononucleosis and has a very high titre of antibodies 7
recognising EBV early lytic antigens (see ref. 12). The human serum was used at a 8
concentration of 1/10,000. 9
RESULTS 10
Some Burkitt’s lymphoma (BL)-derived cell lines – despite having a mutant p53 – were 11
shown to undergo cell cycle arrest and apoptosis after DNA damage by different 12
genotoxic agents (31). For this reason, these cells are excellent tools for the study of B 13
cells lacking wild-type p53 function and the effects EBV has on cell cycle and cell 14
death regulation, since the virus does not appear to target p53 (1, 2, 22). A series of 15
experiments were performed to characterize further the effect of latent EBV on the p53-16
independent responses of BL-derived cell lines to damage induced by irradiation and to 17
the anti-microtubule poisons nocodazole and taxol that specifically target mitosis. 18
EBV protects BL41/B95.8 cells from dying after aberrant mitosis following 19
recovery from �amma-irradiation. In order to investigate the effect of EBV on the 20
response of BL41 cells to �amma-irradiation, both BL41 and BL41/B95.8 cells grown 21
to similar densities were irradiated (850 rads) and then examined periodically by 22
microscopy and their DNA content was analysed by flow cytometry after staining with 23
propidium iodide (Fig. 1a and b). These analyses revealed that BL41 cells accumulated 24
with 4N DNA content and apparently normal interphase nuclei approximately 24 hours 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

8
after irradiation, consistent with the cells being blocked in G2/M. Similar responses in 1
p53-negative B cells have been reported previously (19). EBV-positive BL41/B95.8 2
cells also accumulated with apparently normal nuclei and 4N DNA content. This 3
indicated that EBV is unable to overcome the G2/M arrest after irradiation and is 4
consistent with the results from experiments using EBV-immortalized LCLs (5, 21). 5
Three days after irradiation, both BL41 and BL41/B95.8 populations adapted, recovered 6
from the G2/M-associated arrest and progressed through mitosis. In both cases this 7
produced many morphologically aberrant nuclei (Fig. 1c, upper panels). The EBV-8
negative BL41 cells subsequently died rapidly. In contrast the EBV infected 9
BL41/B95.8 cells survived with grossly abnormal nuclei; cells with multiple nuclei and 10
micronuclei were both abundant (Fig. 1c, lower panels and 1d). Similar experiments 11
were performed using the EBV-negative BL line Ramos and its EBV-positive pair 12
Ramos/AW. Ramos cells behaved in a very similar manner to BL41 and were dead 13
within 5 days. In contrast, Ramos/AW survived, generally with morphologically 14
abnormal nuclei (data not shown). Since Ramos/AW carries an EBV genome with a 15
deletion that removes the EBNA2 gene and part of the EBNA-LP coding sequence, this 16
suggested that a restricted subset of EBV gene expression was necessary for survival. 17
To summarize, these results showed that EBV appears not to overcome the p53-18
independent transient accumulation of cells at G2/M following irradiation. However, 19
after cells adapt and recover from this arrest it appears that a restricted pattern of EBV 20
expression may suppress a mitotic checkpoint and rescue cells from death during 21
aberrant mitosis. 22
EBV overcomes a metaphase arrest in cells exposed to the microtubule disrupting 23
agent nocodazole. The preceding series of experiments suggested that latent EBV has 24
activities that interfere with mitotic checkpoint function. We therefore wanted to 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

9
explore further the effect of latent EBV on other events associated with arrest in mitosis, 1
in particular the spindle assembly checkpoint that is activated in pro-metaphase by 2
microtubule poisons. 3
Microtubule-depolymerizing agents such as nocodazole lead to disruption of mitotic 4
spindle assembly. Because the kinetochores of condensed chromosomes cannot attach 5
to a functional spindle, this activates a checkpoint that results in a metaphase arrest. In 6
normal mammalian cells this M-phase arrest can be transient, there is slippage from 7
metaphase and abnormal progression through the remainder of the cell cycle. 8
Both BL41 and BL41/B95.8 cells were grown to similar densities, treated with 50 ng/ml 9
nocodazole for 24 hours, stained with PI and analysed by flow cytometry. As expected, 10
nocodazole was able to induce an accumulation of BL41 cells with a 4N DNA content. 11
Many BL41/B95.8 cells treated in a similar manner also accumulated with 4N DNA 12
(Fig. 2a). Although it was evident that most cells in both populations accumulate with 13
4N DNA, sub-populations containing more than 4N DNA were also revealed. This 14
indicated that cells were either completing abnormal mitosis but not cytokinesis or 15
endoreduplicating DNA from a pseudo-G1. Interestingly, this >4N population was 16
significantly greater in BL41/B95.8 cells (approximately 24% compared with <10% in 17
BL41 cells), which indicates that EBV is interfering with the mitotic checkpoint 18
activated by nocodazole. 19
Analysis of nuclear morphology in DAPI-stained cells 24 hours after nocodazole 20
treatment was consistent with this interpretation. Nocodazole disrupts assembly of the 21
mitotic spindle, therefore the response of most cells is to arrest during pro-metaphase 22
when condensed chromosomes line up to form the metaphase plate. The percentage of 23
metaphase-arrested cells in a population is known as the mitotic index. Low speed 24
cytospins were prepared from both BL41 and BL41/B95.8 cells treated for 24 hours 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

10
with nocodazole and stained with DAPI. Figs. 2b and c show BL41 cells in a typical 1
experiment with metaphase-arrested morphology (condensed chromosomes can be seen 2
as ‘spreads’). The mitotic index of this population of cells was determined to be about 3
43%. In contrast, staining BL41/B95.8 cells after similar treatment revealed very few 4
cells with condensed chromosomes indicating that the majority were not arrested in 5
metaphase. The mitotic index of this population was judged to be about 10% and it was 6
noted that many of the cells were multinucleated or had developed morphologically 7
abnormal nuclei (see examples in Figs 2b and c, right panels). These cells with grossly 8
abnormal nuclei probably represent the population with 4N or greater DNA content 9
observed in the cell cycle profile of BL41/B95.8 cells after 24 hours (Fig. 2a). Together, 10
these results suggest that EBV latent gene expression in BL cells interferes with the 11
activation or execution of the spindle assembly checkpoint and thus prevents metaphase 12
arrest. 13
EBV-negative BL41 cells die after the mitotic arrest induced by nocodazole or 14
taxol, while EBV-positive BL41/B95.8 become polyploid and develop aberrant 15
nuclei. To further characterise the response of BL cells to nocodazole, BL41 and 16
BL41/B95.8 cells were analysed 72 hours after treatment. Microscopy and flow 17
cytometry were performed after DAPI or trypan blue staining and propidium iodide 18
staining respectively. Fig. 3a shows that after 72 hours, there was a significant increase 19
in the sub-G1 population of BL41 cells indicating that most these cells were undergoing 20
an apoptosis-like cell death. Cellular morphology – as revealed by DAPI and counts of 21
trypan blue-excluding viable cells – also indicated that most of the BL41 cells had died 22
after 72 hours, since only debris could be seen at this time (see Figs. 3b and c for 23
representative examples). The cell cycle profile of BL41/B95.8 cells 72 hours after 24
treatment with nocodazole was markedly different from the profile of BL41 cells. In 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

