Embed Size (px)
Citation preview

CH.VYSHNAVIMSC MLT 1st YEAR

DEFINITION
CLASSIFICATION

Haemolytic anaemia is a condition in which red blood cells are destroyed and removed from the bloodstream before their normal lifespan is over.
Haemolytic anaemia are the anaemia's that result due to increase in the rate of red cell destruction.
About 1%blood is destructed ever day in normal adult

RBC’s normally survive 120 days .
Bone marrow has the capacity to increase erythropoiesis 6 - 8 times than normal.
Therefore Hemolytic Anemia may not be seen until the red cell lifespan is less than 30 days.
Anemia is the result of premature destruction of red cells exceeding the erythropoietic capacity of the bone marrow

When blood cells die, the body's bone marrow makes more blood cells to replace them. However, in haemolytic anaemia, the bone marrow can't make red blood cells fast enough to meet the body's needs.
An increase in destruction and production of erythrocytes can result in a compensated haemolytic state without anaemia being present so a called compensated haemolytic disease
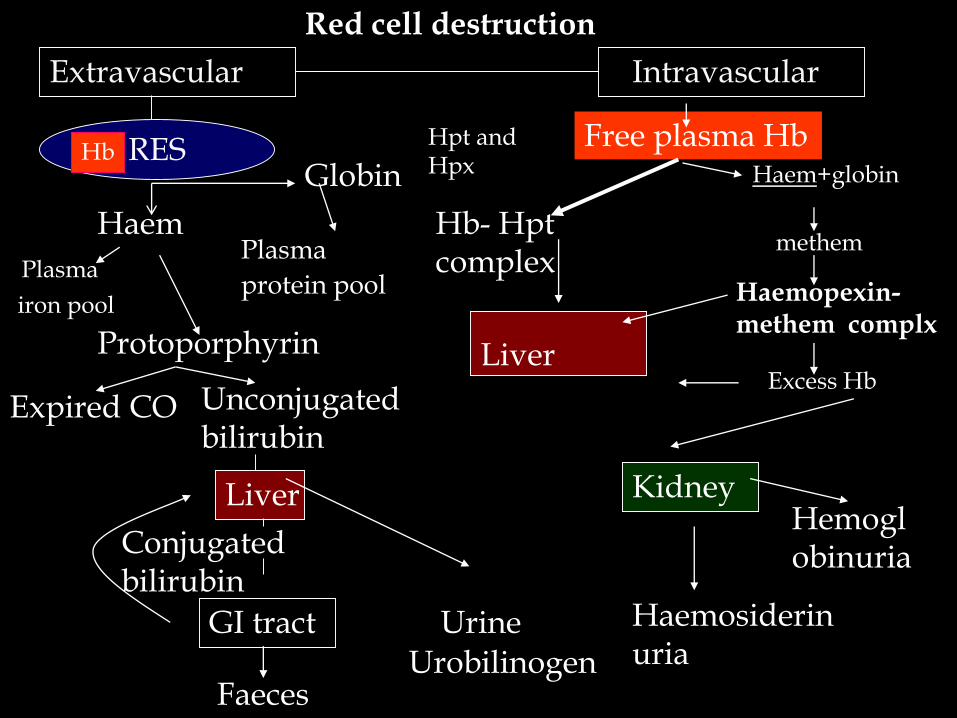
Red cell destruction
Extravascular Intravascular
RES
Haem
Globin
Plasma
iron pool
Plasma
protein pool
Protoporphyrin
Expired CO Unconjugated bilirubin
Liver
Conjugated bilirubin
GI tractUrobilinogen
Faeces
Urine
Free plasma Hb
Hb- Hpt complex
Liver
Hpt and Hpx
Haemopexin-methem complx
Excess Hb
Kidney
Haemosiderinuria
Haem+globinHb
methem
Hemoglobinuria

Hemolytic anemias are classified in a variety of ways
1. Location of hemolysis
INTRA VASCULAR (vascular space)
EXTRA VASCULAR(reticuloendothelial system)

2) Source of defect causing hemolysis:
Intra corpuscular defect
Extra corpuscular defect
3) Mode of onset:
Hereditary
Acquired

Hereditary Acquireda) Red cell Membrane defect
spherocytosiselliptocytosis
a) Red cell Enzyme deficiencyG-6-PD deficiencyPK deficiency
a) Hemoglobin abnormalities
Deficient globin synthesis Thalassemias
Structurally abnormal globins [hemoglobinopathies] sickle cell anemiaUnstable haemoglobins
a) Paroxysmal nocturnal hemoglobinuria
a) Infections

