Embed Size (px)
Citation preview

INCIDENTAL FINDINGS THROUGHOUT MULTIGENE PANEL TESTING
IN CANCER GENETICS
Cours international : médecine génomique, du diagnostic à la thérapie
Aurélie Fabre 21/10/2016
Institut Pasteur de Tunis

SOMMAIRE
I. L’ECHELLE DU SEQUENCAGE
II. TERMINOLOGIE / DEFINITIONS
III. LES VARIANTS ET LEUR CLASSIFICATION
IV. L’EXEMPLE DES BASES DE DONNEES FRANCAISES MMR
V. ASPECTS ETHIQUES ET LEGAUX

GENE(S) SPECIFIQUE(S)
PANEL DE GENES ± SPECIFIQUES
EXOME (WES)
GENOME (WGS)
Gène(s) clairement associé(s) avec le
diagnostic
Gènes ± associés avec le diagnostic
± 22 000 gènes (1,6% du génome)
Tout l’ADN
quelques dizaines de kilobases (Kb)
plusieurs dizaines de Kb
180 000 exons, 52 Mb
3,2 Gb
I. L’ECHELLE DU SEQUENCAGE
BIG DATA : de plus en plus de données générées, à stocker et à interpréter

II.1 TERMINOLOGIE
• « Primary findings » : découvertes d’une altération pathogène sur un gène connu comme associé au diagnostic clinique (prescription d’analyse)
« pathogenic alterations in a gene or genes that are relevant to the diagnostic indication for which the sequencing was ordered »
• « Incidental findings » : découvertes d’altération(s) pathogène(s) sur un (ou des gènes) a priori sans rapport avec le diagnostic clinique mais d’intérêt médical (liste de gènes d’intérêt clinique analysés délibérément)
« the results of a deliberate search for pathogenic or likely pathogenic alterations in genes that are not apparently relevant to a diagnostic indication for which the sequencing test was ordered » « but of medical value for patient care »
NB : l’American College of Medical Genetics, créée en 1991, écrit des « standards and guidelines » pour la génétique clinique depuis 1993

ACMG : 56 gènes d’intérêt clinique (23 « cancer », 25 « pathologies cardio-vasculaires », 8 autres)
II.2 TERMINOLOGIE

• GENES « ACTIONABLE » : gènes pour lesquels il existe des recommandations spécifiques de prise en charge lorsqu’une altération pathogène est retrouvée
II.3 TERMINOLOGIE
« defined as having deleterious mutation(s) whose penetrance would result in specific, defined medical recommendation(s) both supported by evidence and, when implemented, expected to improve an outcome(s) in terms of mortality or the avoidance of significant morbidity »
114 gènes « actionable » 2013 (52/56 en commun ACMG) méthode de classification des variants

II.4 TERMINOLOGIE
Définitions plus récentes :
– « Primary findings » = découvertes primaires
– « Secondary findings » = découvertes secondaires sur les gènes « actionable »
– « Incidental findings » = découvertes fortuites anticipées ou non

II.4 TERMINOLOGIE
Privilégier le terme de « secondary findings » ou d’ « additional findings »
s’agissant des altérations découvertes sur les gènes « actionable »

III. LES VARIANTS ET LEUR CLASSIFICATION
III.1 Fréquence des altérations
III.2 Définitions
III.3 Les variants de signification inconnue (UV)
III.4 L’interprétation des variants
III.5 Travaux ACMG/AMP 2015
III.6 Les bases de données généralistes
III.7 Les bases de données locus-spécifiques

- Analyses primaires ou panels spécifiques : plus de 10% des personnes testées présentent une altération qui modifie la prise en charge (découvertes primaires) - Analyse d’exome ou de génome : découvertes secondaires dans les 56 gènes « actionable » retrouvés chez 1 à 5,6 % des personnes testées selon les études :
- Xue et al. 2012 (179 génomes) : 3,35%
- Dorschner et al. 2013 (1000 exomes) : 1,2-3,4% - Olfson et al. 2015 (1092 génomes) : 1,10% - Amendola et al. 2015 (6503 exomes) : 1,41% - Gambin et al. 2015 (11068 exomes) : 5,6% - Yavarna et al. 2015 (149 exomes) : 2%
COMMENT DETERMINER QU’UNE ALTERATION EST PATHOGENE OU NON ?
III.1 Fréquence des altérations