11
contrast to the BL41 cells – although the sub-G1 population in BL41/B95.8 cells has 1
slightly increased by 72 hours – most of the cells had a 4N or greater DNA content. 2
Consistent with this, 72 hours after the addition of nocodazole, microscopy showed 3
viable BL41/B95.8 cells were abundant (50% or more survived) and many had grossly 4
abnormal or fractured nuclei (Figs. 3b and c). This indicated that these cells continued 5
to synthesise DNA in the presence of nocodazole and became polyploid or aneuploid. 6
Similar experiments were performed using taxol, which causes aberrant microtubule 7
polymerization, prevents spindle assembly and kinetochore attachment and so also 8
activates the checkpoint. Taxol induced a metaphase arrest in the BL41 cells, but – like 9
nocodazole – failed to trigger a similar arrest in the EBV-positive BL41/B95.8 cells 10
(data not shown). As a result, after 72 hours, numerous 4N and >4N cells with abnormal 11
nuclear morphology were seen in the EBV-positive population, whereas mass cell death 12
was seen in the EBV-negative BL41 population (Figs. 3b and c). 13
Latent infection with P3HR1 also increases cell survival after treatment with 14
spindle poisons. In order to test whether the responses associated with the spindle 15
assembly checkpoint described above were restricted to B95.8-infected cells, similar 16
experiments were performed on Ramos BL-derived cells latently infected with P3HR1 17
virus. The responses of P3HR1-infected BL cells to both nocodazole and taxol were 18
very similar to cells carrying latent B95.8 virus. Ramos/AW cells that carry latent 19
P3HR1 (and express only EBNA1, truncated EBNA-LP, EBNAs 3A, 3B and 3C 20
proteins and the EBER and BART RNAs), do not arrest in metaphase nor die, but rather 21
they progress through mitosis and develop gross nuclear abnormalities. In contrast, the 22
parental EBV-negative Ramos cells initially arrested in metaphase then died in a similar 23
manner to BL41 (Fig. 4 and data not shown). 24
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

12
A subpopulation of EBV-positive BL41 and Ramos cells survive for up to 3 weeks 1
and proliferate. In order to determine the long-term fate of cells that were still viable 2
after 72 hours exposed to nocodazole, the lines were treated with nocodazole, washed 3
and re-suspended in freshly conditioned medium. By this time all the EBV-negative 4
BL41 and Ramos cells were dead. However, depending on the cell line and experiment, 5
20-70% of the EBV-positive converted cells were viable after washing and re-6
suspending in fresh medium. On further incubation (with feeding every 4-5 days) it was 7
revealed that in both BL41/B95.8 and Ramos/AW cultures viable cells survived for at 8
least 3 weeks and some – presumably those containing a less severely damaged diploid 9
genome – were able to resume proliferating and hence the number of viable cells 10
increased (see Fig. 5 for examples). 11
Latent infection with P3HR1-like viruses increases BL survival after treatment 12
with spindle poisons. Since BL41, BL41/B95.8, Ramos and Ramos/AW have all been 13
grown in culture for many years they may have developed clonal idiosyncrasies. In 14
order to reinforce the hypothesis that EBV gene expression is responsible for the 15
phenotypes described above and to confirm that the limited pattern of viral proteins 16
associated with P3HR1 is sufficient, newly established lines produced with a 17
recombinant virus were investigated. The EBV-negative BL lines BL41 and BL31 were 18
infected with a recombinant hygromycin-resistant strain of EBV from which the 19
EBNA2 open reading frame had been deleted to produce a P3HR1-like virus (14). 20
EBV-positive converts of BL41 and BL31 were produced by selection in hygromycin 21
and – like BLs latently infected with P3HRI virus – each convert expresses EBNA1 and 22
the EBNA3 proteins but not EBNA2 nor LMP1 (14). Unlike P3HR1-infected cells, the 23
EBNA2-knock-out (KO) converts express full length EBNA-LP. The parental lines 24
(BL41 and BL31) and the converts (BL41+E2KO and BL31+E2KO) were treated with 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

13
nocodazole and analysed as described above. After 24 hours exposure to nocodazole, 1
metaphase-arrested cells were counted to establish the mitotic index of each cell line. 2
While the mitotic index of the parental cells was consistently about 40%, that of the 3
E2KO-converts was consistently about 10% (Fig 6a). Seventy-two hours after initial 4
exposure to nocodazole the parental BL41 and BL31 cells were all dead and only cell 5
fragments remained. In contrast the EBNA2-KO converts were viable with 4N or 8N 6
DNA content (see for example Fig. 6b and d). As anticipated the phenotype of these 7
cells was similar to that of BL41/B95.8 and Ramos/AW and therefore consistent with 8
the restricted pattern of EBV latent gene expression associated with P3HR1 impairing 9
the spindle checkpoint and rescuing the cells from programmed cell death in mitosis. 10
Similar experiments performed on BL cell lines (Dante and Mutu-I) that exhibit the 11
Type-I pattern of latency (EBNA1, EBERs and BARTS) showed their response to 12
nocodazole to be similar to the EBV-negative BL cells. EBNA1, EBERs and BARTs do 13
not reduce the mitotic index nor rescue cells from death in mitosis (Fig. 6c and e and 14
data not shown). It is important to note that Mutu cells that had ‘drifted’ in culture to 15
produce the full latency-III pattern of EBV gene expression (Mutu-III) were fully 16
protected from the lethal effects of nocodazole (Fig. 6c & e). This shows that Mutu cells 17
per se are not intrinsically more susceptible for reasons unrelated to EBV gene 18
expression. 19
Recently, Rickinson and colleagues described Burkitt’s lymphoma biopsy material and 20
newly established cell lines (Ava-BL, Oku-BL and Sal-BL) that carry EBV genomes 21
including similar deletions to P3HR1 and consequently express a P3HR1-like pattern of 22
EBV proteins (13, 14). The responses of low passage Ava-BL, Oku-BL and Sal-BL 23
cells to nocodazole were determined and found to be very similar to those obtained with 24
cells infected with B95.8 or P3HR1 – that is, the cells exhibited a relatively low mitotic 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

