Embed Size (px)
Citation preview

Pbl 2 – mbod Asem mustafa shadid

Objectives1• Define anemia and it's genetic bases.
2• Describe thalassemia and it's genetic bases.
3• What is the molecular defects and pathophysiology of thalassemia
4• Describe the diagnostic test that detect genetic mutation of thalassemia
5• Overview of pre-marital screening test.

Define anemia and it's genetic bases

Define anemia and it's genetic
bases
• it is usually defined as a decrease in the amount of red blood cells (RBCs) or hemoglobin in the blood.
• It can also be defined as a lowered ability of the blood to carry oxygen.
• There are three main types of anemia, that due to blood loss, that due to decreased red blood cell production, and that due to increased red blood cell breakdown.
• Causes of decreased production include iron deficiency, a lack of vitamin B12, thalassemia and a number of neoplasms of the bone marrow.
• Causes of increased breakdown include a number of genetic conditions such as sickle cell anemia, infections like malaria .

Anemia and Your Genes
• Some people are born with genetic abnormalities that can cause certain types of anemia, including sickle cell anemia, thalassemia, and Fanconi anemia.
• Many people think of anemia as something that happens because of outside factors, like a poor diet .
• Among the types of anemia that can be inherited are:
1. Sickle-cell anemia. People with sickle-cell anemia have a gene that causes the blood protein hemoglobin to form abnormally. As a result, red blood cells are produced in a sickle shape.
2. Thalassemia. occurs when your body is unable to produce enough hemoglobin, which functions to carry oxygen throughout the body.
3. Fanconi anemia. This type of anemia stems from an inherited blood disorder that prevents the bone marrow from producing an adequate supply of new blood cells for the body.

Describe thalassemia and it's genetic bases.

Describe thalassemia and it's genetic bases.
• Each type of thalassemia is a result of the body’s inability to make enough red blood cells and hemoglobin.
• Hemoglobin A, which is the normal type of hemoglobin, is made up of two alpha globin and two beta globin protein chains.
• Genes from each parent are needed to make each of these protein chains.
• If there is a defect in either protein chain, alpha (in which the defect is in the alpha globin protein) or beta (in which the defect is in the beta globin protein) thalassemia can occur.
• Both alpha and beta thalassemia are further classified into these subtypes:
• Thalassemia major• Thalassemia minor

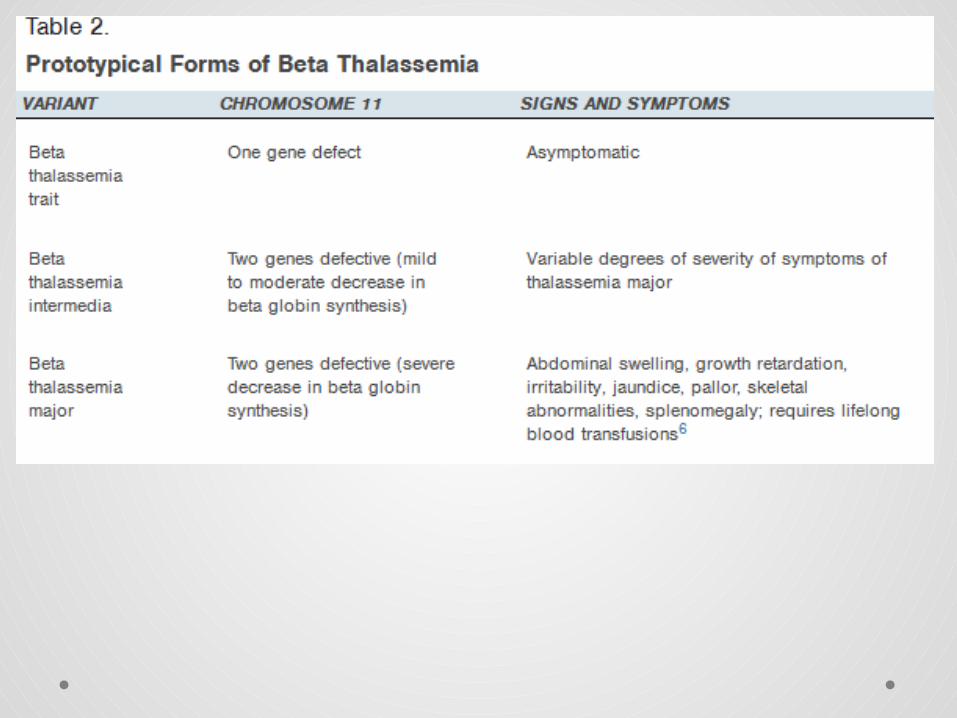
• If you inherit the defective hemoglobin gene from both parents, the more severe form of this blood disorder, thalassemia major, occurs.
• Major Thalassemia: is the homozygous state wherein both parents are carriers of the mutated gene. The individual has two copies of the thalassemia gene. Individuals with thalassemia have severe anemia requiring lifelong blood transfusions and other medical procedures for survival.
• Thalassemia minor occurs when you receive the defective gene from only one parent. If you have this form of the disease, you do not have symptoms but are a carrier of the blood disorder.
• • Minor Thalassemia: is the heterozygous state wherein one of the parents is a carrier of the mutated gene. The individual has only one copy of the thalassemia gene along with one normal beta-globin chain gene. Individuals with thalassemia minor do not require any treatment except nutritional supplements and lead a fully normal life.

