Embed Size (px)
Citation preview

PART-II

Contents
1.Benign and malignant tumours
2. Salivary gland lesions
3. fibro-osseous lesions
4. Infections in oral cavity

ORAL LICHEN PLANUS MALIGNANT POTENTIAL ?ORAL LICHEN PLANUS MALIGNANT POTENTIAL ?
• Frequency of malignant transformation of Oral Lichen Planus
varies between 0.3% to 3%.
• The forms that commonly undergo malignant transformation are
the erosive and atrophic formserosive and atrophic forms.

• The risk of malignant transformation of OLP to eidermoid carcinoma was higher, mostly in women & those infected with hepatitis C hepatitis C virus.virus.
• It is due to chronic inflammation in OLP which generates a cytokine-based microenvironment that affects cell survival, growth, proliferation & differentiation;
This may contribute to cancer initiation, promotion & progression.• Transformation is favoured by an altered expression of apoptosis-
regulating proteins, such as the p53 protein.
skin showing weak p53

• PCNA & p53 PCNA & p53 expression are used to assesses malignant transformation
• The IHC expression of bax, caspase-3 & p21 proteins in oral mucosa
• Anti apoptotic mechanisms & cell cycle interruption mechanisms could cause malignant transformation.

NEUROFIBROMANEUROFIBROMA
Most common peripheral nerve neoplasm Schwann cells and perineural fibroblasts. Localised, diffuse or plexiform. Diffuse and plexiform – NF1 Skin, oral mucosa – buccal mucosa and tongue.
NEUROFIBROMATOSIS TYPE 1NEUROFIBROMATOSIS TYPE 1
• Von Recklinghausen’s disease of the skin.
• Autosomal dominant trait.
• NF1 gene, Chr-17q11.2
• Tumour suppressor protein- neurofibromin

Neurofibromin belongs to a family of proteins that serve as
negative regulators of the ras oncogene
• The new research identified specific location of gene
• Hypothesis is that most patients with NF1 are born with one
defective allele for the gene in every cell. when a postnatal
somatic mutation arises in the second allele in a cell derived
from neural crest, a neurofibroma begins growing. This
hypothesis can now be tested, prenatal diagnostic tests can be
developed, and the molecular basis for NF1 can be pursued.
Discovery of genetic defect causing neurofibromatosis

• Translocation breakpoint region (TBR) gene, recently discovered at the neurofibromatosis type 1 locus and found to be interrupted by deletions and a t(17;22) translocation, have been sequenced.
• Sequencing revealed that one mutant allele contains a T----C transition changing a leucine to a proline; another NF1 allele harbors a C----T transition changing an arginine to a stop codon. These results establish the TBR gene as the NF1 gene and provide a description of a major segment of the gene.

Recent Concepts in Salivary Gland LesionsRecent Concepts in Salivary Gland Lesions

Salivary gland tumours are morphologically heterogenous group of
tumours with greatest diversity
Histogenic concepts Histogenic concepts
Various theories to explain the histogenic concepts
• Basal reserve cell theory
• Pluripotent unicellular reserve cell theory,
• Semipluripotent bicellular reserve cell theory,
• Multicellular concepts
• .Sreeja et al. Taxonomy of Salivary Gland Neoplasm Journal Clinical and Diagnostic Research, 2014; volume8 (3): 291-293.

Basal reserve cell theory:
Basal cells of both excretory and intercalated duct responsible for
differentiation of the functional units and thus capable of forming
tumor.
Pluripotent unicellular reserve cell theory:
It states that “Basal cells of excretory duct responsible for
development of all remaining salivary gland units”
C.Sreeja et al. Taxonomy of Salivary Gland Neoplasm Journal Clinical and Diagnostic Research, 2014; volume8 (3): 291-293.
Pluripotent unicellular reserve cell theory:

Semi pluripotent bi-cellular reserve cell theory:Semi pluripotent bi-cellular reserve cell theory:
• Most commonly accepted theory introduced by Eversole in 1971
• According to this theory, only basal cells of the excretory duct and the
progenitor cells of the intercalated duct are capable of cell division, while
the acinar unit and striated duct are considered to be terminally
differentiated cell and therefore have a minimal capacity of cell division.
Multi cellular Theory:
• Dardick et al. suggested that all types of cells in the normal salivary
gland are capable of cell division. According to this theory,
differentiated cells at all levels of the gland, including the acinar cells and
striated duct can give rise to a neoplasm

