Embed Size (px)
DESCRIPTION
The Validation Master Plan is a a valuable opportunity to provide an overview of your company’s validation process, including organization structure, content, and planning.
Citation preview

Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
For more documentation and validation master plans, go to http://www.ivtnetwork.com
Use the promo code SLIDE1 for a 10% discount on membership!

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 2 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
Glossary
2.0 PURPOSE AND INTRODUCTION ......................................................................................................3 2.1 INTRODUCTION ......................................................................................................................... 3 2.2 PURPOSE ..................................................................................................................................... 3
3.0 SCOPE.....................................................................................................................................................4
4.0 RESPONSIBILITIES ..............................................................................................................................4 4.1 SITE QUALITY ASSURANCE VALIDATION ......................................................................... 5 4.2 SITE ENGINEERING VALIDATION ........................................................................................ 6 4.3 SITE MANUFACTURING TECHNICAL SUPPORT .............................................................. 6 4.4 SITE MANUFACTURING .......................................................................................................... 7 4.5 SITE ENGINEERING ................................................................................................................. 8 4.6 QUALITY CONTROL ................................................................................................................. 8 4.7 REGULATORY AFFAIRS ......................................................................................................... 9
5.0 DEFINITIONS, ABREVIATIONS AND REFERENCES ...................................................................9 5.1 DEFINITIONS LIST .................................................................................................................... 9 5.2 ABBREVIATION LIST .............................................................................................................. 12 5.3 REFERENCES .......................................................................................................................... 12
6.0 MATERIALS ..........................................................................................................................................12
7.0 PROCEDURE .......................................................................................................................................13 7.1 SPRINGFIELD, NY FACILITY OVERVIEW ......................................................................... 13 7.2 VALIDATION MASTER PLAN GOVERNANCE .................................................................. 13 7.3 VALIDATION APPROACH ...................................................................................................... 15 7.4 VALIDATION PROCEDURES ................................................................................................ 17 7.5 VALIDATION SUPPORT SYSTEMS ..................................................................................... 18 7.6 KEY SOPs GOVERNING AND SUPPORTING THE ALLSTON LANDING VALIDATION
PROGRAM ................................................................................................................................ 21 7.7 IDENTIFICATION AND DESCRIPTION OF MANUFACTURING SYSTEMS AND
EQUIPMENT PRESENTLY IN USE AT THE SPRINGFIELD, NY FACILITY AND ASSOCIATED COMPLETED VALIDATION ........................................................................ 21
7.8 VALIDATION MASTER PLAN REVIEW ............................................................................... 21
8.0 ATTACHMENTS...................................................................................................................................21 8.1 Attachment A: Process Flow Diagrams ................................................................................ 21 8.2 Attachment B: Key SOPs Governing and Supporting the Springfield, NY Facility
Validation Program .................................................................................................................... 21 8.3 Attachment C: Process Equipment List and Associated Validation Protocol Numbers
..................................................................................................................................................... 21
9.0 REVISION HISTORY...........................................................................................................................27 9.1 Revision History Table ............................................................................................................. 27

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 3 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
2.0 PURPOSE AND INTRODUCTION 2.1 INTRODUCTION The validation program at the Springfield, NY Facility is intended to satisfy current domestic and international regulations, guidelines and policies for drugs, biologics and APIs to verify the equipment, systems, utilities, software and processes are properly designed, installed, and function as intended in a consistent and controlled manner. The validation program complies with Corporate Quality Standards and Polices (Ref. SOP-XXXX – Validation Program). The revision of the Site Validation Master Plan (SVMP) comes from the validation philosophy changes resulting from the implementation of the “validation lifecycle” approach at Springfield, NY Facility. The changes made in this revision of the Springfield, NY Facility SVMP bring the plan into alignment with the new lifecycle approach. The Springfield, NY Facility has developed the site validation governance document SOP-XXXX, “Validation Project Life Cycle Approach at the Springfield, NY Facility”, which defines the validation philosophy to be used for validation at Springfield, NY Facility. SOP-XXXX uses a risk-based approach with emphasis on verification and qualification that is focused on the high risk attributes of facilities, utilities, equipment, and processes. This SVMP introduces use of subordinate VMPs (sub-VMPs) that are categorized into eight areas and address the full scope of Validation requirements applicable to the Springfield, NY Facility site in support of SVMP. 2.2 PURPOSE The purpose of validation is to assure that the facility, manufacturing process, and supporting services are capable of supporting the manufacture of pharmaceutical product that consistently meets its predetermined quality attributes. For this reason, each facet of the Springfield, NY Facility site related to CGMP manufacture, monitoring, storage, and testing will be validated. The Validation program is integral to design, construction / fabrication, and CGMP Operation of the facility. Validation testing is intended to proactively establish CGMP compliance prior to initiating manufacture of commercial product for distribution. In addition, the sustenance operations at Springfield, NY Facility, such as quality change control, calibration program, preventive maintenance, and periodic requalification of critical equipment and processes ensure that manufacturing equipment and processes stay under the validated state. The purpose of this SVMP is to:
• Define the requirements and validation approach for validation of systems, equipment, and processes at Pharma Co., Inc. Corporation’s Springfield, NY Facility.
• To list or reference the documentation supporting the current validated state of the Springfield, NY Facility.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 4 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
This SVMP explains the governance structure in place that will achieve, demonstrate and maintain acceptable standards of validation for systems, equipment, and processes used in the manufacture of drug product at the Springfield, NY Facility. If the methodologies and rationale presented herein are not followed, the rationale behind the alternate approach must be documented appropriately. This document is designed to accomplish the following objectives:
• Identify the key Standard Operating Procedures governing and supporting the validation program presented in SOP-XXXX.
• Describe the manufacturing systems, equipment, and processes presently in use at the Springfield, NY Facility.
• Itemize the equipment and utilities used in the Springfield, NY Facility manufacturing systems and link them to completed validation studies as evidence of a validated state.
3.0 SCOPE
This Site Validation Master Plan applies to all verification, qualification, and validation activities that support commercial manufacturing at Pharma Co., Inc. Corporation’s Springfield, NY Facility. This VMP is a high-level document that refers to various supporting documents. Validation program elements include:
• Validation/qualification of facilities/utilities/equipment and corresponding automation/controls
• Validation of manufacturing processes • Cleaning validation • Sterilization and depyrogenation • Enterprise Computer systems • QC Laboratory equipment and methods • Interval based requalification
The commercial products that are currently manufactured, packaged, or stored at the Springfield, NY Facility are listed in Table 7.1-1. The equipment within the scope of this Validation Master Plan are listed in Attachment D, Process Equipment List and Associated Validation.
4.0 RESPONSIBILITIES
This SVMP will be approved by the heads of the departments (or their designees) listed in this section.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 5 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
The validation activities in Springfield, NY Facility are the responsibility of the Validation, Engineering, Manufacturing, MTS, and Quality Assurance groups as shown in Figure 1.
The responsibilities outlined below align with the Validation Lifecycle Approach that is presented in SOP-XXXX. 4.1 SITE QUALITY ASSURANCE VALIDATION
• Generate, review, and maintain approve SVMP
• Review and approve the sub-VMPs and Project VPs
• Assist with the generation of User Requirements (URs) document
• Review and approve the URS document
• Assist with the performance of risk assessments
• Review and approve Risk Assessments documents
• Review and approve Critical Aspects (CAs) list and acceptance criteria, as applicable
• Review and approve the Process Control Strategy
• Review and approve the Operational Control Strategy
• Review and approve Commissioning and Qualification and Validation Plans
• Review and approve Qualification Protocols and Reports for facilities, utilities, equipment, and processes that have CAs
• Review and approve Computerized Systems’ Protocols and Reports
• Review and approve Performance Qualification (PQ) Protocols and Reports

