Embed Size (px)
Citation preview

Moderator : Dr. D. Devi ,
Asso prof, Dept of paediatrics, SMCH
Presented by: Dr. Samiul Ahsan Hussain
PGT, Paediatrics

INTRODUCTION
The hallmark of neurodegenerative disease is regression and progressive deterioration of neurologic
function with loss of speech , vision, hearing or locomotion a/w
seizure, feeding difficulties and impairment of intellect.

Neuroregressive /neurodegenerative disorders are a
group of heterogeneous diseases which results from
specific genetic, biochemical defect, chronic viral
infection, toxic substances
Involves both the gray matter and white matter

Dementia, used for neurodevelopmental regression in
children, is associated with loss of memory, ability to
think, understand and recognize along with personality
changes or distressing behaviour

Gray matter & White matter
Striations seen in white mater

Contains mostly myelinated axons
Appears pinkish white to the naked eye (myelin is
composed largely of lipid tissue veined with capillaries)
A 20 year-old male has a 176,000 km of myelinated
axons in his brain while that of a female is 149,000 km
connect various grey matter areas (the locations of nerve
cell bodies) of the brain to each other, and carry nerve
impulses between neuron
White matter

Major component of the CNS having a grey –brown color(due
to capillary blood vessels & neurinal cell bodies)
Consists of
neuronal cell bodies( in contrast to white matter)
neuropil (dendrites and unmyelinated axons )
glial cells (astroglia and oligodendrocytes) & capillaries.
Gray matter

The grey matter includes regions of the brain involved in
muscle control,
sensory perception such as seeing and hearing,
memory, emotions, and speech.
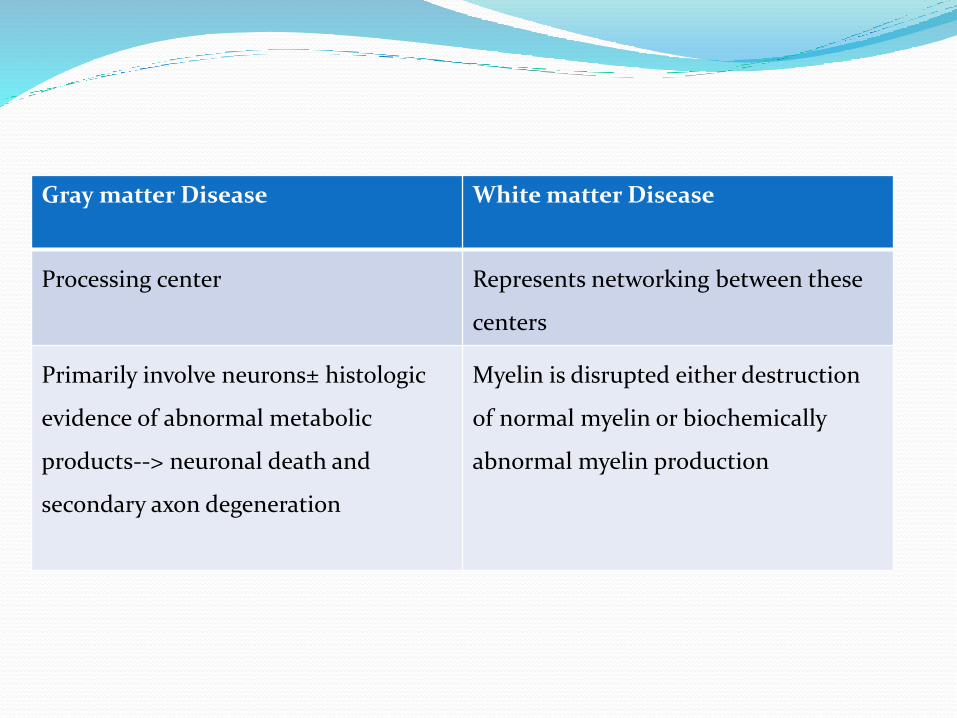
Gray matter Disease White matter Disease
Processing center Represents networking between these
centers
Primarily involve neurons± histologic
evidence of abnormal metabolic
products--> neuronal death and
secondary axon degeneration
Myelin is disrupted either destruction
of normal myelin or biochemically
abnormal myelin production