Immune haemolytic anaemia(antibody-mediated destruction)
Autoimmune haemolytic anaemiaWarm antibody auto immune haemolytic anaemia
Cold antibody auto immune haemolytic anaemia
Paroxysmal cold haemoglobinuria
Alloimmune haemolytic anaemiaHaemolytic disease of the newborn
Haemolytic transfusion reactions : mismatched blood transfusion

Microangiopathic haemolytic anaemia
Disseminated intra vascular coagulation(DIC)
Hemolytic uremic syndrome
Thrombotic thrombocytopenic purpura
Sequestration(hyperspleenism & Spleenomegaly)
Drugs and chemicals

Physical/chemical trauma
Thermal injury/burns
March haemoglobinuria
Infections and infectious agents
Malaria
Clostridium infections
Bartenellosis
Cholera
Animal venoms

Symtoms of HemolysisPaleness of skinFatigueFeverConfusionLightheadednessDizziness

Lab findings of Hemolytic Anemia
Hemoglobinuria & hemoglbinemia
Reticulocytosis
Peripheral smear- anisocytosis , poikilocytosis
Haptoglobin &hemopexin -decreased
Indirectbilirubin - increased.

Increased serum and stored iron.
Increased activity of bone marrow
(erythroid hyperplasia)
Urine & feacal urobilinogen - elevated