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 6 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
• Review and approve Process Validation (PV) and Continued Verification Protocols and Reports
• Review and approve Qualification and Validation Protocol Deviations
• Approve the use of vendor documentation to verify functionality of facility, utility, and equipment
• Reviewing and approving the Site’s Equipment Requalification related documents
4.2 SITE ENGINEERING VALIDATION
• Review and approve SVMP
• Generate, review, approve, and maintain sub-VMP for Facilities, Equipment (including QC Lab storage and process equipment, such as refrigerators, autoclaves and incubators) and Utilities, Cleaning Process, Sterilization and Depyrogenation Process, and Computerized Systems
• Lead/generate, review and approve Site Project VP for new products or projects
• Maintain Validation Schedule per the Project Execution Plan (PEP) and Project VP and CCR timelines
• Assist with the generation of URS document
• Review and approve URS document
• Lead/perform risk assessments using CA list
• Review and approve the Operational Control Strategy
• Review and approve Risk Assessment document
• Generate, review, and approve IQ, OQ, PQ Protocols and Reports for facilities, utilities, equipment, and processes
• Generate, review, and approve PQ Protocols and Reports
• Generate, review, and approve Protocol Deviations
• Review and approve Computer Systems’ Protocols and Reports
4.3 SITE MANUFACTURING TECHNICAL SUPPORT
• Review and approve the SVMP, sub-VMPs, and Project VPs, as appropriate
• Generate, review, approve, and maintain sub-VMP or PV
• Assist with the generation of URS document
• Review and approve URS of document
• Review and approve Risk Assessment document, as applicable
• Review and approve Validation Plans, as applicable

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 7 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
• Review approve and assist in updating the Process Control Strategy
• Generate, review, and approve the Operational Control Strategy
• Lead /perform Technical Transfer activities for product transfer
• Lead/perform process development / engineering runs
• Lead. generate, review, and approve PV and Continued Verification Protocols and Reports
• Execute PV Protocols
• Generate, review, and approve PV Protocol Deviations
• Create and update Manufacturing Process Descriptions and associated Manufacturing Flow Diagrams (MFDs)
• Generate process related sections of the Project VPs
• Review and approve Project VPs that include PV
• Lead/perform risk assessments for PV
• Create and update Manufacturing History File (MHF)
4.4 SITE MANUFACTURING
• Assist with the performance of risk assessments
• Assist with the generation of URS document
• Review and approve URS document
• Assist with the generation of the SVMP, sub-VMPs, and Project VPs, as applicable
• Review and approve the SVMP, sub-VMPs, and Project VPs, as applicable
• Review and approve the Process Control Strategy
• Review and approve the Operational Control Strategy
• Assist Site Engineering with generation of project schedule
• Generate manufacturing SOPs for use during validation
• Review and approve IQ, OQ, PQ, PV, and Continued Verification Protocols and Reports for facilities, utilities, equipment, and processes as applicable
• Prepare schedule in support of IQ, OQ, PQ, PV, and Continued Verification Protocols execution
• Ensure equipment/facilities/materials/personnel availability and technical support on the operation of the equipment during execution of qualification and validation activities

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 8 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
• Ensure manufacturing procedures are developed for maintaining the validated equipment in its validated state throughout its lifecycle
• Review and approve Protocol Deviations affecting process equipment
• Manage and execute the Site’s Equipment Requalification program
4.5 SITE ENGINEERING
• Assist with the performance of risk assessments
• Lead the generation of URS document
• Review and approve URS document
• Generate Functional Specifications based on URS
• Assist with the generation of the SVMP, sub-VMPs, and Project VPs, as applicable
• Review and approve the SVMP, sub-VMPs, and Project VPs, as applicable
• Generate, review, approve, and maintain sub-VMP for Computer Systems
• Review and approve the Operational Control Strategy
• Generate, review, and approve Computer Systems Protocols and Reports
• Review and approve Verification (including IQ and OQ) and PQ Protocols and Reports for facilities, utilities, equipment, and processes as applicable
• Participate in execution of Verification Protocols, as necessary
• Review and approve Protocol Deviations affecting facilities, utilities, and Computer Systems
• Review and approve the Site’s Equipment Requalification documents related to facilities and utilities
4.6 QUALITY CONTROL
• Assist with the generation of the SVMP, sub-VMPs, and Project VPs, as applicable
• Review and approve the SVMP and sub-VMPs, as applicable
• Generate, review, approve, manage execution of, and maintain sub-VMPs for QC Laboratories
• Review and approve Site Project VP for new products when QC testing is required
• Receive and use transferred validated methods
• Review and approve method validation protocols and reports

