UNDERSTANDING THE OSTEOIMMUNOLOGICAL ROLE OF
ROQUIN IN THE MAINTENANCE OF BONE HOMEOSTASIS
BAY SIE LIM BCOM, BSC, BMEDSCI (HONS)
This thesis is submitted to fulfil the requirements of University of Western Australia for
the degree of
DOCTOR OF PHILOSOPHY
2014
The work presented in this thesis was completed at the University of Western Australia, School of Pathology and Laboratory Medicine, Queen Elizabeth II Medical Centre,
Nedlands, Western Australia

TABLE OF CONTENTS
Table of Contents
Declaration 1
Scientific Abstract 2
Acknowledgements 7
Abstracts and Awards 9
Abbreviations 10
List of Figures 17
Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional
Regulations of Bone Homeostasis 20
1.1. Introduction 21
1.2. Bone cells 22
1.2.1. Osteoclasts 22
1.2.2. Osteoblasts 24
1.2.3. Osteocytes 27
1.2.4. Osteolining cells 29
1.3. Formation and maintenance of bone 31
1.3.1. Osteogenesis 31
1.3.2 Bone modelling and remodelling 32
1.4. Conclusion 36
Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune
Systems 41
2.1. Introduction 42
2.2. Osteoclasts and inflammatory bone destruction 42

2.3. Osteoclast and innate immune cell lineages: RANK and TLRs downstream
signalling cascades 43
2.4. Role of inflammatory hematopoietic cytokines as a double-edged sword in
bone modeling and remodeling 46
2.4.1. Tumor necrosis factor (TNFs) 46
2.4.2. Interleukin (IL)-1 48
2.4.3. IL-6 48
2.4.4. IL-8 50
2.4.5. IL-17 51
2.4.6. IL-21 52
2.4.7. Other interleukins 52
2.4.8. Interferons (IFNs) 54
2.4.9. Transforming growth factor (TGF)-β 55
2.5. The orchestration of adaptive immune cells by innate immune cells,
osteoclastogenic RANKL and its antagonist OPG 57
2.6. Delineation of T cell and osteoclastogenic T cell candidates 59
2.7. The immunomodulatory role of bone cells in establishing the haematopoietic
stem cell (HSC) niche 63
2.8. Conclusion 65
Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an
Osteoimmune Mouse Model 68
3.1. General hypothesis and aims 69
3.2. Screening of ENU-induced mutant mouse lines: the sanroque mutation 70
3.2.1. The ubiquitin proteasome system (UPS) and Roquin E3 ligase 70
3.2.2. Sanroque mutant mice exhibits low bone mass phenotype 71
3.2.3. Sanroque mice display a lupus-like autoimmune disease 71

3.3. Specific hypothesis and aims 71
Chapter 4: Materials and Methods 77
4.1. Materials 78
4.1.1. Chemical reagents 78
4.1.2. Commercially purchased kits and molecular products 80
4.1.3. Other products and consumables 82
4.1.4. Tissue culture materials and reagents 83
4.1.5. Cytokines 84
4.1.6. Antibodies 84
4.1.7. Oligonucleotide primers 86
4.1.8. Solutions 89
4.1.9. Equipment 92
4.1.9.1. Centrifugation 94
4.1.10. Software 94
4.2. Animals and bone phenotyping methods 95
4.2.1. The generation of sanroque mutant mouse line 95
4.2.2. Radiographic screening of sanroque mutant mice 96
4.2.3. Microquantitative computed X-ray tomography (MicroCT) analysis of
wildtype and sanroque mutant mice 96
4.2.4. Fluorochrome labelling of wildtype and sanroque mutant mice 97
4.2.5. Histological analysis of wildtype and sanroque mutant mice 97
4.2.5.1. Tissue fixation, decalcification (for hindlimbs), processing, embedding
and sectioning 97
4.2.5.2. Haematoxylin and eosin staining 99
4.2.5.3. TRAP staining 99
4.2.5.4. Goldner Trichome staining on non-decalcified bone samples 100

4.2.5.4.1. Processing, embedding and sectioning of undecalcified bone
samples 100
4.2.5.4.2. Goldner Trichrome staining 101
4.2.5.5. Bone histomorphometric analysis 102
4.2.6. TRAP staining of mouse calvariae 102
4.2.7. Immunofluorescence staining of mouse spleens 103
4.2.7.1. Embedding of sample in Optimal Cutting Temperature (OCT) 103
4.2.7.2. Immunofluorescence staining 103
4.3. Isolation, culture and in vitro studies of primary cell lines 104
4.3.1. Isolation and culture of bone marrow cells 104
4.3.1.1. Generation and maintenance of bone marrow macrophages (BMMs)
104
4.3.1.2. MTS cell proliferation assay 105
4.3.1.3. Osteoclast culture 105
4.3.1.3.1. In vitro osteoclastogenesis assay 106
4.3.1.3.2. In vitro bone resorption assay 106
4.3.1.3.3. In vitro osteoclast cathepsin K release assay using bovine bone
powder 107
4.3.2. Isolation of osteoblastic bone cells from long bones and calvariae 108
4.3.2.1. Osteoblast culture and mineralisation assay 109
4.3.2.2. Osteoblasts and BMMs co-culture experiment 109
4.3.3. Flow cytometry analysis of bone marrow cell niche 110
4.3.3.1. Immunostaining of bone marrow cells 110
4.3.3.2. Flow cytometer and analysis 111
4.3.4. Cryopreservation of cells for long term storage 112
4.4. Molecular biology techniques 112
4.4.1. RNA extraction and quantification 112

4.4.1.1. General handling procedures 112
4.4.1.2. Extraction of total RNA 112
4.4.1.2.1. RNA extraction from bone tissue 112
4.4.1.2.2. RNA extraction from cell culture 113
4.4.1.3. Quantification of RNA concentration 113
4.4.2. Reverse Transcription Polymerise Chain Reaction (RT-PCR) 114
4.4.2.1. Reverse Transcription (RT) 114
4.4.2.2. Polymerase Chain Reaction (PCR) amplification 114
4.4.2.3. DNA agarose gel electrophoresis 115
4.4.3. Quantitative Real-Time PCR (Q-PCR) and Livak’s Equation 116
4.4.4. Protein expression analysis using Sodium Dodecyl Sulfate-PolyAcrylamide
Gel Electrophoresis (SDS-PAGE) and western blotting 117
4.4.4.1. Protein extraction from mouse organs 117
4.4.4.2. Protein extraction from cultured cells 117
4.4.4.3. Quantification of protein concentration 118
4.4.4.4. Separation of proteins by SDS-PAGE 118
4.4.4.5. Protein transfer onto nitrocellulose blotting membrane 120
4.4.4.6. Western blotting 120
4.5. Statistics and data presentation 121
Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice 122
5.1. Introduction 123
5.2. Experimental results 124
5.2.1. Roquin gene expression is regulated during osteoblastogenesis and
osteoclastogenesis and is highly expressed in hematopoietic organs 124
5.2.2. Sanroque mutation alters trabecular and cortical bone morphology 124
5.2.3. Sanroque mutant mice have increased osteoclast numbers in vivo 126

5.2.4. Bone mineral apposition rate in sanroque mutant mice is reduced despite
unchanged histological osteoblast parameters 126
5.2.5. RANKL and osteoclast gene markers are highly expressed in sanroque
bone 127
5.3. Discussion 127
Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast
Biology 141
6.1. Introduction 142
6.2. Experimental results 142
6.2.1. Reduced bone mineralisation by sanroque mutant COB cultures 143
6.2.2. Increased calcium deposition by mutant sanroque LBOB cultures 143
6.2.3. Alkaline phosphatase activity level is altered throughout osteogenesis in
sanroque LBOB cell culture 144
6.2.4. Mutation of Roquin affects the expression levels of osteoblast gene markers
during differentiation of sanroque LBOBs 145
6.2.5. Mutant LBOB from sanroque mutant mice have reduced capability to
support osteoclastogenesis in vitro 146
6.3. Discussion 147
Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology 161
7.1. Introduction 162
7.2. Experimental results 163
7.2.1. Osteoclastogenesis is accelerated in sanroque mutant mice 163
7.2.2. Sanroque mutation elevates the expression of osteoclast marker genes 164
7.2.3. Sanroque mutant osteoclasts demonstrate increased bone lytic activity in
vitro 164
7.2.4. Mutation in Roquin gene alters key RANKL-signalling pathways 165

7.2.5. Ubiquitination of TRAF6 in sanroque BMMs appears unaltered 167
7.2.6. Blood serum from sanroque mutant mice contains proliferative- cytokines
and sanroque BMMs have increased proliferation in response to M-CSF 168
7.3. Discussion 169
Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic
Phenotype of Sanroque Mutant Mice 187
8.1. Introduction 188
8.2. Experimental results 189
8.2.1. The putative osteoclast progenitor population present within the bone
marrow of sanroque mutant mice is increased. 190
8.2.2. CD4 T cells of sanroque mutant mice expressed less CD70 191
8.2.3. Splenocytes of sanroque mutant mice comprised a higher number of
common myeloid progenitors and follicular helper T cells that express RANKL
192
8.3. Discussion 194
Chapter 9: General Overview and Future Directions 213
9.1. Overview of thesis and general discussion 214
9.2. Future Directions 220
9.3. Conclusion 223
Chapter 10: References 225
Appendix 276

1
DECLARATION
This is to certify that all the work contained herein was performed by myself, except
where indicated otherwise.
_____ ____
Bay Sie LIM
W/Prof. Jiake XU
Dr. Jennifer TICKNER

2
SCIENTIFIC ABSTRACT
The bone organ system is a dynamic mineralized tissue that is continuously being
broken down and rebuilt in a process known as bone remodelling. The remodelling
process is an important metabolic activity involved in the maintenance of the
homeostasis and architecture of bone while performing its versatile functions. The bone
remodelling process is a coupled activity between the catabolic effects of bone-
resorbing osteoclasts and the anabolic effects of bone-forming osteoblasts.
The remodelling cycle begins when there is an external mechanical stimulus, which
initiates accessory cells including osteoblasts to secrete RANKL. RANKL binds onto
the cell surface receptor RANK displayed on osteoclast precursors that originate from
the haematopoietic stem cells. The RANKL-RANK interaction drives
osteoclastogenesis and cell fusion to form large multinucleated osteoclasts. Once
mature, osteoclasts begin to secrete proteolytic enzymes to resorb the bone matrix. The
bone erosion by osteoclasts is a crucial process in remoulding the bone tissue.
To prevent osteoclasts from continuously resorbing bone, osteoblasts, which are derived
from mesenchymal stem cells, also secrete regulatory molecules such as OPG. OPG
acts as a decoy receptor that binds onto RANKL to prevent RANKL-RANK interaction.
Such interactions allow osteoblasts to modulate the bone remodelling cycle, balancing
bone resorption and bone formation. Mature osteoblasts are the primary matrix-
producing cells that build and mineralise bone. As a result, some become entombed
within their own matrix and differentiate into osteocytic cells with long dendritic
processes to maintain cell-cell contact amongst themselves and with bone cells residing
on the surfaces of bone. Osteocytes are generally recognised as the mechanotransducers
of bone tissue, responding to mechanical stimuli and generating signals that play
important roles in regulating the bone remodelling cycle.
The cellular coupling among these bone cells to achieve bone homeostasis is complex
and heavily regulated by local and systemic factors. With the emerging field of
osteoimmunology, many studies have implied that both innate and adaptive immune
systems are major components of bone remodelling, and vice versa. The dialogue
between the skeletal and immune systems is inevitable as stem cells, which give rise to
progenitors that constitute both the bone organ system and immune system, are

3
generated within the bone marrow reservoir.
The co-regulation and intimate relationship between the immune and skeletal system is
highlighted in many inflammatory diseases. Periodontal disease, rheumatoid arthritis,
osteomyelitis and lupus are associated with both local and systemic bone destruction,
which inherently leads to bone fractures and significant bone deformities. However, the
mechanisms by which these pathological conditions arise remain poorly understood.
Beyond these observations, recent studies have also reported that the immune system
might play a role in facilitating bone formation. Osteoimmunological interactions thus
play a significant role in the maintenance of bone homeostasis and are fundamental
mechanisms in bone pathology.
To gain insights into the molecular genetics and mechanisms of osteoimmunology, we
have employed a chemical (ENU) mutagenesis and phenotypic assessment. Through
systematic X-ray screening, we identified a mouse line that displayed reduced bone
mass in comparison to wildtype littermates. The sanroque mutant mouse line carries a
single point (M199R) mutation in Roquin (Rc3h1) gene, which encodes for a RING-
type E3 ubiquitin ligase. E3 ubiquitin ligases, along with E1 and E2 family of enzymes,
are responsible for driving the ubiquitin proteasome system (UPS).
UPS is the process of ubiquitination of molecular products mainly for the purpose of
proteolysis to maintain protein homeostasis. The ubiquitination process marks the
molecular product of interest to be recognised by the proteasome for degradation. UPS
is reported to be involved in transcription factor activation, DNA repair, translational
control, kinase activation, histone activity and endocytosis. The abrogation of the UPS
enzymes, particularly E3s, has been implicated in the pathogenesis of many diseases
such as Parkinson’s, leprosy, osteosarcoma and autoimmune diseases. UPS and E3s are
also implicated in the maintenance of bone homeostasis and in the development of bone
cells.
Roquin gene encoding for Roquin E3 ubiquitin ligase is most abundantly expressed in
haematopoietic organs such as the bone and spleen. Within these organs haematopoietic
stem cells that give rise to common myeloid and lymphoid progenitors express high
levels of Roquin gene. Thus, it is not surprising that the sanroque mutant mice were
discovered to display changes in their immune system resulting in an autoimmune
condition consistent to systemic lupus erythematosus (SLE). The sanroque mutation

4
resulted in the dysregulation of a recently characterised T cell subset, the follicular
helper T cells (TFH), which are crucial in assisting the maturation of B cells within the
germinal centre. The sanroque mutant mice have increased numbers of germinal centres
and developed lymphomegaly and splenomegaly, both of which are key symptoms of
autoimmune disorders.
Q-PCR analysis revealed that Roquin gene expression is regulated throughout
osteoblast and osteoclast differentiation, whereby Roquin expression is elevated
throughout osteoblastogenesis. In contrast, the gene expression of Roquin is repressed
throughout osteoclastogenesis. Such regulation of Roquin expression in bone cells
highlights the potential regulatory role of the gene in bone biology.
The association of Roquin in the maintenance of bone homeostasis was clearly evident
when a low bone mass phenotype was observed in preliminary X-ray screening of
sanroque mice, which was then confirmed by microCT. MicroCT data emphasised the
reduction in both cortical and trabecular bone parameters in sanroque mice, both of
which are hallmarks of osteopenia, as compared to wildtype littermates. The low bone
mass phenotypes persisted in both genders of sanroque mutant mice. Therefore, the
sanroque mutation results in systemic bone loss in both male and female mice.
Histological analysis verified the reduced bone mass phenotype of sanroque mutant
mice as shown in microCT analysis. TRAP-stained long bone sections and whole
calvaria revealed an increase in osteoclast surface in sanroque mutant bone ex vivo. In
keeping with the osteopenic phenotype, in vivo calcein and alizarin red dual labelling
showed a reduction in bone mineral apposition rate in sanroque mutant mice. These
observations indicate that osteoclast and osteoblast coupling in vivo is affected by
sanroque mutation, resulting in dysregulation of bone homeostasis and reduced bone
mass.
To gain insight into the molecular mechanism of the pathogenesis of bone loss in
sanroque mutant mice, RNA isolated from bone tissues was subjected to reverse
transcription and Q-PCR analysis. Results showed elevated RANKL:OPG ratio in the
bone of sanroque mice relative to wildtype mice. Consistently, the expression of
osteoclast gene markers such as TRAP and cathepsin K were elevated in sanroque
mutant mice, whereas the expression of the osteoblast gene marker osteocalcin is
reduced in comparison to wildtype mice. This corresponds with the lower bone mass of

5
sanroque mutant mice, whereby increased RANKL:OPG in favour of RANKL usually
equates with increased osteoclastogenesis and bone resorption.
Additionally, flow cytometry analysis demonstrated that the sanroque mutation resulted
in skewed populations of stem cells and abnormal bifurcation of common progenitors
that give rise to dendritic cells, macrophages and osteoclast precursors. The constitutive
RANKL expression was accompanied by a significant expansion of putative osteoclast
progenitors within the bone marrow of sanroque mice in comparison to that of wildtype
littermates.
To gain insight in the cellular mechanism in the pathogenesis of bone loss in sanroque
mutant mice, flushed bone marrow macrophages (BMMs) were stimulated in vitro with
RANKL and M-CSF over the course of 5 days. Consistent with the observed increase in
osteoclast progenitors and osteoclast surface ex vivo, we also observed enhanced
osteoclastogenesis of BMMs derived from sanroque mutant mice, accompanied by
enhanced RANKL-mediated MAPK signalling.
The function and resorption activity of mature osteoclasts was examined by culturing
BMMs-derived osteoclasts on bovine bone discs. Microscopic analysis showed a
significant increase in the area resorbed by sanroque BMM-derived osteoclasts. This
observation is further supported by the increased cathepsin K released upon stimulation
of mature osteoclasts with bovine bone powder, suggesting an overzealous resorption
activity by sanroque BMM derived osteoclasts.
Previous Q-PCR analysis has also shown the upregulation of Roquin expression
throughout osteoblastogenesis, suggesting that the sanroque mutation could have an
effect on osteoblasts. Consistent with the reduction in MAR in vivo, sanroque calvarial-
osteoblasts formed less mineralised bone nodules as demonstrated by the apparent
reduction in alizarin red staining. This confirms the defective anabolic machinery of
osteoblasts in sanroque mutant mice.
The capability of osteoblasts to support osteoclastogenesis was also elucidated by
performing co-culture experiments between calvaria-derived osteoblasts and BMMs.
Upon stimulation with vitamin D3, sanroque calvaria-derived osteoblasts do not
produce as much osteoclastogenic factors as evidenced by the reduction in the number
of TRAP-positive osteoclasts. This observation suggests that the enhanced numbers of

6
osteoclast progenitors within the bone marrow niche in sanroque mice is not
contributed by osteoblastic production of osteoclastogenic cytokines, but by another
extrinsic factor or cell type.
During chronic immune activation, RANKL and osteoclastogenic factors expressed by
activated immune cells drive pathogenic osteoclast differentiation. Studies have
categorised many of these activated immune cells, such as TH17, and coined them
osteoclastogenic cell subsets. While elucidating the role of Roquin gene in the
pathogenesis of lupus, Vinuesa and colleagues (2005) reported that the sanroque
mutation resulted in elevated RANKL expression in dysregulated TFH that comprised
the immune system of sanroque mutant mice. Thus, the increased number of
dysregulated TFH residing in the germinal centres could possibly contribute to the
increased expression of RANKL in sanroque mutant mice.
To test the aforementioned hypothesis, spleens were cryopreserved, sectioned and
stained with antibodies specific for RANKL and cellular markers of TFH. Confocal
microscopy revealed an obvious increase in staining of RANKL in the sanroque spleen
sections in comparison to the wildtype. Co-localisation of TFH cellular markers and
RANKL was observed particularly within the marginal zones of the spleen from
sanroque mutant mice. To validate the increase in RANKL staining as observed in
confocal microscopy, spleen tissues were collected and subjected to protein isolation.
Western Blot analysis showed an increase in RANKL protein levels in the spleens of
sanroque mutant mice. Collectively, these results indicate that mutation in Roquin gene
resulted in elevated RANKL level in the spleen and that TFH could potentially be
categorised as an osteoclastogenic subset that drive pathogenic osteoclastogenesis.
In summary, sanroque mutant mice displayed reduced trabecular and cortical bone
volume due to an imbalance in bone homeostasis that favours bone resorption. A defect
in osteoblasts contributes to the reduced bone mass as they form less bone in vivo and in
vitro. Although Q-PCR analysis suggests that Roquin may negatively regulate
osteoclastogenesis, Roquin was demonstrated to play a crucial role in regulating the
expression level of RANKL, which in turn regulates osteoclastogenesis. In conclusion,
Roquin is an important regulator of osteoimmunological interaction and could serve as a
potential therapeutic target for the treatment of inflammatory-related systemic bone
loss.

7
ACKNOWLEDGEMENTS
This thesis is dedicated to my Popo and Ah Kong, both of whom while unable to be
with me as I finish this thesis, their memory has accompanied me throughout this
journey, and I hope I have made them proud of me.
My deepest appreciation and sincere gratitude for their guidance and support, is
extended to W/Prof. Jiake Xu and Dr. Jennifer Tickner, you both have been great
mentors throughout my journey in completing this thesis.
I am extremely grateful to W/Prof. Jiake Xu for the opportunity to undertake such an
interesting project. Your positivity, compassion and understanding have provided me
with the motivation to go through challenging times. The enthusiasm that you have for
the research is admirable and I am thankful for the knowledge you have imparted to me
throughout the years.
I wish to thank Dr. Jennifer Tickner, for your patience and guidance throughout the
years, which has enabled me to successfully get through the highs and lows that come
with doing scientific research. You are like the compass that has helped to point me in
the right direction as I navigated through my research.
I would also like to give a special thanks to Dr. Jacky Chim, for teaching me so many of
the vital laboratory techniques that I acquired today. I also appreciate your suggestions
and encouragement during challenging times. I would like to extend my thanks to
A/Prof. Nathan Pavlos for his continuous support and scientific advice. Your knowledge
in the bone field and broad outlook often taught me to think from a different
perspective.
I am also indebted to W/Prof. Wendy Erber and A/Prof. Senta Walton for their
generosity. In spite of having such busy schedules, they have shared their very valuable
time to help me with my project. I am grateful towards Irma Larma for the fun times
during training and also for helping me with the flow cytometry experiments. I would
like to acknowledge Euphemie Landao, Alysia Buckley, A/Prof. Paul Rigby and Tracey
Lee-Pullen as well, for their in-depth training and knowledge in histology, confocal
microscopy and flow cytometry. To Dr. Sarah Rae and Melanie Sultana, thank you for
having me in your lab and your contribution in my research.

8
I am also truly blessed to have the support and assistance from many of my awesome
colleagues. To Benjamin Ng, Dian Astari Teguh, Gaurav Jadhav, Jacob Kenny, Jin Bo,
Lin Zhou, Ryan Ming Li Yang, Vincent Kuek, William Chundawan and Dr. Jasreen
Kular, thank you for your endless encouragement, concern, support, assistance and
above of all, for the fun times, food and laughter that we shared. Your jokes and
friendly banter have helped to brighten up the sometimes-stressful situations I find
myself in. My special thanks and gratitude goes toward Dr. Pei Ying Ng for being such
a wonderful and caring housemate. I have enjoyed your cooking and company, and I
hope that we will be able to continue this friendship when I get to Singapore.
The completion of this thesis would not have been possible without the love, faith and
support from my loved ones. I am truly grateful to Ma and Pa for the unconditional love
and sacrifices that you both have made in order to give me the best in life. Your
immeasurable faith in me has given me inspiration and support. To my precious Bay
Gie and Betty, I must say I am the luckiest sister in the world to be blessed with two
pretty angels as my partners in crime. Thank you for making me smile and being there
for me.
To my dearest Sze Chieh, you filled my heart with so much love and I feel truly blessed
to have you in my life. I look forward to spending the rest of my life with you.

9
ABSTRACTS AND AWARDS
1. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2012) “Roquin is a
novel regulator of bone homeostasis.” Sir Charles Gardner Hospital Medical
Research Month Symposium, Perth, Western Australia. (Oral Presentation)
2. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2013) “Roquin is a
novel regulator of bone homeostasis.” 19th Annual Australian and New Zealand
Orthopaedic Research Society Conference, Sydney, NSW. (Oral Presentation)
3. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2013) “Roquin is a
novel regulator of bone homeostasis.” 23rd Annual Australia and New Zealand
Bone & Mineral Society Scientific Meeting, Sydney, NSW. (Oral Presentation)
4. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2014) “Roquin is a
novel regulator of bone homeostasis.” UWA School of Pathology and
Laboratory Medicine Thesis Seminar, Perth, Western Australia. (Oral
Presentation)
5. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2014) “Roquin is a
novel regulator of bone homeostasis.” Australian Society for Medical Research
Medical Research Week Symposium, Perth, Western Australia. (Oral
Presentation) WA Department of Health Platinum Award
6. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2014) “Roquin is a
novel regulator of bone homeostasis.” 24rd Annual Australia and New Zealand
Bone & Mineral Society Scientific Meeting, Auckland, NZ (Poster Presentation)
7. Lim B, Chim SM, Landao E, Tickner J, Pavlos NJ , Xu J (2013) “Roquin is a
novel regulator of bone homeostasis.” 20th Annual Australian and New Zealand
Orthopaedic Research Society Conference, Adelaide, SA. (Oral Presentation)
PhD Student Award

10
ABBREVIATIONS
AC Adenylyl cyclase
ALP Alkaline phosphatase
AP-1 Activator protein 1
APC Adenomatous polyposis coli protein
APC Allophycocyanin
APCs Antigen presenting cells
APS Ammonium persulfate
Atp6v0d2 d2 isoform of vacuolar (H+) ATPase (v-ATPase) V0 domain
BALP Bone alkaline phosphatase
Bcl B cell lymphoma
BMMs Bone marrow macrophages
BMPR BMP receptor
BMPs Bone morphogenetic proteins
BRU Bone remodelling unit
BSA Bovine serum albumin
BV/TV Bone volume per tissue volume
c-Cbl Casitas B-lineage lymphoma
c-fms Colony-stimulating factor 1
cAMP Cyclic adenosine monophosphate
CatK Cathepsin K
CBFA1 Core-binding factor alpha 1
CD Cluster of differentiation
CIA Collagen-induced arthritis
CMCA Centre for Microscopy, Characterisation and Analysis
COB Calvaria-isolated osteoblast
CT-1 Cardiotrophin-1
Ct.Ar Cross sectional cortical area
Ct.BV Cortical bone volume
Ct.Th Cortical thickness
CTLA-4 Cytotoxic T-lymphocyte protein 4

11
CTR Calcitonin receptor
CTX C-telopeptide of type I collagen
Cx 43 Connexin
CXCL CXC motif ligand
CXCR CXC chemokine receptor
DAG Diacylglycerol
DAMPs Damage-associated molecular patterns
DC Dendritic cell
DC-STAMP Dendritic cell-specific transmembrane protein
DD Death domain
DKK Dickkopf-related protein
DMEM Dulbecco's-Modification of Eagle's Medium
DMSO Dimethyl sulphoxide
DNA Deoxyribonucleic acid
DPX Distyrene plasticizer xylene
Dsh/Dvl Dishevelled
EDTA Ethylenediamine tetraacetic acid
ENU N-ethyl-N-nitrosourea
FBS Foetal bovine serum
FC Flow cytometry
FcRγ Fc-receptor common γ-subunit
FITC Fluorescein isothiocyanate
For Forward
FSC Forward scatter
Fz Frizzled
g Gram
G-CSF Granulocyte colony stimulating factor
GJIC Gap junctional intracellular communication
GM-CSF Granulocyte macrophage colony stimulating factor
Gp Glycoprotein
GSK3 Glycogen synthase kinase-3β
GST Glutatione S-transferase
H&E Haematoxylin and eosin

12
HECT Homologous to E6-AP carboxyl terminus
HSC Haematopoietic stem cell
ICOS Inducible T-cell co-stimulator
ICOSL ICOS ligand
IFNs Interferons
IGFs Insulin like growth factors
IKK IĸB kinase
IL Interleukin
IP3 inositol 1,4,5 triphosphate
IPTG Isopropyl-β-thiogalactopyranoside
IRAK IL-1R-associated kinases
ITAM Immunoreceptor tyrosine based activation motif
JAK Janus kinase
JNK c-Jun N-terminal kinase
kg Kilogram
Kif3a Kinesin family member 3A
kV Kilovolt
LAP Latency-associated peptide
LAT Linker of activation of T cells
LBOB Long bone-isolated osteoblast
LCS Lacunocanalicular system
LDL Low-density lipoprotein
LIF Leukaemia inhibitory factor
LRP LDL receptor-related protein
LTBP Latent TGF-β-binding protein
M-CSF Macrophage colony stimulating factor
M.Ar Marrow area
mA Milliampere
MafB V-maf musculoaponeurotic fibrosarcoma oncogene homolog B
MAPK Mitogen-activated kinases
MAPKs Mitogen activated protein kinases
MAR Mineral apposition rate
Mb Megabases

13
MCP Monocyte chemoattractant protein
MEA Methoxyethyl acetate
mg Milligram
MHC Major histocompatibility complex
MicroCT Microcomputed tomography
MITF Micro-opthalmia associated transcription factor
ml Millilitre
MLO-Y4 Murine long bone osteocyte Y4
mm Millimetre
mM Millimolar
MMA Methyl methacrylate
MMLV-RT Moloney leukaemia virus reverse transcriptase
MMPs Metalloproteinases
mRNA messenger RNA
MSC Mesenchymal stem cell
Mϕ Macrophage
N.Ob Number of osteoblast
N.Ob/BS Number of osteoblast per bone surface
N.OC/BS Number of osteoclast per bone surface
NCBI National Center for Biotechnology Information
NF-ĸB Nuclear factor kappa B
NFAT Nuclear factor of activated T cells
ng Nanogram
NIH National Institutes of Health
NKT Natural killer T cell
Oc.S Osteoclast surface
Oc.S/BS Osteoclast surface per bone surface
OCn Osteocalcin
OCP Commited osteoclast progenitors
OCT Optimal cutting temperature
OPG Osteoprotegerin
OPn Osteopontin
OS Osteoid surface

14
OS/BS Osteoid surface per bone surface
OSM Oncostatin M
Osx Osterix
PAMPs Pathogen-associated molecular patterns
PBS Phosphate buffered saline
PC Polycystin
PE Phycoerythrin
PI3K Phosphoinositide 3-kinase
PINP Procollagen type I N-terminal propeptide
PMSF Phenylmethylsulfonyl fluoride
PPRs Pattern recognition receptors
PTH Parathyroid hormone
Q-PCR Quantitative realtime PCR
RA Rheumatoid arthritis
RANK Receptor activator of NF-ĸB
RANKL RANK ligand
RBC Red blood cell
Rev Reverse
RGD Arginine-glycine-aspartic acid
RING Really interesting new gene
RIPA Radio immunoprecipitation assay buffer
Rn18S 18S ribosomal RNA
RNA Ribonucleic acid
RORγt RAR-related orphan receptor gamma
RT-PCR Reverse transcription polymerase chain reaction
S1P Sphingosine 1-phosphate
SA Spondylarthritis
san sanroque
SBE SMAD-binding element
SDF Stromal cell-derived factor
SDS Sodium dodecyl sulphate
SDS-Page Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
SEM Standard error of the mean

15
Sema4D Semaphorin-4D
SLE Systemic lupus erythematosus
SMADs Sma and Mad related proteins
SMI Structure model index
SOST Sclerostin
SSC Side scatter
STAT Signal transducer and activator of transcription
Syk Spleen tyrosine kinases
TAE Tris-acetate EDTA
TAK1 TGF-β-activated kinase 1
Tb.N Trabecular number
Tb.Sp Trabecular separation
Tb.Th Trabecular thickness
TBS Tris buffered saline
TCF/LEF Transcription factor lymphoid enhancer binding factor 1/T cell-
specific transcription factor
TCR T cell receptor
TEMED Tetramethylethylenediamine
TFH Follicular helper T cell
TGF Transforming growth factor
TH T helper
TLRs Toll-like receptors
TNF Tumor necrosis factor
TNFRSF TNF receptor super family
TRACP-5b Tartrate-resistant acid phosphatase 5b
TRAF TNF-receptor associated factor
TRANCE TNF-related activation-induced cytokine
TRAP Tartrate-resistant acid phosphatase
Tregs Regulatory T cells
TRF T cell-replacing factor
Tt.Ar Total cross sectional area
UPS Ubiquitin proteasome system
UTR Untranslated region

16
UV Ultraviolet
v/v Volume/volume
w/v Weight/volume
WB Western blot
Wnts Wingless type proteins
WP White pulp
WT Wildtype
ZA Zoledronic acid
α-MEM α-Modification of Eagle's Medium
μg Microgram
μl Microlitre
μm Micrometre
μM Micromolar

17
LIST OF FIGURES
Page
Figure 1.1 The different types of bone cells involved in the regulation of bone
microenvironment
38
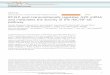
Figure 1.2 RANK/RANKL signalling pathway during osteoclastogenesis 39
Figure 1.3 The bone remodelling cycle 40
Figure 2.1 Lineage bifurcation model between dendritic cells, macrophages and
osteoclasts
66
Table 2.1 The effect of cytokines in modulating the bone and immune system 67
Figure 3.1 Mapping of missense mutation in the conserved Rc3h1gene 73
Figure 3.2 The basic steps of substrate modification by the ubiquitin
proteasome system
74
Figure 3.3 Mutant sanroque mice appeared normal but exhibited low bone
phenotype
75
Figure 3.4 Mutant sanroque mice displayed lymphomegaly and splenomegaly 76
Figure 5.1 Tissue distribution of Roquin (Rc3h1) mRNA expression 131
Figure 5.2 Expression level of Roquin throughout osteoblast (OB) and
osteoclast (OC) differentiation
132
Figure 5.3 Mutant sanroque mice display reduced bone mass 133
Figure 5.4 Histological analysis showed mutant sanroque mice have reduced
trabecular bone mass
135
Figure 5.5 Increase osteoclast number in trabecular bone of sanroque mutant
mice
136
Figure 5.6 Increased TRAP staining on the surface and in sections of sanroque
mutant calvaria
137
Figure 5.7 Osteoblast number and osteoid surface are unaffected in the
trabecular bone of sanroque mutant mice
138
Figure 5.8 Decreased in vivo bone mineral apposition rate in sanroque mutant
mice
139
Figure 5.9 Sanroque mutant bones expressed higher levels of osteoclast gene
markers and increased RANKL:OPG ratio
140

18
Figure 6.1 Osteoblasts isolated from calvariae (COBs) of sanroque mutant
mice have reduced capability to mineralise in vitro
152
Figure 6.2 Enhanced mineralisation in sanroque mutant osteoblasts isolated
from long bones (LBOBs)
154
Figure 6.3 Mutant LBOBs of sanroque mutant mice displayed atypical alkaline
phosphatase (ALP) activity levels throughout osteogenesis
157
Figure 6.4 Q-PCR analyses of osteoblast gene markers expression levels during
osteogenesis
158
Figure 6.5 Coculture systems between LBOBs and bone marrow macrophages
(BMMs)
159
Figure 7.1 Osteoclast differentiation is increased in sanroque BMM culture 174
Figure 7.2 BMMs derived from sanroque mutant mice have increased capacity
to form osteoclasts at lower doses of RANKL
175
Figure 7.3 The mutation in Roquin alters the expression of osteoclast gene
markers.
173
Figure 7.4 Sanroque mutant osteoclasts resorbed more bone 177
Figure 7.5 Increased cathepsin K released by sanroque mutant osteoclasts 178
Figure 7.6 Increased activation of MAPK signalling cascade during RANKL
stimulation in sanroque osteoclast precursors
179
Figure 7.7 Increased protein levels of transcription factor NFATc1, osteoclast
markers DC-Stamp and D2 subunits during osteoclastogenesis in
sanroque BMMs
181
Figure 7.8 FLAG-tagged TRAF6 immunoprecipitates of HEK293 cells
transfected with wildtype and mutant Roquin (M119R) plasmids
showed no difference in ubiquitination level by immunoblotting
with anti-HA
183
Figure 7.9 No difference in ubiquitination of TRAF6 in sanroque BMMs upon
RANKL stimulation
185
Figure 7.10 Blood serum from sanroque mutant mice contains proliferative-
inducing cytokines and sanroque BMMs exhibit increased cell
proliferation in response to M-CSF
186
Figure 8.1 The bone marrow population of sanroque mutant mice appeared
dysregulated
200

19
Figure 8.2 Increased macrophage and reduced dendritic cell population in
sanroque bone marrow
201
Figure 8.3 Significant expansion of putative osteoclast progenitors in sanroque
bone marrow
202
Figure 8.4 No difference in the percentage of CD4 and CD8 T cell population
between the bone marrow of sanroque mutant mice and wildtype
204
Figure 8.5 Expression of CD70 in T, B and DCs 205
Figure 8.6 Increased follicular helper T cell in sanroque mutant mice 208
Figure 8.7 Localisation of RANKL and TFH in spleen 209
Figure 8.8 RANKL protein and gene expressions are elevated in sanroque
spleens
211
Figure 8.9 Increased common myeloid progenitors in sanroque spleen 212
Figure 9.1 The hypothetical model depicting the possible cellular mechanism
that resulted in systemic bone loss in sanroque mutant mice
224
Supp. Fig. 1 Female mutant sanroque mice display reduced bone mass 277
Supp. Fig. 2 Left shift is observed in peripheral blood smears from sanroque
mutant mice
278

CHAPTER ONE
THE BIOLOGY OF BONE MULTICELLULAR UNIT
AND
TRANSCRIPTIONAL REGULATIONS OF BONE
HOMEOSTASIS

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
21
1. CHAPTER 1: THE BIOLOGY OF BONE MULTICELLULAR UNIT AND TRANSCRIPTIONAL REGULATIONS OF BONE HOMEOSTASIS
1.1. Introduction
Bone is a specialized connective tissue that, collectively with cartilage, makes up the
skeleton (Meghji, 1992). The calcified tissue is composed of 5 – 10% water, 60 – 70%
inorganic (mineral) components, with organic component comprising the rest of the
tissue. The major component of the mineral phase is hydroxyapatite crystal. The
organic phase is comprised of concentric layers of type I collagen matrix, a variety of
non-collagenous proteins, and bone cells (Feng, 2009; Lee et al., 2001; Morgan et al.,
2008).
The architecture and composition of bone tissue have evolved to dynamically carry out
several major functions (Bab et al., 1994). Bone tissue provides mechanical support and
attachment sites for soft tissues such as muscles for locomotion. Other purposes of bone
include protecting vital organs and housing the brain and the spinal cord (Feng, 2009;
Harada et al., 2003; Meghji, 1992). The bone represents the primary reservoir of
calcium and other mineral ions such as phosphate, sodium and magnesium (Duplomb et
al., 2007; Rubin et al., 1999). Several studies have indicated that bone is a major
component of the immune system as immune cells form in the bone marrow (Arron et
al., 2000b; Carlsten, 2005; Morgan et al., 2008; Takayanagi, 2005; Wein et al., 2005;
Wu et al., 2008b). This is inevitable as it is also the principal site of haematopoiesis,
where it houses pluripotent bone marrow stem cells (Horowitz et al., 1992).
To fulfil a lifelong execution of its versatile functions while maintaining structural
integrity, the bone tissue is continuously being broken down and rebuilt in a process
known as bone remodelling. Bone remodelling is a process coupled between the
catabolic effects of bone resorbing osteoclasts and anabolic effects of bone forming
osteoblasts (Henriksen et al., 2009; Horowitz et al., 1992). Under normal states of bone
homeostasis, an ideal bone remodelling process serves to remove bone mass where the
mechanical loads are low, while forming bone at sites where mechanical stimuli are
transmitted repeatedly. Bone therefore has the capability to maintain itself, depending
on the external mechanical and physiological stimuli from the systemic environment.
The cellular coupling between osteoclastic and osteoblastic cells is complex and is
heavily coordinated by several regulatory systems, both systemic and local, to keep
both remodelling and resorption processes synchronised (Martin et al., 2006; Morgan et
al., 2008). The accentuation of one or the other process eventually leads to bone

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
22
fragility and/or clinical diseases of the skeleton, such as osteopenia, osteoporosis,
osteopetrosis, periodontitis, arthritis and tumour-induced osteolysis (Hill, 1998; Karsdal
et al., 2007). While these abnormalities are known to associate with an increased risk
for fracture and causing considerable suffering, little is known of the mechanisms
responsible for the dysfunctional bone remodelling that characterises them. This is not
surprising, since we have yet to fully understand the complexity of “normal” bone
remodelling and how the process is so highly coordinated and balanced.
However, the aim to further understand the biochemical and molecular mechanisms of
bone cells to adapt within their mechanical environment is achievable via novel
techniques such as gene ‘knockout’, silencing and transgenic experiments. Molecular
biology and the analysis of the skeletal phenotype of mouse models replicating genetic
mutations in humans has led to a better understanding of the role of factors that govern
bone cell biology, and ultimately, bone homeostasis. Understanding the functions of the
cells within bone, and how they communicate with each other during bone remodelling
is essential to the development of therapeutic options for bone diseases.
1.2. Bone cells
1.2.1. Osteoclasts
Osteoclasts are motile macrophage-like cells that undergo mitosis without cytokinesis
and become multinucleated (Alberts, 2008). The multinucleated osteoclasts serve as
bone-resorbing cells by eroding bone, enabling the tissue to be remodelled during
growth and/or in response to stresses throughout life. Bone resorption is an obligatory
event during bone growth, tooth eruption, fracture healing and the maintenance of blood
calcium level (Vaananen et al., 2000).
Multinucleated osteoclasts are derived from hematopoietic stem cells and are created by
the differentiation of monocyte/macrophage precursor cells at or near the bone surface
(Boyle et al., 2003; Edwards et al., 2011). Hence, osteoclasts share a common pathway
to that of monocyte/macrophages and dendritic cells and the bone resorption process
utilizes similar cellular machinery as phagocytosis (Morgan et al., 2008; Vaananen et
al., 2000).
Depicted in Figure 1.1, the initial step of osteoclastogenesis is the determination of the
stem cell precursor induced by the erythroblast transformation specific (ets)
transcription factor family member PU.1 encoded by Spi-1 gene. At this stage, the cells

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
23
acquire the colony-stimulating factor 1 (c-fms) receptor that binds to macrophage
colony stimulating factor (M-CSF) to stimulate cell survival and proliferation (Fuller et
al., 1993; Zhang et al., 1994). The initiation of cell proliferation is induced by the
phosphorylation of several kinases, importantly Src, Grb2 and Phosphoinositide 3-
kinase (PI3K), which leads to the activation of cyclin D. M-CSF activates Casitas B-
lineage lymphoma (c-Cbl) for the ubiquitination and degradation of the pro-apoptotic
Bim to ensure the survival of osteoclasts (Akiyama et al., 2003; Blair et al., 2008;
O'Brien, 2009). With the appearance of proto-oncogene c-fos and Receptor Activator of
NF-ĸB (RANK), the progenitors are destined to form osteoclasts (Blair et al., 2008).
Around 1997, several independent laboratories identified the essential osteoclastogenic
factor Receptor Activator of Nuclear Factor Kappa B Ligand (RANKL, also called
TRANCE) (Anderson et al., 1997; Matsuo et al., 2008; Wong et al., 1997a; Wong et
al., 1997b). It is an established fact that RANKL, a trans-membrane glycoprotein and a
member of the Tumor Necrosis Factor (TNF)-α superfamily of cytokines, is essential
for efficient osteoclast differentiation. Numerous cell types such as stromal cells,
osteoblast precursors, mature osteoblasts, osteocytes, chondrocytes and lymphocytes
express RANKL. RANKL has two receptors: RANK, and osteoprotegerin (OPG). The
actions of RANKL are opposed by a soluble TNF-receptor OPG. OPG serves as a
decoy receptor by binding to RANKL and limiting the signalling that induces
osteoclastogenesis (Blair et al., 2008; Miyamoto et al., 2003; O'Brien, 2009; Simonet et
al., 1997; Wada et al., 2006). OPG thus inhibits formation of osteoclasts. OPG-/- mice
exhibit severe osteoporosis due to lack of OPG and excessive osteoclastic bone
resorption (Mizuno et al., 1998).
The binding of RANKL to the extracellular domain of RANK activates TNF receptor-
associated factor (TRAF) adaptor or docking proteins and is considered the key
preliminary step in RANK signalling (Boyle et al., 2003). TRAF-2, -5, and particularly
TRAF-6, are all shown to bind to RANK. TRAF6 then phosphorylates IKK (IĸB
kinase), which ubiquitinates IĸB to liberate the transcription factor NF-ĸB and allow its
translocation to the nucleus for the transcription of osteoclast specific genes. RANK-
RANKL interaction also induces at least other four distinct signalling cascades – c-Jun
N-terminal kinase (JNK), mitogen activated protein kinases (MAPKs) including
extracellular signal-regulated kinase (ERK) and p38, and Src pathways to ensure the
survival and maturation of preosteoclast cells (Boyle et al., 2003; Duplomb et al., 2008;