Differentiatingfeatures
White matterdisorders
Gray matterdisorders
Age of onset Usually late(childhood) Usually early(infancy)
Head size May have megaenchepaly Usually microcepaly
Seizures Late , rare Early, severe
Cognitive functions Initially normal Progressive dementia
Peripheral neuropathy Early demyelination Late, axonal loss
Spasticity Early, severe Later, progressive
Reflexes Absent(neuropathy) or exaggerated(long tracts)
Normal or exaggerated
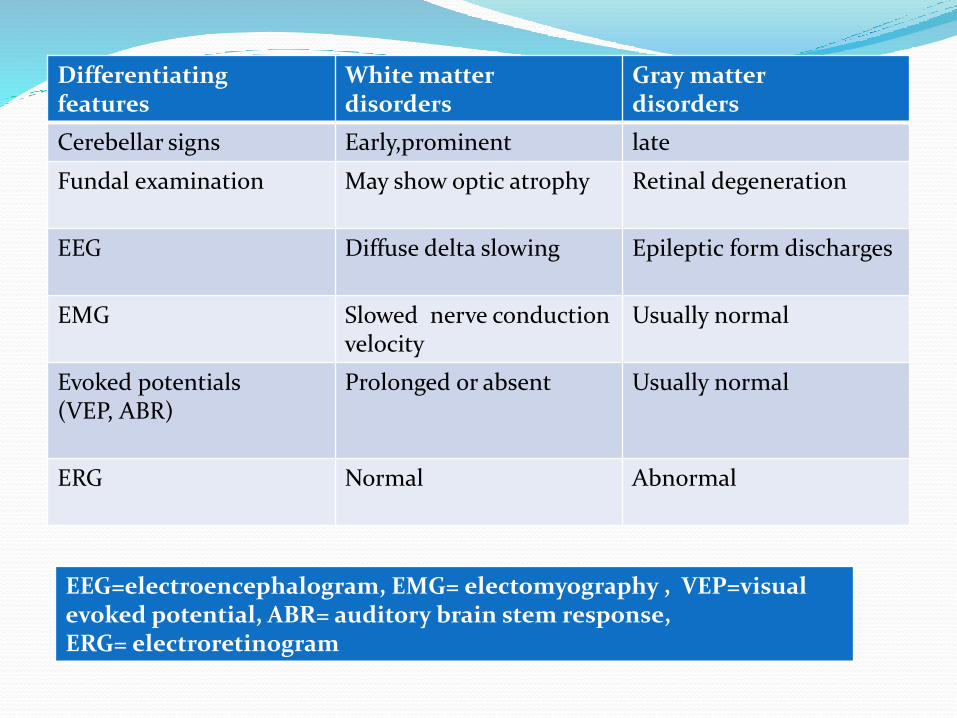
Differentiatingfeatures
White matterdisorders
Gray matterdisorders
Cerebellar signs Early,prominent late
Fundal examination May show optic atrophy Retinal degeneration
EEG Diffuse delta slowing Epileptic form discharges
EMG Slowed nerve conduction velocity
Usually normal
Evoked potentials(VEP, ABR)
Prolonged or absent Usually normal
ERG Normal Abnormal
EEG=electroencephalogram, EMG= electomyography , VEP=visualevoked potential, ABR= auditory brain stem response,ERG= electroretinogram

Gray matter: fits, decrease HMF
EEG: early abnormality
MRI Brain: cortical atrophy
White matter: blindness ,Gait disturbances ,Motor signs-
Spasticity ,optic atrophy ,ataxia , papilledema
EEG: late abnormality
MRI Brain: Demyelination
Nerve conductance + Evoke potentials
Classification