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 9 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
• Generate, review and approve validation protocols and reports for laboratory test instrumentation
• Review and approve laboratory equipment validation protocols and reports storage and process equipment, such as refrigerators, autoclaves and incubators
• Review the PQ and PV protocols for analytical and microbiological testing requirements, as applicable
• Perform testing required for validation samples, as applicable
• Review the PQ and PV Final Reports and verify the accuracy of testing data, as applicable
• Review and approve Protocol Deviations affecting QC test results and equipment
4.7 REGULATORY AFFAIRS
• Assist with the review of SVMP, sub-VMPs, and validation requirements to ensure compliance with regulatory filings
• Update regulatory filings as necessary based on approved validation data
• Assess the impact of validation activities on current approved product filings
• Provide filing dates as part of validation activity scheduling
• Incorporate pertinent validation data into annual reports
5.0 DEFINITIONS, ABREVIATIONS AND REFERENCES
5.1 DEFINITIONS LIST
Cleaning Validation (CV)
Documented evidence that a cleaning process is consistently and effectively reducing potential product and/or cleaning agent residues to pre-determined acceptable limits.
Cleaning Verification
Documented evidence that equipment is cleaned to pre-determined specifications and may be released for use.
Clean in Place (CIP)
Introduction of cleaning solution and/or water rinses into equipment that is fixed in place, for purposes of removing potential product and/or cleaning agent residues.
Clean out of Place (COP)
Cleaning of portable or disassembled equipment/parts involving the use of a cleaning station in a remote designated location.
Commissioning
A well-planned, documented, and managed engineering approach to the startup and turnover of facilities, systems, and equipment to the System Owner that results in a safe and functional environment that meets established design requirements and stakeholder expectations.
Concurrent Validation
Validation of equipment, systems or processes while the equipment, system or process is in current use for clinical or commercial production.
DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 10 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
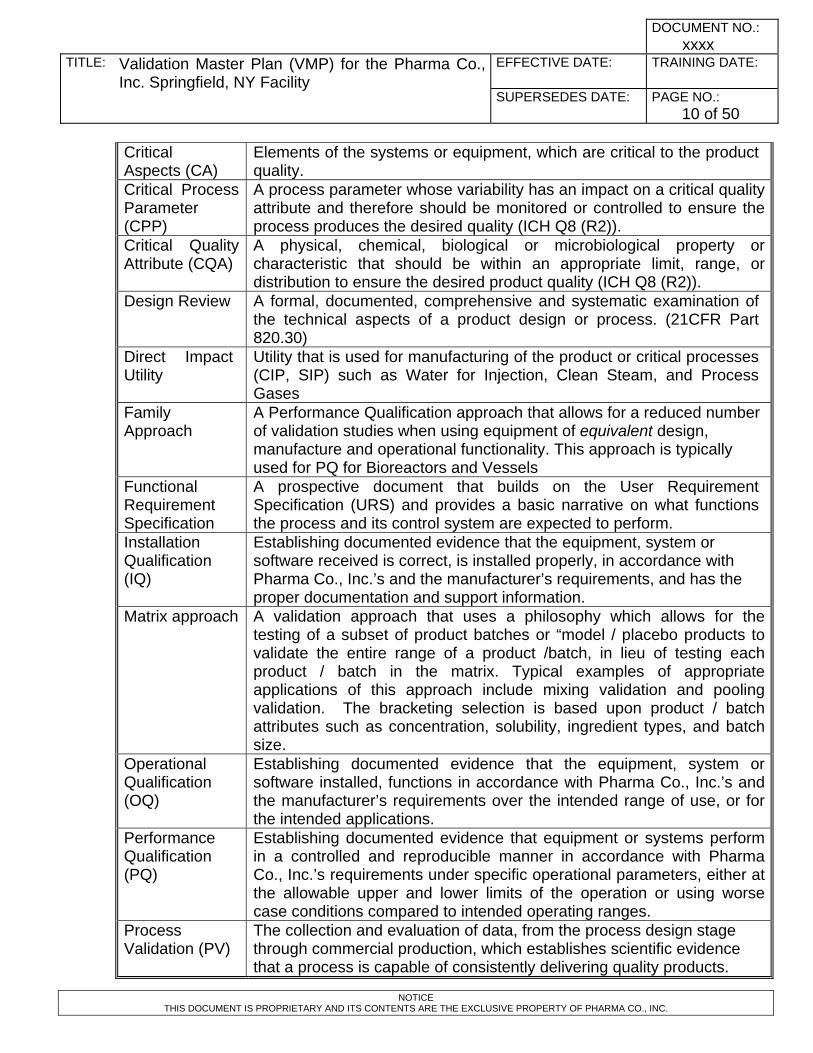
Critical Aspects (CA)
Elements of the systems or equipment, which are critical to the product quality.
Critical Process Parameter (CPP)
A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality (ICH Q8 (R2)).
Critical Quality Attribute (CQA)
A physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality (ICH Q8 (R2)).
Design Review A formal, documented, comprehensive and systematic examination of the technical aspects of a product design or process. (21CFR Part 820.30)
Direct Impact Utility
Utility that is used for manufacturing of the product or critical processes (CIP, SIP) such as Water for Injection, Clean Steam, and Process Gases
Family Approach
A Performance Qualification approach that allows for a reduced number of validation studies when using equipment of equivalent design, manufacture and operational functionality. This approach is typically used for PQ for Bioreactors and Vessels
Functional Requirement Specification
A prospective document that builds on the User Requirement Specification (URS) and provides a basic narrative on what functions the process and its control system are expected to perform.
Installation Qualification (IQ)
Establishing documented evidence that the equipment, system or software received is correct, is installed properly, in accordance with Pharma Co., Inc.’s and the manufacturer’s requirements, and has the proper documentation and support information.
Matrix approach A validation approach that uses a philosophy which allows for the testing of a subset of product batches or “model / placebo products to validate the entire range of a product /batch, in lieu of testing each product / batch in the matrix. Typical examples of appropriate applications of this approach include mixing validation and pooling validation. The bracketing selection is based upon product / batch attributes such as concentration, solubility, ingredient types, and batch size.
Operational Qualification (OQ)
Establishing documented evidence that the equipment, system or software installed, functions in accordance with Pharma Co., Inc.’s and the manufacturer’s requirements over the intended range of use, or for the intended applications.
Performance Qualification (PQ)
Establishing documented evidence that equipment or systems perform in a controlled and reproducible manner in accordance with Pharma Co., Inc.’s requirements under specific operational parameters, either at the allowable upper and lower limits of the operation or using worse case conditions compared to intended operating ranges.
Process Validation (PV)
The collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality products.
DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 11 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
(Guidance for Industry – Process Validation: General Principles and Practices, FDA, January 2011, Rev 1).
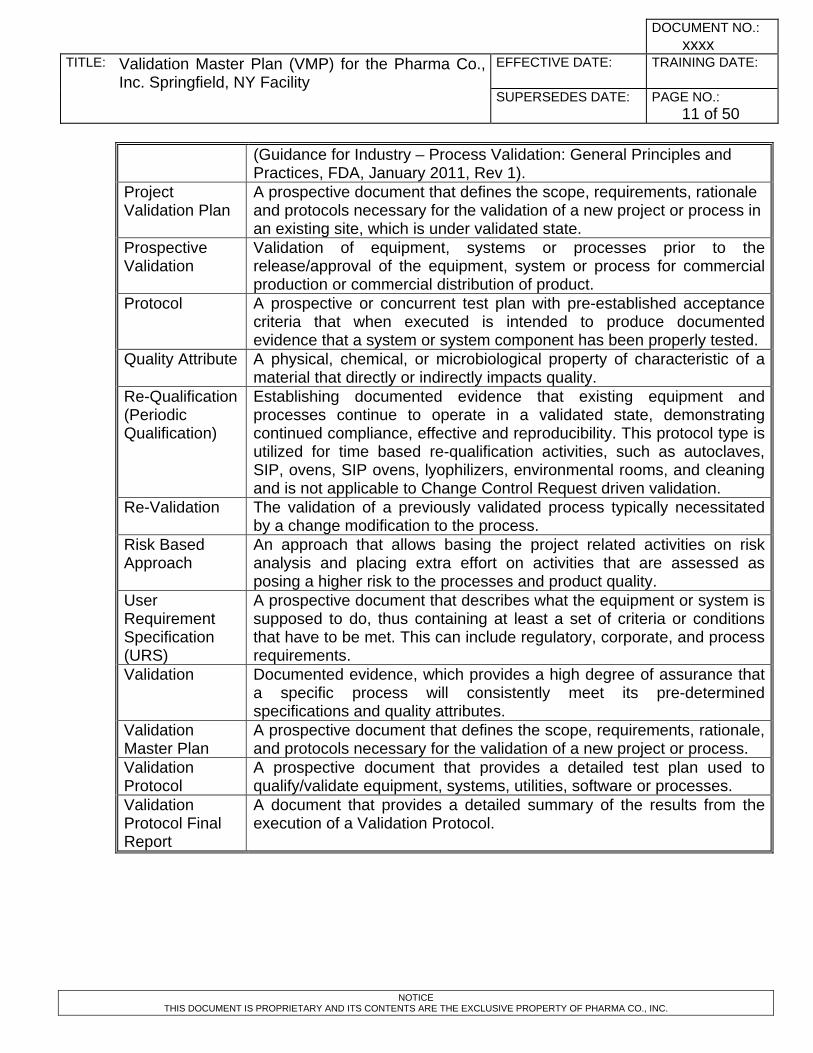
Project Validation Plan
A prospective document that defines the scope, requirements, rationale and protocols necessary for the validation of a new project or process in an existing site, which is under validated state.
Prospective Validation
Validation of equipment, systems or processes prior to the release/approval of the equipment, system or process for commercial production or commercial distribution of product.
Protocol A prospective or concurrent test plan with pre-established acceptance criteria that when executed is intended to produce documented evidence that a system or system component has been properly tested.
Quality Attribute A physical, chemical, or microbiological property of characteristic of a material that directly or indirectly impacts quality.
Re-Qualification (Periodic Qualification)
Establishing documented evidence that existing equipment and processes continue to operate in a validated state, demonstrating continued compliance, effective and reproducibility. This protocol type is utilized for time based re-qualification activities, such as autoclaves, SIP, ovens, SIP ovens, lyophilizers, environmental rooms, and cleaning and is not applicable to Change Control Request driven validation.
Re-Validation The validation of a previously validated process typically necessitated by a change modification to the process.
Risk Based Approach
An approach that allows basing the project related activities on risk analysis and placing extra effort on activities that are assessed as posing a higher risk to the processes and product quality.
User Requirement Specification (URS)
A prospective document that describes what the equipment or system is supposed to do, thus containing at least a set of criteria or conditions that have to be met. This can include regulatory, corporate, and process requirements.
Validation Documented evidence, which provides a high degree of assurance that a specific process will consistently meet its pre-determined specifications and quality attributes.
Validation Master Plan
A prospective document that defines the scope, requirements, rationale, and protocols necessary for the validation of a new project or process.
Validation Protocol
A prospective document that provides a detailed test plan used to qualify/validate equipment, systems, utilities, software or processes.
Validation Protocol Final Report
A document that provides a detailed summary of the results from the execution of a Validation Protocol.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 12 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
5.2 ABBREVIATION LIST
API Active Pharmaceutical Ingredient ASME American Society of Mechanical Engineers CFR Code of Federal Regulations
cGMP current Good Manufacturing Practices CIP Clean-In-Place
CPP Critical Process Parameters CQA Critical Quality Attributes CA Critical Aspects EMA European Medicines Agency FDA Food and Drug Administration
GAMP Good Automated Manufacturing Practices ICH International Conference on Harmonization IQ Installation Qualification
