
©2014 Waters Corporation 1
USP Modernization Initiative in the Context of Method Transfer and Working Globally

©2014 Waters Corporation 2
Method Transfer Environment
How operate today – Work Globally – With Partners – Use CMOs and CROs – Elevates importance of instrument and methods transfer
Aged Instrumentation and Standardization – Need to replace older HPLCs – Many of this older units are in QC laboratories – Looking at ability to standardize and reduce variability – Pressures to “Modernize” and utilize newer technology
Need for Efficiency – New regulatory pressures – Require “lean” laboratory operation

©2014 Waters Corporation 3
HPLC install base, legacy methods, moderization, utilization...

©2014 Waters Corporation 4
Guidance Documents Important to Technology Transfer
US-FDA Guidance for Industry; – PAC-ATLS: Post Approval Changes – Analytical Testing Laboratory Sites (1998) – Comparability protocols (February 2003) – Changes to an Approved NDA or ANDA (April 2004) – Changes to an Approved NDA or ANDA; Specifications – Use of Enforcement Discretion for
Compendial Changes (November 2004) – CMC Postapproval Manufacturing Changes Reportable in Annual Reports (March 2014) – Process validation Guideline (2011) – Analytical Procedures and Methods Validation for Drugs and Biologics (February 2014)
European Variation guideline ISPE Technology Transfer Guide World Health Organization - WHO Annex 7 Technology Transfer ICH
– Q1A-E Stability Testing of New Drug Substances and Products – Q2 (R1): Validation of Analytical Procedures: Text and Methodology – Q8 Pharmaceutical Development – Q9 Quality Risk Management – Q10 Pharmaceutical Quality Systems – Q11 Product Life Cycle Management/applies to API
US Pharmacopeia – Chapter <1224>, <1225>, <1226> – Chapter <621> (specific to chromatography)

©2014 Waters Corporation 5
Method Transfer Approaches
Use existing method and/or monograph
Adjust method within USP Chapter <621> or EP <2.4.46> guidelines
o These modifications of parameters are allowed only when the chromatogram improvement is still within the stated system suitability factors
o Within these allowed limits, the change of method is only regarded as an adjustment of the method
Make changes to an approved method and report as minor change in annual report to FDA or EMA – Validation required; Possibly submit monograph
Re-develop Method(s)
©2014 Waters Corporation 6
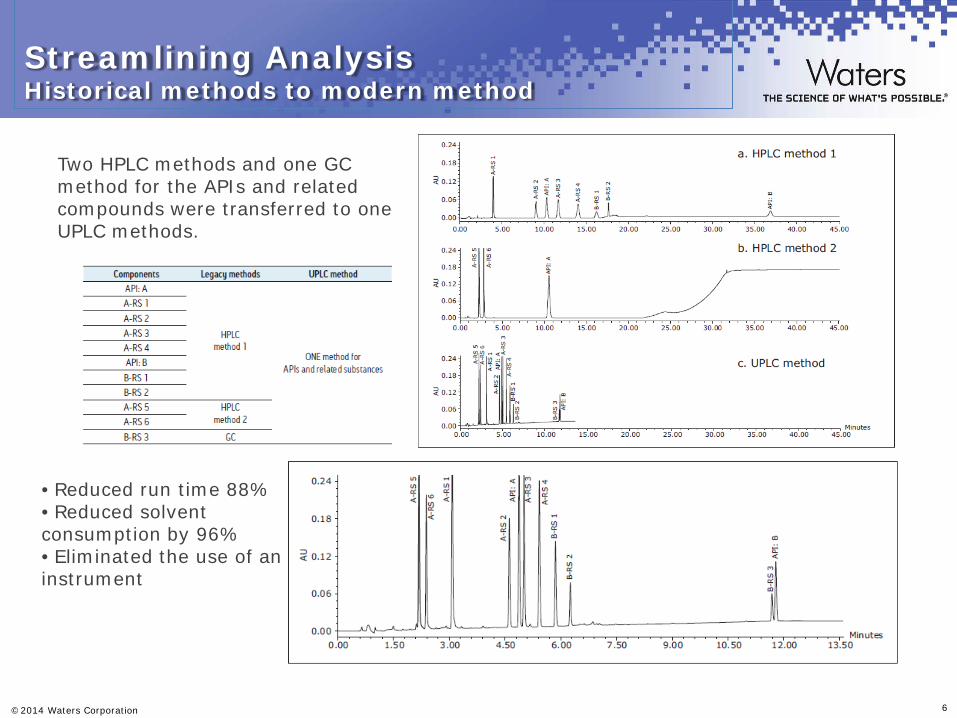
Streamlining Analysis Historical methods to modern method
• Reduced run time 88% • Reduced solvent consumption by 96% • Eliminated the use of an instrument
Two HPLC methods and one GC method for the APIs and related compounds were transferred to one UPLC methods.