Basal Ganglia :Dystonia,Involantary movements
Spinocerebellar degeneration: Ataxia
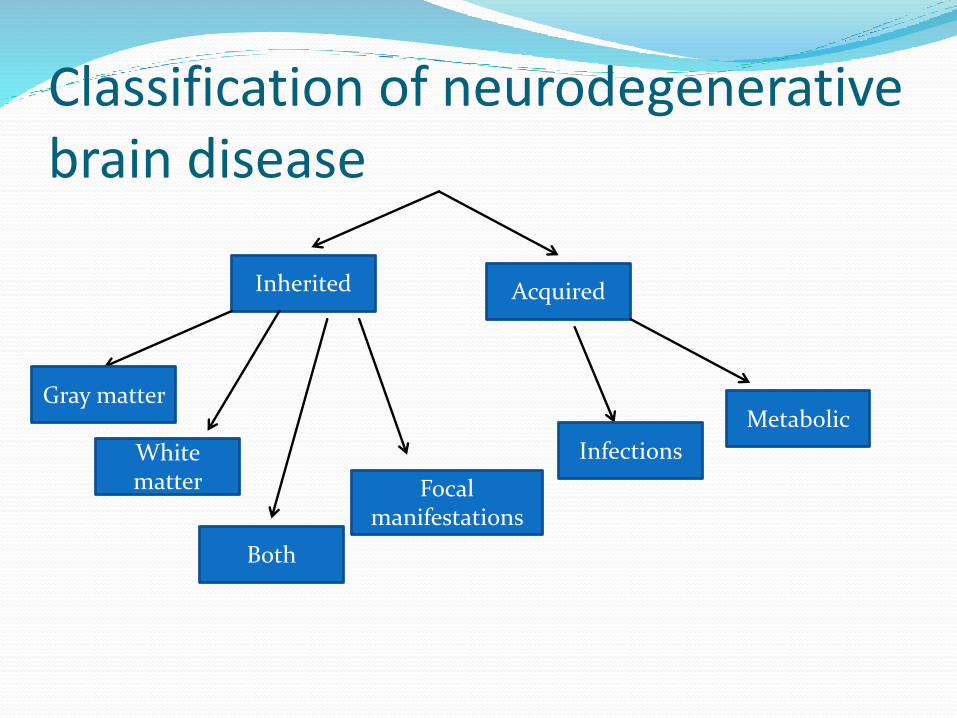
Classification of neurodegenerative brain disease
Inherited Acquired
Focal manifestations
Both
White matter
Gray matter Metabolic
Infections

Acquired causes
Infections
SSPE
Progressive Rubella
Syndrome
Chronic HIV
Metabolic
Chronic lead poisoning
Hypothyroidism
Vit B12 & E deficiency
Drugs (anticonvulsant)

Inherited causes Gray matter involvement:seizure,dementia, visual loss, intellectual
impairment. Spike & sharp waves in EEG
A. Gray matter involvement with visceromegaly
GM1 Gangliosides-Infantile , generalized , juvenile
Sandholf disease (GM2)
Niemann pick Disease( Sphingolipid storage disease)
Sialidosis
MPS
Gaucher disease( Sphingolipid storage disease)

B. Gray matter diseases involvement without visceromegaly
Rett Syndrome
Neuronal curoid lipofuscinosis
Menke’s kinky hair disease