A. Benign epithelial tumorsA. Benign epithelial tumors• Pleomorphic adenoma• Myoepithelioma• Basal cell adenoma• Oncocytoma • Canalicular adenoma• Warthins tumors • Cystadenoma • Papillary cyst adenoma• Mucinous cyst adenoma• Benign sebaceous neoplasm• Sebaceous adenoma• Sebaceous lymphadenoma • Ductal papilloma• Intraductal • Inverted ductal • Sialadenoma papilliferum
B. Malignant epithelial tumorsB. Malignant epithelial tumorsMucoepidermoid carcinomaAcinic cell carcinomaAdenoid cystic carcinomaPolymorphous low grade adenocarcinoma Epithelial myoepithelial carcinomaClear cell carcinomaBasal cell adenocarcinoma Oncocytic carcinomaMyoepithelial carcinomaAdenocarcinoma NOSCarcinoma ex pleomorphic adenomaMetastatising pleomorphic adenomaCarcinosarcoma Salivary duct carcinomaCyst adenocarcinoma Low grade cribriform cystadenocarcinoma Sialoblastoma Malignant sebaceous tumor
2005 WHO classification of Salivary gland tumours 2005 WHO classification of Salivary gland tumours
Epithelial tumoursEpithelial tumours

II. Mesenchymal tumors Benign • Haemangioma • Haemangiopericytoma • Lipoma • Neurofibroma • Schwanomma Malignant• Fibrosarcoma• Malignant fibrous histiocytoma• Liposarcoma• Secondary tumours
III. Malignant Lymphoma IV. Metastatic tumor
C. Sreeja, Tanveer Shahela TAXONOMY OF SALIVARY GLAND NEOPLASM JCDR/2014

WARTHINS TUMOUR PATHOGENESISWARTHINS TUMOUR PATHOGENESIS

• Up to now no agreement has been reached regarding the pathogenesis of this disease.
• The theory is that tumour arises due to entrapment of salivary gland tissue (heterotropic) in paraparotid or intraparotid lymph nodes during the embroyogenesis during the development from
• The aberrant salivary gland tissues in the lymph node of parotid glands are the basis for the tumourogenesis and probably smoking may act as a promoter
• This theory, however, does not explain when aberrant salivary gland tissue develops in the embryo/infants, why Warthin’s tumour does not occur until middle/old age .

A chromosomal translocation with the generation of a novel
fusion- oncogene is an important feature Current information from molecular studies suggests that a
recurrent t(11;19) and associated CRTC1-MAML2 fusion oncogene and supports a clonal origin.
• Initial step was mitochondrial dysfunction within salivary duct cells resulting in oncocytic change. This tissue may either proliferate in a reactive polyclonal fashion, or following a t(11;19) and CRTC1-MAML2 generation, undergo a truly neoplastic clonal expansion.
t(11;19) and the CRTC1- MAML 2 fusion oncogene• Translocation results in the fusion of exon 1 of the CRTC1 gene on
chromosome 19p13 with exons 2–5 of the MAML2 gene on chromosome 11q21 to generate a novel CRTC1-MAML2 fusion oncogene
• Such cells undergo further, as of yet undefined genetic alterations which could predispose to malignant transformation
Iain David O’Neill J Oral Pathol Med (2009) 38: 145–149

Sjogrens syndrome (sicca syndrome)
• Primary sjogrens syndrome characterised by persistent dryness of mouth and eyes due to functional impairment of lacrimal gland and salivary glands with absence of systemic autoimmune disease.
• Secondary sjogrens syndrome: Secondary sjogrens syndrome: associated with connective tissue disorder along with dry eyes and dry mouth.
Etiology:• Genetic , hormonal, infectious, and immunological. Histological featuresHistological features:• Intense lyphocytic infiltration of gland replacing acinar
structures • Proliferation of ductal epithelium and myoepitheliumto
form epimyoepithelial islands• Atrophy of glands.