ISO International Standards Organization ISPE International Society of Pharmaceutical Engineers KPP Key Process Parameter MFD Manufacturing Flow Diagram OQ Operational Qualification PEP Project Execution Plan PQ Performance Qualification PV Process Validation QA Quality Assurance QC Quality Control RQ Re-Qualification SIP Steam-In-Place SME Subject Matter Expert SOP Standard Operating Procedure
SVMP Site Validation Master Plan UR User Requirement
VMP Validation Master Plan
5.3 REFERENCES
• See Attachment B for Key regulatory references and Corporate and site SOPs Governing and Supporting the Springfield, NY Facility Validation Program.
6.0 MATERIALS
All materials needed to perform the functions referenced in this document will be found in the individual referenced documents.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 13 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
7.0 PROCEDURE
7.1 SPRINGFIELD, NY FACILITY OVERVIEW
The Pharma Co., Inc., and is located at: 1 Main Street, Springfield, NY. Pharma Co., Inc.’s Springfield, NY facility is a multi-product manufacturing facility for xxxx products. Manufacturing operations performed at the Springfield, NY facility include bulk drug substance activities, testing, labeling, and packaging. Drug substance manufacture for Product A is performed at this facility (see Attachment A for Process Flow Diagram.) Packaging and labeling operations are performed at this facility for Product A. Quality Control (QC) testing for Product A may be performed in qualified laboratories at Springfield, NY Facility, and approved vendor sites. Table 7.1-1 outlines the manufacturing activities performed at the Springfield, NY Facility for each product. Table 7.1-1: Manufacturing Activities Performed at Springfield by Product
Product Activities Performed at Springfield, NY Facility Product A Manufacturing, formulation, testing, labeling, and packaging
7.2 VALIDATION MASTER PLAN GOVERNANCE The Validation Program incorporates evaluation and testing to establish:
• Manufacturing & Laboratory Facility Installation • Direct Impact Utility Installation, Operation, & Performance • Manufacturing Equipment Installation, Operation, & Performance • Validation of Computer Systems & Software • Validation of Manufacturing Equipment Cleaning Processes and Cycles • Qualification of Laboratory Equipment and Instruments • Validation of Analytical, Physical, & Microbiological Test Methods • Transfer / Development of Manufacturing Processes • Qualification of Environmental Controls and Clean Rooms • Validation of Manufacturing Processes • Assessment and pre-determination of the longevity of CGMP equipment
It is not intended that the SVMP will describe the requirements in detail for execution of the various Validation Program elements. The detailed requirement description may be found in sub-VMPs, Standard Operating Procedures, Corporate Level Standards or Practices, or relevant protocols.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 14 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
This SVMP will be revised at a minimum once per year to reflect the changes made to the validated facilities, utilities, equipment, and processes at Springfield, NY Facility. In addition, a periodic SVMP Summary Report (issued annually, at minimum) will document the summary of changes to the validated systems and corresponding validation documents. The Process Equipment List and associated document numbers will be updated with the new document numbers and titles. In order to address the full scope of Validation requirements applicable to the Springfield, NY Facility site, validation activities have been categorized into eight areas, which will be executed by corresponding sub-VMPs, to be created, and as listed below:
• Sterilization and Depyrogenation Master Plan for Springfield, NY Facility • Cleaning Validation Master Plan for Springfield, NY Facility • Process Validation Master Plan for Springfield, NY Facility • Facility, Equipment, and Utility Validation Master Plan for Springfield, NY
Facility • Computer System Validation Master Plan for Springfield, NY Facility • Requalification Validation Master Plan for Springfield, NY Facility • QC Laboratory Equipment and Instrumentation Qualification Master Plan for
Springfield, NY Facility • Test Method Validation Master Plan for Springfield, NY Facility
The eight sub-VMPs will supplement the overarching SVMP, and will be governed by corresponding Corporate Quality Operations Standards. The Sub-VMPs will present the specific validation requirements and projects within each of these validation groupings, along with the necessary details on the scope, validation approach, roles and responsibilities, required deliverables (equipment, systems, processes to be validated and corresponding protocols and documentation, URS, SOPs, test methods, etc.), and acceptance criteria. The projected schedule and required resources will be covered by the Project Execution Plan (PEP) and Project VP. Standard content of the Sub-VMPs will include:
• Policy statement and description of Validation Methodology to be followed. • Listing of applicable governance policy and procedures describing program
requirements. • Listing of current approved documentation supporting the validated state for the
program addressed. • Execution plan for the specific validation program addressed (based on calendar
year) Upon approval of the Sub-VMP, annual updates will be issued to describe the current year’s project plan status. Record of the annual review will be attached to the Sub-VMP.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 15 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
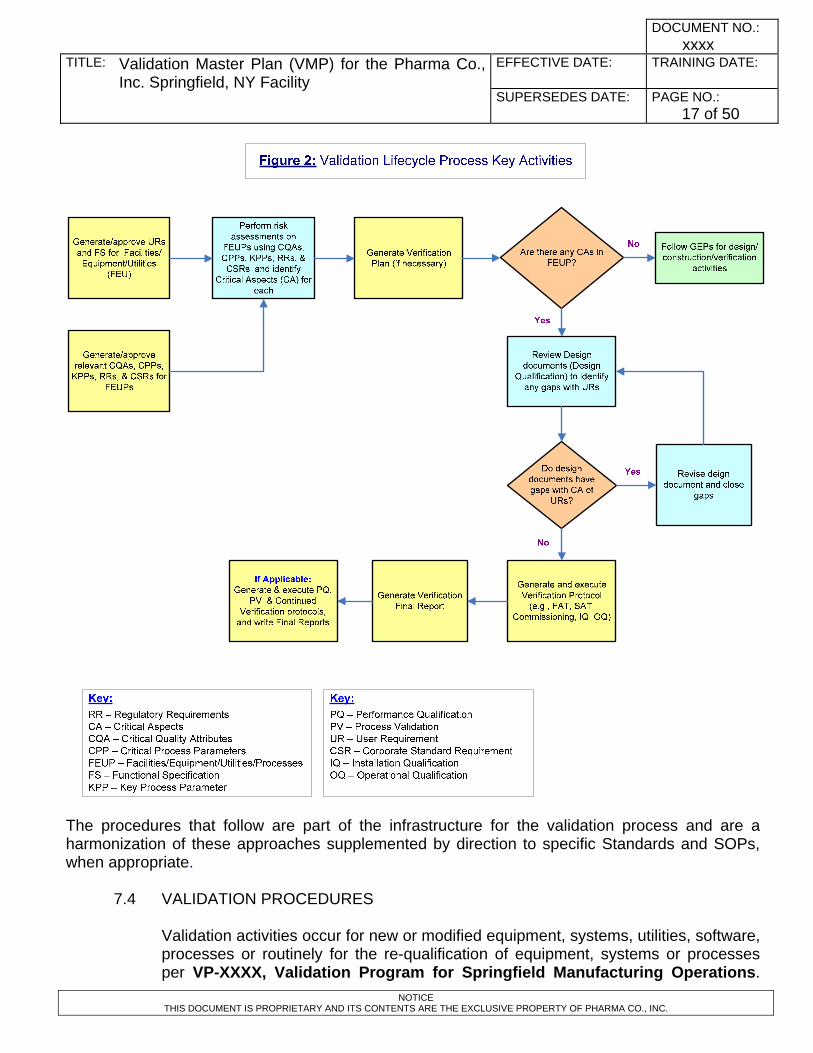
7.3 VALIDATION APPROACH Validation of facilities, equipment, computer systems, and processes is performed at the Springfield, NY Facility for production, monitoring, control, evaluation, and storage of CGMP products. As indicated in the INTRODUCTION section, this site VMP is based on the Validation Life Cycle approaches presented at the site level through governing document SOP-XXXX. SOP-XXXX focuses extensively on the early phases of a project (risk assessment, requirement specification, design, construction, and engineering). The Validation Lifecycle Process Key Activities shown in Figure 2 depicts the use of risk based approach that drives the verification and qualification activities, which are within the scope of this VMP. The Life Cycle Approach may be summarized as follows: Facility Qualification will entail verifying that construction and utility installation is according to design, and that the process requirements are met. Equipment & Instrument Verification will verify and document that installation is according to design (Installation Qualification), meets functional requirements (Operational Qualification), and user requirements (Performance Qualification). All computer and automated control systems employed in the facility will also be validated. Process demonstration, or engineering runs, for new processes/products will be executed utilizing the proposed manufacturing processes. The process demonstration phase will be based on documented process descriptions developed for the product to be transferred to the Springfield, NY Facility site. To support process demonstration, the test instruments will be qualified, and test methods will be validated and / or formally transferred to the site Quality Control / Microbiology Laboratories. During the demonstration phase, raw material supply, equipment settings, process parameters, batch record, product storage, standard operating procedures, and testing will be evaluated to assure satisfactory function. During the demonstration phase, evaluations specific to processing and product quality may also be undertaken. After successful demonstration, confirmatory production performance qualification and continued verification protocols will be executed to verify and document that the manufacturing processes are capable of consistently yielding product that meets predetermined quality attributes when run at prescribed operating settings and within acceptable operating ranges. Stability testing will also be conducted to support product expiration dating intervals. Cleaning validation will be performed for process contact surfaces to assure that the cleaning operation reduces product and cleaning agent to acceptable levels. Subsequent to initial validation activities, the facilities, equipment, systems, and processes are to be maintained in the validated state through implementation of site procedures, including calibration, preventive maintenance, requalification, work