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
24
Sundaram et al., 2007; Xu et al., 2009a) (Figure 1.2).
As the preosteoclasts differentiate, PU.1 also interacts with micro-opthalmia associated
transcription factor (MITF), which targets genes that encode essential molecules in
normal osteoclast machinery, such as Tartrate-resistant acid phosphatase (TRAP) and
carbonic anhydrase (Luchin et al., 2000; So et al., 2003). This is followed by
multinucleation brought by cell fusion of the mononuclear preosteoclasts (Miyamoto et
al., 2003). Studies have shown that dendritic cell-specific transmembrane protein (DC-
STAMP) and the d2 isoform of vacuolar (H+) ATPase (v-ATPase) V0 domain
(Atp6v0d2) are essential for the fusion of osteoclasts. The deficiency of DC-STAMP
and Atp6v0d2 did not affect osteoclast differentiation into TRAP positive cells in vivo,
however the maturation into large multinucleated cells was impaired (Kukita et al.,
2004; Lee et al., 2006; Yagi et al., 2005; Yagi et al., 2007).
It is also reported that stimulation of RANKL leads to the activation of
calcineurin/NFAT (nuclear factor of activated T cells) signalling pathway and
upregulation of the transcription factor NFATc1 expression throughout osteoclast
maturation (Figure 1.2). An in vitro study has demonstrated that constitutively active
calcineurin-independent NFATc1 mutant expression is adequate to induce
osteoclastogenesis of RAW264.7 cells (Hirotani et al., 2004). The activation of RANK
initiates the recruitment of spleen tyrosine kinases (Syk) by DAP12 or FcRγ, both of
which are immunoreceptor tyrosine based activation motif (ITAM)-harbouring
adapters. Syk activates phospholipase Cγ to release Ca2+ from intracellular stores. This
event triggers the calmodulin-regulated phosphatase calcineurin to dephosphorylate the
transcription factor NFATc1. NFATc1 undergoes nuclear translocation and, together
with c-fos, magnifies the expression of critical osteoclast genes (Blair et al., 2008;
Mocsai et al., 2004; Sundaram et al., 2007). These findings thus indicated that
calcineurin is an essential downstream effector of RANKL signalling pathway and that
NFATc1 is involve in regulating osteoclastogenesis and cell function in response to
RANKL stimulation.
1.2.2. Osteoblasts
Osteoblasts are the primary cells responsible for bone formation. They originate from
mesenchymal stem cells (Figure 1.1), which have the potential to differentiate into other
musculoskeletal tissues such as cartilage, fat, muscle, ligament and tendon (Pittenger et

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
25
al., 1999). The commitment of these stem cells to the osteoblast lineage is highly driven
by the growth factors wingless type proteins (Wnts) and bone morphogenetic proteins
(BMPs) (Clarke, 2008; Fakhry et al., 2013; Rosen, 2009).
The significance of the Wnt family of signalling proteins in the maintenance of bone
homeostasis is well documented. Mutations in genes encoding intracellular Wnt
pathway proteins lead to diverse conditions such as osteoporosis pseudoglioma
syndrome (Gong et al., 2001) and high bone mass disorders (Boyden et al., 2002; Little
et al., 2002). The main consequence of Wnt signalling is the activation of β-catenin
dependent transcription. In the absence of Wnt receptor activation, cytoplasmic β-
catenin remains phosphorylated by the axin scaffold proteins, adenomatous polyposis
coli protein (APC), glycogen synthase kinase-3β (GSK3) and casein kinase I.
Phosphorylation of cytoplasmic β-catenin results in its ubiquitination by E3 ubiquitin
ligase β-TrCP, and subsequent proteasomal degradation (Aberle et al., 1997; Su et al.,
2008). In contrast, binding of Wnt proteins to the Frizzled (Fz)/low-density lipoprotein
(LDL) receptor-related protein (LRP) complex at the cell surface activates Dishevelled
(Dsh/Dvl), which inhibits the phosphorylation of β-catenin. This circumvents the
degradation pathway to allow the stabilisation and accumulation of β-catenin in the
cytoplasm and nucleus. Nuclear β-catenin then interacts with the transcription factor
lymphoid enhancer binding factor 1/T cell-specific transcription factor (TCF/LEF) to
mediate gene transcription (Huber et al., 1996).
BMPs are multifunctional growth factors that belong to the transforming growth factor
β (TGF-β) superfamily (Rosen, 2009). They were first identified based on their ability
to initiate ectopic bone formation (Urist, 1965; Wozney et al., 1988) and this unique
feature has allowed for their successful use as therapeutic agents for bone regeneration.
BMPs bind to two types of serine-threonine receptors, BMP receptor (BMPR) type I
and type II. Binding of BMP to BMPR-II leads to the recruitment of BMPR-I to form
an activated heteromeric complex. This complex then phosphorylates and activates
intracellular signal-transducing molecules of the TGF-β superfamily termed Smad
proteins. Receptor-regulated Smads 1, 5 and 8 bind to a co-Smad 4 and then translocate
to the nucleus to initiate transcription (Chen et al., 2012; Fujii et al., 1999). The
survival of mesenchymal stem cells within the bone marrow microenvironment is
maintained by BMP activity (Solmesky et al., 2009; Yang et al., 1999). Endogenously
produced BMPs are crucial survival factors for human MSCs and these findings

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
26
highlight the involvement of BMP signalling in maintenance of the skeletal stem cell
niche in adult bone.
Once skeletal stem cells are committed to the osteoblast lineage, the helix-loop-helix
proteins Twist and Id ensure the proliferation of preosteoblasts to continue, albeit with
reduced plasticity. The osteoblast precursor cells eventually stop proliferating and begin
secreting type I collagen – the basic building block of bone. These cells also produce
non-collagenous proteins, such as osteocalcin (small calcium binding protein specific to
bone that is carboxylated by vitamin K) and alkaline phosphatase, which are crucial for
mineral deposition. Proteoglycans that modulate collagen fibril formation, and
glycoproteins that regulate mineralization and matrix organization are also produced by
osteoblast precursors. Also several arginine-glycine-aspartate-(RGD)-containing-
glycoproteins that are important in osteoclast matrix interactions and osteoblast
attachment are also secreted by the precursors. These precursors resemble active
osteoblasts, however they lack some of the characteristics including the ability to
mineralise bone (Blair et al., 2008; Kular et al., 2012; Ling et al., 2005; Zittermann,
2001).
Preosteoblasts then mature into mineralization competent cuboidal osteoblasts, marking
the final stage of osteoblast differentiation. Cuboidal osteoblasts have a large eccentric
nucleus with prominent rough endoplasmic reticulum and Golgi areas, all of which are
attributes of typical secretory cells (Lian et al., 2001; Lucht, 1972; Tasat et al., 2007).
They no longer divide and can be distinguished from preosteoblasts by the upregulation
of bone markers such as bone sialoprotein, osteocalcin, BMPR-I, vitamin D3 receptor
and type I collagen (Franz-Odendaal et al., 2006). Besides their role in bone formation,
at this stage the osteoblastic cells are involved in the recruitment and maintenance of
osteoclasts by expressing M-CSF, RANKL and OPG (Simonet et al., 1997; Takahashi
et al., 1991; Udagawa et al., 1999).
Mature osteoblasts have one of the following four fates – (1) they may undergo
apoptosis, those that survive (2) coalesce into the heterogeneous population osteolining
cells or (3) eventually become encased and trapped within the matrix to become
osteocytes, or in some scenarios, (4) transdifferentiation into cells that are capable of
chondrogenesis (Redlich et al., 2004). The proportion of osteoblasts following each
fate varies in all mammals and is not conserved among different types of bone. The age

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
27
of the mammal may also influence the number of osteoblasts that transform into
osteocytes (Franz-Odendaal et al., 2006). In human cancellous bone, 65% of the
osteoblasts undergo apoptosis and only ~30% transform into osteocytes (Parfitt, 1990).
1.2.3. Osteocytes
Once assumed to be an inert cell, the osteocyte has gained much attention in the recent
years as one of the central responder to various stimuli involved in the maintenance of
skeletal and mineral homeostasis. Their importance is highlighted by the fact that
osteocytes are the most ubiquitous cellular component of bone situated within the bone
matrix. Derived from mature osteoblasts, the prospective cuboidal cells undergo drastic
morphological changes and differentiate into stellate-shaped osteocytes with multiple
dendritic conduits (Dudley et al., 1961; Palumbo, 1986; Palumbo et al., 1990).
Osteocytes form a functional multicellular syncytium by maintaining a complex
network of long filipodial conduits composed of actin filament (Tanaka-Kamioka et al.,
1998). These dendritic processes radiate out of their confined lacunar space via
microscopic canals (canaliculi), to regulate and communicate with neighbouring
osteocytes and with other bone cells and residential cells (Cao et al., 2011; Neve et al.,
2012; Talmage, 1970). The gap junction channel, an intercellular hemichannel
constructed from transmembrane proteins where tips of the cytoplasmic processes meet,
is enriched with connexin 43 (Cx43) (Doty, 1981; Jones et al., 1993; Kamioka et al.,
2007). This gap junction protein allows the diffusion of molecules with a molecular
weight of less than 1kDa and has been reported to play a protective role against
osteocyte apoptosis (Kar et al., 2013; Plotkin et al., 2002). In vitro experiments using
the MLO-Y4 osteocyte-like cell line show that fluid flow significantly increased the
length of the dendritic extensions and redistribution of Cx43 (Cheng et al., 2001).
Furthermore, gap junctional intracellular communication (GJIC) in MLO-Y4 cells is
amplified when they are subjected to low intensity vibration, suggesting that gap
junctions may serve as channels for signals generated by osteocytes in response to
mechanical strains to mediate cellular communication (Uzer et al., 2014).
It has also been reported that osteocytes display protuberances that project from the
body. Osteocytes retain centrosomes which give rise to primary cilia, and the MLO-Y4
cell line has been found to express transcripts for glycoproteins polycystin (PC) 1 and
PC2, and the ciliary proteins Tg737 and Kif3a (Uzbekov et al., 2012; Xiao et al., 2006).

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
28
These projections of primary cilia on the cell surface are perturbed by bone fluid that
flows through the lacuna-canalicular system. In vitro immunohistochemistry studies
have indicated that the translation of the mechanical stimulus into intracellular
responses by the organelle relies on a molecular mechanism involving membrane-
bound enzyme adenylyl cyclase (AC) 6 and intracellular cyclic adenosine
monophosphate (cAMP) (Kwon et al., 2010; Malone et al., 2007).
Another study revealed that osteocytes in the lacunocanalicular system (LCS) of intact
mouse tibiae displayed intracellular calcium oscillation in response to in situ dynamic
loading. The frequency of calcium spikes correspond to the load magnitude, tissue
strain and sheer stress in the LCS (Jing et al., 2014). These aforementioned cellular
structures of osteocytes and their responses to mechanical strains are some of their
features that lead scientists to hypothesise the stellate cells are the key regulators of
bone mechanotransduction. As mechanosensors of bone, the osteocytic network detects
local mechanical stimulations and in response converts them into biochemical signals
by expressing membrane-bound and releasing soluble factors that coordinate the
function of other bone cells, thus orchestrating the synchronisation of bone cell activity
(Iolascon et al., 2013).
The direct and indirect regulation of osteoblast and osteoclast activity by osteocytes has
been reported. Mechanically stimulated MLO-Y4 cells have been reported to regulate
osteoblast anabolic activity via gap junctions in which osteocytic-osteoblastic cell
contact is essential (Taylor et al., 2007). In vivo studies employing mechanical loading
and unloading experiments in several mouse models with osteoblast/osteocyte-specific
loss of Cx43, cilial Kif3a and AC-6 resulted in altered bone formation (Chung et al.,
2006; Grimston et al., 2008; Lee et al., 2014; Lloyd et al., 2012; Temiyasathit et al.,
2012; Zhang et al., 2011b). Additionally, transgenic osteocyte-ablated mice are resistant
to unloading-induced bone loss, albeit displaying an osteoporotic phenotype
accompanied by osteoblastic dysfunction and microfractures (Tatsumi et al., 2007).
In vivo fracture healing studies revealed osteoblast/osteocyte-specific ablation of Cx43
led to a decrease in GJIC and subsequent reduction in bone mineralisation, recruitment
of osteoclasts and eventually delayed bone formation and healing (Loiselle et al., 2013).
Regions of osteocyte apoptosis due to microdamage have been shown to coincide with
increased local bone turnover and targeted bone resorption (Cardoso et al., 2009; Clark

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
29
et al., 2005; Gu et al., 2005; Noble et al., 1997; Verborgt et al., 2000). Further research
demonstrated that apoptotic osteocytes regulate the recruitment and differentiation of
osteoclast progenitors through the neighbouring non-apoptotic osteocytes that have
responded to the apoptosis cues (Al-Dujaili et al., 2011; Kennedy et al., 2012). All of
these data support the hypothesis that functional osteocytes are crucial bone
mechanotransducers and osteocytes communicate with other bone cells via GJIC and
other factors in response to physical stimuli.
1.2.4. Osteolining cells
Residing on quiescent (non-remodelling) bone surfaces, osteolining cells form a cellular
layer that separates bone tissue from the marrow. Osteolineage lining cells display a
thin, flat nuclear profile and contain a few cell organelles including mitochondria,
microfilaments, free ribosomes and rough endoplasmic reticulum. The cells are also
reported to display cell processes and are connected with one another via gap junctions
(Miller et al., 1987).
Originally thought to be of endothelial origin, these cells are negative for the expression
of endothelial cell marker and are reported to be positive for osteoblastic markers such
as alkaline phosphatase, osteocalcin and osteonectin (Hauge et al., 2001). Despite the
fact that they express osteoblastic gene markers and are hypothesised to be terminally
differentiated osteoblasts, the origin and the role of osteolining cells are still highly
debatable. While exhibiting a non-mitotic and dormant cell-morphology that is hardly
engaged in the maintenance of bone homeostases (Menton et al., 1984), experimental
evidence has suggested many functions of the osteolining cells, particularly when they
are “re-activated” in response to bone remodelling.
Early SEM studies reported the presence of a fluid compartment between osteolining
cells and bone tissue, which extends into the bone matrix itself and around osteocytes in
the lacunae (Baud, 1968). Osteolining cells were postulated to maintain this space.
Furthermore, it has been documented that osteolining cells possess numerous
protoplasmic extensions that extend into bone and make contact with cell processes of
osteocytes (Talmage, 1969; Talmage, 1970; Talmage et al., 1970), implicating their
possible role in supporting osteocytic mechanotransduction.
Osteolining cells have been considered as potential osteogenic precursor cells especially
in an event that initiates rapid bone formation. An event such as mechanical loading

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
30
was proposed to increase bone formation by driving osteoprogenitor cells in the bone
marrow to progress through the cell cycle and differentiate into osteoblasts at the bone
surfaces. In rats, the proliferation, differentiation and recruitment of osteoprogenitors to
the bone surfaces require 72 hours post-loading to establish. However, endocortical
osteoblast surface was observed to be significantly increased at 48 hours after loading
stimulus, suggesting that there is an immediate response from existing cells on the bone
surface (Turner et al., 1998). This observation is also supported by Chow et al., who
have documented the morphological evolution of osteolining cells towards osteoblastic
cells after mechanical stimulation (Chow et al., 1998).
A similar immediate increases in bone forming surface is also observed upon
administration of parathyroid hormone (PTH) (Cho et al., 2014; Dempster et al., 2001;
Gunness-Hey et al., 1984; Hamann et al., 2014; Jilka et al., 1999), and since no cell
proliferation and recruitment of osteoblasts was noted on the bone surface, it has also
been proposed that the anabolic effect of PTH is likely to be due to the activation and
differentiation of pre-existing osteolining cells towards osteoblastic cells (Dobnig et al.,
1995; Leaffer et al., 1995; Onyia et al., 1995). Ablation of bone marrow cells using
high dose radiation is reported to be associated with a transient increase in bone
formation due to activation of osteoblastic lining cells. This is accompanied by an
increase in bone resorption leading to an increased bone turnover rate (Turner et al.,
2013).
Osteolining cells are also implicated in the phagocytosis of collagen fibrils and
mineralised bone particles (Takahashi et al., 1986). Osteolining cells are described to
display catabolic activities by secreting matrix metalloproteinases (MMPs) to degrade
bone matrix (Dierkes et al., 2009; Everts et al., 2002). The capability of these
osteolining cells to have both anabolic and catabolic activities indicates that these cells
are a population of different types of cells that serve multiple functions in maintaining
bone homeostasis. Another subpopulation of osteolining cells is the residential
macrophages (Hume et al., 1984). These resident osteal antigen F4/80 macrophages
found intercalated among osteolining cells are termed osteomacs. They are implicated
in supporting bone mineralisation in vitro and the maintenance of bone homeostasis
through regulation of osteoblast maturation and function in vivo (Chang et al., 2008).
Sharing the same common precursor as osteoclasts, osteomacs are TRAP negative and
localised to sites of bone modelling in vivo, where they formed a distinctive canopy

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
31
structure over mature osteoblasts (Alexander et al., 2011; Chang et al., 2008; Cho et al.,
2014). Osteomacs participate in intramembranous healing and are required for the
deposition of type I collagen and bone mineralisation in tibial injury model (Alexander
et al., 2011). The depletion of these osteal macrophages lead to a reduction of bone
mass and attenuates the anabolic effect of PTH further supporting their positive role in
the maintenance of bone homeostasis (Cho et al., 2014).
1.3. Formation and maintenance of bone
1.3.1. Osteogenesis
Osteogenesis is sculpted by three distinct lineages – the somites, lateral plate mesoderm
and cranial neural crest. The somites and the lateral plate mesoderm give rise to axial
and limb skeleton respectively, whilst the cranial neural crest generates the branchial
arch, craniofacial bones and cartilage. There are two modes of osteogenesis:
intramembranous and endochondral ossification (Gilbert, 2000b). In both processes, the
initial bone formation is made up of disorganised woven collagen fibres, termed
primary bone, which is then replaced by highly organised concentric lamellae to form
architecturally strong secondary bone (Shapiro, 2008; Teti, 2011).
The process of direct condensation of mesenchymal cells that eventually differentiate
into bone-forming osteoblasts to produce flat bones (e.g. skull, scapula and ileum) is
termed intramembranous ossification. The process of intramembranous ossification is
driven by activation of the transcription factor Core-binding factor alpha 1 (CBFA1) by
bone morphogenetic proteins (BMPs) (Ducy et al., 1997; Yamaguchi et al., 2000).
Intramembranous ossification begins with the proliferation and condensation of neural
crest-derived stem cells into compact nodules. Some of these stem cells undergo
angiogenesis to develop capillaries; others undergo osteoblastogenesis to secrete pre-
bone matrix (osteoid) that is made up of collagen-proteoglycan and is able to bind
calcium salts (Gilbert, 2000a; Steele et al., 1988). As the osteoid calcifies, woven bone
spicules emanate out from the region where ossification began. The mesenchymal cells
enveloping the calcified spicules differentiate into a membrane termed periosteum.
Stem cells on the inner surface of the periosteum undergo osteoblastogenesis to secret
more osteoid matrix directly into the condensed mesenchyme, entrapping late-stage
osteoblasts which will evolve into osteocytes. The cycle continues to form many layers
of bone proceeding outward from the nodule centre (Gilbert, 2000a; Teti, 2011).

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
32
The formation of endochondral bone, which includes the long bones and vertebrae, can
be divided into five stages. Endochondral ossification begins with the condensation and
differentiation of mesenchymal stem cells into a template composed of type II collagen
produced by chondrocytes. These early stages are driven by the transcription factor
SOX-9 (Allen et al., 2014). During the next phase, the chondrocytes proliferate rapidly,
expanding the cartilage template. A subset of the chondrocytes stop dividing and
become hypertrophic. The cartilage then undergoes vascular invasion and
mineralization. During this process, the large hypertrophic chondrocytes undergo
apoptosis, leaving space that will become bone marrow spaces that are loosely
compartmentalised. As the cartilage cells die, osteoprogenitors are recruited and
differentiate into osteoblasts. The osteoblasts begin to secrete osteoid to replace the
partially degraded cartilage. Some cartilage is retained at the ends of the bone at the
surface lining the joints to provide low-friction support (articular cartilage), and in the
growth plates, where further proliferation of chondrocytes allows bones to grow in
length (Blair et al., 2008; Boyce et al., 2008; Gilbert, 2000a).
1.3.2. Bone modelling and remodelling
Osteogenesis and skeletal growth require both bone modelling and remodelling. Bone
modelling is characterized by a process of either bone formation or resorption so as to
“re-shape” pre-existing bone with a net increase in bone mass. The mechanism involves
either activation-formation or activation-resorption which occurs primarily throughout
childhood. Activation is signalled by local tissue strain and involves the recruitment of
progenitor cells that differentiate into mature osteoblasts or osteoclasts. Once the
appropriate cell population is activated, the processes of formation or resorption take
place until sufficient bone mass is added or removed to normalize local strains (Allen et
al., 2014; Teti, 2011).
In contrast, bone remodelling is defined as a process of renewing and maintaining bone
in which osteoblast and osteoclast activities occur sequentially in a coupled manner.
The cycle of activation, resorption of old bone and formation of new bone, occurs
throughout life and is tightly balanced and heavily orchestrated by cell activities (Allen
et al., 2014; Teti, 2011).
The bone remodelling unit and osteoblast-osteoclast communication
The process of bone remodelling is executed by temporary anatomic structures

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
33
incorporating a cohort of cells known as the bone remodelling unit (BRU) (Figure 1.3)
(Frost, 1973; Parfitt et al., 1996). With the progression in bone research, the BRU no
longer merely consists of osteoclasts, osteoblasts and osteocytes. Other key cells
including osteolining cells, osteomacs and vascular endothelial cells have also been
reported to be associated with the BRU (Andersen et al., 2009; Chang et al., 2008;
Dierkes et al., 2009; Lafage-Proust et al., 2010; Parfitt, 2001; Pettit et al., 2008;
Rasmussen et al., 1973). Bone is a highly vascularised organ and networks of blood
capillaries have been observed at the site of the BRU (Chim et al., 2013).
Vascularisation and angiogenesis are prerequisites for bone formation and remodelling.
They serve multiple purposes including efficient exchange of matrix constituents and
minerals, and the recruitment of bone cell progenitors via blood capillaries to establish
the modelling and remodelling site (Andersen et al., 2009; Kristensen et al., 2013;
McClugage et al., 1973; Shapiro, 2008). The blood supply also allows the BRU to
become accessible to immune cell infiltration and subsequent interaction with bone
cells. It has been demonstrated that there is significant crosstalk between bone and
immune cells, this has led to the development of the field of osteoimmunology, and will
be discussed in detail in Chapter 2. The BRU represents the anatomical basis for
intercellular communication and coupling of bone remodelling. The bone remodelling
cycle occurs in five stages: activation, resorption, reversal, formation and quiescence
(Allen et al., 2014).
Activation
The activation stage commences with the detection of a signal such as mechanical strain
by osteocytes or osteocyte apoptosis, and the recruitment of osteoclast progenitors to
the bone surface. Detecting the mechanical stimuli, osteolining cells form a
“remodelling canopy”, while allowing the interaction between pre-existing osteoblasts
and recruited osteoclast progenitors underneath (Allen et al., 2014; Clarke, 2008;
Raggatt et al., 2010; Rifkin et al., 1979). The signals of M-CSF and RANKL presented
by osteoblasts are necessary for osteoclastogenesis. Other chemokines, such as
monocyte chemoattractant protein (MCP)-1, also known as CCL2, is released by
osteoblasts and is a candidate recruiter of osteoclast precursors. The expression of
MCP-1 receptors on osteoclast precursors is induced by RANKL, suggesting that
osteoblasts are capable of enhancing MCP-1-dependent recruitment through RANKL
(Li et al., 2007a). Another possible chemokine involved in the recruitment of osteoclast

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
34
precursors is the stromal cell-derived factor (SDF)-1, produced by bone vascular
endothelial and marrow stromal cells, that binds to the chemokine receptor CXCR4
expressed on osteoclast precursors to induce MMP9 expression (Sundaram et al., 2007).
Another form of activation is inflammation, particularly in the context of the pathology
of inflammatory arthritis, whereby inflammatory cytokines secreted by immune cells
are also known to be osteoclastogenic. T cells have been reported to secrete RANKL
and T helper 17 cells have been identified as the osteoclastogenic T cell subset.
Resorption
Upon attachment onto the matrix, differentiated mature osteoclasts create an isolated
microenvironment beneath the cell. The “sealing zone” is where osteoclasts pump in
hydrogen ions to establish an acidic and optimal condition for various collagenolytic
enzymes (Raggatt et al., 2010). One of the most potent collagenolytic cysteine
proteinases that degrades collagen at an acid optimum pH is cathepsin K (Everts et al.,
2005; Morko et al., 2009). Cathepsin K expression is promoted by RANKL (Corisdeo
et al., 2001) and cathepsin K deficiency due to loss of function mutation led to
osteopetrotic bone disease (Gelb et al., 1996). Another collagenase highly expressed in
mature osteoclasts is matrix metalloproteinase-9 (MMP9) (Sundaram et al., 2007).
Osteoclasts also express high levels of lysosomal acid phosphatase TRAP, the enzyme
marker of osteoclasts (Akisaka et al., 1989). TRAP is able to dephosphorylate bone-
associated proteins, and mice deficient for the enzyme exhibit a mild osteopetrotic
phenotype (Andersson et al., 2003; Ek-Rylander et al., 1994; Hayman et al., 1996). The
dissolution of matrix by these enzymes results in the formation of scallop-shaped
resorption pits termed Howship’s lacunae on the surface of trabecular bones and
Haversian canals in cortical bone (Clarke, 2008; Eriksen, 1986; Hattner et al., 1965).
As resorption proceeds, more osteoclasts can be recruited to the remodelling sites to
either support existing osteoclasts or replace those that have undergone apoptosis (Allen
et al., 2014). However, the differentiation of osteoclastic cells is heavily regulated
through OPG expression by osteoblasts to avoid excessive bone resorption (Mizuno et
al., 1998; Morony et al., 1999). To circumvent the antagonist effect of OPG, osteoclasts
have substantial expression of Sema4D, a member of the semaphorin family that
includes both secreted and membrane-associated molecules that interact with plexin and
neuropilins as their primary receptors. Targeted ablation of Sema4D in osteoclasts did

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
35
not lead to dysfunction of osteoclasts; however, it resulted in an increased trabecular
bone mass due to increased number and activity of osteoblasts, suggesting that Sema4D
is an osteoclast-derived inhibitor of osteoblast differentiation and function (Negishi-
Koga et al., 2011). Notably, T cells also produced Sema4D, demonstrating another
possible local source of factors that inhibit osteoblastogenesis (Bougeret et al., 1992;
Zhang et al., 2013b). Osteoclasts also express Sphingosine 1-phosphate (S1P), which
has both stimulatory and inhibitory effects on osteoblasts depending on the source of
osteoblast precursors and cell differentiation (Ishii et al., 2009; Kikuta et al., 2013;
Pederson et al., 2008).
Reversal
The reversal phase is characterised by the termination of osteoclast-mediated bone
resorption and the initiation of bone formation. Following resorption, osteoclasts
undergo apoptosis or migrate to another surface, leaving the resorption pit filled with
debris of undigested demineralised collagen matrix and a myriad of cells. Osteolining
cells and phagocytic macrophages are implicated in the removal of demineralised debris
and the “reconditioning” of the remodelling site (Everts et al., 2002; Takahashi et al.,
1986). Without the removal of this debris, bone formation by osteoblasts does not
proceed (Allen et al., 2014; Hauge et al., 2001). Among the osteolining cells are the
residential osteal macrophages, which are required to prime the area into an osteogenic
environment for osteoblast function (Alexander et al., 2011; Chang et al., 2008; Cho et
al., 2014). An immediate source of osteoblasts is thought to be present among the
osteolining cells, while osteoblast precursors are recruited from the bone marrow. It has
been proposed that the recruitment of osteoblast precursors could be initiated by bone
matrix-derived factors, such as insulin like growth factors (IGFs) I and II, BMPs and
TGF-β, which were pre-embedded within the matrix by osteoblasts and released during
osteoclast resorption (Shinar et al., 1993; Tang et al., 2009; Xian et al., 2012).
Different hypothetical models of direct osteoclast-osteoblast coupling mechanism have
been proposed. The gp130 family of cytokines have been reported to stimulate
osteoclastogenesis and osteoblast proliferation or differentiation and using mice models,
it was concluded that resorption alone was insufficient to promote coupled bone
formation and that active osteoclasts are the likely source of coupling molecules
(Martin et al., 2005; Sims et al., 2004). Gp130 signalling cytokine, cardiotrophin-1

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
36
(CT-1), is detected by resorbing osteoclasts and was shown to stimulate osteoblast
differentiation in vitro and bone formation in vivo (Walker et al., 2008). Another
protein family that has been implicated in osteoclast-osteoblast coupling is the
Ephrin/Eph family, which has a capacity for bidirectional signalling. EphrinB2 is
expressed by osteoclasts and in vivo and ex vivo studies of genetically modified mice
suggested that osteoclast-derived ephrinB2 acts through its receptor EphB4, which is
expressed on osteoblasts, to support forward signalling of osteoblast differentiation.
Upon direct contact, the reverse signalling by osteoblast-derived Eph4 suppresses the
formation of osteoclast precursors (Irie et al., 2009; Matsuo et al., 2012; Zhao et al.,
2006).
There is also evidence suggesting that immune cells participate in the regulation of
osteoblast formation and function (Li et al., 2007b). Supporting their possible role in
the reversal phase, B-lymphocytes have also been shown to inhibit osteoclast activity by
expressing OPG and TGF- β in vivo and in vitro (Horowitz et al., 2005; Horowitz et al.,
2004; Li et al., 2007b; Weitzmann, 2013; Weitzmann et al., 2000). Innate immune
cells, such as activated macrophages is also reported to secrete OSM for the
maintenance of osteoblast differentiation (Guihard et al., 2012; Sims et al., 2014).
Formation
Bone formation is a slow phase and can take 3 to 4 months to complete. During the
bone formation stage, mature osteoblasts lay down osteoid, which serves as a template
that eventually becomes mineralised. At the finale of bone remodelling cycle, the newly
formed bone surface is in a state of quiescence while the matrix within the remodelling
unit will continue to mineralise (Allen et al., 2014).
1.4. Conclusion
Although it is important to dissect a complex system by labelling and categorising
different cells whilst identifying their “unique” roles, the skeletal system must be also
be comprehended as an integration of multiple cellular organisations. While the
OPG/RANKL/RANK signalling pathway is essential for the intimate crosstalk between
anabolic osteoblasts and catabolic osteoclasts, and is considered the central axis
controlling bone remodelling, different cell types within the bone remodelling
compartment also play a crucial role in maintaining the crosstalk. The skeletal system
has been shown to be not only regulated by the immune system, but also by the kidney,

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
37
brain and central nervous system, indicating the complexity of bone homeostasis and
underlying pathology of many bone diseases.
With the many contributors to the regulation of bone homeostasis, future investigation
is essential to understand the complex intercellular communication during bone
remodelling, and how the body compensates in certain pathological conditions, with the
ultimate aim to design interventions that can prevent bone loss while maintaining bone
quality.

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
38
Figure 1.1 The different types of bone cells involved in the regulation of bone
microenvironment. Initially thought to be static, the bone tissue is dynamically
orchestrated by different types of cells and other systems. Osteoclasts and osteoblastic
cells originate from the hematopoietic and mesenchymal stem cells respectively.
Recently characterised bone cells include the bone lining cells and osteomacs.
Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
39
Figure 1.2 RANK/RANKL signalling pathway during osteoclastogenesis. RANKL
stimulation is crucial in driving the formation and function of osteoclasts. The
interaction between RANKL and RANK initiate the recruitment of TRAF6 to initiate
several signaling cascades, including NF-κB, NFATc1/calcineurin and MAPK
pathways. The differentiation of osteoclasts is also supported M-CSF which induce the
expression of RANK in pre-osteoclasts.

Chapter 1: The Biology of Bone Multicellular Unit and Transcriptional Regulations of Bone Homeostasis
40
Figure 1.3 The bone remodelling cycle. The remodelling unit represents cellular
communication that governs the coupling between bone resorption and formation. This
cycle is supported by other systems, such as the blood vessels, which carries nutrients
and other cell types. Immune system also inevitably interacts with bone cells as their
haematopoiesis is spawned with the marrow cavity. For instance, activated CD4+ T
cells are reported to stimulate the production of RANKL by stromal cells, activated
macrophages secrete OSM for the maintenance of osteoblast differentiation, and B
lymphocytes express OPG and Wnt1b, which favours bone formation in vitro.

CHAPTER TWO
OSTEOIMMUNOLOGY – INTERACTIONS OF THE
SKELETAL AND IMMUNE SYSTEMS

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
42
2. CHAPTER 2: OSTEOIMMUNOLOGY – INTERACTIONS OF THE SKELETAL AND IMMUNE SYSTEMS
2.1. Introduction
The bone organ system is a living mineralized tissue that is continuously being broken
down and rebuilt in a process known as bone remodelling. The bone remodelling
process is a coupled activity between the catabolic effects of bone-resorbing osteoclasts
and the anabolic effects of bone-forming osteoblasts. This coupling activity between
these two bone cells is tightly regulated so as to achieve microarchitecturally strong
bone, whereby the imbalance of this delicate regulation often lead to bone and joint
diseases.
With the emerging field of osteoimmunology, many studies have indicated that both
innate and adaptive immune systems are major components of bone remodelling. The
dialogue between the skeletal and immune systems is inevitable as haematopoietic and
mesenchymal stem cells are housed within the bone marrow, and these gives rise to
progenitors of bone cells and lymphocytes that constitute the immune system. The
intimate relationship between the immune and skeletal system is highlighted in many
inflammatory diseases such as periodontal disease (Kayal, 2013), rheumatoid arthritis
(RA) (Spector et al., 1993), spondylarthritis (SA) (Goldring, 2013), Cherubism (Mukai
et al., 2014), Crohn’s disease (Gordon, 2006), osteomyelitis (Ferguson et al., 2006) and
systemic lupus erythematosus (SLE) (Tang et al., 2013a), all of which are associated
with both local and systemic bone loss with accompanying increased risk of bone
fractures and deformities. Beyond these observations, recent data further demonstrate
that the immune system may have pro-anabolic functions. Osteoimmunological
interactions thus play a significant role in the maintenance of bone homeostasis and are
fundamental mechanisms in bone pathology.
Likewise, it is also important to acknowledge the role of the skeleton in fostering
haematopoiesis and the maturation of immune lineages. In view of this, the overlapping
roles of the essential cytokines, receptors, common signalling transduction and
molecular pathways in the osteoimmune system, and how the dysregulation of these
shared pathways can lead to clinical manifestation of bone diseases are discussed below.
2.2. Osteoclasts and inflammatory bone destruction
One of the most well described osteoimmune diseases is RA, which is characterised by
progressive erosion of bone surrounding inflamed joints (Gough et al., 1994; Spector et
al., 1993). Histology studies of tissue sections from sites of erosion in RA patients and

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
43
animal models have revealed multinucleated cells that are phenotypically similar to
bone-resorbing osteoclasts in the bone resorption lacunae (Bromley et al., 1984; Leisen
et al., 1988). Mononuclear cells isolated from the synovial fluid obtained from patients
with RA are reported to be able to differentiate into functional resorbing osteoclasts
(Danks et al., 2002). The osteoclastogenic property of synovial fluid collected from RA
patients is also shown to support the formation of numerous TRAP positive cells
capable of lacunar resorption in vitro (Adamopoulos et al., 2006). Consistent with the
aforementioned observations, mice that are unable to form functional osteoclasts are
protected from arthritis and subsequent development of bone erosion despite exhibiting
levels of synovial inflammation comparable to the wildtype littermates (Pettit et al.,
2001; Redlich et al., 2002).
The expression of receptor activator of NF-κB ligand (RANKL), the key differentiation
factor that drives osteoclastogenesis, was detected specifically in the synovium and at
sites of articular bone erosion of patients with RA (Gravallese et al., 2000; Pettit et al.,
2006). The receptor for RANKL is RANK (previously known as the tumour necrosis
factor (TNF) receptor super family member (TNFRSF) 11a), a type I trans-membrane
protein that is highly homologous to CD40 (a co-stimulatory protein expressed on
antigen presenting cells involved in their activation). RANKL-deficient and RANK-
deficient mice models are both osteopetrotic due to impeded osteoclastogenesis, thus
indicating that RANK-RANKL interaction are crucial to initiate osteoclast
differentiation and the maintenance of osteoclast resorptive activity (Dougall et al.,
1999; Fuller et al., 1998; Kong et al., 1999b). The decoy receptor osteoprotegerin (OPG
or TNFRSF11b) prevents the binding of RANKL to RANK and inhibits
osteoclastogenesis. Rescue assays using inhibitors of osteoclastogenesis such as OPG
have been shown to successfully prevent bone erosion in a collagen-induced arthritis
(CIA) model (Romas et al., 2002). This indicates that RANKL and osteoclasts play a
pathological role in the progression of inflammatory arthropathies (Francois et al.,
2006; Neidhart et al., 2009).
2.3. Osteoclast and innate immune cell lineages: RANK and TLRs downstream
signalling cascades
One of the hallmarks of RA pathology is characterised by the perpetual production of
pro-inflammatory cytokines accompanied by massive infiltration of inflammatory

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
44
immune cells, including T and B cells, into the synovium. Inflammation is considered
as a nonspecific immune response orchestrated by dendritic cells, neutrophils,
monocytes, macrophages, natural killer cells, eosinophils, basophils and mast cells. All
of these effector cells, except for natural killer cells, originate from the haematopoietic
stem cell-derived common myeloid progenitors that also give rise to osteoclast
precursors.
Generation of macrophages and osteoclast precursors from the common myeloid
progenitors is mediated by a lineage specific cytokine termed macrophage-colony-
stimulating factor (M-CSF, also known as CSF-1) and the myeloid master regulator
PU.1. M-CSF directly induces the expression of PU.1, which in turn stimulate the
expression of macrophage colony-stimulating factor 1 receptor (M-CSF1R, also called
c-fms or CD115) (DeKoter et al., 1998; Mossadegh-Keller et al., 2013). M-CSF is
important for the survival and differentiation of osteoclast progenitors. Osteopetrotic
PU.1 deficient mice do not develop macrophage and mature osteoclasts, suggesting that
the transcription factor is crucial in regulating the initial stages of myeloid
differentiation (Tondravi et al., 1997). The induction of PU.1 by M-CSF is restricted by
MafB, which is selectively expressed in mature monocytes and macrophages (Sarrazin
et al., 2009). MafB also negatively regulates osteoclast differentiation (Kim et al.,
2007a).
In the presence of M-CSF, RANK-RANKL interaction induces osteoclastogenesis of
osteoclast precursors and initiates the recruitment of tumour necrosis factor receptor-
associated factors (TRAFs), including TRAF2, 5 and 6, to activate Src kinases and
mitogen-activated kinases (MAPK) (Kobayashi et al., 2001). Simultaneously, RANK
activation results in the phosphorylation of two adaptor proteins associated with distinct
Ig-like receptor, the immunoreceptor tyrosine-based activation motif) in DAP12 and Fc-
receptor common γ-subunit (FcRγ). Ig-like receptor signals are co-stimulatory signals
for RANK during osteoclast differentiation (Kim et al., 2012).
TRAF6 associates with transforming growth factor-β-activated kinase (TAK1) and
activates JUN N-terminal kinase (JNK) and AP-1 (Qi et al., 2014). TRAF6 also induces
nuclear translocation of the transcription factor NF-κB through IκB ubiquitination and
degradation to initiate the transcription of osteoclast specific genes targets (Chen, 2005).
The stimulation of RANKL/RANK interaction also leads to the activation of

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
45
calcineurin/nuclear factor of activated T cells cytoplasmic 1 (NFATc1) signalling
pathway and along with c-Fos, the nuclear translocation of dephosphorylated NFATc1
amplifies the expression of osteoclast-associated genes (Gohda et al., 2005).
Given the right stimuli, macrophage and myeloid dendritic cells are able to form
osteoclasts in vitro, suggesting that they originate from a subset of common myeloid
progenitor cells. This cell subset can be identified as B220- cKit/CD117+ CD115/c-fms+
CD11blo in the bone marrow niche (Jacome-Galarza et al., 2013; Jacquin et al., 2006).
The differentiation of these common progenitors into macrophages, myeloid dendritic
cells and osteoclast can be considered a dynamic lineage bifurcation (Figure 2.1).
Recent findings have identified CD27 as a marker of the subset that discriminates
osteoclast/dendritic precursors (CD27hi) from more osteoclast-committed progenitors
(CD27lo). The differentiation of B220- cKit/CD117+ CD115/c-fms+ CD11blo CD27hi to
B220- cKit/CD117+ CD115/c-fms+ CD11blo CD27lo is tightly regulated by the
expression of ligand CD70 on activated immune cells, whereby constitutive engagement
of CD27/CD70 favours the differentiation towards dendritic cells (Xiao et al., 2013).
The same common myeloid precursors differentiate into dendritic cells in the presence
of granulocyte-macrophage-colony-stimulating factor (GM-CSF) and interleukin (IL)-4
through the down-regulation of a c-Fos oncoprotein, a member of the AP-1 transcription
factor complex (Grigoriadis et al., 1994; Miyamoto et al., 2001). In vitro experiments
revealed that GM-CSF inhibits osteoclastogenesis and stimulates dendritic cell
development through the suppression of c-Fos. Comparatively, the expression of c-Fos
inhibits dendritic differentiation, thus indicating that early c-Fos expression acts as a
switch in the lineage determination of osteoclast and dendritic cells (Miyamoto et al.,
2001).
Macrophage differentiation is marked by the transcription of various genes including
F4/80, Mac1 (CD-11b), MOMA-2 and major histocompatibility complex (MHC) class
II molecules. The expression of CD11c and also MHC class II indicate dendritic cell
differentiation. MHC class II are a family of glycoproteins present only on antigen-
presenting immune cells. The functional manifestation of the innate immunity by
antigen presenting cells requires the activation of pattern recognition receptors (PPRs)
and is greatly increased by signalling through the TNFR family member CD40 (Caux et
al., 1994). PPRs are activated by (1) pathogen-associated molecular patterns (PAMPs)