Variant = modification par rapport à la séquence de référence • Variants clairement pathogènes (mutations) :
Non sens (codon stop prématuré) Insertions ou délétions entraînant un décalage du cadre de lecture Introniques dans les sites canoniques d’épissage (+/- 1 ou 2) Remaniements de grande taille
• Variants clairement non pathogènes (polymorphismes) : fréquence > 1% dans la population générale (cf bases de données de fréquence)
• Variants de signification inconnue (VSI, Unknown Variant, Variation of
Uncertain Significance) Faux sens Silencieux Introniques Petites insertions ou délétions en phase
III.2 Définitions

198 patientes testées avec un panel de 42 gènes de prédisposition au cancer :
2,1 UV par patiente
III.3 Les variants de signification inconnue (UV)
Intérêt des bases de données d’expertise pour l’interprétation des variants primaires et secondaires, et en particulier des UV

III.4 L’interprétation des variants
« On the other hand, when there is a positive incidental finding, the Working Group recommended that laboratories review
available literature and databases at the time of the sequence interpretation to insure there is sufficient support for
pathogenicity before reporting a variant. The Working Group recognized that there is no single database currently
available that represents an accurately curated compendium of known pathogenic variants, nor is there an automated
algorithm to identify all novel variants meeting criteria for pathogenicity. Therefore, evaluation and reporting of positive
findings in these genes may require significant manual curation. »
Mise à jour régulière des recommandations de l’ACMG

Groupe de travail (créé en 2013) : cliniciens + biologistes (ACMG, Association for Molecular Pathology, College of American Pathologists)
EXPERTISE, CONSENSUS
Terminologie standard Système de classification consensuel (combinaison de différents
niveaux de preuve)
enquête auprès de 100 laboratoires de séquençage US et Canada
Système de classification à 5 niveaux : “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, et “benign”
Critères d’évaluation des données d’interprétation, différents niveaux
de preuve : “strong”, “moderate”, “supporting”, “not used”
Règles pour combiner ces critères et conclure sur le niveau de pathogénicité du variant
III.5 Travaux ACMG/AMP 2015 - méthode

Recommandations :
- ne plus utiliser les termes « mutation » ou « polymorphisme » mais seulement le terme « variant »
- y adjoindre l’interprétation associée selon le niveau de pathogénicité : (i) pathogenic,
(ii) likely pathogenic, (iii) uncertain significance, (iv) likely benign, or (v) benign. NB : classification IARC (International Agency for Research on Cancer) = (i) not pathogenic, (ii) likely not pathogenic, (iii) uncertain, (iv) likely pathogenic, (v) definitely pathogenic
III.5 Travaux ACMG/AMP 2015 - terminologie

- Données de fréquence allélique
- Données de prédiction in silico
- Tests fonctionnels
- Analyses de ségrégation
- Données de novo (trios)
- Co-occurrence(s) -cis / -trans
- Autres ressources (BDD, experts)
III.5 Travaux ACMG/AMP 2015 critères de classification et règles

III.5

III.5 Variant non sens
Variant de novo
Hot spot mutationnel
Co-ségrégation
Fréquence > 5%
Individus porteurs sains

- Bases en ligne (nomenclature, méthode de classification utilisée, mises à jour, population utilisée pour données de fréquence, etc.) - Données publiées (« overlap », case reports sur membres d’une même famille, etc.) - Outils de prédiction in silico
- prédictions exactes dans 65-80 % des cas pour les variants faux-sens - peu de spécificité pour les variants faux-sens : plus de « faux délétère » - outils de prédiction des défauts d’épissage ont une bonne sensibilité (90-100%) mais une spécificité plus faible (« faux délétère ») - recours à plusieurs outils de prédiction pour confirmer le niveau de preuve mais = une seule donnée d’interprétation (similarité dans les algorithmes) - Système d’évaluation non adapté à l’interprétation des variants somatiques, des variants pharmacogénétiques et des variants dans des gènes impliqués dans des maladies multigéniques non-mendéliennes ou dans des gènes candidats * Approche plus stringente que d’autres donc plus de variants « uncertain significance » mais évite les « faux pathogenic » avec les conséquences associées pour le patient et sa famille
III.5 Travaux ACMG/AMP 2015 - précautions

