Embed Size (px)
Citation preview

LETTERdoi:10.1038/nature13975
Towards a therapy for Angelman syndrome bytargeting a long non-coding RNALinyan Meng1*, Amanda J. Ward2*, Seung Chun2, C. Frank Bennett2, Arthur L. Beaudet1 & Frank Rigo2
Angelman syndrome is a single-gene disorder characterized by intell-ectual disability, developmental delay, behavioural uniqueness, speechimpairment, seizures and ataxia1,2. It is caused by maternal deficiencyof the imprinted gene UBE3A, encoding an E3 ubiquitin ligase3–5. Allpatients carry at least one copy of paternal UBE3A, which is intactbut silenced by a nuclear-localized long non-coding RNA, UBE3Aantisense transcript (UBE3A-ATS)6–8. Murine Ube3a-ATS reductionby either transcription termination or topoisomerase I inhibition hasbeen shown to increase paternal Ube3a expression9,10. Despite a clearunderstanding of the disease-causing event in Angelman syndromeand the potential to harness the intact paternal allele to correct thedisease, no gene-specific treatment exists for patients. Here we de-veloped a potential therapeutic intervention for Angelman syndromeby reducing Ube3a-ATS with antisense oligonucleotides (ASOs). ASOtreatment achieved specific reduction of Ube3a-ATS and sustainedunsilencing of paternal Ube3a in neurons in vitro and in vivo. Par-tial restoration of UBE3A protein in an Angelman syndrome mousemodel ameliorated some cognitive deficits associated with the dis-ease. Although additional studies of phenotypic correction are needed,we have developed a sequence-specific and clinically feasible methodto activate expression of the paternal Ube3a allele.
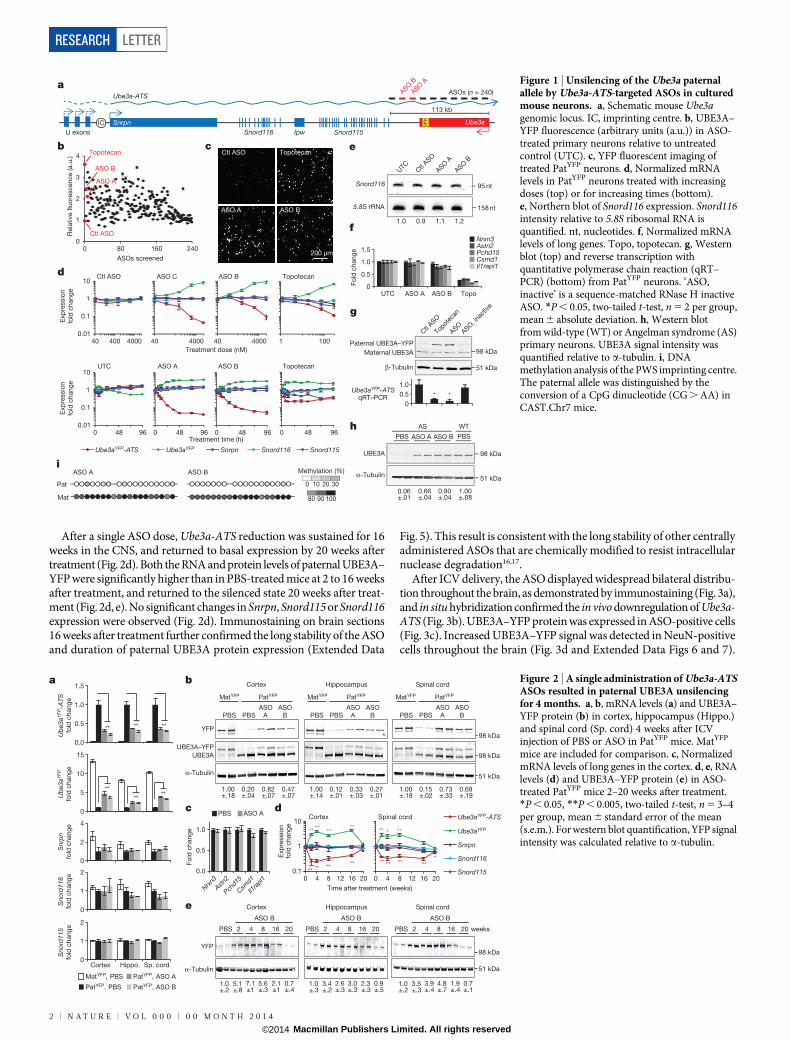
Phosphorothioate-modified chimaeric 29-O-methoxyethyl (29-MOE)DNA ASOs (n 5 240) were designed complementary to a 113 kilobasepair (kb) region of mouse Ube3a-ATS downstream of the Snord115 clus-ter of small nucleolar RNAs (snoRNAs) (Fig. 1a). After nuclear hybrid-ization of the ASO to the target RNA, RNase H cleaves the RNA strandof the ASO–RNA heteroduplex, resulting in subsequent RNA degra-dation by exonucleases11. A high-throughput imaging screen identifiedASOs that unsilenced the Ube3a paternal allele. Primary neurons fromUbe3a1/YFP (PatYFP) knock-in mice12 were cultured and treated withASO (15mM, 72 h), and we determined the fold increase of paternalyellow fluorescent protein (YFP)-tagged UBE3A (UBE3A–YFP) signalin NeuN (also known as Rbfox3)-positive cells (Fig. 1b). The non-targetingcontrol ASO (Ctl ASO) had no effect on fluorescence (0.966 0.01) whereasthe positive control topoisomerase I inhibitor (topotecan, 300 nM) in-creased fluorescence (3.61 6 0.00). ASO A and ASO B resulted in anincrease in paternal UBE3A–YFP fluorescence of 2.11 6 0.02 and 2.476 0.03, respectively (Fig. 1c). ASOs modulated RNA expression in a dose-dependent manner with greater than 90% reduction of Ube3aYFP-ATS(Fig. 1d, top) within 48 h of treatment (Fig. 1d, bottom).
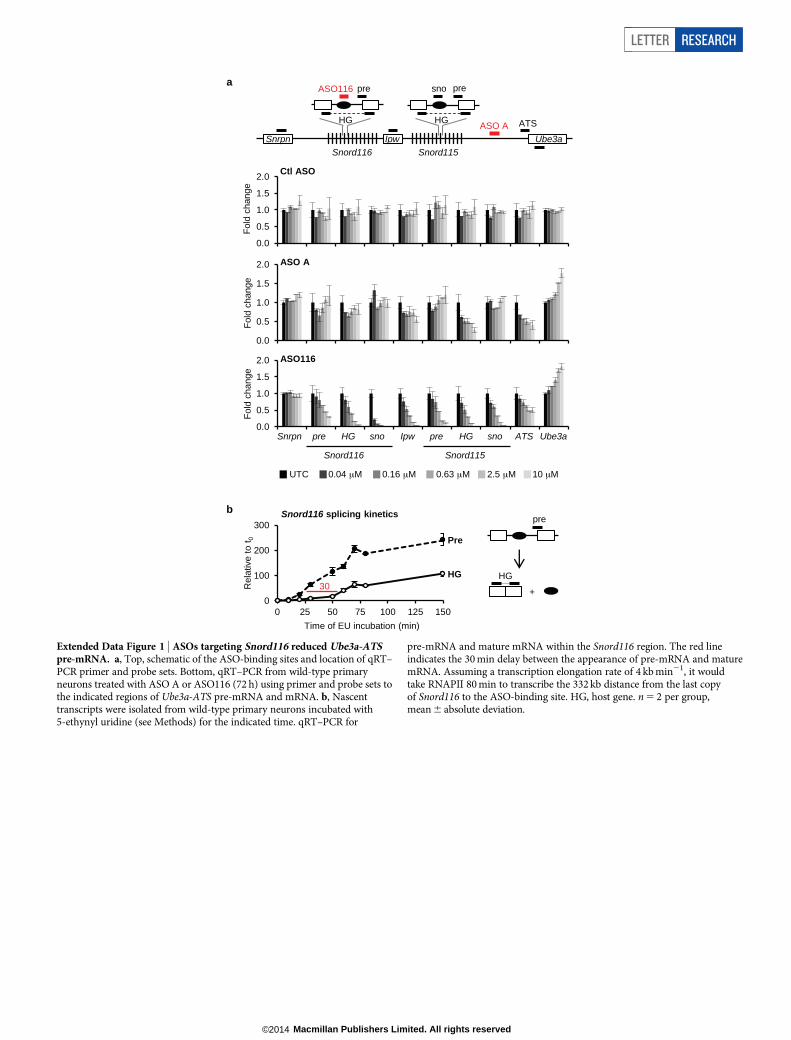
Snrpn, Snord116 and Snord115 are processed from the same precur-sor transcript as Ube3a-ATS (Fig. 1a) and are critical genes in Prader–Willi Syndrome (PWS)13. Their expression was not affected by increasingthe dose or time of ASO treatment (Fig. 1d, e). The ability to down-regulate Ube3a-ATS without affecting Snord116 expression can be attrib-uted to a fast rate of Snord116 splicing (approximately 30 min) relativeto the length of time required for transcription of the 332 kb region be-tween Snord116 and the ASO-binding site (approximately 80 min) (Ex-tended Data Fig. 1). While Ube3a-ATS ASOs did not affect expressionof mature Snord116 or its precursor, ASOs designed directly to Snord116
strongly reduced Snord116 and the entire Ube3a-ATS precursor tran-script (Extended Data Fig. 1).
ASO treatment (10mM, 24 h) specifically reduced Ube3a-ATS(1,000 kb) without affecting expression of five other long genes (Nrxn3,1,612 kb; Astn2, 1,024 kb; Pcdh15, 828 kb; Csmd1, 1,643 kb; Il1rapl1,1,368 kb), whereas topotecan (300 nM, 24 h), which acts by impairingtranscription elongation14, strongly inhibited their expression (Fig. 1f).