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 16 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
order review, and change control. The validated manufacturing processes will be assessed regularly to assure that it operates within specified critical parameter ranges and the product produced continues to meet critical quality attribute specifications. As necessary validation activities can and will be repeated for facilities, equipment, instruments or processes that have been modified, replaced, or otherwise altered to a state outside of the originally validated operating parameters. The validated state of the critical processes, such as cleaning, sterilization, and storage rooms, will be requalified periodically.
NOTES about the Validation Life Cycle Process: • The activities in this process are executed by a cross functional team, which
include Subject Matter Experts (SMEs) from the impacted stakeholder departments. The responsibilities of SMEs from various functional areas are listed in Section 4.0 of this VMP.
• The order of activities in the Life Cycle Process follows the sequence shown in Figure 2.
• The requirement documents (user, functional, regulatory, etc) are generated by contributions from stakeholder groups.
• Risk assessment is conducted to identify Critical Aspects (CA) of the equipment and processes.
• Relevant documents are generated and executed per the Verification Plan, such as FAT, SAT, commissioning, IQ, OQ, and approved prior to starting the next activity.
• Content of the verification protocols will depend upon the risk assessment as delineated in the Verification Plan.
DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 17 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
The procedures that follow are part of the infrastructure for the validation process and are a harmonization of these approaches supplemented by direction to specific Standards and SOPs, when appropriate.
7.4 VALIDATION PROCEDURES Validation activities occur for new or modified equipment, systems, utilities, software, processes or routinely for the re-qualification of equipment, systems or processes per VP-XXXX, Validation Program for Springfield Manufacturing Operations.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 18 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
Prospective validation is required for all new facilities, equipment, systems, utilities, software, or processes that are necessary for the manufacture, testing, and storage/distribution of commercial product and for clinical material. The steps followed for a validation project are referred to as the Validation Life Cycle. Concurrent validation can be used for PV if the manufactured product must be used for commercial needs in which case, the PV test results must meet all of acceptance criteria defined in the protocol as well as any other QA, corporate, and regulatory requirements. For concurrent validation, the details of the conditions that have to be met prior to the release of the batch for commercial use must be included in the PV protocol. The verification (commissioning, IQ, OQ, etc), PQ, and PV protocols are generated and executed per the SOPs XXXX. The verification / qualification / validation results are summarized in corresponding final reports. The final reports are written in accordance with procedure SOP-XXXX and are reviewed and approved by the same departments that reviewed and approved the initial protocol.
7.5 VALIDATION SUPPORT SYSTEMS
7.5.1 Re-Qualification Program
The Re-Qualification (RQ) process is governed by SOP-XXXX. The purpose of this standard is to provide the mandatory compliance requirements for the routine requalification practices to be utilized for equipment and processes used in the manufacture of commercial and clinical material. The intent of requalification is to ensure critical equipment and processes continue to perform within defined limits and operate in a similar manner as originally validated. This document is intended to provide direction to all applicable Pharma Co., Inc. sites to ensure validation requalification programs comply with current regulations and guidance documents authored by regulatory agencies and industry organizations worldwide. Detailed re-qualification requirements for equipment, systems and processes at Pharma Co., Inc. facilities, which include the Springfield, NY Facility, are governed by SOP-XXXX. This procedure is intended to define the time-based re-qualification program for equipment, systems, and processes at Pharma Co., Inc. facilities. Specifically, the procedure will identify the equipment, systems and processes requiring routine re-qualification, the required frequency and the type of testing and documentation required. In addition, the Steam-in-Place requalification schedule at Pharma Co., Inc.’s Springfield, NY Facility is governed by SOP-XXXX. This requalification plan identifies the equipment / systems group into families requiring Steam-in-Place