Red cell Membrane defect

• The most common defect of red cell membrane due to deficiency of cytoskeleton protein
- SPECTRIN
- ANKYRIN
- BAND 3
• Deficient of membrane protein causes change of shape (round, no central pallor)
• Hereditary spherocytosis is caused by a genetic defect.
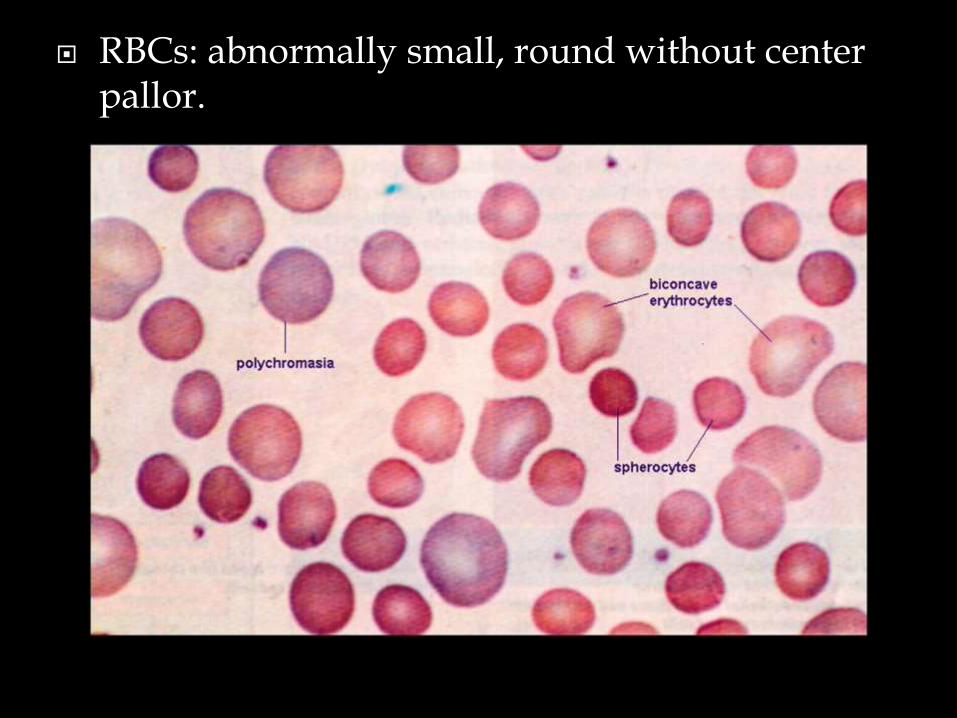
RBCs: abnormally small, round without center pallor.
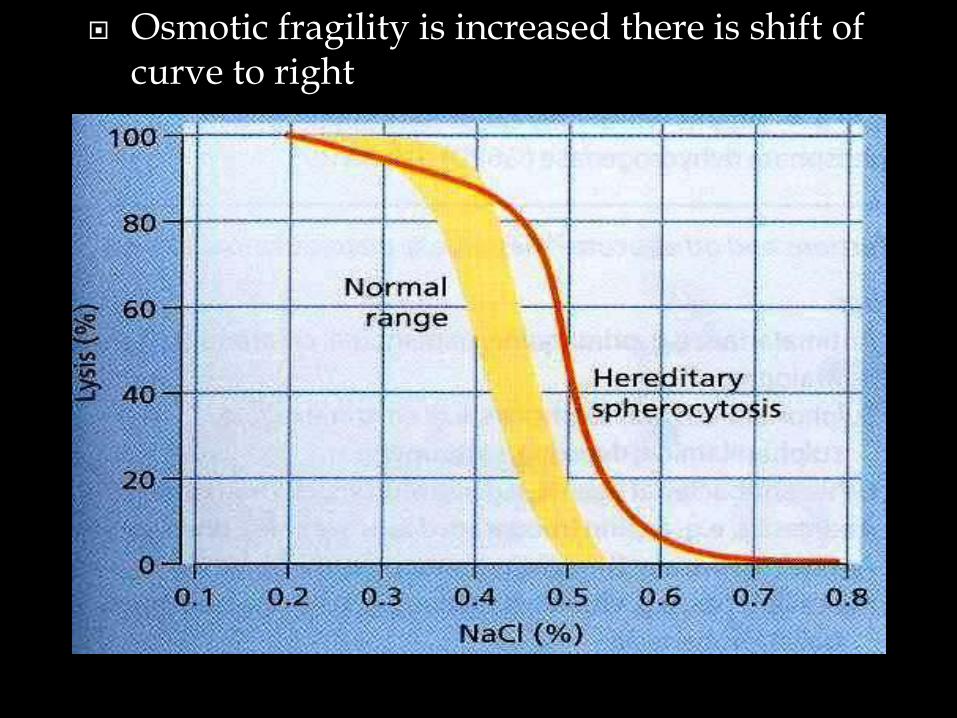
Osmotic fragility is increased there is shift of curve to right
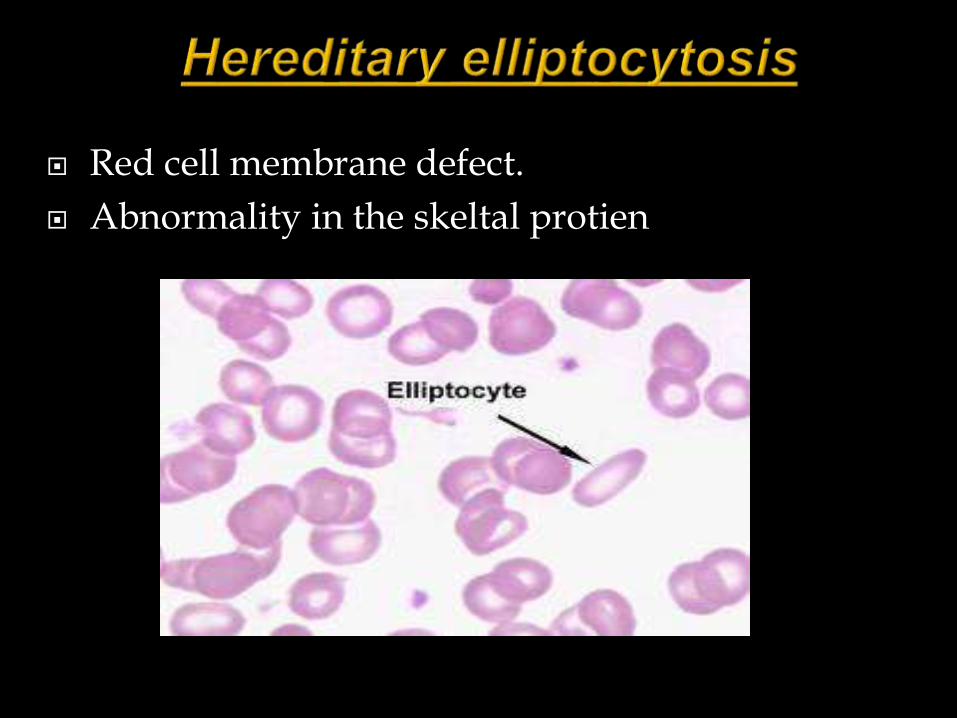
Red cell membrane defect.
Abnormality in the skeltal protien

Causes :
1. Defective spectrin chain
2. Abnormality in the integral protiens
3. Deficiency or defect in ‘band 4.1’
4. Abnormally permeable to Na
Cells become elliptical due to the stress after entering the microcirculation.
Normal cells remains without any change.