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
46
that are expressed on pathogens, and/or (2) damage-associated molecular patterns
(DAMPs) which are released by apoptotic and/or necrotic cells from surrounding
injured tissues. DAMPs are proposed to activate a subset of PPRs known as Toll-like
receptor (TLRs), which are expressed in both macrophage and dendritic cells, during
sterile inflammation in RA joint (Midwood et al., 2009; Park et al., 2004).
Interestingly, TLR engagement is reported to inhibit the expressions of c-fms and
RANK, thus attenuating osteoclastogenesis and favouring the maturation of APCs (Ji et
al., 2009). The activation of most TLRs leads to the initiation of MyD88-dependent
pathway, which involves the IL-1R-associated kinases (IRAK)-1 and -4, TRAF-6, and
MAPK. It culminates in the activation of AP-1 and NF-κB signalling cascades, which
results in the transcription and production of proteases, chemokines and pro-
inflammatory cytokines such as TNF-α, IL-1, IL-6 and interferons (IFNs), most of
which are capable of expediting osteoclast differentiation (Piccinini et al., 2010).
2.4. Role of inflammatory hematopoietic cytokines as a double-edged sword in
bone modeling and remodeling
Previous studies reveal that the equilibrium between pro- and anti-inflammatory
cytokines determines the fine balance between the osteoclastogenic effects of RANKL
and inhibitory effects of its decoy receptor OPG. Several cytokines involved in the
regulation of inflammation behave as a double-edged sword in modulating bone
remodeling and the progression of inflammatory bone diseases including TNFs,
interleukins, IFNs and TGF-β superfamily members (Table 2.1).
2.4.1. Tumor necrosis factor (TNFs)
Early observations regarding inflammation related bone loss led to the discovery of the
osteoclastogenic potential of tumour necrosis factors (TNFs), which are readily detected
within the RA and sacroiliitis tissue (Bertolini et al., 1986; Di Giovine et al., 1988;
Francois et al., 2006; Thomson et al., 1987). TNF proteins are mainly produced by
macrophages, but also by other lymphoid and non-lymphoid cells. TNFs exert their
biological roles via interaction with their associated membrane receptors, TNF-receptor
type 1 (TNF-R1) and TNF-R2. TNF-R1 is commonly expressed in most tissues and
contains a cytoplasmic interaction domain called death domain (DD), which recruits
other DD-containing proteins and stimulates apoptosis. TNF-R1 is a potent activator of
gene expression via the indirect recruitment of TRAF proteins. Expressed mainly by the

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
47
immune cells, TNF-R2 also plays a major role in the lymphoid system. TNF-R2 directly
recruits TRAF2 and activates NF-ĸB, JNK, and p38-MAPK signalling pathways to
induce gene expression and also crosstalks with TNF-R1 (Wajant et al., 2003).
TNF-α is reported to stimulate the expression of c-fms, the receptor of M-CSF in
osteoclast precursors, thereby supporting the expansion of the cell population (Yao et
al., 2006). The addition of TNF-α per se fails to induce the differentiation of osteoclast
precursors, however the addition of the cytokine enhances osteoclastogenesis of
preosteoclasts primed with minimal level of RANKL that is insufficient to induce
osteoclast differentiation (Lam et al., 2000). Furthermore, the secretion of TNF-α by
neutrophils and activated macrophages in response to injury promotes the expression of
RANKL by osteoblastic and stromal cells to induce osteoclast differentiation and
resorption (Hofbauer et al., 1999; Thomson et al., 1987). In RA, the increased TNF-α
leads to an elevated expression of Dickkopf-related protein 1 (DKK1), a natural
inhibitor of Wnt protein and Wnt/β-catenin pathway that are crucial to activate
osteoblasts and bone formation (Diarra et al., 2007). The pro-osteoclastogenic activity
of TNFs is supported by the ability of the cytokine to inhibit osteoblast differentiation
and induce apoptosis on osteoblastic cells via Fas-ligand (FasL)/Fas signalling
(Bertolini et al., 1986; Centrella et al., 1988; Gilbert et al., 2000; Jilka et al., 1998;
Kawakami et al., 1997).
FasL, a member of TNF superfamily, interacts with its receptor Fas to mediate caspase-
activated apoptosis. Fas-FasL signalling is important in the elimination of cytotoxic
CD8+ T cells and the regulation of self-reactive T cells (Waring et al., 1999). Both
precursor and mature osteoclasts express Fas and FasL. Treatment of M-CSF/RANKL-
treated osteoclast precursors and mature osteoclasts with Fas-acting antibody increased
osteoclast formation and apoptosis respectively (Wu et al., 2003). Mice models with
spontaneous mutation in the coding for FasL and Fas genes display lymphoproliferative
disorder and autoimmunity (Ramsdell et al., 1994; Roths et al., 1984; Watanabe-
Fukunaga et al., 1992). The bone mass of these mice models are reported to be
contradictory, where by one group detailed a decrease in FasL-deficient mice, while
another found it to be increased (Katavic et al., 2003; Wu et al., 2003). The role of
Fas/FasL system in arthritic joint destruction also remained controversial whereby
different groups reported both anti- and proinflammatory roles in mice with CIA

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
48
(Ratkay et al., 1993; Tu-Rapp et al., 2004). These findings however reinforced the
notion that general immune disorder directly or indirectly affects normal bone
homeostasis.
2.4.2. Interleukin (IL)-1
The ability of TNFs to stimulate osteoclast formation is mediated by the acute phase
protein IL-1 that is also detected in the synovial fluid (Fontana et al., 1982; Hopkins et
al., 1988; Wei et al., 2005). In vitro, IL-1 is a potent stimulator of bone resorption
(Gowen et al., 1986). IL-1 is implicated in TNF-mediated systemic bone loss in hTNFtg
mouse model with spontaneous arthritis and the abrogation of IL-1 rescued the animal
from severe bone loss (Ness et al., 1991). The osteoclastogenic role of TNF and IL-1 is
further demonstrated with experiments whereby the administration of their respective
inhibitors into mouse models is shown to inhibit osteoclastogenesis and bone resorption
in vivo (Elliott et al., 1994; Kitazawa et al., 1994; Matsuno et al., 2002; Redlich et al.,
2004).
The expression of IL-1 receptor, type I (IL-1RI) is upregulated by RANKL via c-Fos
and NFATc1. The over-expression and c-Fos-mediated induction of IL-1RI receptor in
murine bone marrow macrophages (BMMs) enables IL-1 alone to induce
osteoclastogenesis via the activation of NF-κB, JNK, p38, and ERK. IL-1/IL-1RI
interaction also strongly activates microphtalmia-associated transcription factor (MITF),
which then upregulates osteoclast-specific genes (Kim et al., 2009). IL-1, along with
TNF, is also implicated in the induction of IL-6 gene expression (Zhang et al., 1990).
2.4.3. IL-6
It has been established that the serum level of IL-6 is elevated in RA and SA patients
(Abdel Meguid et al., 2013; Gratacos et al., 1994; Hirano et al., 1988; Karlson et al.,
2009; Sack et al., 1993). IL-6 is a pro-inflammatory cytokine produced by both immune
and non-immune cells that strongly induce local synovial leukocyte accumulation and
antibody production. Fibroblasts isolated from the synovium of RA patients are capable
of inducing IL-6 expression on endothelial cells in vitro, and aid in the adherence of
neutrophils to the endothelium. Thus, IL-6 has been implicated in the shift from acute to
chronic inflammation by facilitating the transition from neutrophil to mononuclear cell
recruitment, which is a hallmark of acute inflammation (Kaplanski et al., 2003).

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
49
IL-6 exerts its activity via a cell surface receptor protein complex comprising of an IL-6
receptor subunit (IL6R, also referred to as IL-6Rα, gp80 or CD126) and IL-6 signal
transducer glycoprotein (gp) 130 (also denoted as IL-6β or CD130). IL-6R is expressed
mainly in hepatocytes and leukocyte populations (such as monocytes, neutrophils, T
cells and activated B cells), while the latter transmembrane protein molecule gp130 is
expressed ubiquitously (Hibi et al., 1990; Zola et al., 1992). Initially identified as the
signal-transducing component of IL-6R, it is now apparent that gp130 acts as a shared
receptor subunit and signal transducer of other members of the IL-6 family of cytokines,
including oncostatin M (OSM) and leukaemia inhibitory factor (LIF). IL-6, OSM and
LIF have been shown to regulate the development of osteoclasts and bone resorption by
modulating the production of RANKL by osteoblasts and synovial cells, which express
IL-6 receptor (Hashizume et al., 2008; Ishimi et al., 1990; Liu et al., 2005; Palmqvist et
al., 2002). In addition to membrane-bound receptor, a soluble form of IL-6R (sIL-6R) is
found in many body fluids including the synovial fluid (Desgeorges et al., 1997). The
sIL-6R/IL-6 complex is also capable of activating cells via interaction with membrane
bound gp130.
The recognition of IL-6 by its receptor leads to the dimerization of gp130 and triggers
two intracellular signalling cascades: the JAK/STAT pathway, which is mediated by
YxxQ motif of gp130, and MAPK pathway, which is regulated via the cytoplasmic
Y759 residue of gp130 (Abe et al., 2001; Fukada et al., 1996; Heinrich et al., 1998).
F759 mice, which contain a single amino acid substitution in gp130 (Y759F), displayed
enhanced STAT3 activation and develop spontaneous RA-like arthritis (Atsumi et al.,
2002). The translocation of these transcription factors to the nucleus independently
induces the expression of target genes for several physiological processes. For instance,
the JAK/STAT pathway is implicated in controlling immune response and activates
RANKL expression by synovial cells (Hashizume et al., 2008). The MAPK pathway is
reported to induce matrix metalloproteinase (MMP) production by chondrocytes to
enhance proteoglycan degradation in the pathogenesis of osteoarthritis (Hashizume et
al., 2010).
IL-6 is also documented to facilitate the recruitment of osteoclast precursors by
upregulating the expression of sphingosine-1-phosphate (S1P) receptor 2 (S1PR2) on
osteoclast precursors in CIA mouse model. S1P, a bioactive lipid molecule, controls the

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
50
migration of osteoclast precursors and the administration of IL-6 receptor antibody in
the mouse model significantly inhibits inflammatory bone loss (Tanaka et al., 2014). IL-
6 deficient mice display a reduction in synovial infiltration and are resistant to arthritic
induction (Alonzi et al., 1998; Boe et al., 1999; Nowell et al., 2009; Ohshima et al.,
1998). Equivalently, the suppression of immunoinflammatory cascade by targeting IL-6
has been shown ameliorate human chronic inflammatory arthritis (Schoels et al., 2013).
2.4.4. IL-8
A recent study observed the newly describe member of type III IFN family (IFN-λ) IL-
29 is overexpressed in blood and synovium of RA patients and triggers the production
of both IL-6 and IL-8 by RA synovial fibroblasts (Xu et al., 2013). IL-8 is a chemokine
(chemoattractant cytokines) that is capable of promoting the recruitment, proliferation,
and activation of leukocytes, especially neutrophils (Harada et al., 1994). IL-8
production by various types of somatic cells in response to injury, infection and
inflammation is induced by TNF-α and GM-CSF (Kurt-Jones et al., 2002; Perng et al.,
2012; Yasumoto et al., 1992; Yoshida et al., 1997). Indeed the serum level of IL-8 is
elevated in inflammatory diseases such as RA, osteoarthritis and osteomyelitis
(Fullilove et al., 2000; Kaneko et al., 2000; Troughton et al., 1996).
IL-8 is observed to stimulate RANKL production by osteoblastic stromal cells while
directly enhancing osteoclastogenesis and bone resorption to exacerbate bone
destruction common in metastatic bone disease (Bendre et al., 2002; Bendre et al.,
2005; Bendre et al., 2003; Kamalakar et al., 2014). The effect of IL-8 is associated with
the cell surface expression of receptor of IL-8 (CXCR1) on osteoclasts and their
precursors (Bendre et al., 2003). Sequentially, IL-8 production by osteoclasts is
stimulated by RANKL, suggesting autocrine signalling to amplify osteoclastogenesis
(Bendre et al., 2003; Kopesky et al., 2014; Rothe et al., 1998).
Other chemokines that are implicated in bone remodelling include chemokine (C-X-C)
motif ligand 1 (CXCL1, previously known as growth-regulated oncogene (GRO)-α),
CXCL9 (formerly called monokine induced by gamma interferon (MIG)), C-C motive
ligand 2 (CCL-2, previously referred to as monocyte chemoattractant protein (MCP)-1),
CCL3 (previously known as macrophage inflammatory protein (MIP)-1α), CCL4 (MIP-
1β), CCL5 (also known as regulated on activation, normal T cell expressed and secreted
(RANTES)), all of which are reported to be localised within arthritic bone biopsies

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
51
(Borzi et al., 1999; Koch et al., 1994; Koch et al., 1995a; Koch et al., 1995b; Konig et
al., 2000; Lisignoli et al., 2002; Lisignoli et al., 1999).
2.4.5. IL-17
IL-17 is a family of proinflammatory cytokines secreted by a specialized subset of
activated CD4 T cells, called T helper type 17 (TH17) (Korn et al., 2009; Moseley et
al., 2003). The receptor of IL-17, IL-17R is ubiquitously expressed, suggesting the
possibility that IL-17 targets a wide variety of cell types (Lee, 2013). IL-17/IL-17R
interaction mediates the recruitment of TRAF6 and activation of NF-κB and MAPK,
leading to the production of proinflammatory cytokines and subsequent myeloid cell
recruitment during inflammation (Awane et al., 1999; Schwandner et al., 2000; Shalom-
Barak et al., 1998).
The osteoclastogenic potential of IL-17, which was abundantly detected in synovial
fluids of RA patients, was reported by Kotake et al. (1999) to stimulate
osteoclastogenesis in an osteoblast-dependent manner (Kotake et al., 1999). The role of
IL-17 in bone remodeling was supported by numerous in vivo and in vitro studies. One
in vitro study revealed that, in combination with TNF-α, IL-17 stimulates bone
resorption (Van bezooijen et al., 1999). The treatment of experimental arthritic mice
with anti-IL-17 antibody dramatically rescued joint inflammation and prevented
cartilage and bone destruction (Koenders et al., 2005; Lubberts et al., 2004).
Most reports suggest that IL-17 stimulate osteoclastogenesis in a co-culture system,
where by IL- 17 induces RANKL expression in osteoclast-supporting cells, such as
osteoblasts (Huang et al., 2009; Koenders et al., 2005; Kotake et al., 1999; Sato et al.,
2006; Zhang et al., 2011a). However, the direct effect of IL-17 on osteoclast precursors
remained controversial. One study demonstrated the ability of IL-17 to directly induce
osteoclastogenesis in human monocytes (Yago et al., 2009). In contrast, IL-17 inhibited
osteoclast formation from RAW264.7 cells (Kitami et al., 2010). A recent study
reported that in the presence of Vitamin D3 (1,25(OH)(2)D(3)), IL-17 inhibits
osteoclastogenesis in mouse osteoblasts-BMM coculture by stimulating the production
of GM-CSF by osteoblast (Balani et al., 2013). The direct effect of IL-17 on
osteoclastogenesis remained controversial, likely due to the fact that multiple factors,
such as the species, culture conditions and source of cells, vary in the experimental
settings involved in the studies above. Nevertheless, these studies highlight the effect of

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
52
IL-17 in the maintenance of bone homeostasis.
2.4.6. IL-21
IL-21 is a pleiotropic cytokine primarily produced by natural killer T (NKT) cells,
follicular helper T cell (TFH) and TH17 cells (Parrish-Novak et al., 2000; Spolski et al.,
2014). The receptors for IL-21 are IL-21 receptor (IL-21R) 2, IL-21R3 and common
cytokine receptor γ-chain, γc (Asao et al., 2001; Ozaki et al., 2000; Parrish-Novak et
al., 2000). IL-21R is expressed on myeloid cells and other cell types, allowing IL-21 to
exert its effect on other systems (Spolski et al., 2014). IL-21/IL-21R downstream
signalling activates JAK/STAT, MAPK and PI3K pathways (Zeng et al., 2007).
A study implicating IL-21 in bone remodelling and progression of bone destruction in
RA was reported by Kwok et al. (2012). IL-21 level was upregulated in the synovium,
synovial fluid and serum of patients with RA and in serum of mice with CIA. Befitting
to its osteoclastogenic property, IL-21 induced RANKL in CD4 T cells and augments in
vitro osteoclastogenesis (Kwok et al., 2012). Nonetheless, further investigations are
required to support this observation and rescue experiment by administrating IL-21
antibody into CIA animal models may provide an insight in the potency of the
interleukin in exacerbating the progression of inflammatory arthropathies.
2.4.7. Other interleukins
IL-4 and IL-13, produced by TH2, mast cells and basophils, are implicated in
pathological conditions such as asthma and allergy. They share a receptor IL-4Rα to
activate many common signalling pathways, particularly the JAK/STAT pathways
(Jiang et al., 2000; Kelly-Welch et al., 2003). Skeletal analysis of IL-4 and IL-13
knockout mice revealed a reduction in cortical bone mineral content and bone strength,
implicating both IL-4 and IL-13 in the regulation of bone homeostasis (Silfversward et
al., 2007). The depletion of IL-4 and IL-13 does not alter fracture healing in mice,
suggesting that both cytokines may not exert their effects on osteoblastic bone
formation (Silfversward et al., 2008). In vitro investigation demonstrated that IL-4
exerts its function on osteoclast by dose-dependently inhibits RANKL-induced bone
resorption. The anti-osteoclastogenic property of IL-4 affects osteoclast function by
inhibiting NF-κB and Ca2+ signalling, and the formation of actin-ring crucial for
osteoclast migration and resorption (Mangashetti et al., 2005).

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
53
IL-5 was first reported as a T cell-replacing factor (TRF) that plays a role in eosinophils
and B cell differentiation towards immunoglobulin-secreting cells (Adachi et al., 1998;
Takatsu et al., 1988). IL-5 is a member of the haematopoietic growth factor family of
cytokines and has significant functional homology to GM-CSF and IL-3, thus they share
a common β-chain in their receptors. IL-5 interaction with its receptor IL-5R activates
multiple signalling cascades, including the JAK/STAT pathway (Adachi et al., 1998;
Huang et al., 2013; Takatsu et al., 1988). Mice with T cells that over-expressed IL-5
demonstrated heterotrophic ossification within the spleen. The calcified tissue sections
revealed both woven and lamellar bone with higher growth and mineralisation rate
when comparison to the skeletal bone. This study suggests that IL-5 overexpression
mediates bone modelling by mobilising osteogenic progenitors and/or the inhibition of
recruited osteoclasts (Macias et al., 2001). Miyamoto et al. (2001) further showed that
IL-5 does not inhibit osteoclastogenesis (Miyamoto et al., 2001), suggesting that IL-5
may play a regulatory role on osteoblastic cells instead.
Another interleukin implicated in bone remodelling is IL-10. The expression of IL-10
and its receptor are detected in chondrocytes, synovial fibroblasts and mesenchymal
stem cells (Iannone et al., 2001; Ritchlin et al., 2001; Zheng et al., 2008). IL-10
knockout mice display elevated serum proinflammatory cytokines and develop bone
abnormalities with shortened proliferating zone of the growth plates, lower bone
mineral density with reduced bone formation and mechanical fragility when compared
with wildtype (Cohen et al., 2004; Dresner-Pollak et al., 2004; Jung et al., 2013).
Treatment with IL-10 increased tibial lengths in organ culture and induced chondrocyte
proliferation and hypertrophic differentiation in vitro. Molecular study showed that IL-
10 utilise activated STAT-3, Smad 1/5/8 and MAPK pathways to induce the expression
of bone morphogenetic protein (BMP)-2 and 6, indicating that IL-10 is a pro-osteogenic
cytokine (Jung et al., 2013).
IL-10-producing induced osteoblast-like cells generated by Fujioka et al. (2014) was
shown to strongly inhibit osteoclastogenesis from RANKL-stimulated RAW264.7 cells
(Fujioka et al., 2014). Previous studies have demonstrated IL-10 potently inhibits
osteoclastogenesis, potentially acting upon osteoclast progenitors and by suppressing
the expression of NFATc1, c-Fos and c-Jun (Hong et al., 2000; Mohamed et al., 2007;
Xu et al., 1995).

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
54
2.4.8. Interferons (IFNs)
Type I IFNs (IFN-α and IFN-β) are secreted by infected cells and activate surveillance
immune cells such as macrophages and dendritic cells. They share the same receptor
and upon the engagement between type I IFNs and IFN-α receptor (IFNAR), canonical
type I IFN signalling activates JAK/STAT pathway to induce antiviral, antimicrobial
and inflammatory programmes (Ivashkiv et al., 2014; Zhang et al., 2008). In vitro
osteoclast activity assay revealed that IFN-α impairs osteoclast differentiation and bone
resorption (Avnet et al., 2007). Optimal induction of IFN-α requires IFN-β (Deonarain
et al., 2000), and RANKL induces the expression of IFN-β in osteoclast progenitors to
function as an auto-regulator to inhibit osteoclastogenesis by interfering with RANKL-
induced expression of c-Fos. This negative-feedback regulatory mechanism for bone
homeostasis was identified in osteopenic mice deficient in IFN-β signalling
(Takayanagi et al., 2002b).
Early in vitro studies have reported the inhibitory actions of type II IFN-γ on bone
resorption (Gowen et al., 1986; Peterlik et al., 1985). IFN-γ inhibits the RANKL/RANK
signalling cascade by accelerating the degradation of recruited TRAF6 via the
ubiquitin/proteasome system (Takayanagi et al., 2000). Mice lacking a functional IFN-γ
receptor exhibit bone destruction by overzealous osteoclasts and are more susceptible to
CIA (Manoury-Schwartz et al., 1997; Takayanagi et al., 2000; Vermeire et al., 1997).
In vivo administration of IFN-γ to tumour-bearing Tax+IFN-γ-/- mice prevented tumour-
associated bone loss and hypercalcemia, further highlighting the critical role of IFN-ɣ in
maintaining bone integrity (Xu et al., 2009b).
In contrast, in vivo administration of IFN-γ into a Cyclosporin A-induced osteopenic
mouse model failed to rescue the bone phenotype and further increased bone resorption
(Mann et al., 1994). IFN-γ treatment of a mouse model and patients with congenital
osteopetrosis showed an opposite outcome, whereby the therapy resulted in an increased
bone turnover and ameliorate the disease (Key et al., 1995; Rodriguiz et al., 1993).
Such opposing observations could be because the anti-osteoclastogenic activity of IFN-
γ appears to mediate its affects during the bifurcation of myeloid progenitors by
complementing the down regulation of RANK and c-fms by TLRs (Ji et al., 2009).
RANKL on the other hand, induces a broad resistance to cellular effects of IFN-γ
(Huang et al., 2003). Such observation is further validated by Gao et al. who reported

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
55
that direct targeting of IFN-γ on osteoclast precursors blunts osteoclastogenesis, but
stimulates T cell secretion of RANKL and TNF-α to indirectly stimulate osteoclast
formation and promote bone resorption (Gao et al., 2007). Therefore, the fate of
osteoclast precursors relies heavily on the balance of cytokines and is dictated by the
first cytokine they encountered.
2.4.9. Transforming growth factor (TGF)-β
Another cytokine that is abundantly expressed throughout the haematopoietic and bone
system is transforming growth factor-β (TGF-β). TGF-β is secreted by many cell types
and is implicated in many cellular processes such as cellular proliferation and
differentiation, embryonic development, wound healing, haematopoiesis, immune and
inflammatory cell regulation as well as suppression, and also bone development and
remodelling (Centrella et al., 1994). Secreted TGF-β exists in at least three homologous
isoforms: TGF-β1, TGF-β2 and TGF-β3 and their expression levels vary depending on
the tissue (Akhurst et al., 2012; Millan et al., 1991; Schmid et al., 1991). They share a
receptor complex and induce similar signalling pathways to execute different functions
as demonstrated by the distinct and partially overlapping phenotypes between knockout
mice (Dickson et al., 1995; Kulkarni et al., 1993; Millan et al., 1991; Pelton et al.,
1991; Proetzel et al., 1995; Runyan et al., 1992; Sanford et al., 1997; Shull et al., 1992).
TGF-β ligands are synthesised as a large TGF-β complex. The latent complex consists
of mature dimeric TGF-β associated with its latency-associated peptide (LAP) and a
latent TGF-β-binding protein (LTBP). Once activated via proteolytic cleavage, active
TGF-β dimers interact with extracellular domains of TGF-β receptors, which are well-
defined serine-threonine kinases. The specific type II receptors (such as TGF-β receptor
type II (TβRII)) undergo autophosphorylation, which then transphosphorylate type I
receptors (such as TβRI). This event is reported to propagate the specific Sma and Mad
related proteins (SMADs)-dependent canonical pathway and the SMADs-independent
pathways (Derynck et al., 2003).
In the canonical pathway, the phosphorylation of the receptors facilitates the
intracellular SMADs (R-SMADs: SMAD1, SMAD2, SMAD3, SMAD5 and SMAD8)
to form heteromeric complexes with a common mediator SMAD (co-SMAD: SMAD4).
R-SMAD/co-SMAD complexes translocate to the nucleus and preferentially associate
with the genomic SMAD-binding element (SBE). Transcriptional activation by the

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
56
TGF-β/SMAD pathway is regulated by the antagonistic inhibitory SMADs (I-SMADs:
SMAD 6 and SMAD7), and nuclear proteins SKI and SNO. In the non-SMAD
pathways, the phosphorylated tyrosines of the intracellular receptor domains are capable
of activating Erk through-Ras, Raf, and the downstream MAPK cascades to regulate
gene transcription. The activated TGF-β receptor complex is also able to transmit signal
through other factors, such as TRAF4 or TRAF6, TGF-β-activated kinase (TAK) 1,
phosphoinositide 3 kinase (PI3K)-AKT, JNK and NF-κB (Akhurst et al., 2012; Zhang,
2009).
The role of TGF-β in bone homeostasis is demonstrated by mice lacking TGF-β1 gene,
which exhibit decreased bone mass and elasticity (Geiser et al., 1998). TGF-β1 is
important for the induction of bone mesenchymal stem cells to coordinate bone
formation (Tang et al., 2009). Inhibition of TGF-β receptor signalling in transgenic
mice expressing cytoplasmic truncated TβRII leads to decreased remodelling and aged-
dependent increase in trabecular bone mass with lower levels of osteolysis due to
decreased bone resorption by osteoclasts (Filvaroff et al., 1999). In contrast, the
overexpression of osteoblast-specific TGF-β2 in transgenic mice resulted in an
osteoporotic phenotype due to increased osteoblastic matrix deposition, however this
was coupled with defective bone mineralisation and accompanied by increased
osteoclastic bone resorption (Erlebacher et al., 1996). In humans, TGF-β3 serum levels
were highly correlated with serum osteocalcin levels, suggesting a systemic regulation
of the cytokine in bone homeostasis (Hering et al., 2001).
The osteoimmunoregulatory effects of TGF-β are highlighted in the synovial fluid from
RA patients, which contain high levels of both latent and active TGF-β. Additionally,
arthritic synovial tissues isolated from the same patients are reported to express TGF-β
(Brennan et al., 1990). TGF-β1 is critical in the prevention of autoimmunity, as TGF-
β1-knockout and mutant mice succumb to rapid wasting due to excessive inflammatory
response (Kulkarni et al., 1993; Shull et al., 1992). In arthritic rat model, the systemic
administration of TGF-β1 was described to reduced infiltration of immune cells, thus
preventing the development of polyarthritis (Brandes et al., 1991). On the contrary,
human fibroblast-like synoviocytes isolated from RA and OA patients are reported to
produce higher levels of TNF-α, MIP-1, IL-1 and IL-8, all of which are pro-
inflammatory cytokines, upon TGF-β1 treatment (Cheon et al., 2002). Synovial TGF-β1

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
57
serum level is associated with the polymorphisms at the TGF-β1 genes, which may
contribute to the progression of joint destruction (Kim et al., 2004).
Interestingly, the severity of RA and OA in human patients correlates with the
upregulation of TGF-β1 (Pohlers et al., 2007). This appears to be consistent with the
anabolic role of TGF-β1, whereby the progression of arthritis is often marked by, but
not limited to, the formation of osteophytes (bone spurs along the joint margins) due to
hyper-modelling at the damaged cartilaginous area. The dual role of TGF-β is further
confounded by other studies which revealed that TGF-β directly acts on RANKL- and
M-CSF-dependent-BMMs to promote osteoclastogenesis via TRAF6/TAK1 pathway
(Fox et al., 2000; Kaneda et al., 2000; Kim et al., 2005; Yasui et al., 2011), whereas
inhibits osteoclastogenesis by stimulating osteoprotegerin and repressing RANKL
production by bone marrow stromal cells and osteoblasts respectively (Quinn et al.,
2001; Takai et al., 1998). These contrasting effects indicate that TGF-β influence
several aspects of osteoblast and osteoclast biology.
2.5. The orchestration of adaptive immune cells by innate immune cells,
osteoclastogenic RANKL and its antagonist OPG
All of the aforementioned cytokines plays a bridging role in the recruitment,
development, expansion and activation of the more specific adaptive immune response.
Correspondingly, B cell-subsets and T cells mediate the adaptive immune system.
Developmentally, B cells originate from the bone marrow, while T cells are generated
in the thymus where circulating lymphoid progenitor cells from the bone marrow are
recruited to acquire cognate receptors and functional reactivity.
The potent stimulator of osteoclastogenesis RANKL, previously known as the TNF
ligand super family member 11 (TNFSF11), and its receptor RANK were first described
to be expressed on antigen-presenting dendritic cells and to play a role in T cell
expansion (Anderson et al., 1997; Bachmann et al., 1999; Wong et al., 1997b). Deletion
of RANKL or RANK resulted in not only an osteopetrotic bone phenotype, but also the
absence of all peripheral and mesenteric lymph nodes (Dougall et al., 1999; Kong et al.,
1999b). The absence of gross lymph nodes in these mouse models is accompanied by
dramatic decreases in B cell development and number. Paradoxically, mouse model
lacking RANK expression on B cell showed normal B cell development with no defects
in antibody production, class switch recombination and somatic hypermutation (Perlot

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
58
et al., 2012). RANKL-deficient mice also display impaired thymocyte development,
which is not observed in RANK-deficient mice, thus indicating that RANKL regulates
not only osteoclastogenesis, but also early thymocyte development at the stage of pre-T
cell receptor (TCR) expression (Kim et al., 2000; Kong et al., 1999b).
OPG, the decoy receptor of RANKL historically attributed to osteoblasts that inhibits
osteoclastogenesis, is upregulated in B cell and dendritic cell through the activation of
CD40 (Yun et al., 1998). Mature B cells are the source of a large proportion of OPG in
bone marrow, and knockout mice lacking B cells were found to be osteoporotic (Li et
al., 2007b). The role of OPG in the regulation of B cell development and function is
emphasised in OPG-deficient mice, which are also osteoporotic due to overzealous
osteoclastogenesis resulting in a high incidence of fractures (Bucay et al., 1998). The
ability to sustain antigen response is compromised in OPG-deficient mice due to the
perturbed B cell maturation. Functional assays revealed the hyper responsiveness of
pro-B cells in OPG-deficient mice, however with less efficacy in antibody isotype
switch and production, in comparison to those of wild-type control (Yun et al., 2001).
In sum, these studies demonstrate that RANKL/RANK/OPG axis regulates the
development and activation of adaptive immune cells.
A further crosslink between the innate and adaptive immune system is that macrophages
and dendritic cells serve as antigen presenting cells (APCs), which are required to
activate T-dependent responses. After the recognition by TLRs and phagocytosis by
APCs, the antigen is processed by proteolysis and displayed by MHC class II molecules
on the cell surface. The interaction between TCR/CD3 complex with peptide-MHC
complex (pMHC) and co-stimulation of T cell surface CD28 with CD80/CD86 are
required for T cell activation. The continuous co-stimulation of T cell is tightly
regulated by T cell surface molecules such as cytotoxic T-lymphocyte protein 4 (CTLA-
4), which interrupts CD80/CD86-CD28 interactions and provides a feedback signal to
APCs (Walunas et al., 1996). CTLA-4 has been reported to directly inhibit osteoclast
differentiation (Axmann et al., 2008). Such an observation is anticipated since
osteoclast precursors are derived from common progenitors with APCs, and osteoclasts
have been reported to display APC-like features and express cell surface MHC class I
and II, CD80 and CD86 (Li et al., 2010). Interestingly, CD80/CD86 deficient mice
display decreased bone mass due to increased osteoclast numbers. In vitro studies

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
59
further demonstrated that CD80-/CD86- monocytic-derived osteoclasts are resistant to
physiological inhibition by CTLA-4, indicating that the engagement of CD80/CD86 by
CTLA-4 is important during the inhibition of osteoclastogenesis (Bozec et al., 2014).
The recognised general signalling pathways initiated by the dynamic interaction
between TCR and pMHC have been reviewed comprehensively (Au-Yeung et al., 2009;
Cheng et al., 2011; Salmond et al., 2009; Smith-Garvin et al., 2009). Briefly, proximal
downstream signalling cascade of TCR is mediated by the associated CD3 (γ, δ, and ε)
and ζ chains, which contain conserved motifs of the immune-receptor tyrosine-based
activation motif (ITAM) sequences. The tyrosines within these motifs are
phosphorylated by Src family kinases p56lck (Lck) or p59fyn (Fyn) present at the
cytoplasmic tails of CD4 or CD8, co-receptors that co-engaged with the TCR. The
phosphorylation of ITAM leads to the recruitment and activation of Zap70 tyrosine
kinase. Zap70 proceeds to phosphorylate adaptor proteins Linker of Activation of T
cells (LAT) and SH2 domain-containing leukocyte phosphoprotein of 76kDa (SLP-76),
which results in downstream production of inositol 1,4,5 triphosphate (IP3) and
diacylglycerol (DAG) to perform cell signalling. IP3 releases Ca2+ from the
endoplasmic reticulum and the elevation of intracellular Ca2+ activates calcineurin,
calmodulin and the transcription factor NFAT. DAG initiates two major pathways, Ras
and PKC-θ signalling, which triggers MAPK and NF-κB pathways respectively. The
aforementioned signalling cascades eventually lead to the activation and regulation of
transcription factors in activated T cells.
2.6. Delineation of T cell and osteoclastogenic T cell candidates
T cells are a part of the adaptive immunity that can be subdivided based on the
expression of co-receptors, CD4 helper T cells and CD8 cytotoxic T cells. CD4 T cells
assist the maturation of B cells as well as the activation of CD8 T cell and other immune
cells. Activated CD8 T cells terminate virally infected cells and tumour cells through at
least three mechanisms: granular exocytosis of cytotoxic molecules, Fas/FasL-induced
apoptosis and secretion of TNF-α. Distinctively, the generation and differentiation of
CD4 T cells are driven by the interaction between cytokine-secreting APCs that are
delivered to naïve T cells. Activated CD4 T cells can differentiate and trans-
differentiate into TH1, TH2, TH17, Foxp3+ regulatory T cell (Tregs) and TFH. Other
TH cells in the immune system that are still not well characterised include TH9 and

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
60
TH22, which secrete IL-9 and IL-22 respectively.
The secretion of IL-12 and IFN-γ by innate immune cells initiates TH1 responses
against intracellular pathogen. These cytokines activate JAK/STAT pathway to induce
the transcription factor T-bet and turn on the IFN-γ gene. The secretion of IFN-γ in turn
activates macrophages and influences B cell maturation, and amplifies their own
responsiveness to IL-12. Correspondingly, parasitic infections favour the development
of TH2-mediated humoral responses. TH2 targeting extracellular pathogen requires IL-4
to activate STAT4. STAT4 promotes the expression of transcription factor GATA-3,
which stimulates the production of IL4, 5 and 13 to inhibit TH1 polarisation and
macrophage activation while stimulating B cells. TH1 and TH2 regulate each other
whereby IFN-γ inhibits TH2 polarisation and concomitantly IL-4 hinders TH1. The
homeostasis between the populations of these two cell types is critical in preventing
pathological consequences.
TH17 is a T helper subset specialised in the protection against extracellular bacterial
and fungal pathogens. The generation of IL-17 to induce local inflammation is governed
TGF-β and IL-6, which signal through STAT3 to upregulate the expression of RAR-
related orphan receptor gamma (RORγt), the master regulator of this lineage (Ivanov et
al., 2006). TGF-β also induces the Tregs-specific transcription factor Foxp3 via the
phosphorylation of SMAD2/3 proteins and the formation of a complex with SMAD4.
However in the presence of IL-6, TGF-β antagonistically inhibits the generation of
Tregs and induces TH17 cells that are highly pro-inflammatory. Tregs are responsible
for maintaining tolerance to self-antigens to prevent autoimmunity and the reciprocal
proportion of TH17 versus Tregs population is clearly demonstrated to be critical for
mediating self-tolerance and preventing tissue inflammation. An imbalance favouring
TH17 differentiation eventually leads to inflammatory disorders and autoimmune
diseases (Bettelli et al., 2006; Mucida et al., 2007). Autoimmune diseases are therefore
the outcome of the imbalance between Treg and TH17 cells, whereby TGF-β is the key
to the delicate balance between immune activation (immunity) and immune non-
responsiveness (tolerance).
CD4 T cells expressing C-X-C chemokine receptor type 5 (CXCR5) are phenotypically
distinct from TH1 and TH2 cells and are named TFH (Breitfeld et al., 2000; Chtanova
et al., 2004; Schaerli et al., 2000). The differentiation of CD4 T cells towards TFH is

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
61
coordinated by the transcription factor B-cell lymphoma 6 (Bcl6) and is mediated by
STAT3, IL-21, IL-6 and B cell expression of ICOS ligand (ICOSL) (Akiba et al., 2005;
Nurieva et al., 2008; Nurieva et al., 2009; Rasheed et al., 2006; Vogelzang et al., 2008).
TFH express IL-21 to directly regulate B cell proliferation and class switching, and
potentially provide an autocrine action with its concomitant expression of IL-21R
(Chtanova et al., 2004; Nurieva et al., 2008; Rasheed et al., 2006; Vinuesa et al., 2005a;
Vogelzang et al., 2008). The dysregulation of TFH is implicated in many autoimmune
diseases that are partially mediated by autoantibodies, thus highlighting its role in
humoral immunity as professional B cell helper.
Early osteoimmunology studies have shown that activated T cells express RANKL to
directly induce osteoclastogenesis (Horwood et al., 1999; Kong et al., 1999a; Kong et
al., 1999b; Kotake et al., 2001; Weitzmann et al., 2001; Wong et al., 1997b). In this
scenario, TH1 was previously associated with bone destruction, while its antagonist
TH2 was described to attenuate the progression. However, these subsets of activated T
cells also produce cytokines that dampened RANKL-signalling, for example TH1
secrete IFN-γ and TNF, while TH2 produce IL-4, IL-5, IL-10 and IL-13, some of which
are anti-osteoclastogenic (Pappalardo et al., 2013; Takayanagi et al., 2000). Beyond the
TH1/TH2 paradigm, TH17 cells emerged as the osteoclastogenic subset in view of their
pro-inflammatory nature. Indeed the expression of IL-17 uniquely produced by TH17 is
elevated in classic autoimmune diseases such as RA (Kotake et al., 1999; Leipe et al.,
2010; van Hamburg et al., 2011). IL-17 is capable of triggering local inflammation and
the production of inflammatory cytokines. Befitting to its osteoclastogenic descriptions,
TH17 cells express significant amounts of membrane-bound RANKL and do not
produce large amounts of IFN-γ in comparison to TH1 (Sato et al., 2006).
Their role in osteoclastogenesis and the development of arthritis is evident in IL-17-/-
mice, which are protected from CIA (Nakae et al., 2003). Accordingly, local injection
of IL-17 into the knee joint of an arthritis mouse model shows that IL-17
overexpression exacerbates CIA and the neutralisation of IL-17 abates the disease
(Burchill et al., 2003; Lubberts et al., 2002). It is important to note however, that their
osteoclastogenic nature is largely indirect via the influence of IL-17 to stimulate the
production of inflammatory cytokines and RANKL expression on synovial fibroblasts
and mesenchymal cells (Kotake et al., 1999; van Hamburg et al., 2011). Consequently,

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
62
therapeutic preventions to counteract the differentiation and infiltration of TH17 cells
into the inflammatory joint are of potentially great clinical importance. Clinical trials on
pharmaceutical inhibitors of IL-17 and cytokines favouring TH17 cell differentiation
and expansion such as IL-6, IL-23 and TGF-β, have shown promising results in the
treatment of inflammatory systemic bone loss such as RA and AS (D.Baeten et al.,
2011; Genovese et al., 2010; McInnes et al., 2014).
The neutralisation of IL-17 and IL-6 switched off the generation of TH17 leading to
preferential differentiation to Tregs (Nardelli et al., 2004; Nishihara et al., 2007). On
account of the fact that Tregs attenuate the pro-inflammatory behaviours of TH17, the
elimination of Tregs in vivo is shown to aggravate arthritis while the supplementation of
Tregs decreased the severity of the disease (Frey et al., 2005; Morgan et al., 2005). The
anti-catabolic effects of Tregs on bone appeared to be via their aptitude to directly
inhibit osteoclastogenesis (Kim et al., 2007b; Zaiss et al., 2010). The suppression of
osteoclast formation and activity is supported by the engagement CTLA-4 via cell-cell
contact (Zaiss et al., 2007). Nevertheless, it was recently revealed that under arthritic
conditions, the stability of the transcription factor Foxp3, which is indispensable for the
anti-inflammatory function of Tregs, is compromised under arthritic conditions
(Komatsu et al., 2014). Komatsu and colleagues reported that synovial fibroblast-
derived IL-6 mediates the loss of Foxp3 expression in Tregs leading them to undergo
transdifferentiation into TH17, resulting in accumulation of IL-17 in inflamed joints
(Komatsu et al., 2014). The conversion towards TH17 suggests the plasticity of Tregs
cells in contributing to the pathogenesis of RA and hence, emphasising the importance
of inflammatory cytokines as potent stimulators that drive progression of inflammatory
bone diseases.
Similarly, dysregulation of TFH tipping the balance of the immune system towards
favouring inflammation mediates autoimmune pathologies. Circulating TFH-like CD4+
CXCR5+ T cells were recently documented to be elevated in patients with autoimmune
disorders such as RA and SLE. The increased frequency of TFH correlates with high
levels of IL-21, Bcl6 expression, autoantibody titres and tissue damage (Ma et al., 2012;
Simpson et al., 2010; Terrier et al., 2012). Several mouse models have provided
insights into the role of TFH in autoimmunity. San Roque is an ENU-induced mutant
mouse line, which exhibits a spontaneous deregulation of the germinal centres in the

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
63
spleen, and generation of circulating autoantibodies associated with an increased in
TFH and IL-21 secretion (Vinuesa et al., 2005a). The single point mutation in the
Roquin-1 (Rc3h1) gene resulted in SLE-like disease and the abrogation of TFH in the
mutant mice ameliorated the pathology, demonstrating that the dysregulation of TFH
cell function mediates the disease (Vinuesa et al., 2005a). Roquin-1 and its paralog
Roquin-2 appear to be crucial in directing the degradation of inducible T-cell
costimulator (ICOS) mRNA (Pratama et al., 2013). ICOS expression on T cells
provides co-stimulation for T cell responses and is critical for TFH cell survival,
germinal centre reactions and generation of B-cell memory (Akiba et al., 2005;
Bossaller et al., 2006; Dong et al., 2001a; Dong et al., 2001b; Mak et al., 2003). Thus,
the disruption of the repressive activity by the mutation in Roquin gene resulted in
increased ICOS expression and accumulation of lymphocytes (Pratama et al., 2013; Yu
et al., 2007).
Interestingly, microarray analysis performed by Vinuesa and colleague revealed that the
TFH isolated from San Roque mice expressed higher level of RANKL in comparison to
WT TFH (Vinuesa et al., 2005a). These observations raised the possibility of TFH to
emerge as a potential osteoclastogenic T cell subset, by directly or indirectly facilitating
osteoclastogenesis, to expedite systemic bone loss in autoimmune disorders.
2.7. The immunomodulatory role of bone cells in establishing the
haematopoietic stem cell (HSC) niche
The bone organ system is heavily integrated with the lymphatic network, harbouring
haematopoietic progenitors with multilineage potential, and is the site of B cell
development and maturation. Within the bone marrow cavity, bone cells share their
microenvironment with HSCs, thus inevitably participating in haematopoiesis and the
modulation of immune cells. Accumulating evidence indicates that cells of the
osteoblast lineage are important components in the maintenance of the HSC niche
(Calvi et al., 2003; Taichman et al., 1994; Taichman et al., 1996; Visnjic et al., 2004).
Taichman et al. (1994) documented the first direct evidence that human osteoblasts
support basal haematopoiesis by constitutive expression of granulocyte colony-
stimulating factor (G-CSF), GM-CSF, IL-6 and TNF-α (Taichman et al., 1994). HSCs
are present in close proximity to endosteal surfaces where osteoblasts and bone lining
cells reside in vivo, and accordingly, osteoblasts are able to support the maintenance of

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
64
an immature haematopoietic phenotype of bone marrow cells in an in vitro co-culture
system (Taichman et al., 1996). An increase in osteoblastic cells directly boosts the
number of HSCs in vivo via the Notch signalling pathway (Calvi et al., 2003).
Conversely, osteoblast deficiency leads to severe alteration of haematopoiesis, resulting
in the lost of lymphoid, erythroid and myeloid progenitors in transgenic mice with
induced-conditional ablation of the osteoblast lineage (Visnjic et al., 2004).
In addition to their role in HSC niche maintenance, osteoblasts are also documented to
support the microenvironment for sustaining B cells development. In vitro experiment
revealed that bone marrow differentiation towards B cells requires cell-cell contact with
osteoblasts. The selective ablation of osteoblast in transgenic mice severely depleted B
cell lineages, preceding the decline in HSCs (Zhu et al., 2007). Furthermore,
osteoblastic cells express CXCL12 and IL-7, which are crucial factors for B cell
development (Jung et al., 2006; Nagasawa et al., 1996; Zhu et al., 2007). Mice lacking
heterotrimeric G protein α subunit (Gsα), which is required in early osteoblast lineage,
exhibit a B lymphocytopenia due to attenuated IL-7 expression with other mature
hematopoietic lineages being not significantly affected (Wu et al., 2008a).
B cell development in the bone marrow is also modulated by osteoclasts. Osteopetrotic
mice with defective osteoclast bone resorption exhibit a reduction in B cell population.
Zoledronic acid (ZA)-induced osteopetrosis similarly resulted in B lymphocytopenia
within the bone marrow of ZA-treated mice. The aberrant B cell development is the
consequence of decreased expressions of CXCL12 and IL-7 associated with reduced
osteoblastic engagement by inactive osteoclasts (Mansour et al., 2011).
The absence of sclerostin (SOST), a secreted protein expressed by osteocytes, was
recently reported to cause an adverse affects on B cell survival. SOST is well
characterised to induce osteoblast apoptosis and effectively preventing osteoblast
activity and maturation into osteocytes. Mice with the deletions of Sost-coding region
display increased mineralised bone due to increased osteoblast activity, resulting in
reduced bone marrow cavity. Strikingly, the bone marrow of Sost-knockout mice is
depleted of B cells, and this is supplemented by a reduction in CXCL12 (Cain et al.,
2012). Collectively, these observations reinforce the role of bone cells in supporting
normal B cell development, and drastic alteration of the bone microenvironment is
therefore disadvantageous for haematopoiesis.