14
index, survived the treatment and developed aberrant nuclei (Fig. 6a, c and e). It would 1
appear that, as with latency-III, the restricted pattern of EBV gene expression found in 2
Ramos/AW, E2-KO converts, Ava-BL, Oku-BL and Sal-BL is sufficient to impair the 3
spindle assembly checkpoint and rescue cells from programmed cell death during 4
aberrant mitosis. 5
Proteolytic cleavage of poly (ADP-ribose) polymerase (PARP) indicates caspase-6
dependent cell death. To further characterise the fate of the BL cells responding to 7
microtubule poisons, we asked whether the cell death seen in BL41 and BL31 was 8
associated with caspase activity and whether this was suppressed by latent EBV. Cells 9
were treated with nocodazole for up to 72 hours and samples taken periodically for 10
western blot analysis. Blots were probed to show caspase-specific cleavage of PARP 11
and also for tubulin as a protein loading control. Consistent with the death of these 12
metaphase-arrested cells involving activation of caspases, full length PARP was 13
completely cleaved in EBV-negative BL41, BL31 and also the Latency-I Dante and 14
Mutu-I cells, but remained largely or completely unaffected in the EBNA2-KO converts 15
and Mutu-III (Figure 7a). Similarly the PARP in the cells from the newly isolated BL 16
lines (Ava, Oku and Sal) that carry P3HR1-like virus remained largely or entirely intact 17
after the cells were exposed to nocodazole. 18
Rescue from nocodazole-induced cell death is not dependent on EBV lytic gene 19
expression. We showed previously that sometimes activators of the EBV lytic 20
programme are able to induce caspase activity and apoptosis, but that EBV lytic gene 21
expression protects the cells containing replicating virus from cell death (12). It has also 22
been reported that some cytotoxic agents (for instance irradiation) can induce the EBV 23
lytic cycle in some cells (33). In order to determine whether lytic gene products were 24
involved in the rescue from mitotic death seen here, various lines judged to be resistant 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

15
were treated with nocodazole for up to 72 hours and protein extracts were western 1
blotted for evidence of EBV lytic activity. Fig. 7b shows that nocodazole failed to 2
consistently induce the expression of the lytic-switch protein BZLF1 or various EBV 3
early antigens recognised by the human serum EE in a selection of EBV-positive BL 4
cells used in this study. Extracts were also probed with a monoclonal antibody raised 5
against the EBV Bcl2-homologue BHRF1 (Fig. 7b and c). In the BL41, BL31 and 6
Ramos EBV-positive lines this protein was neither constitutively expressed nor induced 7
by nocodazole. However, it can be seen that in BLs Sal and Oku, BHRF1 was 8
constitutively expressed at a low level and that this was slightly induced after 72 hours. 9
This was surprising because using the very sensitive serum EE and the BZLF1 mAb we 10
calculated that <1.5% of the Sal and Oku cells were in the lytic cycle (see Fig. 7, 11
legend). This suggests that in these two lines BHRF1 may be expressed as a latent gene. 12
We conclude that EBV lytic activity is not generally required to rescue cells from death 13
in aberrant mitosis, but that BHRF1 could be making a minor contribution to survival in 14
the unusual BL lines Sal and Oku. 15
Bim could be a determinant of sensitivity to nocodazole. It has been shown that the 16
BH3-only pro-apoptotic protein Bim may be a major determinant of sensitivity to 17
spindle-disrupting drugs in transformed epithelial cells (27). Bim is also particularly 18
important in the regulation of apoptosis during B cell development and its regulation by 19
cMyc can play an important role in the pathogenesis of BL (8, 11, 26). Furthermore a 20
recent report indicated that EBV-converted BL41 and Ramos cells express lower levels 21
of Bim than the parental lines (7). Protein extracts from the matched pairs of EBV-ve 22
and EBV+ve cells previously screened for caspase activity were western blotted and 23
probed for expression of the Bim proteins (Fig. 7d). Consistent with Bim playing a role 24
in the mitotic death of the EBV-negative cells and EBV causing Bim down-regulation, 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

16
levels of all three isoforms of Bim were significantly higher in BL31 and BL41 cells 1
than the EBV-converted partners. Mutu-III also expressed significantly lower levels of 2
Bim than the EBNA1, EBER and BARTs-only Mutu-I. These data extend the report 3
that latent EBV infection can lead to a down-regulation of Bim in B-lymphocytes and 4
confirm that the reduction of expression does not require EBNA2 nor the latent 5
membrane proteins (7). 6
DISCUSSION 7
After experiments with ionizing radiation suggested that EBV might interfere with the 8
regulation of mitosis, a study was performed in which the spindle assembly checkpoint 9
was specifically triggered by exposing cells to microtubule-disrupting agents. This has 10
very clearly demonstrated that EBV-negative BL41, Ramos and BL31 and EBV-11
positive BL lines Dante and Mutu-I that express only EBNA1, EBERs and BARTs are 12
all very sensitive to the lethal actions of nocodazole and taxol. In each case a transient 13
arrest in metaphase was followed by high levels of caspase-associated cell death that 14
probably occurs during aberrant mitosis following adaptation and slippage from 15
metaphase (24, 29, 32). Since the precise distinction between apoptosis and mitotic 16
catastrophe remains unclear and somewhat controversial (4, 6, 17), here this process is 17
described as caspase-dependent mitotic cell death. 18
When the EBV-negative BL lines are latently infected with EBV – in established cell 19
lines such as BL41/B95.8 or Ramos/AW or in newly converted lines produced using 20
EBNA2-knockout recombinant EBV – the cells exhibit an impaired metaphase arrest 21
and are rescued from cell death. Newly isolated BL-lines (Ava, Sal and Oku) carrying 22
P3HR1-like EBV showed a similar response. The consistency of these responses 23
suggested that clonal variations that are independent of EBV infection are very unlikely 24
to play a significant role in the observed phenotypes. 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

