Embed Size (px)
Citation preview



•Producción defectuosa de hemoglobina
•Disminución en la producción y un aumento de la destrucción de los glóbulos rojos.
•Es un trastorno hereditario.
Nombres alternativos:Talasemia; Anemia mediterránea; Anemia de Cooley

HemoglobinaEstructura tetrámero de dos pares de
cadenas – globinas- ; cada una contiene un grupo prostético denominado Heme.

ESTRUCTURA DEL HEM
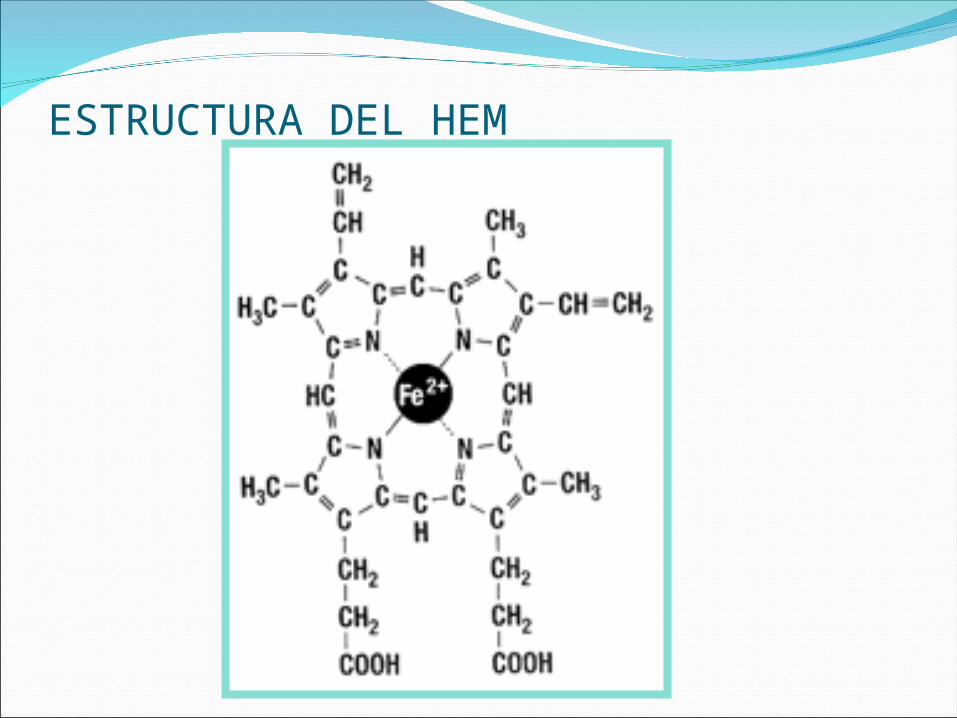
ESTRUCTURA DEL HEM
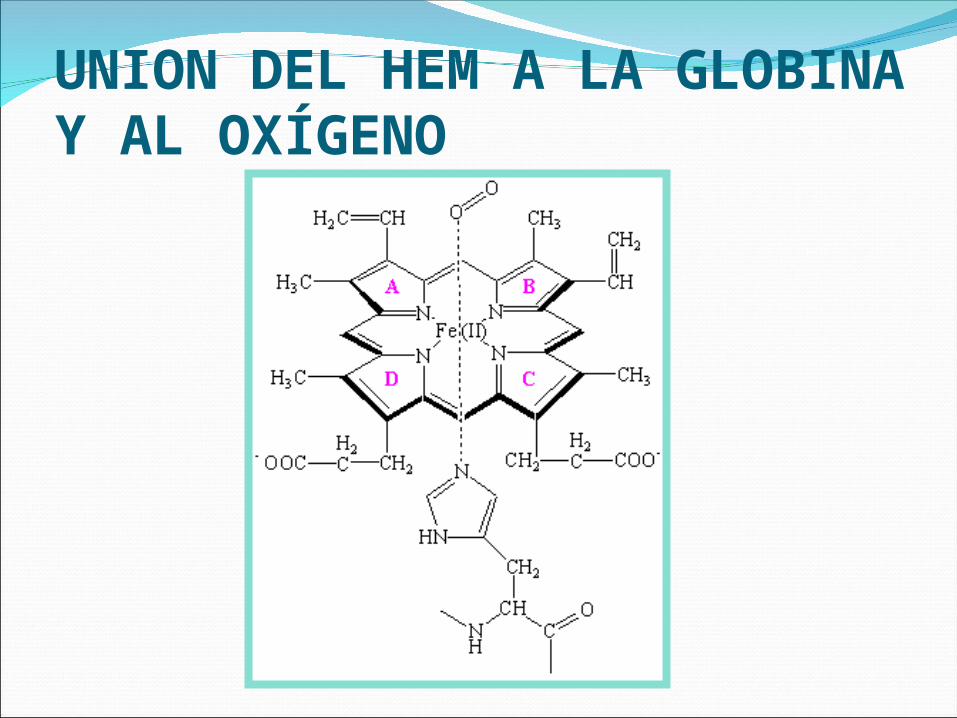
UNION DEL HEM A LA GLOBINA Y AL OXÍGENO