Primary neurons from PatYFP mice treated with ASO (10mM, 72 h) ortopotecan (300 nM, 72 h) resulted in biallelic UBE3A protein express-ion due to unsilencing of the paternal allele (Fig. 1g). Additionally, ASOtreatment of primary neurons from Ube3aKO/1 (Angelman syndrome)mice15 achieved 66–90% of wild-type levels of UBE3A protein (Fig. 1h).ASO treatment (10mM) did not affect DNA methylation at the PWSimprinting centre (Fig. 1i). A sequence-matched ASO that was renderedunresponsive to RNase H by complete modification with 29-MOEnucleotides (ASO, inactive) did not affect paternal UBE3A expression,indicating that reduction of the antisense transcript is required forpaternal Ube3a unsilencing (Fig. 1g).
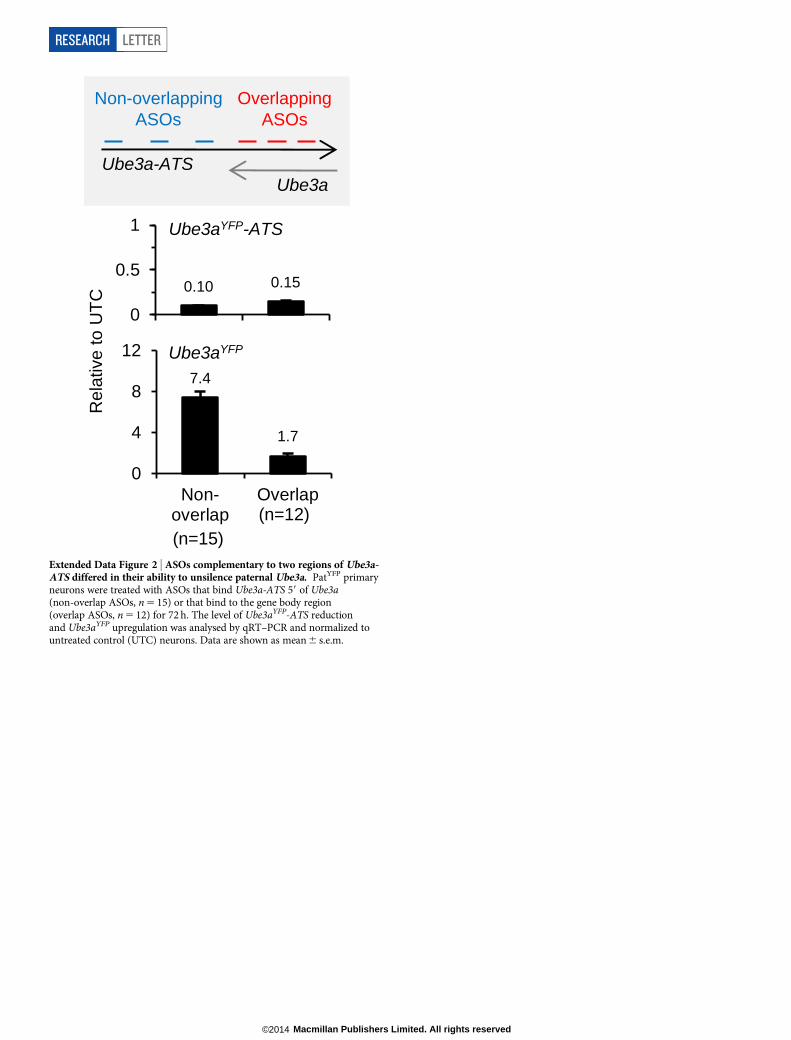
Although reduction of the antisense transcript was required, addi-tional studies indicated that it was not sufficient for paternal Ube3a un-silencing. ASOs complementary to the region of Ube3aYFP-ATS upstreamof Ube3a (non-overlapping ASOs, n 5 15) upregulated Ube3aYFP RNA7.4 6 0.6 fold relative to untreated control neurons (Extended Data Fig. 2).ASOs complementary to the region of Ube3aYFP-ATS located within theUbe3a gene body (overlapping ASOs, n 5 12) only upregulated Ube3aYFP
RNA 1.76 0.2 fold. Because both non-overlapping and overlapping ASOsreduced Ube3aYFP-ATS to a similar level, a mechanism independentof the presence of the long non-coding RNA may have a role in Ube3asilencing.
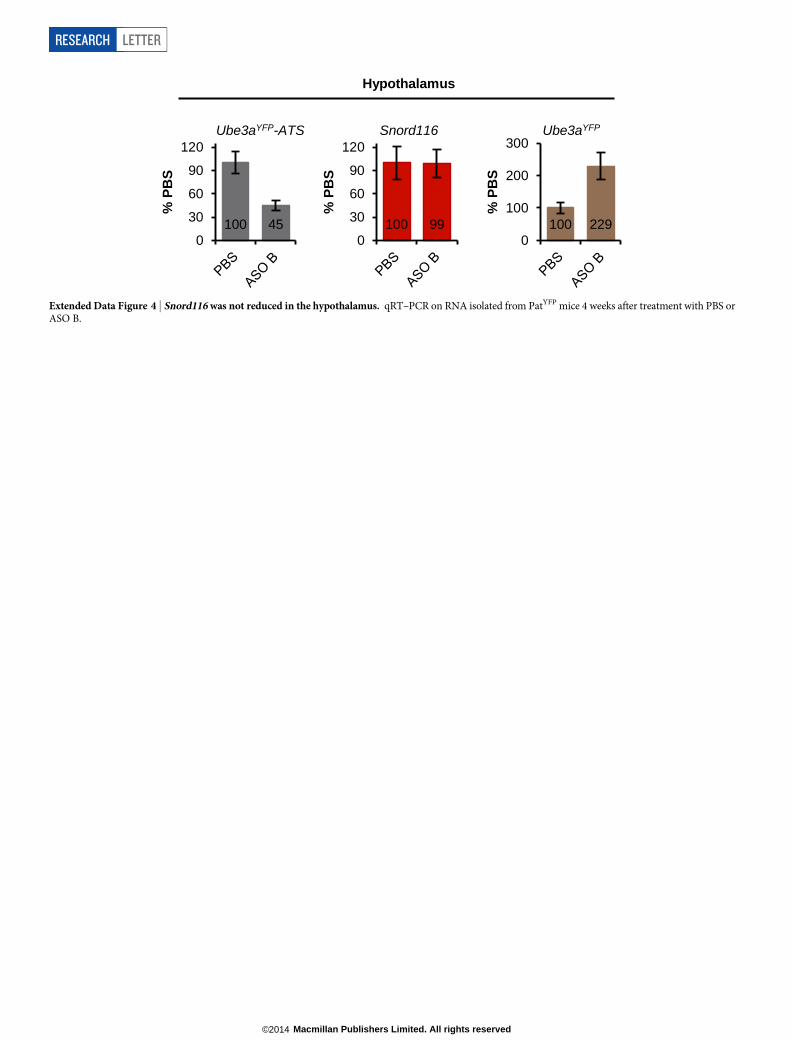
Next, we tested whether central nervous system (CNS) administra-tion of Ube3a-ATS ASOs unsilenced paternal Ube3a in vivo. A singleintracerebroventricular (ICV) injection of ASO was administered intothe lateral ventricle of adult PatYFP mice. The ASO treatment was gener-ally well tolerated, despite transient sedation after surgery. No significantchanges in body weight, expression of AIF1 (marker for microgliosis),or expression of GFAP (marker for astrocytosis) were observed 1 monthafter treatment (Extended Data Fig. 3). Four weeks after treatment, ASOA and ASO B reduced Ube3a-ATS RNA by 60–70% and upregulatedpaternal Ube3aYFP RNA two- to five-fold in the brain and spinal cord(Fig. 2a). However, compared with Ube3aYFP/1 (MatYFP) mice, ASOtreatment did not fully unsilence the paternal allele. Ube3aYFP RNA inASO-treated PatYFP mice was 30–40% of the level in MatYFP mice (Fig. 2a).Western blot quantification showed that UBE3A–YFP protein wasupregulated in the cortex (82 6 7%), hippocampus (33 6 3%) and thor-acic spinal cord (73 6 33%) in ASO-A-treated PatYFP mice comparedwith MatYFP mice (Fig. 2b). No significant downregulation of Snrpn,Snord116, Snord115, or the sentinel long genes was observed, includingany Snord116 reduction in the hypothalamus (Fig. 2a, c and ExtendedData Fig. 4).
*These authors contributed equally to this work.
1Department of Molecular and Human Genetics, Baylor College of Medicine, and Texas Children’s Hospital, Houston, Texas 77030, USA. 2Department of Core Antisense Research, Isis Pharmaceuticals,Carlsbad, California 92010, USA.
0 0 M O N T H 2 0 1 4 | V O L 0 0 0 | N A T U R E | 1
Macmillan Publishers Limited. All rights reserved©2014
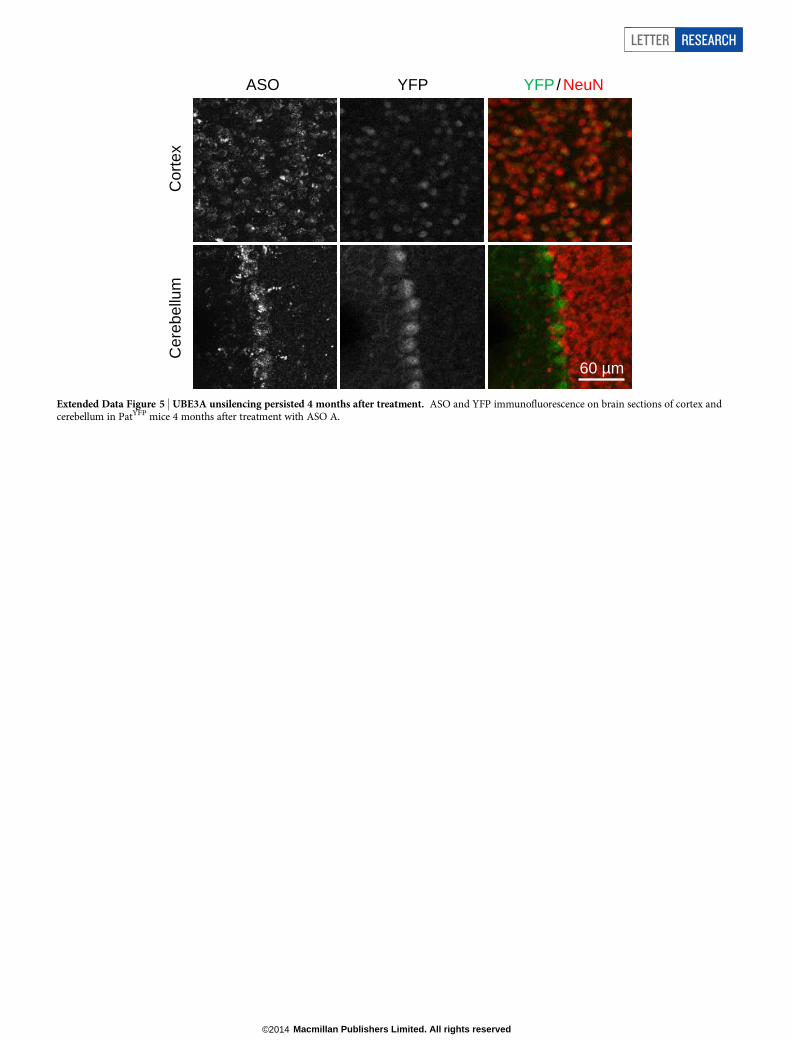
After a single ASO dose, Ube3a-ATS reduction was sustained for 16weeks in the CNS, and returned to basal expression by 20 weeks aftertreatment (Fig. 2d). Both the RNA and protein levels of paternal UBE3A–YFP were significantly higher than in PBS-treated mice at 2 to 16 weeksafter treatment, and returned to the silenced state 20 weeks after treat-ment (Fig. 2d, e). No significant changes in Snrpn, Snord115 or Snord116expression were observed (Fig. 2d). Immunostaining on brain sections16 weeks after treatment further confirmed the long stability of the ASOand duration of paternal UBE3A protein expression (Extended Data
Fig. 5). This result is consistent with the long stability of other centrallyadministered ASOs that are chemically modified to resist intracellularnuclease degradation16,17.