Revised Classification criteria

Mammary analoge of secretory carcinomaMammary analoge of secretory carcinoma
Is a recently described entity the characteristic t(12;15)
(p13;q25) with ETV6-NTRK3 translocation found in
secretory carcinomas of the breast
Clinical features:Clinical features: Occurs in parotid gland Mean age is 47 yrs, often males are affected than females.
Andre Pinto; Vania Nosé Mammary Analog Secretory Carcinoma of Salivary Gland Among Its MimicsMod Pathol. 2014;27(1):30-37.

HISTOPATHOLOGIC FEATURESHISTOPATHOLOGIC FEATURES
• Similar to secretory carcinoma of breast• Tumour cells exhibit bland vesicular nuclei surrounded by slightly
granuar or vaculoted cytoplasm • Arranged in solid , tubular , microcystic or macrocystic structures.• Large cystic spaces may exhibit papillary infolding of tumour cells
with hobnail appearance.• Immunoreactivity for s- 100 protein, vimentin, mammoglobin.• Chromosomal translocation – fluorescent in situ hybridization using the
ETV6 break apart probe

Current Concepts in Fibro-osseous LesionsCurrent Concepts in Fibro-osseous Lesions

• The term fibroosseous lesion refers to a diverse process in which normal mature bone is replaced firstly by a variably cellular fibroblastic stroma within which pathological ossification and/or calcification then occurs.
• Diseases with quite variable etiologies and pathogenesis but share similar and overlapping histological features that make precise categorization difficult.
• Updated classification schemes for Fibro-osseous lesions of the oral and maxillofacial region : a review IOSR-JDMS vol 13,issue 2 ,2014

EVERSOLE 2008 : Classification of benign fibro-osseous EVERSOLE 2008 : Classification of benign fibro-osseous lesions of the craniofacial complex lesions of the craniofacial complex (working classification)
I. Bone dysplasias a. Fibrous dysplasia i. Monostotic ii. Polyostotic iii. Polyostotic with endocrinopathy (McCune-Albright) iv Osteofibrous dysplasia b. Osteitis deformans c. Pagetoid heritable bone dysplasias of childhood d. Segmental odontomaxillary dysplasiaUpdated classification schemes for Fibro-osseous lesions of the oral and maxillofacial region : a
review IOSR-JDMS vol 13,issue 2 ,2014

II. Cemento-osseous dysplasiasa. Focal cemento-osseous dysplasiab. Florid cemento-osseous dysplasia
III. Inflammatory/reactive processesa. Focal sclerosing osteomyelitisb. Diffuse sclerosing osteomyelitisc. Proliferative periostitis
IV. Metabolic Disease:IV. Metabolic Disease: hyperparathyroidism
V. Neoplastic lesions (Ossifying fibromas) a. Ossifying fibroma NOS b. Hyperparathyroidism jaw lesion syndrome c. Juvenile ossifying fibroma i. Trabecular type ii. Psammomatoid type

Before 6th week of development
At 6th week of development After 6th week of development