Genética:
HERENCIA AUTOSOMICA RECESIVA (herencia mendeliana).
• Se manifiesta al momento que ambos miembros del par génico afectados (genes recesivos). Homocigotos para alelo recesivo son encontrados en el genotipo del individuo.
• Los portadores sanos poseen un alelo recesivo del par de alelos en su genotipo, el cual no es manifestado hasta el cruce de transmisión a su siguiente generación

•Las talasemias alfa son causadas por la eliminación de uno o más genes de la cadena de globina alfa
•Las talasemias beta son causadas por una mutación en la cadena de la globina beta.
•Las talasemia alfa se presentan más comúnmente en habitantes del sudeste de Asia y China
•Las talasemias beta se presentan en personas de origen mediterráneo y, en menor grado, en individuos chinos, otros asiáticos y negros.

Se da como resultado de un defecto en la síntesis de las cadenas globínicas a nivel de transcripción, y procesamiento del RNA y traducción de la proteína.
Esta produce la Deleción de grandes segmentos de ADN
que incluye genes completos o pequeños segmentos; siendo este el causal de la talasemia alfa y en el caso de la talasemia beta se ha dado por algún tipo de mutación específica.
Al manifestarse esta afecta proteínas enzimáticas y en igual
proporción a hombres como mujeres. Esta dependiendo del alelo afectado se generan de modo
diferente.

•Los dos tipos principales son:
Talasemia Alfa :la cadena alfa es afectada Talasemia Beta : la cadena beta es afectada
•Las talasemias también se categorizan por el número de alelos que están defectuosos:
•Talasemia Menor - un alelo anormal (portador / rasgo talasémico / heterocigoto)
•Talasemia Mayor - dos alelos anormales (Enfermo / homocigoto)

•Microcitosis e hipocromía eritrocitaria.
•Disminución mínima de la hemoglobina.
•HCM (hemoglobina corpuscular media).
•VCM (volumen corpuscular medio).
Severa:
•Anemia grave con una tasa en la síntesis de globinas irregular. •Vida fetal exceso de globina alfa (Hemoglobina Bart).
•Vida adulta deficiencia de globina alfa y exceso de globinas beta (Hemoglobina H).
Talasemia Alfa: Benigna:
Talasemia Beta:
Menor: Mayor:•Primeros meses de vida anemia hemolítica y hepatoesplenomegalia (Indicación de transfusión sanguínea).•Prominencia del cráneo y sobrecrecimiento de la región maxilar con facies mongoloides.•Aumento en los niveles de hierro sérico lo cual produce afecciones cardiaca como ICC, endocrinas como hipotiroidismo e hipoparatiroidismo y afección hepática (Cirrosis).
•Anemia moderada (No tienen indicada transfusión sanguínea).•Aumento de glóbulos rojos con hipocromía y Microcitosis.
•Asintomático en la mayoría de los casos.•Aumento en los niveles de hemoglobina A.

Prevención •Asesoría genética en familias con talasemia conocida.
•Evaluación prenatal
La muestra de vellosidad corionica (CVS) normalmente se realiza entre las semanas 10 y 12 del embarazo. la amniocentesis usualmente se practica entre las semanas 15 y 18 del embarazo.

•Un frotis de sangre periférica muestra glóbulos rojos pequeños con forma anormal
Una electroforesis hemoglobínica muestra hemoglobina anormal
•Esta enfermedad puede alterar también los resultados de los siguientes exámenes:Índices de glóbulos rojos sanguíneos (tamaño, contenido de la hemoglobina)
•Cuadro hemático.
•El diagnóstico genético preimplantacional (DGP).
•Es el análisis de embriones obtenidos in vitro para evitar la transmisión de enfermedades y/o alteraciones genéticas. Este análisis se realiza antes del establecimiento del embarazo e implica el uso combinado de técnicas de reproducción asistida y de genética molecular.(PCR)
•Transfusiones de sangre regulares.
•El trasplante de medula ósea.
•Terapia de quelación.(desferrioxamina)

•NANDA RITA. Electroforesis de hemoglobina. Department of Medicine, Section of Hematology/Oncology, University of Chicago Medical Center, Chicago, IL. Review provided by VeriMed Healthcare Network. 2007.• http://www.emea.europa.eu/humandocs/PDFs/EPAR/exjade/H-670-PI-ro.pdf
•FRANK A. GRECO, Porfirinas en sangre. The Lahey Clinic, Burlington, MA. Review provided by VeriMed Healthcare Network.2005.
•VARGAS DE LOS MONTEROS, FERNÁNDEZ-NOVOA GARCÍA. Diagnóstico de las enfermedades monogénicas y cromosómicas. Vox paediatrica, 7,1 (114-117), 1999.






























