Embed Size (px)
Citation preview

Sample to Insight
PCR – From Setup to CleanupA beginner’s guide with useful tips and tricks!

2 PCR – From Setup to Cleanup 10/2015
PCR
The invention of the polymerase chain reaction (PCR) by K. Mullis and co-workers in 1985 revolutionized molecular biology
and molecular medicine. Major research areas, such as biomarker discovery, gene regulation and cancer research, are
challenging today’s PCR technologies with more demanding requirements. These include the need for increased throughput,
higher assay sensitivity and reliable data analysis. Assay development and evaluation, reproducibility of data and time to
result are still major problems encountered by researchers.
Guidelines for PCR
PCR amplification is performed routinely and thousands of PCR protocols have been developed,
yet researchers still encounter technical difficulties with PCR experiments and often fail to obtain
specific amplification products. Although there are several different challenges (e.g., smearing, low
yield and nonspecific amplification), there are two main reasons for PCR failure or poor results: the
specificity of the reaction and template secondary structure.
PCR is both a thermodynamic and an enzymatic process. Successful PCR requires amplification
and detection under optimal conditions and each reaction component can affect the result. The
annealing step is critical for high PCR specificity. When primers anneal to the template with high
specificity, this leads to high yields of specific PCR products and increases the sensitivity of the
amplification reaction. However, due to the high primer concentration in the reaction, primers
will also hybridize to non-complementary sequences with mismatches. If the primers anneal
to the template sequence with low specificity, amplification of nonspecific PCR products and
primer–dimers may occur. Factors critical in successful PCR include primer design and the reaction
chemistry used.
PCR primer design
Optimal primer sequences and appropriate primer concentrations are essential for maximal
specificity and efficiency in PCR. The PCR section of the QIAGEN Protocols and Applications
Guide provides more information about primer design.
The following points should be considered when designing PCR primers and are common to all
types of PCR:
• The melting temperature (Tm) can be calculated according to the formula:
2°C x (A+T) + 4°C x (G+C)
• Avoid complementarity in the 2–3 bases at the 3’ end of the primer pairs

PCR – From Setup to Cleanup 10/2015 3
• Avoid mismatches between the 3’ end of the primer and the template
• Avoid runs of 3 or more Cs or Gs at the 3’ end of the primer
• Avoid complementarity within primers and between the primer pair
• Avoid a T as ultimate base at the 3’ end
• Ensure primer sequence is unique for the template sequence
• Use a concentration of 0.1–1.0 μM of each primer. For many applications, a primer
concentration of 0.2 μM is sufficient
Lyophilized primers should be dissolved in a small volume of distilled water or TE to make a
concentrated stock solution. Prepare small aliquots of working solutions containing 10 pmol/μl to
avoid repeated thawing and freezing. Store all primer solutions at –20°C. Primer quality can be
checked on a denaturing polyacrylamide gel; a single band should be seen.
PCR conditions
The primer and Mg2+ concentration in the PCR buffer and annealing temperature of the reaction
may need to be optimized for each primer pair for efficient PCR. In addition, PCR efficiency can
be improved by additives that promote DNA polymerase stability and processivity or increase
hybridization stringency, and by using strategies that reduce nonspecific primer–template
interactions. Use of high-purity reagents is also essential for successful PCR, especially for
amplification of rare templates, for example, single copy genes in genomic DNA or pathogenic
viral DNA sequences in genomic DNA isolated from an infected organism.
Inclusion of control reactions is essential for monitoring the success of PCR reactions. Wherever
possible, a positive control known to contain the target sequence should be included to check
that the PCR conditions used can successfully amplify the target sequence. As PCR is extremely
sensitive, requiring only a few copies of target template, a negative control containing no template
DNA should always be included to ensure that the solutions used for PCR have not become
contaminated with the template DNA.
Primer annealing specificity and PCR buffers
In PCR, annealing occurs between the primers and complementary DNA sequences in the template.
Primer annealing must be specific for successful amplification. Due to the high concentration
of primers necessary for efficient hybridization during short annealing times, primers can
anneal to non-complementary sequences. Amplification of products from nonspecific annealing
competes with specific amplification and may drastically reduce the yield of the specific product.
Tip: PCR setup should be performed in a separate area from PCR analysis to ensure that reagents used for PCR do not become contaminated with PCR products. Similarly, pipets used for analysis of PCR products should never be used for setting up PCR.

4 PCR – From Setup to Cleanup 10/2015
The success of PCR largely depends on maintaining a high ratio of
specific to nonspecific annealing of the primer molecules. Annealing is
primarily influenced by the components of the PCR buffer (in particular
the cations) and annealing temperature. Special cation combinations
can maintain high primer annealing specificity over a broad range
of annealing temperatures. This eliminates the need for optimization
of annealing temperatures for each individual primer–template system
and also allows the use of non-ideal PCR assays with different primer
annealing temperatures.
A balanced combination of cations promotes specific primer annealing
Cations in commonly used PCR buffers bind to the negatively charged
phosphate groups on the DNA backbone and thereby neutralize these
negative charges. This weakens the electrorepulsive forces between the
DNA template and primer molecule leading to more stable hybridization
of the primer. Most commercially available PCR buffers contain only one
monovalent cation, K+, which stabilizes both specific and nonspecific
primer annealing. This often results in smearing and nonspecific DNA
amplification, which leads to lower product yields. QIAGEN® has found
that the balanced combination of K+ and NH4+ used in all QIAGEN
PCR buffers provided with all QIAGEN PCR enzyme and master mix kits
strongly increases primer annealing specificity. The improved specificity
is caused by ammonium ions destabilizing the weak hydrogen bonds at
mismatched bases (Figures 1 and 2).
Discover more about optimized PCR conditions with QIAGEN’s PCR Kits.
Annealing temperature
The optimal primer annealing temperature is dependent on the base
composition (i.e., the proportion of A, T, G and C nucleotides), primer
concentration and ionic reaction environment. Therefore, annealing
temperature needs to be optimized for each primer pair.
Tip: There is no need to calculate annealing temperature when using
QIAGEN PCR kits, as they work over a wide temperature range!
Figure 1. Animation on QIAGEN’s unique PCR buffer system. Watch the video on www.qiagen.com/PCR-video.
Figure 2. NH4+ and K+ cations in QIAGEN PCR buffers
increases specific primer annealing. K+ binds to the phosphate groups (P) on the DNA backbone, stabilizing the annealing of the primers to the template. NH4
+, which exists both as the ammonium ion and as ammonia under thermal-cycling conditions, can interact with the hydrogen bonds between the bases (B), destabilizing principally the weak hydrogen bonds at mismatched bases. The combined effect of the two cations maintains the high ratio of specific to non-specific primer–template binding over a wide temperature range.
NH3 + H+
DestabilizationNH4
+
Stabilization
K+
P– B H
H
H
H
K+
P– B
Stabilization
K+
P–B
K+
P–B
Strong hydrogen band (e.g., between A and T nucleotides)
Weak hydrogen band (e.g., between C and T nucleotides)

PCR – From Setup to Cleanup 10/2015 5
Magnesium ion concentration
Magnesium ions are a critical DNA polymerase cofactor necessary for
enzyme activity. Mg2+ binds to DNA, primers and nucleotides contained
in the amplification reaction. The Mg2+ concentration is generally
higher than that of dNTPs and primers, and some optimization may be
necessary for different template and primer concentrations. A higher than
optimal concentration of Mg2+ can stabilize nonspecific binding and is
often indicated by decreased yields of specific PCR products and the
appearance of background smear or other PCR artifacts.
PCR additives
Various PCR additives or enhancers are available for improving PCR
results. It is claimed that these reagents relieve secondary DNA structure
(e.g., hairpin loops in GC-rich regions or in long amplification products),
lower template melting temperature, enhance enzyme processivity,
stabilize DNA polymerases or prevent attachment of polymerases to
plasticware. Commonly used PCR additives include dimethyl sulfoxide
(DMSO), bovine serum albumin (BSA) and glycerol.
Q-Solution®
This innovative PCR additive facilitates amplification of difficult templates
by modifying the melting behavior of DNA. Use of this unique reagent
will often enable or improve suboptimal PCR. Unlike DMSO and other
PCR additives, Q-Solution is used at a defined working concentration
with any primer—template system and is not toxic (Figure 3).
Loading dyes
Many QIAGEN’s PCR kits are supplied with CoralLoad® PCR Buffer,
which has all of the advantages of QIAGEN PCR Buffer, but can also be
used to directly load the PCR reaction onto an agarose gel without the
need for an additional gel loading buffer. CoralLoad PCR Buffer provides
the same high PCR specificity and minimal reaction optimization as the
Tip: Q Solution — QIAGEN’s PCR enhancer for difficult templates — is found in: QIAGEN Multiplex PCR kits, QIAGEN OneStep RT-PCR Kit, Type-it® kits, QIAGEN Fast Cycling PCR kits, QIAGEN LongRange PCR Kit, HotStar HiFidelity PCR Kits, and with all standard DNA polymerases.