III.6 Les bases de données généralistes
Human Gene Mutation Database (HGMD) : extraction et compilation de données de mutations de la littérature (27 000 anomalies / 4 860 gènes dans la version publique ; > 179 000 / 7 189 gènes dans la version professionnelle). Uniquement variants pathogènes, pas de curation
ClinVar : compilation de données de variants OMIM, GeneReviews, UniProt, dbSNP, soumission par des laboratoires de diagnostic, des chercheurs, des groupes d’expertise, intégration de données d’autres bases de données (173 000 enregistrements dont 85 642 avec des critères d’évaluation associés). Pas de curation, pas d’arbitrage

III.7 Les bases de données locus-spécifiques
Human Genome Variation Society (HGVS) Universal Mutation Database (UMD) Leiden Open Variation Database (LOVD) Gen2Phen Autres...
Spécifiques d’un seul gène Enregistrent les données de tous les variants soumis + littérature Données associées / curation / expertise / classification +/- précises +/- faciles d’utilisation Libre accès en ligne pour la majorité
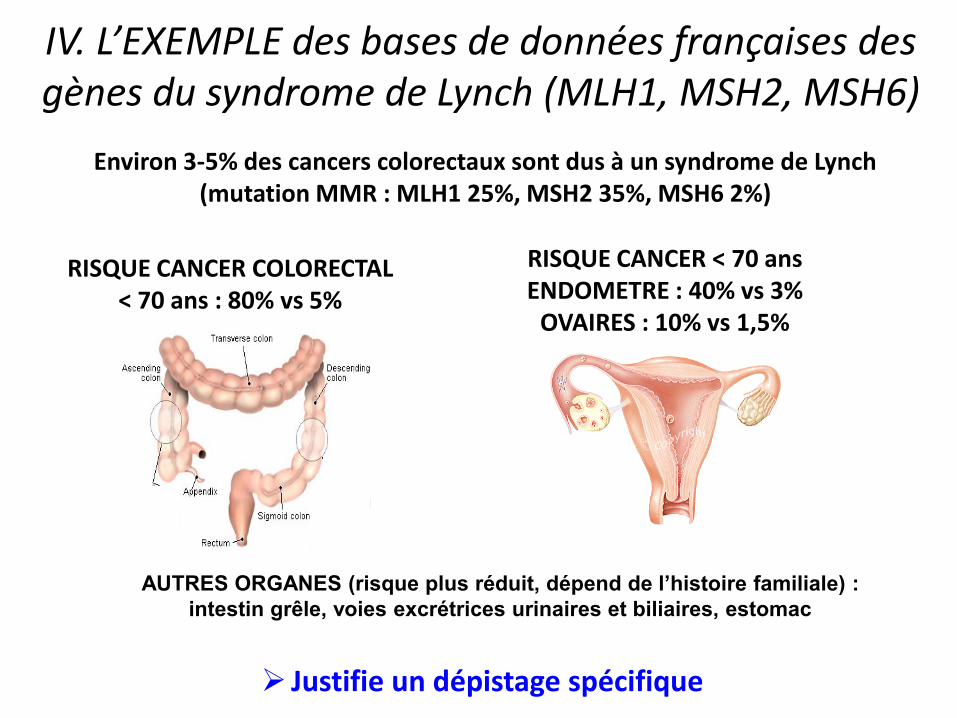
IV. L’EXEMPLE des bases de données françaises des gènes du syndrome de Lynch (MLH1, MSH2, MSH6)
RISQUE CANCER < 70 ans ENDOMETRE : 40% vs 3%
OVAIRES : 10% vs 1,5%
RISQUE CANCER COLORECTAL < 70 ans : 80% vs 5%
AUTRES ORGANES (risque plus réduit, dépend de l’histoire familiale) :
intestin grêle, voies excrétrices urinaires et biliaires, estomac
Environ 3-5% des cancers colorectaux sont dus à un syndrome de Lynch (mutation MMR : MLH1 25%, MSH2 35%, MSH6 2%)
Justifie un dépistage spécifique