RED CELL ENZYME DEFICIENCIES

G-6-PD catalyses the conversion of g-6-phosphate into ribulose 5 phosphate
A deficiency of this enzyme reduces the ability of RBCs to with strand the effect of various oxidising drugs/agents, resulting in haemolytic anaemia
Fava beans High fevers Severe infections Anti malarials-primaquine /quinine and choloroquine
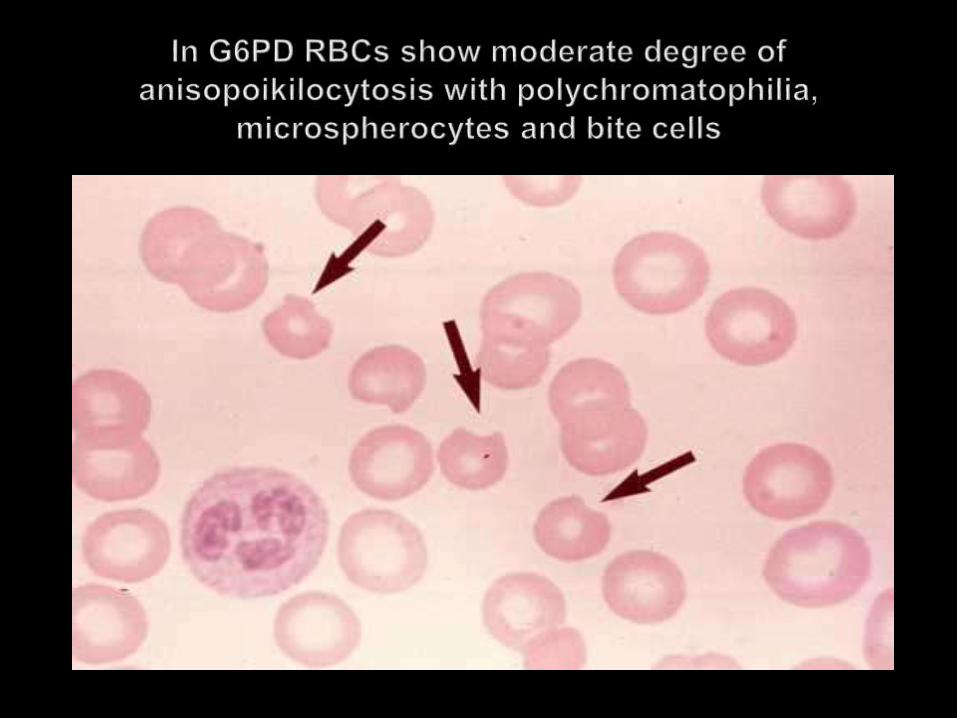
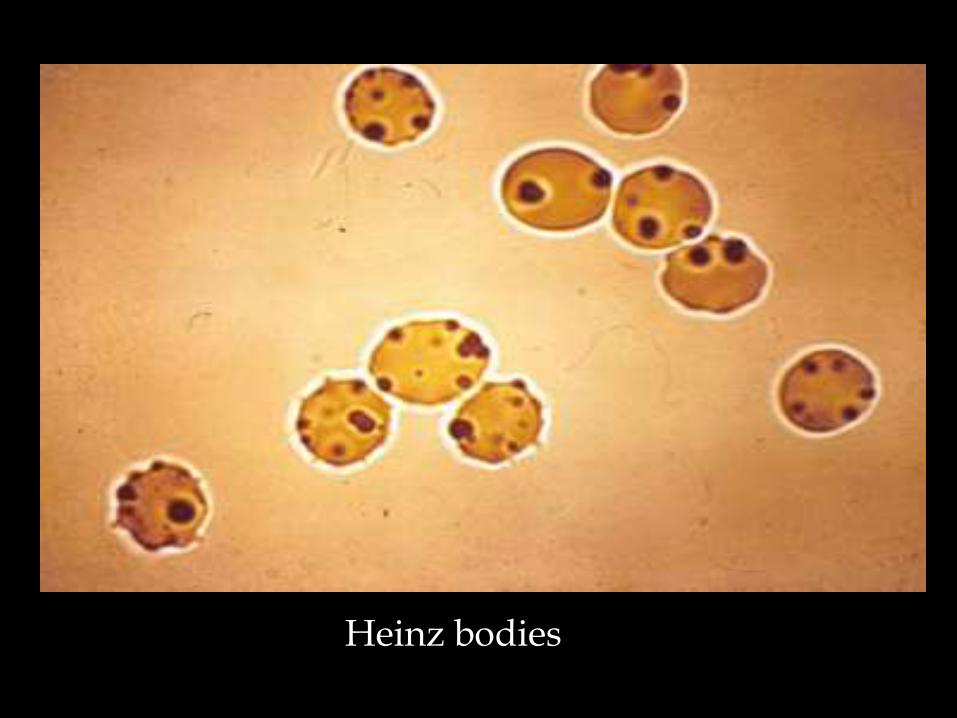
Heinz bodies