G protein coupled receptors
• G protein- coupled receptors link the binding of an extracellular Ligand, such as a growth factor or hormone, to the activation of the associated G protein and intracellular signal generation.
Fibrous dysplasia
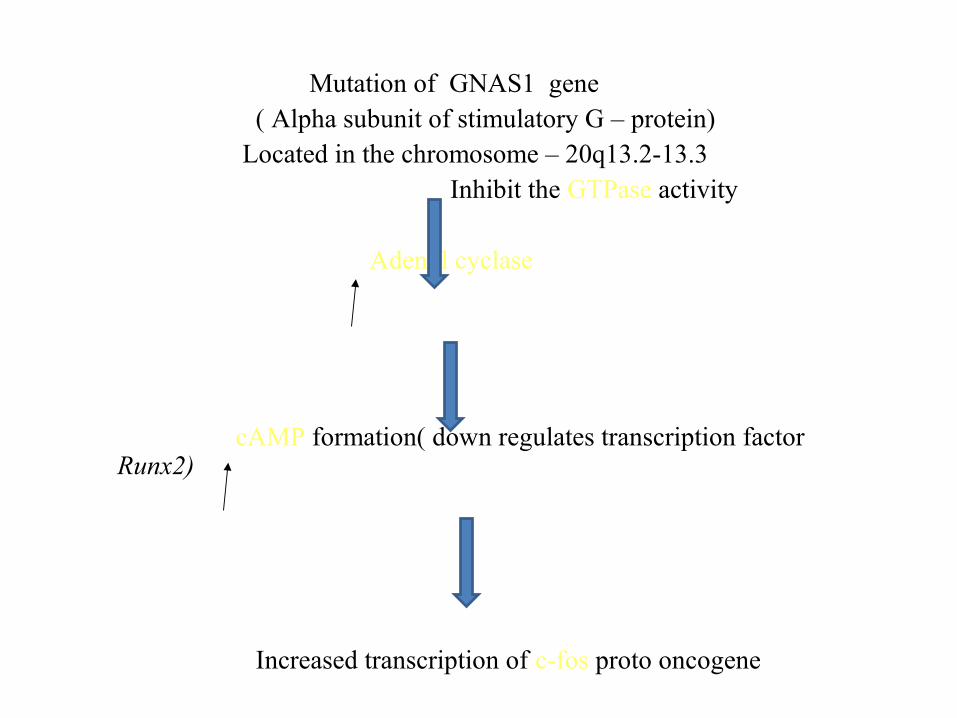
Mutation of GNAS1 gene ( Alpha subunit of stimulatory G – protein) Located in the chromosome – 20q13.2-13.3 Inhibit the GTPase activity Adenyl cyclase
cAMP formation( down regulates transcription factor Runx2)
Increased transcription of c-fos proto oncogene
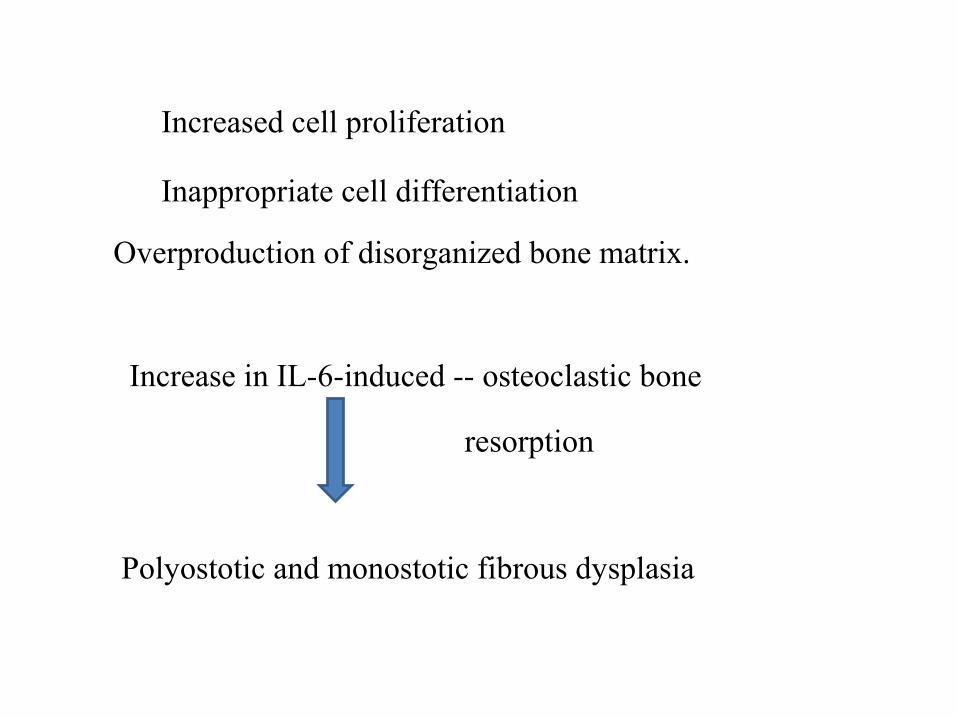
Increased cell proliferation
Inappropriate cell differentiation
Overproduction of disorganized bone matrix.
Increase in IL-6-induced -- osteoclastic bone
resorption
Polyostotic and monostotic fibrous dysplasia

OSSIFYING FIBROMA (cemento ossifying fibroma, OSSIFYING FIBROMA (cemento ossifying fibroma, cementifying fibroma )cementifying fibroma )
A well demarcated , encapsulated, expansile intraosseous lesion of the jaw.
Same progenitor cell produces different materials.
Pathogenesis :Pathogenesis : Fibro-osseous neoplasm arising from ectopic multipotent periodontal ligament cells
• Histiogenesis include a reparative reaction to trauma, abnormal reparative process, a neoplastic process, a developmental defect.