6 PCR – From Setup to Cleanup 10/2015
conventional QIAGEN PCR Buffer. Additionally, it contains two marker
dyes — an orange dye and a red dye — that facilitate estimation of
DNA migration distance and optimization of agarose gel run time (see
Figure 7 on page 17). The buffer ensures improved pipetting visibility
and enables direct loading of PCR products onto a gel, for enhanced
convenience. Since the PCR results are colored for convenience, for some
sensitive downstream applications, it may be necessary to clean up the
DNA (see pages 24—34 for more information).
Figure 3. Influence of Q-Solution on PCR success. A 4.8 kb fragment was amplified in standard reactions using TopTaq® DNA Polymerase with or without Q-Solution. M: marker. Specific amplification was achieved only in reactions containing Q-Solution.
M with Q-Solution without Q-Solution
– 4.8 kb

PCR – From Setup to Cleanup 10/2015 7
Guidelines for degenerate primer design and use
PCR primer sequences are often deduced from amino acid sequences if the exact nucleotide sequence of their target is
unknown. However, because of the degeneracy of the genetic code, the deduced sequences may vary at one or more
positions. A common solution in these cases is to use a degenerate primer, which is a mixture of similar primers that have
different bases at the variable positions. Using degenerate primers can lead to difficulties optimizing PCR assays: within
a degenerate primer mixture only a limited number of primer molecules are complementary to the template; the melting
temperature (Tm) of primer sequences may vary significantly; and the sequences of some primers can be complementary to
those of others. For these reasons, amplification conditions that minimize nonspecific primer–template and primer–primer
interactions are required. The following guidance may help when designing and using degenerate primers are required.
Primer sequence:
• Avoid degeneracy in the 3 nucleotides at the 3’ end, i.e., if
possible use Met- or Trp-encoding triplets at the 3’ end
• To increase primer–template binding efficiency, reduce degeneracy
by allowing some mismatches between the primer and template,
especially towards the 5’ end, but not the 3’ end
• Try to design primers with less than 4-fold degeneracy at any given
position
Primer concentration:
• Begin PCR with a primer concentration of 0.2 μM
• In case of poor PCR efficiency, increase primer concentrations in
increments of 0.25 μM until satisfactory results are obtained
Enzymes used in PCR
Several types of thermostable DNA polymerases are available for use in
PCR, providing a choice of enzymatic properties. Taq DNA polymerase,
isolated from the eubacterium Thermus aquaticus, is the most commonly
used enzyme for standard end-point PCR. The robustness of this enzyme
allows its use in many different PCR assays.
However, as this enzyme is active at room temperature, it is necessary to
perform reaction setup on ice to avoid nonspecific amplification. QIAGEN solution: QIAGEN DNA Taq Polymerase — for standard and specialized PCR applications

8 PCR – From Setup to Cleanup 10/2015
A number of modifications of the original “PCR polymerase” — Taq
DNA polymerase — are now available for different downstream
application needs, such as hot-start, single-cell, high-fidelity or multiplex
PCR. With an average error rate of 1 in 10,000 nucleotides, Taq DNA
polymerase and its variants are less accurate than thermostable enzymes
of DNA polymerase family B. However, due to its versatility, Taq DNA
polymerase is still the enzyme of choice for most routine applications and
when used with a stringent hot-start, is suitable for several challenging
PCR applications.
Hot-start DNA polymerase
When amplification reaction setup is performed at room temperature,
primers can bind nonspecifically to each other, forming primer–dimers.
During amplification cycles, primer–dimers can be extended to produce
nonspecific products, which reduces specific product yield. For more
challenging PCR applications, the use of hot-start PCR is crucial for
successful specific results. To produce hot-start DNA polymerases, Taq
DNA polymerase activity can be inhibited at lower temperatures with
antibodies or with chemical modifiers that form covalent bonds with
amino acids in the polymerase. The chemical modification leads to
complete inactivation of the polymerase until the covalent bonds are
broken during the initial heat activation step.
High-fidelity DNA polymerase
Unlike standard DNA polymerases (such as Taq DNA polymerase),
high-fidelity PCR enzymes generally provide a 3’ to 5’ exonuclease
activity for removing incorrectly incorporated bases. High-fidelity
PCR enzymes are ideally suited to applications requiring a low error
rate, such as cloning, sequencing and site-directed mutagenesis.
However, if the enzyme is not provided in a hot-start version, the 3’ to 5’
exonuclease activity can degrade primers during PCR setup and the early
stages of PCR. Nonspecific priming caused by shortened primers can
result in smearing on a gel or amplification failure — especially when
using low amounts of template. It should be noted that the proofreading
function often causes high-fidelity enzymes to work more slowly than
QIAGEN solution: HotStarTaq® Plus DNA Polymerase — for fast and highly specific amplification in all applications
QIAGEN solution: HotStar HiFidelity Polymerase Kit — the only high-fidelity kit producing sticky ends for simple UA/ TA cloning procedures for highly sensitive and reliable
high-fidelity hot-start PCR

PCR – From Setup to Cleanup 10/2015 9
other DNA polymerases. In addition, the A-addition function required
for direct UA- or TA-cloning is strongly reduced, resulting in the need for
blunt-end cloning with lower ligation and transformation efficiency.
Optimizing 3’ to 5’ exonuclease activity
Taq DNA Polymerase introduces more errors into the PCR product while
copying the template than do so-called proofreading DNA polymerases.
Once a mismatch occurs during synthesis, Taq DNA polymerase will
either extend the mismatched strand or fall off the template strand,
leading to mutated or incomplete PCR products, respectively. Although
this does not generally affect PCR efficiency when amplifying shorter
PCR fragments, amplification of longer PCR products can be significantly
impaired by mismatches introduced during DNA synthesis.
PCR cycling
In theory, each PCR cycle doubles the amount of amplicon in the
reaction. Therefore, 10 cycles multiply the amplicon by a factor of
~1000 and so on.
Each PCR cycle consists of template denaturation, primer annealing and
primer extension. If the temperatures for annealing and extension are
similar, these two processes can be combined. Each stage of the cycle
must be optimized in terms of time and temperature for each template
and primer pair combination.
After the required number of cycles has been completed (see the PCR
section of the QIAGEN Protocols and Applications Guide for more
information about cycle numbers), the amplified product may be
analyzed or used in downstream applications.
Tip: Proofreading DNA polymerases contain an inherent 3’ to 5’ exonuclease activity that removes base-pair mismatches. Adding a small amount of a proofreading DNA polymerase to the PCR mixture therefore significantly improves the amplification efficiency of longer PCR products.
Figure 4. High-resolution analysis of multiplex PCR samples PCR products were generated using the QIAGEN Multiplex PCR Kit according to the standard protocol. PCR samples (13 µl) were analyzed [A] on a 2% agarose gel and [B] and [C] using the QIAxcel system with the QIAxcel DNA High Resolution gel cartridge and the preinstalled OM500 method. [B] The gel image produced by the QIAxcel system shows much higher resolution than the agarose gel. [C] Each sample lane can be visualized individually in electropherogram form. Lane 7 is shown.
QIAGEN solution: QIAGEN LongRange PCR Kit — for sensitive and accurate long-range PCR up to 20 kb
A B
C
1 2 3 4 5 6 7 8 9 10 NTC 1 2 3 4 5 6 7 8 9 10

