Embed Size (px)
Citation preview

Statistics for K-mer Based Splicing Event Analysis
Data Learner Miner Practitioner
Ruofei Du, Hao Li, Hui Miao, Shangfu Peng

Alternative Splicing Events
Image from: "Alternative Splicing Event" Wikipedia: The Free Encyclopedia. Wikimedia Foundation, Inc. 2 Apr. 2014. <http://en.wikipedia.org/wiki/Alternative_splicing>
● Alternative splicing is used to describe
any case in which a primary transcript
can be spliced in more than one pattern
to generate multiple and distinct
mRNAs.
● 5 traditional basic modes; most
common: exon skipping.
● It is a widespread mechanism for
generating protein diversity and
regulating protein expression.

● Improve
understanding of
cell
differentiation
and classify
disease types
Image from: Sammeth, Michael, Sylvain Foissac, and Roderic Guigó. "A General Definition and Nomenclature for Alternative Splicing Events." PLoS Computational Biology 4.8 (2008): e1000147.
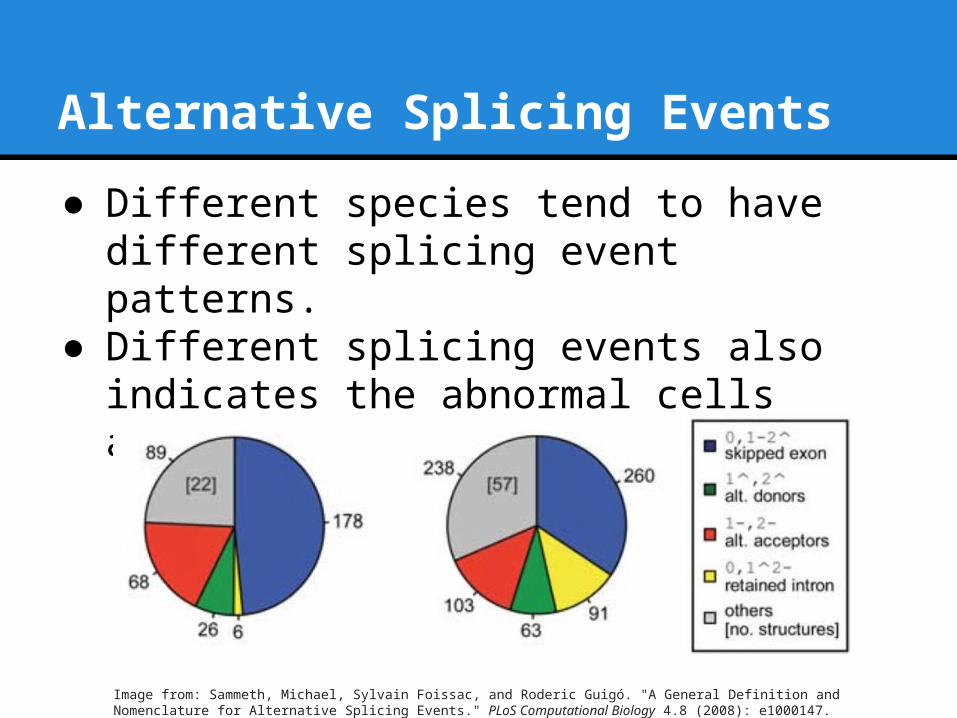
Alternative Splicing Events
● Different species tend to have different splicing event patterns.
● Different splicing events also indicates the abnormal cells activities, such as cancer
Image from: Sammeth, Michael, Sylvain Foissac, and Roderic Guigó. "A General Definition and Nomenclature for Alternative Splicing Events." PLoS Computational Biology 4.8 (2008): e1000147.

Abundance Estimation for Alternative Splicing Events
● Given RNA-Seq samples, estimate the abundance and the relative proportion of every alternative transcription path
Image from: Hu, Yin, et al. "DiffSplice: the genome-wide detection of differential splicing events with RNA-seq." Nucleic acids research 41.2 (2013): e39-e39.

Abundance Estimation for Isoforms
● The Standard Paradigmo Read alignment step can be very computationally
intensive.
● Sailfisho Far faster than the standard paradigm
o Replace the step of read mapping with the much
faster and simpler process of k-mer counting
Sailfish: Alignment-free Isoform Quantification from RNA-seq Reads using Lightweight Algorithms Rob Patro, Stephen M. Mount, and Carl Kingsford. Manuscript Submitted (2013) http://www.cs.cmu.edu/~ckingsf/class/02714-f13/Lec05-sailfish.pdf