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 19 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
sterilization procedures as part of a validated process. This requalification plan also specifies and justifies the requalification frequency of the family group and the family group members. The validated family groups then require requalification according to SOP-XXXX. The re-qualification program assures that critical equipment, systems, and processes continue to perform within prescribed limits and are operating in a similar manner as originally validated, thereby demonstrating a continued state of control. The re-qualification approach is to perform similar testing utilized during the initial validation, however at an attenuated level deemed sufficient to evaluate the performance of the equipment, system or process, and detect potential adverse changes.
7.5.2 Metrology Program
Calibration policies and procedures are governed by SOP-XXXX, Metrology Program. The Metrology Program covers the services provided by the Pharma Co., Inc. Metrology Department to Pharma Co., Inc. Corporation. This includes, but is not limited to, all Manufacturing and Quality Control operations at Pharma Co., Inc.’s Springfield, NY Facility. Services by the Pharma Co., Inc. Metrology Department may also be provided to Pharma Co., Inc.’s subsidiaries and to joint projects involving other companies having partnerships with Pharma Co., Inc. and its subsidiaries. In order for equipment to be included in this program, it must be used to measure, gauge, test, inspect or otherwise examine a quantitative value to a known degree of accuracy. If such equipment is used, it must be identified as a Critical, Non-Critical, or Reference Use Only instrument. Instrumentation requiring calibration must be current prior to and remain current during protocol execution and throughout the Validation Program.
7.5.3 Change Control Program
The Corporate Quality Operations Standard for Change Control is documented as SOP-XXXX. The Change Control Program to ensure that equipment within the Springfield, NY Facility maintains its validated state is governed by SOP-XXXX. These program requirements apply to all Pharma Co., Inc. functional groups involved in the manufacture and testing of products (both commercial and clinical including material/ assays used to demonstrate product comparability) at the Springfield, NY Facility.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 20 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
New equipment, both within Quality Control and Manufacturing intended to be validated, is covered under this Change Control System.
7.5.4 Maintenance Program
The process used for developing and optimizing an effective asset maintenance program is governed by SOP-XXXX, Springfield, NY Facility Maintenance Management Program / Work Order System Procedure. The procedure applies to new and in-service assets based on risks and operational reliability while assuring employee safety. The process calls for utilizing operational and maintenance experience relative to 21 CFR Part 210/211 and quality management systems that must be compliant with CGMP regulations. The process to determine / evaluate Part 11 compliance and security risks is included. The maintenance program targets critical production equipment, QC Laboratory equipment and test instrumentation, plant utilities and environmental areas with significant risk to product quality. Process steps for developing / optimizing the maintenance program are described in the program. Additionally, instructions for work order management and integration with the Computerized Maintenance Management System (CMMS) are also described.
7.5.5 Training Program
The general definitions, responsibilities, methodologies for assignments, assessments, documentation of training, and integration with the LMS is governed by SOP-XXXX Learning Management System. This document outlines the activities required to document training in compliance with CGMPs and other applicable regulatory requirements and ensures personnel performing work affecting product quality will be competent on the basis of appropriate education, training, and experience. This document applies to the recording of training for full-time, part-time and non-employees (temporary/contractors) and either concurrent or subsequent entry of records into the LMS database, where applicable. If a Business Unit has its own local training procedures, that procedure may take precedence. Training is further defined in SOP-XXXX, to establish a set of initial, minimal regulatory training requirements for personnel having the ability to impact medical device or therapeutic product quality. This document applies to all full-time, part-time, and temporary employees at all levels whose tasks affect the design, production, testing, handling and distribution of products, or affect the environment, process or systems in which products or data are produced, controlled or managed.