17
Which EBV gene product is responsible for this mitotic phenotype? EBNA2 and the 1
latent membrane proteins (LMPs) can be excluded as candidates because these are not 2
expressed in any of the P3HR1-converted cells, in the EBNA2-knockout lines, or the 3
newly established Ava-, Oku- and Sal-BLs (14, 31). This is because EBNA2 is required 4
as a transactivator of the LMP genes. Since EBNA2, LMP1 and LMP2 have all been 5
shown to promote cell survival, this is a rather surprising observation (9, 16, 23). 6
EBNA-LP is truncated in the P3HR1-converted cells and is expressed at a very low 7
level in the nocodazole-resistant Oku-BL, arguing against EBNA-LP playing a 8
significant role (14). BLs with a latency-I pattern of EBV gene expression (Mutu-I and 9
Dante) are very sensitive to nocodazole – relative to a latency-III expressing derivative 10
– so it is unlikely that EBNA1, the EBERs or BARTs play any role in this resistance. 11
Nocodazole failed to consistently induce lytic gene expression so the various anti-12
apoptotic EBV lytic proteins cannot be involved in most cases. We did note that Sal and 13
Oku BL cells expressed low levels of BHRF1. Here it is possible that BHRF1 may 14
contribute to survival. The only full-length EBV-encoded factors that are common to all 15
the nocodazole-resistant cells are EBNA3A, EBNA3B and EBNA3C. Thus in principle 16
one or more of this family of nuclear proteins is likely to be responsible for this striking 17
phenotype. Nevertheless we cannot formally exclude the role of EBV latent gene 18
products that have yet to be identified or the possibility that entry of a small number of 19
cells into the lytic cycle may exert paracrine effects on the main population. 20
There appear to be two components of the resistance to spindle poisons seen in these 21
EBV-carrying cells. There is suppression of metaphase arrest leading to a reduced 22
mitotic index and escape from caspase-associated cell death leading to nuclear 23
abnormalities (summarised in Fig. 8). It is presently unclear to what extent these 24
phenomena are linked and precisely which EBV function(s) are involved. Recent 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

18
reports of cells with a weakened or completely ablated spindle assembly checkpoint 1
incurring much less mitotic catastrophe/apoptosis than their checkpoint-proficient 2
counterparts are consistent with the hypothesis that EBV need only target the 3
checkpoint (18, 29, 32) and our previous observation that EBNA3C can suppress the 4
spindle checkpoint in U2OS cells is also consistent with this hypothesis (22). 5
One or more of the EBNA3 genes – whether expressed from a recombinant EBV or the 6
Ava-, Oku- and Sal-strains of EBV – mediate survival of cells exposed to ionomycin or 7
anti-IgM (14), however, it should be noted that even complete latent EBV gene 8
expression does not always enhance cell survival. For instance, LCLs are induced to 9
undergo apoptosis by various DNA-damaging agents (1, 2, 21, 31) and LCLs and DG75 10
BL cells expressing the EBNA3 proteins are induced to die by a histone deacetylase 11
inhibitor (15, 25). It is evident that the EBNA3 proteins do not suppress apoptosis per 12
se. Nevertheless, since the level of the pro-apoptotic factor Bim is a determinant of 13
sensitivity to taxol in breast epithelial cells and can influence the pathogenesis of BL (8, 14
11, 27), it is probable that the down-regulation of Bim levels by EBV plays a role in the 15
development of resistance to spindle poisons in the BL cells studied here. The data 16
shown are consistent with one or more of the EBNA3 proteins reducing Bim expression 17
and increasing cell survival by this, and perhaps other means. Systematic deletion of 18
each of the EBNA3 genes in recombinant EBV and the ‘conversion’ of nocodazole-19
sensitive BL lines should cast further light on these issues and may distinguish between 20
cell cycle and cell survival activities. 21
A weakened or compromised spindle assembly checkpoint is frequently seen in cancer 22
cells and because it can result in the passage of a damaged genome to progeny cells it 23
may directly contribute to tumorigenesis [reviewed in (24, 29, 32)]. Since even after 24
exposure to 50ng/ml nocodazole for 72 hours, some EBV-carrying Ramos and BL41 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

19
cells survive in culture for up to 3 weeks and resume proliferating (Fig. 5), we suggest 1
that this could be the case when lymphomas carry latent EBV and express the EBNA3 2
proteins. It is of practical significance that the presence of these proteins appears to 3
dramatically modulate the sensitivity of BL cells to agents used in anticancer therapy. 4
Two reports have led to suggestions that microtubule-disrupting agents may be highly 5
suitable for the clinical management of a subset of lymphomas, including BL. El-Deiry 6
and colleagues showed that silencing the polo-like kinase Snk/Plk2 gene leads to a 7
significant increase in mitotic catastrophe or apoptosis in cells exposed to taxol-like 8
drugs. They therefore suggested that this knowledge could be of therapeutic value when 9
using taxanes or other microtubule-disrupting agents on tumours (4). More recently, 10
Crook and colleagues reported that Snk/Plk2 is often silenced by promoter methylation 11
in several different B cell malignancies. More specifically they showed that this 12
methylation-dependent silencing occurs at very high frequency in BL (n=24/25). On 13
this basis they suggest that BL may be a particularly good target for taxane-like drugs 14
(28). 15
The data we have presented here support this speculation. The EBV-negative BL and 16
Type-I latency BL cells examined were indeed very sensitive to nocodazole and taxol. 17
However, it is clear from recent studies (13, 14) that a minor but significant sub-18
population of BLs may carry deletion mutant EBV that express the EBNA2-knockout 19
pattern of genes that includes the EBNA3 family of proteins. It is not known how 20
frequently such tumours arise, but when they occur it is likely that they will have a 21
significantly reduced sensitivity to cytotoxic therapies based on the disruption of 22
microtubules or agents that damage DNA. 23
ACKNOWLEDGEMENTS 24
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

20
We are very grateful to Gemma Kelly (Birmingham) for the low passage Ava-BL, Oku-1
BL, Sal-BL, BL41-EBNA2-KO and BL31-EBNA2-KO cells and to Carol McDonald 2
and Paul Farrell for the anti-BZLF1 antibodies and Akata protein samples. We thank 3
Jenny O’Nions and Paul Farrell for helpful comments on the manuscript. This work was 4
partly supported by grants from the Wellcome Trust and the Portuguese Foundation for 5
Science and Technology (grant ref: SRFR/BD/2730/2000) and MRC PhD studentships. 6
REFERENCES 7
1. Allday, M. J., G. J. Inman, D. H. Crawford, and P. J. Farrell. 1995. DNA 8
damage in human B cells can induce apoptosis, proceeding from G1/S when p53 9
is transactivation competent and G2/M when it is transactivation defective. 10
Embo J 14:4994-5005. 11
2. Allday, M. J., A. Sinclair, G. Parker, D. H. Crawford, and P. J. Farrell. 12
1995. Epstein-Barr virus efficiently immortalizes human B cells without 13
neutralizing the function of p53. Embo J 14:1382-91. 14
3. Bornkamm, G. W., and W. Hammerschmidt. 2001. Molecular virology of 15
Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci 356:437-59. 16
4. Burns, T. F., P. Fei, K. A. Scata, D. T. Dicker, and W. S. El-Deiry. 2003. 17
Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in 18
paclitaxel (taxol)-exposed cells. Mol Cell Biol 23:5556-71. 19
5. Cannell, E. J., P. J. Farrell, and A. J. Sinclair. 1998. Cell cycle arrest 20
following exposure of EBV-immortalised B-cells to gamma irradiation 21
correlates with inhibition of cdk2 activity. FEBS Lett 439:297-301. 22
6. Castedo, M., J. L. Perfettini, T. Roumier, A. Valent, H. Raslova, K. 23
Yakushijin, D. Horne, J. Feunteun, G. Lenoir, R. Medema, W. 24
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