K-mer
● A fixed sized (K) sequence
Sailfish: Alignment-free Isoform Quantification from RNA-seq Reads using Lightweight Algorithms Rob Patro, Stephen M. Mount, and Carl Kingsford. Manuscript Submitted (2013) http://www.cs.cmu.edu/~ckingsf/class/02714-f13/Lec05-sailfish.pdf
A
C
G
T
AA AC AG AT
CA CC CG CT
GA GC GG GT
TA TC TG TT
● A string of length N contains N-K+1 k-mers
● One can build K-mer index to represent a string
7-mer iD N
ATTCGAC 1 1
TTCGACA 2 1
TCGACAG 3 1
...
1-mer 2-mer

Sailfish Workflow
● Indexingo Build K-mer index for known
isoform transcripts
Sailfish: Alignment-free Isoform Quantification from RNA-seq Reads using Lightweight Algorithms Rob Patro, Stephen M. Mount, and Carl Kingsford. Manuscript Submitted (2013)
● Quantificationo Counts the number of times
each K-mer occurs in the reads.
o Estimating abundances via an EM algorithm

Sailfish Workflow: Indexing
● Perfect Hashing
http://www.cs.cmu.edu/~ckingsf/class/02714-f13/Lec05-sailfish.pdf
Domain(K-mer) Range([0,|D|-1])
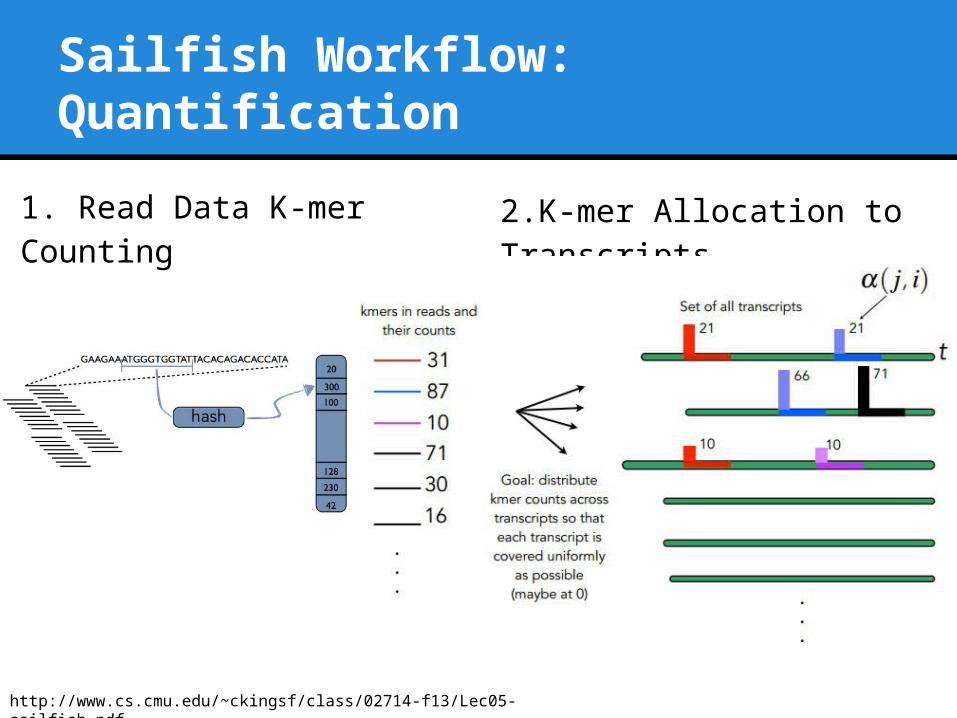
Sailfish Workflow: Quantification
2.K-mer Allocation to Transcripts
http://www.cs.cmu.edu/~ckingsf/class/02714-f13/Lec05-sailfish.pdf
1. Read Data K-mer Counting

Our Proposal
● We propose to investigate the scalable statistic method using k-mer and k-mer index to estimate abundance of alternative splicing events.
● We will focus on the most frequent event type:Exon Skipping Evento other event types can
be extended naturally
Shen, Shihao, et al. "MATS: a Bayesian framework for Flexible Detection of Differential Alternative Splicing from RNA-Seq Data." Nucleic Acids Research 40.8 (2012): e6
(1) (2) (3)

● Variables for abundance:
● Build k-mer index for a specific gene: e.g. A B C D E
● On reads part, aggregated k-mer counts like Sailfish
● Use EM to do maximum likelihood estimation
Class I: Each exon i
Class II: Each exon-exon junction (non-spliced)
Class III: Each spliced junction
Initial Idea
Exon A, B, C, D, E
Non-spliced junction AB, BC, CD, DE
Spliced junction AC, BD, CE

Advantage
● Do not require to know the Isoform space.
● Replace the step of read mapping, and provide a faster
approach for splicing event analysis.

Thank you

Questions
1. The drawback of the straightforward method: get the Pi of each Isoform using
EM first, and then calculate the frequency of events.
2. Why we have to use EM, why not solve equations?
3. Require to know the frequency of the five events?





