Pyruvate kinase deficiency is an autosomal recessive disorder which causes congenital chronic hemolytic anemia
In pyruvate kinase deficiency ATP is not formed
ATP is required for maintaining cell permeability.
Potassium is lossed result in dehydration and hemolysis.
The peripheral blood film shows normocytic normochromic anemia and increased reticulocytes
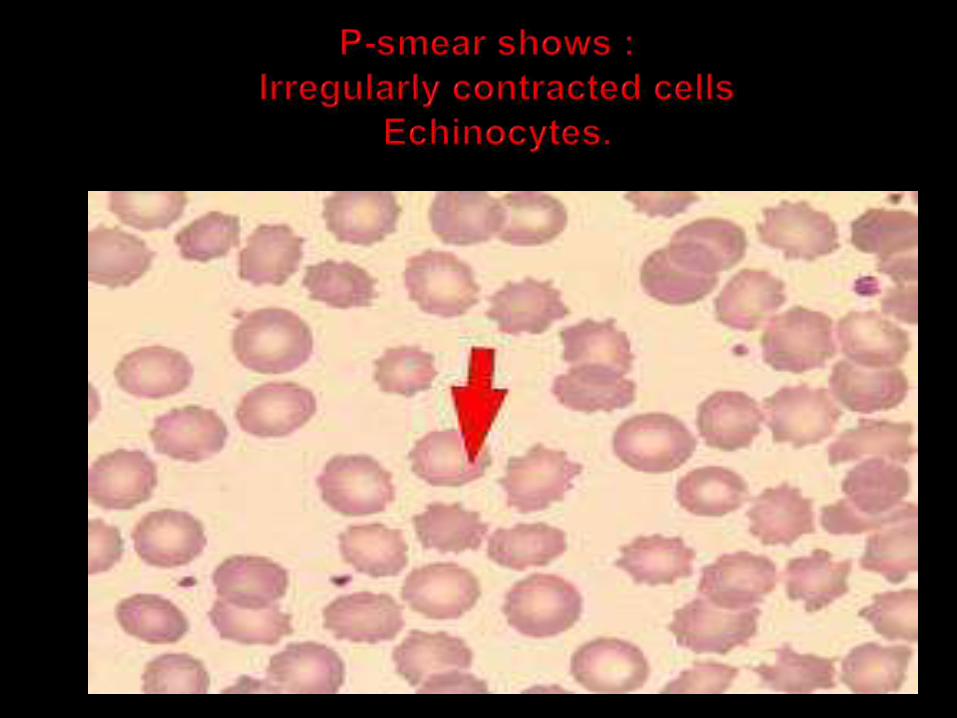