21
Vainchenker, and G. Kroemer. 2004. Mitotic catastrophe constitutes a special 1
case of apoptosis whose suppression entails aneuploidy. Oncogene 23:4362-70. 2
7. Clybouw, C., B. McHichi, S. Mouhamad, M. T. Auffredou, M. F. 3
Bourgeade, S. Sharma, G. Leca, and A. Vazquez. 2005. EBV infection of 4
human B lymphocytes leads to down-regulation of Bim expression: relationship 5
to resistance to apoptosis. J Immunol 175:2968-73. 6
8. Dang, C. V., A. O'Donnell K, and T. Juopperi. 2005. The great MYC escape 7
in tumorigenesis. Cancer Cell 8:177-8. 8
9. Dirmeier, U., R. Hoffmann, E. Kilger, U. Schultheiss, C. Briseno, O. Gires, 9
A. Kieser, D. Eick, B. Sugden, and W. Hammerschmidt. 2005. Latent 10
membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation 11
with control of apoptosis. Oncogene 24:1711-7. 12
10. Gualandi, G., L. Giselico, M. Carloni, F. Palitti, P. Mosesso, and A. M. 13
Alfonsi. 2001. Enhancement of genetic instability in human B cells by Epstein-14
Barr virus latent infection. Mutagenesis 16:203-8. 15
11. Hemann, M. T., A. Bric, J. Teruya-Feldstein, A. Herbst, J. A. Nilsson, C. 16
Cordon-Cardo, J. L. Cleveland, W. P. Tansey, and S. W. Lowe. 2005. 17
Evasion of the p53 tumour surveillance network by tumour-derived MYC 18
mutants. Nature 436:807-11. 19
12. Inman, G. J., U. K. Binne, G. A. Parker, P. J. Farrell, and M. J. Allday. 20
2001. Activators of the Epstein-Barr virus lytic program concomitantly induce 21
apoptosis, but lytic gene expression protects from cell death. J Virol 75:2400-10. 22
13. Kelly, G., A. Bell, and A. Rickinson. 2002. Epstein-Barr virus-associated 23
Burkitt lymphomagenesis selects for downregulation of the nuclear antigen 24
EBNA2. Nat Med 8:1098-104. 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

22
14. Kelly, G. L., A. E. Milner, R. J. Tierney, D. S. Croom-Carter, M. Altmann, 1
W. Hammerschmidt, A. I. Bell, and A. B. Rickinson. 2005. Epstein-Barr virus 2
nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A, -3
3B, and -3C expression in Burkitt's lymphoma cells and with increased 4
resistance to apoptosis. J Virol 79:10709-17. 5
15. Krauer, K. G., A. Burgess, M. Buck, J. Flanagan, T. B. Sculley, and B. 6
Gabrielli. 2004. The EBNA-3 gene family proteins disrupt the G2/M 7
checkpoint. Oncogene 23:1342-53. 8
16. Lee, J. M., K. H. Lee, C. J. Farrell, P. D. Ling, B. Kempkes, J. H. Park, and 9
S. D. Hayward. 2004. EBNA2 is required for protection of latently Epstein-Barr 10
virus-infected B cells against specific apoptotic stimuli. J Virol 78:12694-7. 11
17. Mansilla, S., W. Priebe, and J. Portugal. 2006. Mitotic catastrophe results in 12
cell death by caspase-dependent and caspase-independent mechanisms. Cell 13
Cycle 5:53-60. 14
18. Nitta, M., O. Kobayashi, S. Honda, T. Hirota, S. Kuninaka, T. Marumoto, 15
Y. Ushio, and H. Saya. 2004. Spindle checkpoint function is required for 16
mitotic catastrophe induced by DNA-damaging agents. Oncogene 23:6548-58. 17
19. O'Connor, P. M., J. Jackman, D. Jondle, K. Bhatia, I. Magrath, and K. W. 18
Kohn. 1993. Role of the p53 tumor suppressor gene in cell cycle arrest and 19
radiosensitivity of Burkitt's lymphoma cell lines. Cancer Res 53:4776-80. 20
20. O'Nions, J., and M. J. Allday. 2004. Deregulation of the cell cycle by the 21
Epstein-Barr virus. Adv Cancer Res 92:119-86. 22
21. O'Nions, J., and M. J. Allday. 2003. Epstein-Barr virus can inhibit genotoxin-23
induced G1 arrest downstream of p53 by preventing the inactivation of CDK2. 24
Oncogene 22:7181-91. 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

23
22. Parker, G. A., R. Touitou, and M. J. Allday. 2000. Epstein-Barr virus 1
EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear 2
division divorced from cytokinesis. Oncogene 19:700-9. 3
23. Portis, T., and R. Longnecker. 2004. Epstein-Barr virus (EBV) LMP2A 4
mediates B-lymphocyte survival through constitutive activation of the 5
Ras/PI3K/Akt pathway. Oncogene 23:8619-28. 6
24. Rieder, C. L., and H. Maiato. 2004. Stuck in division or passing through: what 7
happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 8
7:637-51. 9
25. Sculley, T. B., M. Buck, B. Gabrielli, P. G. Parsons, and K. G. Krauer. 2002. 10
A histone deacetylase inhibitor, azelaic bishydroxamic acid, shows cytotoxicity 11
on Epstein-Barr virus transformed B-cell lines: a potential therapy for 12
posttransplant lymphoproliferative disease. Transplantation 73:271-9. 13
26. Strasser, A. 2005. The role of BH3-only proteins in the immune system. Nat 14
Rev Immunol 5:189-200. 15
27. Sunters, A., S. Fernandez de Mattos, M. Stahl, J. J. Brosens, G. 16
Zoumpoulidou, C. A. Saunders, P. J. Coffer, R. H. Medema, R. C. 17
Coombes, and E. W. Lam. 2003. FoxO3a transcriptional regulation of Bim 18
controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem 19
278:49795-805. 20
28. Syed, N., P. Smith, A. Sullivan, L. C. Spender, M. Dyer, L. Karran, J. 21
O'Nions, M. Allday, I. Hoffmann, D. Crawford, B. Griffin, P. J. Farrell, and 22
T. Crook. 2006. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is 23
a frequent event in B-cell malignancies. Blood 107:250-6. 24
29. Tao, W. 2005. The mitotic checkpoint in cancer therapy. Cell Cycle 4:1495-9. 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