10 PCR – From Setup to Cleanup 10/2015
Amplification of long PCR products
Amplification of PCR products longer than 3–4 kb is often compromised
by nonspecific primer annealing, suboptimal cycling conditions and
secondary structures in the DNA template. Lengthy optimization is often
necessary, by varying factors such as cycling conditions, primer and
dNTP concentrations and special additives. Our LongRange PCR kits
overcome the need to develop specific long-range PCR protocols by
providing a dedicated and highly reliable solution for amplification of
extremely long PCR product (up to 40 kb DNA).
Optimizing cycling conditions
While depurination is usually not a problem in standard PCR, it can
significantly influence the amplification of longer PCR fragments. This
is because longer templates are proportionally more depurinated than
shorter ones. For this reason, very short denaturation steps of only
10 seconds give higher yields and no background smearing compared
to denaturation steps of 30 seconds or 1 minute (which leads to PCR
failure). Extensive depurination is also observed during the final extension
step. Therefore, using a lower extension temperature of 68°C instead of
72°C dramatically improves yield of longer amplification products.
The PCR section of the QIAGEN Protocols and Applications Guide
provides more information about cycling conditions for longer PCR
products.
Successful multiplex PCR
Multiplex PCR allows simultaneous amplification of multiple DNA targets
in the same reaction. This increases throughput, reduces reagent costs
and conserves precious sample material. Multiplex PCR offers many
advantages for end‐point PCR applications, including genotyping from
genomic DNA and gene expression analysis from mRNA derived
cDNA. It also permits co‐amplification of an internal positive control
with sequences of interest in the same reaction, allowing sample
preparation and PCR to be monitored and the absence of inhibitors to
be confirmed. The internal control can either be an endogenous gene
QIAGEN Multiplex PCR Kit – for highly specific and sensitive multiplex PCR without optimization requirements.

PCR – From Setup to Cleanup 10/2015 11
(e.g., a housekeeping gene) or an exogenous nucleic acid spiked into
the reaction.
QIAGEN’s multiplex PCR chemistry enables unlimited success with as
many targets as you like and is provided in an easy‐to‐use master mix
format. The master mix contains a novel PCR buffer and pre‐optimized
concentrations of HotStarTaq DNA Polymerase, MgCl2 and dNTPs. The
master mix also contains Factor MP, which further facilitates multiplex
amplification by increasing and stabilizing the local concentration of
primers at the template. These pre‐optimized reagents conveniently
remove the need to optimize reaction conditions or cycling parameters.
Template
Primer
Destabilizationof mismatchedprimer by NH4
+
Template
Primer Stabilizationby Factor MP
Nonspeci�c primer annealing
Speci�c primer annealing
Figure 5. Unique Type‐it Microsatellite PCR Buffer promotes stable and efficient annealing. A. NH4
+ ions prevent nonspecific primers from annealing to the template. Synthetic Factor MP, an innovative PCR additive, increases the local concentration of primers at the template. B. Together with K+ and other cations, synthetic Factor MP stabilizes specifically bound primers, allowing efficient primer extension by HotStarTaq Plus DNA Polymerase.

12 PCR – From Setup to Cleanup 10/2015
Capillary electrophoresis – an advanced alternative
Capillary electrophoresis offers an excellent, non‐hazardous alternative to traditional gel analysis of DNA and RNA, while
streamlining your workflow and reducing time to results. Nucleic acid molecules are size separated by applying a current
to a gel‐filled capillary and detected as they migrate past a detection spot. The signal data is then transmitted through a
photomultiplier tube and converted into an electropherogram for further analysis.
The QIAxcel® Advanced System — for effortless DNA and RNA analysis
The QIAxcel Advanced System fully automates sensitive, high‐resolution
capillary electrophoresis of up to 96 samples per run (Figure 6). DNA
fragment analysis of 12 samples can be performed in as little as
3 minutes. The QIAxcel Advanced offers a high degree of flexibility
by providing four different ready-to-run gel cartridges that match most
analysis requirements (see Table 1). These cartridges also allow samples
to be analyzed with a minimum of hands-on interaction, reducing
handling errors and eliminating the need for tedious gel preparation.
Only a few, simple steps are required to operate the system: load the
gel cartridge, fill and load the buffer tray, load your samples in 96‐well
plates or in PCR tubes or strips and select the process profile. Within
minutes of starting a run, you can see the first results appearing in real
time on the computer screen. Comprehensive data reports are easily
generated, saved or exported to meet your needs, facilitating integration
of the system in your daily routine.
With a resolution down to 3–5 bp for fragments smaller than 500 bp,
the QIAxcel Advanced System ensures greater accuracy than slab‐gel
methods. Sample consumption is less than 0.1 μl per run, saving
precious sample for downstream analysis. Used in combination with
proven QIAGEN PCR kits, this system provides an all‐in‐one solution for
reproducible, standardized PCR fragment analysis, ensuring significant
time and cost savings.
A B
Gel matrix with dyePositivecharge
Capillary
Nucleic acid with dye
LED light source
DetectorPhoto-
multiplier
QIAxcel ScreenGel Software
Negativecharge
Figure 6. An innovative principle. A. QIAxcel gel cartidge. B. The capillary electrophoresis principle is similar to that used in sequencers. Nucleic acid molecules are size separated by applying a current to a gel‐filled capillary and detected as they migrate toward the positively charged terminus. The signal data pass through a photomultiplier and are converted to an electropherogram and gel image by QIAxcel ScreenGel Software.
* Best resolution between 1 – 3 kb. † Run time depends on method used.
QIAxcel Kit Analyte Size range 100 bp – 500 bp 500 bp – 1 kb 1 kb – 5 kb 5 kb – 10 kb
Run time/
12 samples†
QX DNA High Resolution Kit DNA 15 bp – 10 kb 3 – 5 bp 50 bp 200 – 500 bp 1 – 1.5 kb 7 – 20 min
QX DNA Screening Kit DNA 15 bp – 5 kb 20 – 50 bp 50 – 100 bp 500 bp – 5 min
QX DNA Fast Analysis Kit DNA 15 bp – 3 kb 50 – 100 bp 100 – 250 bp 250 bp – 1 kb* – 3 – 5 min
QX RNA QC Kit v2.0 RNA 15 bp – 6 kb – – – – 10 min
Table 1. QIAxcel Kit speci�cations
Best resolution

PCR – From Setup to Cleanup 10/2015 13
Frequently asked questions
How can I tell if I have primer–dimers in my PCR reaction?
In non-quantitative endpoint PCR, primer–dimers will appear as a more or less faint smear on an
agarose gel, below the product band of interest.
Must I use CoralLoad Gel loading dye when using QIAGEN’s HotStarTaq Plus DNA Polymerase?
No. HotStarTaq Plus DNA Polymerase is supplied with conventional QIAGEN PCR Buffer and
CoralLoad PCR Buffer in separate vials. Both buffers minimize nonspecific amplification products,
primer–dimers and background.
CoralLoad PCR Buffer has all of the advantages of QIAGEN PCR Buffer, but can also be used to
directly load the PCR reaction onto an agarose gel without the need to add a gel loading buffer.
How comparable is CoralLoad gel loading dye contained in various QIAGEN PCR Kits to Sigma Red?
CoralLoad gel tracking dye contained in Taq, HotStarTaq Plus, TopTaq DNA Polymerase and
TopTaq Master Mix Kits separates into 2 fragment-size dependent colors (orange and red) when
loaded onto an agarose gel. Sigma Red buffer only has one color which is harder to visualize.