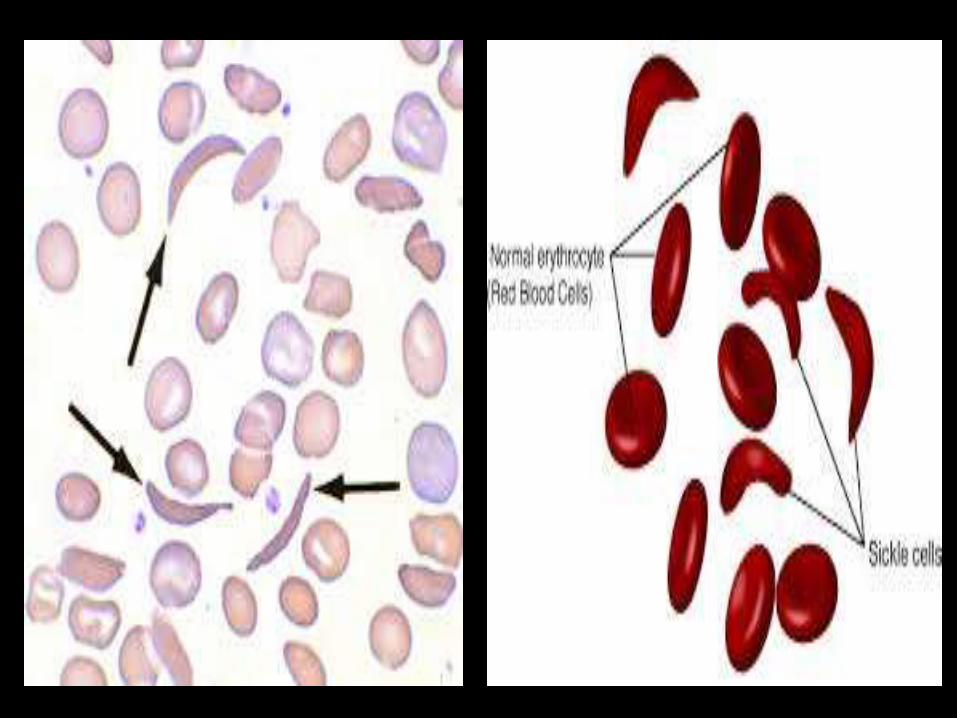
Hemoglobin abnormalities
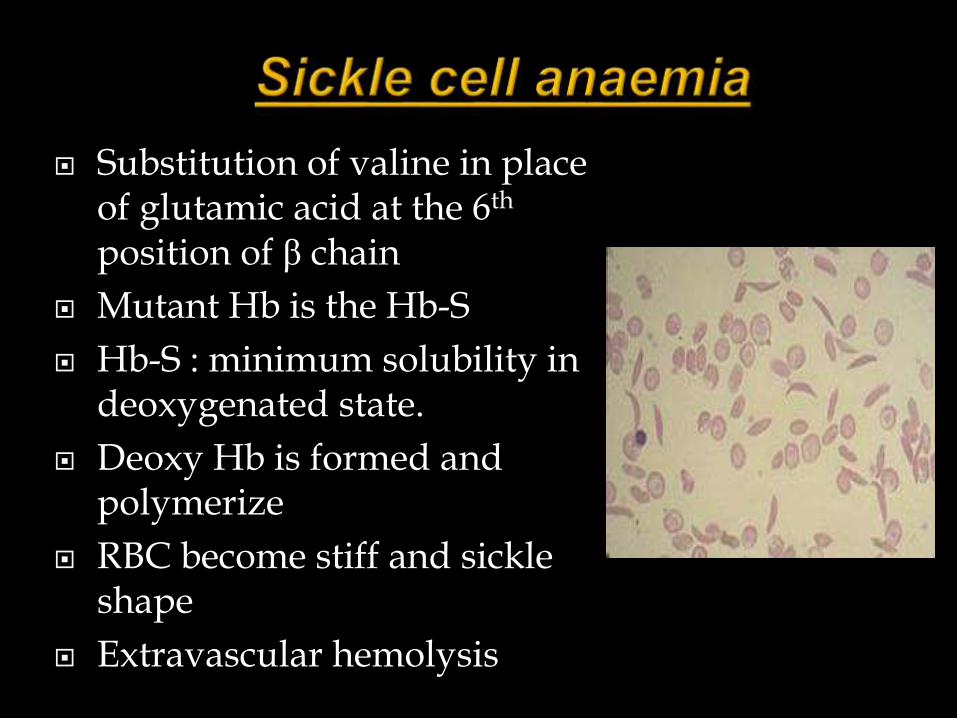
Substitution of valine in place of glutamic acid at the 6th
position of β chain
Mutant Hb is the Hb-S
Hb-S : minimum solubility in deoxygenated state.
Deoxy Hb is formed and polymerize
RBC become stiff and sickle shape
Extravascular hemolysis

Hb electrophoresis
Sickling test
HPLC: high –performance liquid chromatography is useful for confirmation of diagnosis

The thalassemia syndrome is a heterogeneous group of inherited disorder in which one or more globin chain synthesis is either absent or reduced.
Thalassemia syndrome results in microcytichypochromic anemia due to decreased production of hemoglobin with erythroid hyperplasia of the bone marrow.
The imbalance of globin synthesis causes an excess of the normally produced globin chain. This results in damage to red blood cells or their precursors and hemolysis

Thalassemia syndrome is mainly of two types,
1. Alpha thalassemia
2. Beta thalassemia
β-THALASSEMIA
-Thalassemia major(homozygous)
-Thalassemia minor(trait)
-Thalassemia intermedia
α-THALASSEMIA
-Hydrops foetalis
-Hb-H disease
-α-thalassemia trait

THALASSEMIA ASOCIATED WITH OTHER HAEMOGLOBINOPATHIES
-Hb-S thalassemia
-Hb-E thalassemia
-Hb-D thalassemia
-HPFH(Hereditary Persistence of Foetal Hb)