Spasticity , optic atrophy, ataxia ,peripheral neuropathy .Seizure , dementia
are the late manifestations. Slow waves in EEG
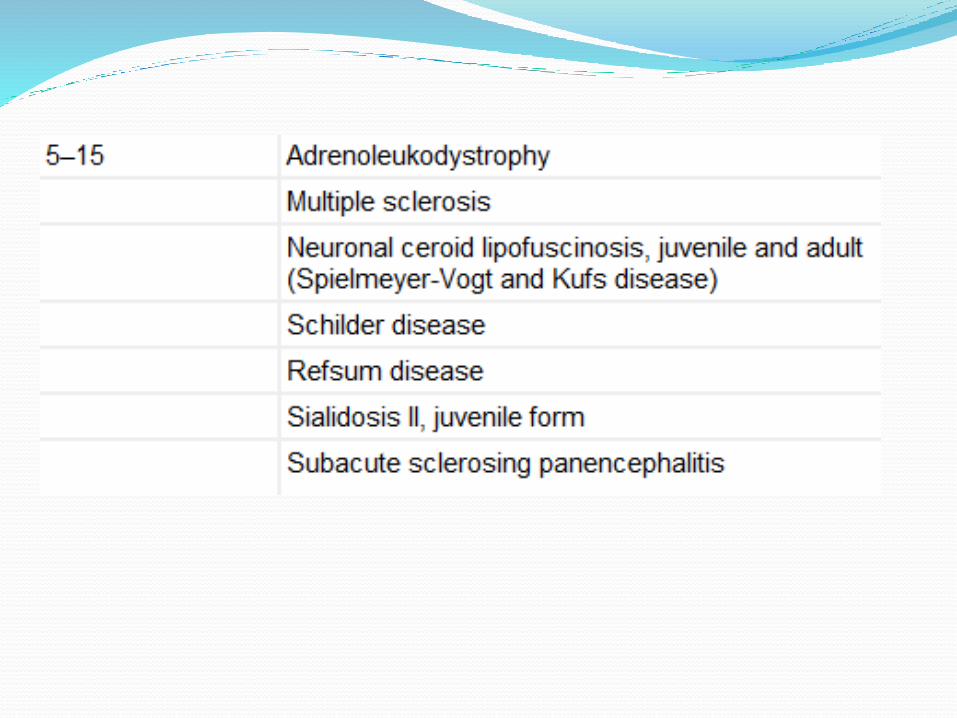
A. Leukodystrophies
Metachromatic leukodystrophy
Krabbe disease
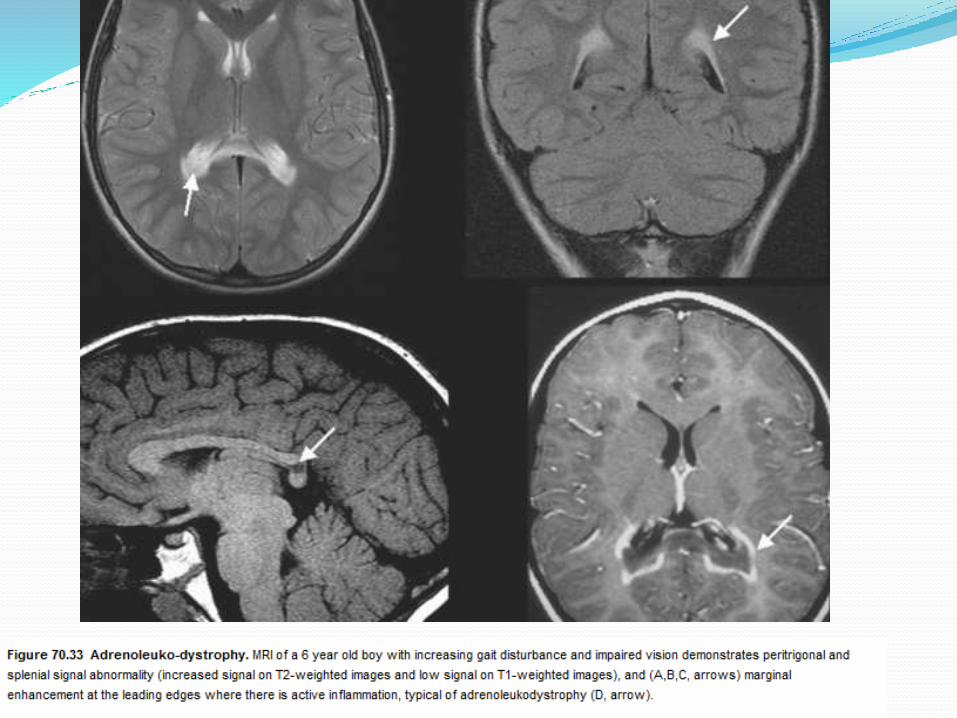
Adrenoleukodystrophy
Alexander disease ,
Cannavan disease,
P.Merzbacker disease
White matter involvement

B. Acquired causes/ Demyelinating
Multiple sclerosis
Schilder’s disease
Devic disease

Basal Ganglia
Wilson's disease
Dystonia muscular
Deformans
Huntington’s Disease
Spinocerebellar
Friedrich’s Ataxia
Ataxia Telangiectasia

Classification according to age


An approach to a child with regression of milestones
Divide the babies according to age.
Look for organomegaly.