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 21 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
7.6 KEY SOPs GOVERNING AND SUPPORTING THE ALLSTON LANDING VALIDATION PROGRAM
See Attachment B for a full list of key SOPs and standards used to support the Springfield, NY Facility Validation Program. The site SOPs are written, revised, and controlled per SOP-XXXX, Documentation System and Control.
7.7 IDENTIFICATION AND DESCRIPTION OF MANUFACTURING SYSTEMS AND EQUIPMENT PRESENTLY IN USE AT THE SPRINGFIELD, NY FACILITY AND ASSOCIATED COMPLETED VALIDATION
See Attachment C for a Process Equipment List and Associated Validation Protocol Numbers.
7.8 VALIDATION MASTER PLAN REVIEW
This document must be reviewed, updated and approved on an annual basis to reflect current philosophy, standards and Good Manufacturing Practices. A “VMP Review Schedule” must be generated for the time of the year, when the Site VMP and sub-VMPs will be reviewed and a VMP Review Summary Report generated.
8.0 ATTACHMENTS
8.1 Attachment A: Process Flow Diagrams 8.2 Attachment B: Key SOPs Governing and Supporting the Springfield, NY Facility
Validation Program 8.3 Attachment C: Process Equipment List and Associated Validation Protocol Numbers

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 22 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
8.1 Attachment A: Product Process Flow Diagrams