14 PCR – From Setup to Cleanup 10/2015
Comments and suggestions
Little or no product or product is multi-banded
Pipetting error or missing reagent Repeat the PCR. Check the concentrations and
storage conditions of reagents, including primers
and dNTP mix.
Suboptimal PCR cycling conditions Using the same cycling conditions, repeat the
PCR using Q-Solution.
Primer concentration not optimal or primers
degraded
Repeat the PCR with different primer concentrations
from 0.1–0.5 μM of each primer (in 0.1 μM
increments). In particular, when performing highly
sensitive PCR, check for possible degradation of
the primers on a denaturing polyacrylamide gel.
Problems with starting template Check the concentration, storage conditions and
quality of the starting template. If necessary,
make new serial dilutions of template nucleic
acid from stock solutions and repeat the PCR.
Mg2+ concentration not optimal Perform PCR with different final concentrations of
Mg2+ from 1.5–5.0 mM (in 0.5 mM increments)
using a 25 mM MgCl2 solution.
Enzyme concentration too low Use 2.5 units of Taq DNA Polymerase per
100 μl reaction.
PCR troubleshooting

PCR – From Setup to Cleanup 10/2015 15
Comments and suggestions
Incorrect annealing temperature or time Decrease annealing temperature by 2°C
increments. Annealing time should be between
30 and 60 s. Difficulties in determining the
optimal annealing temperature can often be
overcome by performing touchdown PCR.
Incorrect denaturation temperature or time Denaturation should be at 94°C for 30–60 s.
Ensure that a prolonged initial denaturation is
performed as described in the protocols.
Hot start may be necessary Try using the hot-start procedure supplied with
QIAGEN Taq DNA Polymerase, or for greater
specificity, try using HotStarTaq Plus DNA
polymerase.
Primer design not optimal Review primer design
PCR overlaid with mineral oil when using a
thermal cycler with a heated lid
When performing PCR in a thermal cycler
using a thermal cycler with a heated lid that is
switched on, do not overlay the PCR samples
with mineral oil, as this may decrease the yield
of PCR product.
PCR troubleshooting

16 PCR – From Setup to Cleanup 10/2015
Comments and suggestions
Product is smeared
Hot start may be necessary Try using the hot-start procedure supplied with
QIAGEN Taq DNA Polymerase, or for greater
specificity, try using HotStarTaq DNA Polymerase.
Too much starting template Check the concentration and storage conditions
of the starting template. Make serial dilutions
of template nucleic acid from stock solutions.
Perform PCR using serial dilutions.
Carry-over contamination If the negative-control PCR (without template
DNA) shows a PCR product or a smear,
exchange all reagents. Use disposable tips
containing hydrophobic filters to minimize cross-
contamination. Set up all reaction mixtures
in an area separate from that used for DNA
preparation or PCR product analysis.
Enzyme concentration too high Use 2.5 units of Taq DNA Polymerase per
100 μl reaction.
Too many cycles Reduce the number of cycles in increments of
3 cycles.
Mg2+ or primer concentration/design not
optimal or primers degraded
See above section for these problems.
PCR of long fragments from genomic DNA Follow the specific protocols provided for
amplification of long PCR products.
PCR troubleshooting

PCR – From Setup to Cleanup 10/2015 17
Agarose gel analysis
Agarose gel analysis enables quick and easy quantification of DNA, especially for small DNA fragments (such as PCR
products). As little as 20 ng DNA can be detected by agarose gel electrophoresis with ethidium bromide staining. The DNA
sample is run on an agarose gel alongside known amounts of DNA of the same or a similar size. The amount of sample
DNA loaded can be estimated by comparison of the band intensity with the standards either visually or using a scanner or
imaging system. Be sure to use standards of roughly the same size as the fragment of interest to ensure reliable estimation
of the DNA quantity, since large fragments interchelate more dye than small fragments and give a greater band intensity.
CoralLoad PCR Buffer, included in most QIAGEN PCR kits, can be used to
directly load the PCR reaction onto an agarose gel — separate addition
of a gel loading buffer is not required. CoralLoad PCR Buffer provides
the same high PCR specificity and minimal reaction optimization as the
conventional QIAGEN PCR Buffer. Additionally, it contains two marker
dyes — an orange dye and a red dye — that facilitate estimation of DNA
migration distance and optimization of agarose gel run time (Figure 7).
The buffer ensures improved pipetting visibility and enables direct
loading of PCR products onto a gel, for enhanced convenience. More
precise agarose gel quantification can be achieved by densitometric
measurement of band intensity and comparison with a standard curve
generated using DNA of a known concentration. In most experiments the
effective range for comparative densitometric quantification is between
20 and 100 ng.
CoralLoad PCR Buffer is found in the following QIAGEN kits:
• QIAGEN Multiplex PCR Plus Kit
• QIAGEN Fast Cycling PCR Kit
• QIAGEN LongRange PCR Kit
• HotStarTaq Plus DNA Polymerase
• HotStarTaq Plus Master Mix Kit
• Taq DNA Polymerase
• Taq PCR Core Kit
• TopTaq DNA Polymerase
• TopTaq Master Mix Kit
Figure 7. CoralLoad PCR Buffer. A. The buffer contains gel-tracking dyes for easier pipetting, B. enabling immediate gel loading of PCR samples and easy visualization of DNA migration.
Tip: The amount of DNA used for densitometric quantification should fall within the linear range of the standard curve.
A B

18 PCR – From Setup to Cleanup 10/2015
Running an agarose gel
Agarose gel electrophoresis allows analysis of DNA fragments between 0.1 and 25 kb (e.g.,
genomic DNA digested with a frequently cutting restriction endonuclease), while pulse-field gel
electrophoresis enables analysis of DNA fragments up to 10,000 kb (e.g., undigested genomic
DNA or genomic DNA digested with rare cutting restriction endonucleases). The amount of
genomic DNA loaded onto a gel depends on the application, but in general, loading of too much
DNA should be avoided as this will result in smearing of the DNA bands on the gel.
Gel loading buffer must be added to the samples before loading and serves three main purposes:
• To increase the density of the samples to ensure that they sink into the wells on loading
• To add color to the samples through use of dyes such as bromophenol blue or xylene cyanol,
facilitating loading
• To allow tracking of the electrophoresis due to co-migration of the dyes with DNA fragments
of a specific size
Molecular-weight markers should always be included on a gel to enable analysis of DNA fragment
sizes in the samples.
Preparation of samples
1. Add 1 volume of gel loading buffer to 6 volumes DNA sample and mix.
Samples should always be mixed with gel loading buffer prior to loading on a gel.
Ensure that no ethanol is present in the samples, as this will cause samples to float out of the wells
on loading.
Tip: Do not use sample volumes close to the capacity of the wells, as samples may spill over
into adjacent wells during loading.
Tip: Be sure that all samples have the same buffer composition. High salt concentrations,
for example in some restriction buffers, will retard the migration of the DNA fragments.

PCR – From Setup to Cleanup 10/2015 19
Agarose gel electrophoresis
1. Apply samples in gel loading buffer to the wells of the gel.
Prior to sample loading, remove air bubbles from the wells by rinsing them with electrophoresis
buffer.
2. Connect the electrodes so that the DNA will migrate towards the anode (positive electrode).
3. Turn on the power supply and run the gel at 170 V with a switch interval of 5–40 s until the
dyes have migrated an appropriate distance. This will depend on the size of DNA being
analyzed, the concentration of agarose in the gel and the separation required.
Tip: Make sure that the entire gel is submerged in the electrophoresis buffer.
Tip: To load samples, insert the pipet tip deep into the well and expel the liquid slowly.
Take care not to break the agarose with the pipet tip.
Tip: Once samples are loaded, do not move the gel tray/tank as this may cause samples
to float out of the wells.
Tip: Be sure to always include at least one lane of appropriate molecular-weight markers.
Tip: Electrophoresis apparatus should always be covered to protect against electric shocks.
Tip: Avoid use of very high voltages which can cause trailing and smearing of DNA bands
in the gel, particularly with high-molecular-weight DNA.
Tip: Monitor the temperature of the buffer periodically during the run. If the buffer becomes
heated, reduce the voltage.
Tip: Melting of an agarose gel during the electrophoresis is a sign that the buffer may have
been incorrectly prepared or has become exhausted during the run.
Tip: For very long runs, e.g., overnight runs, use a pump to recycle the buffer.