©2014 Waters Corporation 7
US FDA Guidelines for method change
Changes are allowed to registered methods – FDA - Allowing implementation of new techniques while maintaining
standards – FDA looking to allow for changes in methods but not methodology
FDA recognizes that there needs to be balance – Allow for some changes to be more routine
o Minor changes, Moderate changes, Major changes – Minor (annual report – no fees), Moderate (supplement), Major (Prior
approval supplement)
Exercise of Enforcement Discretion – Notification that FDA intends to exercise enforcement discretion and does
not intend to take action to enforce compliance with the compendial changes requirement as stated in 21 CFR 314.70(c)(2)(iii) if manufacturers submit such changes in their annual reports.
– FDA intends to develop further guidance to clarify the requirements of 21 CFR 314.70(c)(2)(iii).
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM077097.pdf http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM122871.pdf http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070544.pdf

©2014 Waters Corporation 8
US FDA New Guidance March 2014
The revised 13 page document covers changes to both NDAs and ANDAs Appendix A and Appendix B describe the types of changes that can now be submitted
in the Annual Report. – Appendix A contains those types of changes that can be submitted in the Annual Report if the
change has minimal potential to have an adverse effect on product quality – Appendix B restates the annual reportable changes found in the various Scale-up and Post-
approval Changes (SUPAC) Guidances. FDA reminds the industry to continue to pay attention to the older “Changes to an
Approved NDA and ANDA” Guidance and also notes that data supporting the change should be adequately presented to support the applicant’s determination that the change did not require the submission of a supplement.
http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm217043.pdf

©2014 Waters Corporation 9
FDA’s 21st Century Quality Vision and Advancing Regulatory Science

©2014 Waters Corporation 10
FDA’s 21st Century Quality Vision
FDA’s Center for Drugs Redoubles Effort to Achieve 21st Century Quality Vision and Foster Continuous Process Improvement Jun 25th, 2013 FDA doubles down on its effort to achieve the vision laid out under its Pharmaceutical Quality for the 21st Century initiative a decade ago of a “maximally efficient, agile, flexible pharmaceutical manufacturing sector that can reliably produce high-quality drugs without extensive regulatory oversight.” Delivering the keynote speech at the generic drug CMC conference cosponsored by the Generic Pharmaceutical Association (GPhA) and FDA in early June 2013, CDER Director Janet Woodcock reviewed:
● The “historical trajectory of FDA quality regulation” ● Where the agency has succeeded and fallen short in realizing its 21st century vision ● What the obstacles are ● What the agency is now doing across the CMC and GMP arenas organizationally and policy-wise to overcome them.
Woodcock stated candidly, “we did not achieve a regulatory system that would enable the vision that we put forth, and we knew that. We knew we had not gotten there. And therefore what we are going to do now is make another attempt to do this.” Source: International Pharmaceutical Quality Pulications
http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/RegulatoryScience/UCM268225.pdf

©2014 Waters Corporation 11
Application of novel science and technologies

©2014 Waters Corporation 12
Categories of Post Approval Changes
Classification of Post Approval Changes
FDA EMA Major Change Type II Variation Substantial potential to have an adverse effect on the identity, strenth, quality, purity, or potency of a drug product.
Significant impact on the quality, safety or efficacy of a medicineal product.
Prior Approval Supplement (PAS) Prior Approval Procedure
PDUFA V goal date - 4 months Validation +(30, 60, 90) CHMP + 15 days to review and approve
Moderate Change Type IB Variation Moderate potential to have an adverse effect on the identity, strenth, quality, purity, or potency of a drug product.
Minor Variation which is neither a Type IA nor a Type II Variation nor an Extension
CBE 30 - Submission at least 30 days before ditribution of the post change product
Notification Procedure Validation +30 days
CBE-0 Distribution can occcure whe the FDA recieves the supplement
Minor Change Type IA, IAin Variation Minimal potential to have an adverse effect on the identity, strenth, quality, purity, or potency of a drug product.
Minimal impact, or no impact on the quality, safety or efficacy of the medincal product
Annual Report Notification Procedure 30 days