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
65
2.8. Conclusion
Inflammatory bone loss highlights the abundance of overlapping molecules and
regulatory pathways shared between the bone organ and immune system. It is now
established that there exists a constant dialogue between these systems in both health
and disease. Within the past four decades, the field of osteoimmunology has made
significant advances in enriching our understanding of the interactions between immune
and bone cells within the bone microenvironment. While the regulation of osteoclast
biology has been widely studied and demonstrated to be a promising target in treatment
of inflammatory bone loss, accumulating evidence shows that to tackle inflammatory
bone diseases requires more than just targeting the RANKL/RANK/OPG pathway. The
use of genetically modified mice will greatly facilitate our understanding of the
molecular basis regulating cell lineage determination and interactions of immune and
bone cells. It is hoped that better understanding leads to the development of strategic
and efficient therapies to prevent and reverse human diseases that affect both systems.

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
66
Figure 2.1 Lineage bifurcation model between dendritic cells, macrophages and
osteoclasts. Differentiation between macrophages, osteoclasts and dendritic cells from
common myeloid progenitors (Lin- CD117+ CD11b+ CD115+ cells). The differentiation
of macrophages osteoclasts is supported by M-CSF. RANKL is required for
osteoclastogenesis, which is inhibited by GM-CSF through reduction of c-Fos
expression. In contrast, dendritic cell maturation induced by GM-CSF and is inhibited
by the expression of c-Fos or M-CSF.

Chapter 2: Osteoimmunology – Interactions of the Skeletal and Immune Systems
67
Osteoclast
Osteoblast/
Stromal Cells T cells
IFNs Osteoclastogenesis
Bone resorption
RANKL
TH1 expansion
TH2 expansion
IL-1 Osteoclastogenesis
Resorption
IL-10 Osteoclastogenesis BMP2
IL-17 Resorption RANKL
IL-21 ? ? RANKL
TH17 expansion
IL-4/
IL-13
Osteoclastogenesis
Actin-ring formation
Resorption
= TH2 expansion
IL-5 = Bone formation
IL-6 Recruitment of
osteoclast precursors RANKL
Foxp3
Tregs expansion
TH17 and TFH
expansion
IL-8 Osteoclastogenesis
Resorption RANKL
TGF-β Osteoclastogenesis
Bone formation
OPG
RANKL
Foxp3
Tregs expansion
TNFs c-fms expression
Osteoclastogenesis
RANKL
DKK1
Wnt/β-catenin
Apoptosis
Table 2.1 The effect of cytokines in modulating the bone and immune system.
Cytokines highlighted above are involved in the regulation of immune system and
behave as a double-edged sword to modulate bone remodelling. Whether IL-21 directly
regulates osteoclastogenesis and/or osteoblastogenesis remained to be elucidated.

CHAPTER THREE
HYPOTHESIS AND AIMS
SANROQUE MUTANT MOUSE AS AN OSTEOIMMUNE
MOUSE MODEL

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
69
3. CHAPTER 3: HYPOTHESIS AND AIMS – SANROQUE MUTANT MOUSE AS AN OSTEOIMMUNE MOUSE MODEL
3.1. General hypothesis and aims
The bone organ tissue is continuously being broken down and rebuilt in a dynamic
process referred to as bone remodelling. The remodelling process is the coupled activity
between the catabolic effects of bone resorbing osteoclasts and the anabolic effects of
bone forming osteoblasts. The bone remodelling cycle begins when there is an external
stimulus, which initiates osteoblasts to recruit osteoclast precursors by expressing
RANKL. RANKL binds onto the cell surface receptor RANK displayed on osteoclast
precursors to drive osteoclastogenesis and cell fusion, forming multinucleated
osteoclasts. Once osteoclasts mature, they begin to secrete proteolytic enzymes such as
cathepsin K to degrade the bone matrix. To prevent osteoclasts from continuously
resorbing bone, osteoblasts also secrete reversal cues, such as osteoprotegerin that acts
as a decoy receptor by binding onto RANKL. Such cues allow osteoblasts to modulate
the bone remodelling cycle to favour bone formation.
The coupled activity between these two cell types is tightly regulated to achieve a
micro-architecturally strong bone. The dysregulation of the homeostasis within this
bone microenvironment eventually leads to bone diseases. However, the underlying
molecular and cellular mechanisms regulating the state of homeostasis remains poorly
resolved. Understanding this homeostatic mechanism is further complicated by the fact
that the bone microenvironment is supported by, and supports, many other systems and
cell types. Immune cells, which are hematopoietically generated within the marrow
cavity, inevitably infiltrate into the microenvironment and interact with bone cells and
vice versa. The co-regulation and dialogue between these two systems is termed
osteoimmunology, and is highlighted in many inflammatory diseases such as
rheumatoid arthritis, spondylarthritis, periodontal disease, osteomyelitis and lupus.
All of these aforementioned osteoimmune diseases caused significant discomfort and
dilapidating deformities. However, the mechanism(s) by which these conditions occur is
still largely unknown. Therefore, the underlying hypothesis for this project is that there
are factors implicated in regulating the cross talk between the bone organ and immune
system that are yet to be identified. Based on this hypothesis, the aim of this study was
to identify novel factors that play a significant role in the maintenance and regulation of
the osteoimmune system through a chemically induced (ENU) mutagenesis phenotypic
screen.

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
70
3.2. Screening of ENU-induced mutant mouse lines: the sanroque mutation
Systematic screening of the ENU-mutagenesis library led us to identify a mouse line
named sanroque, which carried a single point M199R mutation in the Roquin (Rc3h1)
gene (Figure 3.1). The T to G substitution in the mutant mice resulted in a non-
conservative Methionine to Arginine codon change in mRNA transcript. The consensus
of the encoded Roquin protein conforms to the structure of RING-type E3 ubiquitin
ligase family, which is one of the enzymatic components that drive the ubiquitin
proteasome system (UPS).
3.2.1. The ubiquitin proteasome system (UPS) and Roquin E3 ligase
The main function of the UPS is the ubiquitination (also referred to as ubiquitylation) of
molecular products for the purpose of proteolysis to maintain protein homeostasis. The
ubiquitination process marks the protein of interest and signals the proteasome to
degrade and recycle the protein. Besides protein degradation, ubiquitination is also
reported to be involved in transcription factor activation, DNA repair, translational
control, kinase activation, histone activity and endocytosis (Pickart et al., 2004). The
UPS is an energy-dependent enzymatic process driven by three enzymes: E1 ubiquitin
activating enzyme, E2 ubiquitin conjugating and E3 ubiquitin ligase (Figure 3.2).
E1, along with ATP, is responsible for the activation of ubiquitin to initiate the
ubiquitination process. E1 enzyme then transfers the activated ubiquitin to E2. E2 acts
as a platform and forms a complex with an E3. E3 recognises both E2 and the protein
that needs to be tagged and catalyses the transfer of activated ubiquitin onto the protein.
E3s are therefore primarily responsible for conferring specificity to ubiquitination and
can be defined by one of the three domains: Homologous to E6-AP Carboxyl Terminus
(HECT), Really Interesting New Gene (RING) and RING-related domains, and A20-
type zinc finger. Roquin E3 ligase is characterised to have the RING domain (Figure
3.1).
The dysfunction of the UPS enzymes, especially E3s, has been implicated in the
dysregulation of biological activities leading to diseases such as, but not limited to,
Parkinson’s, leprosy, osteosarcoma and autoimmune diseases (Ardley et al., 2004;
Cohen, 2014; Severe et al., 2013). UPS and E3s also play a major role in bone
homeostasis and in the development of bone cells (Shu et al., 2013; Zhang et al.,
2013a).

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
71
3.2.2. Sanroque mutant mice exhibits low bone mass phenotype
While there was no apparent abnormality in the gross body phenotype of the mutant
mice in comparison to the wildtype littermates (Figure 3.3 A), X-ray imaging revealed
that the hindlimbs of sanroque mice have reduced bone mass in comparison to wildtype
hindlimbs. The reduction in bone mass of the sanroque mice was consistently observed
in the vertebrae and tail, indicating systemic bone loss when compared to wildtype
controls (Figure 3.3 B – D).
3.2.3. Sanroque mice display a lupus-like autoimmune disease
Vinuesa and colleagues (2005) were the first to characterise the sanroque mutant mouse
line, which develop high titres of autoantibodies and displayed a severe autoimmune
disease consistent to Systemic Lupus Erythematosus (SLE). The mutation in the Roquin
gene led to the dysregulation of T cell maturation, causing the formation of excessive
numbers of follicular helper T cells (TFH) and germinal centres. Consequently, the
sanroque mutant mouse line exhibited enlarged lymph nodes and spleens (Figure 3.4).
The loss of function mutation disrupted a repressor of ICOS, which is a crucial co-
stimulator receptor for TFH, and resulted in the excessive production of the cytokine
interleukin-21. Roquin gene thus plays a regulatory role in preventing self-reactive T
cells from providing assistance to B cell maturation and production of self-reactive
antibodies.
3.3. Specific hypothesis and aims
In view of the susceptibility of sanroque mice to SLE-like autoimmunity, it is
worthwhile to mention that the effect of SLE disease per se contributes to systemic bone
loss and the deterioration of the patients’ bone mineral density (BMD), microstructure
and strength (Tang et al., 2013a). Consistent with this report, preliminary X-ray
screening revealed the low bone mass phenotype of the sanroque autoimmune mouse
line when compared to their wildtype counterparts. However, there is also a possibility
whereby the autoimmunity is developed due to a compromised bone microenvironment
that consequently resulted in the abnormal development of myeloid cell lineages. Which
of these disease development models are correct remains to be answered.
Interestingly, microarray analysis performed by Vinuesa and colleague revealed that the
TFH isolated from sanroque mice expressed higher level of RANKL in comparison to

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
72
wildtype TFH (Vinuesa et al., 2005a). These observations raised the possibility of TFH
to emerge as a potential osteoclastogenic T cell subset, by directly or indirectly
facilitating osteoclastogenesis, to expedite systemic bone loss in autoimmune disorders.
Hence we hypothesise that Roquin is an important regulator of bone homeostasis and
sanroque mice emerge as a useful model to gain osteoimmunological insight into
systemic bone loss in autoimmune diseases. The specific aims of this project are:
1. To characterise the bone phenotype of the sanroque mutant mice.
2. To elucidate the pathology of systemic bone loss by understanding the cellular
and molecular mechanisms by which Roquin regulates bone homeostasis.

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
73
Figure 3.1 Mapping of missense mutation in the conserved Rc3h1gene. (A) The T to
G substitution in roquin mRNA resulted in a non-conservative Methionine to Arginine
codon change (M199R) of the encoded protein. (B) The primary structure of Roquin
illustrating the M199R in the conserved ROQ domain. (C) The RING finger domain of
Roquin aligned perfectly with other RING-type E3 ubiquitin ligases. Figures are
adapted from Vinuesa et al., 2005.
(B)
(C)
(A)
Wildtype
sanroqueENU-induced mutation
Wildtype
sanroque

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
74
Figure 3.2 The basic steps of substrate modification by the ubiquitin proteasome
system. E1 activates ubiquitin and transfers the activated ubiquitin to E2. E2 serves as a
platform to facilitate the ligation of ubiquitin and substrate by E3. The ubiquitinated
substrate is now marked and recognised by the proteasome for degradation. The
degraded products can be recycled for substrate synthesis.

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
75
Figure 3.3 Mutant sanroque mice appeared normal but exhibited low bone
phenotype. (A) Phenotypic gross body comparison of 99 day old male wildtype and
sanroque mutant mice. The mutant mice appeared normal in comparison to wildtype
littermates. X-ray images of (B) hindlimbs, (C) vertebrae and (D) tails from 100 day old
male wildtype and sanroque mutant mice highlight the decreased bone mass of
sanroque mice.
(A)
(B)
A)
Wild
type
sa
nroq
ue
Wildtype Wildtype sanroque
Wildtype Wildtype sanroque sanroque(C)
(D)

Chapter 3: Hypothesis and Aims – Sanroque Mutant Mouse as an Osteoimmune Model
76
Figure 3.4 Mutant sanroque mice displayed lymphomegaly and splenomegaly. (A)
Post-mortem dissection revealed grotesquely enlarged spleens and (B) lymph nodes of
the mutant mice. Image (B) was taken from Vinuesa et al., 2005.
(A) (B) A)
Wild
type
sa
nroq
ue
sanroque

CHAPTER FOUR
MATERIALS AND METHODS

Chapter 4: Materials and Methods
78
4. CHAPTER 4: MATERIALS AND METHODS
4.1. Materials
4.1.1. Chemical reagents
All chemical reagents involved in all experiments conducted in this thesis were of
analytical grade unless indicated otherwise and were purchased from the following
manufacturers:
Description Manufacturer
1α, 25-dihydroxyvitamin D3 Sigma-Aldrich, USA
2-Ethoxyethanol Sigma-Aldrich, USA
2-Mercaptoethanol 14.3M Sigma-Aldrich, USA
30% Acrylamide/Bis solution Bio-Rad, USA
Acetone Scot Scientific, Australia
Agarose powder Promega Corp., Madison,
Wi., USA
Alizarin Red S Monohydrage MP Biochemicals Inc, USA
Ammonium Persulfate (APS) Sigma-Aldrich, USA
Baxter water Baxter Healthcare Pty Ltd,
Australia
Bovine Serum Albumin Sigma-Aldrich, USA
Bromophenol Blue Sigma-Aldrich, USA
Chloroform ThermoFisher Scientific,
Australia
ClearMount Mounting Solution Invitrogen, Australia
Dexamethasone Sigma-Aldrich, USA
Dimethyl Sulphoxide (DMSO) BDH Laboratory Supplies,
England
Distyrene Plasticizer Xylene (DPX) Sigma-Aldrich, USA
Eosin Y disodium salt Sigma-Aldrich, USA
Ethanol (100%) BDH Laboratory Supplies,
England
Ethidium bromide (2,7-diamino-10-ethyl-9-
phenylphenanthridium bromide;
homidium bromide)
Sigma Chemical Co., USA

Chapter 4: Materials and Methods
79
Ethylenediamine tetraacetic acid (EDTA) BDH Laboratory Supplies,
England
Formaldehyde Sigma-Aldrich, USA
Glacial acetic acid BDH Laboratory Supplies,
England
Glutaraldehyde solution Fisher Scientific, USA
Glycerol BDH Laboratory Supplies,
England
Glycine Sigma-Aldrich, USA
Goat Serum Sigma-Aldrich, USA
Haematoxylin Sigma-Aldrich, USA
Hydrochloric acid BDH Laboratory Supplies,
England
IPTG (Isopropyl-β-thiogalactopyranoside) dioxane free Promega Corp., Madison,
Wi., USA
Isopropanol ThermoFisher Scientific,
Australia
L-ascorbic acid Sigma-Aldrich, USA
Liquid nitrogen (N2) Air Liquid, Australia
Methanol BDH Laboratory Supplies,
England
Methoxyethyl acetate (MEA) Sigma-Aldrich, USA
Methyl methacrylate (MMA) Sigma-Aldrich, USA
Mono-sodium phosphate (NaH2PO4) Sigma-Aldrich, USA
Naphthol AS-MX phosphate Sigma-Aldrich, USA
Paraformaldehyde Sigma-Aldrich, USA
PMSF (Phenylmethylsulfonyl fluoride) Roche Diagnostic,
Germany
Prolong Gold Antifade reagent Invitrogen, Australia
Propan-2-ol (Isopropanol) BDH Laboratory Supplies,
England
Protease Inhibitor Cocktail Roche Diagnostic,
Germany

Chapter 4: Materials and Methods
80
Skim Milk Powder Standard supermarket
brand
Sodium acetate (CH3COONa) Sigma-Aldrich, USA
Sodium chloride (NaCl) BDH Laboratory Supplies,
England
Sodium deoxycholate (C24H39NaO4) BDH Laboratory Supplies,
England
Sodium dodecyl sulphate (SDS) BDH Laboratory Supplies,
England
Sodium hydroxide (NaOH) BDH Laboratory Supplies,
England
Sodium Iodate (NaIO3) Sigma-Aldrich, USA
Sodium Orthovanadate Sigma-Aldrich, USA
Sodium phosphate dibasic (Na2HPO4) Sigma-Aldrich, USA
Sodium phosphate monobasic (NaH2PO4) Sigma-Aldrich, USA
Sodium tartrate dihydrate (C4H4Na2O6·2H2O) AJAX chemicals, Australia
TEMED (N. N, N', N'-tetra-methyl-ethylenediamine) Bio-Rad, USA
Tissue-Tek® OCT compound for cryostat sectioning Sakura Finetek Inc, Usa
Toluidine Blue Sigma-Aldrich, USA
Triton X-100 Sigma-Aldrich, USA
Trizma Base, Tris Sigma-Aldrich, USA
Trizma Hydrochloride Sigma-Aldrich, USA
Tween-20 MP-Biochemicals, France
Xylene Hurst, Scientific, Australia
β-glycerophosphate Sigma-Aldrich, USA
β-mercaptoethanol Sigma Chemical Co., USA
4.1.2. Commercially purchased kits and molecular products
Description Manufacturer
1 kb DNA ladder Promega Corp., Madison,
Wi., USA
100 bp DNA ladder Promega Corp., Madison,

Chapter 4: Materials and Methods
81
Wi., USA
10X Reaction Buffer, 1 ml of 200mM Tris-HCl, pH 8.3,
20mM MgCl2
Sigma-Aldrich, USA
5X First Strand Buffer Invitrogen, USA
7-AAD BD Biosciences, USA
Amplification Grade DNAse I (1 u/μl) Sigma-Aldrich, USA
Anti-Mouse Ig κ/Negative Control (FBS) Compensation
Particles Set (#552843)
BD Biosciences, USA
Anti-Rat and Anti-Hamster Ig κ/Negative Control
Compensation Particles Set (#552845)
BD Biosciences, USA
BD Pharm LyseTM red cell lysis buffer (10X) BD Biosciences, USA
BD Stabilizing Fixative 3X Concentrate (#338036) BD Biosciences, USA
Bio-Rad Protein Assay Bio-Rad, USA
Blue/Orange Loading Dye, 6X Promega Corp., Madison,
Wi., USA
CellTiter 96® AQueous One Solution Cell Proliferation
Assay
Promega Corp., Madison,
Wi., USA
DAPI (4', 6-diamidino-2-phenylindole), dilactate Biotium, USA
DNase I Stop Solution Kit, Amplification Grade Sigma-Aldrich, USA
dNTPs – dATP, dTTP, dCTP and dGTP nucleotides Promega Corp., Madison,
Wi., USA
Goat serum (for immunofluorescence, blocking) Sigma-Aldrich, USA
GoTaq® Polymerase Green Master Mix Promega Corp., Madison,
Wi., USA
GoTaq® Real-Time qPCR Master Mix Promega Corp., Madison,
Wi., USA
LIVE/DEAD® Fixable Aqua Dead Cell Stain Kits
(L34957)
Invitrogen, USA
MMLV Reverse Transcriptase 10000u (200u/μl) Promega Corp., Madison,
Wi., USA
MMLV-RT (Moloney Murine Leukaemia Virus Reverse
Transcriptase)5X Buffer
Promega Corp., Madison,
Wi., USA
Oligo dT Sigma-Aldrich, USA

Chapter 4: Materials and Methods
82
RNAsin® RNAse Inhibitior Promega Corp., Madison,
Wi., USA
RNEasy Mini Kit QIAGEN Gmbh, Australia
TRIZOL® Invitrogen, USA
Western LightningTM Ultra Chemiluminescence Substrate
Kit
PerkinElmer, USA
Zombie UVTM Fixable Viability Kit (#423107) BioLegend, USA
4.1.3. Other products and consumables
Description Manufacturer
Acrodisc Syringe Filters with Supor Membrane, sterile –
0.2 μm, 0.8 μm
Pall Corp, USA
BD 1 mL Syringe Luer-LokTM Tip – sterile BD Biosciences, USA
BD PrecisionGlideTM Needles 25G×11/2" BD Biosciences, USA
Coverslips – round 5 mM, 22×22 mm2 and 22×40 mm2 Knittle Glaser, Germany
Carbon Steel Surgical Blade – sterile Swann Morton LTD, UK
Filter Paper Whatmans International,
UK
Glass Slides – Star Frost Knittle Glaser, Germany
HyBondTM-C 45 Micron Nitrocellulose Membrane Amersham Biosciences,
UK
iCycler iQTM Optical Tape Bio-Rad, USA
iCycler iQTM PCR Plates, 96 wells and 384 wells Bio-Rad, USA
Parafilm Laboratory Film American National
CanTM, USA
Petri Dishes Sarstedt, Germany
PCR Tubes: 0.2 ml thin-walled tubes Unimed Australia Pty Ltd,
Australia
Transfer pipettes Sarstedt, Germany
50 ml centrifuge tubes BD Biosciences, USA
Cling Film Glad, AUS
Diamond Wafering Blade, high concentration (for Buehler, USA

Chapter 4: Materials and Methods
83
isoMet® Low Speed Saw)
Centrifuge Tubes – sterile, 0.5 ml, 1.5 ml and 2.0 ml Sarstedt, Germany
4.1.4. Tissue culture materials and reagents
Description Manufacturer
1α, 25-dihydroxyvitamin D3 Sigma-Aldrich, USA
Biocoat Collagen I cellware – 6 well plate Beckton Dickson Labware,
USA
Cell Scrapers Sarstedt, Germany
Collagenase Type II Sigma-Aldrich, USA
Cryogenic vials (2 ml) Sarstedt, Germany
D-MEM (Dulbecco's-Modification of Eagle's Medium)-
GlutaMaxTM-1
Gibco, Life Technologies,
Australia
Falcon Cell Strainer – 100 μm BD Biosciences, USA
FBS (Foetal Bovine Serum) Gibco, Life Technologies,
Australia
Hanks' Balanced Salt Solution Gibco, Life Technologies,
Australia
L-Glutamax Gibco, Life Technologies,
Australia
Mr. FrostyTM Freezing Container Thermofisher, USA
Penicillin-Streptomycin Gibco, Life Technologies,
Australia
Serological pipettes – 5 ml, 10 ml, 25 ml Sarstedt, Germany
Tissue Culture Flask – 25 cm2, 75 cm2 Nunc, Denmark
Tissue Culture Plate – 6 wells, 12 wells, 24 wells, 96
wells
Sarstedt, Germany
TrypLETM Express Cell Dissociation Reagent (trypsin
replacement)
Gibco, Life Technologies,
Australia
α-MEM (α-Modification of Eagle's Medium) Gibco, Life Technologies,
Australia

Chapter 4: Materials and Methods
84
4.1.5. Cytokines
Description Manufacturer
Murine M-CSF (Macrophage Colony
Stimulating Factor
Generated by Cell Signalling Group,
UWA School of Pathology and Laboratory
Medicine using CMG14-12 cells
conditioned-media as previously described
(Takeshita et al., 2000) (Takeshita et al.,
2000).
Murine RANKL (Receptor Activator of
Nuclear factor Kappa-B Ligand)-GST
Produced by Cell Signalling Group, UWA
School of Pathology and Laboratory
Medicine as previously described (Xu et
al., 2000) (Xu et al., 2000).
4.1.6. Antibodies
Antibody/Probe Manufacturer Dilution
AlexaFluor®700 Rat Anti-Mouse CD8a BioLegend, USA 1 in 200 (FC)
Anti-Mouse IgG Peroxidase Conjugate
(A9917)
Sigma-Aldrich, USA 1 in 5000 (WB
2° Ab)
Anti-Mouse IκB-α (C-21) Santa Cruz
Biotechnology, USA
1 in 1000 (WB
1° Ab)
Anti-Mouse NFaTc1 (#556602) BD Biosciences, USA 1 in 1000 (WB
1° Ab)
Anti-Mouse p38 (#9212) Cell Signalling, USA 1 in 1000 (WB
1° Ab)
Anti-Mouse Phospho-ERK (E-4) Santa Cruz
Biotechnology, USA
1 in 1000 (WB
1° Ab)
Anti-Mouse Phospho-JNK (#92521) Cell Signalling, USA 1 in 1000 (WB
1° Ab)
Anti-Mouse Phospho-p38 (#9211) Cell Signalling, USA 1 in 1000 (WB
1° Ab)
Anti-Mouse SAPK/JNK (#9252) Cell Signalling, USA 1 in 1000 (WB
1° Ab)

Chapter 4: Materials and Methods
85
Anti-Mouse β-actin (JLA20) Calbiochem (EMD
Millipore), USA
1 in 5000 (WB
1° Ab)
Anti-Rabbit IgG Peroxidase Conjugate
(A0545)
Sigma-Aldrich, USA 1 in 5000 (WB
2° Ab)
APC Rat Anti-Mouse CD115, CSF-1R
(#135509)
BioLegend, USA 1 in 200 (FC)
APC Rat Anti-Mouse CD11b (#17-0112) eBioscience, USA 1 in 100 (FC)
APC Rat Anti-Mouse CXCR5 (#560615) BD Biosciences, USA 1 in 100 (IF), 1
in 25 (FC)
APC-H7 Rat Anti-Mouse CD4 (#560181) BD Biosciences, USA 1 in 100 (FC)
Brilliant VioletTM 421 Rat-Anti Mouse
CD117, c-Kit (#562609)
BD Biosciences, USA 1 in 100 (FC)
BUV Rat Anti-Mouse CD45R/B220
(#563793)
BD Biosciences, USA 1 in 100 (FC)
BV421 Rat Anti-Mouse CD70 (#562908) BD Biosciences, USA 1 in 100 (FC)
BV510 Rat Anti-Mouse CD11b
(#562950)
BD Biosciences, USA 1 in 200 (FC)
BV650 Rat Anti-Mouse CD11b
(#563402)
BD Biosciences, USA 1 in 200 (FC)
BV711 Rat Anti-Mouse CD117, c-Kit
(#563160)
BD Biosciences, USA 1 in 200 (FC)
FITC Hamster Anti-Mouse CD3ε
(#561827)
BD Biosciences, USA 1 in 2000 (FC)
FITC Rat Anti-Mouse CD45R/B220
(#553087)
BD Biosciences, USA 1 in 1000 (FC)
FITC Rat Anti-Mouse CD62L (#553150) BD Biosciences, USA 1 in 100 (FC)
FITC Rat Anti-Mouse Ly-6A/E, Sca-1
(#122505)
BioLegend, USA 1 in 1000 (FC)
Goat Anti-Rabbit FITC conjugate Life Technologies,
USA
1 in 50 (IF)
Goat Anti-Rat DyLight 549 ThermoFisher
Scientific, Australia
1 in 200 (IF)
PE Hamster Anti-Mouse CD279 BD Biosciences, USA 1 in 100 (FC)

Chapter 4: Materials and Methods
86
(#551892)
PE Rat Anti-Mouse CD265, RANK
(#119805)
BioLegend, USA 1 in 500 (FC)
PE Rat Anti-Mouse CD265, RANK (#12-
6612)
eBioscience, USA 1 in 500 (FC)
PE Rat Anti-Mouse CD279, PD-1
(#109103)
BioLegend, USA 1 in 100 (FC)
PE-Cy7 Hamster Anti-Mouse CD3ε (#25-
0031)
eBioscience, USA 1 in 2000 (FC)
PE-Cy7 Hamster Anti-Mouse/Rat/Human
CD27 (#124215)
BioLegend, USA 1 in 100 (FC)
PE-Cy7 Rat Anti-Mouse CD25 (#25-
0251)
eBioscience, USA 1 in 100 (FC)
PE-Cy7 Rat AntiMouse/Human
CD45R/B220 (#25-0452)
eBioscience, USA 1 in 100 (FC)
PE-CyTM7 Hamster Anti-Mouse CD3ε
(#552774)
BD Biosciences, USA 1 in 2000 (FC)
PE-DazzleTM594 Hamster Anti-Mouse
CD11c (#117347)
BioLegend, USA 1 in 100 (FC)
PerCP-CyTM5.5 Rat Anti-Mouse CD4
(#561115)
BD Biosciences, USA 1 in 100 (FC)
PerCP-CyTM5.5 Rat Anti-Mouse CD44
(#560570)
BD Biosciences, USA 1 in 200 (FC)
Rabbit Anti-Mouse RANKL Santa Cruz
Biotechnology, USA
1 in 500 (IF)
Rat Anti-Mouse CD4 Life Technologies,
USA
1 in 100 (IF)
V450 Rat Anti-Mouse CD11b (#560455) BD Biosciences, USA 1 in 100 (FC)
V450 Rat Anti-Mouse CD25 (#561257) BD Biosciences, USA 1 in 100 (FC)
4.1.7. Oligonucleotide primers
Oligonucleotide primer sequences for PCR and Real-Time PCR amplifications were
designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) which

Chapter 4: Materials and Methods
87
was developed by National Center for Biotechnology Information (NCBI) based on
Primer3 (http://primer3.ut.ee/), and screened through NCBI-BLAST to ensure
specificity (Ye et al., 2012). The primers were purchased from Geneworks, Australia
and were shipped in freeze-dried form. The primers were then reconstituted in sterile
and nuclease-free ddH2O at a final concentration of 100 µM according to
manufacturer’s instructions. Aliquots of resuspended oligos were stored at -20 °C and
placed on ice during usage.
Primers Sequence (5' - 3') Amplicon size (bp)
Tumor necrosis factor receptor
superfamily, member 11a,
NFKB activator RANK
(NM_009399.3)
For: AGC TCA GCA TCC
CTT GCA G
Rev: GGA AGA GCT GCA
GAC CAC AT
178
Tumor necrosis factor (ligand)
superfamily, member 11
Tnfsf11, RANKL
(NM_011613.3)
For: TCT GTT CCT GTA
CTT TCG AGC GCA G
Rev: TGT TGC AGT TCC
TTC TGC ACG GC
198
Tartrate resistant acid
phosphatase 5
TRAP (NM_001102404.1)
For: CAG CAG CCA AGG
AGG ACT AC
Rev: ACA TAG CCC ACA
CCG TTC TC
190
SRY (sex determining region Y)-
box 9
Sox9 (NM_011448.4)
For: AAG CTC TGG AGG
CTG CTG AAC G
Rev: TTC GGC CTC CGC
TTG TCC GT
150
Secreted phosphoprotein,
osteopontin
SPP1, OPn (NM_001204201.1)
For: TCT GAT GAG ACC
GTC ACT GC
Rev: TCT CCT GGC TCT
CTT TGG AA
349
Sclerostin
Sost (NM_024449.5)
For: CAG ACC ATG AAC
CGG GCG GAG
Rev: CAC TGG CCG GAG
CAC ACC AAC
167

Chapter 4: Materials and Methods
88
Rn18s
(NR_003278.3)
For: ACC ATA AAC GAT
GCC GAC T
Rev: TGT CAA TCC TGT
CCG TGT C
216
RING CCCH (C3H) domains 1
Rc3h1 (NM_001024952.2)
For: TAC CAT CGG CTC
CTA CAT TGG
Rev: GTC CAA CCT CTG
CTC TCT CG
115
Osterix, Sp7 transcription factor
Osx (NM_130458.3)
For: TTC CCT CAC TCA
TTT CCT GG
Rev: GTA GGG AGC TGG
GTT AAG GG
348
Osteoprotegerin
OPG (NM_011613.3)
For: GCC GTG CAG AAG
GAA CTG CAA C
Rev: GGT GTG CAA ATG
GCT GGG CCT
127
Osteocalcin
Bglap, OCn (NM_001037939.2)
For: GCG CTC TGT CTC
TCT GAC CT
Rev: ATA GAT GCG TTT
GTA GGC GG
440
Nuclear factor of activated T
cells, cytoplasmic,
calcineurin dependent 1 NFATc1
(NM_001164109.1)
For: CAA CGC CCT GAC
CAC CGA TAG
Rev: GGC TGC CTT CCG
TCT CAT AGT
392
Dentrocyte expressed seven
transmembrane protein
DC-Stamp (NM_001289506.1)
For: TGT GCT TGT GGA
GGA ACC TAA
Rev: GGC CAC AAA GCA
ACA GAC TC
163
Cathepsin K
Ctsk (NM_007802.4)
For: CCA GTG GGA GCT
ATG GAA GA
Rev: AAG TGG TTC ATG
GCC AGT TC
162
Calcitonin receptor For: CGG ACT TTG ACA 186

Chapter 4: Materials and Methods
89
CTR (NM_001042725.1) CAG CAG AA
Rev: CAG CAA TCG ACA
AGG AGT GA
Alkaline phosphatase
Alpl, ALP (NM_001287172.1)
For: AAC TGC TGG CCC
TTG ACC CCT
Rev: TCC TGC CTC CTT
CCA CCA GCA
186
4.1.8. Solutions
Solutions and buffers were prepared using Milli-Q double distilled water (ddH2O). HCl
and NaOH were used to adjust the pH unless stated otherwise.
Solution Composition and preparation
Ammonium
persulphate
10% (w/v) ammonium persulphate dissolved in ddH2O, stored
at 4 °C.
Ascorbic acid 10% (w/v) stock dissolved in 1× PBS and filter-sterilised. A
1:200 working stock was used.
Bromophenol blue 1% (w/v) stock dissolved in ddH2O. Stirred before use to
resuspend any precipitates.
Collagenase type II
solution
2 mg/ml collagenase type II in serum free α-MEM, filter-
sterilised and warmed to 37 °C prior to use.
Decalcifying
solution (EDTA-G)
0.4 mM EDTA, 0.3 mM NaOH and 15% (v/v) glycerol
dissolved in ddH2O. Adjusted to pH 7.3 and stored at 4 °C.
DNA loading buffer 6× stock composition: 0.25% xylene cyanol, 0.25%
bromophenol blue, 30% glycerol.
Stored at 4 °C and dilute 1 in 6 with DNA samples prior to
loading.
dNTPs: dATP,
dCTP, dGTP and
dTTP
Equal volumes of 100 mM stock of each constituent was mixed
to yield a 25 mM combined stock of dNTP, stored at -20 °C.
EDTA 14% (w/v) EDTA, initially adjusted to pH 8.0 to dissolve
EDTA then readjust to pH 7.4.

Chapter 4: Materials and Methods
90
Eosin-Phloxine
staining solution
Eosin stock solution: 1% (w/v) Eosin Y in ddH2O.
Phloxine stock solution: 1% (w/v) Phloxine B in ddH2O.
Working solution (per litre): 100 ml 1% eosin, 100 ml 1%
phloxine B, 780 ml, 95% ethanol and 4 ml glacial acetic acid.
Flow cytometry
wash buffer
1× PBS containing 1% FBS and 0.1% (w/v) sodium azide, store
at 4 °C.
Glutaraldehyde 25% stock diluted with ddH2O to yield 2.5% working solution.
Goldner's trichrome
stain solution A
0.075% (w/v) Ponceau 2R, 0.025% (w/v) acid fuchsin, 0.01%
(w/v) azophloxine and 0.2% (v/v) acetic acid dissolved in
ddH2O.
Goldner's trichrome
stain solution B
2% (w/v) Orange G and 4% (w/v) phosphomolybdic acid
dissolved in ddH2O. The solution was left to stand at room
temperature for a few days to allow the sedimentation of
precipitates and then filtered before use.
Goldner's trichrome
stain solution C
0.2% (w/v) Light green and 0.2% (v/v) acetic acid dissolved in
ddH2O.
Haematoxylin
(Gill's)
Stock solution: 250 ml ethylene glycol, 750 ml ddH2O, 6 g
haematoxylin, 0.6 sodium iodate (NaIO3), 80 g aluminium
sulphate (Al2(SO4)3) and 20 ml glacial acetic acid, stir and leave
to mix for 1 hour.
Dilute 1 in 5 and filter prior to use.
Infiltration reagent
for histology plastic
embedding
10 g plasticiser, 1 g Perkadox, 0.01 g Tinogard® TT
(Scavenger) dissolved in 89 g methyl methacrylate (MMA).
Neutral Buffered
Formaline
10% (v/v) formaldehyde solution, 45 mM Na2HPO4 and 33 mM
NaH2PO4 in ddH2O.
Paraformaldehyde 4% (w/v) Paraformaldehyde in ddH2O heated to 70 °C until
dissolved. Aliquots stored at -20 °C.
Phosphate buffered
saline (PBS)
10× PBS stock solution in ddH2O: 70 mM disodium hydrogen
phosphate (Na2HPO4), 30 mM monosodium phosphate
(NaH2PO4) and 1.3 M sodium chloride (NaCl).
1× PBS: 1 in 10 dilution of 10× PBS stock solution and
calibrated to pH 7.4.

Chapter 4: Materials and Methods
91
PMSF Working stock was prepared by dissolving 17.6 mg/ml in
isopropanol. Aliquots stored at -20 °C.
Radio
Immunoprecipitatio
n Assay Buffer
(RIPA) lysis buffer
50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.1%
SDS and 1% sodium deoxycholate. Adjusted to pH 7.5 and
stored at 4 °C.
Prior to protein isolation from cells or tissue samples, 100
µg/ml PMSF, 1× protease inhibitor, 1 mM sodium
orthovanadate and 500 µg/mL DNase I were promptly added to
the amount of lysis buffer required.
SDS-PAGE
running buffer
10× stock solution in ddH2O: 25 mM Trizma base, 1.92 M
glycine and 1% (v/v) SDS.
1× working solution: 1 in 10 dilution of 10× stock solution in
ddH2O.
SDS-PAGE sample
loading buffer
4× stock solution dissolved in ddH2O: 240 mM Tris-HCl (pH
6.8), 8% (w/v) SDS, 40% (v/v) glycerol, 0.04% (w/v)
bromophenol blue and 5% (v/v) β-mercaptoethanol.
Stored at 4 °C.
SDS-PAGE
separating gel
buffer
1.5 M Tris-HCl (pH 8.8) and 0.4% (w/v) SDS prepared in
ddH2O.
SDS-PAGE
stacking gel buffer
1 M Tris-HCl (pH6.8), 0.4% SDS (w/v) dissolved in ddH2O.
Sodium
deoxycholate
solution (SDS)
10% (w/v) solution dissolved in ddH2O.
Sodium
orthovanadate
(Na3VO4)
0.25 M of Na3VO4·14H2O dissolved in ddH2O and is pH to 10
with HCl, yielding a yellow solution. Boil solution until it turned
colourless and cool on ice until the solution reaches room
temperature. Readjust to pH 10 and repeat boiling/cooling steps a
total of 3-5 times. Stock is stored at -20 °C.
Tris-acetate EDTA
(TAE)
1 L 50× stock solution in ddH2O: 2 M Trizma base, 5.71% (v/v)
glacial acetic acid and 50 mM EDTA.
1× TAE working solution: dilute 1 in 50 in ddH2O prior to use.

