Embed Size (px)
Citation preview

MASCOTNixon Mendez
Department of Bioinformatics

Basics
Simple MS – molecular weight of peptide mixture.
MS/MS (Tandem MS) – sequence structural information by
recording the fragment ion spectrum of peptide.

BASICS
Tandem MS
Mass spectrometry

PURPOSE OF MS
Elemental composition.
Masses of particles of molecules.
Identify unknown compounds.
Isotopic Composition.

INTRODUCTION
Mascot is a software package from Matrix Science
(www.matrixscience.com) that interprets mass spectral data into
protein identities.
It uses mass spectrometry data to identify proteins from primary
sequence databases.

INTRODUCTION
The experimental mass values are then compared with calculated
peptide mass by applying cleavage rules to the entries in a
comprehensive primary sequence database.
If unknown protein is present, we will get precise entry otherwise pull
out those entries which exhibit the closest homology(related species).

Database
Eg. Protein databases -
Non-redundant NCBI,
Swiss-Prot,
IPI, etc.
Peak Lists
In silico digest
820.7
842.5
1012.6
1296.6
1555.7
……...
Algorithm
compares peak
lists
Gel separation – 1D or 2D
Excise
Spot
Trypsin
Digest
Protein Peptides699.0 1159.2 1619.4 2079.6 2539.8 3000.0
Mass (m/z)
1.6E+4
0
10
20
30
40
50
60
70
80
90
100
% Intensity
4700 Reflector Spec #1 MC=>TR[BP = 1479.9, 15779]
1479.8824
1439.8967
1567.8276
1163.7000
2045.1273
927.5582
1881.0223
1724.9272
1305.7888
1730.7723
1399.7751
1249.6954
1895.0386
1283.7881
1433.8074
1554.7437
1640.0277
841.5205
2555.2903
1763.7820
1687.8691
2262.0557
1516.7135
1014.6827
1590.8619
1081.5479
1121.5520
2458.3052
1195.6243
789.5378
898.5428
2493.3501
Mass spectrum (MS)
Peak List820.7
842.5
1012.6
1296.6
1555.7
……...
Reports Protein
Identification
Database searching

Algorithm used..
• Program MASCOT is based on the MOWSE algorithm; this program also
evaluates a possibility of random matching of experimental and
theoretical peptide masses.
• The Algorithm MOWSE (Molecular Weight Search) is more selective and
sensitive.

Two Mascot Choices
Matrix Sciences offers two choice for users:
A free, open access web-based system for occasional (1-10) queries.
A locally installed version for heavy use or highthroughput MS (100’s
queries/day)

MASCOT Home Page

MASCOT SEARCH STRATEGIES
Mascot has three main search modes:
Peptide Mass Fingerprint(based on a list of peptide mass values).
Sequence Query (based on one or more peptide mass values
associated with information such as partial sequence information).
MS/MS Ion Search (based on raw MS/MS data from one or more
peptides).

PEPTIDE MASS FINGERPRINT
• It is possible to identify the protein from available mass spectrum of the peptide mixture
resulting from the digestion of a protein by specific enzyme.
• This method is useful for identifications of protein with already known sequences.
• Requires enzymes of great specificity.
• Mascot looks for the highest scoring set of peptide matches which are within a contiguous
stretch of sequence less than or equal to the specified protein molecular weight.

SEQUENCE QUERY
• The sequence query, in which one or more peptide molecular masses are combined with sequence,
composition and fragment ion data
• It is potentially the most powerful search.
• The source of the information is MS/MS spectrum.
• The sequence query mode of Mascot supports both standard and error tolerant sequence tags.

MS/MS IONS SEARCH
• The MS/MS ions search accepts data in the form of peak lists containing mass and intensity
pairs.
• The high level of specificity of an MS/MS ions search means that it is not essential to choose
an enzyme.
• Obtaining matches to a number of peptides from a single protein provides a very high level of
confidence that the result is correct.

PEPTIDE MASS FINGERPRINT


Parameters used in database
searching
Database searched
Taxonomy
Enzyme
Missed cleavages
Fixed versus variable modifications (PTMs)

SCORING SCHEMES
PROBABILITY BASED SCORING
Mascot incorporates a probability based implementation of the Mowse
algorithm
The total score is the absolute probability that the observed match is
a random event.
Advantages :
Different types of matching (peptide masses and fragment ions) can
be combined in a single search.
Scores from different searches and on different databases can be
compared.
Search parameters can be optimised more readily by iteration.