After ICV delivery, the ASO displayed widespread bilateral distribu-tion throughout the brain, as demonstrated by immunostaining (Fig. 3a),and in situ hybridization confirmed the in vivo downregulation of Ube3a-ATS (Fig. 3b). UBE3A–YFP protein was expressed in ASO-positive cells(Fig. 3c). Increased UBE3A–YFP signal was detected in NeuN-positivecells throughout the brain (Fig. 3d and Extended Data Figs 6 and 7).
0 48 96
641788
158 nt
95 nt
51 kDa
98 kDa
e
Ube3a
Ipw
Ube3a-ATS
Snord116 Snord115Snrpn
ASOs (n = 240)ASO B
UTC
Ctl
ASO
Ctl
ASO
ASOASO
, ina
ctive
Topot
ecan
ASO A
ASO B
ASO A
IC
a
bU exons
113 kb
0
0.5
1.0
1.5
UTC ASO A ASO B Topo
Fo
ld c
han
ge
Paternal UBE3A–YFP
Maternal UBE3A
β-Tubulin
Ube3aYFP-ATSqRT–PCR * *
0
0.5
1.0
f
g
0.06±.01
PBS
UBE3A
α-Tubulin
0.66±.04
0.90±.04
1.00±.08
ASO A ASO B PBS
AS WTh
Nrxn3Astn2Pchd15Csmd1Il1rapl1
51 kDa
98 kDa
Snord116
5.8S rRNA
Ube3aYFPUbe3aYFP-ATS Snrpn Snord116 Snord115
d
0 48 960.01
0.1
1
10
0 48 96
Ctl
0 48 96
592401UTC ASO A ASO B Topotecan
Treatment time (h)
0.01
0.1
1
10
40 400 4000
Exp
ressio
n
fold
ch
an
ge
Exp
ressio
n
fold
chang
e
Ctl
40 4000
592401
40 4000
592483
1 100
Topotecan
Treatment dose (nM)
Ctl ASO ASO C ASO B Topotecan
cCtl ASO Topotecan
ASO BASO A
200 μm
iASO A
Pat
Mat
ASO B
0 10 20
80 90 100
Methylation (%)
30
YF
P
0
1
2
3
4
0 80 160 240
Rela
tive fl
uo
rescen
ce (a.u
.)
ASOs screened
1.0 0.9 1.1 1.2
Topotecan
ASO B
ASO A
Ctl ASO
Figure 1 | Unsilencing of the Ube3a paternalallele by Ube3a-ATS-targeted ASOs in culturedmouse neurons. a, Schematic mouse Ube3agenomic locus. IC, imprinting centre. b, UBE3A–YFP fluorescence (arbitrary units (a.u.)) in ASO-treated primary neurons relative to untreatedcontrol (UTC). c, YFP fluorescent imaging oftreated PatYFP neurons. d, Normalized mRNAlevels in PatYFP neurons treated with increasingdoses (top) or for increasing times (bottom).e, Northern blot of Snord116 expression. Snord116intensity relative to 5.8S ribosomal RNA isquantified. nt, nucleotides. f, Normalized mRNAlevels of long genes. Topo, topotecan. g, Westernblot (top) and reverse transcription withquantitative polymerase chain reaction (qRT–PCR) (bottom) from PatYFP neurons. ‘ASO,inactive’ is a sequence-matched RNase H inactiveASO. *P , 0.05, two-tailed t-test, n 5 2 per group,mean 6 absolute deviation. h, Western blotfrom wild-type (WT) or Angelman syndrome (AS)primary neurons. UBE3A signal intensity wasquantified relative to a-tubulin. i, DNAmethylation analysis of the PWS imprinting centre.The paternal allele was distinguished by theconversion of a CpG dinucleotide (CG . AA) inCAST.Chr7 mice.
98 kDa
51 kDa
98 kDa
** ** ** ***
** * **
0.1
1
10
0 4 8 12 16 20
a b
MatYFP, PBS PatYFP, ASO A
PatYFP, ASO B
Ub
e3aY
FP-A
TSfo
ld c
hang
e
Ub
e3aY
FP
fold
chang
e
Sno
rd11
6fo
ld c
hang
e
Sno
rd11
5fo
ld c
hang
e
Snr
pn
fold
chang
e
Cortex Hippo. Sp. cord
0.0
0.5
1.0
Nrxn3Astn
2
Pchd1
5
Csmd1
Il1ra
pl1
Fo
ld c
hang
e
PBS ASO A
Cortex Hippocampus Spinal cord
ASO B
weeks
YFP
α-Tubulin
c
e
Exp
ressio
nfo
ld c
hang
e
d
2 4 8 16 20PBS
ASO B
2 4 8 16 20PBS
ASO B
2 4 8 16 20PBS
Spinal cordCortex
Time after treatment (weeks)
** ** ****
** ****
**
0.1
1
10
0 4 8 12 16 20
Ube3aYFP
Ube3aYFP-ATS
Snrpn
Snord116
Snord115
0.0
0.5
1.0
1.5
0
5
10
15
0
2
4
0
1
2
0
1
2
PatYFP, PBS
** ** **
**
**
1.0±.2
5.1±.8
7.1±1
5.6±.3
2.1±1
0.7±.4
1.0±.3
3.4±.2
2.6±.3
3.0±.3
2.3±.3
0.9±.5
1.0±.2
3.5±.3
3.9±.4
4.8±.7
1.9±.4
0.7±.1
**
98 kDa
51 kDa
PBS PBSASO
B
MatYFP PatYFP MatYFP PatYFP MatYFP PatYFP
ASOA
YFP
α-Tubulin
PBS PBSASO
BASO
A PBS PBSASO
BASO
A
Cortex Hippocampus Spinal cord
UBE3A–YFP
1.00±.18
0.20±.04
0.82±.07
0.47±.07
1.00±.14
0.12±.01
0.33±.03
0.27±.01
1.00±.18
0.15±.02
0.73±.33
0.68±.19
UBE3A
Figure 2 | A single administration of Ube3a-ATSASOs resulted in paternal UBE3A unsilencingfor 4 months. a, b, mRNA levels (a) and UBE3A–YFP protein (b) in cortex, hippocampus (Hippo.)and spinal cord (Sp. cord) 4 weeks after ICVinjection of PBS or ASO in PatYFP mice. MatYFP
mice are included for comparison. c, NormalizedmRNA levels of long genes in the cortex. d, e, RNAlevels (d) and UBE3A–YFP protein (e) in ASO-treated PatYFP mice 2–20 weeks after treatment.*P , 0.05, **P , 0.005, two-tailed t-test, n 5 3–4per group, mean 6 standard error of the mean(s.e.m.). For western blot quantification, YFP signalintensity was calculated relative to a-tubulin.
RESEARCH LETTER
2 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 4
Macmillan Publishers Limited. All rights reserved©2014

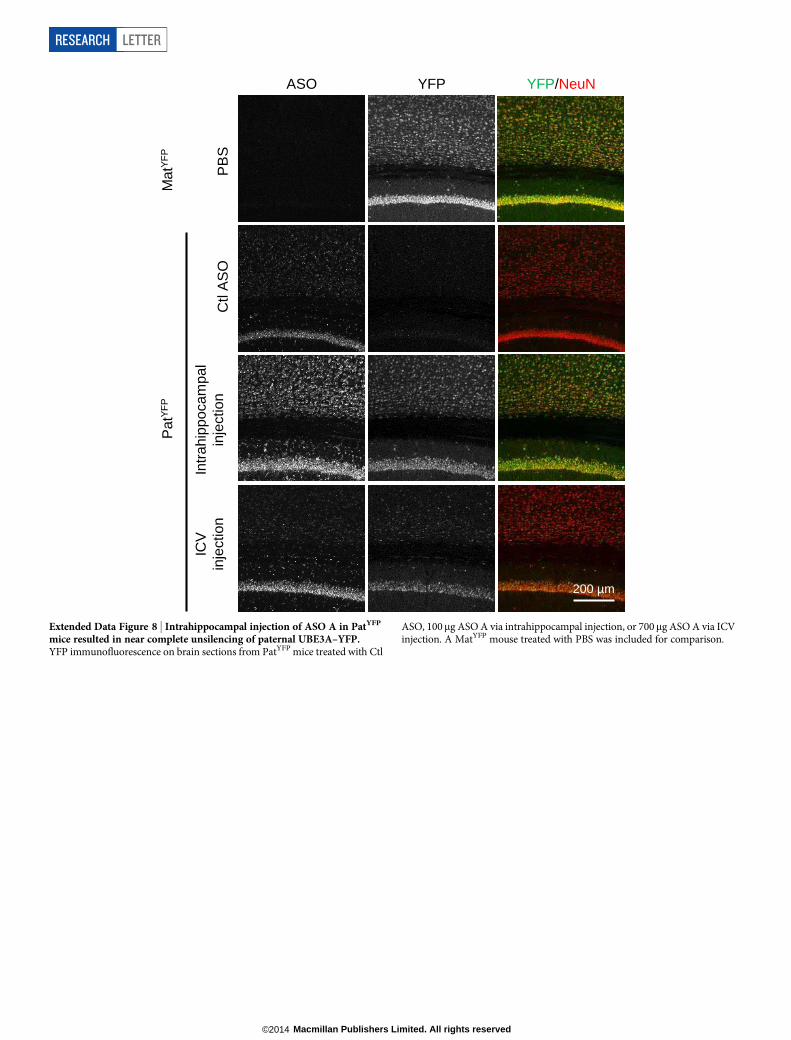
However, paternal unsilencing was not complete compared with thematernal UBE3A–YFP level, consistent with the western blot analysis.To further increase the concentration of ASO in the brain, intrahip-pocampal delivery of ASO A was performed in PatYFP mice and com-plete unsilencing of UBE3A–YFP was observed near the injection site(Extended Data Fig. 8).