IV. L’EXEMPLE des bases de données françaises des gènes du syndrome de Lynch (MLH1, MSH2, MSH6)
Consultations de génétique
18 laboratoires
de diagnostic
Pas de variant ou polymorphismes
Variant pathogène
UV
~70%
~10%
~20%
DatabasesDatabases UMDUMD--MLH1MLH1 UMDUMD--MSH2MSH2 UMDUMD--MSH6MSH6
0
500
1000
1500
2000
2500
3000
2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013
2003-2013 : 22 758 analyses des gènes MMR (MLH1, MSH2, MSH6)
Source : rapport d’activité d’oncogénétique en France - INCa 2013
Nombre d’analyses des gènes MMR
depuis 1995
Accès libre

- Prédiction in silico (UMD-Predictor,
HSF3, SIFT)
- Analyse de ségrégation (LOD-score)
- Phénotype clinique
- Statut MMR dans les cellules
tumorales (microsatellites, IHC)
- Test fonctionnel (littérature)
- Test d’épissage (minigène, RT-PCR)
- Co-occurrence
- Fréquence allélique (dbSNP, ExAC, …)
Critères de classification des bases MMR
IV.1 Algorithme de classification des bases MMR

IV.2 Algorithme de classification des bases de données polypose (APC, MUTYH)
Algorithme spécifique en fonction du gène, de la pathologie
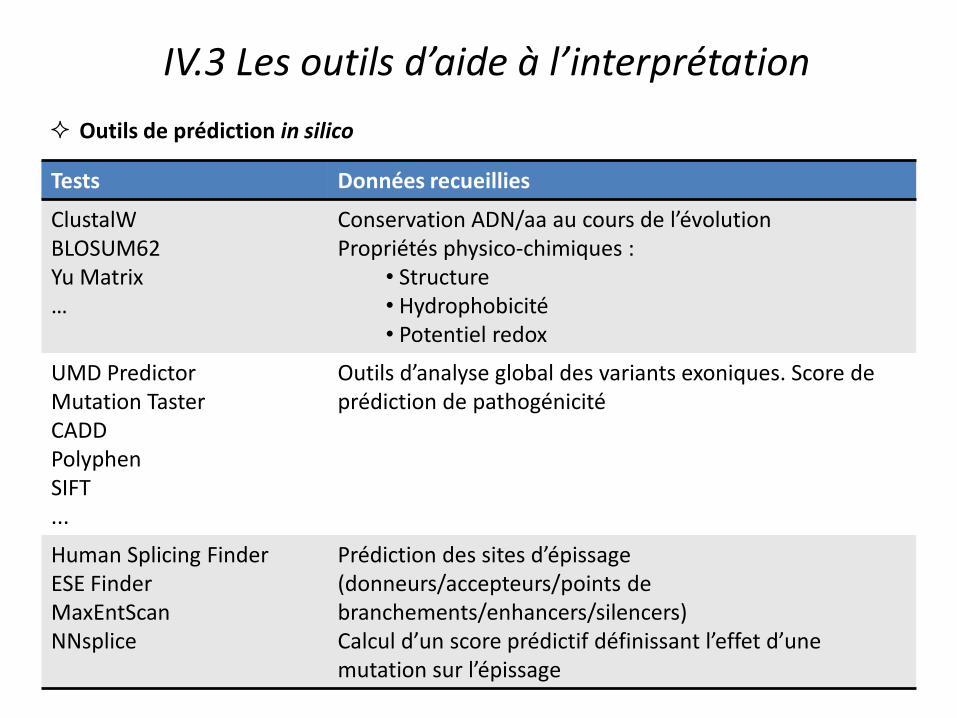
Tests Données recueillies
ClustalW BLOSUM62 Yu Matrix …
Conservation ADN/aa au cours de l’évolution Propriétés physico-chimiques :
• Structure • Hydrophobicité • Potentiel redox
UMD Predictor Mutation Taster CADD Polyphen SIFT ...
Outils d’analyse global des variants exoniques. Score de prédiction de pathogénicité
Human Splicing Finder ESE Finder MaxEntScan NNsplice
Prédiction des sites d’épissage (donneurs/accepteurs/points de branchements/enhancers/silencers) Calcul d’un score prédictif définissant l’effet d’une mutation sur l’épissage
IV.3 Les outils d’aide à l’interprétation
Outils de prédiction in silico