24
30. Thorley-Lawson, D. A., and A. Gross. 2004. Persistence of the Epstein-Barr 1
virus and the origins of associated lymphomas. N Engl J Med 350:1328-37. 2
31. Wade, M., and M. J. Allday. 2000. Epstein-Barr virus suppresses a G(2)/M 3
checkpoint activated by genotoxins. Mol Cell Biol 20:1344-60. 4
32. Weaver, B. A., and D. W. Cleveland. 2005. Decoding the links between 5
mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell 6
death. Cancer Cell 8:7-12. 7
33. Westphal, E. M., W. Blackstock, W. Feng, B. Israel, and S. C. Kenney. 8
2000. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and 9
sodium butyrate in vitro and in vivo: a potential method for treating EBV-10
positive malignancies. Cancer Res 60:5781-8. 11
34. Young, L. S., R. Lau, M. Rowe, G. Niedobitek, G. Packham, F. Shanahan, 12
D. T. Rowe, D. Greenspan, J. S. Greenspan, A. B. Rickinson, and et al. 13
1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 14
transactivator protein in oral hairy leukoplakia. J Virol 65:2868-74. 15
FIGURE LEGENDS 16
FIG. 1. EBV does not prevent radiation-induced G2/M arrest but protects cells from 17
dying as they subsequently enter mitosis with damaged DNA. BL41 and BL41/B95.8 18
cells were exposed to �����irradiation (850 rads) and harvested after treatment. 19
Nuclear morphology was observed by microscopy and flow cytometric analysis of 20
cellular DNA content was performed at the times indicated. (a & b) After 24 hours, both 21
BL41 and BL41/B95.8 have normal nuclear morphology and are largely arrested with 22
4N DNA content. (c) After 72 hours both populations recovered from the G2/M arrest, 23
completed mitosis and abnormal nuclei appeared in both populations. EBV-negative 24
BL41 cells then died between 3 and 5 days; in contrast the EBV infected BL41/B95.8 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

25
cells survived with increasing multi- and micronucleation. (d) Enlarged view of aberrant 1
nuclei 5 days after irradiation. 2
FIG. 2. Impaired metaphase arrest in EBV-positive BL41/B95.8 cells treated with 3
nocodazole (Noc). Cells were left untreated or treated with nocodazole (at a 4
concentration of 50 ng/ml) for 24 hours. (a) Cells were harvested for flow cytometry. 5
An accumulation of cells with 4N DNA content was seen in both EBV-negative BL41 6
and EBV-positive BL41/B95.8 cells. In the EBV-positive population, there was a 7
significantly greater increase in proportion of cells with > 4N DNA content (23.8% 8
compared with 9.7% in BL41). (b & c) DAPI staining 24 hours after treatment with 9
nocodazole showed that while EBV-negative BL41 cells are arrested at metaphase with 10
condensed chromosomes (metaphase spreads, examples indicated by white arrows), 11
EBV-positive BL41/B95.8 cells generally showed few chromosome spreads suggesting 12
that the EBV-positive cells have an impaired checkpoint. BL41/B95.8 cells with 13
multiple or fractured nuclei were observed (indicated with long white arrows). 14
Quantification of both metaphase and interphase cells 24 hours after treatment with 15
nocodazole showed a high percentage of BL41 arrested in metaphase (43%) and only a 16
very small percentage of interphase cells. In contrast the majority of BL41/B95.8 cells 17
had interphase nuclei and almost no cells were obviously arrested at metaphase (<10%). 18
FIG. 3. EBV-positive BL cells survive treatment with nocodazole or taxol. Cells were 19
treated with either nocodazole (Noc) or taxol (both at a concentration of 50 ng/ml) and 20
harvested at the times indicated. (a) The cell cycle profiles of the BL41 EBV-negative 21
cells and BL41/B95.8 EBV-positive cells were very different 72 hours after treatment 22
with nocodazole or taxol. A significant increase in the sub-G1 population indicated 23
apoptosis or mitotic catastrophe in the EBV-negative BL41 cells. In contrast, EBV-24
positive BL41/B95.8 cells showed only modest increases in the sub-G1 population and 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

26
most of the cells had a 4N or >4N DNA content after treatment. (b) The percentage of 1
viable cells surviving after treatment with nocodazole or taxol (Tax) was determined. 2
Viable cells that excluded trypan blue were expressed as a percentage of the starting 3
population. (c) DAPI staining confirmed that EBV-negative BL41 cells died, since after 4
72 hours only cell debris was seen. In contrast the EBV infected BL41/B95.8 cells 5
survived treatment with nocodazole or taxol and many cells became multinucleated or 6
developed micronuclei (example indicated with white arrow). 7
FIG. 4. P3HR1-positive BL cells also survive treatment with nocodazole or taxol. (a) 8
Cell cycle profiles of the EBV-negative Ramos cells and P3HR1-EBV-positive cells 9
Ramos/AW are very different 72 hours after treatment with either nocodazole (Noc) or 10
taxol. A significant increase in the sub-G1 population was observed in EBV-negative 11
Ramos cells indicating apoptosis had occurred. In contrast, EBV-positive Ramos/AW 12
cells show only a slight increase in the sub-G1 population and most of the cells had 13
developed 4N or > 4N DNA content. (b) The percentage of viable cells surviving after 14
treatment with nocodazole or taxol (Tax) was determined. Viable cells that excluded 15
trypan blue were expressed as a percentage of the starting population. (c) DAPI 16
staining showed that EBV-negative Ramos cells die and 72 hours after the treatment 17
with either nocodazole or taxol only cell debris was seen. In contrast the EBV-infected 18
Ramos/AW cells survived treatment and many cells became multinucleated or 19
developed micronuclei. 20
FIG. 5. EBV-positive cells can recover from exposure to nocodazole. BL41 and 21
BL41/B95.8 (a) and Ramos and Ramos/AW (b) were treated with nocodazole 22
(50ng/ml) for 72 hours. The cells were then washed, re-suspended in conditioned 23
medium (without nocodazlole) and viable cells were counted. The cells were then 24
cultured for a further 18 days and samples removed, stained with trypan blue and 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