20 PCR – From Setup to Cleanup 10/2015
Pulse-field gel electrophoresis
1. Apply samples in gel loading buffer to the wells of the gel.
2. Connect the electrodes so that the DNA will migrate towards the anode (positive electrode).
3. Turn on the power supply and run the gel at 170 V with a switch interval of 5–40 s until the
dyes have migrated an appropriate distance. This will depend on the size of DNA being
analyzed, the concentration of agarose in the gel and the separation required.
Tip: Pulse-field gel electrophoresis uses high voltages, so TBE buffer, which has greater
buffering capacity than TAE buffer, should be used.
Tip: Prior to sample loading, remove air bubbles from the wells by rinsing them with
electrophoresis buffer.
Tip: Make sure that the entire gel is submerged in the running buffer.
Tip: To load samples, insert the pipet tip deep into the well and expel the liquid slowly. Take
care not to break the agarose with the pipet tip.
Tip: Once samples are loaded, do not move the gel tray/tank as this may cause samples
to float out of the wells.
Tip: Be sure to always include at least one lane of appropriate molecular-weight markers.
Tip: Electrophoresis apparatus should always be covered to protect against electric shocks.
Tip: Monitor the temperature of the buffer periodically during the run. If the buffer becomes
overheated, reduce the voltage.
Tip: Melting of an agarose gel during the electrophoresis is a sign that the buffer may have
been incorrectly prepared or has become exhausted during the run.
Tip: For very long runs, e.g., overnight runs, use a pump to recycle the buffer.

PCR – From Setup to Cleanup 10/2015 21
Visual analysis of the gel
Staining
To allow visualization of the DNA samples, agarose gels are stained with an appropriate dye,
such as ethidium bromide or SYBR® Green. SYBR Green is considered to be less hazardous, with
respect to mutagenicity, than ethidium bromide.
Addition of ethidium bromide prior to electrophoresis — add ethidium bromide at a concentration
of 0.5 μg/ml to the melted and subsequently cooled agarose, that is, just before pouring the gel.
Mix the agarose–ethidium bromide solution well to avoid localized staining.
Addition of ethidium bromide after electrophoresis — soak the gel in a 0.5 μg/ml solution of
ethidium bromide (in water or electrophoresis buffer) for 30–40 minutes.
Tips for handling ethidium bromide:
• Stock solutions of ethidium bromide (generally 10 mg/ml) should be stored at 4°C in a dark
bottle or bottle wrapped in aluminum foil
• Rinse the gel with buffer or water before examining it to remove excess ethidium bromide
• Staining buffer can be saved and re-used
Visualization
DNA stained with SYBR Green or ethidium bromide displays increased fluorescence compared
to the dye in solution. This means that illumination of a stained gel under UV light (254–366 nm)
allows bands of DNA to be visualized against a background of unbound dye. The gel image can
be recorded by taking a Polaroid photograph or using a gel documentation system.
Note: Ethidium bromide is a powerful mutagen and is very toxic. Wear gloves and take appropriate safety precautions when handling. Use of nitrile gloves is recommended as latex gloves may not provide full protection. After use, ethidium bromide solutions should be decontaminated as described in commonly used manuals.

22 PCR – From Setup to Cleanup 10/2015
Tip: UV light can damage the eyes and skin. Always wear suitable eye and face protection when working with a UV light source.
Tip: UV light damages DNA. If DNA fragments are to be extracted from the gel, use a lower intensity UV source if possible and minimize exposure of the DNA to the UV light.
Frequently asked questions
Why does my DNA sample float out of the slot when loading it onto an agarose gel?
DNA fragments purified with the QIAGEN DNA cleanup systems, (e.g., the QIAquick® PCR
Purification Kit, the MinElute® Reaction Cleanup Kit, the QIAEX® II Gel Extraction Kit, etc.) may
float out of the loading wells of agarose gels due to residual ethanol carried over from the wash
step with Buffer PE (despite the addition of glycerol-containing loading buffer).
Use any of the following options to remove residual ethanol from the eluate:
• Re-purify the sample using a QIAquick or MinElute column or QIAEX II resin
• Incubate the eluate at 56°C for 10 min to evaporate the ethanol
• Dry down the sample in a vacuum centrifuge, and resuspend the pellet in a small volume of
sterile water
How should gels be cast so that optimal resolution is achieved?
Gels should be cast 3–4 mm thick for optimal resolution of DNA fragments. We also recommend
the use of a thin comb (1 mm) to obtain sharper DNA bands.
How much DNA should be loaded per well of an agarose gel?
The amount of DNA per well is variable. The least amount of DNA that can be detected with
ethidium bromide is 10 ng. DNA amounts of up to 100 ng per well will result in a sharp, clean
band on an ethidium-bromide–stained gel.
What voltage should be used to run an agarose gel?
We recommend running agarose gels at 4–10 volts/cm under horizontal electrophoretic
conditions. Higher voltage may result in band streaking, while lower voltage may result in reduced
mobility of small (<1000 bp) DNA and diffusion.

PCR – From Setup to Cleanup 10/2015 23
What buffer conditions give the best resolution for agarose gel electrophoresis?
We recommend the use of 1x TBE buffer for small DNA fragments (<1000 bp) when DNA recovery
is not necessary. Gels made using TBE buffer give sharper bands than gels made using TAE buffer.
For large DNA fragments of >15000 bp, we recommend the use of 1x TAE buffer. However,
since TAE buffer has a lower buffering capacity, it may be necessary to change the buffer when
performing electrophoresis for an extended period of time.
Why do I have wavy DNA bands on my agarose gel?
Wavy DNA bands on an agarose gel can be caused by dried agarose on the comb teeth. Check
the comb teeth for residual dried agarose prior to casting the gel and clean if necessary.
Can I store agarose gel slices containing DNA for gel extraction at a later point?
Yes. Cut out the slice of agarose containing the DNA fragment of interest, and store it at 4°C in a
microcentrifuge tube sealed with Parafilm®.
How do I separate PCR fragments that are small and very close in size on an agarose gel?
The concentration of the agarose gel for separation of multiplex PCR products should be
appropriate for the overall size of products generated and can be adjusted for resolving small size
differences between PCR fragments. For optimal results, we recommend the use of 1x TAE buffer
for preparation and running of the gel. Use the general guidelines listed in the table for choosing
the percentage of agarose.
* Efficient separation of PCR products differing in size by about 20 bp is usually possible using standard molecular-biology– grade agarose. For separation of fragments that differ in size by less than 20 bp, we recommend using high-resolution agarose, for example MetaPhor® agarose (FMC Bioproducts). For more information, visit www.cambrex.com.
Please see the QIAGEN Multiplex PCR Handbook for additional information and for details on successful multiplex PCR using the QIAGEN Multiplex PCR Kit.
Maximum size of fragments Concentration of agarose
>200 bp
2000 bp 1.3%>100–200 bp 1000 bp 1.4–1.6%
>50–100 bp 750 bp 1.7–2.0%
20–50 bp 500 bp 2.5–3.0%
<20bp* 250 bp 3.0–4.0%
Minimum difference in sizeof PCR products

24 PCR – From Setup to Cleanup 10/2015
Does QIAGEN have protocols for multiple extractions of DNA fragments from agarose gels?
Yes. Follow the Supplementary Protocol “High-throughput gel extractions using the QIAquick 96
PCR Purification Kit” (QQ03). Contact QIAGEN Technical Services for this protocol.
My sample does not give a fluorescent signal. How do I know whether this is because the PCR did
not work or because the target is not expressed?
Use a control sample in which the gene of interest is definitely expressed. PCR products that span
the region to be amplified in the real-time experiment can also be used as a positive control.
Check by agarose gel electrophoresis that the amplification reaction was successful. The quality
of the starting template and the integrity of the reagents can be determined by amplifying a
housekeeping gene, such as GAPDH or HPRT.
What is the QIAxcel Advanced System?
The revolutionary QIAxcel Advanced System is a fully automated one-step capillary electrophoresis
system that replaces traditional, labor-intensive gel analysis of DNA and RNA.
• Rapid analysis of up to 96 samples without manual intervention
• Safety and convenience with ready-to-use gel cartridges
• Robust results for nucleic acid concentrations as low as 0.1 ng/μl
• Standardized and accurate analysis with a resolution down to 3–5 bp
• User-friendly analysis software that supports 21 CFR Part 11 compliance
For a virtual demonstration of the software, features and applications of the QIAxcel Advanced
System visit: www.qiagen.com/QIAxcelAdvancedDemo
What is the shelf life of the QIAxcel cartridges?
All QIAxcel DNA and RNA Kits can be stored for a guaranteed minimum time of 135 days after
delivery. QIAxcel DNA and RNA ready-to-use cartridges are reusable and can process up to
2400 samples.
What is the minimum sample volume required for the QIAxcel Advanced?
For the QIAxcel System and QIAxcel Advanced, the minimum sample volume required to guarantee
sample injection in each channel is 10 μl. Robust results can be achieved with sample concentration
as low as 0.1 ng/μl of nucleic acid; you can dilute your sample without loss of signal quality.