Chapter 4: Materials and Methods
92
Tris buffered saline
(TBS)
10× stock solution in ddH2O: 0.5 M Trizma base and 1.5 M
NaCl.
1× TBS: 1 in 10 dilution of stock solution in ddH2O.
TBS-Tween 1× TBS with addition of 0.1% (v/v) Tween-20.
Toluidine blue dye
solution
1% (w/v) toluidine blue dissolved in in ddH2O.
TRAP stain
(complete)
5 mg Naphthol AS-MX dissolved in 250 μl 2-ethoxyethanol with
the addition of 40 mg Fast Red-Violet LB salt dissolved in TRAP
stain solution A.
Aliquots are stored at -20 °C, thawed at 37 °C and filtered before
use.
TRAP stain
solution A
100 mM sodium acetate trihydrate, 50 mM sodium tartrate
dihydrate and 0.22% (v/v) glacial acetic acid. Adjusted to pH 5.0.
Triton X-100/PBS 0.1% (v/v) 100% Triton X-100 in 1× PBS. Filtered and stored at
4 °C.
Weigert's iron
haematoxylin
Solution A: 1% (w/v) haematoxylin and 96% (v/v) ethanol in
ddH2O.
Solution B: 4 ml 30% iron chloride and 1 ml 37% HCl mixed in
ddH2O.
Equal parts of solution A and solution B were mixed to make a
working solution.
Western blot
membrane
stripping buffer
62.5 mM Tris-HCl and 2% (w/v) SDS dissolved in ddH2O and
calibrated to pH 6.7 prior to addition of 100 mM 2-
mercaptoethanol.
Western blot
transfer buffer
25 mM Trizma base, 192 mM glycine and 10% (v/v) methanol in
ddH2O.
β-
glycerophosphate
0.4 M stock dissolved in 1× PBS, filter-sterilised and aliquots
stored at -20 °C.
4.1.9. Equipment
Description Manufacturer
Wide Mini-Sub® Cell GT Bio-Rad, USA

Chapter 4: Materials and Methods
93
Verity 96-well Thermal Cycler Applied Biosystems, USA
Tissue-Tek III Embedding Console Miles Scientific, USA
Tissue Processor – TP1020 Leica Microsystems, Germany
Tissue Cryostat Processor – CM1900 Leica Microsystems, Germany
Thermomixer Comfort Eppendorf AG, Germany
Skyscan 1174 Compact MicroCT Scanner Skyscan, Belgium
Shimadzu Balance ELB300 Walker Scientific, Australia
Senographe DS Digital Mammography Machine GE Healthcare, UK
Rotofix Hettich Zentrifugen HD Scientific Supplies Pty Ltd,
Australia
Rotary Tube Mixer with disc Ratek Instruments, Australia
Removable (UVTP) Gel Tray and Gel Caster Bio-Rad, USA
Ratek Platform Mixer Ratek Instruments, Australia
Power-Pac 300 Bio-Rad, USA
POLARstar OPTIMA Bio-Rad, USA
Olympus CK30 Micrscope Olympus Optical Co Ltd, Japan
Nikon CoolPix S4 Digital Camera Nikon Corp., Japan
Neubauer Improved Cell Counting Chamber Laboroptic Ltd, UK
Nanodrop® ND-1000 UV-Vis
Spectrophotometer V3.3
Thermo Scientific, USA
Movable 7×10 cm2 and 10×10 cm2 UV Perspex
Gel Casting Tray;
8 and 15 well comb for agarose gels
Bio-Rad, USA
Model 680 Microplate Reader Bio-Rad, USA
MiniSpin® and MiniSpin® Plus microcentrifuge Eppendorf AG, Germany
Mini-sub® Cell GT Bio-Rad, USA
Mini Trans-Blot Electrophoretic Transfer Cell Bio-Rad, USA
MilliQ Reagent Water System Millipore Corporation, USA
Microcentrifuge Tubes: 1.5 ml Sarstedt, Germany
Microcentrifuge Tubes: 0.5 ml & 2.0 ml TRACE Biosciences, Australia
Micro One TOMY Centrifuge Quantum Scientific, Australia
Life Technologies ViiA™ 7 Real-Time PCR
System
Life Technologies, USA

Chapter 4: Materials and Methods
94
Letiz Labourlux S Microscope Leitz Wetzlar, Germany
ImageQuantTM LAS-4000 Mini Imager GE Healthcare Life Sciences, USA
isoMet® Low Speed Saw Buehler, USA
Hot air oven Memmert, Germany
Epson Perfection 3490 Photo Scanner Seiko Epson Corporation, Japan
Eppendorf MasterCycle® Personal Thermal
Cycler
Brinkmann, Canada
Eclipse TE2000S Fluorescent Microscope Nikon Instruments Inc., USA
Clemco FCS Fume Cupboard Clemco Industristies Corp., USA
Chiltern Vortex Mixer MT17 Scot Scientific, Australia
Centrifuge 5415D Eppendorf AG, Germany
Biophotometer Plus Eppendorf AG, Germany
Biological Safety Cabinet Class II Gelman Sciences, USA
Automatic CO2 incubator Forma Scientific, USA
4.1.9.1. Centrifugation
Micro-centrifugations (0.5 mL, 1.5 mL and 2.0 mL tubes) at room temperature were
carried out in MiniSpin or MiniSpin Plus microcentrifuge. Micro-centrifugations at 4 ˚C
were carried out using the Eppendorf Centrifuge 5415D. PCR (0.2 mL thin-walled
tubes) centrifugations at room temperature were performed using MicroOne
microcentrifuge. Centrifugations of 15 ml and 50 ml centrifuge tubes were performed
using Hettich Rotofix 32 centrifuge.
4.1.10. Software
Description Supplier
BD FACSDiva™ BD Biosciences, USA
Bioquant Osteo 14.1.6 Bioquant Image Analysis Corp,
USA
CT Analyser 1.10.1.0 Skyscan, Belgium
Dataviewer 1.4.3.0 Skyscan, Belgium
FlowJo FlowJo LLC, USA
ImageJ 1.48 National Institutes of Health, USA

Chapter 4: Materials and Methods
95
Microsoft Research Image Composite Editor
(ICE)
Microsoft, USA
NIS-Elements Basic Research Nikon Instruments Inc, USA
NIS-Elements C Nikon Instruments Inc, USA
NRecon Version 1.5.1.14 Skyscan, Belgium
4.2. Animals and bone phenotyping methods
4.2.1. The generation of sanroque mutant mouse line
The sanroque mutant mice on which this study is based on, were generated by the
Australian Phenomics Facility (APF) of the Australian National University (ANU),
Canberra, Australia. The mutant mice were generated using N-ethyl-N-nitrosourea
(ENU)-induced mutagenesis, which resulted in single-base substitutions at an estimated
rate of 1 per 0.5 megabases (Mb) in the germline (Jun et al., 2003; Nelms et al., 2001;
Vinuesa et al., 2005a). Briefly, normal C57BL/6 male mice 8–15 weeks old (generation
0; G0 mice) were treated with super-mutagenic ENU (100 mg/kg) three times 1 week
apart and were mated with wildtype C57BL/6 female mice. Individual first generation
male progeny (G1) were used as founders of inbreeding pedigrees by breeding with
wildtype C57BL/6 females and then with several G2 daughters. Phenotypic-screening is
performed on G3 offspring with a subset of mutations that would reach homozygosity.
The mapping and sequencing of sanroque mice were also performed by APF. For
mapping of recessive mutations an intercross breeding approach was employed whereby
the mutant C57BL/6.sanroque mice were outcrossed to a mapping strain (CBA/Ca)
mice to produce F1 carrier mice. The F1 mice were intercrossed and 25% of the
generated F2 progeny will be homozygous for the recessive mutation. Using MIT
microsatellite panel, the mutation was mapped to a single locus on distal chromosome 1
in F2 mice. The chromosomal location of sanroque is localised at 1-Mb chromosomal
interval containing 11 predicted genes. Resequencing of mRNA and exons in genomic
DNA revealed that, of the 11 annotated transcription units in the sanroque interval, only
one mRNA contained the mutation in sanroque mice relative to the B6 parental
controls. The single T G nucleotide substitution resulting in a non-conservative Met
199 Arg codon change was identified in the mouse homolog 1 Roquin (Rc3h1) gene.
F6 to F8 wildtype and sanroque mutant littermates of the age of 80 to 100 days old were

Chapter 4: Materials and Methods
96
used in this study. When required, the mice were imported from APF and quarantined
for one week at the Biomedical Research Facility, UWA, Western Australia. Once
cleared from quarantine, the mice were shipped to the School of Pathology and
Laboratory Medicine, UWA and animal handling procedures were performed in
accordance with protocols approved by the UWA Animal Ethics and the ANU Animal
Ethics committees.
4.2.2. Radiographic screening of sanroque mutant mice
Radiographic assessment of forelimbs, hindlimbs, spines and tails of wildtype and
sanroque mutant mice was carried out using the Senographe DS digital mammography
system at the Sir Charles Gairdner Hospital, Perth. The samples were imaged using the
following settings:
Sample Voltage (kV) Current (mA)
Forelimbs and hindlimbs 22 12.5
Spines 25 14.0
Tails 25 10.0
4.2.3. Microquantitative computed X-ray tomography (MicroCT) analysis
of wildtype and sanroque mutant mice
The mouse distal femur or proximal tibia was fixed in neutral buffered formalin for 24
hours, washed in PBS, and then stored in 70% ethanol. The fixed tissue was wrapped in
moistened tissue and placed in an X-ray transparent tube to be scanned using a Skyscan
1174 microCT scanner (Skyscan, Belgium) at a voltage of 50 kV and a current of 0.8
mA with a 0.5 mm aluminium filter. The resolution (pixel size) was set to 6.03 µm and
scanning angular rotation was 180˚ at the angular increment of 0.4˚. Datasets of
tomographic sections were reconstructed using the cone beam CT reconstruction
software (NRecon) facilitated by the Feldkamp algorithm and segmented into binary
images using adaptive local thresholding using CT analyser software (CTAn, Skyscan).
Bone volume analysis of the trabecular bone was performed using analysis software
(CTAn, Skyscan). The secondary spongiosa 0.5 mm distal to the proximal growth plate
at a height of 1 mm was selected for analysis within a conforming volume of interest
(excluding cortical bone). Morphometric parameters including trabecular bone volume

Chapter 4: Materials and Methods
97
(BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.n) and trabecular
separation (Tb.sp) were measured. Assessment of the mid-diaphysis cortical volume
was performed by similar methods in a region 3.3 mm from the growth plate, with a
height of 1 mm. The cortical bone volume (Cortical BV) and cortical thickness were
calculated.
MicroCT analysis was performed in the same settings for each biological pair of all age
group and sexes of mice. For 3D images, the CT files were saved as .p3g 3D models in
CTan (Skyscan) and were generated using CTVol program (Skyscan).
4.2.4. Fluorochrome labelling of wildtype and sanroque mutant mice
Fluorochrome labelling using calcein was performed to assess in vivo bone formation
rates. Three biological pairs of wildtype and sanroque mutant mice received an
intraperitoneal injection of calcein (5 mg/kg) (day 0), followed by a second
intraperitoneal injection of alizarin red (30 mg/kg) 4 days later, and another calcein
injection on day 8. The mice were sacrificed on day 10 and the hind limbs were
dissected out of the mice. The limbs were fixed in 70% ethanol and were exported to
our collaborator, Dr. Jerry Feng, in China to be processed for plastic embedding.
Briefly, the samples were dehydrated at room temperature under vacuum through 3
changes of 95% ethanol for 4 hours each step. This is followed by 4 changes of 100%
ethanol, 4 hours per change. The samples were then immersed in freshly prepared
embedding solution of 84% (v/v) methalmethacrylate, 14% (v/v) dibutylphthalate, 1%
(v/v) polyethylene glycol 400 and 0.7% (w/v) benzoyl peroxide, and vacuumed. The
infiltration was carried out at room temperature for 3 – 4 days. The infiltrated samples
was subsequently incubated at 37 °C for 2 – 3 days until the plastic resin polymerised.
The samples were then sectioned at 5 µm thickness. The calcein labelling was
visualised by confocal microscopy. The distance between the labels at 3 points along
the cortical bone were measured using ImageJ (NIH) software.
4.2.5. Histological analysis of wildtype and sanroque mutant mice
4.2.5.1. Tissue fixation, decalcification (for hindlimbs), processing, embedding
and sectioning
Tissues or organs of interest were dissected out and fixed in 10% NBF on a Rotary
Tube Mixer (Ratek) overnight. The samples were then washed twice with 1× PBS and

Chapter 4: Materials and Methods
98
left on the Rotary Tube Mixer overnight to completely wash the fixative solution. The
samples were then transferred into 70% ethanol and stored at 4 °C until ready for
processing (or decalcification of bone, if required).
For decalcification of bone tissues, all soft tissues were thoroughly removed, and the
tibia and femur were separated gently. The bones were washed once with 1× PBS
before the decalcification process. The decalcification process were carried out by
immersing the tissues in 14% EDTA (pH 7.4) and incubated at 37 °C. Fresh EDTA was
changed daily for 5 days. Once the bone tissues became soft and flexible, the tissues
were washed with 1× PBS and were stored in 70% ethanol until they are ready to be
processed. The decalcified bones were placed in sample cassettes and processed using
the Leica TP1020 tissue processor over 24 hours according to the following steps:
Station Reagent Time (minutes)
1 70% Ethanol 30
2 95% Ethanol 30
3 95% Ethanol 45
4 100% Ethanol 60
5 100% Ethanol 60
6 100% Ethanol 60
7 50% Ethanol/50% Xylene 60
8 Xylene 90
9 Xylene 120
10 Paraffin wax at 60 °C 90
11 Paraffin wax at 60 °C 120
12 Paraffin wax at 60 °C 120
The processed samples were then paraffin-embedded and cut into 5 µm sections by Mr.
Jacob Kenny from The School of Pathology and Laboratory Medicine, UWA. In brief,
the samples were placed in Tissue-Tek thermal console (Miles Scientific) to be paraffin-
embedded and were cooled using Tissue-Tek cryo console (Miles Scientific) for 15 to
20 minutes. Once set, the embedded samples were sectioned using Leica 2035
microtome and the 5 µm sections were floated on a 37 °C water bath and lifted onto a
clean glass slides. The sections were set onto the glass slides by incubating the samples

Chapter 4: Materials and Methods
99
in a hot air oven at 60 °C for at least one hour.
4.2.5.2. Haematoxylin and eosin staining
Once the sections were set, the slides were dewaxed, hydrated, stained and dehydrated
by immersing the samples into the following series of solutions:
Step Solution Time (minutes)
1 Xylene 5
2 Xylene 5
3 100% Ethanol 3
4 100% Ethanol 3
5 95% Ethanol 3
6 80% Ethanol 3
7 70% Ethanol 3
8 Gently running tap water 3
9 Gill's Haematoxylin 5
10 Gently running tap water 3
11 1% Eosin 0.5
12 Scott's water 1
13 70% Ethanol 1
14 80% Ethanol 1
15 95% Ethanol 1
16 100% Ethanol 2
17 100% Ethanol 2
18 Xylene 5
19 Xylene 5
20 Xylene 5
Haematoxylin and eosin stain nucleic acid in blue and protein in shades of pink
respectively. The stained slides were mounted on coverslips with DPX mounting
medium.
4.2.5.3. TRAP staining
TRAP solutions were warmed up to 37 °C and filtered prior to staining. The slides were
dewaxed and hydrated as listed above (steps 1 – 8). Excess liquid was removed from the

Chapter 4: Materials and Methods
100
slide without letting the section dry out. Filtered pre-warmed TRAP solution was
applied and the slides were incubated at 37 °C. The slides were checked every 30
minutes to monitor the development of the stain. After approximately 2 hours, with
observation of magenta-stained osteoclasts, the slides were rinsed in ddH2O before
counterstaining with a quick immersion in Gill’s Haematoxylin solution. The slides
were rinsed in ddH2O to remove excess stain, followed by a 1 minute immersion in
Scott’s tap water. If the counterstain appeared too intense, a 3 second dip into 1% acid
alcohol was performed to wash off the stain, and further adjusted using Scott’s tap water
until the desired contrasting colour was achieved. The sections were washed briefly in
running tap water and were mounted with Clearmount aqueous mounting medium
(Invitrogen). The mounted slides were left to set in a hot air oven at 60 °C for 45
minutes.
4.2.5.4. Goldner Trichome staining on non-decalcified bone samples
4.2.5.4.1. Processing, embedding and sectioning of undecalcified bone
samples
Tibia and femurs were collected and fixed as previously described in section 4.2.5.1.
For plastic embedding, the decalcification step was omitted and the fixed samples
underwent a series of dehydration process in the Leica TP1020 tissue processor
according to the following steps:
Station Reagent Time (minutes)
1 50% Ethanol 120 120
2 70% Ethanol 120
3 80% Ethanol 120
4 96% Ethanol 120
5 100% Ethanol 180
6 100% Ethanol 180
7 Xylene 60
8 Xylene 720
Excess xylene was gently wiped off and the samples were immersed in the infiltration
reagent (described in section 4.1.8) for 3 – 4 days in vacuum condition at 4 °C. The
samples were then transferred into moulds and were embedded in fresh infiltration

Chapter 4: Materials and Methods
101
reagent. The moulds were sealed tightly and polymerisation was catalysed by
incubating the samples at 29 °C for at least 3 days until the samples were fully
polymerised. The embedded samples were sectioned using the Leica RM2255
Automated Microtome at a thickness of 5 µm. The sections were placed directly on the
glass slides and were gently straightened using a soft artist painting brush dipped in
30% ethanol. A layer of plastic sheet was overlaid onto the straightened sections and the
glass slides were stacked onto one another. To facilitate section adherence onto the
glass, the stacked glass slides were clamped and left to set at 60 °C overnight. The
processing, embedding and sectioning of undecalcified bone samples were performed
with the assistance of Miss Euphemie Landao (Centre for Orthopaedic Research, School
of Surgery, UWA).
4.2.5.4.2. Goldner Trichrome staining
The samples were de-plasticised, rehydrated, stained and dehydrated according to the
following protocol:
Step Solution Time
1 Methoxyethyl acetate (MEA) 20 minutes
2 MEA 20 minutes
3 MEA 20 minutes
4 Xylene 10 minutes
5 Xylene 10 minutes
6 100% Ethanol 10 seconds
7 100% Ethanol 10 seconds
8 96% Ethanol 10 seconds
9 80% Ethanol 10 seconds
10 70% Ethanol 10 seconds
11 50% Ethanol 10 minutes
12 ddH2O 15 seconds
13 ddH2O 15 seconds
14 Acid alcohol 3 seconds
15 ddH2O 15 seconds
16 Solution A 5 minutes
17 1% Aqueous acetic acid 15 seconds

Chapter 4: Materials and Methods
102
18 Solution B 3 minutes
19 1% Aqueous acetic acid 15 seconds
20 Solution C 5 minutes
21 1% Aqueous acetic acid 5 minutes
22 100% Ethanol 10 seconds
Goldner Trichrome stained the mineralised bone green, osteoid (unmineralised bone)
orange/red, nuclei bluish-grey and cartilage purple.
4.2.5.5. Bone histomorphometric analysis
Stained tibia and femur sections were scanned at 40× magnification using ScanScope®
XT System (Quorum Technologies) at the Centre for Microscopy, Characterisation and
Analysis (CMCA), UWA. Bone histomorphometric analysis on mouse tibia or femur
sections was conducted using BioQuant Osteo software (Bioquant Image Analysis).
Trabecular bone (secondary spongiosa) was analysed within the region of interest of 1
mm in height, set to 0.5 mm below the growth plate. Three independent sections were
analysed per bone sample to yield the following parameters:
Parameter Stained sections
Trabecular number (BV/TV) H&E or Goldner Trichrome
Trabecular thickness (Tb.Th) H&E or Goldner Trichrome
Trabecular number (Tb.N) H&E or Goldner Trichrome
Trabecular separation (Tb.Sp) H&E or Goldner Trichrome
Osteoid surface (OS) Goldner Trichrome
Osteoid surface per bone surface (OS/BS) Goldner Trichrome
Number of osteoclast per bone surface (N.Oc/BS) Goldner Trichrome
Number of osteoblast (N.Ob) Goldner Trichrome
Number of osteoblast per bone surface (N.Ob/BS) Goldner Trichrome
Osteoclast surface (Oc.S) TRAP
Osteoclast surface per bone surface (Oc.S/BS) TRAP
4.2.6. TRAP staining of mouse calvariae
Soft tissues surrounding the calvariae dissected from euthanized wildtype and sanroque
mutant mice were thoroughly cleaned using a scalpel. The calvariae were then

Chapter 4: Materials and Methods
103
extensively washed in 1× PBS and fixed as dictated in section 4.2.5.1. Prior to TRAP
staining, the calvariae were decalcified until they became soft and malleable.
Decalcified calvariae were then extensively washed with 1× PBS and briefly dried on a
paper towel before placing individual calvaria in each well of a 12-well plate. The
samples were then incubated in filtered pre-warmed TRAP stain at 37 °C for
approximately 1 hour. Once apparent magenta-staining was observed, the TRAP
solution was removed and the calvariae were once again washed extensively with 1×
PBS and stored at 4 °C. The superficial surface of the calvaria was photographed using
a Nikon Coolpix S9600 digital compact camera.
4.2.7. Immunofluorescence staining of mouse spleens
4.2.7.1. Embedding of sample in Optimal Cutting Temperature (OCT)
Isolated spleen was gently blotted dry on tissue to remove excess blood and was left to
immerse and equilibrate in OCT (Tissue-Tek) media in a mould for 5 – 10 minutes.
During tissue equilibration isopentane was pre-chilled in a liquid nitrogen bath. The
mould containing the equilibrated OCT-immersed spleen was floated on the surface of
the isopentane until the OCT solidified. Frozen blocks containing samples were stored
at -70 °C until they were ready to cut. Prior to cutting, the blocks were placed into
CM1900 Tissue Cryostat Processor (Leica) to equilibrate for at least 30 minutes. The
blocks were cut into 5 µm sections and allowed to adhere onto charged slides. The
slides were left to air-dry at room temperature for at least 30 minutes prior to fixing. If
the slides were not required to be fixed immediately, the samples were stored at -70 °C
with desiccants to remove humidity and when required for staining were subsequently
air-dried at room temperature for 15 minutes prior to fixing. The slides were fixed in
acetone for 15 minutes at - 20 °C. Excess acetone was gently removed and the slides
were left to air-dry for another 15 minutes. The fixed samples were stained
immediately.
4.2.7.2. Immunofluorescence staining
The tissue section on each slide was isolated by drawing the perimeter with a PAP pen
and the slides were rehydrated in 1× PBS with the addition of 1% serum (1× PBS/1%
serum solution) for 10 minutes at room temperature. The serum used for blocking was
from the same species in which the secondary antibody was raised. Samples were then
blocked with 10% serum in 1× PBS for 30 minutes at room temperature. After blocking,

Chapter 4: Materials and Methods
104
the slides were gently rinsed with 1× PBS and incubated for 60 minutes with primary
antibodies diluted in 1× PBS/1% serum solution. The slides were washed with three
changes of 1× PBS for 3 minutes each wash followed by 45 minutes incubation with
secondary antibodies diluted in 1× PBS/1% serum solution. Subsequently, the
antibodies were washed away with three changes of 1× PBS. For nuclear staining, the
samples were incubated with DAPI diluted in 1× PBS for 15 minutes. The samples were
once again washed three times with 1× PBS and excess liquid was thoroughly removed.
Prolong Gold Antifade (Invitrogen) was used to mount the samples with coverslip and
the samples were protected from light until ready to be viewed under fluorescence
microscope.
4.3. Isolation, culture and in vitro studies of primary cell lines
All cells were cultured in plates or flasks as indicated in a humidified water-jacketed
incubator with 5% CO2, 95% air at 37 °C. Cultured cells were handled using aseptic
techniques in UV sterilised biological safety cabinets (Class II).
4.3.1. Isolation and culture of bone marrow cells
Bone marrow cells were isolated from long bones (tibia, femur and humerus) of
euthanized wildtype and sanroque mutant mice. The long bones were dissected out
from the mice and placed in ice-cold complete α-MEM (α-MEM supplemented with
10% FBS, 2 mM L-glutamine, 100 U/ml Penicillin and 100 µg/mL Streptomycin).
Muscle was removed using a sterile scalpel and the epiphyses were cut off to expose the
marrow cavity. Bone marrow cells were flushed from the bone marrow cavity
repeatedly with complete α-MEM (pre-warmed at 37 °C) using a syringe and 23G
needle in a sterile petri dish. Flushed long bones were set-aside in sterile 1× PBS for
osteoblastic bone cell isolation where required (see 4.3.2). The bone marrow cells were
collected and filtered through a 100 µm sterile cell strainer to remove loose bone and
soft tissue fragments. The bone marrow cells were pelleted by centrifugation at 350 g
for 5 minutes at room temperature and resuspended in complete α-MEM.
4.3.1.1. Generation and maintenance of bone marrow macrophages (BMMs)
Bone marrow macrophages (BMMs) were generated by culturing isolated bone marrow
cells in 10 ml of complete α-MEM supplemented with 1 in 20 dilution of murine M-
CSF conditioned media (α-MEM/M-CSF) in 75-cm2 tissue culture flasks. When cells
were 90% confluent, they were washed with 1× PBS and trypsinised using TrypLETM

Chapter 4: Materials and Methods
105
Express for 5 – 10 minutes at 37 °C, occasionally tapping the flask. BMMs adhere
strongly to the plastic, hence scraping was required to detach the cells. Complete α-
MEM was added to deactivate the enzyme and the cell suspension was pelleted by
centrifugation at 350 g for 5 minutes. The cell pellet was then resuspended in α-
MEM/M-CSF and a cell count was performed using a Neubauer counting chamber.
BMMs were then plated into culture dishes at the appropriate cell density for
downstream experiments.
4.3.1.2. MTS cell proliferation assay
Cell proliferation assay on BMMs using CellTiter 96® AQueous One Solution Cell
Proliferation Assay kit (Promega) containing tetrazolium compound [3-(4,5-
dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt; MTS] was performed according to manufacturer’s instructions. Briefly,
6×103 BMMs were seeded in two 96 well plates, one to be measured at basal level and
the other to be measured after treatment. Once the cells adhered and after 24 hours of
treatment, 20 μl of thawed CellTiter 96® AQueous One Solution Reagent is added into
each well of the 96-well plate containing the cell culture in 100 μl medium. Repeat
pipetting was performed to ensure that the solution is well mixed and the plate was
incubated at 37 °C for 1–4 hours in a humidified, 5% CO2 atmosphere. The reaction
was stopped by the addition of 25µl of 10% SDS into each well. The plate was
protected from light at room temperature before recording the absorbance at 490 nm
using a 96 well plate reader.
4.3.1.3. Osteoclast culture
Cell culture plate
Cell density
6-well plate 5×104/2.0 ml/well
12-well plate 3×104/1.5 ml/well
96-well plate 6×103/0.1 ml/well
For osteoclastogenesis, seeded BMMs were cultured in α-MEM/M-CSF and left to
settle overnight to allow cell attachment onto the plate. The following day, BMMs were
stimulated with 25 ng/ml RANKL-GST and the culture media (α-MEM/M-
CSF/RANKL) was replaced every two days until multinucleated osteoclast-like cells

Chapter 4: Materials and Methods
106
are observed at day 5 – 7 post-stimulation with RANKL. Unstimulated cells served as a
negative control.
4.3.1.3.1. In vitro osteoclastogenesis assay
Osteoclastogenesis assays were carried out using BMMs cultured as described above in
96-well plates. Once stimulated to form multinucleated osteoclasts, the cells were gently
washed once with 1× PBS and fixed with 4% paraformaldehyde for 15 minutes at room
temperature, followed by 3 gentle washes with 1× PBS. The cells were subsequently
stained with filtered TRAP stain and left to incubate at 37 °C for 30 minutes. The TRAP
stained cells were washed with 1× PBS three times and the plates were stored at 4 °C
until they are ready to be imaged and counted.
The osteoclast cultures were visualised using Eclipse TE2000S (Nikon) microscope and
NIS-Elements Basic Research Nikon software. Serial images covering the entire well
were taken at 40X magnification and then stitched together using Image Composite
Editor (ICE) (Microsoft) to form a single image of each well. The total number of
TRAP stained, magenta-coloured osteoclasts with more than three nuclei were tallied
for each well.
4.3.1.3.2. In vitro bone resorption assay
Bone resorption assays were performed using multinucleated osteoclasts seeded onto
bovine bone discs. Bone discs were produced from bovine bone that was cleaned
thoroughly to remove excess tissue and marrow. The bone was sliced using an IsoMet®
low speed saw (Buehler) into 0.70 mm sections. Bone slices were soaked in ddH2O to
soften the bone matrix and then circular 5 mm bone discs were generated with using a 5
mm diameter hole punch. Bone discs were cleaned by sonicating for 5 minutes to a total
of 10 times in MilliQ ddH2O, replacing the water each time. Bone powder generated
from the action of the low speed saw was collected and reserved for cathepsin K release
experiments (4.3.1.2). After sonication, the bone discs were stored in 70% ethanol at 4
°C. When the bone discs were required, they were thoroughly air dried in a sterile
biological safety cabinet (Class II). The dried and sterile bone discs were washed three
times with serum free α-MEM and then incubated for at least 2 hours in α-MEM/M-
CSF at 37 °C. Before placing the bone discs into the 96-well plate for cell seeding, the
bone discs were washed a further three times with α-MEM/M-CSF.

Chapter 4: Materials and Methods
107
To generate mature multinucleated osteoclasts for bone resorption assays, BMMs were
cultured in 6-well collagen-coated plates and stimulated as described previously. Instead
of maintaining the culture until large osteoclasts were formed, cells were dissociated at
approximately day 3 when small osteoclasts began to form. The cells were incubated
with cell dissociation solution (Sigma) for 10 minutes and observed every 5 minutes
under the microscope. Once the cells began to lift off, remaining adherent cells were
gently scraped off and the cell suspension pelleted by centrifugation at 350 g for 5
minutes. The cells were then reconstituted with 1.5 ml of α-MEM/M-CSF/RANKL
culture media and 100 µl aliquots were seeded onto individual bone discs. Aliquots of
cells were seeded in wells with no bone discs to allow visual monitoring of the
differentiation of the cells.
Osteoclasts were left to settle and resorb the bone discs for 72 hours. Cells on bone
discs were gently washed, fixed, stained and imaged as described above. Osteoclast
numbers were counted on each bone discs, and then osteoclasts were gently removed
using cotton buds (soaked in 1% (v/v) bleach) to expose the underlying resorbed bone.
The cleaned bone discs were then stained with toluidine blue for 5 minutes to stain the
resorption pits dark blue, while the non-resorbed bone surface remained pale blue. The
discs were then washed extensively with 1× PBS and serial images of the whole bone
discs were taken and stitched as previously described. The area of resorption was
measured using ImageJ (NIH).
4.3.1.3.3. In vitro osteoclast cathepsin K release assay using bovine bone
powder
Bovine bone powder was collected as described above. The bone powder was collected
in distilled water as a by-product of cutting multiple bone slices. The powder was
pooled and centrifuged at maximum speed for 5 minutes at room temperature. The
supernatant was discarded and the bone powder was reconstituted in 70% ethanol. The
suspension was vigorously vortexed to ensure thorough sterilisation and the bone
powder was again pelleted by centrifugation. The pelleted bone powder was then
collected and spread in a sterile petri dish and left to dry in a sterile class II biological
safety cabinet. The sterile bone powder was collected and placed in a 50 ml centrifuge
tube to be reconstituted and extensively washed with sterile 1× PBS. The suspension
was vigorously mixed to break up clumps of powder and then centrifuged. The

Chapter 4: Materials and Methods
108
supernatant was discarded and the pelleted bone powder was then washed again with
vortexing in 1× PBS and the wash process was repeated 3 times. The bone powder was
reconstituted in sterile α-MEM and left to agitate on a platform shaker for at least 2
hours to ensure that all bone powder clumps were completely dispersed. The bone
powder suspension was subsequently centrifuged and washed a further three times with
sterile α-MEM. The bone powder was subsequently resuspended 50% v/v with sterile α-
MEM and stored at 4 °C until required. When required, the bone powder suspension
was mixed thoroughly, the appropriate amount aliquoted, centrifuged and resuspended
50% v/v in α-MEM/M-CSF/RANKL culture media.
BMMs were seeded in a 6-well plate at the cell density cited above and were maintained
in α-MEM/M-CSF/RANKL culture media. Once large multinucleated osteoclasts were
formed, 100 µl of 50% v/v bone powder suspension in α-MEM/M-CSF/RANKL culture
media was gently and evenly distributed drop-wise into each well to stimulate cathepsin
K enzymatic release. The cells were stimulated at 4, 8 and 12 hours and cultured media
was collected followed by cell protein extraction as described in 4.4.4.2. The cultured
media and cell proteins were separated by SDS-PAGE gel electrophoresis and analysed
by western blotting using the appropriate antibody as explained in sections 4.4.4.3 –
4.4.4.6.
4.3.2. Isolation of osteoblastic bone cells from long bones and calvariae
Primary osteoblast bone cell isolation procedure from long bones and calvariae was
modified from (Bakker et al., 2012). Flushed long bones were collected and pooled
together as described above. To isolate primary osteoblastic bone cells from adult
mouse calvaria, calvaria from euthanized mice were dissected from the skull using
scissors. Sutures and other tissues were removed by gentle scraping the calvaria. Both
flushed long bones and cleaned calvariae undergo similar processing hereafter. Briefly,
samples were washed with 1× PBS and chopped into small fragments (~1 mm2). The
bone fragments were then incubated in collagenase II solution at 37 °C with gentle
shaking for 2 hours and occasional vigorous shaking by hand. An equal amount of
DMEM containing 15% FCS, 2 mM L-glutamine, 100 U/ml Penicillin and 100 µg/mL
Streptomycin (complete DMEM) was added to inhibit further collagenase digestion, and
the bone fragments were rinse three times with complete DMEM. The bone fragments
were then transferred into 25-cm2 tissue culture flasks containing 5 ml complete DMEM

Chapter 4: Materials and Methods
109
at a density of about 20 – 30 fragments per flask. Adult mouse osteoblastic bone cells
were observed migrating from the bone fragments after 3 – 5 days. The culture medium
was replaced and the flasks were gently agitated every 3 days to prevent crowding
around the bone fragments and encourage cell migration. Bone fragments were
maintained for approximately 10 – 14 days until migrating cells reached subconfluency,
upon which cells were trypsinised and used for experiments. Bone fragments were
discarded.
4.3.2.1. Osteoblast culture and mineralisation assay
Cell culture plate Cell density
12-well plate 2.0 ×105/1.0 ml/well
24-well plate 1.0 ×105/0.5 ml/well
48-well plate 0.5×105/0.5 ml/well
For osteoblastogenesis assays, trypsinised primary osteoblastic cells were seeded into
12- well plates and the cells were maintained in complete DMEM until they are
confluent (approximately 7 – 10 days post cell-seeding). Once confluent, the cells were
stimulated with mineralisation media consisting of complete DMEM supplemented with
10 nM dexamethasone, 2 mM β-glycerophosphate and 50 µg/ml ascorbate and medium
was replaced every 3 days. Bone nodules were observed at day 10 – 14. At day 21the
cells were washed once with 1× PBS and fixed with 2.5% glutaraldehyde solution for
10 minutes followed by three washes with 1× PBS. For staining of the calcium within
mineralised nodules using alizarin red S, the fixed cells were post-fixed with 70%
ethanol three times and left to dry completely. An aliquot of 300 µl of 1% alizarin red
solution was placed into each well and left to stain for 2 minutes before destaining with
three washes of 50% ethanol. The plates were left to air dry completely and stored at
room temperature.
To analyse the area of mineralised nodules, the plates were scanned at 600 dpi using an
Epson Perfection 3490 Photo scanner in TIFF format. ImageJ software was used to
measure the mineralised area.
4.3.2.2. Osteoblasts and BMMs co-culture experiment
To investigate the capability of wildtype and sanroque mutant osteoblasts to support

Chapter 4: Materials and Methods
110
osteoclastogenesis, isolated osteoblastic cells were seeded in 12-well plates and allowed
to settle overnight in complete DMEM. The following day, the medium was removed
and BMMs were seeded directly on the osteoblast monolayer and the coculture system
was maintained in α-MEM. The isolated osteoblastic cells were stimulated to produce
RANKL via the supplementation of complete α-MEM with 10 nM 1α, 25
dihydroxyvitamin D3, and half-medium changes with fresh α-MEM/1α, 25
dihydroxyvitamin D3 carried out every two days. Upon the observed presence of
multinucleated cells, the cells were gently washed once with 1× PBS prior to fixation
with 4% paraformaldehyde for 15 minutes at room temperature. The fixed cells were
washed three times with 1× PBS and the osteoclasts formed were TRAP stained and
counted as previously described.
4.3.3. Flow cytometry analysis of bone marrow cell niche
4.3.3.1. Immunostaining of bone marrow cells
Immunostaining of cell surface markers using antibodies was performed based on the
protocol established by BD Bioscience. Briefly, bone marrow cells were extracted by
flushing the cavity of long bones of euthanized mouse with complete α-MEM as
described above. The collected bone marrow cells were pelleted by centrifugation at
350 g for 5 minutes. To minimise background signal, red blood cells (RBCs) lysis was
performed by resuspending the bone marrow cells in 1 ml of 1× BD Pharm LyseTM red
cell lysis buffer incubating for 5 minutes, occasionally tapping the samples gently.
Samples were then diluted with 9 ml of 1× PBS to terminate the RBC lysis and the cells
were pelleted to remove the supernatant. The cells were then resuspended in 1× PBS
and a cell count was conducted. The cells were once again centrifuged and resuspended
in ice-cold 1× PBS at a concentration of 1× 106 cells/ml.
Cell viability staining was then performed (with either LIVE/DEAD® Fixable Aqua
Dead Cell Stain Kits (Invitrogen) or Zombie UVTM Fixable Viability Kit (BioLegend))
according to manufacturer’s recommendations for 30 minutes at room temperature. If
the cells were not fixed, live/dead staining was assessed by the addition of 7-AAD (BD
Bioscience) just before running the samples through the flow cytometry machine.
Following staining with the fixable viability dyes cells were then washed with ice-cold
1× PBS and centrifuged. The following steps were then carried out on ice. The cells
were resuspended in 50 µl ice-cold wash buffer and the cell suspension was gently

Chapter 4: Materials and Methods
111
mixed with 50 µl of diluted antibody master mix in wash buffer and left to incubate for
30 minutes on ice in the dark. After the incubation, the cell suspension was diluted with
500 µl of wash buffer and cells pelleted at 350g for 5 minutes. Cells were washed twice
more by resuspension in 100 µl of wash buffer followed by pelleting at 350g..
If the cells were to be fixed, the pelleted cells were then resuspended in 400 µl of 1×
BD Stabilising Fixative (BD Biosciences) (~106 cells in 0.5 ml) and were stored at 4 °C
until required for flow cytometric analysis the following day (fixed cells were analysed
within 24 hours). If the cells were to be analysed on the same day and were not required
to be fixed, the cells were resuspended in 400 µl of wash buffer after the third washing
step and were stained with 7-AAD as previously mentioned.
Antibodies involved in this study were titrated to appropriate concentrations to
minimise background staining. Single stained controls were performed using
Compensation Particles Set (BD Biosciences) according to manufacturer’s instructions.
Unstained and Fluorescence Minus One (FMO) controls were also prepared using bone
marrow cells and were treated the same way as test samples. The FMO controls were set
up to facilitate identification and gating of cells in the context of data spread due to the
multiple fluorochromes in the flow panel (Maecker et al., 2006). An FMO control
contains all the fluorochromes in the designed panel, except for the one that is being
analysed and hence a 4-colour panel would require 4 separate FMO controls. FMO
setups were particularly important as many of the flow panels designed for this study
involved more than 4 fluorochromes whereby, despite using the appropriate antibody
concentrations, the major source of background staining tends to be fluorescence spill
over.
4.3.3.2. Flow cytometer and analysis
The FACSCalibur, FACSCanto II and LSR Fortessa TM SORP (BD Biosciences) at the
Centre for Microscopy, Characterisation and Analysis (CMCA) of UWA were used for
flow cytometric studies. FACSDiva (BD Biosciences) software was used for application
setup, data acquisition, and data compensation. The live and single mononuclear cells
were gated using Forward Scatter (FSC) and Side Scatter (SSC) plots and at least
100,000 events were recorded. Acquired and compensated data was then imported into
FlowJo software (Three Star) for analysis.