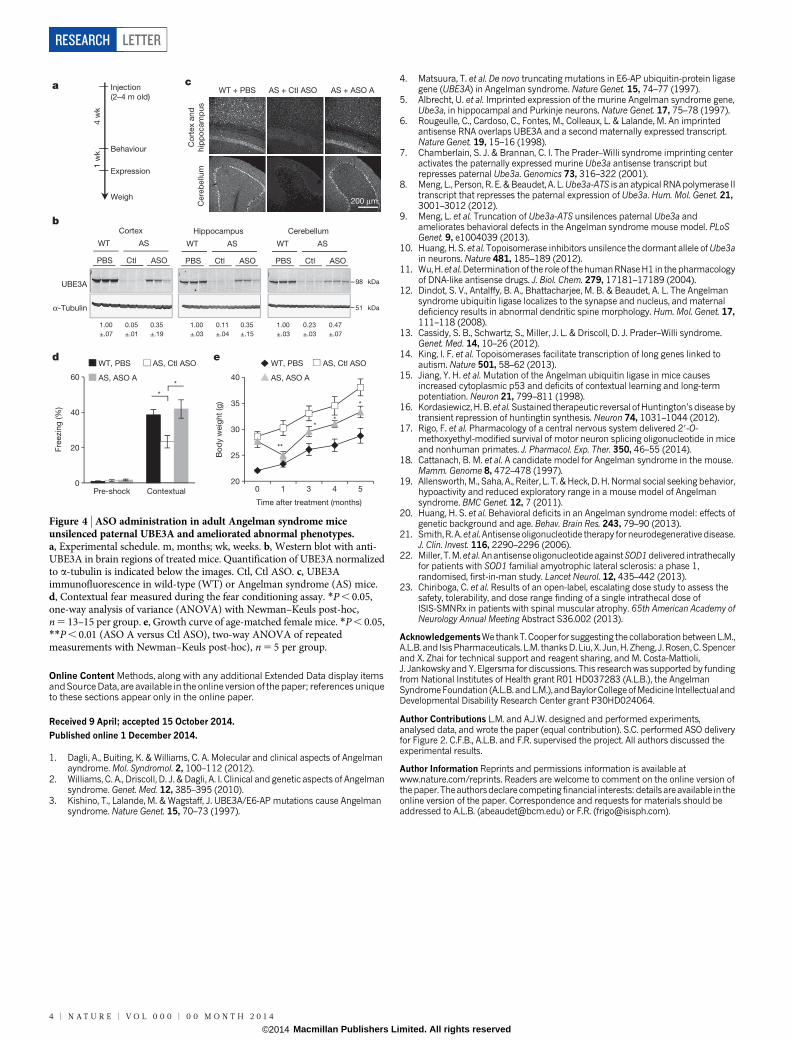
On the basis of the ability of ASO A to upregulate UBE3A, it was cho-sen for assessment of phenotypic correction in Angelman syndrome mice.Angelman syndrome mice phenocopy the impaired motor coordina-tion and memory deficit observed in patients with the disease15. Theyhave additional phenotypes including obesity, hypoactivity and decreasedmarble burying behaviour18–20. Sex-matched Angelman syndrome litter-mates at 2–4 months of age were treated with ASO A or non-targetingcontrol ASO (Ctl ASO). To determine the ability of ASO A to correctexpression and behaviours relative to wild-type levels, a group of PBS-treated wild-type mice was included. After a single ICV injection, Angelmansyndrome mice treated with ASO A showed reduction of Ube3a-ATSand partial restoration of UBE3A protein in the cortex (35 6 19%), hip-pocampus (35 6 15%) and cerebellum (47 6 7%) compared with wild-type mice (Fig. 4b and Extended Data Fig. 9). UBE3A immunofluorescencealso showed partial restoration of UBE3A protein in these brain regions(Fig. 4c and Extended Data Fig. 9). Four weeks after treatment, the micewere subjected to behavioural tests. A reversal of contextual freezingcomparable to normal behaviour was observed in ASO-A-treatedAngelman syndrome mice (analysis of variance (ANOVA), F(2,39) 5
5.242, P , 0.01), indicating that the memory impairment was reversed
(Fig. 4d and Extended Data Fig. 9). However, there was no differencebetween mice treated with ASO A or Ctl ASO in open field, marbleburying and accelerating rotarod tests (Extended Data Fig. 9). Com-plete phenotypic reversal may require treatment before a critical devel-opmental window, a longer recovery time for rewiring of neural circuits,or a higher UBE3A induction level. Body weight was measured in a setof female mice that were injected at 3 months of age and followed for5 months (Fig. 4e and Extended Data Fig. 9). The obesity phenotype inAngelman syndrome mice was corrected 1 month after treatment, andbody weight remained significantly decreased compared with controlASO-treated mice for 5 months.
The genomic organization and regulation at the imprinting controlcentre is highly conserved between mouse and human. Therefore, ASO-mediated reduction of UBE3A-ATS is expected to restore UBE3A mes-senger RNA and protein in Angelman syndrome patient neurons. It isbelieved that maternal deficiency of UBE3A causes the majority of phe-notypic findings in Angelman syndrome, and it is reasonable to expectthat all patients with the condition, regardless of exact genotype, wouldbenefit enormously from restored UBE3A expression. ASO therapy hasbeen tested for neurological diseases in non-human primates and humanclinical trials via intrathecal administration, with no serious adverseevents16,21–23. The well-tolerated delivery, broad tissue distribution andlong duration of action indicate that ASOs may be a viable therapeuticstrategy for CNS diseases and highlights the potential of an ASO drugfor Angelman syndrome.
ASO YFP
PB
S
MatY
FP
PatY
FP
Ctl A
SO
A
SO
B
AS
O A
YFP/NeuN
Cortex and hippocampus Cerebellum
ASO YFP
d
a
c CA1 pyramidal neurons Cerebellum Purkinjie neurons
YFP/NeuN
Ub
e3a-
ATS
YFP ASO/YFP
PBS
ASO B
YFP ASO/YFP
b Cortex Hippocampus
PBS ASO A PBS ASO A
50 μm
50 μm
200 μm 200 μm
Figure 3 | Widespread distribution of paternal UBE3A unsilencingthroughout the brain. a–d, Imaging of brain sections 4 weeks after treatmentwith Ctl ASO or ASO A and/or B in PatYFP mice, as labelled. a, ASOimmunofluorescence on whole brain coronal sections. b, In situ hybridization
of Ube3a-ATS. c, High-magnification staining of UBE3A–YFP and ASO.d, Immunofluorescence for ASO, UBE3A–YFP and NeuN in criticalbrain regions. MatYFP mice treated with PBS were included forexpression comparison.
LETTER RESEARCH
0 0 M O N T H 2 0 1 4 | V O L 0 0 0 | N A T U R E | 3
Macmillan Publishers Limited. All rights reserved©2014
Online Content Methods, along with any additional Extended Data display itemsandSourceData, are available in the online version of the paper; references uniqueto these sections appear only in the online paper.
Received 9 April; accepted 15 October 2014.
Published online 1 December 2014.
1. Dagli, A., Buiting, K. & Williams, C. A. Molecular and clinical aspects of Angelmanayndrome. Mol. Syndromol. 2, 100–112 (2012).
2. Williams, C. A., Driscoll, D. J. & Dagli, A. I. Clinical and genetic aspects of Angelmansyndrome. Genet. Med. 12, 385–395 (2010).
3. Kishino, T., Lalande, M. & Wagstaff, J. UBE3A/E6-AP mutations cause Angelmansyndrome. Nature Genet. 15, 70–73 (1997).
4. Matsuura, T. et al. De novo truncating mutations in E6-AP ubiquitin-protein ligasegene (UBE3A) in Angelman syndrome. Nature Genet. 15, 74–77 (1997).
5. Albrecht, U. et al. Imprinted expression of the murine Angelman syndrome gene,Ube3a, in hippocampal and Purkinje neurons. Nature Genet. 17, 75–78 (1997).
6. Rougeulle, C., Cardoso, C., Fontes, M., Colleaux, L. & Lalande, M. An imprintedantisense RNA overlaps UBE3A and a second maternally expressed transcript.Nature Genet. 19, 15–16 (1998).
7. Chamberlain, S. J. & Brannan, C. I. The Prader–Willi syndrome imprinting centeractivates the paternally expressed murine Ube3a antisense transcript butrepresses paternal Ube3a. Genomics 73, 316–322 (2001).
8. Meng, L., Person, R. E. & Beaudet, A. L. Ube3a-ATS is an atypical RNA polymerase IItranscript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 21,3001–3012 (2012).
9. Meng, L. et al. Truncation of Ube3a-ATS unsilences paternal Ube3a andameliorates behavioral defects in the Angelman syndrome mouse model. PLoSGenet. 9, e1004039 (2013).
10. Huang, H. S. et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3ain neurons. Nature 481, 185–189 (2012).
11. Wu, H.et al. Determination of the role of thehumanRNase H1 in the pharmacologyof DNA-like antisense drugs. J. Biol. Chem. 279, 17181–17189 (2004).
12. Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B. & Beaudet, A. L. The Angelmansyndrome ubiquitin ligase localizes to the synapse and nucleus, and maternaldeficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17,111–118 (2008).
13. Cassidy, S. B., Schwartz, S., Miller, J. L. & Driscoll, D. J. Prader–Willi syndrome.Genet. Med. 14, 10–26 (2012).
14. King, I. F. et al. Topoisomerases facilitate transcription of long genes linked toautism. Nature 501, 58–62 (2013).
15. Jiang, Y. H. et al. Mutation of the Angelman ubiquitin ligase in mice causesincreased cytoplasmic p53 and deficits of contextual learning and long-termpotentiation. Neuron 21, 799–811 (1998).
16. Kordasiewicz,H. B. et al. Sustained therapeutic reversal of Huntington’s disease bytransient repression of huntingtin synthesis. Neuron 74, 1031–1044 (2012).
17. Rigo, F. et al. Pharmacology of a central nervous system delivered 29-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in miceand nonhuman primates. J. Pharmacol. Exp. Ther. 350, 46–55 (2014).
18. Cattanach, B. M. et al. A candidate model for Angelman syndrome in the mouse.Mamm. Genome 8, 472–478 (1997).
19. Allensworth, M., Saha, A., Reiter, L. T. & Heck, D. H. Normal social seeking behavior,hypoactivity and reduced exploratory range in a mouse model of Angelmansyndrome. BMC Genet. 12, 7 (2011).
20. Huang, H. S. et al. Behavioral deficits in an Angelman syndrome model: effects ofgenetic background and age. Behav. Brain Res. 243, 79–90 (2013).
21. Smith, R. A.et al.Antisense oligonucleotide therapy for neurodegenerative disease.J. Clin. Invest. 116, 2290–2296 (2006).
22. Miller, T.M.et al.AnantisenseoligonucleotideagainstSOD1 delivered intrathecallyfor patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1,randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013).
23. Chiriboga, C. et al. Results of an open-label, escalating dose study to assess thesafety, tolerability, and dose range finding of a single intrathecal dose ofISIS-SMNRx in patients with spinal muscular atrophy. 65th American Academy ofNeurology Annual Meeting Abstract S36.002 (2013).
Acknowledgements WethankT.Cooper for suggesting the collaboration between L.M.,A.L.B. and IsisPharmaceuticals. L.M. thanksD. Liu, X. Jun,H. Zheng, J.Rosen,C. Spencerand X. Zhai for technical support and reagent sharing, and M. Costa-Mattioli,J. Jankowsky and Y. Elgersma for discussions. This research was supported by fundingfrom National Institutes of Health grant R01 HD037283 (A.L.B.), the AngelmanSyndrome Foundation (A.L.B. and L.M.), andBaylorCollegeofMedicine Intellectual andDevelopmental Disability Research Center grant P30HD024064.
Author Contributions L.M. and A.J.W. designed and performed experiments,analysed data, and wrote the paper (equal contribution). S.C. performed ASO deliveryfor Figure 2. C.F.B., A.L.B. and F.R. supervised the project. All authors discussed theexperimental results.
Author Information Reprints and permissions information is available atwww.nature.com/reprints. Readers are welcome to comment on the online version ofthepaper. Theauthorsdeclare competing financial interests: details are available in theonline version of the paper. Correspondence and requests for materials should beaddressed to A.L.B. ([email protected]) or F.R. ([email protected]).
WT, PBS AS, Ctl ASO
AS, ASO A
d e
Pre-shock Contextual
Co
rtex a
nd
hip
po
cam
pus
Cere
bellu
m
WT + PBS AS + Ctl ASO AS + ASO Ac
200 μm
98 kDa
51 kDa
b
WT AS
Ctl ASO
α-Tubulin
1.00
±.07
PBS
UBE3A
0.05
±.01
0.35
±.19
1.00
±.03
0.11
±.04
0.35
±.15
1.00
±.03
0.23
±.03
0.47
±.07
Cortex
WT AS
Ctl ASOPBS
Hippocampus
WT AS
Ctl ASOPBS
Cerebellum
0
20
40
60
Fre
ezin
g (%
)
WT, PBS AS, Ctl ASO
AS, ASO A*
*
a4 w
kInjection
(2–4 m old)
Behaviour
Expression
Weigh
1 w
k
20
25
30
35
40
0 1 3 4 5
Bo
dy w
eig
ht
(g)
Time after treatment (months)
**
*
**
Figure 4 | ASO administration in adult Angelman syndrome miceunsilenced paternal UBE3A and ameliorated abnormal phenotypes.a, Experimental schedule. m, months; wk, weeks. b, Western blot with anti-UBE3A in brain regions of treated mice. Quantification of UBE3A normalizedto a-tubulin is indicated below the images. Ctl, Ctl ASO. c, UBE3Aimmunofluorescence in wild-type (WT) or Angelman syndrome (AS) mice.d, Contextual fear measured during the fear conditioning assay. *P , 0.05,one-way analysis of variance (ANOVA) with Newman–Keuls post-hoc,n 5 13–15 per group. e, Growth curve of age-matched female mice. *P , 0.05,**P , 0.01 (ASO A versus Ctl ASO), two-way ANOVA of repeatedmeasurements with Newman–Keuls post-hoc), n 5 5 per group.
RESEARCH LETTER
4 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 4
Macmillan Publishers Limited. All rights reserved©2014

METHODSAnimals. All the animals of wild-type, Ube3aKO/1 (ref. 15), and Ube3a1/YFP (ref. 12)genotypes were kept on C57BL/6 background and housed under standard condi-tions in a pathogen-free mouse facility. All animal procedures were performed inaccordance with National Institutes of Health guidelines and approved by the In-stitutional Animal Care and Use Committee at Baylor College of Medicine and IsisPharmaceuticals. To generate Angelman syndrome mice, Ube3aKO/1 mice wereborn to mothers who carry the mutation on their paternal chromosome. Wild-type and Angelman syndrome littermates were housed in the same cage wheneverpossible.Oligonucleotide synthesis. Synthesis and purification of all chemically modifiedoligonucleotides was performed as previously described24. The 29-MOE gapmer ASOsare 20 nucleotides in length, wherein the central gap segment comprising ten 29-deoxynucleotides is flanked on the 59 and 39 wings by five 29-MOE modified nucle-otides. Internucleoside linkages are purely phosphorothioate (ASO B) or interspersedwith phosphodiester (ASO A), and all cytosine residues are 59-methylcytosines. TheRNase H inactive ASO consists of 20 29-MOE modified nucleotides. The sequencesof the ASOs are as follows: Ctl ASO (in vitro), 59-CCTTCCCTGAAGGTTCCTCC-39; Ctl ASO (in vivo), 59-CTCAGTAACATTGACACCAC-39 (ref. 25); ASO A, 59-GATCCATTTGTGTTAAGCTG-39; ASO B, 59-CCAGCCTTGTTGGATATCAT-39; ASO116, 59-CAGAGTTTTCACTCATTTTG-39.Primary neuron culture and ASO treatment. Primary cultures of hippocampaland cortical neurons were established as previously described8 from P0–P2 off-spring of wild-type C57BL/6 or Ube3a1/YFP mice. Four days after plating, half ofthe medium was replaced and the ASO (10mM) or topotecan (300 nM) was addedto the culture medium for 72 h, unless otherwise noted. Arabinofuranosyl cytidine(Sigma) was used to inhibit glial proliferation.Immunofluorescence. Primary neurons were fixed with 4% paraformaldehyde(PFA) for 1 h and washed in PBS. For in vivo samples, mice were anaesthetized andperfused with PBS and 4% PFA. Brain tissue was fixed with PFA overnight and de-hydrated in 30% sucrose. Coronal sections of 35mm were prepared and stained aspreviously described9. The following antibodies were used: anti-GFP (ab13970, Abcam,1:1,000), anti-NeuN (MAB377, Millipore, 1:1,000 dilution), anti-UBE3A (A300-352A, Bethyl Laboratories,1:500), and anti-ASO (Isis, 1:10,000)26. For the high-throughput in vitro ASO screen, the plates were imaged with ImageXpressUltra