©2014 Waters Corporation 13
Europe EMA Variation (Change) categories
IA: Minor variations – Do not require prior approval for implementation – Require notification
o IA: require notification within 12 months of implementation o IAIN: require immediate notification
IB: Minor variations that are not IA/IAIN nor II nor extension – Must be notified to the National Competent Authority/European Medicines
Agency (‘the Agency’) by the Marketing Authorization Holder (MAH) before implementation, but do not require a formal approval
II: Major variations – Require formal approval prior to implementation
http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500003981.pdf

©2014 Waters Corporation 14
Europe EMA Variation for drug substances
©2014 Waters Corporation 15
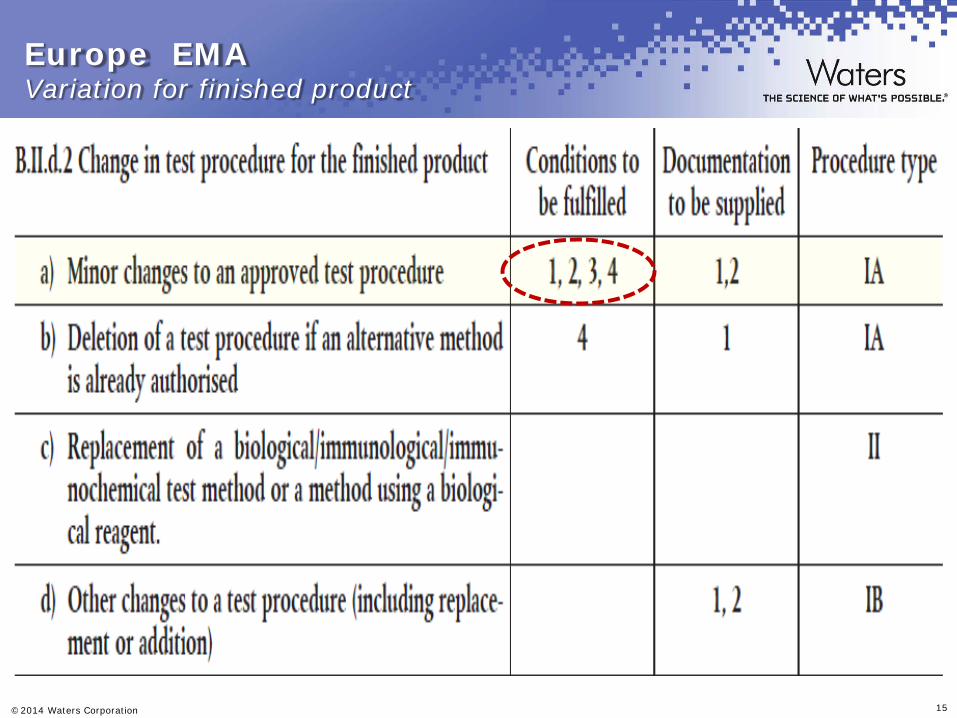
Europe EMA Variation for finished product

©2014 Waters Corporation 16
Europe EMA Conditions for Variation

©2014 Waters Corporation 17
European Medicines Agency (EMA) Current guidelines for method change
Use new Post Approval Change Management Protocol (for EU only) – principale: declare in PAC MP the expected future changes
(strategy), that will then be later considered as minor changes (results)
– introduce in a new Application, or as variation (type II) in an existing file
– easier in a centralized procedure
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/04/WC500125400.pdf

©2014 Waters Corporation 18
USP and Method Transfer
USP Monographs USP Chapter <621> USP Chapters <1224>, <1225>, <1226>
– <1224>Transfer of Analytical Procedures – <1225> Elements Recommended for the Transfer of Analytical Procedures – <1226> Verification of Compendial Procedures

©2014 Waters Corporation 19
USP Standards: Monographs
USP – USP creates the official public monographs for prescription and over-the counter medications in the United
States in conjunction with the pharmaceutical industry and others. – Monographs for these products are published in the United States Pharmacopeia and National Formulary
(USP–NF). – The current USP–NF comprises more than 4,500 monographs and are named in the U.S. Federal Food, Drug,
and Cosmetic Act as the official compendia of the nation. – USP–NF standards are also used in more than 130 countries
What is a Monograph? – A written document, or standard, that describes an item (e.g., a finished drug, a drug ingredient, or food
chemical). – A monograph published in any USP compendium provides:
• the name of a substance; its definition; package, storage, and labeling requirements; • and information on tests needed to ensure the substance is of the appropriate strength, quality, and
purity.
Importance of Monographs – Monographs give scientists, governments, manufacturers, and others a public standard by which to judge
an article’s quality. – The existence of a public standard for substances consumed in the global marketplace is a key element of
the safety nets that help maintain and improve public health. What is a public standard?
– Public standards are developed in an open and collaborative process, seeking informed input from independent experts with a wide variety of backgrounds – healthcare, regulatory, industry, academia, and others.
– This is different from private standards, which may be developed by a manufacturer and not made available for outside assessment and commentary.
– The transparency of USP’s open process provides a high level of public assurance that the standard had been developed using a broadly representative body of science.