27
counted at weekly intervals. Samples were also taken at the beginning of the experiment 1
and after 14 days, stained with propidium iodide and analyzed by flow cytometry. 2
FIG. 6. BL cells carrying P3HR1-like EBV have an impaired spindle checkpoint and are 3
rescued from cell death induced by nocodazole. (a) Cells were treated with nocodazole 4
(Noc, 50 ng/ml) for 24 hours and the Mitotic Index was determined. Spreads of 5
condensed metaphase chromosomes were scored as arrested cells and expressed as a 6
percentage of the total number of DAPI-stained cells counted in three separate fields (a 7
minimum of 300 cells was counted in each experiment). The results of a typical 8
experiment are shown graphically with the mean and standard deviation of three fields. 9
(b and c) The percentage of viable cells surviving after treatment with nocodazole was 10
determined. Viable cells that excluded trypan blue were expressed as a percentage of the 11
starting population. (d and e) 72 hours after the addition of nocodazole, cells were 12
analyzed by flow cytometry and microscopy. EBV-negative BL41 and BL31 and EBV-13
positive, latency-I Dante and Mutu-I (clone 179) were all dead and only cell debris was 14
visible. The EBV-positive EBNA2-KO converts, Ava-BL, Oku-BL, Sal-BL and Mutu-15
III appeared largely viable with a significant 4N or greater DNA content. 16
FIG. 7. (a) Nocodazole induces proteolytic cleavage of PARP in EBV-negative and 17
Latency-I BL cells but not in cells infected with P3HR1-like EBV. Cells were treated 18
with nocodazole (Noc) and at the times indicated, samples were taken and protein 19
extracts prepared. Proteins were resolved by SDS-PAGE (7.5%) and western 20
immunoblotted with rabbit anti-PARP and a mouse anti-����a-tubulin mAb. Full-21
length (113kDa) PARP and the 89kDa product of caspase-mediated cleavage are both 22
indicated by arrows. (b) Rescue from cell death is not dependant on EBV lytic gene 23
expression. Protein extracts from the nocodazole-treated cells were separated by SDS-24
PAGE (12.5%) and western immunoblotted with mouse anti-BZLF1 and anti-BHRF1 25
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

28
mAbs or the human serum EE. Akata BL cells were left untreated (-) or treated with 1
anti-Ig [+ and (+)] to stimulate the expression of EBV lytic proteins. The induced 2
population was approximately 15%, as judged by anti-BZLF1 immunofluorescence 3
staining (Carol McDonald, personal communication) and the track labeled (+) shows 4
anti-Ig induced Akata extracts diluted to 1/10 with EBV-negative BL31 extract. (c) 5
BHRF1 is neither expressed nor induced by nocodazole in BL41/B95.8 nor Ramos/AW. 6
(d) Bim levels correlate with sensitivity to nocodazole. Protein extracts from untreated 7
BL cells were separated by SDS-PAGE (12.5%) and western immunoblotted using a 8
rabbit anti-Bim polyclonal antibody. The three Bim isoforms (Bim-EL, Bim L and Bim-9
S) are indicated. Gamma-tubulin was used as a control for equal loading in all these 10
western blots. 11
FIG. 8. A Summary of the responses to activators of the spindle assembly checkpoint in 12
BL cells. Schematic diagram showing the responses of EBV-positive and EBV-negative 13
BL-derived cells to mitotic spindle damaging agents, nocodazole and taxol. EBV-14
negative (-ve) BL cells and those expressing the latency I pattern of EBV genes (Lat-I) 15
largely arrest in pro-metaphase and subsequently die by caspase-dependent mitotic 16
catastrophe or apoptosis. In contrast, EBV-positive cells expressing either the full 17
spectrum of latent EBV genes (Lat-III) or the P3HR1-like pattern (∆EBNA2) may fail 18
to arrest in pro-metaphase, survive aberrant mitosis, develop abnormal nuclei and 19
sometimes become polyploid. EBV-positive cells sometimes recover sufficiently to 20
resume proliferation. 21
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

28
mAbs or the human serum EE. Akata BL cells were left untreated (-) or treated with1
anti-Ig [+ and (+)] to stimulate the expression of EBV lytic proteins. The induced2
population was approximately 15%, as judged by anti-BZLF1 immunofluorescence3
staining (Carol McDonald, personal communication) and the track labeled (+) shows4
anti-Ig induced Akata extracts diluted to 1/10 with EBV-negative BL31 extract. (c)5
BHRF1 is neither expressed nor induced by nocodazole in BL41/B95.8 nor Ramos/AW.6
(d) Bim levels correlate with sensitivity to nocodazole. Protein extracts from untreated7
BL cells were separated by SDS-PAGE (12.5%) and western immunoblotted using a8
rabbit anti-Bim polyclonal antibody. The three Bim isoforms (Bim-EL, Bim L and Bim-9
S) are indicated. Gamma-tubulin was used as a control for equal loading in all these10
western blots. 11
FIG. 8. A Summary of the responses to activators of the spindle assembly checkpoint in12
BL cells. Schematic diagram showing the responses of EBV-positive and EBV-negative13
BL-derived cells to mitotic spindle damaging agents, nocodazole and taxol. EBV-14
negative (-ve) BL cells and those expressing the latency I pattern of EBV genes (Lat-I)15
largely arrest in pro-metaphase and subsequently die by caspase-dependent mitotic16
catastrophe or apoptosis. In contrast, EBV-positive cells expressing either the full17
spectrum of latent EBV genes (Lat-III) or the P3HR1-like pattern ("EBNA2) may fail18
to arrest in pro-metaphase, survive aberrant mitosis, develop abnormal nuclei and19
sometimes become polyploid. EBV-positive cells sometimes recover sufficiently to20
resume proliferation.21
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 1.
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
BL41/B95.8BL41
24h 4N
2N
0h4N
2N
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
2N
4N
4N
2N
72h
5D
a. b.
c. d.
50 m 50 m
50 m
BL41/B95.8BL41
24h
0h
BL41/B95.8BL41
BL41/B95.8
5D
50 m
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

b.a.
c.
Figure 2.
BL
41/B
95.8
-Noc 0h +Noc 24h
200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
BL
41
2N
8N (23.8%)
4N
4N
2N
4N 4N
8N (9.7%)
BL41 BL41/B95.8
BL41 BL41/B95.8
Mitotic Index- 43% Mitotic Index- < 10%
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
BL
41
/B9
5.8
BL
41
2N
4N
2N
4N
0ha.
1000 0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
+Noc 72h
8N 4N
8N
Figure 3.
+Taxol 72h
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
8N 4N
<G1 <G1
b.
c. BL41/B95.8 BL41
+N
oc
72h
+T
axo
l 72h
0
20
40
60
80
100
120
0 24 48 72
hours
% v
iab
le c
ell
s
BL41+Noc
BL41/B958+Noc
BL41+Tax
BL41/B958+Tax
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