confocal system (Molecular Device) and then further processed with the MetaXpresssoftware (Molecular Device). Typically 200–800 cells were scored per well and thesignal intensities were averaged and normalized to untreated control cells. For tissuesections, images were taken using a confocal microscope (Leica).qRT–PCR. Total cellular RNA was isolated from cultured neurons and mouse tis-sue using the RNeasy kit (Qiagen). For preparation of mouse tissue, samples werefirst lysed using FastPrep Lysing Matrix Tubes (MP-Biomedicals) in RLT buffercontaining 1% b-mercaptoethanol. On-column DNase digestion was performedfor all samples. For qRT–PCR, approximately 10 ng RNA was added to EXPRESSOne-Step SuperScript qRT–PCR Kit (Life Technologies) with Taqman primer andprobe sets or EXPRESS One-Step SYBR GreenER Kit (Life Technologies) with SYBRprimer sets (see Extended Data Table 1 for sequences). All quantification was per-formed by the relative standard curve method and normalized to total RNA byRibogreen or to the housekeeping genes Gapdh.DNA methylation analysis. Primary neuron cultures were derived from the F1hybrid of CAST.chr7 male and C57BL/6 female mice and treated with ASO (10mM,72 h). Genomic DNA was then extracted and processed for bisulphite sequencingof the PWS imprinting centre at the Snrpn DMR1 region (Snrpn promoter andexon 1) as previously described8.Northern blot. Total RNA was isolated from ASO-treated primary neurons (10mM,72 h) by TRIzol (Life Technologies) according to the manufacturer’s protocol. Threemicrograms total RNA was separated on an 8% polyacrylamide-7M urea gel, andthen transferred by semi-dry transfer (12 V, 30 min) to GeneScreen plus hybridiza-tion transfer membrane (Perkin Elmer). The northern probes were 59-end labelledwith ATP Gamma 32P (Perkin Elmer) using T4 polynucleotide kinase (New EnglandBiolabs), and then hybridized to the membrane at 42 uC for 30 min. After washingmembrane in wash buffer (23 SSC containing 0.1% SDS), the membrane was ex-posed to a PhosphorImager and quantified. The oligonucleotide probe sequencesused were Snord116 59-TTCCGATGAGAGTGGCGGTACAGA-39 and 5.8S rRNA59-TCCTGCAATTCACATTAATTCTCGCAGCTAGC-39.Western blot. Cultured neurons and mouse tissue were homogenized and lysed inRIPA buffer (Sigma-Aldrich) containing EDTA-free cOmplete Protease InhibitorCocktail (Roche). Protein concentration of the supernatant was determined by theDC protein assay (Bio-Rad). Ten to forty micrograms of protein lysate was separatedon a precast 4–20% Bis-Tris gel (Life Technologies) and transferred by iBlot (LifeTechnologies). The following primary antibodies were diluted in Odyssey block-ing buffer: anti-UBE3A (611416, BD Biosciences, 1:500), anti-GFP (NB600-308,Novus Biologicals, 1:500), anti-b-tubulin (T9026, Sigma, 1:20,000) and anti-a-tubulin
(T5168, Sigma, 1:8,000). Following primary antibody incubation, membranes wereprobed with goat anti-rabbit IRDye 680LT (LiCor) or goat anti-mouse IRDye 800CW(LiCor) and imaged and quantified using the LiCor Odyssey system.ASO in vivo administration. Lyophilized ASOs were dissolved in sterile PBS with-out calcium or magnesium and quantified by ultraviolet spectrometry. The ASOswere then diluted to the desired concentration required for dosing mice and ster-ilized through a 0.2mm filter. Mice were anaesthetized with 2% isoflurane and placedin a stereotaxic frame (David Kopf Instruments). After exposing the skull, a needle(Hamilton, 1701 RN 10 ml micro syringe, needle 26 s/299/2) was used to penetratethe skull at 0.2 mm posterior and 1.0 mm lateral to the bregma, and lowered to adepth of 3.0 mm, to deliver PBS or ASO (ASO A, 700mg; ASO B, 500mg) at a rateof approximately 1ml per 30 s. The needle was left in place for 5 min, slowly with-drawn and the incision was sutured. For intrahippocampal injection, the coord-inate of 22.0 mm anterior, 1.5 mm lateral and 22.0 mm dorsal to the bregma wasused.Fluorescence in situ hybridization. Tissue preparation and RNA fluorescence insitu hybridization (FISH) was carried out by the RNA In-Situ Hybridization Coreat Baylor College of Medicine, as previously described9. Primers for DNA templatesynthesis are 59-ATTTAGGTGACACTATAGAAGCGAAGATGAGTCAGTTTGGTTTT-39 and 59-TAATACGACTCACTATAGGGAGATTCTGAGTCTTCTTCCATAGC-39. The T7 promoter was used to generate the Ube3a-ATS probe.Behavioural tests. Three groups of age- and sex-matched littermates were gen-erated, and mice were randomly assigned to each treatment group. At 2 to 4 monthsof age, Angelman syndrome mice received a single 700mg dose of non-targetingcontrol ASO or ASO A. Wild-type mice injected with an equal volume of PBS wereincluded as controls. Four weeks after treatment, a battery of behavioural tests wasperformed by an experimenter blind to the genotype and treatment group using aprotocol previously described9 in the Neurobehavioural Core at Baylor College ofMedicine. The open field and marble burying tests were performed on day 1, theaccelerating rotarod test was performed on day 2 and 3, and the fear conditioningtest was performed on day 4 and 5. Mice were acclimated to the test room for30 min before each behaviour test.
For the open field assay each mouse was placed in the centre of a clear Plexiglas(40 3 40 3 30 cm) open-field arena (Versamax Animal Activity Monitor, AccuScanInstruments) and allowed 30 min to explore. Overhead lighting was ,800 lx insidethe field, and the white noise was at ,60 dB. Mouse activity was recorded andquantified.
For the marble burying test, each mouse was placed in a standard mouse cagecontaining 20 small (1.5–2 cm) clean black marbles on top of 4 inches of corn cobbedding, forming 4 rows of 5 columns. After a period of 30 min exploration, themouse was removed from the cage and the number of marbles buried at least 50%was recorded.
For the accelerating rotarod, the test was performed with a rotating rod systemthat rotates from 4 to 40 r.p.m. within 5 min (model 7650 Rota-rod, Ugo Basile).Mice were placed on the rotating rod and the time until falling off or losing balance(mice not walking on the rod for two consecutive turns) was recorded. For twoconsecutive days, four trials were performed per day with at least a 30 min intervalbetween trials.