IV.3 Les outils d’aide à l’interprétation
Bases de données de fréquence spécifiques : Exome Sequencing Project, 1000 Genomes Project, HapMap, CEU, etc.
Bases de données de fréquence intégrées : dbSNP, Exome Aggregation Consortium (> 60 000 exomes), Kaviar

%
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
MLH1
MSH2
MSH6
Anomalie d’épissage : 27%
Fonction MMR altérée : 3%
IV.4 Données utilisées pour le classement des variants MMR

Variants différents
(nb d’enregistre-
ments)
Variants tronquants
Variants non
tronquants
Class 1 Neutral
Class 2 Likely
Neutral
Class 3 VUS
Class 4 Likely Causal
Class 5 Causal
APC 720 (3717) 545 175 32 21 100 2 20
MUTYH 109 (1767) 33 76 8 3 52 2 11
MLH1 568 (3063) 248 320 63 25 157 24 51
MSH2 638 (3231) 238 400 84 31 208 34 43
MSH6 443 (2106) 171 272 60 30 164 13 5
TOTAL 2478 (13884) 1235 1243 247 (20%) 110 (9%) 681 (55%) 75 (6%) 130 (10%)
CLASSEMENT DES VARIANTS NON TRONQUANTS
IV.5 Classement des variants MMR / APC / MUTYH
CLASSEMENT DES VARIANTS NON TRONQUANTS

IV.6 Exemple

IV.7 Plus-value de l’interprétation des variants MMR par un réseau de spécialistes
- Réseau national d’experts - Algorithme consensuel de classification
- Collecte systématique de données en rapport avec la pathologie + données in silico,
de fréquence, bibliographiques, etc. - Sessions régulières (2 fois / an) de revue des variants pour classification - Requêtes spécifiques auprès des contributeurs pour des informations
complémentaires (phénotype clinique, fonction MMR, analyses de ségrégation, tests minigènes, RT-PCR, etc.)
- Etudes épidémiologiques (ex : Bonadona et al., JAMA 2011)
INTEGRATION DE L’ENSEMBLE DES DONNEES PAR UN CURATEUR POUR HOMOGENEISATION ET PROPOSITION DE CHANGEMENT DE CLASSE
BASES DE DONNEES ROBUSTES UTILISABLES EN PRATIQUE POUR LE CONSEIL GENETIQUE

« Based upon these considerations, the Working Group did not favor offering the patient a preference as to whether or not to receive the minimum list of incidental findings described in these recommendations. We recognize that this may be seen to violate existing ethical norms regarding the patient’s autonomy and “right not to know” genetic risk information. However, in selecting a minimal list that is weighted toward conditions where prevalence may be high and intervention may be possible, we felt that clinicians and laboratory personnel have a fiduciary duty to prevent harm by warning patients and their families about certain incidental findings and that this principle supersedes concerns about autonomy, just as it does in the reporting of incidental findings elsewhere in medical practice. The Working Group therefore recommended that whenever clinical sequencing is ordered, the ordering clinician should discuss with the patient the possibility of incidental findings, and that laboratories seek and report findings from the list described in the Table without reference to patient preferences. Patients have the right to decline clinical sequencing if they judge the risks of possible discovery of incidental findings to outweigh the benefits of testing. »
V. Aspects légaux et éthiques LE POINT DE VUE AMERICAIN
(ACMG)

Décembre 2013 : rapport « Anticipate and Communicate: Ethical Management of Incidental and Secondary Findings in the Clinical, Research, and Direct-to-Consumer Contexts : 17 recommendations for the ethical and professional management of such findings. »
Etablir un consentement éclairé et informer sur les possibilités de découvertes fortuites et
secondaires
Communiquer sur les risques associés à ces découvertes et les possibilités de prise en charge (formation préalable du clinicien pour information préalable au patient)
Privilégier les tests diagnostiques plus sélectifs pour minimiser le risque de découvertes
fortuites et secondaires
Questions en suspens : Est-ce que les anomalies sur les gènes « actionable » doivent être recherchées
systématiquement ?
Est-ce que les découvertes fortuites et secondaires doivent être systématiquement communiquées au patient ?
S’il y a une prise en charge spécifique associée à une découverte secondaire, qui en est responsable ?
V. Aspects légaux et éthiques LE POINT DE VUE AMERICAIN
(USA Bioethics Commission 5/05/2014)