Genetics Mutations in HRPT2 gene mapped to the chromosomal
region 1q24-q32 encodes for parafibromin protein . It is a 531 amino acid putative tumour suppressor
protein. Mutation HRPT2 ( HYPERPARATHYROIDISM
JAW TUMOUR SYNDROME)
PARAFIBROMIN is associated with Wnt signaling pathway by interaction with beta catenin, in the absence of b catenin , progenitor cells differentiate into fibroblasts instead of osteoblasts.

HISTOPATHOLGY
• Ossifying fibroma shows three histologic patterns and some demonstrate a mixture of these patterns.
• Ossifying fibroma
• Cementifying fibroma
• Storiform
• The common ‘‘ossifying’’ form shows, with small irregular osteoid trabeculae that are typically rimmed by osteoblasts. The stromal element is hypercellular and the fibroblastic cells are devoid of atypical cytologic features.
Roy Eversole , Lan Su Benign et al Fibro-Osseous Lesions of the Craniofacial Complex. A Review

• Storiform form type of ossifying fibroma Storiform form type of ossifying fibroma (NOS) is typified by streaming of the fibroblastic stromal elements in a pinwheel configuration similar to benign fibrous histiocytoma.
• Calcifications that appear like dystrophic bone
Cementifying types also contain more typical osseous trabeculae in addition to the cemental structures which are ovoid or droplet in shape.

Juvenile ossifying fibroma, juvenile aggressive ossifying fibroma
It is a controversial lesion . Juvenile ossifying fibroma (JOF) is a fibro-osseous neoplasm
described as an actively growing lesion consisting of a cell-rich fibrous stroma, containing bands of cellular osteoid without osteoblastic lining, together with trabeculae of more typical woven bone. Small foci of giant cells may also be present. The lesion is non-encapsulated but well demarcated from surrounding bone (WHO).
Resemblance between ossicles and the cementum spheres of the odontogenic ossifying fibroma,
The lesion has occasionally been mislabeled as cemento-ossifying fibroma, implying an odontogenic origin which, as mentioned above, is rather unlikely in extra- gnathic bone

Trabecular Juvenile Ossifying FibromaTrabecular Juvenile Ossifying Fibroma• Microscopically, TrJOF is unencapsulated and shows
infiltration of the surrounding bone.
• The stroma is cell-rich, with spindle or polyhedral cells that produce little collagen .
• Cellular, immature osteoid strands that may be long and slender or plump . These structures have been likened to paint brush strokes.

• Irregular mineralization takes place at the center of the strands. Maturation to lamellar bone is not observed. Local aggregates of osteoclastic giant cells are invariably present in the stroma
• Mitotic activity of the stromal cells may be present but is never numerous.
• Cystic degeneration and aneurysmal bone cyst formation has been reported in a few cases

Psammomatoid Juvenile Ossifying FibromaPsammomatoid Juvenile Ossifying Fibroma• Affects predominantly the extragnathic craniofacial bones,
particularly centered on the periorbital, frontal, and ethmoid bones.
• Multiple round uniform small ossicles (psammomatoid bodies) embedded in a relatively Cellular stroma composed of uniform, stellate, and spindle shaped cells .
• The psammomatoid bodies are basophilic and bear superficial resemblance to dental cementum, but may have an osteoid rim.
• Mitotic activity is extremely rare. Cystic degeneration and aneurysmal bone cyst formation has been

• Confusion between TrJOF and PsJOF is mainly due to the use of the term ‘‘Juvenile ossifying fibroma’’ to describe these two entities, in spite of clear differences in microscopic appearance. The former is Trabecular, the later is psammomatoid. In addition PsJOF have a definite site predilection for periobital bones while TrJOF favors the gnathic bones. TrJOF also has a younger average age of incidence.
• Surgical excision is the treatment of choice, although recurrence even after definitive surgery is not unusual. Recurrence rates of have been reported

Histocytosis X
Proliferative disorder of histiocyte like cells that are accompanied by
lymphocytes, plasma cells and multinucleated giant cells
Histiocytes are langerhans cells found in the epidermis, lymphnodes, and
bone marrow.
Controvesy either as non neoplastic or true neoplasm.
Recently recognised monoclonal proliferation – neoplastic process.
BRAF mutations have been shown .