For contextual fear conditioning, on the training day, each mouse was placed ina test chamber. After 2 min of free exploration (baseline/pre-shock freezing), themouse received an auditory tone (2,800 Hz, 85 db, 30 s) followed by a foot-shock(0.7 mA, 2 s). The training was repeated once. The mouse remained in the chamberfor one additional min (post-shock freezing) and then was returned to the homecage. Twenty-four hours after training, mice were returned to the same test cham-ber for 5 min and tested for freezing in response to the training context (contextualfreezing). Afterwards, the environmental settings of the test chamber were dras-tically altered and the mice were placed back in the modified context. They wereallowed 3 min of free exploration, and then the auditory tone was presented for3 min to test the fear response to the cue (cued freezing). Freezing frequency wasanalysed with FreezeFrame software (San Diego Instruments).Isolation of nascent RNA. Nascent RNA was isolated using the Click-iT NascentRNA Capture Kit (Life Technologies), according to the manufacturer’s protocol.In brief, wild-type primary neurons were incubated with 5-ethynyl uridine (EU,0.5 mM) for 0 to 150 min at which time total RNA was isolated by TRIzol. Five mi-crograms total RNA was biotinylated with 0.5 mM biotin azide, and 500 ng bio-tinylated RNA was precipitated on streptavidin beads. Nascent EU-containing RNAcaptured on the beads was used for SuperScript VILO cDNA synthesis (Life Tech-nologies) followed by qPCR.
24. Swayze, E. E. et al. Antisense oligonucleotides containing locked nucleic acidimprove potency but cause significant hepatotoxicity in animals. Nucleic AcidsRes. 35, 687–700 (2007).
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

25. Lagier-Tourenne, C. et al. Targeted degradation of sense and antisense C9orf72RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl Acad.Sci. USA 110, E4530–E4539 (2013).
26. Butler, M., Stecker, K. & Bennett, C. F. Cellular distribution of phosphorothioateoligodeoxynucleotides in normal rodent tissues. Lab. Invest. 77, 379–388(1997).
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014
0.0
0.5
1.0
1.5
2.0
0.0
0.5
1.0
1.5
2.0
ASO A ATS
pre
Snrpn pre HG sno Ipw pre HG sno ATS Ube3a
Snrpn Ube3a Ipw
HG
ASO116
Snord116 Snord115
0.0
0.5
1.0
1.5
2.0
Snord116 Snord115
pre
HG
sno a
Fol
d ch
ange
F
old
chan
ge
Fol
d ch
ange
Ctl ASO
ASO A
ASO116
b
Time of EU incubation (min)
pre
HG
+
Snord116 splicing kinetics
UTC 0.16 μM 0.63 μM 2.5 μM 10 μM 0.04 μM
0
100
200
300
0 25 50 75 100 125 150
Rel
ativ
e to
t 0
Pre
HG 30
Extended Data Figure 1 | ASOs targeting Snord116 reduced Ube3a-ATSpre-mRNA. a, Top, schematic of the ASO-binding sites and location of qRT–PCR primer and probe sets. Bottom, qRT–PCR from wild-type primaryneurons treated with ASO A or ASO116 (72 h) using primer and probe sets tothe indicated regions of Ube3a-ATS pre-mRNA and mRNA. b, Nascenttranscripts were isolated from wild-type primary neurons incubated with5-ethynyl uridine (see Methods) for the indicated time. qRT–PCR for
pre-mRNA and mature mRNA within the Snord116 region. The red lineindicates the 30 min delay between the appearance of pre-mRNA and maturemRNA. Assuming a transcription elongation rate of 4 kb min21, it wouldtake RNAPII 80 min to transcribe the 332 kb distance from the last copyof Snord116 to the ASO-binding site. HG, host gene. n 5 2 per group,mean 6 absolute deviation.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014
0.10 0.15
0
0.5
1 Ube3aYFP-ATS
Ube3aYFP
Rel
ativ
e to
UT
C
Ube3a Ube3a-ATS
Overlapping ASOs
Non-overlapping ASOs
7.4
1.7
0
4
8
12
Non-overlap
Overlap
(n=15) (n=12)
Extended Data Figure 2 | ASOs complementary to two regions of Ube3a-ATS differed in their ability to unsilence paternal Ube3a. PatYFP primaryneurons were treated with ASOs that bind Ube3a-ATS 59 of Ube3a(non-overlap ASOs, n 5 15) or that bind to the gene body region(overlap ASOs, n 5 12) for 72 h. The level of Ube3aYFP-ATS reductionand Ube3aYFP upregulation was analysed by qRT–PCR and normalized tountreated control (UTC) neurons. Data are shown as mean 6 s.e.m.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014
0.8
0.9
1.0
1.1
1.2
0 1 2 3 4
Rel
ativ
e to
wk
0
Time post-treatment (wk)
PBSASO A
15
17
19
21
23
25
0 1 2 3 4
Bod
y w
eigh
t (g
)
Time post-treatment (wk)
PBS1PBS2PBS3PBS4A1A2A3A4
0.8
0.9
1.0
1.1
1.2
0 1 2 3 4
Rel
ativ
e to
wk
0
Time post-treatment (wk)
PBSASO B
12
14
16
18
20
22
0 1 2 3 4
Bod
y w
eigh
t (g
)
Time post-treatment (wk)
PBS1PBS2PBS3PBS4B1B2B3B4
0
50
100
PBS ASO B
% B
ody
wei
ght
at
trea
tmen
t
0
50
100
PBS ASO A
% B
ody
wei
ght
at
trea
tmen
t
a b
*
0.0
0.5
1.0
1.5
0.0
0.5
1.0
1.5
CTX HIP SC
Exp
ress
ion
fold
cha
nge
Aif1
CTX HIP SC
Exp
ress
ion
fold
cha
nge
Aif1
PBS ASO A
c d
PBS ASO B
Whole brain Lateral ventricle Hippocampus Cerebellum
GF
AP
IHC
PBS
ASO A
AIF
1 IH
C
PBS
ASO A
100 μm 2 mm
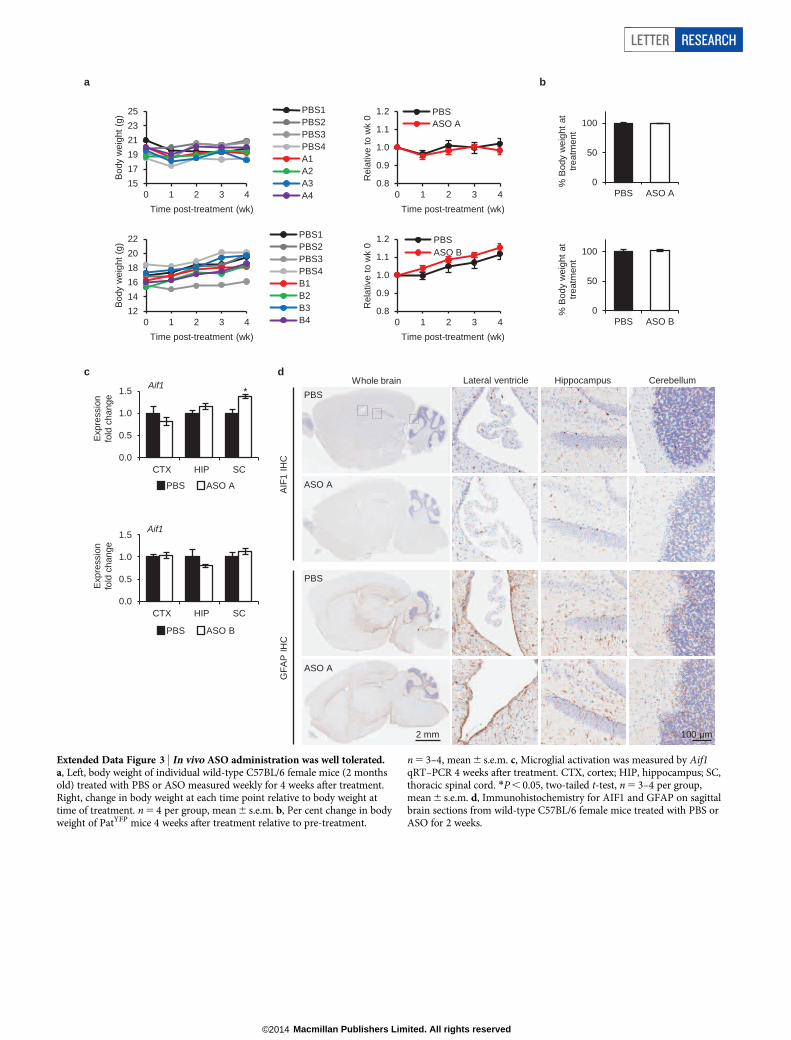
Extended Data Figure 3 | In vivo ASO administration was well tolerated.a, Left, body weight of individual wild-type C57BL/6 female mice (2 monthsold) treated with PBS or ASO measured weekly for 4 weeks after treatment.Right, change in body weight at each time point relative to body weight attime of treatment. n 5 4 per group, mean 6 s.e.m. b, Per cent change in bodyweight of PatYFP mice 4 weeks after treatment relative to pre-treatment.
n 5 3–4, mean 6 s.e.m. c, Microglial activation was measured by Aif1qRT–PCR 4 weeks after treatment. CTX, cortex; HIP, hippocampus; SC,thoracic spinal cord. *P , 0.05, two-tailed t-test, n 5 3–4 per group,mean 6 s.e.m. d, Immunohistochemistry for AIF1 and GFAP on sagittalbrain sections from wild-type C57BL/6 female mice treated with PBS orASO for 2 weeks.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014
100 45 0
30
60
90
120
% P
BS
100 229 0
100
200
300
% P
BS
100 99 0
30
60
90
120
% P
BS
Ube3aYFP-ATS Snord116 Ube3aYFP
Hypothalamus
Extended Data Figure 4 | Snord116 was not reduced in the hypothalamus. qRT–PCR on RNA isolated from PatYFP mice 4 weeks after treatment with PBS orASO B.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014
ASO YFP YFP/NeuN
Cor
tex
Cer
ebel
lum
60 µm
Extended Data Figure 5 | UBE3A unsilencing persisted 4 months after treatment. ASO and YFP immunofluorescence on brain sections of cortex andcerebellum in PatYFP mice 4 months after treatment with ASO A.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014
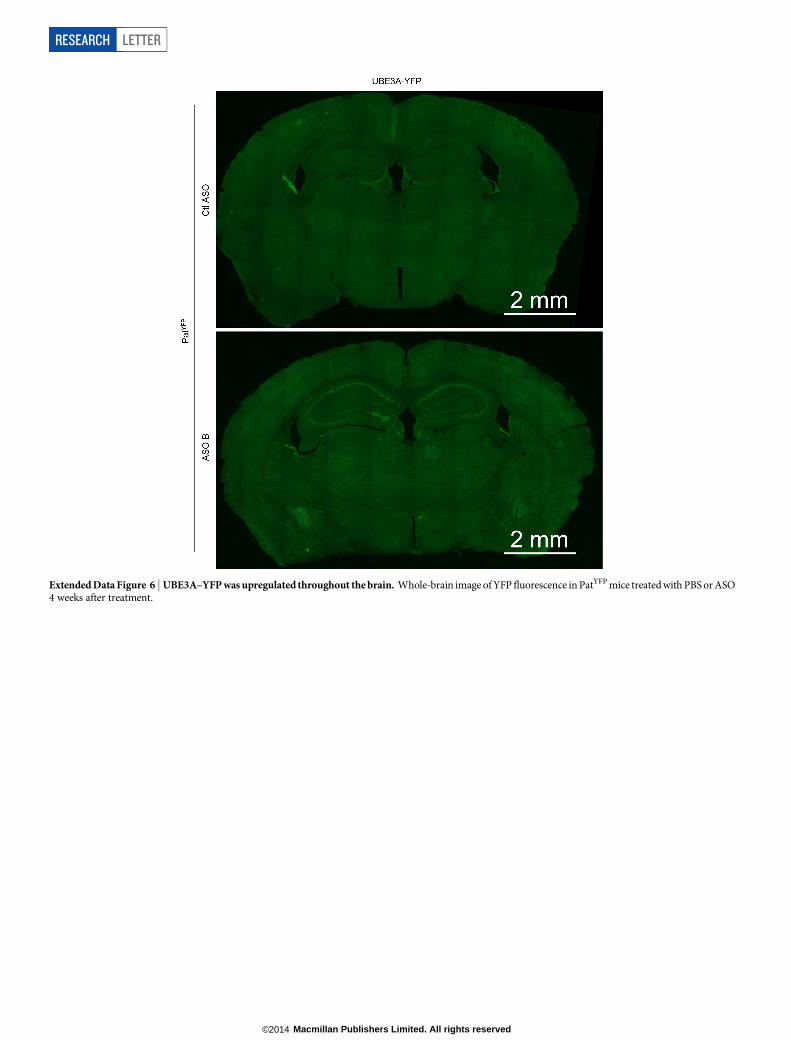
Extended Data Figure 6 | UBE3A–YFP was upregulated throughout the brain. Whole-brain image of YFP fluorescence in PatYFP mice treated with PBS or ASO4 weeks after treatment.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 7 | Imaging of unsilenced UBE3A–YFP in specificbrain regions. a–l, Immunofluorescence for ASO, UBE3A–YFP and NeuN4 weeks after treatment in MatYFP or PatYFP mice of the amygdala
(a), hippocampus CA2 and CA3 layers (b), dentate gyrus (c), striatrum(d), thalamus (e), hypothalamus (f), medulla (g), third ventricle (h), motorcortex (i), somatosensory cortex (j), auditory cortex (k) and visual cortex (l).