<2 year with hepatomegaly
Jaundice, vomiting, lethargy, irritability, and convulsions
hypoglycemiaand lactic acidosis/
cirrohosis
Typical facies OTHER
Fructose intolerance /Galactosemia
GSD TYPE 1 t0 4
MPS/Zellwegersyndrome
TSD/NPD/
GD Type 2
biochemglycogensynthesikz6.jpg
vongierke-glucose-
metabolism.jpg

Typical faciesMPS Zellweger syndrome
Diagnosis is usually made between 6 and 24 mo of age with evidence of hepatosplenomegaly, coarse facial features, corneal clouding, large tongue, prominent forehead, joint stiffness, short stature, and skeletal dysplasia .
• Typical facial appearance (high forehead, unslanting palpebral fissures, hypoplastic supraorbital ridges, and epicanthal folds ),
• severe weakness and hypotonia, neonatal seizures, and eye abnormalities (cataracts, glaucoma, corneal clouding, brushfield spots, pigmentaryretinopathy, and nerve dysplasia).
• More than 90% show postnatal growth failure

Difficulty in feeding, FTT, Cherry red spot,
hypotonia, death by 3yr
• loss of motor skills, increased startle reaction, cherry red spots .
• norma until 4–5 mo of age when decreased eye contact and an exaggerated startle response to noise (hyperacusis) are noted.
increased tone, strabismus, . Failure to
thrive and stridor caused by laryngospasm are
typical
NEIMANN–PICK DISEASE
Tay-Sachs disease Gaucher disease
Gucher cell, glucocerebrosida
se
Vacuolated histocytes,
sphingomyelinase
CRS, Hexoseaminida
se, Mutation analysis

< 2 yr without hepatomegaly

KRABBE DISEASE
• The infantile form of Krabbe disease is rapidly progressive and patients present in early infancy with irritability, seizures, and hypetonia.
• Optic atrophy is evident in the 1st yr of life, and mental development is severely impaired.
• MRI: diffuse demyelination of cerebral hemisphere• Delayed motor nerve conduction velocity• Increase CSF protein• Beta Galactosidase• krabbe disease.jpg

RETT SYNDROME
• Development normal until 1 yr of age, when regression of language and motor milestones and acquired microcephaly become apparent
• The hallmark of Rett syndrome is repetitive hand-wringing movements and a loss of purposeful and spontaneous use of the hands; these features may not appear until 2–3 yr of age.
• Autistic behavior is a typical finding in all patients.• Generalized tonic-clonic convulsions occur • Feeding disorders and poor weight gain are common

MAPLE SYRUP URINE DISEASE
• This form has the most severe clinical manifestations. Affected infants who are normal at birth develop poor feeding and vomiting in the 1st wk of life; lethargy and coma may ensue within a few days.
• Physical examination reveals hypertonicity and muscular rigidity with severe opisthotonos. Periods of hypertonicity may alternate with bouts of flaccidity.

PHENYLKETONURIA
• The affected infant is normal at birth.
• Mental retardation may develop gradually and may not be evident for the 1st few months.
• Older untreated children become hyperactive, with purposeless movements, rhythmic rocking, and athetosis

2-5 yearsMyoclonus,
Myoclonic epilepsy,Ataxia,
Raggaed red fibre in muscle
hypotonic extremitabsent deep
tendon reflexes,Inability to walk
Ataxia/Involuntary movements/infections / cancer Dysarthria
MERRF MLD AT

Acquired causes

SSPE
The initial clinical manifestations include personality changes, aggressive behavior, and impaired cognitive function. Myoclonic seizures soon dominate the clinical picture. Later, generalized tonic-clonic convulsions, hypertonia, and choreoathetosis become evident, followed by progressive bulbar palsy, hyperthermia, and decerebrate postures.
• Chronic lead poisoning
• loss of short-term memory or concentration, depression, nausea, abdominal pain, loss of coordination, and numbness and tingling in the extremities.] Fatigue, problems with sleep, headaches, stupor, slurred speech, and anemia are also found in chronic lead poisoning.
• A "lead hue" of the skin with pallor• ]Burton line• Children with chronic poisoning may refuse to play or may
have hyperkinetic or aggressive behavior disorders.