Paroxysmal nocturnal hemoglobinuria is a rare , acquired , potentially life-threatening disease.
Characterized by complement-induced intra vascular hemolytic anemia(anemia due to destruction of red blood cells in the blood stream),red urine(due to the appearance of hemoglobin in the urine)
It is the only hemolytic anemia caused by an intrinsic defect in the cell
It may develop on its own or in the context of other bone marrow disorders such as aplastic anemia


The gold standard is flow cytometry for CD55 and CD59 on red blood cells. Based on the levels of these cell proteins, erythrocytes may be classified as type I, II, or III PNH cells. Type I cells have normal levels of CD55 and CD59; type II have reduced levels; and type III have absent levels.The fluorescein-labeled proaerolysin (FLAER) test is being used more frequently to diagnose PNH on WBCs (neutrophil and monocyte).

Immune haemolytic anaemia Immune haemolytic anaemia is a group of anaemia's in
which premature red cell destruction is mediated by antibodies that bind to red cells.
The antibodies may be either allo or auto antibodies
The haemolysis is caused by extracorpuscular mechanism
The site of haemolysis may be extravascular or intravascular

Alloimmune hemolytic anemia
Hemolytic disease of new born
Hemolytic transfusion reaction
Autoimmune hemolytic anemia
Warm antibody type (IgG antibodies active at 37Oc)
Primary(idiopathic)
Secondary
• Autoimmune disorders(systemic lupus erythematosus

In alloimmune hemolytic anemia allo-antibodies are present either in serum or bound to red cells
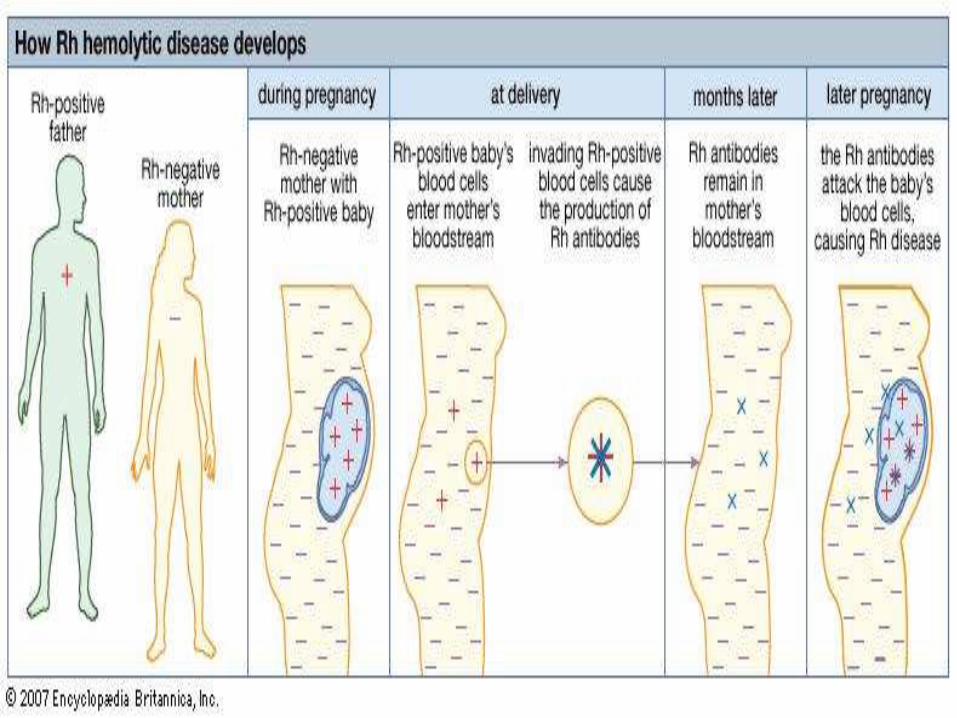
Hemolytic disease of the new born(HDN)
This is an immune hemolytic anemia which affects the fetus and newborn baby

It is due to maternal foetal blood group incompatibility.
HDN develops when mother lacks an antigen that expressed by the foetal red cells
HDN may be due to incompatibility of Rh type or ABO group.

Autoimmune hemolytic anemia(AIHA)are a group of disorders in which antibodies against self-antigens on the RBC membrane cause premature destruction of RBCs.
Interaction of the autoantibody with the red cell antigen is dependent on the temperature, i.e. warm or cold type.

Warm antibody type
Warm antibody type is the most common type(50to 70%) of immunohemolytic anaemia
This can be further divided into idiopathic (primary)or secondary to drug exposure or predisposing diseases.
The antibodies are mainly of IgG type.
The antibodies combine with red cell antigen at 34°c (warm antibody).