• Other forms of thalassemia include:• Hb S/beta thalassemia. The affected person has thalassemia
minor and inherits one gene for sickle cell disease.
• E-beta thalassemia. The affected person has thalassemia minor and a defective gene that produces a variant form of hemoglobin called hemoglobin E.
• The thalassemias are classified according to which chain of the hemoglobin molecule is affected. In α-thalassemias, production of the α globin chain is affected, while in β-thalassemia, production of the β globin chain is affected.
• Both α- and β-thalassemias are often inherited in an autosomal recessive manner.
• For the autosomal recessive forms of the disease, both parents must be carriers for a child to be affected.
• If both parents carry a hemoglobinopathy trait, the risk is 25% for each pregnancy .

What is the molecular defects and pathophysiology
of thalassaemia

Pathophysiology• Normally, the majority of adult hemoglobin (HbA) is composed of
four protein chains, two α and two β globin chains arranged into a heterotetramer.
• In thalassemia, patients have defects in either the α or β globin chain, causing production of abnormal red blood cells . note that : (In sickle-cell disease, the mutation is specific to β globin).
• The thalassemias are classified according to which chain of the hemoglobin molecule is affected.
• The β globin chains are encoded by a single gene on chromosome 11.
• α globin chains are encoded by two closely linked genes on chromosome 16.

Hemoglobin• Hemoglobin consists of an iron-containing heme ring and four globin
chains: two alpha and two nonalpha.
• The composition of the four globin chains determines the hemoglobin type.
• Fetal hemoglobin (HbF) has two alpha and two gamma chains (alpha2 gamma2).
• Adult hemoglobin A (HbA) has two alpha and two beta chains (alpha2 beta2), whereas hemoglobin A2 (HbA2) has two alpha and two delta chains (alpha2 delta2).
• At birth, HbF accounts for approximately 80 percent of hemoglobin and HbA accounts for 20 percent.
• The transition from gamma globin synthesis (HbF) to beta globin synthesis (HbA) begins before birth. By approximately six months of age, healthy infants will have transitioned to mostly HbA, a small amount of HbA2, and negligible HbF .

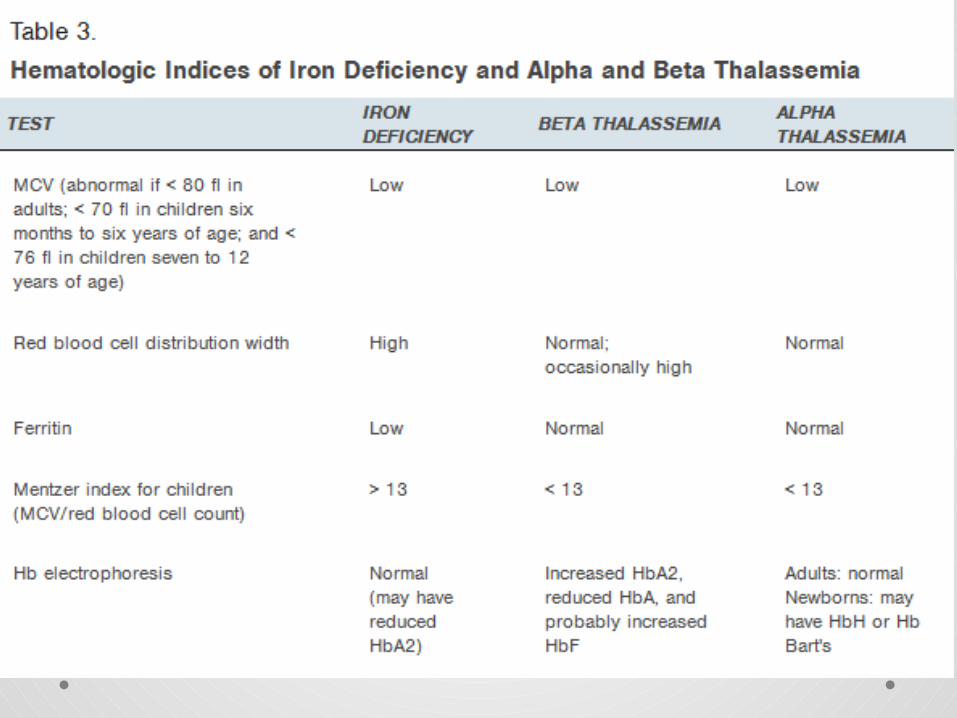
ALPHA THALASSEMIA• Alpha thalassemia is the result of deficient or absent synthesis of alpha globin
chains, leading to excess beta globin chains. therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns.
• Alpha globin chain production is controlled by two genes on each chromosome 16 . The α-thalassemias involve the genes HBA1and HBA2 .
• Deficient production is usually caused by a deletion of one or more of these genes
1. A single gene deletion results in alpha thalassemia silent carrier status, which is asymptomatic .
2. The two-gene deletion causes alpha thalassemia trait (minor) with microcytosis and usually no anemia.
3. The three-gene deletion results in significant production of hemoglobin H (HbH), which has four beta chains (beta4).
4. The four-gene deletion results in significant production of hemoglobin Bart's (Hb Bart's) .