PCR – From Setup to Cleanup 10/2015 25
Cleanup
DNA cleanup
One of the most common tasks in molecular biology is cleaning up nucleic acids from varied matrices to remove buffer salts,
enzymes or other substances that may affect downstream applications, such as cloning, sequencing, microarray analysis or
amplification. There are 3 main areas of DNA cleanup:
• Cleanup from enzymatic reactions, e.g., PCR
• Nucleotide removal
• Gel extraction and cleanup
A number of cleanup kits are available for different starting samples and downstream application needs. QIAquick and
MinElute Kits contain a silica membrane assembly for binding of DNA in high-salt buffer and elution with low-salt buffer
or water. The purification procedure removes primers, nucleotides, enzymes, mineral oil, salts, agarose, ethidium bromide
and other impurities from DNA samples (Figure 8, next page). Silica-membrane technology eliminates the problems and
inconvenience associated with loose resins and slurries. Specialized binding buffers are optimized for specific applications
and promote selective adsorption of DNA molecules within particular size ranges.
Cleanup solutions from QIAGEN:
• QIAquick PCR Purification Kit — for purification of up to 10 μg PCR products >100 bp. DNA
of up to 10 kb is purified using a simple and fast bind-wash-elute procedure and an elution
volume of 30–50 μl.
• QIAquick Gel Extraction Kit — for purification of DNA fragments from gels (up to 400 mg
slices) or enzymatic reactions. DNA ranging from 70 bp to 10 kb is purified using a simple
and fast bind-wash-elute procedure and an elution volume of 30–50 μl.
• QIAquick Nucleotide Removal Kit — for up to 10 μg oligonucleotide (17–40mers) and DNA
(40 bp to 10 kb) cleanup from enzymatic reactions. The process uses just three easy steps!
• MinElute PCR Purification Kit — for purification of up to 5 μg PCR products (70 bp to 4 kb) in
low elution volumes using a very simple procedure.
• MinElute Gel Extraction Kit — for purification of DNA fragments of 70 bp – 4 kb from up to
400 mg gel slices. The spin columns are designed to allow elution in very small volumes (as
little as 10 μl), delivering high yields of highly concentrated DNA.
• MinElute Reaction Cleanup Kit — for cleanup of up to 5 μg DNA (70 bp to 4 kb) from
enzymatic reactions. The kit enables very low elution volumes and uses a fast procedure with
easy handling.