©2014 Waters Corporation 20
How are monographs developed? – The process begins with manufacturers, who
submit a draft monograph to USP. – USP scientific staff review the draft monograph
and the data, conduct laboratory tests, and then prepare proposed monograph.
– The proposed monograph is sent to a group of expert scientific volunteers (USP’s Council of Experts [CoE] and its Expert Committees) for their scientific opinion of the relative merits of the proposal.
– If approved by the CoE, the proposed monograph is published in the Pharmacopeial Forum (PF).
• PF is a peer-reviewed journal that provides interested parties, such as pharmaceutical and dietary supplement manufacturers, government agencies and others, the opportunity to review and comment as the CoE develops and/or revises USP–NF standards— and is the working vehicle of the CoE.
• Public comments are received and reviewed by the appropriate USP Expert Committee.
– Once the comments are accepted and incorporated, the new monograph is published in the USP–NF.
USP Standards: Monographs

©2014 Waters Corporation 21
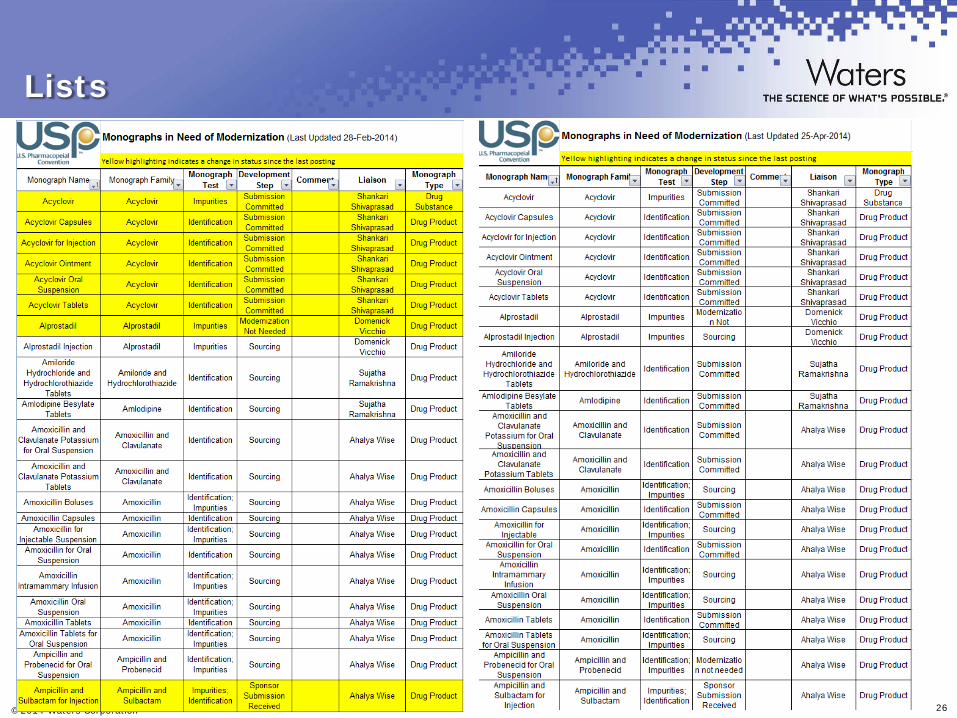
USP Monograph Modernization Initiative
Driver is to maintain up-to-date standards to support USP’s commitment to public health
Need for modernization – Aging of monographs, decades old in some cases – Content does not reflect current expectation for procedure and
acceptance criteria – Complaints from manufactures – General lack of specificity
Benefits – Strengths the public standards – Moves from non-specific to specific procedures – Considers practical factors (removes unnecessary test, eliminates
safety/environment issues – Increases consistency across monographs