Chapter 4: Materials and Methods
112
4.3.4. Cryopreservation of cells for long term storage
Monolayer cells reaching subconfluency were trypsinised and pelleted as described
above. The cell pellet was then resuspended in 92% (v/v) FBS and 8% (v/v) DMSO and
aliquoted as 1 ml cell suspension per sterile cryovial. As a rule of thumb, a confluent
75-cm2 tissue culture flasks was divided into 4 cryovials. The samples were frozen at -
80 °C in an isopropanol equilibrated Mr. FrostyTM Freezing Container (Thermofisher,
USA), which is designed to achieve an optimal cryopreservation at the cooling rate of
approximately -1 °C/minute. Cryopreserved samples were subsequently transferred to
liquid nitrogen for long-term storage.
4.4. Molecular biology techniques
4.4.1. RNA extraction and quantification
4.4.1.1. General handling procedures
All buffers, PCR-grade nuclease-free water, microfuge tubes, micropipette tips and
equipment were sterilized by autoclaving or filtration prior to extractions. General
aseptic techniques were employed when handling samples and RNAs. RNAs were
stored in sterile Nuclease-free water at -80˚C. Upon removing from the freezer, RNAs
were allowed to thaw on ice. Caution was taken to avoid keeping RNAs on ice for a
long period of time so as to prevent RNA degradation.
4.4.1.2. Extraction of total RNA
4.4.1.2.1. RNA extraction from bone tissue
Total RNA was extracted from the long bones of wildtype and sanroque mutant mice
using TRIzol reagent (Invitrogen) according to manufacturer’s instructions. Briefly,
cleaned bone (with no muscle tissue attached) was snap-frozen using liquid nitrogen
and was immediately homogenised using a mortar and pestle. The snap-freezing and
grinding process was repeated several times until the tissue was completely
homogenised to powder form. An appropriate amount of TRIzol (500 μl/50 mg tissue)
was then added to the powdered tissue and thoroughly mixed by pipetting before
transferring the lysed tissue mixture into a 1.5 ml microfuge tube.
The mixture was spun down at 12000 g for 10 minutes at 4 ˚C to eliminate debris and
the supernatant was transferred to a new tube. Following 5 minutes incubation at room
temperature, chloroform (0.2 ml/1 ml TRIzol) was added to the supernatant. The
mixture was mixed vigorously by hand, incubated for 2 minutes at room temperature

Chapter 4: Materials and Methods
113
and spun down at 12000 g for 15 minutes at 4 ˚C. The transparent upper phase
containing RNA was carefully transferred to a nuclease-free 1.5 microcentrifuge tube
and 100% isopropanol (0.5 ml/1 ml TRIzol) was added. The mixture was gently mixed
and left to incubate at room temperature for 10 minutes. The sample was then
centrifuged at 12000 g for 10 minutes at 4 ˚C. The supernatant was removed to isolate
the RNA pellet and 75% ethanol (1 ml/1ml TRIzol) was added to wash the pellet. The
sample was briefly vortexed, and then centrifuged at 7500 g for 5 minutes at 4 ˚C. The
supernatant was discarded and the RNA pellet was left to air dry for 5 minutes. 30 – 50
μl of nuclease-free water was directly added to resuspend the RNA pellet and the
sample was incubated at 55 ˚C for 10 minutes to thoroughly dissolve the RNA. The
RNA was ready for immediate downstream application, or stored at -80 ˚C until future
use.
4.4.1.2.2. RNA extraction from cell culture
Total RNA was isolated from cells in culture using TRIzol reagent (Invitrogen)
according to manufacturer’s protocol as mentioned above. Briefly, cells cultured in 6 or
12-well plates were washed once with 1× PBS and directly lysed by adding TRIzol (1
ml and 0.5 ml in 6 and 12-well plate respectively). The cells were scraped and mixed by
pipetting repeatedly prior to transferring into nuclease-free 1.5 ml microcentrifuge tubes
and the RNA isolation and wash was carried out as described above. 20 μl of nuclease-
free water was used to resuspend the pelleted RNA isolated from cell cultures.
4.4.1.3. Quantification of RNA concentration
The concentration of RNA was determined spectrophotometrically by Nanodrop® ND-
1000 UV-Vis Spectrophotometer V3.3 located in WAIMR, WA. Each RNA sample was
diluted 1:4 in nuclease-free water and measured against a nuclease-free water blank.
Two microlitres of the blank or the diluted sample were placed on the lower
measurement pedestal of Nanodrop® and were measured at 260nm and 280nm. The
measurements were repeated three times using separate drops of the sample.
The quality of RNA was determined by the ratio of the readings at 260nm and 280nm
(A260/A280), which provides an estimate of the purity of RNA with respect to
contaminants that absorb UV light, such as protein. Pure RNA has an A260/A280 ratio
of 1.5 – 2.0 and partially dissolved RNA has a ratio of <1.6 (Fleige et al., 2006;
Wilfinger et al., 1997). All purified RNA samples measured for experiments in this

Chapter 4: Materials and Methods
114
thesis had A260/A280 ratios higher than 1.8.
4.4.2. Reverse Transcription Polymerise Chain Reaction (RT-PCR)
4.4.2.1. Reverse Transcription (RT)
Reverse transcription was carried out in a 20 μl volume, with 1000 ng RNA extracted
from bone tissue and cell culture as the starting template. All procedures were
completed on ice unless stated otherwise.
Reagent Volume
RT 1*
RNA 1 μg
dNTP (20 mM) 2 ul
Oligo dT (100 μM) 0.25 μl
Nuclease-free water x μl
*Heated at 75 ˚C for 3 minutes before adding RT2.
RT 2
5× annealing buffer 4 μl
Moloney Leukaemia Virus (MMLV)-Reverse
Transcriptase 0.5 μl
Rnase inhibitor 0.5 μl
Total volume 20 μl
The RT1 master mix, placed in a thin-walled, nuclease-free PCR tube, was spun briefly
to collect all reagent/solution at the bottom of the tube prior to heating in a PCR
machine at 75 ˚C for 3 minutes to disrupt secondary structures. The reaction mixtures
were returned on ice to promote primer annealing and this was followed by the addition
of RT2 master mix. The reaction mixture was gently mixed, spun briefly and incubated
at 42 ˚C for 1 hour to facilitate cDNA transcription. Inactivating the enzyme at 92 ˚C for
10 minutes then terminated the process. Synthesized cDNA was stored at -20 ˚C. As a
negative control, the RNA template was replaced with nuclease-free water to check for
genomic contamination.
4.4.2.2. Polymerase Chain Reaction (PCR) amplification
Polymerase chain reaction (PCR), to amplify the house-keeping gene 18S and other

Chapter 4: Materials and Methods
115
gene markers, was carried out to ensure the presence and the quality of cDNA, and the
specificity of the primers before proceeding with real-time PCR. Where possible
primers used in this study were designed to be specific to the sequence of intron-intron
junctions of cDNA to avoid the possibility of amplifying any possible genomic
contaminants in the cDNA stock.
Reagent Volume
cDNA 1 μl
Forward primer (20 μM) 0.5 μl
Reverse primer (20 μM) 0.5 μl
2× GoTaq® Green Master Mix 10 μl
Nuclease-free water 8 μl
Total volume 20 μl
All the reaction mixes were prepared on ice. Each reaction was mixed to distribute the
ingredients evenly and was briefly centrifuged before placing into a thermal cycler.
Both non-template (negative) PCR and negative RT controls were also prepared to
identify possible contamination that might have occurred during both RT and PCR
steps.
The standard PCR cycling profiles comprised of an initialization step carried out at 94
˚C for 5 minutes, followed by denaturation at 94 ˚C for 40 seconds, annealing at 60 ˚C
for 40 seconds (all primers were designed so that the annealing temperature can be
carried out at 60˚C), and elongation at 72 ˚C for 40 seconds. The cycle of denaturation,
annealing and elongation was repeated 30 times prior to the final extension step at 72 ˚C
for 10 minutes, and was terminated and left at 4 ˚C indefinitely. The generated DNA
amplicons were loaded onto an agarose gel for electrophoresis to verify the size of the
fragments or stored at -20 ˚C.
4.4.2.3. DNA agarose gel electrophoresis
Agarose gel electrophoresis was performed using 1.5% (w/v) agarose gels prepared by
melting a agarose powder (1.5 g) in 1× TAE buffer (100 ml) in a microwave oven.
Ethidium bromide or Sybr Safe DNA stain (6 μl) was added to the molten agarose gel
and the mixture swirled gently to ensure uniform distribution. The UVTP gel tray (Bio-

Chapter 4: Materials and Methods
116
Rad) was fitted into the Gel Caster (Bio-Rad) and the well-forming comb(s) were
placed, prior to pouring the gel mixture into the tray.
The agarose was left to solidify at room temperature. The solidified gel, along with the
tray, was placed in a Mini-Sub Cell (Bio-Rad). 1× TAE buffer was poured into the cell
until the gel was completely submerged in the buffer and the comb was gently removed.
DNA ladder (3 μl), and samples and controls (9 μl) were loaded into individual wells
and the electrophoresis was carried out at 90 V for 30 minutes. For the purpose of the
experiments in this thesis, all primers were designed to amplify amplicons that were
below 500bp in size, thus only a 100bp (260 ng) DNA ladder (Promega) was used.
Separated DNA bands were visualised using ImageQuant LAS-4000 (GE) with
fluorescence via transilluminator (312 nm).
4.4.3. Quantitative Real-Time PCR (Q-PCR) and Livak’s Equation
Following primer optimisation using PCR as described above, real-time PCR was
performed using the same cDNA with the addition of the following ingredients and the
reactions were carried out in either 96 or 384-well PCR plate.
Reagent Volume
cDNA 1.5 μl
Forward primer (100 μM) 0.15 μl
Reverse primer (100 μM) 0.15 μl
SYBR Green PCR Master
Mix 7.5 μl
Nuclease-free water 5.7 μl
Total volume 15 μl
The SYBR Green PCR Master Mix (QIAGEN) contained HotStar Taq Plus DNA
Polymerase, QuantiFast SYBR Green PCR buffer, a mix of dNTPs, SYBR Green I dye,
which emits a fluorescence signal upon binding double-stranded DNA, and ROX dye
for the normalization of fluorescent signals. To reduce pipetting error, a master mix
sufficient for three reactions (to yield 3 CT values) and negative control was prepared
for every single primer set. Each master mix was mixed to distribute the ingredients
evenly and briefly centrifuged before distributing into the corresponding wells. The
real-time PCR was carried out using the ViiA™ 7 Real-Time PCR System (Life

Chapter 4: Materials and Methods
117
Technologies) with the following sequence: 95 ˚C for 10 minutes (activation) followed
by 40 cycles of denaturation at 95 ˚C for 15 seconds, annealing step at 60 ˚C for 20
seconds and an extension step at 72 ˚C for 20 seconds with a final elongation phase at
72 ˚C for one minute. Melt-curve analysis was also carried out to identify non-specific
products. Three CT values (threshold cycle) were obtained to deduce the mean CT value,
which was then used in Livak’s calculation as listed below to derive fold change values.
The experiments carried out in this thesis adopted the Livak’s ∆∆CT or comparative CT
method to analyse the relative fold change in gene expression of the samples and with
18S ribosomal RNA (Rn18S) serving as the internal housekeeping gene (Livak et al.,
2001; Pfaffl, 2001). Equation 1 shows the final form of Livak’s equation for a gene of
interest (GI):
Fold change = 2-∆∆CT
where ∆∆CT = ∆CT of sample A – ∆CT of sample B
∆CT = CT of GI – CT of Rn18S
CT = Mean of CT triplicate
4.4.4. Protein expression analysis using Sodium Dodecyl Sulfate-
PolyAcrylamide Gel Electrophoresis (SDS-PAGE) and western
blotting
4.4.4.1. Protein extraction from mouse organs
Approximately 5 mg of tissue from organs of interest was dissected out and rinsed
briefly with ice-cold 1× PBS. The rinsed tissue was placed in a pre-chilled mortar and
100 µl of ice-cold RIPA Lysis Buffer was added. The tissue was homogenised using the
pestle and the resulting lysate was transferred into a pre-chilled 1.5 ml microcentrifuge
tube. The lysate was passed through a 23G needle and syringe repeatedly to further lyse
the cells. The samples were left to incubate on ice for another 20 minutes. Cell debris
and intact cells were pelleted by centrifugation of the lysates at 12,000 g at 4 °C for 20
minutes. The clear supernatant was retained and transferred to fresh 1.5 ml
microcentrifuge tubes and stored at -20 °C (-80 °C for longer storage) until required.
4.4.4.2. Protein extraction from cultured cells
Monolayers of cultured cells in 6-well plates were washed once with ice-cold 1× PBS
and lysed directly by adding 200 µl of ice-cold RIPA Lysis Buffer per well and

Chapter 4: Materials and Methods
118
incubating on ice for 20 minutes. Cell lysis was assisted by scraping the cells with
pipette tips. The resulting lysates were collected and transferred to 1.5 ml
microcentrifuge tubes. Cell debris and intact cells were pelleted by centrifugation as
described above and the clear supernatant was retained and stored at -20 °C (-80 °C for
longer storage) until required.
4.4.4.3. Quantification of protein concentration
Protein concentrations were determined based on the method established by (Bradford,
1976) using the Bio-Rad Protein Assay kit. A bovine serum albumin (BSA) standard
curve was prepared by serially diluting 10 mg/ml BSA stock to generate 6 protein
standards of known concentration (0.05 mg/ml, 0.1 mg/ml, 0.2 mg/ml, 0.3 mg/ml, 0.4
mg/ml and 0.5 mg/ml). Using a 96-well microplate, 10 ul of each of the protein
standards and protein lysates of interest were aliquotted into each well containing 200 µl
of Bio-Rad Protein Assay dye reagent, which was diluted 1 in 5 with ddH2O and filtered
through Whatman filter paper. The samples were mixed gently and incubated at room
temperature for 5 – 10 minutes followed by an absorbance reading at 595 nm using the
Bio-Rad Microplate reader. Each protein standard and sample was assayed in duplicate
and triplicate respectively and RIPA lysis buffer served as the negative control. The
protein concentration was extrapolated from the BSA standard curve.
4.4.4.4. Separation of proteins by SDS-PAGE
Using the protein concentrations readings, an equal amount of protein (20 µg) per
sample was denatured by adding 4× SDS-PAGE sample loading buffer and heating to
99 °C for 5 minutes using the Thermomixer comfort (Eppendorf). After a brief
centrifugation, denatured protein samples were separated by SDS-PAGE using the
Mini-PROTEAN® 3 Cell Electrophoresis System (Bio-Rad). An acrylamide gel with
1.5 mm thickness consisting of a separating gel (10%) and a stacking gel was prepared
as described below:
Reagent Volume
30% Acrylamide/bis
solution 2.5 ml
1.0M Tris-HCl, pH 6.8 1.875 ml
10% SDS 75 μl
MilliQ H2O 2.97 ml

Chapter 4: Materials and Methods
119
10% APS* 25 μl
TEMED* 6 μl
Total volume 7.5 ml
*APS and TEMED were added last to initiate polymerisation.
Once mixed, the separating gel solution was immediately poured into a pre-assembled
glass plate sandwich aligned in a gel caster. The top surface of the separating gel was
overlaid with 20% ethanol and the gel allowed to polymerise at room temperature. The
ethanol was removed by inversion prior to the addition of 4% stacking gel solution,
prepared as follows:
Reagent Volume
30% Acrylamide/bis solution 0.5 ml
1.0M Tris-HCl, pH 6.8 375 ml
10% SDS 30 μl
MilliQ H2O 2.062 ml
10% APS* 30 μl
TEMED* 3 μl
Total volume 3 ml
*APS and TEMED were added last to initiate polymerisation.
The stacking gel solution was poured to overlay the separating gel and a 1.5 mm 15-
well comb was immediately inserted into the solution to create the sample wells. The
gel was left to set at room temperature for approximately 20 minutes after which the
comb was removed. Once the well comb was removed, the wells were gently rinsed
with ddH2O to remove air bubbles. The gel was then assembled into the Mini-
PROTEAN® 3 Cell Electrophoresis System filled with 1× SDS-PAGE running buffer
to cover the inner chamber and the electrode wire in the tank.
20 µl of denatured proteins and a pre-stained protein molecular weight ladder were
loaded into each well and the gel was electrophoresed at 100 V for approximately 2
hours at room temperature until the dye front of the 4× SDS-PAGE sample loading
buffer was observed dispersing from the bottom of the gel. The separated proteins were
then transferred onto a nitrocellulose membrane for detection of proteins using
antibodies specific to the target proteins of interest.

Chapter 4: Materials and Methods
120
4.4.4.5. Protein transfer onto nitrocellulose blotting membrane
Glass plates were disassembled from the electrophoresis apparatus and were gently
separated using a spatula to expose the gel. The stacking gel was removed and discarded
while the separating gel with protein samples was left to equilibrate in western blot
transfer buffer. Pieces of filter paper and a HyBondTM nitrocellulose membrane were
measured and cut to the dimensions of the gel. The membrane, filter paper and scotch
fibre pads were soaked in the transfer buffer.
The Mini Trans-Blot Electrophoretic Transfer Cell cassette was used to arrange the
“protein transfer sandwich” according to manufacturer’s instruction. The sandwich was
prepared while being submerged in the transfer buffer with the following arrangement:
With the negative (black) side of the cassette facing upward, pre-wetted scotch fibre pad
was laid on the positive (clear or white) side of the cassette in the transfer buffer. This
was followed with three sheets of the filter paper, the pre-wetted nitrocellulose
membrane, followed by the separating gel. The sandwich was completed with another 3
pieces of filter paper on top of the gel followed by the fibre pad, thus the gel is facing
facing the black terminal and membrane is facing the red terminal.
The sandwich was gently rolled with a 50 ml tube to roll out any trapped bubbles and
the negative side of the cassette was gently pressed down onto the sandwich. The
cassette was locked firmly with the white latch and the sandwich was inserted into the
module. The apparatus were then placed in the Western Transfer tank filled with chilled
transfer buffer. An ice block was placed in the tank to prevent over-heating and the
transfer was left to run at a constant current of 0.03 A overnight.
4.4.4.6. Western blotting
After the transfer, the sandwich was unclamped, and the pads, filter papers and gel were
removed to expose the protein-containing membrane. The membrane was immediately
rinsed in 1× TBS for 5 minutes three times. To reduce non-specific binding of the
primary antibody, the membrane was then blocked by immersion in 1× TBS-Tween
containing 5% (w/v) skim milk for 1 hour with gentle rocking at room temperature. The
membrane was then washed with 1× TBS-Tween for 5 minutes at room temperature. To
detect protein expression, the membrane was probed with primary antibody specific to
the protein of interest, diluted according to dilutions listed in 4.1.6 in 1× TBS-Tween
with 1% (w/v) skim milk, and the membrane was then incubated with gentle rocking for

Chapter 4: Materials and Methods
121
at least 4 hours (or overnight) at 4 °C.
The diluted antibody was removed and kept at -20 °C for future use. The membrane was
again rinsed three times with 1× TBS-Tween for 5 minutes per wash before it was
incubated with secondary HRP-conjugated antibody (specific to the host in which the
primary antibody was raised) diluted in 1% skim milk/1× TBS-Tween solution for 45
minutes at room temperature. The secondary antibody solution was then discarded and
the membrane was washed twice in TBS-Tween for 5 minutes and a further two times
in 1× TBS for 5 minutes.
To visualize protein expression, the Western Lightning Ultra Chemiluminescence
Substrate (Perkin-Elmer) was used according to the manufacturer’s instruction. The
membrane was allowed to incubate with the detection solution for 2 minutes at room
temperature in the dark. The detection solution was removed and the membrane was
imaged using the ImageQuant LAS-4000 luminescent image analyser (GE).
4.5. Statistics and data presentation
Data charts were generated and statistical analysis were performed using Microsoft®
Office Excel (Microsoft) software. All data presented in this study is expressed as mean
± standard error of the mean (SEM). The results are representative of at least three
independent experiments and biological repeats. For a direct comparison between
wildtype sample and sanroque mutant sample, an unpaired Student’s t-test was
performed. Statistical significance was assumed at p-values <0.05.

CHAPTER FIVE
BONE PHENOTYPIC CHARACTERISATION OF
SANROQUE MUTANT MICE

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
123
5. CHAPTER 5: BONE PHENOTYPIC CHARACTERISATION OF SANROQUE MUTANT MICE
5.1. Introduction
Bone homeostasis is dynamically maintained by the bone remodelling cycle, which is a
complementary effect of bone resorbing osteoclasts and bone forming osteoblasts
(Henriksen et al., 2009). To achieve a healthy and strong bone tissue, the bone
remodelling process is meticulously regulated in order to maintain bone homeostasis.
Dysregulation of this coupling activity between osteoclastic bone resorption and
osteoblastic bone formation leads to bone diseases, whereby overzealous resorption
results in osteolysis and osteoporotic bone, and an imbalance favouring bone formation
results in osteopetrosis (Teitelbaum, 2000).
The coupled process is orchestrated by different cytokines and factors released locally
and systemically by different cell types (Martin et al., 2006; Morgan et al., 2008). With
the emerging field of osteoimmunology, many studies have shown that the immune
system plays a major role in bone homeostasis, and vice versa (Arron et al., 2000a;
Arron et al., 2000b). The co-regulation and dialogue between the bone organ and
immune system is highlighted in many inflammatory bone diseases, many of which are
associated with excessive bone resorption resulting in net bone loss. These conditions
can be very debilitating and place an economic burden on the society. Many of the
therapeutic treatments available are not sufficient to eradicate the diseases, and little is
known to prevent the occurrence of aberrant bone remodelling. This is because little is
understood about the mechanisms that regulate normal physiological bone remodelling,
let alone the mechanisms that tipped the bone homeostasis in these autoimmune
conditions. Hence, it is crucial to focus our research on identifying the mechanisms
regulating osteoimmune homeostasis and thus, discovering novel therapeutic targets to
prevent and treat these inflammatory bone diseases.
An effort to identify novel genes regulating bone remodelling and osteoimmunology
was carried out in collaboration with the Australian Phenomics Facility of the
Australian National University by employing an ENU-induced mutagenesis screening
approach as described in Chapter 2. Bone phenotype screening of ENU-induced mutant
mouse lines using X-ray led us to the mutant mouse line that is studied in this thesis,
the sanroque mutant mice, in which the mutation and the pre-existing autoimmune
condition had been previously mapped and characterised (Vinuesa et al., 2005a).
The sanroque mutation lies in the Roquin (Rc3h1) gene that encodes for an E3 ubiquitin

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
124
ligase, resulting in a T to G substitution at the ROQ domain. The E3 ubiquitin ligase
family is one of the enzymes responsible in the enzymatic cascades of the ubiquitin
proteasome system (UPS). UPS has been implicated in many physiological conditions,
including the immune system and bone homeostasis, as the system is crucial in the
maintenance of protein homeostasis.
As highlighted in Chapter 3, the hindlimbs, vertebrae and tails of the sanroque mice
screened using X-ray indicate lower bone mass when compared to wildtype littermates.
This chapter reports the quantitative characterisation of bone phenotype of sanroque
mutant mice.
5.2. Experimental results
5.2.1. Roquin gene expression is regulated during osteoblastogenesis and
osteoclastogenesis and is highly expressed in hematopoietic organs
The mRNA expression of Roquin gene encoding for the Roquin E3 ligase is expressed
in different mouse tissues and cell lines and is most abundantly expressed in
haematopoietic organs such as the bone and spleen (Figure 5.1) (Wu et al., 2009). It is
also interesting to note that haematopoietic stem cells and common myeloid
progenitors, which give rise to osteoclast progenitors, also highly express Roquin gene.
Q-PCR analysis of Roquin gene expression throughout osteoblast and osteoclast
differentiation revealed that Roquin expression is up regulated during the course of
osteoblastogenesis (Figure 5.2 A) but is down regulated during the process of
osteoclastogenesis (Figure 5.2 B). The relative differentiation state of the cells was
assessed by the expression of the marker gene osteocalcin for osteoblasts and calcitonin
receptor for osteoclasts. Such regulation of Roquin expression during differentiation
highlights the potential regulatory role of the gene in bone biology.
5.2.2. Sanroque mutation alters trabecular and cortical bone morphology
To investigate the effect of the mutation in Roquin on the maintenance of bone
remodelling in vivo, the bone architecture of the distal femurs of adult male sanroque
mutant and wildtype littermate mice (average of 100 days old) were quantitatively
analysed using Micro Computed Tomography (µCT) to obtain trabecular and cortical
bone parameters (Bouxsein et al., 2010).
The µCT data revealed that there was a significant reduction in trabecular bone volume

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
125
normalised to tissue volume fraction (BV/TV) in male 100 day old sanroque mutant
mice compared to littermate controls (Figures 5.3 A and B). This was accompanied by a
decrease in trabecular number (Tb.N) and an increase in trabecular separation (Tb.Sp)
(Figures 5.3 C and D). Despite observing no significant change in the trabecular
thickness (Tb.Th), the structure model index (SMI) value is higher in the sanroque
mutant mice, indicating weaker trabecular bone structure in comparison to that of
wildtype mice (Figures 5.3 E and F). The bone phenotype appeared to be largely
consistent between both sexes whereby the changes in trabecular parameters were also
observed in the female sanroque mice; however, no statistically significance change
was observed in SMI in female mice (Supplementary figure 1).
Consistently, the cortical bone volume (Ct.BV) and thickness (Ct.Th) were significantly
reduced in the femurs of the sanroque mutant mice compared to littermate controls
(Figures 5.3 G and H). While there was a trend of reduction in the cross-sectional area
inside the periosteal envelope (Tt.Ar), the cross-sectional cortical bone area (Ct.Ar)
within this region is significantly reduced and thus there is a significant reduction in the
cortical area fraction (Ct.Ar/Tt.Ar) in the sanroque mutant mice compared to the
wildtype mice (Figures 5.3 I, J and K). The marrow area (M.Ar) was also lower in the
mutant femurs, however it did not reach statistical significance (Figure 5.3 L).
Collectively these results indicate that the cortical bone of the sanroque mutant mice is
thinner. This analysis established the decreased bone mass of the sanroque mutant mice.
H&E, Goldner Trichrome and TRAP staining was performed on sections of tibias from
100 day old male sanroque mutant and wildtype mice. Visual examination of the
stained sections showed an obvious reduction of bone mass in the tibia of sanroque
mutant mice (Figures 5.4 A, 5.5 A and 5.6 A). Histomorphometric analysis was
consistent with the μCT results, male sanroque mutant mice showed a significant
reduction in trabecular bone volume normalised to tissue volume (BV/TV) when
compared to wildtype littermate controls (Figure 5.4 B). The reduction in BV/TV
corresponded to the significant decrease in trabecular number (Tb.N) and increase in
trabecular separation (Tb.Sp) in the tibias of sanroque mutant mice (Figures 5.4 C and
D). Again no significance difference in the trabecular thickness (Tb.Dm) of the tibias
was observed (Figure 5.4 E). These data further confirm the osteopenic phenotype of
sanroque mutant mice.

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
126
5.2.3. Sanroque mutant mice have increased osteoclast numbers in vivo
To determine if the bone loss observed in sanroque mutant mice is associated with
increased osteoclastogenesis, histomorphometry quantification of TRAP-positive
osteoclasts along bone surfaces was performed. Compared to littermate controls,
sanroque mutant mice have significantly more osteoclasts per unit bone surface
(N.Oc/BS) (Figure 5.5 A). There is no significant difference in the length of osteoclast
surface, however as there is reduced bone surface in the mutant mice there is a trend of
increase in osteoclast surface normalised to bone surface (Figures 5.5 B and C). The
increase in TRAP staining is also apparent on the macroscopic view of the calvarial
surface of the sanroque mutant mice (Figure 5.6 A). This relates to an increase in
TRAP-positive osteoclasts observed by visual microscopic examination of calvarial
sections of sanroque mutant mice when compared to wildtype control (Figure 5.6 B).
Histological analysis thus revealed that an abnormal increase in osteoclasts might
contribute to the osteopenic phenotype of sanroque mutant mice.
5.2.4. Bone mineral apposition rate in sanroque mutant mice is reduced
despite unchanged histological osteoblast parameters
Additional histomorphometry studies were performed on Goldner Trichrome sections to
analyse osteoblast parameters to further examine the possible cellular mechanism of
bone loss observed in sanroque mutant mice. Quantitative analysis on Goldner
Trichrome sections revealed a non-significant trend of increase in osteoblast number per
unit bone surface (N.Ob/BS) in the tibias of sanroque mutant mice when compared to
that of wildtype littermates (Figure 5.7 A). Whilst there is no significant difference in
osteoid surface (OS), there is a trend of increase in osteoid surface normalised to bone
surface (OS/BS) in the tibia of sanroque mice (Figures 5.7 B and C). Histological
assessment hence revealed no significant aberration in osteoblast activity in sanroque
mutant mice.
To investigate the anabolic activity of osteoblasts, Fluorescence-calcein and Alizarin-
red double labelling was performed to quantify bone formation in vivo. Despite
observing a trend of increase in osteoblast number, the measurements of interlabel
distance revealed a significant reduction in the mineral apposition rate (MAR) in
sanroque mutant mice when compared to wildtype littermates (Figure 5.8). Therefore,
the osteopenic phenotype of sanroque mutant mice may partially be due to abnormality

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
127
in osteoblast activity.
5.2.5. RANKL and osteoclast gene markers are highly expressed in
sanroque bone
Total RNA extracted from the bone tissues of 100 days old wildtype and sanroque mice
was reverse-transcribed in order to assess the expression levels of osteoblast and
osteoclast gene markers. Relative mRNA levels of both wildtype and sanroque bone
analysed using Q-PCR showed a significant increase in the expression level of
osteoclast markers TRAP and Cathepsin K (CatK) in the sanroque mutant (Figure 5.9
A). This data is consistent with the increase in TRAP-positive osteoclasts ex vivo
observed in histological analysis of sanroque mutant mice described above. There is no
statistical significance in the difference in expression levels of osteopontin in the
sanroque mutant bone relative to wildtype bone (Figure 5.9 B). However, expression of
the osteoblast gene marker osteocalcin was significantly reduced, which corresponds
with the observed defect in mineralisation in sanroque mutant mice.
To explore the possible molecular mechanism of bone loss in sanroque mutant bone,
the expression levels of two key cytokines of bone remodelling RANKL and OPG were
also analysed. The expression levels of RANKL and OPG were both increased in the
sanroque bone in comparison to wildtype bone (Figure 5.8 C). However, the increase in
RANKL was higher than that in OPG, hence despite the increased OPG expression
level in sanroque mutant bone, the relative RANKL:OPG mRNA ratio remained
significantly higher (Figure 5.8 D). This correlates with the increase of histologically
quantitated osteoclast number and the elevated expression of osteoclast gene markers in
the sanroque mutant mice. This observation suggests that the elevated RANKL
expression may contribute to the osteoclast abnormality and the osteoporotic-like
phenotype of sanroque mutant mice.
5.3. Discussion
By employing genome-wide, random mutagenesis with the ethylating chemical ENU to
produce and screen genetic variants for osteological phenotypes, we have identified the
low bone mass phenotype of the sanroque coisogenic mutant strain that was previously
characterised as an autoimmune mouse model (Vinuesa et al., 2005a). Genetic
sequencing has mapped the single point M199R mutation in the Roquin gene that
resulted in the loss of function of the E3 ubiquitin ligase Roquin in the sanroque mutant

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
128
mice. To our knowledge the involvement of Roquin gene in bone homeostasis is
unreported to date, thus highlighting the benefits of ENU-mutagenesis phenotypic
screening to mine for novel molecules involved in the regulation of bone homeostasis.
Bio-GPS analysis showed that the Roquin gene is expressed highly in haematopoietic
organs such as the bone marrow and spleen. This suggests that Roquin may play a role
in the maintenance of haematopoiesis. Furthermore, wildtype haematopoietic stem cells
that give rise to common myeloid progenitors, myeloid cells, immune cells and
osteoclast precursors, expressed high level of Roquin gene. Using Q-PCR analysis, it
was revealed that the expression of Roquin gene is suppressed during RANKL-induced
osteoclastogenesis in wildtype cells, as opposed to osteoclast differentiation gene
markers such as calcitonin receptor. Such a trend supports the fact that Roquin is highly
expressed in common myeloid progenitors in which pre-osteoclasts are derived from,
and that Roquin potentially negatively regulates osteoclast differentiation.
By comparison, the gene expression of Roquin is upregulated throughout
osteoblastogenesis in wildtype cells. Given that other upregulated genes, such as
osteocalcin, are known to be involved during the maturation of osteoblasts, it is possible
that Roquin may also play a role in regulating osteoblast function. Notwithstanding the
opposing trends of the gene expression during osteoblast and osteoclast differentiation,
such regulation of Roquin expression indicates the potential role of the gene in bone cell
biology.
The ex vivo bone phenotype characterisation of sanroque mutant mice provides the
firmest definition of function of Roquin in bone homeostasis. MicroCT analysis
highlighted the obvious overall reduction in bone mass of sanroque mutant mice.
Trabecular bone parameters such as trabecular bone volume (BV/TV), trabecular
separation (Tb.Sp) and trabecular number (Tb.N) indicate reduce trabecular bone mass
of the sanroque mutant mice. The structure model index (SMI) was developed to
provide an estimation of the microarchitectural deterioration of cancellous bone. An
aging and/or diseased bone is characterised by a conversion from plate elements to rod
elements, in which an ideal plate and rod structure has an SMI value of 0 and 3
respectively (Hildebrand et al., 1997). Consistently, the higher SMI parameter of the
sanroque mice corresponds to the weaker trabecular structure in comparison to that of
wildtype mice. The reduction in trabecular bone mass is accompanied by a decrease in

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
129
cortical bone mass in the sanroque mutant mice, as highlighted by reduced cortical bone
parameters such as cortical bone volume (Ct.BV), cortical thickness (Ct.Th) and cortical
area fraction (Ct.Ar/Tt.Ar).
Histomorphometric analysis further confirms the reduced bone mass of sanroque
mutant mice and could potentially provide some insights into the possible cellular
mechanisms underlying the osteopenic bone phenotype. The increase in osteoclast
number is evident in the TRAP-stained tibia and calvaria of sanroque mutant mice. This
observation agrees with the decreased bone mass of sanroque mutant mice, as increased
osteoclast numbers are associated with increased bone resorption. The increased
osteoclast number and activity in sanroque mutant bone also correlates with the
increased expression levels of the osteoclast gene markers TRAP and Cathepsin K.
Cathepsin K is a protease expressed predominantly by osteoclasts to support resorption
(Bossard et al., 1996), consequently, increased expression of Cathepsin K could
possibly indicate increased osteoclastic resorption in sanroque mutant mice. Hence,
sanroque mutation resulted in an increase in osteoclast number that potentially
contributed to bone loss.
It was intriguing to note the trend of increase in osteoblast number and osteoid surface
quantitated in Goldner-Trichrome-stained tibia sections of sanroque mutant mice. Such
observation theoretically equates with increased bone formation. Despite such
observation, calcein and Alizarin-red double labelling experiments revealed a reduction
in mineral apposition rate in the sanroque mutant mice, suggesting that the sanroque
mutation resulted in impaired osteoblast matrix deposition and mineralisation in vivo.
These observations implied that sanroque mutation possibly resulted in a condition
similar to osteomalacia, a bone disease characterised by increased amount of osteoid
surface that does not mineralise in a timely manner (Maricic, 2008). Consistently, Q-
PCR analysis also revealed a significant reduction in the expression level of the
osteoblast-specific gene marker osteocalcin, which plays a major role in mineralisation
(Neve et al., 2013), in the bone of sanroque mutant mice.
Collectively, our data suggest that there are abnormalities in both osteoclast and
osteoblast regulation of bone remodelling in the sanroque mutant mice. The bone
homeostasis was disrupted with the balance tipped in favour of bone resorption, and
subsequently increased bone turnover in the sanroque mutant mice. The apparent

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
130
increase in osteoblast number in sanroque mutant bone could possibly be a
compensatory effect in order for the mutant osteoblasts to cope with the defective
mineralisation and increased osteoclast resorption in vivo. Nonetheless, such assumption
has to be validated by measuring serum bone turnover markers, including biochemical
markers of bone resorption, such as serum C-telopeptide of type I collagen (CTX) and
tartrate-resistant acid phosphatise (TRACP-5b), and also bone formation markers, such
as serum procollagen type I N-terminal propeptide (PINP) and bone alkaline
phosphatase (BALP). It is also important to acknowledge the possibility of the existing
autoimmune condition that may contribute to the pathogenesis of systemic bone loss
observed in sanroque mutant mice.
In conclusion, through the bone phenotypic characterisation of sanroque mutant mice,
Roquin has been identified as a novel regulator of bone homeostasis. The sanroque
mutation appears to alter the physiological roles of bone cells in vivo, leading to
compromised bone microarchitecture and reduced bone mass of sanroque mutant mice.

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
131
Figure 5.1 Tissue distribution of Roquin (Rc3h1) mRNA expression. High-
throughput gene expression profiling using BioGPS (http://biogps.org) to investigate the
transcription levels of Roquin in mouse-derived tissues, organs and cell lines. The
expression of Roquin gene is highly elevated in hematopoietic organs such as the spleen
and bone marrow, and in haematopoietic stem cells, common myeloid progenitors and
immune cells.

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
132
Figure 5.2 Expression level of Roquin throughout osteoblast (OB) and osteoclast
(OC) differentiation. (A) Wildtype mouse calvaria-derived osteoblasts were cultured
with mineralising medium for 7, 14, and 21 days to stimulate osteoblastogenesis. (B)
Wildtype mouse derived-BMMs were stimulated with RANKL to drive
osteoclastogenesis for 5 days. RNA isolated from the cells was subjected to RT and Q-
PCR analysis to determine the expression level of Roquin throughout the differentiation
of bone cells. Rn18s was used as the housekeeping gene while osteocalcin and
calcitonin receptor were used as osteoblast and osteoclast marker genes respectively.
The expression levels analysed using Livak’s equation revealed that the expression of
Roquin increased throughout osteoblast differentiation, conversely Roquin expression
decreased throughout osteoclastogenesis. Data are presented as mean ± SEM. * p<0.05;
** p<0.001 comparison against osteoblast day 7 (OB day 7) and osteoclast day 0 (OC
day 0).
(A)
(B)

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
133
Figure 5.3 Mutant sanroque mice display reduced bone mass. (A) Representative 2D
μCT images and morphometric analysis of (B-E) trabecular and (G-L) cortical bone of
tibias from 100 days-old male wildtype and sanroque mice. Data are collected from 4
Wildtype sanroque (A) (A)
Sagi
ttal s
ectio
n Tr
ansa
xial
sect
ion
(B)
(C)
(D) (E)
Wildtype
sanroque
(F)
(I) (G) (H)
(J) (K) (L)

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
134
mice and are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no significance in
comparison to wildtype littermates, n = 4 per group.

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
135
Figure 5.4 Histological analysis showed mutant sanroque mice have reduced
trabecular bone mass. (A) H&E stained sections of 100 day old sanroque and wildtype
decalcified paraffin-embedded tibias. Histomorphometric quantification of (B) BV/TV,
(C) Tb.N, (D) Tb.Sp and (E) Tb.Dm of mice. Data are presented as mean ± SEM. *
p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype littermates. Data
are representative for 3 mice per group.
(B)
Wildtype sanroque (A)
(C)
(D) (E)
Wildtype sanroque

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
136
Figure 5.5 Increase osteoclast number in trabecular bone of sanroque mutant mice.
(A) TRAP stained sections of 100 day old sanroque and wildtype non-decalcified
plastic-embedded tibias. Histomorphometric quantification of (B) number of osteoclasts
per bone surface (N.Oc/BS, OC/mm), (C) osteoclast surface normalized to bone surface
(Oc.S/BS, %) and (D) osteoclast surface (mm). Data are presented as mean ± SEM. *
p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype littermates. Data
are representative for 3 mice per group.
Wildtype sanroque(A)
(B) (C)
(C) sanroque
Wildtype

Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
137
Figure 5.6 Increased TRAP staining on the surface and in sections of sanroque
mutant calvaria. (A) Whole calvariae were fixed and stained for TRAP to identify
osteoclasts on the calvarial surface. (B) Representative images of TRAP stained
sections of 100 day old sanroque and wildtype decalcified paraffin-embedded calvaria.
Original magnifications ×10 (top) and ×20 (bottom).
(A) A)
Wild
type
sa
nroq
ue
Wildtype sanroque
×10
×20
(B)
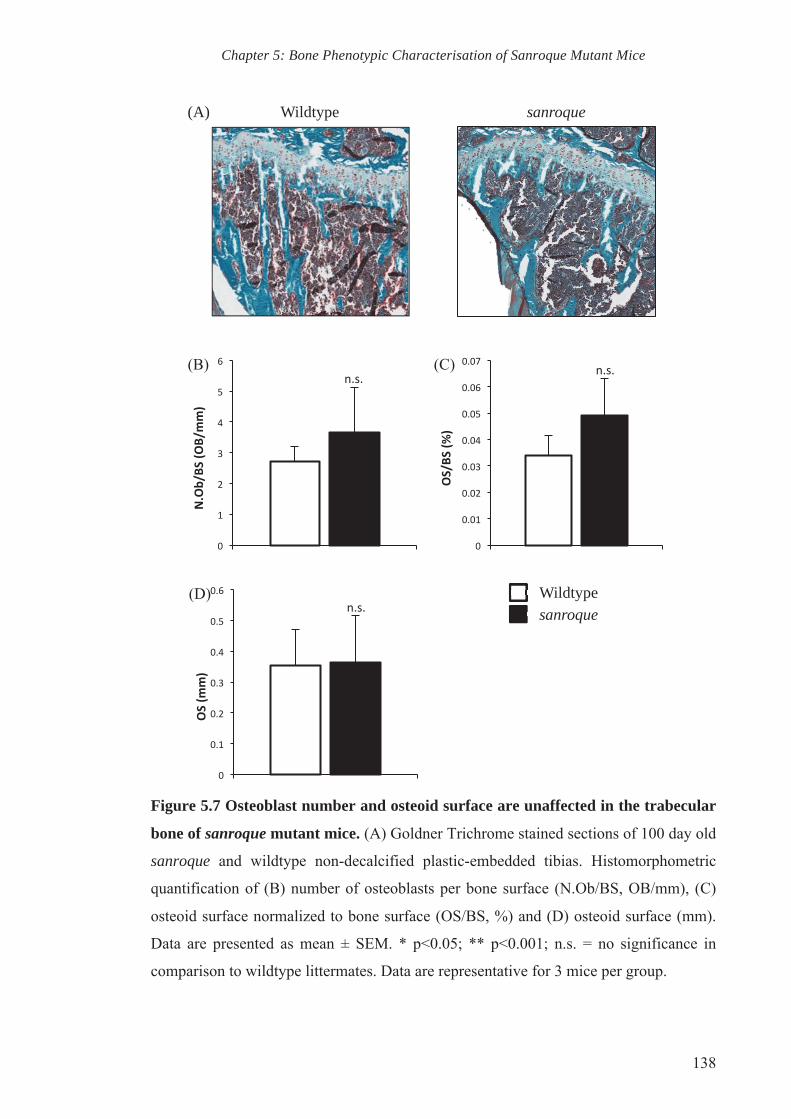
Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
138
Figure 5.7 Osteoblast number and osteoid surface are unaffected in the trabecular
bone of sanroque mutant mice. (A) Goldner Trichrome stained sections of 100 day old
sanroque and wildtype non-decalcified plastic-embedded tibias. Histomorphometric
quantification of (B) number of osteoblasts per bone surface (N.Ob/BS, OB/mm), (C)
osteoid surface normalized to bone surface (OS/BS, %) and (D) osteoid surface (mm).
Data are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no significance in
comparison to wildtype littermates. Data are representative for 3 mice per group.
Wildtype sanroque (A)
(B) (C)
(D) sanroque
Wildtype
Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
139
Figure 5.8 Decreased in vivo bone mineral apposition rate in sanroque mutant
mice. (A) Representative images of calcein and Alizarin-red double labelled tibias of 70
day old male wildtype and sanroque mice. (B) Consistent with the osteoporotic
phenotype, the mineral apposition rate (MAR) in sanroque mutant mice is significantly
reduced. Data are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no
significance in comparison to wildtype littermates, n = 3 per group.
sanroque
Wildtype
Wildtype sanroque (A)
(B)
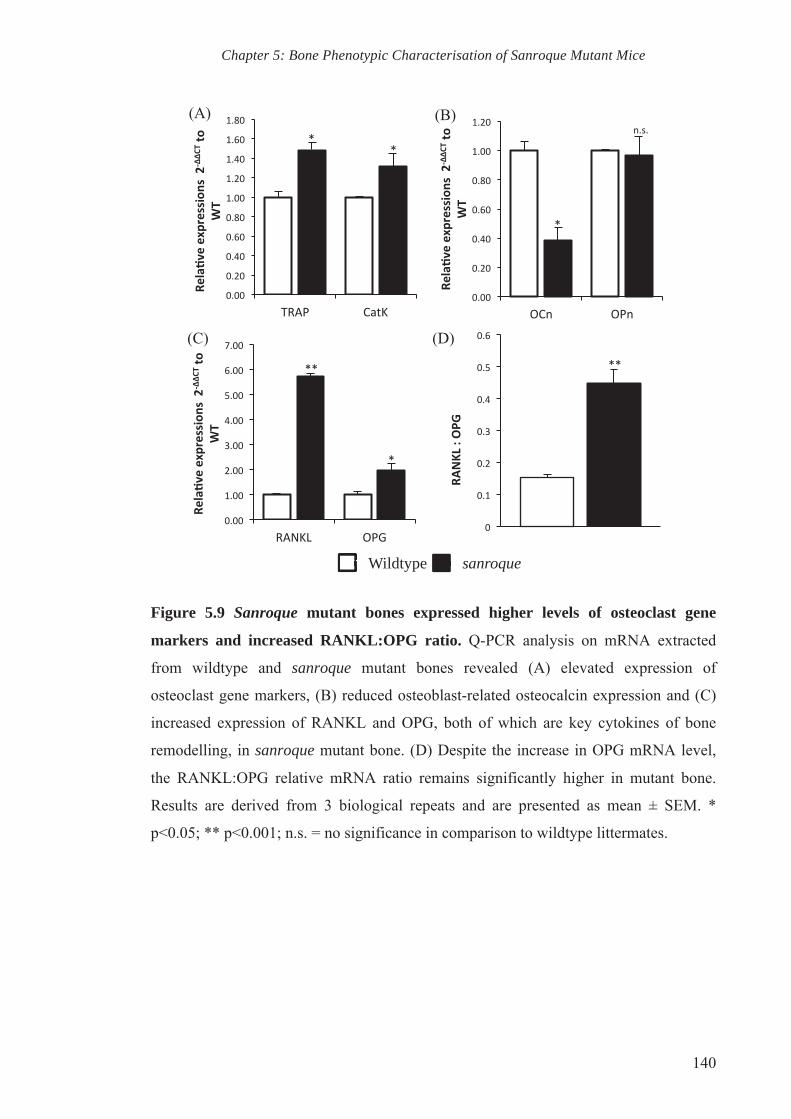
Chapter 5: Bone Phenotypic Characterisation of Sanroque Mutant Mice
140
Figure 5.9 Sanroque mutant bones expressed higher levels of osteoclast gene
markers and increased RANKL:OPG ratio. Q-PCR analysis on mRNA extracted
from wildtype and sanroque mutant bones revealed (A) elevated expression of
osteoclast gene markers, (B) reduced osteoblast-related osteocalcin expression and (C)
increased expression of RANKL and OPG, both of which are key cytokines of bone
remodelling, in sanroque mutant bone. (D) Despite the increase in OPG mRNA level,
the RANKL:OPG relative mRNA ratio remains significantly higher in mutant bone.
Results are derived from 3 biological repeats and are presented as mean ± SEM. *
p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype littermates.
(B)
(D)
Wildtype sanroque
(C)
(A)