26 PCR – From Setup to Cleanup 10/2015
• QIAEX II Gel Extraction Kit — for purification of DNA fragments
(40 bp to 50 kb) from gels and solutions. QIAEX II Suspension is
added to solutions or solubilized agarose gel slices and binds DNA
in the presence of chaotropic salts before washing and elution.
The advantages of silica-membrane technology include:
• Yielding high-purity nucleic acids for use in most downstream
applications
• Fast and inexpensive
• No silica-slurry carry over, no alcohol precipitation
QIAGEN’s QIAquick and MinElute cleanup system use a simple
bind-wash-elute procedure (Figure 9). Sample mixtures are applied to the
spin column (in some applications, this includes a buffer that contains a
pH indicator; allowing easy determination of the optimal pH for DNA
binding — see Figure 10).
Nucleic acids adsorb to the silica membrane in the high-salt conditions
provided by the buffer. Impurities are washed away and pure DNA is
eluted with a small volume of low-salt buffer provided or water, ready to
use in all subsequent applications.
–
–
– –
Maaa bbbM
2000 bp
500 bp
20 mer100 bp
Optimal pH pH too High
Figure 8. Analysis of PCR products before (b) and after (a) purification using the QIAquick PCR Purification Kit. Samples were analyzed on a 1% TAE agarose gel. M: markers.
Figure 10. pH Indicator Dye. Indicator enables easy
checking of the optimal pH. Indicator dye in solubilization
and binding buffers (Buffer QG and Buffer PB) identifies
optimal pH for DNA binding.
Figure 9. QIAquick and MinElute procedure. In addition to spin columns, manual and automated high-throughput kits are also available.
Vacuum
Vacuum
QIAquick and MinElute Procedure
PCR or otherenzymatic reaction orsolubilized gel slice
Pure DNA fragment
QIAcube
From solutions 5minFrom gels 15min
PCR – From Setup to Cleanup 10/2015 27
DNA yield and concentration
DNA yield depends on the following 3 factors:
• Elution buffer volume
• How the buffer is applied to the column
• Incubation time of the buffer on the column
To completely cover the QIAquick/MinElute membrane, use 100–200 μl elution buffer. This ensures
maximum yield, even when not applied directly to the center of the membrane. Elution with
≤50 μl requires the buffer to be added directly to the center of the membrane, and if elution is done
with the minimum recommended volume of 30 μl, an additional 1 minute incubation is required
for optimal yield. DNA will be up to 1.7 times more concentrated if the column is incubated for
1 minute with 30 μl of elution buffer, than if it is eluted in 50 μl without incubation.
The kits vary depending on the type of reaction and DNA fragment size (e.g., PCR products, gel
extraction, enzymatic reactions, nucleotide removal, dye terminator removal) and the required
elution volume.
Tip: Depending on the application, fragment size, etc. it is advisable to use a specialized kit for optimal results. Please consult Tables 2 and 3 to find the kit that best meets your needs.
QIAquick PCR Puri�cation Kit QIAquick Nucleotide Removal Kit QIAquick Gel Extraction Kit
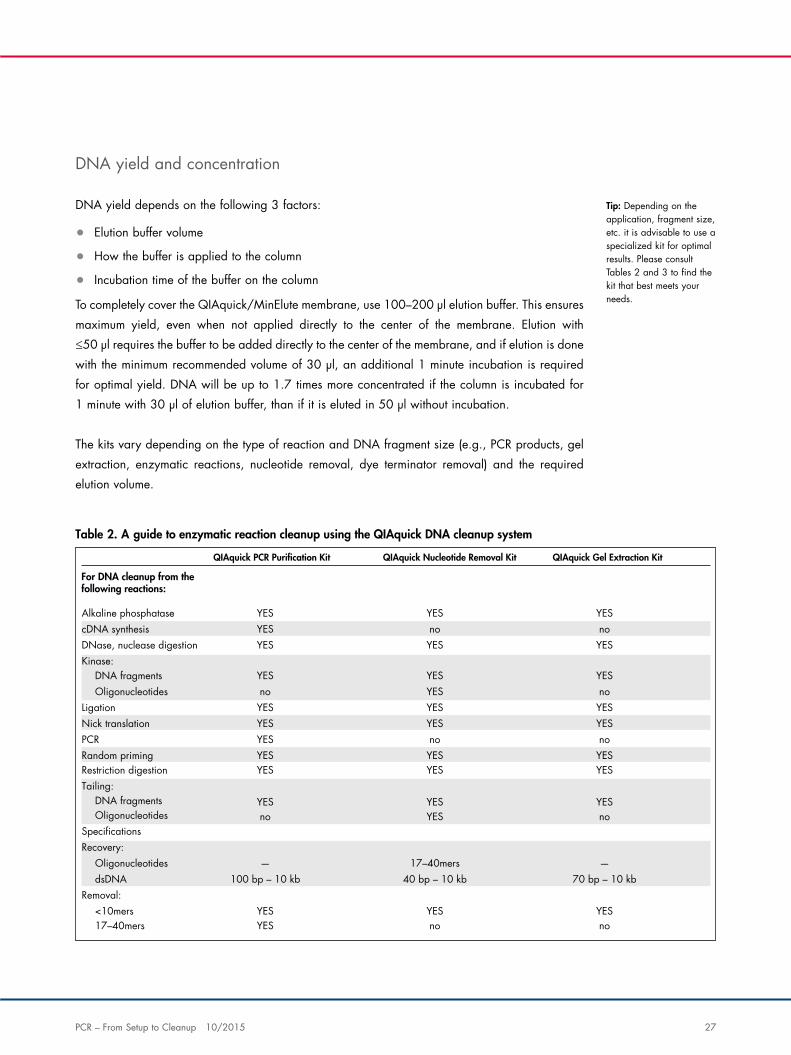
For DNA cleanup from thefollowing reactions:
YES no noYES
cDNA synthesisAlkaline phosphatase YES YES
YESKinase:DNase, nuclease digestion YES YES
no YES noYES
OligonucleotidesDNA fragments YES YES
YES YES YESYES
Nick translationLigation YES YES
YESPCRRandom priming
no noYES YES YES
YES YES YES
YESTailing:Restriction digestion YES YES
DNA fragments
Recovery:Speci�cations
Oligonucleotides no YES no
100 bp – 10 kb 40 bp – 10 kb 70 bp – 10 kb—
dsDNAOligonucleotides 17–40mers —
Removal: <10mers YES YES YES
YES17–40mers no no
Table 2. A guide to enzymatic reaction cleanup using the QIAquick DNA cleanup system
28 PCR – From Setup to Cleanup 10/2015
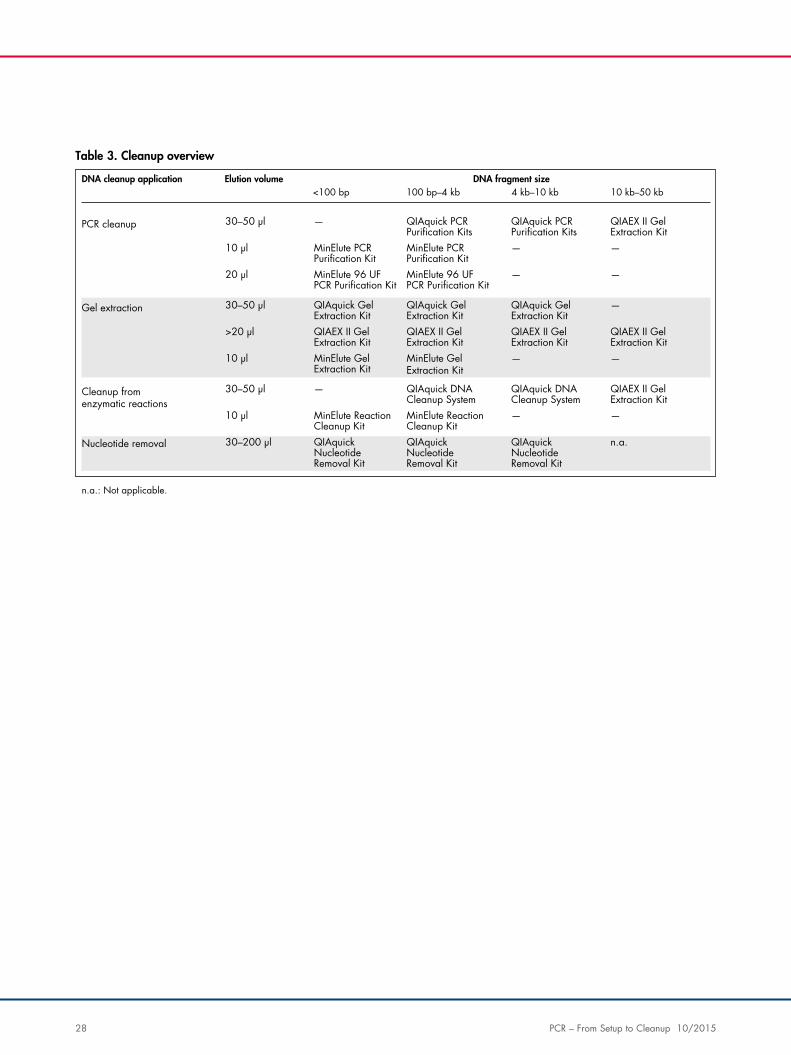
DNA cleanup application Elution volume<100 bp 100 bp–4 kb 4 kb–10 kb 10 kb–50 kb
—
DNA fragment size
30–50 µl
— —10 µl
— —20 µl
PCR cleanup
—30–50 µl
>20 µl
— —10 µl
Gel extraction
—30–50 µl
— —10 µl
QIAquick PCRPuri�cation Kits
QIAquick PCRPuri�cation Kits
QIAEX II GelExtraction Kit
MinElute PCRPuri�cation Kit
MinElute PCRPuri�cation Kit
MinElute 96 UFPCR Puri�cation Kit
MinElute 96 UFPCR Puri�cation Kit
QIAquick GelExtraction Kit
QIAquick GelExtraction Kit
QIAquick GelExtraction Kit
QIAEX II GelExtraction Kit
QIAEX II GelExtraction Kit
QIAEX II GelExtraction Kit
QIAEX II GelExtraction Kit
MinElute GelExtraction Kit
MinElute GelExtraction Kit
QIAquick DNACleanup System
QIAquick DNACleanup System
QIAEX II GelExtraction Kit
MinElute ReactionCleanup Kit
MinElute ReactionCleanup Kit
QIAquickNucleotideRemoval Kit
QIAquickNucleotideRemoval Kit
QIAquickNucleotideRemoval Kit
n.a.30–200 µl
Cleanup fromenzymatic reactions
Nucleotide removal
n.a.: Not applicable.
Table 3. Cleanup overview

PCR – From Setup to Cleanup 10/2015 29
Frequently asked questions
How can I ensure that I have the right pH?
The binding buffer contains a pH indicator, allowing easy determination of the optimal pH for DNA
binding (see Figure 10). Binding buffer PB and binding and solubilization buffer QG are specially
optimized for use with the QIAquick silica membrane. Buffer QG contains an integrated pH
Indicator, while an optional pH Indicator can be added to Buffer PB allowing easy determination of
the optimal pH for DNA binding. DNA adsorption requires a pH ≥7.5, and the pH Indicator in the
buffers will appear yellow in this range. If the pH is >7.5, which can occur if during agarose gel
electrophoresis, the electrophoresis buffer had been used repeatedly or incorrectly prepared, or if
the buffer used in an enzymatic reaction is strongly basic and has a high buffering capacity, the
binding mixture turns orange or violet. This means that the pH of the sample exceeds the buffering
capacity of Buffer PB or QG and DNA adsorption will be inefficient. In these cases, the pH of the
binding mixture can easily be corrected by addition of a small volume of 3 M sodium acetate,
pH 5.0, before proceeding with the protocol.
What is the small band below my fragment of interest on an agarose gel after DNA cleanup using
QIAquick?
Occasionally, DNA fragments eluted from the silica matrix of QIAquick, MinElute or QIAEX II
Kits will contain denatured single-stranded DNA (ssDNA), appearing as a smaller band on an
analytical gel. Under certain conditions, chaotropic agents (present in all silica-based DNA
purification methods) can denature DNA fragments. This is a rare event that may be influenced by
sequence characteristics such as the presence of inverted repeats or A–T-rich stretches.
Because salt and buffering agents promote renaturation of DNA strands, the following tips are
recommended:
• Use the eluted DNA to prepare your downstream enzymatic reaction, but omit the enzyme.
Incubate the reaction mix at 95°C for 2 minutes to reanneal the ssDNA, and allow the tube
to cool slowly to room temperature before adding the enzyme and proceeding
• Alternatively, the DNA can be eluted from the silica-gel membrane or resin in 10 mM Tris
buffer containing 10 mM NaCl. However, the salt concentration of the eluate must then be
taken into consideration in downstream applications.
Can I use the QIAquick PCR Purification Kit for restriction enzyme cleanup?
Yes. The QIAquick PCR Purification Kit has been used to clean up fragments between 100 bp and
10 kb from a wide range of enzymatic reactions, removing salts, buffers, enzymes, nucleotides