Privilégier les panels avec une liste de gènes clairement en rapport avec le diagnostic (« core disease gene lists ») ou si WES ou WGS n’analyser que les gènes d’intérêt (pipeline d’analyse ciblée)
Informer le patient et le clinicien de la possibilité de découvertes fortuites ou secondaires
(consultation de génétique, consentement). Eventuellement lui donner le choix de savoir/ne pas savoir (suppose une logistique d’analyse et de compte-rendu personnalisés, complexe en pratique)
Etablir des comptes-rendus compréhensibles. Y détailler tous les variants pathogènes (classe 5) ou
probablement pathogènes (classe 4) (classe 3 à l’appréciation du laboratoire) Conserver dans une base de données locale tous les variants retrouvés afin de faciliter leur
interprétation future (fréquence, LSDBs) et de modifier les comptes-rendus si besoin
V. Aspects légaux et éthiques LE POINT DE VUE EUROPEEN
(EuroGentest - projet CE pour harmoniser les pratiques des tests génétiques + European Society of Human Genetics)

1994 : premières lois de bioéthique
Janvier 2016 : avis consultatif N°124 - Réflexion éthique sur l’évolution des tests génétiques liée au séquençage de l’ADN humain à très haut débit
Changement d’échelle, problème du « big data » (interprétation, conservation, protection des données)
Risque de passage de la génétique diagnostique à la génétique prédictive
L’analyse de génome est susceptible de bientôt représenter le standard
Déterminisme génétique qui pourrait faire abstraction de la complexité de l’humain
Souci des droits de l’Homme, de l’information préalable et du droit de ne pas savoir
Questionnement sur le coût des analyses et des prises en charge en cas de découvertes secondaires
Problématique de l’incertitude dans l’interprétation des données (laboratoire, clinicien, patient)
Quelle analyse pour quel diagnostic ? Quel rendu de résultat ?
V. Aspects légaux et éthiques LE POINT DE VUE FRANCAIS
(Comité National Consultatif d’Ethique)

V.1 Le consentement éclairé L’exemple de l’APHM

1/ Résultats - Indiquer le(s) variant(s) en nomenclature HGVS (c., p., NG ou NM, exon), son interprétation, la maladie associée, le mode de transmission - Eviter le jargon propre à la génétique médicale ou définir les termes - Dans le cas d’un résultat de séquençage d’exome, de génome, de panel, utiliser une terminologie complémentaire :
• Variant dans un gène clairement identifié comme associé au diagnostic clinique • Variant dans un gène probablement associé au diagnostic clinique • Variant secondaires
V.2 Le compte-rendu d’analyse Travaux ACMG/AMP 2015

2/ Interprétation - Préciser la méthode de classification et les critères d’évaluation du variant - Faire toute recommandation au clinicien permettant de compléter l’interprétation (prélèvement pour test fonctionnel, test des personnes apparentées, etc.)
V.2 Le compte-rendu d’analyse Travaux ACMG/AMP 2015
3/ Méthodologie
Préciser
- la méthode de détection utilisée (PCR ou capture ou amplification génome entier et Sanger ou NGS ou
génotypage, etc.)
- les scores qualité s’il y a lieu
- les limites de la méthode
- Les noms des gènes étudiés, leur RefSeq, la version du génome utilisé pour l’alignement

4/ Accès aux associations, à la recherche et aux essais cliniques Possibilité d’ajouter ce type d’information dans le compte-rendu ou dans un document attaché. Intérêt d’une connexion laboratoire/clinicien particulièrement dans le cas de variant de signification incertaine : demande d’analyse complémentaire chez le patient / sa famille, données d’interprétation (expressivité, pénétrance, etc.) 5/ Mise à jour de l’interprétation • Préciser la politique de mise à jour de la classification des variants 6/ Confirmation • Dans le cas d’un variant « pathogenic » ou « likely pathogenic », une confirmation du résultat sur un
nouveau prélèvement est recommandée (la plupart du temps analyse ciblée par une méthode alternative)
• Dans certains cas, nécessaire de tester les parents
V.2 Le compte-rendu d’analyse Travaux ACMG/AMP 2015

