Embed Size (px)
Citation preview

Lifeng Lin, Tim Looney, Geoffrey M. Lowman, Denise Topacio-Hall, Jian-ping Zheng, Elizabeth Linch, Lauren Miller, Mark Andersen and Fiona Hyland, Thermo Fisher Scientific, South San Francisco, CA, USA, 94080
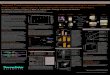
RESULTS
Using 5’-RACE as presumed “truth” we judge the accuracy of our assay using correla9on of V-gene usage.
A) Comparisons of V-gene usage to 5’-RACE library prepara9on strategies give correla9on values ranging
from r = 0.90-0.92; Correla9on in V-gene usage between BIOMED-2 and 5’-RACE are normally in the
range r ~ 0.75-0.80
B) Limit of detection/linearity experiments including 30 plasmid sequences result in expected level of plasmid
representation (linear with input) and high sensitivity at low abundance. Libraries prepared using pooled
plasmids at single known input concentrations (1pg to 0.00001pg = 50,000 to ~5 copies) in a background of
100ng Leukocyte cDNA.
C) Count of input T cells vs detected clones.
Figure 5. Identification of novel polymorphismsINTRODUCTION
TCRβ immune repertoire analysis by next-generation sequencing is emerging as a
valuable tool for research studies of the tumor microenvironment and potential immune
responses to cancer immunotherapy. Generation of insight from immune repertoire
profiling often requires comparative analysis of immune repertoires across research
sample cohorts representing immune responses to defined antigens or
immunomodulatory agents. Here we describe the development of a computational
framework enabling large-scale comparative analysis of immune repertoire data on
cloud-based infrastructure.
TCRβ receptors were amplified from matched peripheral blood and tumor biopsy cDNA
using AmpliSeqTM multiplex primers targeting the Variable gene Framework 1 and
Constant gene to produce an amplicon spanning all three CDR domains. To evaluate
assay performance, we sequenced TCRβ rearrangements from donor peripheral blood
leukocyte (PBL) cDNA that had been spiked with 30 reference rearrangements taken
from literature. Raw data was uploaded to the Ion Reporter data analysis platform for
clonotype annotation and storage to enable rapid downstream comparative analysis of
repertoire features.
We demonstrate the ability to rapidly compare clonotype data across sample cohorts
and find that a subset of clones identified in peripheral blood are also found in matched
tumor samples. Peripheral blood-derived repertoires typically contained 10-100 fold
more distinct clones than found in tumor, with correspondingly higher estimates of
diversity via the Shannon Index. Results from sequencing of spike-in reference
rearrangements indicate that the assay is accurate and sensitive over 5 logs of input
template amount while showing minimal amplification bias. Technical replicates showed
high concordance (r >.96) in the frequency of detected clones, indicating that results
were reproducible and samples were sequenced to an appropriate depth. Comparison
of AmpliSeqTM multiplex PCR-derived data to that produced by 5’ RACE or BIOMED-2
primers revealed the AmpliSeqTM solution to provide comprehensive and unbiased
coverage of the human TCRβ repertoire.
MATERIALS AND METHODSIon AmpliSeqTM primers are designed based on sequences downloaded from IMGT (1)
(http://www.imgt.org/vquest/refseqh.html). Forward primer set was designed to target
the FR1 region of all TRBV loci; reverse primer set was designed from the two TRBC
loci. The resulting amplicon spans across all CDR regions of the mature mRNA
molecule. Every known variable gene allele have at least one perfect matching forward
primer.
TCRB sequences are amplified using non-FFPE RNA from tumor biopsy, peripheral
blood or sorted cells, followed by multiplex sequencing via the Ion S5 530 chip (15-20M
reads). PCR and sequencing errors are eliminated before clone reporting. In some
cases, an individual will possess a plurality of clones that do not match any IMGT
variable gene allele; this may indicate presence of a novel allele. If sufficient clone
support exists, Ion Reporter classifies the sequence as a putative novel variant. As a
last step, putative variants are compared to those found in the Lym1k database (2)
derived from 1000 genomes data.
CONCLUSIONSWe have developed a computational framework to enable rapid analysis of large immune
repertoire datasets derived from AmpliSeq-based sequencing of human TCRβ receptors via
the Ion Torrent S5. The AmpliSeqTM procedure, which features the ability to produce
uniform and reliable results in extremely highly multiplexed PCR, is well suited for immune
repertoire sequencing applications.
REFERENCES1. Lefranc et al. Nucleic Acids Res (2015). 43:D413
2. Yu et al. J Immunol (2017) 198:2202
3. Ye et al. Nucleic Acids Res (2013) 41:W34
ACKNOWLEDGEMENTSThe authors would like to acknowledge the work of all who participated in this program:
Alex Pankov, Grace Lui, Gauri Ganpule, Sonny Sovan, Xinzhan Peng, Larry Fang, Tyler
Stine, Laura Nucci, Rob Bennett, and Jim Godsey.
A computational framework for large-scale analysis of TCRβ immune repertoire sequencing data on cloud-based infrastructure
Thermo Fisher Scientific • 200 Oyster Point Blvd • South San Francisco, CA 94080 • www.lifetechnologies.com
Figure 4 Comparative sequencing
Figure 2. Analysis workflow of Ion Reporter
Annotate the V, D and J gene
for each rearrangement by
comparing to IMGT database
FR1-C multiplex PCR
Report clones and secondary
repertoire features
Report novel alleles
Ion Reporter Workflow
Eliminate PCR and
sequencing errors
Figure 3. Performance Benchmarking
Figure 1. TCRβ AmpliSeq primer locations
Polymorphism within the TCRB variable gene (TRBV) has been linked to chronic autoimmune
diseases. Existing sequencing assays targeting CDR3 region risks missing imporantant
polymorphsims in the CDR1 and CDR2 regions. The AmpliSeq-based sequencing assay
generates sequences of all three CDRs, making the detection of previously unknown
polymorphisms possible.
Example of a non-synonymous IMGT variant. IgBLAST (3) alignment of an allele having two amino acid
substitutions compared to the best matching IMGT allele. This particular allele was detected in our sample
cohort and the Lym1K database derived from 1000 genomes data.
A collection of tools specifically designed to analyze
immune repertoire data has been built into a preset
workflow in the Ion Reporter platform. The V, D and J
gene portions of the reads are identified by comparing
against the IMGT database and the subtypes assigned
accordingly. The frequency of each V-gene type and
clonotype are summarized and ranked in a report table
(D).
Visualizations are generated based on the clonotype
data, including bar-plots of allele frequency (A), heat-
map of V-J pairs (B) and a set of interactive
spectratyping plots by evenness, Shannon-diversity,
largest-clone frequency and number of clones in each
cluster (C).
A
C
C
A
B
B
D
T-cell receptor repertoire generated from different samples
can be analyzed as a cohort using the “Compare Samples”
function in Ion Reporter. A summary table showing the count
of each CDR3 region and its frequency in all samples is
generated. (above)
Questions such as number of shared clones across samples
or different sequencing runs can be easily extrapolated. We
have used this analysis to compare T-cell repertoire between
tumor infiltrating leukocytes and leukocytes drawn from
peripheral blood, identifying sharing clones between the two
sample types, as well as clones unique to tumor. (left) TRADEMARKS/LICENSING For Research Use Only. Not for use in diagnostic
procedures. © 2017 Thermo Fisher Scientific Inc. All rights
reserved. All trademarks are the property of Thermo Fisher
Scientific and its subsidiaries unless otherwise specified