30 PCR – From Setup to Cleanup 10/2015
and primers smaller than 40 nucleotides. Reactions that can be cleaned up with the QIAquick PCR
Purification Kit include restriction digests, random priming, ligase, kinase, phosphatase, nuclease,
nick translation and cDNA synthesis reactions.
Do you have information about the cleanup of single-stranded DNA (ssDNA) with QIAquick
columns?
As a rule of thumb, single-stranded DNA binds to silica with approximately half the affinity of
a double-stranded DNA fragment of the same length under the buffer conditions used in the
QIAquick and MinElute Kits. Even though no systematic experimental data exists, we expect
that recovery of ssDNA fragments of approximately 200 nucleotides and below will not be very
efficient after cleanup using the QIAquick PCR Purification Kit or MinElute PCR Purification Kit.
By comparison, it should be possible to purify fragments longer than 140 nucleotides using the
QIAquick Gel Extraction Kit.
Note that recovery of single strand DNA is influenced to some degree also by factors such as
base composition and secondary structure. It has to be determined empirically by the researcher if
cleanup of single-stranded DNA with QIAquick columns yields satisfactory results.
Are Buffer PB of the QIAquick PCR Purification Kit and Buffer QG of the QIAquick Gel Extraction
Kit interchangeable?
Buffer PB of the QIAquick PCR Purification Kit cannot be used to extract DNA from agarose gels.
However, Buffer QG of the QIAquick Gel Extraction Kit can be used to remove salt and proteins
from enzymatic reactions by adding 3 volumes of Buffer QG and 1 volume of isopropanol to the
reaction and proceeding with step 6 of the Gel Extraction Spin Protocol in the QIAquick Spin
Handbook. See the QIAquick Spin Handbook for a list of reactions which can be cleaned up with
the various QIAquick kits.
I bound an 11 kb DNA fragment to a QIAquick column; is it completely lost?
Larger DNA fragments bind more tightly to the QIAquick columns. It is difficult to predict whether
a DNA fragment larger than 10 kb can be efficiently recovered, because this depends on base
composition as well as fragment size. If the fragment is only a few kb larger than the 10 kb limit,
it can be helpful to heat the elution buffer EB to 60°C and let it incubate on the column for a few
minutes before centrifuging. However, please note that it will become less likely to recover your
sample the larger the fragment size is. As we cannot guarantee recovery of fragments larger than
the maximum cutoff size, we do not recommend purifying such fragments using QIAquick Kits. The
QIAEX II Kit can be used to extract DNA fragments up to 50 kb from agarose or polyacrylamidegels.

PCR – From Setup to Cleanup 10/2015 31
Comments and suggestions
Low or no recovery
Buffer PE did not contain ethanol Ethanol must be added to Buffer PE (concentrate)
before use. Repeat procedure with correctly
prepared Buffer PE.
Inappropriate elution buffer DNA will only be eluted efficiently in the presence
of low-salt buffer (e.g., Buffer EB: 10 mM Tris·Cl,
pH 8.5) or water.
Elution buffer incorrectly dispensed Add elution buffer to the center of the QIAquick
membrane to ensure that the buffer completely
covers the membrane. This is particularly
important when using small elution volumes
(30 μl).
QIAquick Gel Extraction Kit only
Gel slice incompletely solubilized After addition of Buffer QG to the gel slice, mix
by vortexing the tube every 2–3 min during
the 50°C incubation. DNA will remain in any
undissolved agarose.
pH of electrophoresis buffer too high (binding
mixture turns orange or violet)
The electrophoresis buffer has been repeatedly
used or incorrectly prepared, resulting in a
sample pH that exceeds the buffering capacity of
Buffer QG and leads to inefficient DNA binding.
Add 10 μl of 3 M sodium acetate, pH 5.0, to
the sample and mix. The color of the mixture will
turn yellow indicating the correct pH for DNA
binding. Even for binding mixtures with only
small color changes (slight orange color), add
10 μl sodium acetate.
Cleanup troubleshooting

32 PCR – From Setup to Cleanup 10/2015
Cleanup troubleshooting
Comments and suggestions
Gel slice was too large 70–80% recovery can only be obtained from
≤400 mg gel (>400 mg) slice per QIAquick
column. For gel slices >400 mg, use multiple
QIAquick columns.
QIAquick PCR Purification Kit only
Insufficient/no product PCR Estimate DNA recovery by running 10% of
PCR product before and after purification on an
agarose gel.
QIAquick Gel Extraction Kit and QIAquick PCR Purification Kit only
Cloudy and gelatinous appearance of sample
mixture after addition of isopropanol
This may be due to salt precipitation, and will
disappear upon mixing the sample. Alternatively,
the gel slice may not be completely solubilized.
In this case, apply the mixture to the QIAquick
column, centrifuge and then add 0.5 ml Buffer
QG to the column. Let stand for 1 min at room
temperature (15–25°C), and then centrifuge and
continue with the procedure. This additional
wash will solubilize remaining agarose.
Binding mixture turns orange or violet The pH in the sample exceeds the buffer capacity
of Buffer QG or PB respectively. Add 20 μl of
3 M sodium acetate, pH 5.0, to the sample and
mix. The color of the mixture will turn yellow
indicating the correct pH for DNA binding. Even
for samples with slight color changes (orange
color), add 10 μl sodium acetate.

PCR – From Setup to Cleanup 10/2015 33
Comments and suggestions
DNA does not perform well (e.g., in downstream ligation reactions)
Salt concentration in eluate too high Modify the wash step by incubating the column
for 5 min at room temperature after adding
750 μl of Buffer PE, then centrifuge.
Eluate contains residual ethanol Ensure that the wash flow-through is drained from
the collection tube and that the QIAquick column
is then centrifuged at 17,900 x g (13,000 rpm)
for an additional 1 min.
QIAquick Gel Extraction Kit only
Eluate contaminated with agarose The gel slice is incompletely solubilized or
weighs >400 mg. Repeat procedure, including
the optional Buffer QG column-wash step.
QIAquick PCR Purification Kit only
Eluate contains primer–dimers Primer–dimers formed are >20 bp and are not
completely removed. After the binding step,
wash the QIAquick column with 750 μl of a 35%
guanidine hydrochloride aqueous solution (35 g
in 100 ml). Continue with the Buffer PE wash step
and the elution step as in the protocol.
Cleanup troubleshooting

34 PCR – From Setup to Cleanup 10/2015
Cleanup troubleshooting
Comments and suggestions
Eluate contains denatured ssDNA, which
appears as a smaller smeared band on an
analytical gel
Use the eluted DNA to prepare the subsequent
enzymatic reaction but omit the enzyme. To
reanneal the ssDNA, incubate the reaction
mixture at 95°C for 2 min, and allow the tube
to cool slowly to room temperature. Add the
enzyme and proceed as usual. Alternatively,
the DNA can be eluted in 10 mM Tris buffer
containing 10 mM NaCl. The salt and buffering
agent promote the renaturation of DNA strands.
However, the salt concentration of the eluate must
then be considered for subsequent applications.

PCR – From Setup to Cleanup 10/2015 35
Discover our complete range of PCR products at www.qiagen.com.
For up-to-date licensing information and product-specific disclaimers, see the respective QIAGEN
kit handbook or user manual. QIAGEN kit handbooks and user manuals are available at
www.qiagen.com or can be requested from QIAGEN Technical Services or your local distributor.
Trademarks: QIAGEN®, Sample to Insight®, QIAxcel®, QIAEX®, QIAquick®, CoralLoad®, HotStarTaq®, Q-Solution®, TopTaq®, Type-it®, (QIAGEN Group); Parafilm® (Bemis Company, Inc.); MetaPhor® (FMC Bioproducts); SYBR® (Life Technologies Corporation). Registered names, trademarks, etc. used in this document, even when not specifically marked as such, are not to be considered unprotected by law.
PROM-6053-004 © 2015 QIAGEN, all rights reserved.

1096
514
10/2
015





