CHAPTER SIX
IN VITRO INVESTIGATION ON THE ROLE OF ROQUIN IN
OSTEOBLAST BIOLOGY

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
142
6. CHAPTER 6: IN VITRO INVESTIGATION ON THE ROLE OF ROQUIN IN OSTEOBLAST BIOLOGY
6.1. Introduction
The sanroque ENU-induced mutant mouse model was reported to develop an
autoimmune disease consistent to SLE (Vinuesa et al., 2005a; Yu et al., 2007). As
established in the previous chapter, the sanroque mutant mice also display reduced bone
mass consistent with an osteopenic phenotype. The sanroque mutant mice carry a single
point mutation in the Roquin gene, which resulted in the loss of function of an encoded
protein that has a structure similar to an E3 ubiquitin ligase. Q-PCR analysis revealed
that Roquin gene transcription is upregulated throughout osteoblastogenesis, however is
suppressed during osteoclastogenesis.
Histomorphometric characterisation of the bone phenotype of sanroque mutant mice
revealed some insight into the possible cellular mechanisms underlying the reduced
bone mass. While a non-significant trend of increase in osteoblast number was observed
in the sanroque mice ex vivo, this analysis does not provide evidence of the osteoblast
functional defects that may contribute to the low bone mass phenotype. To assess
osteoblast function, double fluorochrome labelling was performed. Quantitative analysis
of the two labels revealed that sanroque mutants have reduced mineral apposition rate,
implying a possible deficiency in bone deposition by sanroque mutant osteoblasts in
vivo.
Consistently, Q-PCR revealed a reduction in osteocalcin gene transcription in the bone
of sanroque mutants. Given that osteocalcin is a terminal marker of osteoblastic
differentiation, this suggests that the sanroque mutation could possibly play a role in
osteoblastic gene expression and bone formation in vivo. In line with these findings, this
chapter investigates the role of Roquin in osteoblast biology through a series of in vitro
experiments.
6.2. Experimental results
Based on the aforementioned observations, we hypothesised that the low bone mass
phenotype of sanroque mutant mice was due to a cell-autonomous defect of osteoblasts.
To assess this, in vitro osteoblastogenesis was carried out on cells isolated from two
sources: the long bones and calvariae of wildtype and mutant mice. Due to the nature of
the bone tissues in which these cells come from, whereby long bones and calvariae are
formed via endochondral and intramembranous ossification respectively, we assumed
that the osteoblasts isolated from these two sources were pre-programmed differently to

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
143
form bone and hence were treated separately rather than being pooled together.
The calvaria isolated osteoblasts (COBs) and long bone isolated osteoblasts (LBOBs)
were cultured in osteogenic media to induce osteoid production and mineralisation.
Since larger numbers of cells are obtainable from the long bones, LBOB cell cultures
were also subjected to RNA and protein extraction for gene expression analysis and
alkaline phosphatase analysis respectively. In addition, the capability of sanroque
mutant LBOBs to support osteoclastogenesis was examined by performing an in vitro
co-culture system between osteoblasts and bone marrow macrophages.
6.2.1. Reduced bone mineralisation by sanroque mutant COB cultures
Osteoblasts isolated from calvariae of 100 days old mice were cultured with osteogenic
medium for 21 days in vitro. Alizarin red staining of mineralisation formed revealed an
obvious reduction in mutant sanroque COB cultures (Figure 6.1 A). Using ImageJ to
examine the percentage area of mineralisation, the analysis confirmed that the bone
formation by sanroque COBs is completely abrogated in comparison to wildtype
control (Figure 6.1 B).
In an attempt to rescue the abated osteoblast function from the calvariae of sanroque
mutant mice, COBs were cultured with osteogenic media in the presence of bone
morphogenetic protein-2 (BMP-2), a strong inducer of osteoblast differentiation. The
addition of BMP2 at a dose of 50 μg/ml significantly elevated the formation of
mineralised nodules by wildtype COBs, emphasising the potency of the cytokine as an
osteoinducer. However, the addition of BMP2 did not improve the abrogated
mineralisation in sanroque mutant COB.
These results supports the reduced mineral apposition rate based on the in vivo double
calcein labelling experiment in the sanroque mutant mice, suggesting that the mutation
in the Roquin gene may result in osteoblast-autonomous defect.
6.2.2. Increased calcium deposition by mutant sanroque LBOB cultures
To determine if the osteopenic phenotype of sanroque mutant long bones is due to cell
autonomous defects in osteoblasts, osteoblasts from long bones of 100 days old mice
were isolated and expanded. LBOBs were cultured in osteogenic medium for 21 days
and alizarin red staining showed more calcium deposition from sanroque mutant
LBOBs than from wildtype controls (Figure 6.2 A). Quantitative analysis of the area of

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
144
mineralisation revealed a significant increase in LBOB cultures from sanroque mutant
mice when compared to wildtype LBOB (Figure 6.2 C).
The addition of the osteoinducer BMP2 into wildtype LBOB cultures significantly
elevated the area of mineralisation (Figure 6.2 B and C). Interestingly, the addition of
BMP2 into sanroque LBOB culture did not significantly increase the area of
mineralisation, which was unexpected (Figure 6.2 B and C). On a closer inspection, the
areas of mineralisation formed by sanroque LBOBs appeared morphologically different
from those observed in wildtype LBOB cell cultures, which formed a distinctly
organised structure with relatively smooth edges. On the other hand, mutant sanroque
LBOBs formed calcified deposits that seemed irregular, and had rough edges (Figure
6.2 B). However, the morphology of the calcified deposits in vivo requires further
investigation with 3D-ultrastructural analysis using immunohistochemistry, confocal
microscopy and μCT. Collectively, these results show that there is a cell-autonomous
mineralisation defect and suggest an acceleration of osteoblast maturation in the long
bone of sanroque mutant mice.
6.2.3. Alkaline phosphatase activity level is altered throughout osteogenesis
in sanroque LBOB cell culture
During osteoblast differentiation, the matrix maturation phase is characterised by
maximal expression of alkaline phosphatase (ALP). To further evaluate the activity of
mutant osteoblasts of sanroque mutant mice, LBOBs were cultured in osteogenic
medium. Cell cultures were stained for ALP (blue staining) at the end point of day 21 to
reveal an increase in ALP staining in sanroque LBOB culture when compared wildtype
control (Figure 6.3 A). The staining however does not provide quantitative evidence of
ALP activity in sanroque mutant LBOB.
To determine ALP activity protein was collected from LBOB at 0, 7, 14 and 21 days
post osteogenic induction to determine ALP activity using colorimetric assay. As
expected, the ALP activity of wildtype LBOBs cultured in osteogenic medium
progressively increased throughout osteoblast differentiation and is significantly
elevated at day 14 and 21 in comparison to day 0. However, sanroque mutant LBOB
showed no significant changes in ALP activity during osteogenic induction (Figure 6.3
B). When compared against wildtype LBOB on day 0, 7 and 14, sanroque LBOB
displayed a trend of increase, albeit not significant, in ALP activity. At day 21, ALP

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
145
activity of sanroque LBOB decreased slightly. When ALP activity throughout
osteoblast differentiation by wildtype and sanroque LBOB is compared on respective
days, the difference however did not reach statistical significance, most likely due to the
large error bars.
6.2.4. Mutation of Roquin affects the expression levels of osteoblast gene
markers during differentiation of sanroque LBOBs
To gain some insight into the abnormal osteoblastic activity of sanroque LBOBs, the
expression of osteoblast gene markers was compared between wildtype and sanroque
mutant LBOBs. RNA was isolated from wildtype and sanroque LBOBs at day 0, 7, 14
and 21 of culture in osteogenic medium and reverse transcribed to obtain cDNA. The
expression levels of osteoblast gene markers alkaline phosphatase, osteocalcin,
osteopontin and osterix were monitored by Q-PCR and normalised against the house
keeping gene Rn18s.
By comparing the expression levels of alkaline phosphatase on each day, the mRNA
level of alkaline phosphatase in non-stimulated sanroque LBOBs (day 0) is significantly
lower in comparison to that of wildtype LBOBs (Figure 6.4 A). Seven days post
osteogenic induction resulted in a significant increase in alkaline phosphatase
expression in sanroque LBOB culture. However there is no significant difference
observed in day 14. The significant increase in the mRNA level of alkaline phosphatase
in sanroque LBOB in comparison to that of wildtype is observed on day 21. This
observation suggests a possible role of Roquin in regulating alkaline phosphatase
mRNA levels.
Wildtype LBOBs also show a significant trend of increase in osteocalcin expression
level throughout osteogenic induction (Figure 6.4 B). Comparative analysis of
osteocalcin expression at day 0 showed a significant reduction in non-stimulated
sanroque LBOBs relative to wildtype controls. Similar to alkaline phosphatase
expression level at day 7, the stimulation with osteogenic media resulted in a significant
increase in the expression level of osteocalcin by sanroque LBOB culture in
comparison to wildtype LBOB. However at day 14 and 21 there is no statistical
difference in osteocalcin expression level of both wildtype and sanroque LBOBs. When
compared at day 0, the sanroque LBOB culture showed a significant decreased in
osteopontin expression level. The expression level then increased to draw the same level

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
146
to wildtype in day 7 and 14. Despite showing no significant difference on day 7 and 14,
the expression level of osteopontin in sanroque mutant LBOBs is significantly higher
on day 21 when compared to wildtype control.
The upregulation of osteoblast gene markers during osteogenesis requires the
transcription factor osterix. The progressive increase in expression levels of alkaline
phosphatase, osteocalcin and osteopontin corresponds to the significant increase of
osterix expression level at day 14 and 21 against respective day 0 in wildtype LBOB
cell culture (Figure 6.4 D). In both wildtype and sanroque LBOB cultures, osteogenic
stimulation induced the expression of osterix as early as day 7. It is interesting to note
that at day 0, osterix expression in non-stimulated sanroque LBOBs is significantly
higher than that in wildtype LBOBs. The comparative analysis of osterix expression
between wildtype and sanroque LBOBs on day 7 and 14 revealed no significant
changes. However at day 21, the level of osterix expression is significantly elevated in
sanroque mutant LBOB culture when compared against wildtype culture.
Collectively, Q-PCR analyses indicate that the expression of osteoblast gene markers in
sanroque mutant LBOB throughout osteogenesis is altered. This could contribute to the
abnormal increase in in vitro mineralisation and altered alkaline phosphatase activity by
sanroque LBOB.
6.2.5. Mutant LBOB from sanroque mutant mice have reduced capability
to support osteoclastogenesis in vitro
As observed in Chapter 5, RANKL gene expression is increased in the long bones of
sanroque mice. As osteoblasts are a major source of RANKL in vivo, an in vitro
coculture system was set up to mimic in vivo osteoblast-osteoclast interactions. LBOBs
grown in monolayers were stimulated with 1,25-dihydroxyvitamin D3 (D3) to induce
RANKL and M-CSF expression. Bone marrow macrophages (BMMs) were then seeded
onto the LBOB monolayer and these factors then stimulate early osteoclast precursors
to differentiate into mature osteoclasts. The experiment was set up in four groups: (1)
wildtype LBOB and wildtype BMMs (WT OB/WT BMM), (2) sanroque LBOB and
sanroque BMM (san OB/san BMM); (3) wildtype LBOB and sanroque BMMs (WT
OB/san BMM), and (4) sanroque LBOB and wildtype BMMs (san OB/WT BMM).
Upon the formation of large multinucleated osteoclasts, the cell cultures were fixed and
stained for tartrate-resistant acid phosphatase (TRAP) activity. TRAP-positive

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
147
osteoclasts with more than 3 nuclei were then tallied.
Osteoclast counts from WT OB/WT BMM and san OB/WT BMM were compared to
explore the ability of sanroque osteoblasts to support osteoclast differentiation of
presumably “normal” BMMs from wildtype mice and the analysis revealed a significant
decline in osteoclast number in san OB/WT BMM cultures (Figure 6.5). This suggests
that sanroque mutant LBOBs have reduced capability to support osteoclastogenesis in
vitro. In contrast to this result, when sanroque BMM were cultured with wildtype
osteoblasts (WT OB/san BMM) in comparison to wildtype osteoblasts and BMM (WT
OB/WT BMM) a significant increase in osteoclasts was observed, suggesting enhanced
osteoclastogenesis by sanroque BMMs. This trend of increased osteoclastogenesis was
also observed when comparing san OB/ WT BMM with san OB/san BMM. Therefore,
osteoclastogenesis of sanroque BMM is not affected by the diminished ability of
sanroque LBOBs to support osteoclastogenesis. Overall, the assay revealed a
dysregulated cell-cell interaction between sanroque mutant LBOBs and BMMs that
may contribute to the osteopenic phenotype.
6.3. Discussion
In this chapter, the cellular mechanism(s) by which Roquin is involved in the
maintenance of bone homeostasis was investigated by primarily addressing the
differentiation and activity of osteoblasts isolated from sanroque mutant mice. The in
vitro experiments documented here have indicated the abnormal osteoblast activities in
sanroque mutant mice, which may contribute to the osteopenic phenotype.
Analysing their ability to form mineralised nodules in vitro provides an insight in the
maturation and activity of osteoblasts. Due to the nature of the bone organ tissues that
the cells originated from, calvaria isolated osteoblasts (COBs) and long bone isolated
osteoblasts (LBOBs) were treated as two distinct osteoblast populations. This is because
bone tissues develop via two different processes: intramembranous ossification to form
most of the craniofacial skeleton and endochondral ossification that is responsible for
formation of the long bones (Ortega et al., 2004). Endochondral ossification requires the
differentiation of mesenchymal cells into chondrocytes to serve as a template for bone
morphogenesis. This process requires the essential osteogenic factor Indian hedgehog
(Ihh), which is not required during intramembranous ossification where mesenchymal
cells are capable of directly differentiating into bone-forming cells (Chung et al., 2004;

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
148
St-Jacques et al., 1999). Different signalling pathways direct these two distinct bone
morphogeneses and hence bone cells from these two sources are pre-programmed
differently. Therefore, bone cells have distinct signalling properties that reflect their
origin and this should be considered in the research of bone biology.
The important differences between calvarial and long bone osteoblasts are especially
obvious in sanroque mutant osteoblast cultures. Mutant COBs and LBOBs responded to
osteogenic induction differently, whereby sanroque COBs failed to formed bone
nodules, whereas sanroque LBOBs displayed higher calcific deposits in comparison to
wildtype LBOB cultures (Figure 6.1 A and 6.2 A). Interestingly, osteogenic medium
stimulates both wildtype COBs and LBOBs to form mineralised nodules. The analysis
of mineralisation formed by sanroque COBs was consistent with the reduction of
BV/TV and MAR in the mutant bone as discussed in the previous chapter. Although it
seems contradictory, the increase bone nodule formation by sanroque LBOB does not
necessarily disagree with the μCT, histology and MAR results discussed above. The
accelerated mineralisation in vitro by sanroque LBOB could possibly be cell intrinsic,
which resulted in the abnormal osteoblastic machinery that affect normal calcium and
mineralisation as observed in bone nodules that are morphologically different from that
of wildtype (Figure 6.2 B). Nevertheless, the morphology of the bone nodules has to be
further confirmed with in vitro bone nodule 3D-ultrastructural analysis using a
combination of techniques such as transmission electron microscopy, X-ray microprobe
and fluorescence-confocal scanning laser microscopy (Bhargava et al., 1988; Xynos et
al., 2000).
Accelerated mineralisation is often associated with the production of immature, woven
bone characterised by poor lamellation and fragile bone, often seen in bone diseases
such as osteomalacia (Corsi et al., 2003; Tranquilli Leali et al., 2009). The increased
SMI of sanroque mutant bone that was picked up during μCT analysis indicates a
weaker bone structure. However, this has to be further validated by measuring the
biomechanical properties of the mutant bone. There is also a possibility of increased
mineralization despite the reduction of bone matrix deposited (MAR) in sanroque long
bones. This could be assessed by analysing parameters related to matrix mineralisation,
such as mineralisation lag time and osteocyte number. Hypothetically, defective
mineralisation should show decreased mineralisation lag time with an increase in

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
149
osteocyte number in vivo (Meury et al., 2010).
The cell autonomous defect of sanroque mutant osteoblasts is further shown by the
response of COBs and LBOBs toward BMP2. In COB cell culture, BMP2 was not able
to rescue to aberrant bone formation by sanroque COBs, whereas the cytokine
significantly elevated the formation of bone by wildtype COBs. The insensitivity
towards the potent osteoinducer BMP2 is also observed in sanroque LBOBs; however,
this could be because osteogenesis of sanroque LBOBs is inherently higher. A potential
reason for this observation is that the BMP2 signalling pathway itself may be altered
due to the sanroque mutation. Several publications have implicated the role of E3
ubiquitin ligases in BMP2 signalling (Zhang et al., 2001; Zhao et al., 2003; Zhu et al.,
1999). Further more, a recent publication has reported that BMP/Smad pathway is being
repressed in osteoblastic cells of SLE patients, leading to impaired osteoblastic
differentiation and may underlie the pathology of osteoporosis in these patients (Tang et
al., 2013b). Thus, the analysis of signalling pathway would be useful in order to
investigate the role of Roquin as an E3 ubiquitin ligase in BMP2 signalling.
Alkaline phosphatase and osteocalcin are both reported to play crucial roles in
mineralisation (Boskey et al., 1998; Orimo, 2010). Consistently, the accelerated
mineralisation by sanroque LBOB is accompanied by a significant increase in the
expression of osteoblast gene markers ALP and OCN within 7 days of osteogenic
induction (Figure 6.4 A and B). Osteopontin expression levels are also increased,
particularly at day 21 post osteogenic induction (Figure 6.4 C). The increase in
expression of transcription factor osterix at day 0 could partially explain the increase in
ALP and OCN in sanroque LBOBs (Figure 6.4 D), and hints at differential in vivo
regulation of osteoblast progenitors in sanroque mice. Q-PCR analysis revealed that
sanroque mutation affects the expression of osteoblast gene markers throughout
osteoblastogenesis, yet a more in-depth gene expression study is still required. The
analysis of other key osteoblast gene markers such as transcription factor Runx2 would
also provide valuable insight into the molecular mechanism regulating
osteoblastogenesis of sanroque mutant osteoblasts. Since sanroque mutant osteoblasts
were insensitive to osteoinduction by BMP2 by, the receptors for BMP2 expression
levels would be worth analysing. It would also be interesting to analyse the expression
of collagen I in order to further elucidate the molecular mechanism through which

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
150
Roquin regulates mineralisation.
The coculture experiments have provided a possible cellular mechanism underlying the
bone loss of sanroque mutant mice. BMMs of sanroque mutant mice showed enhanced
osteoclastogenesis and the result is consistent with the histomorphometric data of
increased TRAP positive osteoclasts in the long bones of sanroque mice. The increase
in osteoclast formation could have been explained by an increase in osteoclastogenic
factor production by sanroque LBOBs. However the co-culture experiments revealed
that sanroque mutant LBOBs have reduced capability to support physiological
osteoclastogenesis, hence indicating that the osteoblasts might not be the contributor of
osteoclastogenic factors in vivo. Since the RANKL:OPG ratio is elevated in long bones
(Chapter 5), this suggests that there might be other cell types that contribute to the
production of osteoclastogenic factors resulting in enhanced osteoclastogenesis in
sanroque mice.
The coculture experiment further revealed that BMMs from sanroque mice have
enhanced capacity to form osteoclasts irrespective of the supporting osteoblast cells. A
possible explanation for the enhanced osteoclastogenesis could be an increase of
putative osteoclast progenitors within the bone marrow of sanroque mutant long bones.
As a result, the pre-priming effect in vivo leads to an increase in osteoclast formation
supported by both wildtype and sanroque LBOBs in vitro. The differences between
activity of osteoblasts and osteoblasts-osteoclast interaction observed in vitro could also
be due to the alteration of cellular composition of the bone marrow in sanroque mutant
mice. Assessing the percentages of stem cells and progenitor cells in the marrow by
flow cytometry would provide an indication of the bone marrow composition. However,
it is also possible that the enhanced osteoclastogenesis is observed due to the existing
autoimmune condition of the mutant mice. As discussed in chapter 2, inflammatory
cytokines have been reported to have both pro- and anti-osteogenic effects on
homeostasis. These inflammatory cytokines indirectly affect the normal signalling
pathways governing the differentiation of bone cells in vivo. When taken out of this
inflammatory milieu, it is expected that they would behave differently in vitro as well.
In summary, this chapter characterises the osteoblast defects of sanroque mutant mice.
Through a series of in vitro experiments, the results have shown the potential role of
Roquin in osteoblast differentiation and function. Osteoblasts of sanroque mutant mice

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
151
showed abnormal mineralisation and reduced capability to support osteoclastogenesis.
However, the molecular mechanism(s) through which Roquin regulates osteoblast
functions remains to be resolved and should be addressed in future studies.
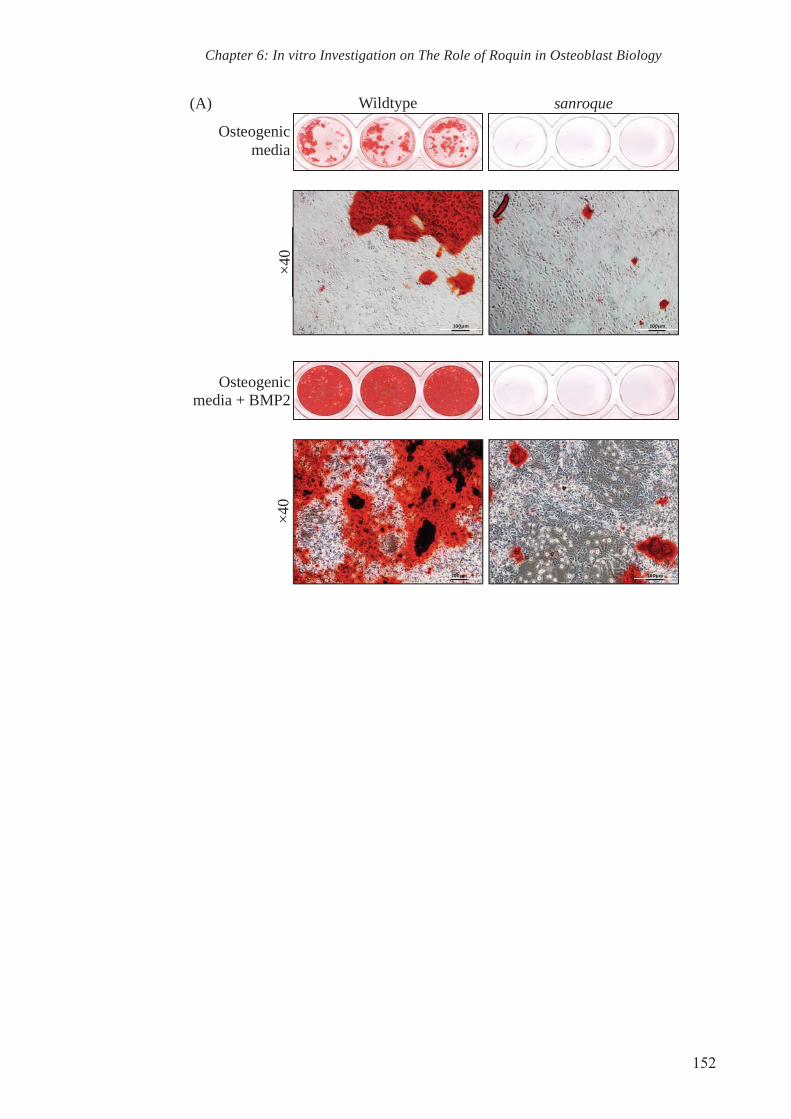
Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
152
Wildtype sanroque (A)
Osteogenicmedia
×40
Osteogenicmedia + BMP2
×40

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
153
Figure 6.1 Osteoblasts isolated from calvariae (COBs) of sanroque mutant mice
have reduced capability to mineralise in vitro. Osteoblasts isolated from calvariae
were cultured with osteogenic medium in the presence of dexamethasone (10 nM), β-
glycerophosphate (2 mM) and ascorbate (50 μg/ml) for 21 days in vitro. Upon the
apparent formation of mineralised areas, cell cultures were fixed with 1%
Glutaraldehyde and stained with alizarin red. (A) Representative images of wildtype and
sanroque mutant COBs at day 21 post-confluence. The reduction of in vitro
mineralisation by mutant sanroque COBs is clearly evident when compared wildtype
COBs culture. While 50 μg/ml of BMP2 further induced wildtype COBs to mineralise,
the cytokine did not rescue the aberrant bone formation by sanroque COB. (B) The
culture plates were scanned and mineralised area was quantitated using ImageJ to reveal
the significant reduction of mineralised nodule formation by sanroque mutant COBs,
with or without BMP2, in comparison to wildtype COBs. Results are representative of 3
independent biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls and to osteogenic
medium only cultures.
sanroque
Wildtype (B)

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
154
(A)
Wildtype sanroque
Osteogenic media
×10
×40

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
155
Figure 6.2 Enhanced mineralisation in sanroque mutant osteoblasts isolated from
long bones (LBOBs). Primary osteoblasts isolated from long bones of 100 days old
mice were cultured in osteogenic medium for 21 days to induce mineralisation in vitro.
(B)
Wildtype sanroque
Osteogenic media + BMP2
×10
×40
×80
sanroque
Wildtype (C)

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
156
(A) Representative images of alizarin red-stained mineralisation formed by wildtype
and sanroque mutant LBOBs. (B) The potent osteoinducer, BMP2, significantly
increased the mineralisation efficiency and the area of mineralisation in wildtype LBOB
cell culture. Interestingly, BMP2 did not have any effect on the area mineralised by
sanroque LBOBs. Observation under higher magnifications revealed disorganised
nodules formed by sanroque LBOB. (C) Quantification of the area of mineralisation
from the images in (A) and (B) using ImageJ. Results are representative of 3
independent biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls and to cell cultures
treated with only osteogenic medium in the presence of dexamethasone (10 nM), β-
glycerophosphate (2 mM) and ascorbate (50 μg/ml).

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
157
Figure 6.3 Mutant LBOBs of sanroque mutant mice displayed atypical alkaline
phosphatase (ALP) activity levels throughout osteogenesis. (A) Representative
images of primary LBOB cells cultured in osteogenic media for 21 days. Cells were
fixed and stained for ALP to reveal a more intense blue staining in sanroque LBOB cell
culture. (B) Colorimetric analysis of ALP activity revealed an abnormal trend of the
enzymatic activity throughout sanroque LBOB maturation as opposed to the steady
increase of ALP activity observed in wildtype LBOB culture. Results are representative
of 3 independent biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls and to respective day
0.
sanroque
Wildtype (B)
×80
Wildtype sanroque (A)
×40

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
158
Figure 6.4 Q-PCR analyses of osteoblast gene markers expression levels during
osteogenesis. Primary LBOB cells were cultured in osteogenic medium and RNA was
collected at day 0, 7, 14 and 21. Osteoblast differentiation was evaluated with Q-PCR
analysis of osteoblast marker genes (A) alkaline phosphatase, (B) osteocalcin, (C)
osteopontin and (D) osterix normalized to 18s ribosomal RNA expression. Results are
representative of 3 independent biological repeats and are presented as mean ± SEM. *
p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype controls.
Wildtype sanroque
(B)
(C) (D)
(A)

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
159
Figure 6.5 Coculture systems between LBOBs and bone marrow macrophages
(BMMs). (A) Representative images of four coculture groups: (1) wildtype LBOB and
wildtype BMMs (WT OB/WT BMM), (2) sanroque LBOB and sanroque BMM (san
OB/san BMM), (3) wildtype LBOB and sanroque BMMs (WT OB/san BMM) and (4)
sanroque LBOB and wildtype BMMs (san OB/WT BMM). LBOBs were stimulated
with 1,25-dihydroxyvitamin D3 (10 nM) to induce RANKL and M-CSF expression.
san OB/WT BMM WT OB/san BMM
WT OB/san BMM san OB/san BMM
(A)
sanroque LBOB/sanroque BMM
(B)
Wildtype LBOB/sanroque BMM
sanroque LBOB/Wildtype BMM Wildtype LBOB/Wildtype BMM
×10
×10

Chapter 6: In vitro Investigation on The Role of Roquin in Osteoblast Biology
160
Osteoclasts were identified by staining for tartrate-resistant acid phosphatase (TRAP).
(B) TRAP-positive osteoclasts with more than 3 nuclei were then tallied. Results are
representative of 3 independent biological repeats and are presented as mean ± SEM. *
p<0.05; ** p<0.001; n.s. = no significance when compared to each coculture system.

CHAPTER SEVEN
THE EFFECT OF SANROQUE MUTATION ON
OSTEOCLAST BIOLOGY

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
162
7. CHAPTER 7: THE EFFECT OF SANROQUE MUTATION ON OSTEOCLAST BIOLOGY
7.1. Introduction
As described in the previous chapter, the mutation in Roquin gene results in perturbed
osteoblast activity in vivo and in vitro, which in turn may contribute to the osteopenic
phenotype of the autoimmune sanroque mouse model. However, multiple observations
indicate abnormal osteoclast activity in sanroque mutant mice. Histomorphometry
studies revealed increased osteoclast number and TRAP staining in sanroque bone
tissues. Q-PCR analysis showed elevated expression of osteoclast gene markers TRAP
and Cathepsin K in the bone of sanroque mutants. Furthermore, the co-culture
experiment showed that sanroque bone marrow macrophages (BMMs) inherently
formed more osteoclasts in comparison to wildtype BMMs culture, regardless of
osteoblast source.
At a glance, these observations correlate well with the increased bone loss due to
increased osteoclast functions seen in many patients of autoimmune diseases. However,
the underlying mechanisms of the overzealous osteoclast activities in these pathological
conditions are complex and remain ill defined. Several osteoimmunology studies have
shown that immune cells and osteoclasts often share co-stimulatory signalling
molecules and pathways (Bozec et al., 2014; Franzoso et al., 1997; Zhang et al., 2013a).
Therefore, molecular mechanisms, such as the ubiquitin-proteasome system (UPS), that
mediate abnormal immune cell functions could also be functioning in osteoclasts and
may contribute to the bone loss in these autoimmune patients.
The systemic regulation of protein turnover by UPS underlies a wide variety of
molecular pathways essential for normal cellular functioning (Ang et al., 2009). UPS
mediates intracellular protein degradation by marking the protein of interest with a
chain of small polypeptides called ubiquitin. As described previously, UPS involves the
coordinated interplay between E1 (ubiquitin-activating), E2 (ubiquitin-conjugating) and
E3 (ubiquitin-ligating enzymes) (Pickart et al., 2004). In the context of osteoclast
differentiation, UPS is critical in mediating RANKL/RANK signalling cascades (Ang et
al., 2009). The ubiquitination of tumour necrosis factor receptor-associated factor 6
(TRAF6), a crucial adaptor of the cytoplasmic domain of RANK, is an essential step in
activating the signalling complex mediating osteoclast differentiation (Takayanagi et
al., 2000). Biochemical studies demonstrate that TRAF6 is in fact an E3 ubiquitin
ligase. Upon stimulation, TRAF6 promotes polyubiquitination of itself and of target

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
163
proteins of the NF-κB signalling pathway (Deng et al., 2000). The ubiquitination and
proteasomal degradation of phosphorylated IκB kinase allows the nuclear translocation
of NF-κB proteins to initiate gene transcription of downstream gene targets (Chen,
2005). NF-κB along with c-Fos, c-Jun, MITF and NFATc1 turn on the expression of
genes such as TRAP, Cathepsin K and DC-Stamp in osteoclasts (Qi et al., 2014).
In addition to TRAF6, an increasing number of E3 ubiquitin ligases have been recently
implicated in osteoclast development and function (Shu et al., 2013; Zhang et al.,
2013a). Roquin is reported to be an E3 ubiquitin ligase (Vinuesa et al., 2005a) and Q-
PCR analysis has shown that the expression of the gene is down regulated throughout
osteoclastogenesis (Chapter 3). Thus, in this chapter the effect of the sanroque
mutation, which resulted in loss of function of the protein Roquin, on osteoclast biology
is examined.
7.2. Experimental results
Initial characterisation of the bone phenotype of sanroque mutant mice revealed an
increase in osteoclast number and osteoclast gene expression markers. Based on these
observations, we hypothesised that the mutation in Roquin gene resulted in increased
osteoclast differentiation and function. To investigate this, bone marrow was extracted
from age and sex matched pairs of wildtype and sanroque mutant mice and a series of
in vitro experiments were then carried out to investigate osteoclast differentiation and
function.
7.2.1. Osteoclastogenesis is accelerated in sanroque mutant mice
To evaluate the formation of osteoclasts in sanroque mutant mice, in vitro
osteoclastogenesis assays were conducted. Bone marrow cells flushed from the long
bones of wildtype and sanroque mutant mice were expanded into bone marrow
macrophages (BMMs) in the presence of M-CSF. BMMs were subsequently stimulated
with RANKL and M-CSF over a time-course of 0, 1, 3 and 5 days to stimulate
osteoclastogenesis. The cells were fixed, TRAP stained and the numbers of
multinucleated osteoclasts generated were quantified. A significant increase in the
number of osteoclasts formed by BMM derived from sanroque mutant mice was
evident at days 3 and 5, indicating enhanced osteoclastogenesis in the mutant mice
(Figure 7.1).

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
164
The sensitivity of the wildtype and sanroque BMM cultures to RANKL was also
assessed by stimulating the cells with 12.5, 25 or 50 ng/ml RANKL for 5 days, while
the concentration of M-CSF was maintained. Quantitative analysis of the TRAP
positive multinucleated osteoclasts showed that RANKL induces a dose-dependent
increase in osteoclast number in both wildtype and sanroque BMM cultures (Figure
7.2). The increased osteoclastogenesis is also apparent in this assay, in which sanroque
mutant BMM generated significantly more osteoclasts in comparison to the number of
wildtype osteoclasts at all RANKL doses. This implies that sanroque mutant osteoclast
precursors are more sensitive to RANKL stimulation, or that there are increased
numbers of osteoclast precursors in the sanroque bone marrow, as they show increased
osteoclast numbers at lower doses of RANKL.
7.2.2. Sanroque mutation elevates the expression of osteoclast marker
genes
Total RNA isolated from RANKL-induced osteoclasts at various time points were
subjected to reverse transcription to yield cDNA. Q-PCR was performed to investigate
if the expression of osteoclast marker genes is altered in sanroque osteoclasts. The
crucial transcription factor NFATc1 involved in osteoclastogenesis is upregulated
during osteoclast differentiation of both wildtype and sanroque BMMs. In comparison
to wildtype osteoclast cultures, sanroque mutant osteoclasts showed a drastic increase
of NFATc1 expression at day 3 and 5 post-RANKL stimulation (Figure 7.3 A).
Concomitantly, the expression DC-Stamp, which encodes for the transmembrane
protein required for cell fusion during osteoclastogenesis, is markedly increased during
osteoclastogenesis of sanroque mutant cultures (Figure 7.3 B). These results suggest
that Roquin may play a regulatory role in the transcription of genes during osteoclast
differentiation upon RANKL stimulation. However, RANK expression levels showed
no significant difference throughout osteoclastogenesis between wildtype and sanroque
mutant cultures, indicating that the expression of RANK was not affected by sanroque
mutation.
7.2.3. Sanroque mutant osteoclasts demonstrate increased bone lytic
activity in vitro
The decreased bone mass of sanroque mutant mice could be associated with increased
osteoclast resorptive activity. To assess the consequence of sanroque mutation on

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
165
osteoclast function, in vitro bone resorption and secretion of the matrix proteinase
cathepsin K by mature osteoclasts was assessed.
To perform the bone resorption assay, wildtype and sanroque mutant BMMs were
cultured on a collagen-coated plate in the presence of M-CSF and stimulated with
RANKL to induce osteoclastogenesis. Upon the apparent fusion of BMMs to generate
small osteoclasts, cells were detached and counted. Similar numbers of small osteoclasts
were then seeded onto bovine bone discs in the presence of M-CSF and RANKL. After
36 hours the cell cultures on the bone discs were then fixed, TRAP stained and
multinucleated osteoclasts were tallied (Figure 7.4 A and C). Subsequently, the
osteoclasts were gently removed to reveal the “pits” formed by osteoclast resorptive
activity. The bone discs were stained with Toluidine blue and representative
microphotographs of bone resorption are shown (Figure 7.4 B). The darker blue areas of
resorptive pits were quantified and the data is presented as resorptive area per osteoclast
(Figure 7.4 D). In comparison to wildtype osteoclasts, each sanroque mutant osteoclast
demonstrated a significant increase in resorbed area indicating an increase in resorptive
activity.
To further evaluate the bone lytic activity of sanroque mutant osteoclasts, BMMs were
cultured in a plastic plate and stimulated to undergo osteoclastogenesis as described
above. Once multinucleated osteoclasts were observed, bovine bone powder was added
into the cell culture to initiate cathepsin K release into the medium. The supernatant and
cell lysates were collected at 4, 8 and 12 hours post addition of bone powder and were
subjected to western blotting. As shown in Figure 7.5, the addition of bone powder to
wildtype osteoclast cultures resulted in the gradual build up of endogenous cathepsin K
and prompted the release of the enzyme into the supernatant in a timely fashion. In
contrast, such a trend is not observed in sanroque osteoclast cultures. The secretion of
cathepsin K into the medium of sanroque osteoclast cultures was significantly increased
at all time points analysed (Figure 7.5). These results indicate that loss of function of
Roquin positively regulates cathepsin K release and bone resorption by sanroque
mutant osteoclasts. Thus, sanroque mutation causes increased numbers of highly active
osteoclasts to form in vitro from the bone marrow of mutant mice.
7.2.4. Mutation in Roquin gene alters key RANKL-signalling pathways
To investigate the possible mechanisms by which sanroque mutation promotes

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
166
osteoclastogenesis in vitro, RANKL/RANK signalling cascades were examined by
immunoblot analysis. BMMs from wildtype and sanroque mutant mice were serum
starved for four hours prior to stimulation with RANKL over a short time course of one
hour (Figure 7.6). Long time course of RANKL stimulation throughout
osteoclastogenesis was also performed (Figure 7.7). Total protein was extracted from
cell cultures at the indicated time points and western blots were performed for proteins
involved in major RANKL induced signalling pathways: ERK and p38 (MAPK
signalling), NF-κB signalling (IκB-α) and NFATc1 (Calcineurin-NFAT signalling),
with β-actin serving as an internal loading control.
As expected, RANKL stimulation rapidly induced the phosphorylation of ERK within
the first 30 minutes, followed by a rapid degradation of the protein in both wildtype and
sanroque mutant BMMs (Figure 7.6 A). Interestingly, semi-quantitative analysis of the
blots revealed significantly increased levels of phosphorylated ERK at all time points of
sanroque BMM cell cultures compared to wildtype (Figure 7.6 B). This suggests that
sanroque mutation augments the RANKL-induced ERK signalling pathway.
Endogenous phosphorylated p38 also accumulates within sanroque BMMs at earlier
time points as indicated by the marked increase in the protein level at 10 minutes post
RANKL-stimulation (Figure 7.6 A and B). Although the levels of phosphorylated p38
are comparable to wildtype over the remaining course of RANKL-stimulation, these
data suggest that sanroque mutation enhances RANKL-induced MAPK signalling
pathways.
Densitometry analysis also showed that RANKL-stimulation resulted in the rapid
degradation of phosphorylated IκB-α within the first 30 minutes, however the level of
the phosphorylated protein dramatically increased at 60 minutes. Such a trend is
observed in both wildtype and sanroque mutant cultures, though the level of
phosphorylated IκB-α of sanroque mutant cultures remained higher throughout the
experiment after the initial 10 minutes of stimulation, in comparison to wildtype control
(Figure 7.6 A and B). This indicates that the regulation of IκB-α phosphorylation and
degradation is altered in sanroque pre-osteoclasts.
Western blot analysis showed that protein expression of NFATc1 is upregulated during
osteoclastogenesis in wildtype cell cultures. The level of NFATc1 is sanroque mutant
precursors started off to be significantly lower than that of wildtype. However the

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
167
expression gradually caught up and by the third day of RANKL stimulation, the level of
NFATc1 in sanroque mutant cell cultures drastically increased in comparison to
wildtype cell cultures (Figure 7.7 A and B). The high levels of endogenous NFATc1 in
sanroque cell cultures persisted throughout until osteoclastogenesis was terminated at
day 4.
DC-Stamp and v-ATPase V0 subunit d2 protein expressions were used as markers of
osteoclast formation (Figure 7.7 A and B). Similar to NFATc1, the protein level of DC-
Stamp in sanroque BMM culture started lower than wildtype at day 0. By day 3, the
spike in NFATc1 coincided with the significant increase in DC-stamp protein
expression during osteoclastogenesis of sanroque BMMs in comparison to that of
wildtype. The expression of v-ATPase V0 subunit d2 protein showed a similar trend to
that of DC-Stamp, however the protein could only be detected after 3 days of RANKL
stimulation. The elevated protein expression levels of NFATc1 and DC-Stamp
throughout sanroque osteoclastogenesis were similar to that of the gene expression
analysed using Q-PCR shown above. Overall, the western blot analysis of long time
course RANKL stimulation supported the enhanced in vitro osteoclastogenesis observed
in sanroque cell cultures.
7.2.5. Ubiquitination of TRAF6 in sanroque BMMs appears unaltered
Data from in vitro investigations thus far have emphasised the increased
osteoclastogenesis in sanroque mutant BMM cultures. The increased osteoclastogenesis
could be due to the altered upstream signalling that contributes to the augmented effect
of RANKL/RANK signalling cascades in sanroque BMM cultures, which then resulted
in increased transcription of osteoclast gene markers. One of the earliest proteins being
recruited upon RANKL-stimulation is TRAF6 (Takayanagi et al., 2000). Bearing in
mind the role of Roquin as an E3 ubiquitin ligase, we set to investigate the
ubiquitination of TRAF6, which is crucial in mediating RANKL/RANK signalling.
As a prelude to understanding the E3 ubiquitin ligase activity of Roquin on TRAF6, we
obtained wildtype and mutant Roquin (M199R) plasmids to conduct in vitro
ubiquitination assay using HEK293 cells (Figure 7.8 A). The GFP-tagged constructs are
transfected in HEK293 cells along with HA-Ubiquitin and FLAG-TRAF6 and the cell
lysates were pulled-down using anti-FLAG and Protein G Sepharose beads (Sigma).
Immunoblotting with anti-HA showed no difference in the ubiquitination of FLAG-

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
168
tagged TRAF6 immunoprecipitates of HEK293 cells transfected with wildtype and
mutant Roquin constructs (Figure 7.8 B). This preliminary result indicates that Roquin
may not play a role in the regulation of TRAF6.
To confirm this in cells that utilise the RANK/TRAF6 signalling pathway, ubiquitinated
TRAF6 was assessed in BMMs of wildtype and sanroque mice. Firstly, BMMs were
incubated for two hours with MG132, a potent protease inhibitor, to prevent the
degradation of ubiquitinated proteins. RANKL was subsequently added in order to
stimulate the ubiquitination of TRAF6. After two hours, protein lysates were collected
and subjected to immunoprecipitation using anti-ubiquitin antibody and Protein G
Sepharose beads (Sigma) to collect all ubiquitinated proteins. The immunoprecipitates
were then subjected to immunoblotting with TRAF6 antibody.
As shown in Figure 7.9, non-stimulated cell cultures showed relatively low levels of
TRAF6 ubiquitination. Stimulation with RANKL only resulted in increased TRAF6
ubiquitination. A similar trend was observed in cell cultures that are pre-treated with
MG-132 (without RANKL stimulation), whereby the protease inhibitor resulted in the
accumulation of ubiquitinated products, including TRAF6. Interestingly, the addition of
MG132 did not appear to further accumulate the amount of ubiquitinated TRAF6 in
MG132-pretreated cell culture stimulated with RANKL. Semi-quantitative densitometry
analysis further showed that the ubiquitination level of TRAF6 in sanroque BMM
seemed relatively normal to that of wildtype in all conditions. This confirms the
previous observation that Roquin does not play a role in the posttranslational
modification of TRAF6, and that the E3 ubiquitin ligase may be involved with other
factors to mediate the increase of osteoclastogenesis observed in sanroque cell culture.
7.2.6. Blood serum from sanroque mutant mice contains proliferative-
cytokines and sanroque BMMs have increased proliferation in
response to M-CSF
During the early stages of osteoclastogenesis, cytokines such as IL-6 and GM-CSF
promote cell proliferation and survival, thereby increasing the osteoclast precursor pool
for subsequent osteoclast formation (Reddy et al., 1998). Therefore, enhanced
osteoclastogenesis could be due to increased proliferation of osteoclast precursors
within the bone marrow. It has been established that serum cytokine levels of
autoimmune patients differ from healthy controls (Kariuki et al., 2010; Lourenco et al.,

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
169
2009). Considering the existing autoimmune condition in sanroque mutant mice, we
speculated that this altered serum cytokine profile could mediate proliferation of
BMMs.
To investigate this, blood serum from wildtype and sanroque mutant mice was collected
via cardiac puncture. Similar number of BMMs from wildtype and sanroque mutant
mice were then cultured in medium supplemented with 10% of the collected serum for
24 hours, with or without the addition of M-CSF, and MTS assay was performed
(Figure 7.10). In cultures with no addition of M-CSF, there were no significant
differences in proliferation between wildtype and sanroque BMM. However, the
different serum types did alter proliferation rates in comparison to cells cultured in 10%
FCS for both wildtype and sanroque BMMs. Interestingly, sera from sanroque mice
induced significantly higher rates of proliferation in both wildtype and sanroque BMMs
than FCS and sera from wildtype mice.
As expected, the addition of M-CSF elevates cell proliferation in both wildtype and
sanroque BMMs. In comparison to wildtype BMMs cultured in all sera, sanroque
BMMs display higher proliferative activity in the presence of M-CSF. In contrast to the
non-M-CSF treated groups, both wildtype and sanroque sera had no significant
influence on proliferation. This is probably because the amount of M-CSF added was
excessive to mask the proliferative effect of the sera. The experiment should be
redesigned to use a serial increase of M-CSF dosage in order to further analyse if the
sera from sanroque mutant mice augments M-CSF-induced BMM expansion.
Nevertheless, these findings show that the blood serum from sanroque mutant mice
contains cytokines which induce cell proliferation and that the sanroque mutation
elevates the proliferative activity of sanroque BMMs upon stimulation with M-CSF.
7.3. Discussion
Given that the balanced activities between osteoclasts and osteoblasts are required for
the maintenance of healthy bone, it is important to analyse both cell types in the
sanroque mutant mice. The aim of this chapter was to investigate the effect of sanroque
mutation on the differentiation and activity of osteoclasts in sanroque mutant mice.
Based on a series of in vitro experiments, this chapter has shown that Roquin regulates
osteoclastogenesis and bone resorption.