V.2 Le compte-rendu d’analyse Travaux ACMG/AMP 2015

CENTRUM MENSELIJK ERFELIJKHEID Patient identification Herestraat 49, 3000 Leuven XXXX XXXX
tel: 016 34 59 03 fax: 016 34 60 60 00000000 erk.#:8-24990-92-996
Gene panel (report)
Indication HCMLQT cardiomyopathies / arrhythmias
Reason of request
The patient has a clinical diagnosis of hypertrophic cardiomyopathy (HCM).
Report Results: This patient is heterozygous for the mutation c.98_99delCA (p.Thr33Argfs*15) in the MYBPC3 gene. The nomenclature of the mutation is based on the transcript NM_000256.3. Conclusion: The patient has a mutation in the gene MYBPC3. This mutation causes a frameshift that leads to an early stop codon causing the production of a truncated protein or no protein production. The mutation has not been previously described in literature, but given the nature of the mutation it has been classified as likely pathogenic. The clinical diagnosis could thus be confirmed at the molecular level. Genetic counseling is recommended. The probability that the first-degree relatives of these patients carry this mutation is 50%. Predictive testing is now possible in the family, but only after genetic counseling.
Background
Cardiomyopathies are a heterogeneous group of structural heart diseases. The various forms of cardiomyopathy are classified on the basis of imaging and electrocardiographic characteristics in four diseases: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), and left ventricular non-compaction cardiomyopathy (LVNC). Hereditary arrhythmogenic heart defects are a heterogeneous group of disorders in which abnormalities in the electrical activity of the heart can lead to arrhythmias. The different types are usually divided into the following conditions: Long QT syndrome (LQTS), Brugada syndrome (BRS), catecholaminergic polymorphic ventricular tachycardia (CPVT), short QT syndrome (SQTS), early repolarization syndrome (ERS), idiopathic ventricular fibrillation (IVF), atrial fibrillation (AF), cardiac conduction defects (CCD), and the sick sinus syndrome (SSS).
The coding exons and flanking introns of the gene panel (target capture HCMLQTv1, Nimblegen) was sequenced by massive parallel sequencing. The analysis and interpretation was done on a subpanel defined below and selected according to the clinical phenotype. If a mutation was found with this method, the result was confirmed on the basis of Sanger sequencing.
In this patient the following genes were analysed: HCM: ACTC1, ACTN2, ANKRD1, CALR3, CASQ2, CAV3, CSRP3, GLA, JPH2, LAMP2, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYLK2, MYOZ2, PLN, PRKAG2, RYR2, TCAP, TNNC1, TNNT2, TNNI3, TPM1, VCL A deletion/duplication analysis has also been performed by MLPA (Salsa P100, MRC Holland) for the gene MYBPC3.
Three annexes complete this report (to be delivered by mail): For the full panel of genes and a description of the technique and the bioinformatics pipeline, see Annex 1. For quality parameters, see Annex 2 . For an overview of the mutations and variants in this patient, see Annex 3 .
V.2 Le compte-rendu d’analyse Travaux EuroGentest / ESHG 2015
Analyse panel

V.2 Le compte-rendu d’analyse Compte-rendu Pitié-Salpêtrière

CONCLUSION
Tendance actuelle : panel de gènes – bon rapport coût/efficacité, se limite +/- aux découvertes primaires
Exome / génome : de moins en moins coûteux, pourraient devenir la norme : possibilité de
ne regarder que les gènes d’intérêt +/- gènes secondaires +/- autres gènes (exhaustivité ou pas)
Pour les variants primaires comme secondaires ou autres : intérêt de bases de données
d’interprétation gène-spécifique (classification adaptée, expertise, mises à jour régulières). Les critères d’évaluation et les niveaux de preuves sont fonction du gène étudié et de la pathologie associée
Intérêt d’une communication inter-laboratoires pour compléter et croiser les données et
conclure (création de bases de données communes, réunions et mises à jour régulières pour établir/adapter les critères de classification et interpréter les variants, etc.)
Nécessité d’informer les personnes par un conseil génétique adapté (consentement, droit
du patient de savoir ou de ne pas savoir)
Compte-rendu détaillé et clair : le clinicien doit considérer l’interprétation du laboratoire en fonction des informations propres à son patient (ex. ségrégation dans la famille) et doit adapter la décision thérapeutique en fonction du contexte clinique et de l’indication du test

MERCI DE VOTRE ATTENTION





