0 200 400 600 800 1000FL2-A
Figure 4.
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
Ram
os/A
WR
am
os
0h
2N
4N
2N
4N
4N 8N
a. +Noc 72h
b.
+Taxol 72h
0 200 400 600 800 1000
FL2-A
4N
8N
Ramos/AWRamos
+N
oc
72h
+T
axo
l 72h
!<G1 <G1
c.
0
20
40
60
80
100
120
0 24 48 72
hours
% v
iab
le c
ells
Ramos+Noc
Ramos/AW+Noc
Ramos+Tax
Ramos/AW+Tax
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 5.
a.
b.
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0d 14dDays
+ Noc:
Days
+ Noc: 0d 14d
BL41/B95.8
Ramos/AW
2N
2N
2N
2N4N
4N
4N4N
0
20
40
60
80
100
120
0 7 14 21 28
days
% v
iab
le c
ells
BL41
BL41/B95.8
0
50
100
150
200
250
300
350
0 7 14 21 28
days
% v
iab
le c
ells
Ramos
Ramos/AW
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
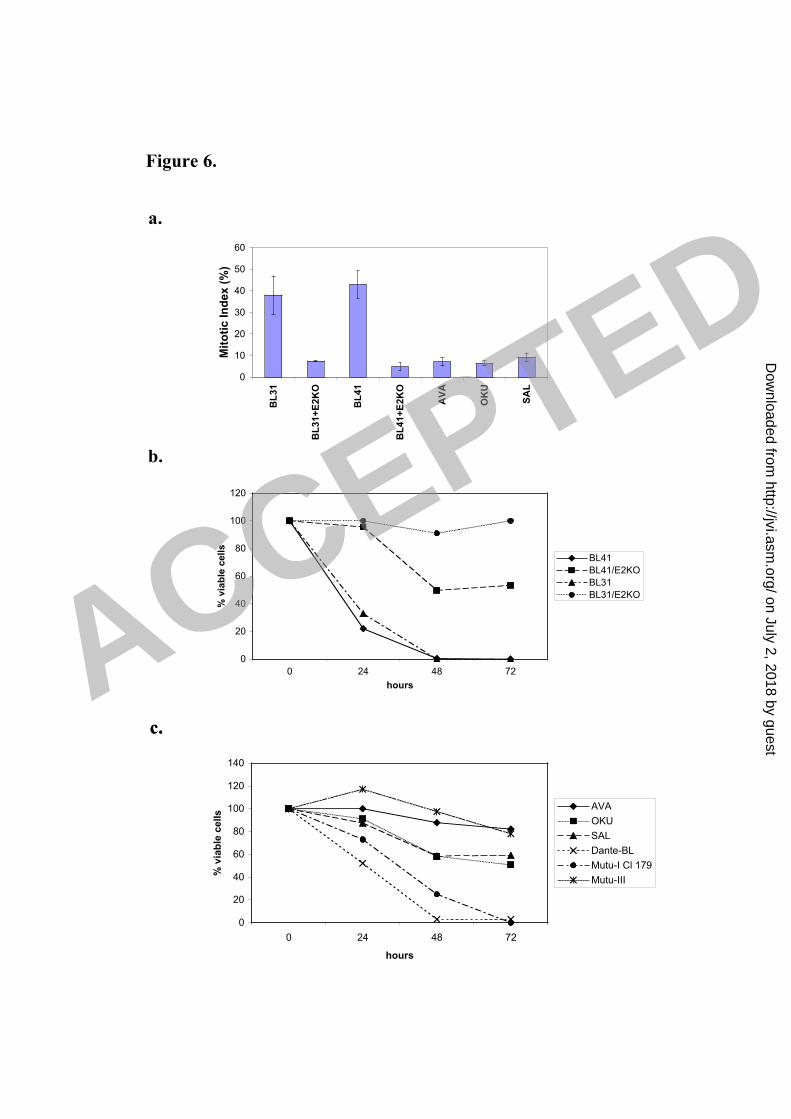
Figure 6.
a.
c.
0
10
20
30
40
50
60
BL
31
BL
31
+E
2K
O
BL
41
BL
41
+E
2K
O
AV
A
OK
U
SA
L
Mit
oti
c I
nd
ex
(%
)
b.
c.
0
20
40
60
80
100
120
0 24 48 72
hours
% v
iab
le c
ell
s
BL41
BL41/E2KO
BL31
BL31/E2KO
0
20
40
60
80
100
120
140
0 24 48 72
hours
% v
iab
le c
ell
s
AVA
OKU
SAL
Dante-BL
Mutu-I Cl 179
Mutu-III
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

BL41
BL41
+E2KO
BL31
+E2KO
BL31
<2N 2N 4N 8N
0h 72h 72hHours
+ Noc:
Figure 6d.
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 6e.
Dante-BL
Mutu-I
Cl 179
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
0 200 400 600 800 1000FL2-A
<2N 2N 4N 8N
0h 72h 72h
Hours
+ Noc:
Ava-BL
Oku-BL
Sal-BL
0 200 400 600 800 1000
FL2-A
0 200 400 600 800 1000
FL2-A
0 200 400 600 800 1000
FL2-A
0 200 400 600 800 1000
FL2-A
Mutu-III
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 7a.
BL31 BL31+E2KO
0 12 24 48 72 0 12 24 48 72Hours
+Noc:
PARP
Cleaved
PARP
Tubulin
0 12 24 48 72 0 12 24 48 72
Hours
+Noc:
PARP
CleavedPARP
Tubulin
BL41+E2KOBL41
0 12 24 48 72
PARP
Cleaved
PARP
Tubulin
Oku-BL
0 12 24 48 72Hours
+Noc:
Ava-BL
0 12 24 48 72
Hours
+Noc:
Sal-BL
PARP
Cleaved
PARP
Tubulin
Dante-BL
Mutu-I Cl 179
0 12 24 48 72
0 12 24 48 72
Hours
+Noc:
PARP
CleavedPARP
Tubulin
0 12 24 48 72
Mutu-III
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 7.
BL31
+E2K
O
Sal
-BL
Oku
-BL
BL41
+E2K
O
0 24 72 0 24 72 0 24 72 0 24 72 + –
Aka
ta
BZLF1
Tubulin
(+)
BZLF1
(mAb)
Early lytic
antigens
BHRF1
0 72 0 72 (+) –+
Aka
ta
BL41
/B95
.8
Ram
os/AW
Tubulin
BHRF1
Hours
+Noc:
Hours
+Noc:
b.
c.
Ram
os/A
W
BL
31+
E2K
O
BL
41+
E2K
O
BL
41/B
95.8
Ram
os
BL
31
BL
41
d.
BL
41
Mu
tu-I
Cl1
79
Mu
tu-I
II
Tubulin
BimSBimL
BimEL
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Figure 8.
Nocodazole/Taxol
†
Mitotic
cell death
EBV-ve EBV-Lat-III
4N
Polyploidy/
Multi/Micronucleation
4N
G1
S
G2
M
Metaphase
arrest
†
Mitotic
cell death
EBV-Lat-I
4N
G1
S
G2
M
Metaphase
arrest
BL cells
EBV- EBNA2
4N
Polyploidy/
Multi/Micronucleation
Impaired
Metaphase
arrest
ACCEPTED
on July 2, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from





