©2014 Waters Corporation 22
Modernization History in the USP
2004-2005 Initial evaluations of monograph status – Many monographs identified as needing revision
2005 Initial Priority List (~700 articles) 2009 Expanded Priority List (~2700)
– Modernization is a subset of USP’s ongoing revision work – Started using the term “modernization” in 2009
Working with FDA Monograph Modernization Task Group (MMTG, Nov 2010-ongoing)
2011 FDA Modernization request w/ priorities Launched modernization “hot topics” web page (Feb 2011) Continued Collaboration with FDA and Industry
– Prioritization – Timing considerations
22
http://ipecamericas.org/sites/default/files/ef12april25-hall.a%232-catherine.sheehan(usp).pdf http://ipecamericas.org/system/files/EF13May1HallB3CatherineSheehan(USP).pdf http://www.usp.org/sites/default/files/events/stakeholder_forums/2013/meeting-1/5-excipient-modernization-2013-06-07.pdf

©2014 Waters Corporation 23
Communications regarding modernization of monographs

©2014 Waters Corporation 24
Communications regarding modernization of monographs
Source: FDA and USP websites

©2014 Waters Corporation 25
US Pharmacopeia (USP) USP Monograph Modernization Initiative
http://www.usp.org/usp-nf/development-process/monograph-modernization
©2014 Waters Corporation 26
Lists

©2014 Waters Corporation 27
Lists comments

©2014 Waters Corporation 28
USP Monograph Modernization Initiative - 2013

©2014 Waters Corporation 29
USP Monograph Modernization Initiative - 2013

©2014 Waters Corporation 30
USP Modernization
Monographs needing update: >2600 Approaches
– Donor model • Sponsored monographs • Alternative source monographs
– Internal Development model • Samples procured from market/sponsor • Reference procedure developed in USP labs
– Development under FDA Cooperative Research and Development
Agreement (CRADA), which includes as one of its objectives the development of procedures to support monograph modernization.

©2014 Waters Corporation 31
Barriers
Sample Procurement – Potential Solutions being identified
Impurity Standards Timing Public Review Timing Reference Standards Pipeline

©2014 Waters Corporation 32
USP and Method Transfer
USP 621 – This chapter defines the terms and procedures used in chromatography – Adjustments of operating conditions can be made to meet system suitability
requirements and these adjustments are listed. – The specific allowed deviations listed include column length, particle size,
and flow rate, etc. – Within these allowed limits, the change of method is regarded as an
adjustment of the method. Although the suitability of an analytical procedure should be verified under actual conditions of use.
USP <1224>, <1225>, <1226> – <1224>Transfer of Analytical Procedures – <1225> Elements Recommended for the Transfer of Analytical Procedures – <1226> Verification of Compendial Procedures

©2014 Waters Corporation 33
What System Adjustments Can be Made in Chromatography <621>?
Variable Allowed Adjustments in USP Chromatography <621>
Particle Size -50% Column Length ±70%
Flow Rate F2=F1 (d22/d12) and ±50% Column ID Any allowed if linear velocity is constant
Injection Volume Any reduction consistent with precision and detection
limits; no increase permitted Column Temperature ±10%
Mobile Phase pH ±0.2 unit
USP 36-NF 31 Currently Official

©2014 Waters Corporation 34
USP 37-NF 32 through First Supplement August 1, 2014
Variable USP 36-NF 31 USP 37-NF 32 Through first supplement
Isocratic Gradient
Particle Size -50% L/dp Ratio Constant or N: -25 to + 50%
No changes allowed
Column Length ±70%
No changes allowed
Flow Rate F2=F1 (d22/d12) and ±50% F2-F1 x[(dc22 x dp1)/dC12 x dp2)] and ±50% Not applicable
Column ID
Any allowed if linear velocity is constant
Any allowed if linear velocity is constant
No changes allowed
Injection Volume
Any reduction consistent with precision and detection limits;
no increase permitted
Can be adjusted as consistent with precision and detection
limits
Can be adjusted as consistent with precision and
detection limits
Column Temperature ±10% ±10%
±10%
Mobile Phase pH ±0.2 unit ±0.2 unit
±0.2 unit
F=Flow rate; d = internal column diameter; dc = column diameter, dp = particle size
©2014 Waters Corporation 35
USP Stimuli Article PF39 (6)
http://www.usp.org/usp-nf/notices/stimuli-article-lifecycle-management-analytical-procedures-posted-comment