Chronic HIV
Onset: 2 month t0 five yr after exosure.Progressive loss of developmental milestones , microcephaly, dementia and spastcity is characteristics
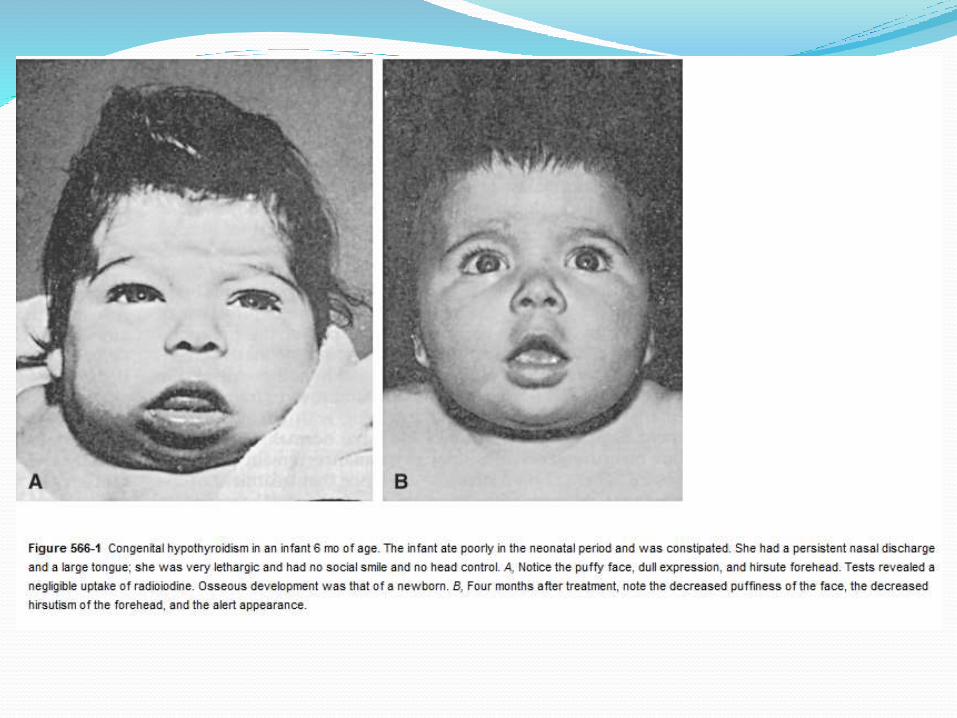
Hypothyrodism
Asymtomatic at birthWide open posterior frontanalare, constipation , jaundice,poor temperature control, and umbilical hernia, large tongue, edema of eyes , hands and feet.

History History of present illness:
Onset/Age of onset
Fits ,Clumsiness or difficulty in gait
Deterioration of HMF
Ataxia or imbalance
Headache,Blindness,Vomiting, deafness
Change in personality and behaviour
Deteriorance in school performance
Increased startle response or hyperacusis

Birth history:
Term/preterm
Postnatal complications
Meningitis
Head trauma
kernicterus

Developmental history:
Detailed development history- decide whether there is delayed
development milestones or regression of milestones
Family history:
H/o of consanguinity
Family history of neurological disorder
Early or unexplained death
Nature of the neurological manifestations should be clarified

Classically , the loss of previously acquired
milestones(regression) marks the onset of most
Neurodegenerative brain disease with subsequent
progressive neurological deterioration

Clinical examination
General physical examination
Dysmorphism: Zellweger syndrome, Neonatal adrenal
leukodystrophy, coarse facial features(MPS)
OFC –microcepaly (gray matter disease)
Megaenchepaly – certain white mater disorder(Cannavan &
Alexander)
Jaundice
Enlarged tongue

Skin & hair ( Hartnup Diseases-pellagra like skin rash, Menkes
disease-kinky hair)
Examination of the spine- for associated complications (scoliosis)
Contractures of joints
Systemic examination:
Hepatosplenomegaly
Chest deformity
Cardiomyopathy

Neurological examination Higher mental function, signs of raised ICP
Speech, memory
Cranial nerves
Gait
Motor system:
Tone-hypo/ hypertonia,Deep tendon reflexes
Motor spasticity
Sensory loss /neuropathy
Abnormal /involuntary movements