For this reason,
We report scores as -10*LOG10(P), where P is the absolute probability.
Probability of 10-20 thus becomes a score of 200.
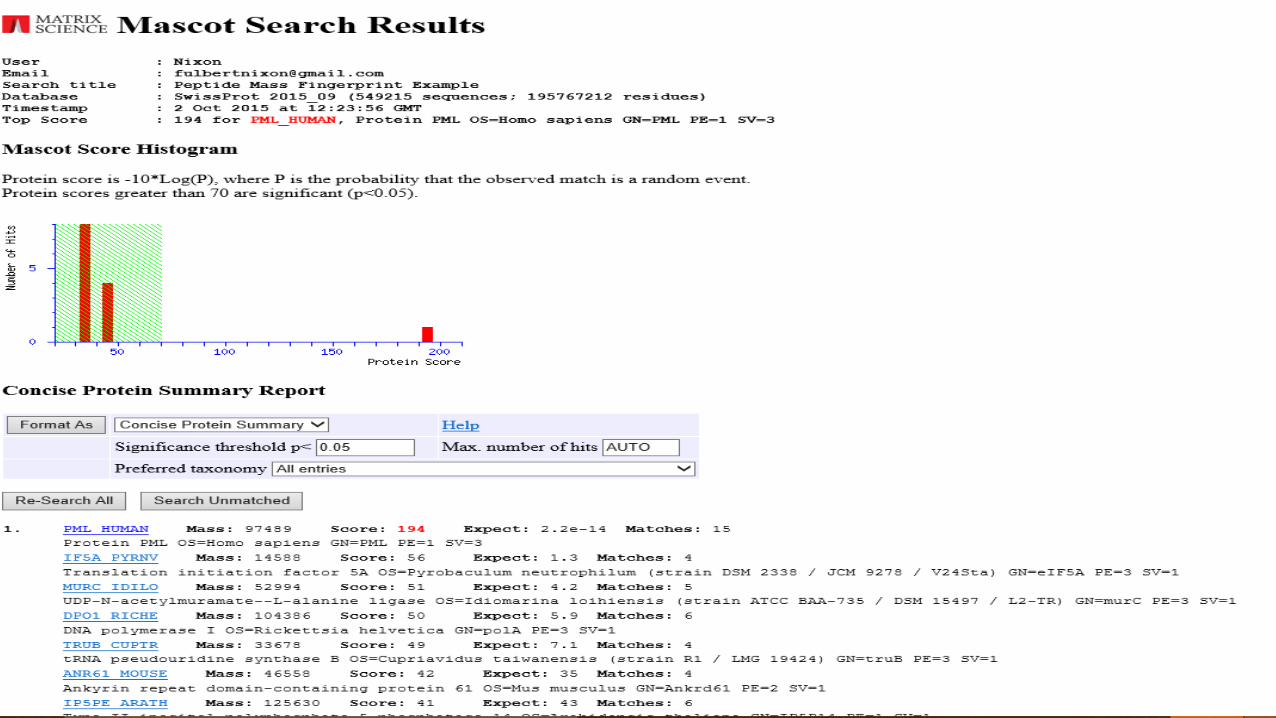
Result

The best result is obtained for PML_HUMAN having a score of 194, we
can confirm it by referring the graph where there is one hit and 194
lies in the significant region having a threshold score of 70 so the
chances of randomness is reduced.
The hits below the threshold region are insignificant.

Result

MS/MS IONS SEARCH

Parameters
Additional parameters within each query are optional, and can be used to
specify:
TITLE for spectrum identification
CHARGE state of the precursor peptide
TOL peptide tolerance
TOLU peptide tolerance units
SEQ for a sequence qualifier (multiple SEQ qualifiers are allowed)
COMP for a composition qualifier (only one COMP qualifier is allowed)
TAG for a sequence tag (multiple TAG qualifiers are allowed)



Result

There is only one hit having a score of 180 that falls in the significant
region.
In MS/MS Ion search the best result is taken by number of queries
matched and the score should be highlighted in bold & red.

Sequence Query

SEQUENCE QUERY

Result

Result

Here we obtain 3 hits for the score 76, which fall in the significant
region.
So, here the best match is selected by the numbers of the queries
matched.
LYSCO_PHACO is the best match for this result.

Thank You





