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014
ASO YFP YFP/NeuN
PB
S
Mat
YF
P
Pat
YF
P
Ctl
AS
O
Intr
ahip
poca
mpa
l in
ject
ion
ICV
in
ject
ion
200 µm
Extended Data Figure 8 | Intrahippocampal injection of ASO A in PatYFP
mice resulted in near complete unsilencing of paternal UBE3A–YFP.YFP immunofluorescence on brain sections from PatYFP mice treated with Ctl
ASO, 100mg ASO A via intrahippocampal injection, or 700mg ASO A via ICVinjection. A MatYFP mouse treated with PBS was included for comparison.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014
0
1000
2000
3000
4000
PBS ctl ASO ASO A
WT AS
Tot
al d
ista
nce
(cm
)
0
100
200
300
400
PBS ctl ASO ASO A
WT AS
Ver
tical
act
ivity
(bea
m c
ount
s)
0
1000
2000
3000
4000
5000
PBS ctl ASO ASO A
WT AS
Ste
reot
ype
(cou
nts)
c d e * NS * NS * NS
0
50
100
150
200
250
300
1 2 3 4 5 6 7 8
Tim
e ba
lanc
ed (
s)
Trials
0
2
4
6
8
10
12
14
PBS ctl ASO ASO A
WT AS
# M
arbl
es b
urie
d
f g*** NS
WT, PBSAS, Ctl ASOAS, ASO A
h
0
20
40
60
80
post-shock cued
% F
reez
ing
* NS
NS NS
WT, PBS AS, Ctl ASO
AS, ASO A i
20
30
40
75 125 175 225 275
Bod
y w
eigh
t (g
)
Age (days)
Tx age
200 µm
Cor
tex
and
hipp
ocam
pus
Cer
ebel
lum
WT + PBS 2 wk post-treatment
AS + Ctl ASO 2 wk post-treatment
AS + ASO A 2wk post-treatment
AS + ASO A 8wk post-treatment
0
0.3
0.6
0.9
1.2
PBS ctl ASO ASO A
WT AS
0
0.3
0.6
0.9
1.2
PBS ctl ASO ASO A
WT AS
Ube3a-ATS Ube3a
Exp
ress
ion
fold
cha
nge Cortex
Hippocampus
Cerebellum
a
b
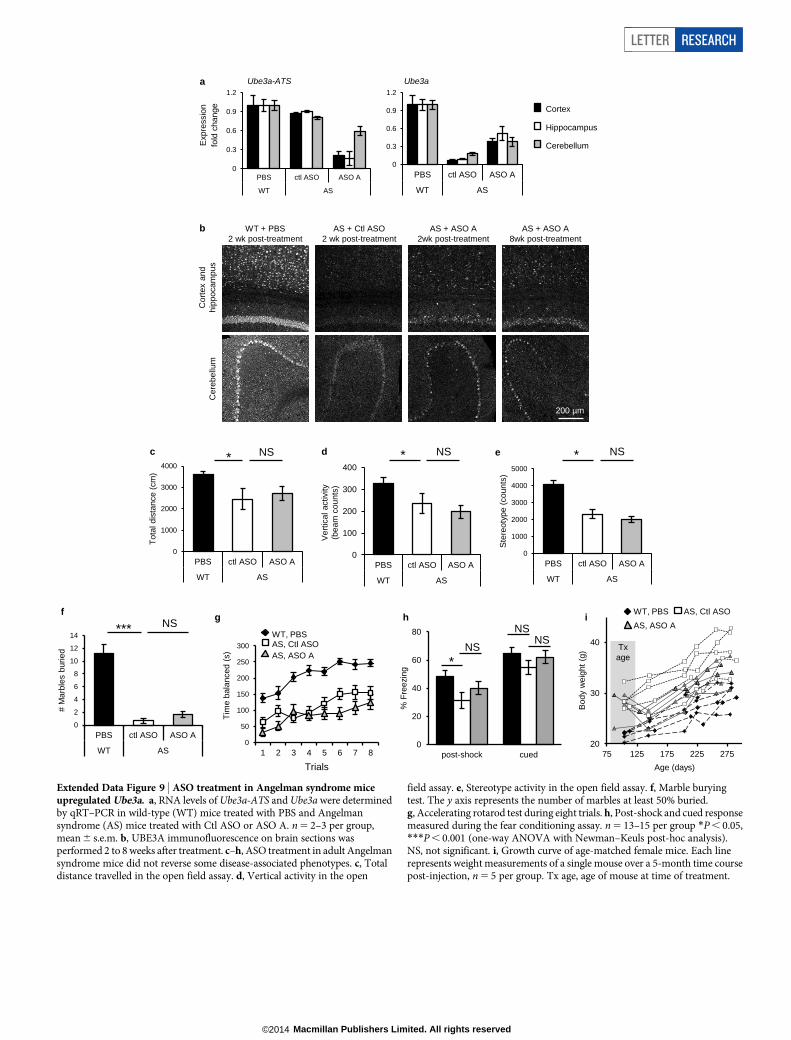
Extended Data Figure 9 | ASO treatment in Angelman syndrome miceupregulated Ube3a. a, RNA levels of Ube3a-ATS and Ube3a were determinedby qRT–PCR in wild-type (WT) mice treated with PBS and Angelmansyndrome (AS) mice treated with Ctl ASO or ASO A. n 5 2–3 per group,mean 6 s.e.m. b, UBE3A immunofluorescence on brain sections wasperformed 2 to 8 weeks after treatment. c–h, ASO treatment in adult Angelmansyndrome mice did not reverse some disease-associated phenotypes. c, Totaldistance travelled in the open field assay. d, Vertical activity in the open
field assay. e, Stereotype activity in the open field assay. f, Marble buryingtest. The y axis represents the number of marbles at least 50% buried.g, Accelerating rotarod test during eight trials. h, Post-shock and cued responsemeasured during the fear conditioning assay. n 5 13–15 per group *P , 0.05,***P , 0.001 (one-way ANOVA with Newman–Keuls post-hoc analysis).NS, not significant. i, Growth curve of age-matched female mice. Each linerepresents weight measurements of a single mouse over a 5-month time coursepost-injection, n 5 5 per group. Tx age, age of mouse at time of treatment.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014
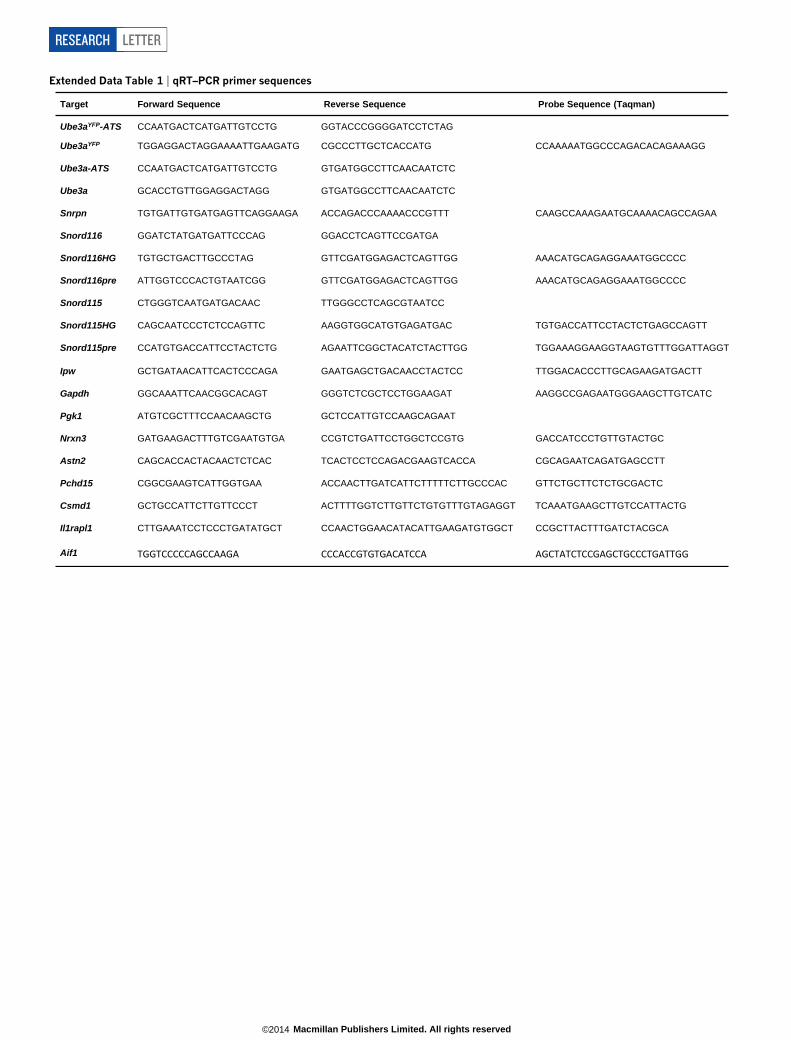
Extended Data Table 1 | qRT–PCR primer sequences
Target Forward Sequence Reverse Sequence Probe Sequence (Taqman)
Ube3aYFP-ATS
CCAATGACTCATGATTGTCCTG GGTACCCGGGGATCCTCTAG
Ube3aYFP TGGAGGACTAGGAAAATTGAAGATG CGCCCTTGCTCACCATG CCAAAAATGGCCCAGACACAGAAAGG
Ube3a-ATS CCAATGACTCATGATTGTCCTG GTGATGGCCTTCAACAATCTC
Ube3a GCACCTGTTGGAGGACTAGG GTGATGGCCTTCAACAATCTC
Snrpn TGTGATTGTGATGAGTTCAGGAAGA ACCAGACCCAAAACCCGTTT CAAGCCAAAGAATGCAAAACAGCCAGAA
Snord116 GGATCTATGATGATTCCCAG GGACCTCAGTTCCGATGA
Snord116HG TGTGCTGACTTGCCCTAG GTTCGATGGAGACTCAGTTGG AAACATGCAGAGGAAATGGCCCC
Snord116pre ATTGGTCCCACTGTAATCGG GTTCGATGGAGACTCAGTTGG AAACATGCAGAGGAAATGGCCCC
Snord115 CTGGGTCAATGATGACAAC TTGGGCCTCAGCGTAATCC
Snord115HG CAGCAATCCCTCTCCAGTTC AAGGTGGCATGTGAGATGAC TGTGACCATTCCTACTCTGAGCCAGTT
Snord115pre CCATGTGACCATTCCTACTCTG AGAATTCGGCTACATCTACTTGG TGGAAAGGAAGGTAAGTGTTTGGATTAGGT
Ipw GCTGATAACATTCACTCCCAGA GAATGAGCTGACAACCTACTCC TTGGACACCCTTGCAGAAGATGACTT
Gapdh GGCAAATTCAACGGCACAGT GGGTCTCGCTCCTGGAAGAT AAGGCCGAGAATGGGAAGCTTGTCATC
Pgk1 ATGTCGCTTTCCAACAAGCTG GCTCCATTGTCCAAGCAGAAT
Nrxn3 GATGAAGACTTTGTCGAATGTGA CCGTCTGATTCCTGGCTCCGTG GACCATCCCTGTTGTACTGC
Astn2 CAGCACCACTACAACTCTCAC TCACTCCTCCAGACGAAGTCACCA CGCAGAATCAGATGAGCCTT
Pchd15 CGGCGAAGTCATTGGTGAA ACCAACTTGATCATTCTTTTTCTTGCCCAC GTTCTGCTTCTCTGCGACTC
Csmd1 GCTGCCATTCTTGTTCCCT ACTTTTGGTCTTGTTCTGTGTTTGTAGAGGT TCAAATGAAGCTTGTCCATTACTG
Il1rapl1 CTTGAAATCCTCCCTGATATGCT CCAACTGGAACATACATTGAAGATGTGGCT CCGCTTACTTTGATCTACGCA
Aif1 TGGTCCCCCAGCCAAGA CCCACCGTGTGACATCCA AGCTATCTCCGAGCTGCCCTGATTGG
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014



![A quantitative comparison of the brain and the inner surface of …subsol/WWW/SMEF.0618.Dumoncel.2.pdf · shape data [Durrleman et al 2015, Beaudet et al 2016, Beaudet et al 2017]](https://img.pdfslide.us/doc/110x75/5e7085eb9ab1e952e3755d1d/a-quantitative-comparison-of-the-brain-and-the-inner-surface-of-subsolwwwsmef0618dumoncel2pdf.jpg)

