Eye examination Optic atrophy(white matter- due to demyelination)
Retinal degeneration(gray matter)- as the retinal
receptors are neuronal cells): Cherry red spot, retinitis
pigmentosa
Cataracts
Telengiectasias
K.F ring

DECIDE REGRESSION AND NOT DELAY
AGE ABOVE 2 YEARS OR LESS THAN 2 YEARS
VISCEROMEGALY
NEUROPATHY
GRAY OR WHITE MATTER DISEASE

Investigations- to identify the underlying diagnosis
& examining the associated complications
Complete Blood picture-pancytopenia, vacuolated
lymphocytes,acanthocytes
ABGs-metabolic acidosis(organic acidopathies, urea cycle
defects, mitochondrial encephalopathies)
S/E (Anion gap), for adrenal
insufficiency(adrenoleukodystrophy)
Ammonia level,LFTs,RFTs

Special tests:
Lactate & pyruvate levels, Lysosomal enzyme level
WBCs, Fibroblast enzyme level
Wilson’s disease-serum ceruloplasmin level, serum copper
Amino acids
Urinary organic acids
Uric acid level

Urine
Reducing substances, Organic acids,24 hr (MPS)
Imaging
Skull & Vertebrae, Long bones
CT/MRI
Biopsy
Skin, Bone marrow, nerve, brain

ROLE OF MRI The abnormalities of metabolic disease are
characteristically bilateral and symmetrical.
Assessment on mri should include analysis of grey and white matter structures.
Calcification is much better assessed on ct.
Inherited hypomyelination (pelizaeus merzbacher)
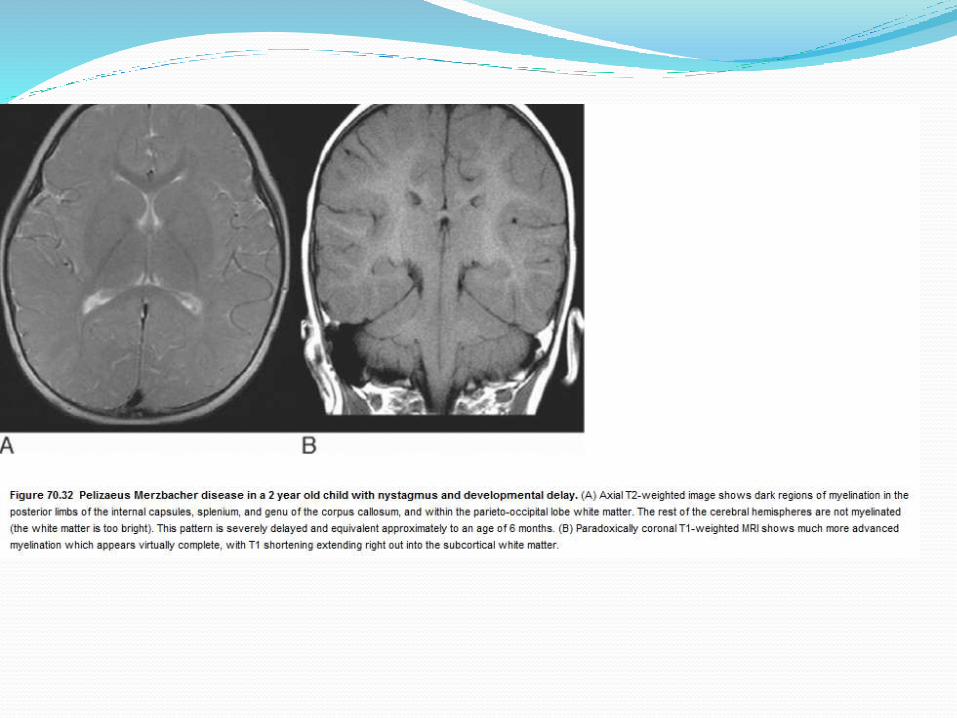

Pathognomonic imaging patterns are seen in
X-linked adrenoleukodystrophy (ALD),
Alexander's disease
Neonatal maple syrup urine disease