Eosinophilic granuloma(langerhans cell histiocytosis)Eosinophilic granuloma(langerhans cell histiocytosis)
• Histiocytic proliferation with abundance of eosinophilic
leukocytes with no intercellular accumulation of lipids
involving single or multiple bones.
C/F:- Older children & young adults with male predilection.
• Asymptomatic , local pain , swelling & tenderness, fever,
malaise.
• Skull, mandible, along with overlying soft tissue.
• Destructive , replaced by soft tissue, well demarcated.
• Soft brown, later fibrous & grayish.

Histologic features
• Histocytic proliferation in the form of sheets.
• Multinucleated gaint cells.
• Initial lesions- large focal collections of histiocytes.
• Mature lesion- Fibrosis & eosinophils are less or completely
absent with only histocytes.

Hand- Schuller christian diseaseHand- Schuller christian disease
• C/F:- Early in life with in 5yrs.
• Triad:- Single or multiple areas of punched out destructive lesions of
skull.
Bilateral exophthalmos.
Diabetes insipidus.
• Facial asymmetry-facial bones with soft tissue swellings
• Otitis media
• Skin lesions – Nodular or papular

Oral manifestations
• Soar mouth with or without ulcerations
• Halitosis, gingivitis, suppuration.
• Loose or sore teeth, precocious exfoliation
• Failure of healing extraction wounds.
R/F:- R/F:- Punched out sharply outlined lesions of skull.
• Lesions of jaws are more diffuse with displacement of teeth

Letterer Siwe disease
• Severe form of Histocytosis X seen in the age below 3yrs.
• A rare usually fatal condition characterized by skin lesions,
bleeding tendency, hepatospleenomegaly, enlarged lymph
nodes and progressive anemia.
• The condition is caused by excessive proliferation of
histiocytes.
• Persistent fever
• Cutaneous maculopapular lesions.
Prognosis:- Poor

Fungal Infections

CHRONIC HYPERPLASTIC CANDIDIASISCHRONIC HYPERPLASTIC CANDIDIASIS
White patch that cannot be removed by scrapping
Represents candidiasis that is superimposed on prexisting leukoplakia.
Sometimes candidal organism alone induces hyperkeratotic lesion

Leukoplakic lesion associated with candidal infection has fine intermingling of red and white areas resulting in speckled leukoplakia
If the lesion resolves by antifungal therapy then it is chronic hyperplastic candidiasis

Immunological Disease Immunological Disease
SARCOIDOSISSARCOIDOSIS
INFLAMMATORY DISEASE OF UNKNOWN ETIOLOGY
Multisystem granulomatoses
cause unknown – improper degradation of antigenic material with
the formation of non caseating granulomatous inflammation.
Antigens include infectious agents
(mycobacterium,propionibacteria, EBV ,HHV-8, Environmental
factorslike wood dust
Genetic predisposition and association with HLA types.
most often in 3rd-5th decade

Histopathology • Granulomatous inflammation• Aggregates of epitheloid histiocytes with rim of lymphocytes.• Intermxied with the histiocytes are scattered langerhans cells or foreign
body type giant cells.• Contain laminated basophilic calcifications,known as schaumann bodies(
degenerations lysosomoes or stellate inclusion sknown as asteroid bodies ( entrapped fragments off collagen
• In lymph nodes HAMAZAKI –WESENBERG BODIES ( large lysosomes)

HUMAN PAPILLOMA VIRUS • The human papilloma virus (HPV) is one of the most common
virus groups in the world to affect the skin and mucosal areas of the body.
• Studies have shown HPVs association with benign, premalignant and malignant lesions.
• Human papillomavirus (HPV) infection with high-risk types 16 and 18 has widely been reported as one of the prominent mechanisms behind the development of cervical squamous cell carcinoma

• Oncogenic potential of HPV is related to products of 2 viral genes E6 & E7.
High risk HPV types express oncogenic proteins that :-
• Inactivate tumour suppresors• Activate cyclins• Inhibit apoptosis• Combat cellular senescence

THANK YOUTHANK YOU
ALL!!ALL!!





