BETA THALASSEMIA• Beta thalassemia is the result of deficient or absent
synthesis of beta globin chains, leading to excess alpha chains.
• Beta globin synthesis is controlled by one gene on each chromosome 11. Beta thalassemias are due to mutations in the HBB gene .
• Beta globin chain production can range from near normal to completely absent, leading to varying degrees of excess alpha globin to beta globin chain production.
• The one gene defect, beta thalassemia trait (minor), is asymptomatic and results in microcytosis and mild anemia.
• Persons with beta thalassemia major are almost never symptomatic at birth because of the presence of HbF, but symptoms begin to develop by six months of age.

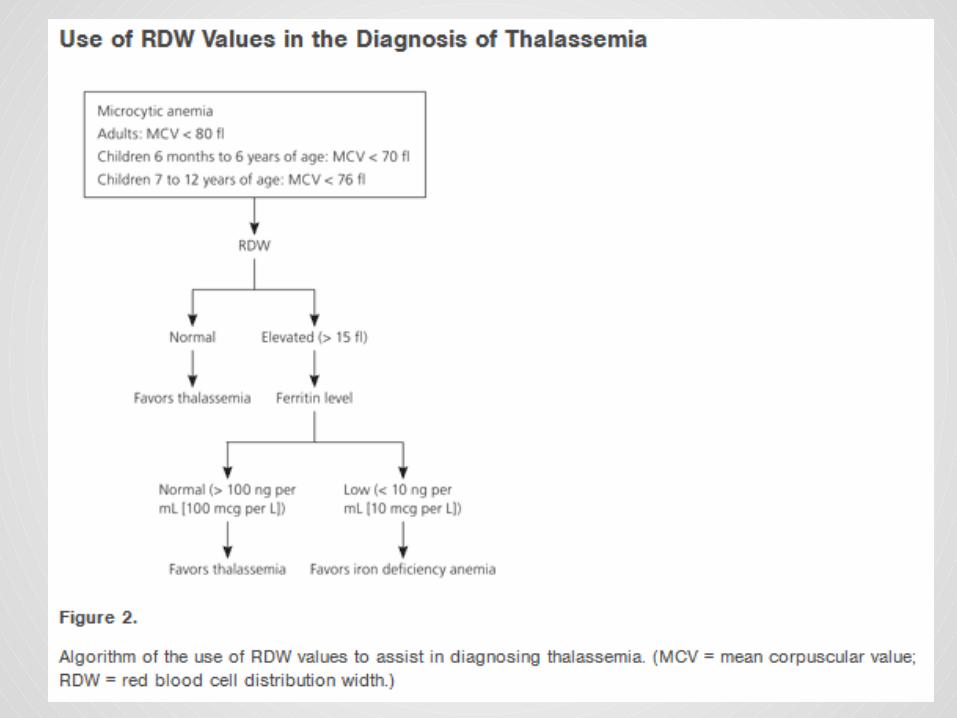
Describe the diagnostic test that detect genetic mutation of thalassemia

Overview of pre-marital screening test.

Definition of Premarital
Screening:
• It is defined as conducting examination for soon-to-be married in order to identify if there is any injury with genetic blood diseases such as sickle-cell anemia (SCA) and Thalassemia, and some infectious diseases such as hepatitis B, C and HIV "Aids".
• This is in order to provide medical consultation on the odds of transmitting these diseases to the other marriage partner or the children in the future, and to give options and alternatives before soon-to-be married with the aim of helping them plan for a hea;thy,sound family.

Objectives of Premarital
Screening:
• • The healthy marriage program is considered a national, communal. Aware, and preventive program aims at :
•• Limiting the spread of some genetic blood diseases: sickle-cell anemia (SCA) and thalassemia, and some infectious diseases: hepatitis B, C and AIDS/HIV.
•• Reducing the financial burdens resulting from the treatment of the injured in terms of the family and community.• Reducing pressure over health institutions and blood banks.• Avoiding the social and psychological problems for families whose children suffer.• Making those seeking such an check-up feel at ease.• Disseminating awareness with regard to the concept of the comprehensive, healthy marriage.

Thank you Asem shadid