©2014 Waters Corporation 36
Lifecycle Management of Analytical Procedures
Published July 2013; Comments close January 2014 The Expert Panel recommends adoption of a lifecycle
approach for the management of analytical procedures. – This approach builds on and enhances the current
information contained in several USP general chapters and ICH guidance documents.
– Adoption of this approach would introduce new concepts to the USP: o The Analytical Target Profile and associated
predefined acceptance criteria o Evaluation of the uncertainty associated with the
analytical procedure o Incorporation of risk analysis strategies o Consideration of the potential effect of changes to an
analytical procedure in the context of the analytical procedure's lifecycle
– The traditional approaches to validation, transfer, and verification would be integrated into the analytical procedure lifecycle process rather than being viewed as separate entities.
– The modern concept of a lifecycle model, which is based on process validation and described in ICH guidelines Q8, Q9, and Q10 (and more recently Q11), and can be applied to analytical procedures
Proposed Chapters: – Concepts addressed in 1224 , 1225, and 1226 would be
revised and compiled into a single new general information chapter, Lifecycle Management of Analytical Procedures 1220
– A new general chapter 220 specifying the basic requirements

©2014 Waters Corporation 37
FDA and EMA on QbD in 2013

©2014 Waters Corporation 38
Supersedes the 2000 draft guidance on Analytical Procedures and Methods Validations
Also when finalized it will replace the 1987 FDA guidance Submitting Samples and Analytical Data for Methods Validations
This new document explicitly mentions biologics in its title.
The objective of the Guideline is to inform applicants about what data are expected by the FDA in marketing authorization dossiers.
The provisions of the Guideline apply to – New drug applications (NDAs), – Abbreviated new drug applications (ANDAs) – Biologics license applications (BLAs), – Variation applications regarding these types of
application, as well as to Type II Drug Master Files. – Cannot be directly used for investigational new drug
applications (INDs) New chapters have been added
– Chapter VIII. Life cycle management of analytical procedures"
– Chapter IX: FDA methods verification A chapter on the verification of analytical methods in FDA's own laboratories
Guidance has become shorter due to references – Cross-references to corresponding 21 CFR paragraphs – Extensive list of essential FDA Guidelines which have to
be considered in this context, references to corresponding USP chapters, and technical literature on statistical topics.
New Guidance 2014

©2014 Waters Corporation 39
Lifecycle Management of an Analytical Procedure
Lifecycle Management involves: – Revalidation – Analytical Method Comparability Studies
o Alternative Analytical Procedures o Analytical Methods Transfer
– Reporting Postmarketing Changes to an Approved NDA, ANDA, or BLA Over the Life Cycle of a validated and implemented procedure
– Trend analysis on method performance should be performed at regular intervals to evaluate the need to optimize the analytical procedure or to revalidate all or a part of the analytical procedure. o If an analytical procedure can only meet the established system suitability requirements with
repeated adjustments to the operating conditions stated in the analytical procedure, the analytical procedure should be reevaluated, revalidated, or amended, as appropriate.
– New information (e.g., a better understanding of product CQAs or awareness of a new impurity) may warrant the development and validation of a new or alternative analytical method.
– New technologies may allow for greater understanding and/or confidence when ensuring product quality.
– In anticipation of life cycle changes in analytics, an appropriate number of samples should be archived to allow for comparative studies.
– If a risk-based evaluation or other drivers lead to changes in an analytical procedure or replacement with a new method
– If the procedure is transferred to a new testing site; revalidation, a new validation exercise, an analytical method comparability study, or a combination of these exercises should be considered.
– Changes to the drug substance or drug product manufacturing process may also warrant analytical procedure revalidation.

©2014 Waters Corporation 40
Method Transfer Approaches
Use existing method and/or monograph
Adjust method within USP Chapter <621> or EP <2.4.46> guidelines
o These modifications of parameters are allowed only when the chromatogram improvement is still within the stated system suitability factors
o Within these allowed limits, the change of method is only regarded as an adjustment of the method
Make changes to an approved method and report as minor change in annual report to FDA or EMA – Validation required; Possibly submit monograph
Re-develop Method(s)

©2014 Waters Corporation 41
Summary
Method transfer decisions need to consider – Regulatory allowances – Business parameters – Technology and knowledge advances – Continuing Quality
Methods going forward: – High quality and robust – Validated – Equivalency or comparable – Able to use lab to lab around the world – Modernized
o Specific ID tests and related impurities/degradants o Updated technology o Run on either a HPLC or UPLC instrument (desired) o MS compatible, if possible
Life cycle management of analytical procedures

©2014 Waters Corporation 42
Recommended

