There IgG bound RBCs bind to Fc receptors on phagocytes (macrophages)
This remove part of red cell membrane with attached antibody and hence known as “partial” phagocytosis.
Progressive loss of membrane converts the red cells to spherocytes.
As in hereditary spherocytosis , the spleen is the major site of removal and destruction of these spherocytes , resulting in moderate splenomegaly..

Antibodies bind to red cell antigens at low temperature(4-18oc).
The antibodies are of IgM type.
It fixes complement and result in intra vascular hemolysis

Clinical features
Skin color turn white and then blue
On rewarming it becomes red
Numbness, tingling and pain
Hemoglobinuria

The autoantibodies are of IgG type and bind to red cells at low temperatures and fix the complement.
Massive, intermittent , acute haemolysis and hemoglobinuria after cold exposure.
Caused by biphasic IgG antibody (Donath-Landsteiner antibody).
It often follows viral infections and patient recover within a month.

Shear damage to red cells as the result of endothelial cell activation/damage.
Hallmark is presence of schistocytes on the peripheral smear.
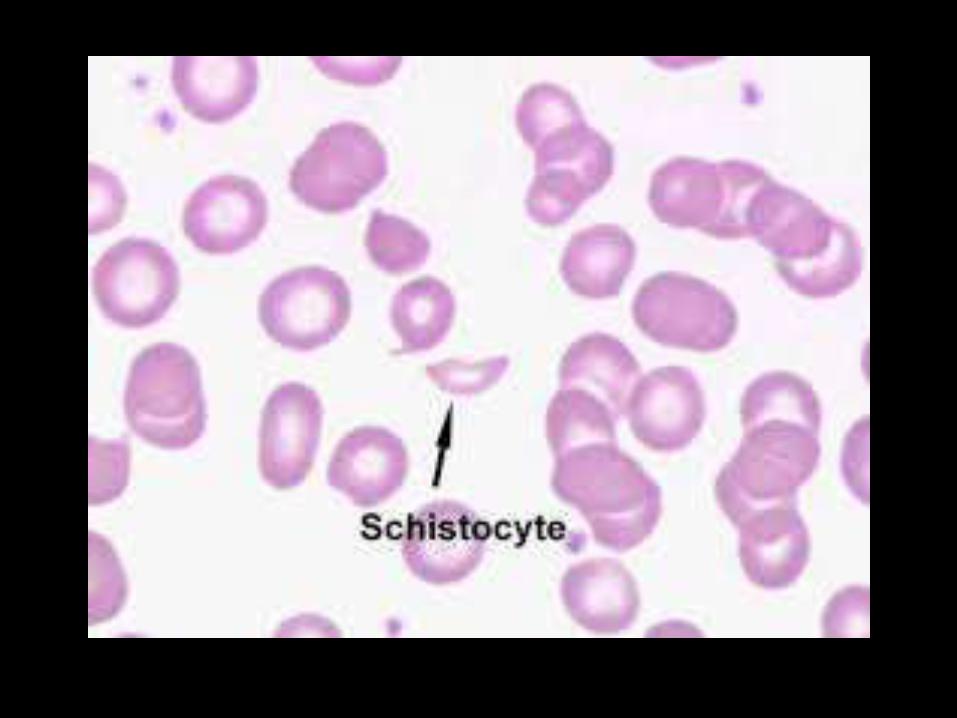

Causes :
- DIC=(DISSEMINATED INTRA VASCULAR COAGULATION)
- TTP=(THROMBOTIC THROMBOCYTOPENIC PURPURA)
- HUS=(HAEMOLYTIC UREMIC SYNDROME)

HAEMOLYTIC UREMIC SYNDROME
Damage to renal glomerular capillaries resulting in agglutination of cells and thrombi formation
THROMBOTIC THROMBOCYTOPENIC PURPURA
Haemolysis is due to the deposition of thrombi in microcirculation
DISSEMINATED INTRA VASCULAR COAGULATION
Activation of coagulation mechanism invivo by thromboplastic substances resulting in the deposition of fibrin in the microvasculature.
RBC fragmentation due to stress produced by fibrin.

Wintrobe’s haematology and clinical pathology
Essentials in Hematology & Clinical pathology-ramdas nayak ,sharada rai , astha gupta
Practiacal pathology- Dr.k. uma chaturvedi, Dr tejinder singh
Text book of haematology by Mckenzie
PARTICAL HEMATOLOGY-dacie and lewis






