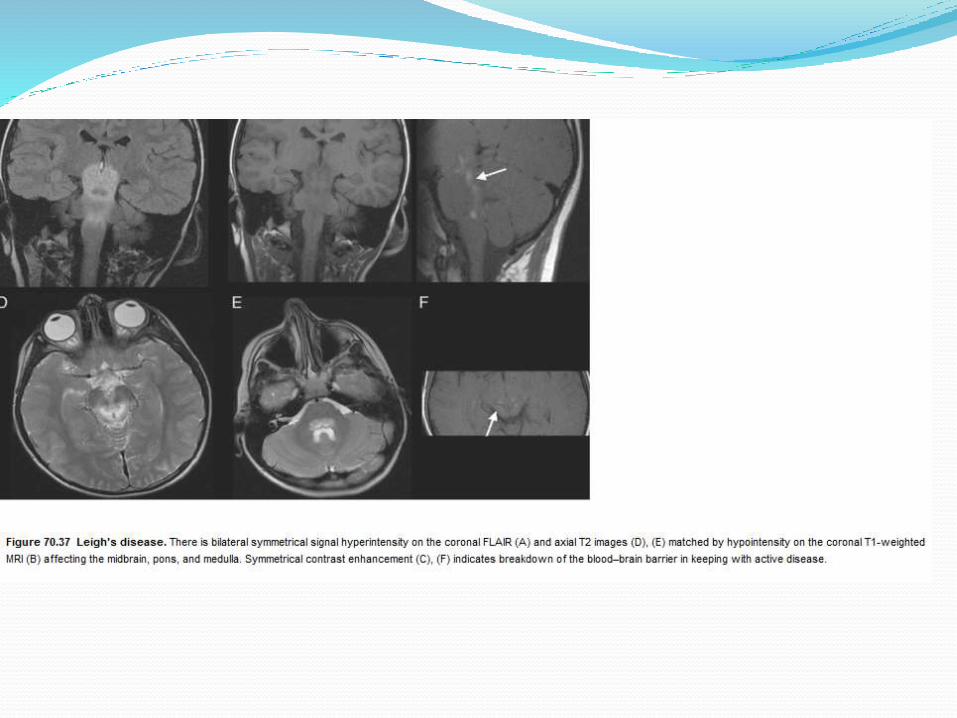
Respiratory chain tend to be multisystem diseases.
In the brain they may result in multiple cerebral infarcts in nonvascular territories.
Leigh's disease : Bilateral typically symmetrical signal change is seen within the brainstem, deep cerebellargrey matter, subthalamic nuclei and basal ganglia

Dystosis multplexElongated (J-
shaped) sella. The vault shows an overall ground-
glass opacity
The ribs are broad, and the clavicles short and broad

Inferior hook (arrowhead) on the body of L2
with a mild kyphosis.
Bilateral hip subluxation with
long femoral necks and coxa
valga
proximal pointing of the second to fifth
metacarpals


Diagnosis
Important for genetic counseling
Outcome
Invariably fatal

Management
Directed towards the treatment of the underlying
disorder, other associated features and complications
Supportive :The treatable complications :
feeding difficulties, Gastoresophageal reflux
spasticity, drooling
skeletal deformities, and recurrent chest infections
epilepsy, sleep disorder, behavioral symptoms
A multidisciplinary approach(pediatrics, neurology, genetics, orthopedics, physiotherapy, and occupational therapy.

Specific treatmentNeurodegenerativedisorders
Specific treatment modality
Krabbe leukodystrophy Bone marrow transplantation
Metachromatic leukodystrophy
Bone marrow transplantation
Adrenoleukodystrophy Glyceryl trioleate and trierucate,steroids for adrenal insufficiency, diet low in VLCFA, bone marrowtransplantation
Mucopolysaccharidosis Bone marrow transplantation,recombinant human α-L-iduronidase
Menkes kinky hair syndrome Copper sulfate

Counseling the families and educating the public about these
potentially preventable disorders is very important.
Neurodegenerative
disorders
Specific treatment modality
Mitochondrial encephalopathies Nicotinamide, riboflavin,
dichloroacetate, L-carnitine, CoQ10
Wilson disease D- penicillamine, trietine, zinc acetate,
liver transplantation
Refsum disease Reduction of phytanic acid intake
Lesch-Nyhan disease Allopurinol
Fabry’s Disease Recombinant human α galactosidase A






