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 23 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
8.2 Attachment B: Key Documents Governing and Supporting the Springfield, NY Facility Validation Program
Reference Documents
• Food and Drug Administration (FDA) – 21 CFR Parts 11, 210 & 211
• European Medicines Agency (EMA) – EudraLex Vol 4
• International Committee on Harmonization (ICH) – Q7, Q8, Q9 & Q10
• International Standards Organization (ISO) - ISO 9001
• FDA Guidance for Industry – Quality Systems Approach to Pharmaceutical cGMP Regulations
• FDA Guidance for Industry – Process Validation: General Principles and Practices – January 2011
• ASTM International – Designation E2500-07, Standard Guide for the Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment
Corporate Standards:
• SOP-XXXX: Change Control
• SOP-XXXX: Quality Manual for the Pharma Co., Inc. Quality System
• SOP-XXXX: Corporate Quality Operations Standard: Risk Management
• SOP-XXXX: Corporate Quality Operations Standard for Equipment and Process Requalification
• SOP-XXXX: Method Development and Validation
• SOP-XXXX: Validation Program
• SOP-XXXX: Laboratory Equipment Qualification
• SOP-XXXX: Cleaning Validation
• SOP-XXXX: Process Validation
• SOP-XXXX: Sterilization and Depyrogenation Validation

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 24 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
General and Administrative SOPs:
• SOP-XXXX: Documentation System and Control
• SOP-XXXX: Generation of Protocols and Final Reports for Validation
• SOP-XXXX: Change Control Procedure
• SOP-XXXX: Metrology Program
• SOP-XXXX: Records Retention
• SOP-XXXX: Springfield, NY Facility Maintenance Management Program / Work Order System Procedure
• SOP-XXXX: Technology Transfer into TMD Manufacturing and Quality
• SOP-XXXX: Guidance for 21 CFR Part 11 Compliance for TMD Computer Systems
• SOP-XXXX: Pharma Co., Inc. Corporate Quality Manual Note: This manual also lists pertinent SOPs for validation
related topics
• SOP-XXXX: Documentation of Employee Training
• SOP-XXXX: Installation Qualification of Laboratory Instruments
• SOP-XXXX: Learning Management System (LMS)– Governance and Documentation
• SOP-XXXX: cGMP & Regulatory Training Requirements for Personnel Engineering SOPs:
• SOP-XXXX: Technical Project Management Process for Springfield, NY Facility Manufacturing Facility
• SOP-XXXX: Project Lifecycle Management Process - Springfield, NY Facility
• SOP-XXXX: Commissioning Procedure for Start-Up of Systems and Equipment at Springfield, NY Facility
• SOP-XXXX: Engineering Document Management Procedure at the Springfield, NY Facility
• SOP-XXXX: Administration of Drawings at Pharma Co., Inc. Springfield, NY Facility
• SOP-XXXX: Project Change Management Procedure at the Springfield, NY Facility

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 25 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
Technical SOPs: • SOP-XXXX: Validation of Autoclaves
• SOP-XXXX: Validation of Dry Heat Ovens used in Sterilization or Depyrogenation Processes
• SOP-XXXX: Validation of Filter Integrity Test Instruments
• SOP-XXXX: Validation of Temperature/Relative Humidity Controlled Warm Rooms/Chambers
Key Validation Department Operational SOPs:
• SOP-XXXX: Master Validation Plan for the Pharma Co., Inc. Laboratory Information Management System
• SOP-XXXX: Standard Operating Procedure Guidelines for Determination of Validation Requirements for Change Control Requests
• SOP-XXXX: Guidelines for Computer Validation Protocols
• SOP-XXXX: Guidelines for Generating, Approving, Amending and Closing-out a Validation Plan at Pharma Co., Inc.
• SOP-XXXX: Guidelines for the Performance of Airflow Pattern Testing for Clean Rooms and Laminar Flow Hoods
• SOP-XXXX: Protocol Execution Procedure at Springfield, NY Facility
• SOP-XXXX: Routine Re-Qualification of Equipment, Systems and Processes
• SOP-XXXX: Cleaning Validation and Changeover Verification
• SOP-XXXX: Process Validation for Springfield, NY Facility
• SOP-XXXX: Process Equipment Steam-in-Place (SIP) Requalification Matrices and Scheduling Plan at Springfield, NY Facility
• SOP-XXXX: Cleaning Master Plan for the Springfield, NY Facility Manufacturing Facility
• SOP-XXXX: Validation Project Execution Life Cycle Approach at the Springfield, NY Facility

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 26 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
8.3 Attachment C: Process Equipment List and Associated Validation Protocol Numbers
EQ # Description Size Location IQ OQ CIP SIP CQ PQ/PV

DOCUMENT NO.: xxxx
TITLE: Validation Master Plan (VMP) for the Pharma Co., Inc. Springfield, NY Facility
EFFECTIVE DATE:
TRAINING DATE:
SUPERSEDES DATE:
PAGE NO.: 27 of 50
NOTICE THIS DOCUMENT IS PROPRIETARY AND ITS CONTENTS ARE THE EXCLUSIVE PROPERTY OF PHARMA CO., INC.
9.0 REVISION HISTORY
9.1 Revision History Table Revision Description of Changes DCR
Number Related
document Date
1 Initial Release xxxx xxxx mm/dd/yyyy
![Understand Critical Components of a Validation Master · PDF fileCONFIDENTIAL [ 1 ] Understand Critical Components of a Validation Master Plan Jorge A. Cordero-Monroig; BSChE, MBA-GM](https://img.pdfslide.us/doc/110x75/5a9df47b7f8b9a4a238cb041/understand-critical-components-of-a-validation-master-1-understand-critical.jpg)




