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
170
Consistent with the increased osteoclast numbers observed in vivo, in vitro
osteoclastogenesis assay revealed the increase in RANKL-induced osteoclast formation
by sanroque mutant BMMs compared to wildtype. RANKL-induced osteoclastogenesis
is driven by the transcription factor NFATc1 (Takayanagi et al., 2002a). Activation of
NFATc1 in turn induces osteoclast fusion via the upregulation of DC-Stamp and v-
ATPase V0 subunit d2 (Kariuki et al., 2010; Kim et al., 2008; Lee et al., 2006). Indeed
the accelerated osteoclastogenesis observed in sanroque cell cultures was supported by
the increased expressions of NFATc1, DC-Stamp and v-ATPase V0 subunit d2 gene
and protein.
Bone resorption assays further showed that the function of sanroque mutant mice is
increased as each mutant osteoclast resorbed a larger area of bone compared to wildtype
osteoclasts. The bone lytic activity was further assessed by measuring the secretion of
lysosomal protease Cathepsin K that is responsible for bone resorption. As shown in
Figure 7.5, the addition of bone particles dramatically increased Cathepsin K secretion
into the supernatant of sanroque mutant osteoclast cultures, consistent with the elevated
expression of Cathepsin K gene expression in sanroque mutant bone. Collectively, these
results showing the increase osteoclastogenesis and lytic activity partially explain the
decrease bone mass of sanroque mutant mice.
Q-PCR analysis revealed the repression of Roquin gene throughout osteoclastogenesis
(Chapter 3), and this implies that the gene may play a negative regulatory role in
osteoclast formation and function. On the other hand, sanroque mutation positively
regulates osteoclastogenesis and bone resorption. The sanroque mutation resulted in
partial loss of function in Roquin (Vinuesa et al., 2005a; Yu et al., 2007), which equates
to the absence of the functional protein at earlier time point of osteoclastogenesis and
this could partially explained the augmented osteoclastogenesis and overzealous lytic
activity.
In an attempt to understand the molecular mechanism(s) by which Roquin regulates
early signalling pathways regulating osteoclastogenesis, canonical RANKL/RANK
signalling cascades were analysed. Upon binding with RANKL, RANK recruits the
adapter protein TRAF6 to activate six major signalling pathways: NFATc1, NF-κB,
Akt/PKB, JNK, ERK and p38, which then direct osteoclast-specific gene expression
leading to differentiation and function (Boyle et al., 2003; David et al., 2002; Feng,

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
171
2005; Gohda et al., 2005; Kobayashi et al., 2001; Li et al., 2002; Xing et al., 2002).
RANKL-stimulation resulted in the posttranslational modification of TRAF6, reportedly
via ubiquitination (Lamothe et al., 2007; Takayanagi et al., 2000). Immunoprecipitation
experiments revealed that the ubiquitination level of TRAF6 in RANKL-stimulated
sanroque BMM culture does not differ from that in wildtype, suggesting that Roquin
does not play a role in the ubiquitination of TRAF6.
One of the most elucidated pathways is the activation of the transcription factor NF-κB,
which is rapidly induced upon the recruitment of TRAF6 during RANKL stimulation.
In non-stimulated conditions, NF-κB dimers are bound to inhibitory IκB proteins and
remain inactive in the cytoplasm. In the canonical signalling pathway, RANKL
stimulation induces the ubiquitination and degradation of IκB proteins via
phosphorylation by IκB kinase (IKK) complex, thus releasing the bound NF-κB dimers
to allow the translocation to the nucleus and subsequent gene transcription
(Oeckinghaus et al., 2011). Interestingly, the levels of IκB-α phosphorylation in
RANKL-stimulated sanroque mutant BMMs remained higher. This indicates that
Roquin may not play a regulatory role in NF-κB canonical signalling pathway and the
result seemed contradictory to the increased osteoclastogenesis observed in sanroque
mutant mice. A possible explanation for this is that Roquin may play a regulatory role
in the non-canonical NF-κB pathway. It is important to note that the non-canonical NF-
κB pathway relies on phosphorylation and ubiquitination of p100 processing instead of
degradation of IκB-α to activate RelB/p52 NF-κB complex (Sun, 2011). It would be
beneficial to investigate the expression levels of proteins involved in the non-canonical
pathway.
Other RANKL-signalling pathways could probably compensate for the reduced
canonical NF-κB signalling observed in sanroque cell cultures. Conversely,
immunoblotting assay showed that the downstream NFATc1 signalling is increased as
mentioned above. The phosphorylation of p38 in sanroque mutant BMMs also occurred
at an earlier time point, indicating enhanced p38/MAPK-mediated signals, which are
required for inducing osteoclast differentiation (Li et al., 2002). The ERK signalling
pathway is also elevated as represented by the increased phosphorylation of ERK1/2 in
sanroque BMM culture. Consistent with the observed increased osteoclastogenesis and
bone resorption by sanroque mutant cell cultures, ERK has been reported to be

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
172
associated with cell survival and positively regulates osteoclast differentiation and bone
resorptive activity (He et al., 2011; Miyazaki et al., 2000). However, other
RANKL/RANK signalling cascades including Akt/PKB and JNK have yet to be
examined. A more in depth analysis is certainly required to identify the exact molecular
pathway through which Roquin regulates RANKL/RANK signalling cascades and
osteoclastogenesis. Attempts at immunoprecipitation with an anti-Roquin antibody in
order to find a potential substrate have been quite challenging, as the only available
antibody in the market to date did not successfully identify the protein on
immunoblotting.
The upregulation of osteoclastogenesis and osteoclast activity could also be initiated by
the increased proliferation of osteoclast precursors (Zhou et al., 2006). As highlighted
by Q-PCR analysis on gene expression throughout osteoclastogenesis, the expression of
Roquin is elevated at day 0 when BMMs were maintained in M-CSF and were not
stimulated with RANKL. This implies that Roquin may have a more prominent role in
regulating the maintenance of BMMs. Bearing in mind that the sanroque mutant mice
exhibit a Lupus-like autoimmune disease, we further postulated that the cytokine levels
in the blood serum are altered and may have an effect on the proliferation of sanroque
BMMs. Intriguingly, culture in the medium supplemented with blood sera collected
from sanroque mutant mice elevated the proliferation rate of both wildtype and
sanroque BMMs. MTS assay further revealed that Roquin deficiency accentuates M-
CSF-induced macrophage proliferation. These data show that sanroque BMMs are
potentially pre-primed to be more sensitive towards M-CSF stimulation, or that they
may have higher number of cells expressing c-fms, the receptor for M-CSF, within the
BMMs population due to the altered cytokines in the blood serum of sanroque mice.
However, such assumption should be further validated using flow cytometry and Q-
PCR analysis to assess the expression level of c-fms in BMMs. The levels of
proliferation-inducing cytokines such as GM-CSF and IL-6, in the blood serum should
also be measured. Investigation of the M-CSF downstream signalling pathway, such as
ERK and AKT, in sanroque mutant BMMs may provide another avenue in determining
the exact molecular pathway in which Roquin is involved.
In summary, this chapter has highlighted the novel role of Roquin in the regulation of
osteoclast formation and function. Through an in vitro approach, the data shown have

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
173
characterised the osteoclast biology of sanroque mutant mice, which showed enhanced
osteoclastogenesis and bone lytic activity. These in vitro observations agreed with the
increased in osteoclast numbers in vivo. Hence, sanroque mutation augments
osteoclastogenesis and the Roquin gene is a negative regulator of osteoclast formation
and function.

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
174
Figure 7.1 Osteoclast differentiation is increased in sanroque BMM culture. BMMs
isolated from sanroque mutant and wildtype mice were cultured in vitro for 5 days in
the presence of M-CSF and RANKL. Cultures were fixed and the indicated time points
and TRAP stained. Significant increase in the number of sanroque BMMs derived
osteoclast at day 3 and 5 is observed. (A) Representative microscopic images of
osteoclast differentiation at day 0, 1, 3 and 5 showing TRAP staining. (B) Quantitative
analysis of OC differentiation. TRAP positive cells with more than 3 nuclei were
categorised as osteoclasts. Results are representative of three independent biological
repeats and are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no significance
in comparison to wildtype controls. (Osteoclast differentiation assay performed by Dr.
Jasreen Kular)
0 1 3 5
Wild
type
sa
nroq
ue
Days of RANKL stimulation (A)
(B)
Wildtype sanroque
(B)(B)
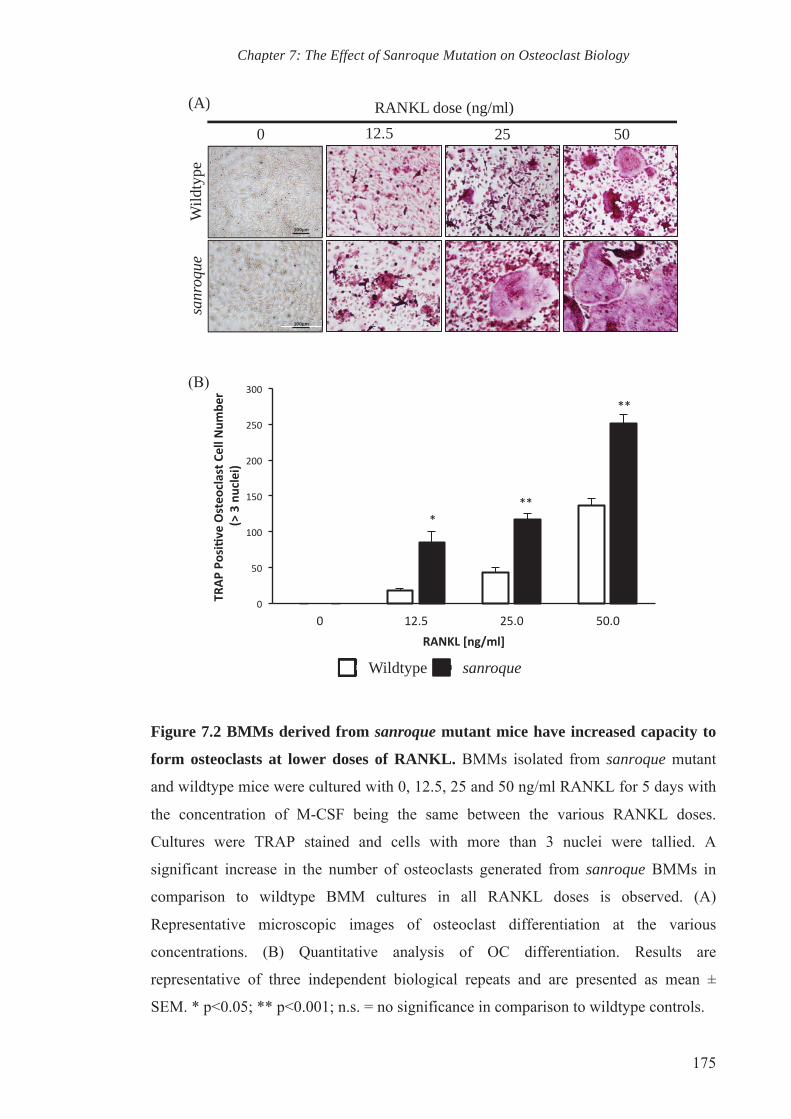
Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
175
Figure 7.2 BMMs derived from sanroque mutant mice have increased capacity to
form osteoclasts at lower doses of RANKL. BMMs isolated from sanroque mutant
and wildtype mice were cultured with 0, 12.5, 25 and 50 ng/ml RANKL for 5 days with
the concentration of M-CSF being the same between the various RANKL doses.
Cultures were TRAP stained and cells with more than 3 nuclei were tallied. A
significant increase in the number of osteoclasts generated from sanroque BMMs in
comparison to wildtype BMM cultures in all RANKL doses is observed. (A)
Representative microscopic images of osteoclast differentiation at the various
concentrations. (B) Quantitative analysis of OC differentiation. Results are
representative of three independent biological repeats and are presented as mean ±
SEM. * p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype controls.
(A)
12.5 25 50
Wild
type
sa
nroq
ue
) RANKL dose (ng/ml)
0
(B)
Wildtype sanroque

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
176
Figure 7.3 The mutation in Roquin alters the expression of osteoclast gene
markers. BMMs isolated from sanroque mutant and wildtype mice were stimulated
with RANKL to induce osteoclastogenesis. RNA was isolated from these cultures on
day 0 (non-stimulated), 3 and 5 and Q-PCR was carried out to determine the expression
levels of (A) NFATc1, (B) DC-Stamp and (C) RANK relative to housekeeping gene
Rn18s. Results are representative of three independent biological repeats and are
presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no significance when the value
of mutant samples was compared to that of wildtype control samples, and in comparison
to respective day 0.
sanroque
Wildtype
(B) (A)
(C)

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
177
Figure 7.4 Sanroque mutant osteoclasts resorbed more bone. BMMs were cultured
with M-CSF and RANKL in collagen-coated plates. Small osteoclasts were trypsinised
and similar number of wildtype and sanroque small osteoclasts were transferred onto
bovine bone discs. Representative microphotographs of (A) TRAP-stained osteoclasts
on bone discs and (B) the pits formed by actively resorbing osteoclasts. (C) TRAP-
stained osteoclasts were tallied and (D) the resorbed area was quantified. Results are
representative of 3 biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls.
Wildtype sanroque
(D) (C)
Wildtype sanroque (A)
(B)

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
178
Figure 7.5 Increased cathepsin K released by sanroque mutant osteoclasts.
Wildtype and sanroque multinucleated osteoclasts were stimulated with bovine bone
particles. (A) Culture medium (supernatant) was collected at 4, 8 and 12 hours post
stimulation and subjected to western blot analysis for the presence of cathepsin K. Cell
lysates were collected to serve as control. (B) Semi-quantitative densitometry analysis
of cathepsin K released in the supernatant relative to cell lysate. Results are
representative of 3 biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls.
sanroque
Wildtype (B)
(A) Wildtype sanroque
4 8 12 4 8 12
Supernatant
Cell lysate
CatK
CatK

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
179
Figure 7.6 Increased activation of MAPK signalling cascade during RANKL
stimulation in sanroque osteoclast precursors. Wildtype and sanroque BMMs were
p-ERK 1/2
Total ERK 1/2
IκB-α
p-p38
Total p38
β-actin
Wildtype
0 5 10 15 30 60
sanroque
0 5 10 15 30 60 RANKL stimulation (min)
sanroque
Wildtype
(B)
(A)

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
180
stimulated with RANKL over the time course of 0, 5, 10, 20, 30 and 60 minutes.
Proteins were extracted and subjected to immunoblotting. Phosphorylation of ERK is
increased in sanroque BMM cultures throughout the course of RANKL stimulation. (A)
Representative western blot images of protein expression involved in RANKL/RANK
signalling cascades with β-actin probed as loading control. (B) Semi-quantitative
densitometry analysis of the protein expressions relative to β-actin. Results are
representative of 3 biological repeats and are presented as mean ± SEM. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls.

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
181
Figure 7.7 Increased protein levels of transcription factor NFATc1, osteoclasts
markers DC-Stamp and D2 subunits during osteoclastogenesis of sanroque BMMs.
Wildtype and sanroque BMMs were stimulated with RANKL over the course of four
days to induce osteoclastogenesis. Proteins were extracted on each day and were
subjected to western blot analysis. Transcription factor NFATc1, DC-Stamp and D2
protein levels were increased in sanroque BMM cultures at day 3 and 4 post RANKL-
stimulation. (A) Representative western blot images of protein expression of involved in
sanroque
Wildtype
(B)
NFATc1
DC-Stamp
v-ATPase V0 subunit d2
β-actin
Wildtype
0 1 2 3 4
sanroque
0 1 2 3 4 RANKL stimulation (day) (A)

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
182
signalling pathway mediating osteoclastogenesis and bone resorption. (B) Semi-
quantitative densitometry analysis of the protein expressions relative to β-actin. Results
are representative of 3 biological repeats and are presented as mean ± SEM. * p<0.05;
** p<0.001; n.s. = no significance in comparison to wildtype controls.

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
183
Figure 7.8 FLAG-tagged TRAF6 immunoprecipitates of HEK293 cells transfected
with wildtype and mutant Roquin (M119R) plasmids showed no difference in
NheI KpnI
5’ LTR (MSCV)
IRES
GFP
Nhe1 Kpn1
NcoI
BamHI 3’ LTR (MSCV)
Unique sites in the MCS
BgIII XhoI SalI
EcoRI Xmn1
Xmn1
Am
pici
llin+
ψ
(A)
Wildtype sanroque
50
150
25
75
37
pcD
NA
3.1
WT
Roq
uin
Kpn1
Mut
ant R
oqui
n (M
199R
)
Construct TRAF6-FLAG
Ubiquitin HA + +
+ +
+ +
+
++ +
+ +
Total extracts IP: FLAG
IB: HA
50
150
25
75
37
IB: TRAF6
(B)
+
pcD
NA
3.1
+
WT
Roq
uin
+
LAG
Mut
ant R
oqui
n (M
199R
)

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
184
ubiquitination level by immunoblotting with anti-HA. (A) Schematic maps for
plasmid adapted from Yu et al. (2007). IRES, internal ribosome entry sequence; LTR,
long terminal repeat; ψ, packaging signal; MCS, multiple cloning site. (B) HEK293
cells were transfected with the indicated plasmids encoding wildtype or mutant Roquin
in the presence of HA-Ub and FLAG-TRAF6. Cells were lysed and immunoprecipitated
with anti-FLAG. Bound proteins were subjected to SDS-PAGE and immunoblotted first
with anti-HA. The membrane was stripped and reprobed with anti-TRAF6. Results are
representative of 2 biological repeats.

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
185
Figure 7.9 No difference in ubiquitination of TRAF6 in sanroque BMMs upon
RANKL stimulation. Analysis of ubiquitination of TRAF6 in the presence or absence
of protease inhibitors MG132, and in stimulated and unstimulated BMM. (A)
Immunoprecipitates with anti-ubiquitin was blotted with anti-TRAF6 antibody. RANKL
stimulation resulted in the ubiquitination of TRAF6. (B) Semi-quantitative densitometry
analysis of TRAF6 ubiquitination relative to total extracts showed no significant
difference between wildtype and sanroque mutant BMM. Results are representative of 3
biological repeats and are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no
significance in comparison to wildtype cell cultures.
sanroque
Wildtype (B)
(A)
IP: Ubiquitin
Total extracts
25
37
50
MG132 RANKL
+ +
+ -
-
+-
-+
++
-- +
- -
Wildtype sanroque
IB: TRAF6

Chapter 7: The Effect of Sanroque Mutation on Osteoclast Biology
186
Figure 7.10 Blood serum from sanroque mutant mice contains proliferative-
inducing cytokines and sanroque BMMs exhibit increased cell proliferation in
response to M-CSF. BMMs from wildtype and sanroque mutant mice were plated and
cultured with medium supplemented with either 10% of foetal calf serum (FCS), blood
serum collected from wildtype (WT sera) or sanroque mutant mice (san sera), in the
absence or presence of M-CSF. Cell proliferation was measured 24 hours after
culturing. Results are representative of 3 biological repeats and are presented as mean ±
SEM. * p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype cell
cultures.
Wildtype sanroque

CHAPTER EIGHT
OSTEOIMMUNOLOGICAL MECHANISM BEHIND THE
OSTEOPOROTIC PHENOTYPE OF
SANROQUE MUTANT MICE

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
188
8. CHAPTER 8: OSTEOIMMUNOLOGICAL MECHANISM BEHIND THE OSTEOPENIC PHENOTYPE OF SANROQUE MUTANT MICE
8.1. Introduction
Inflammation-induced bone loss highlights the abundance of overlapping molecules and
regulatory pathways shared between the bone organ and immune system. Firstly, the
complex dialogue between the bone organ and immune system is unavoidable as stem
cells, which give rise to progenitors to bone cells and immune cells, are generated
within the bone marrow. For instance, with the appropriate stimuli, macrophage and
myeloid dendritic cells are able to form osteoclasts in vitro, suggesting that they
originate from a subset of common myeloid progenitors and that share a developmental
pathway. This subset of common myeloid progenitors can be identified as CD3ε- B220-
cKit/CD117+ CD115/c-fms+ CD11blo cells within the mouse bone marrow niche
(Jacome-Galarza et al., 2013).
Using transgenic mice exhibiting an autoimmune disorder due to constitutive expression
of CD70, Xiao et al. (2013) was able to further discriminate these common myeloid
progenitors into osteoclast/dendritic progenitors (CD27hi) and the more osteoclast-
committed precursors (CD27lo). This dynamic lineage bifurcation of B220-
cKit/CD117+ CD115/c-fms+ CD11blo CD27hi towards B220- cKit/CD117+ CD115/c-
fms+ CD11blo CD27lo is tightly regulated by the expression of the ligand CD70 on
activated immune cells, whereby constitutive engagement of CD27/CD70 is required to
sustain the differentiation towards dendritic cells. As a result, the aforementioned
transgenic mice develop a high trabecular bone mass pathology due to impeded
osteoclastogenesis in a CD27-dependent manner (Xiao et al., 2013).
Secondly, cytokines that play a role in orchestrating immune responses also modulate
bone remodeling and the progression of inflammatory bone diseases. The potent
stimulator of osteoclastogenesis RANKL is expressed by antigen presenting dendritic
cells and is crucial for T cell expansion (Anderson et al., 1997; Bachmann et al., 1999;
Wong et al., 1997b). The deletion of RANKL and RANK resulted in osteopetrotic bone
and dysregulated maturation of immune cells, thus highlighting the dual role of the
RANK/RANKL signalling pathway in bone homeostasis and the immune system
(Dougall et al., 1999; Kim et al., 2000; Kong et al., 1999b; Perlot et al., 2012). Early
osteoimmunology studies have shown that activated T cells are able to express RANKL
to directly induce in vitro osteoclastogenesis (Horwood et al., 1999; Kong et al., 1999a;
Kong et al., 1999b; Kotake et al., 2001; Weitzmann et al., 2001; Wong et al., 1997b). In

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
189
this scenario, T helper (Th) 17 cells emerged as an osteoclastogenic subset in view of its
pro-inflammatory nature. Conforming to its osteoclastogenic properties, Th17 cells also
express significant amounts of membrane-bound RANKL and play a role in the
progression of bone destruction in rheumatic joints (Sato et al., 2006).
OPG, the decoy receptor for RANKL first characterised in osteoblasts as an inhibitor of
osteoclastogenesis, is also reported to play a role in dendritic cell and B cell function
(Yun et al., 1998). OPG-deficient mice are both osteoporotic due to increased osteoclast
activity and have a compromised immune system due to perturbed B cell maturation
(Bucay et al., 1998; Yun et al., 2001). It also appears that B cells abundantly express
OPG that plays a crucial role in the maintenance of bone homeostasis, as knockout mice
lacking B cells exhibit osteoporosis (Li et al., 2007b).
In line with these aforementioned reports, the previous chapter has documented
observations that suggested the blood serum of sanroque mutant mice displayed
different cytokine compositions to that of wildtype controls. Indeed Vinuesa and
colleagues (2010) have reported that the sanroque mutant mice have elevated IL-21,
which contributes to the increase in the number of follicular helper T cells (TFH) and
the pathogenesis of lupus-like autoimmune disease (Vinuesa et al., 2005a).
Furthermore, in vivo and in vitro osteoclastogenesis is elevated in sanroque mutant mice
in comparison to wildtype littermates, suggesting that the mutation may affect
osteoclastogenesis. Following these observations, we set to investigate the population of
common progenitors present within the bone marrow reservoir and the possible in vivo
cellular mechanism which may contribute to the upregulation of putative osteoclast
progenitors in sanroque mutant mice.
8.2. Experimental results
Using flow cytometry, we analysed the profile of several sub-populations of myeloid
cells in the bone marrow of both wildtype and sanroque mutant mice. Briefly, sex and
age-matched wildtype and sanroque mutant mice were euthanized and bone marrow
cells were harvested by flushing the long bone with serum-free α-MEM. Spleens were
also collected and homogenised to obtain single cell suspension. Red blood cells were
lysed prior to immuno-staining with antibodies listed in Chapter 4. The stained single
cell suspensions were then analysed and cell phenotypes were identified with cell
surface markers based on the following gating strategies: non-lymphoids/myeloids (Lin-

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
190
; CD3e- CD45R/B220- CD11b-), haematopoietic stem cells (HSCs; Lin- CD117(cKit)+
Sca1(Ly6A/E)+), mesenchymal stem cells (MSCs; Lin- CD117+ Sca1-), macrophages
(Mϕ; Lin- CD117+ CD11blo CD115(c-fms)+), dendritic cells (DCs: Lin- CD115-
CD11c+), osteoclast/dendritic progenitors (OC/DCP; Lin- CD117+ CD115+ CD11blo
CD27hi), pre-committed osteoclast precursors (pOCP; Lin- CD117+ CD115+ CD11blo
CD27lo CD27lo), committed osteoclast progenitors (OCP; LM- CD117+ CD11blo
CD115+ RANK+), T cells (CD3ε+ CD4+ and CD3ε+ and CD8+ for T helper and T
cytotoxic cells respectively), follicular helper T cells (TFH; CD3ε+ CD4+ CXCR5+
PD1+) and B cells (CD3ε- CD45R/B220+).
8.2.1. The putative osteoclast progenitor population present within the
bone marrow of sanroque mutant mice is increased.
Using antibodies for pan-lymphocyte markers (CD3ε and CD45R/B220) and leukocyte
marker (CD11b) to gate out lymphocyte and myeloid cells, flow cytometric analysis
revealed the very apparent reduction of a lymphocyte population in quadrant 4 (Q3) in
sanroque bone marrow niche in comparison to wildtype (Figure 8.1 A). Further analysis
using a different antibody panel revealed that the missing population within the mutant
bone marrow is CD45R/B220hi, indicating that the differentiation of B cells in sanroque
mutant mice could possible by compromised (Figure 8.1 B).
To provide a rough overview of the stem cell niche of the bone marrow, CD117 and
Sca-1 antibodies were used to further analyse the non-lymphocyte/myeloid (Lin-)
population. As shown in Figure 8.1 C, the Lin- population of sanroque bone marrow
cells were skewed towards Sca1+ and CD117+, indicating that the sanroque mutation is
affecting early differentiation of myeloid stem cells, and possibly haematopoiesis.
By comparison, monocytic CD11b+ only cell population in Q1 is significantly increased
in the bone marrow harvested from sanroque mutant mice compared to that of wildtype
littermates (Figure 8.1 A). On the other hand, the population of non-
lymphocyte/myeloid (Lin-) population in sanroque bone marrow is comparable to the
wildtype. Since common myeloid progenitor cells are a subset of the monocytic Lin-
CD11blo population, we further dissected the population to analyse macrophage and
dendritic cell populations using the pan-macrophage marker CD115 (or c-fms) and pan-
dendritic cell marker CD11c respectively. The analysis revealed that there is a
significant increased in the percentage of macrophages in sanroque bone marrow when

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
191
compared to wildtype (Figure 8.2 A). This suggests that the differentiation of common
myeloid progenitors is skewed towards macrophages, and as a result the percentage of
dendritic cells in the mutant bone marrow is significantly reduced (Figure 8.2B).
As reported, osteoclast precursors reside within CD11blo subset of Lin- CD117+ CD115+
bone marrow cells and express RANK. Flow cytometry indicated that this population is
also significantly higher in sanroque mutant bone marrow that wildtype controls (Figure
8.3 A). RANK+ committed osteoclast precursors are reported to arise from pre-
committed osteoclast precursors (Lin- CD117+ CD115+ CD11blo CD27lo), which have
higher osteoclastogenic potential in comparison to OC/DC common precursors (Lin-
CD117+ CD115+ CD11blo CD27hi). Consistently, flow cytometric analysis of CD27
expression levels on gated committed osteoclast precursors Lin- CD117+ CD115+
CD11blo revealed that the rare subpopulation of pre-committed osteoclast precursors
CD27lo is significantly higher in sanroque mutant bone marrow (Figure 8.3 B). The
OC/DC CD27hi common precursor subpopulation of sanroque bone marrow on the
other hand is comparable, albeit with a non-significant reduction, to that of wildtype.
Perhaps increasing the sample size may change the statistical significance of the
reduced OC/DCP Lin- CD117+ CD115+ CD11blo CD27hi observed in mutant bone
marrow.
Collectively, these data indicate that haematopoiesis of sanroque mutant mice is
dysregulated, resulting in a significant expansion of putative osteoclast progenitors
within the bone marrow reservoir.
8.2.2. CD4 T cells of sanroque mutant mice expressed less CD70
It has been reported that sustained CD27 interaction with its ligand CD70 prevents the
differentiation of OC/DCP into osteoclasts. The expression of CD27 is down regulated
from the cell surface upon contact with CD70-bearing cells thus favouring
differentiation towards DC. Based on this report, we hypothesised that the CD27-CD70
interaction is reduced in sanroque mice.
We first examined the population of CD4 and CD8 T cells and flow cytometric analysis
revealed a trend of increase, albeit not significant, in both populations in sanroque
mutant mice (Figure 8.4). Increasing the sample size would improve the statistical
power of the data. Among the T cell populations in the bone marrow of the wildtype

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
192
control, CD70-bearing cells are mostly CD4+ (Figure 8.5A). Comparative analysis of
CD70 expression between wildtype and sanroque CD4 T cells revealed a significant
down regulation of the ligand for CD27 in mutant CD4 T cells (Figure 8.5 B). The
expression of CD70 in CD8 T cells, B and dendritic cells of sanroque bone marrow is
similar to that the wildtype control (Figure 8.5 C, D and E). Therefore, CD4 T cells are
the major contributor of CD70 in wildtype controls and the down regulation of the
ligand in sanroque mutant mice resulted in a reduction of CD27-CD70 interaction,
leading to the observed decrease in the dendritic cell population and an increase in
osteoclast progenitors in sanroque mutant mice.
8.2.3. Splenocytes of sanroque mutant mice comprised a higher number of
common myeloid progenitors and follicular helper T cells that
express RANKL
The elevated expansion of putative osteoclast progenitors in sanroque mutant mice
could be contributed to by the elevated expression of RANKL in the bone of sanroque
mutant mice, which has been documented in the previous chapter. TFH is a newly
characterised T cell subset that has been implicated in the pathogenesis of
autoimmunity. Preliminary microarray analysis by Vinuesa and colleagues (2005)
showed that sanroque mutant TFH expressed higher levels of RANKL in comparison to
wildtype TFH (Vinuesa et al., 2005a). This prompted us to further postulate that the
mutation in Roquin gene resulted in elevated RANKL in mutant TFH, which then
contributes to the elevated osteoclastogenesis in sanroque mutant mice.
In an attempt to test this hypothesis, we performed Q-PCR on sorted TFH from
wildtype and sanroque mutant spleens. While it was possible to attain a reasonable
number of TFH from sanroque mutant mice, the population of the aforementioned cell
type was so rare (< 1% of CD4 T cells) in the wildtype mice that it was not possible to
isolate sufficient RNA to perform a direct comparison with the expression of RANKL
in mutant TFH (Figure 8.6 A). We also endeavoured to perform cryosectioning and
immunofluorescence staining on non-decalcified bone samples to investigate the
potential localisation of the cell subset that contributed to the abundance of RANKL in
sanroque mutant bone. However, this experiment was unsuccessful due to the lack of
appropriate equipment and facilities in our laboratory.
Since it has been previously shown, and we have confirmed using flow cytometry, that

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
193
most TFH reside within the germinal centres of the spleens, we performed
cryosectioning and immunofluorescence staining on frozen spleens to assess the source
of RANKL in this organ. Briefly, harvested spleens from sex and age matched mice
were immersed and allowed to equilibrate in O.C.T. compound (Tissue-Tek) before
snap freezing in a beaker full of isopentane in a liquid nitrogen bath until frozen. Frozen
spleens were cryosectioned at a thickness of 5 μm and sections were placed on charged
slides before proceeding to blocking steps crucial for immunofluorescence staining.
Sections were probed with antibodies specific for CD4 and CXCR5 to identify TFH,
RANKL and DAPI and visualised under confocal microscopy. Images were captured
around the white pulp (characterised by circular densely populated area) at ×20 and ×30
magnifications while retaining similar voltages and gain settings throughout wildtype
and mutant samples.
As shown in Figure 8.7, RANKL was mainly expressed by cells localised around the
periphery of the white pulp, i.e. the red pulp area of the spleen, while the expression of
CD4 and CXCR5 was spread out in both areas. Consistent to the observation reported
by Vinuesa et al. (2005), the density of CD4 CXCR5 (markers of TFH) double positive
cells is increased due to the dysregulated expansion of the T cell subset in sanroque
mutant mice. Furthermore, we observed co-localisation of RANKL and CD4 CXCR5
double positive T cells (indicated by white arrow) in sanroque mutant spleen. It is also
interesting to note that under the same image settings, RANKL expression appeared
more intense in the sanroque mutant section in comparison to wildtype. Many of these
RANKL-expressing cells also express CD4. This data suggests that not only sanroque
TFH but also other T cells within the mutant spleen expressed high levels of membrane-
bound RANKL.
This observation is supported by western blotting whereby total spleen of sanroque
mutant mice expressed higher levels of RANKL protein when compared to that of
wildtype control (Figure 8.8 A). Q-PCR analysis further showed that both RANKL and
M-CSF gene expression was elevated in sanroque mutant spleen compared to wildtype
(Figure 8.8 B). Although the expression level of OPG was also significantly higher in
sanroque spleens than wildtype spleen, the RANKL:OPG ratio is still significantly
higher in sanroque mutant mice (Figure 8.8 C). The increase in M-CSF and RANKL
may in turn contribute to the increase in common myeloid progenitors in sanroque

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
194
mutant spleen as shown in the flow cytometric analysis of splenocytes (Figure 8.9).
However, more biological repeats are required to validate the flow cytometric studies on
the population of osteoclast precursors in mutant splenocytes.
In summary, sanroque mutation leads to increased RANKL expression not only within
the bone but also the spleen, both of which are organs of haematopoiesis. The increase
in M-CSF and RANKL gene expression in sanroque mice in turn contributes to the
elevated number of common myeloid progenitors and accelerated osteoclastogenesis
respectively.
8.3. Discussion
In an attempt to elucidate the pathogenesis of systemic bone loss during autoimmunity,
we investigated the possible osteoimmunological mechanisms that contribute to the
accelerated osteoclastogenesis in sanroque mutant mice. The novel field of
osteoimmunology highlights that the bone marrow reservoir provides a conducive
environment for immune cells to interact with osteoblasts, osteoclasts and their
precursors (Takayanagi, 2007). RANKL is not solely expressed by osteoblasts, but also
by certain activated CD4 T cells, including the proinflammatory Th17 subset (Sato et
al., 2006). Furthermore, the interaction between RANKL-bearing T cells with osteoclast
precursors is reported to drive osteoclast development and induce bone loss during
chronic immune activation (Kong et al., 1999a; Kotake et al., 2005; Kotake et al., 2001;
Pettit et al., 2001; Sato et al., 2006).
Our data now implicate the TFH cells as a novel T cell osteoclastogenic subset that may
contribute to the induction of bone loss by expressing RANKL during pathological
conditions of chronic autoimmune disease. TFH are the class of effector TH cells that
regulate the in vivo development of antigen-specific B cell immunity (Fazilleau et al.,
2009). As highlighted in sanroque mutant mice, the dysregulation of TFH is implicated
in autoimmune diseases that are partially mediated by autoantibodies, thus affirming its
role in humoral immunity as a professional B cell helper (Simpson et al., 2010; Vinuesa
et al., 2005a; Vinuesa et al., 2005b). TFH express CXCR5 and abundant amounts of IL-
21 to directly regulate B cell proliferation and class switching (Akiba et al., 2005;
Nurieva et al., 2008; Rasheed et al., 2006; Terrier et al., 2012; Vinuesa et al., 2005b;
Vogelzang et al., 2008).

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
195
A recent study has associated IL-21, which is elevated in the serum of sanroque mutant
mice, with the pathogenesis of bone loss in patients with RA and in mice with collagen-
induced arthritis. IL-21 augments osteoclastogenesis by inducing RANKL expression in
CD4 T cells, thus confirming osteoclastogenic activity of the cytokine (Kwok et al.,
2012). As such, aside from TFH we also observed increased co-localisation of RANKL
and CD4 in the spleen of sanroque mutant mice. The increase in RANKL expression
was validated by western blotting and Q-PCR, and this was accompanied with increased
M-CSF expression. This attributed to the elevated expansion of common myeloid
progenitors in the spleen of sanroque mutant mice. The subpopulation of osteoclast
progenitors within the common myeloid progenitors within the spleen has yet to be
determined and should be analysed in the future.
On a side note, it would be crucial to validate the expression of RANKL in TFH using a
more accurate tool such as Q-PCR. Since TFH are activated to proliferate during an
immune response, the rare population of TFH in wildtype mice could be boosted by
vaccination (Ma et al., 2014). In vitro co-culture between TFH and BMMs would also
provide a useful insight into the crosstalk and the possible mechanisms by which the T
cell subset supports osteoclastogenesis.
The crosstalk between immune cells and bone cells in mice is also emphasised by the
CD27 interaction with its ligand CD70. CD27 is a member of TNF-receptor superfamily
of co-stimulators and is constitutively expressed on CD4 and CD8 T cells. Expression
of CD27 is also found on murine haematopoietic stem cells and differentiated
precursors including the earliest lymphocyte precursors as well as common lymphoid
progenitors (Igarashi et al., 2002; Nolte et al., 2005; Serwold et al., 2009; Wiesmann et
al., 2000). The ligand for CD27, CD70, is also expressed on T, B and dendritic cells.
CD27-CD70 signalling plays a crucial role in the regulation of cellular immune
responses, whereby the fine balance between CD27-CD70 interaction is crucial in
preventing autoimmunity (Coquet et al., 2013; Nolte et al., 2009; Wang et al., 2013). In
the context of osteoimmunology, CD70-bearing immune cells infiltrate the bone
marrow to inhibit osteoclastogenesis and favour the differentiation of common myeloid
progenitors towards dendritic cells (Xiao et al., 2013). Our data has suggested that CD4
T cells are a major contributor of CD70-bearing cells in murine bone marrow, which is
significantly reduced in sanroque mice. As a result, the population of dendritic cells in

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
196
sanroque mice is diminished, and the bifurcation of the common myeloid cells was
driven towards the pre-committed osteoclast precursor population. Consistently,
sanroque mutant bone marrow showed a significant expansion of committed osteoclast
precursors.
The relocation of HSCs into the bone marrow at birth establish the bone organ as the
primary site for haematopoiesis in adult mammals (O'Neill et al., 2011). It is interesting
to note that the non-myeloid/lymphocyte population in sanroque mutant mice appeared
atypical in comparison to wildtype, whereby the cells are skewed towards CD117+ and
Sca-1+ population. This preliminary observation indicated that the haematopoiesis in
sanroque mice might be compromised. The abnormality is also apparent in preliminary
analysis of blood films of sanroque mutant mice with an increased number of immature
cells (left-shift) (Supplementary Figure 2, personal communication with W/Prof. Wendy
Erber); however, this requires further validation using appropriate differential counts to
quantify the different types of leukocytes. Additionally, the stem cell population of
sanroque mice also needs to be further dissected with flow cytometry using appropriate
antibodies to determine lineage negative cells and analyse different subpopulations of
hematopoietic stem cells including long term and short term HSCs. Perhaps the
mutation in the E3 ubiquitin ligase Roquin may affect the stem cell niche of sanroque
mutant mice, and the observations reported in this thesis are the downstream effects of
dysregulated differentiation of myeloid and lymphocytic progenitors. Indeed several E3
ubiquitin ligases have been implicated in fine-tuning haematopoiesis (Rathinam et al.,
2008; Rybak et al., 2009; Thompson et al., 2008; Yokomizo et al., 2008).
As previously mentioned, B cells is implicated in contributing a significant amount of
OPG to regulate bone remodelling (Li et al., 2007b). It is also interesting to note the
diminished population of B cells in sanroque mutant bone marrow. This data implies
that the reduction of OPG levels in the sanroque mutant mice is probably due to the
absence of B cells within the bone marrow reservoir. Taken together, the results in this
chapter demonstrate that sanroque mutant mice exhibit diminished CD27-CD70
interaction to favour the differentiation of common myeloid progenitors towards
osteoclast precursors. The mutation in Roquin gene resulted in the augmented
expression of RANKL not only in bone but also in the spleen of sanroque mutant mice.
Using confocal microscopy, much of the RANKL expression observed is contributed by

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
197
CD4 T cells, some of which are TFH, in sanroque mutant spleen. The increased
RANKL expression is compounded by the increase in MCSF expression and the
reduction in OPG level, resulting in enhanced osteoclast differentiation and function,
thus contributing to systemic bone loss observed in sanroque mutant mice.

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
198
(A) Wildtype sanroque
CD
11b
CD3ε/CD45R/B220
yp q Single live cells
Wildtype
sanroque

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
199
(B) Wildtype sanroque Single live cells
CD45R/B220
Wildtype
sanroque
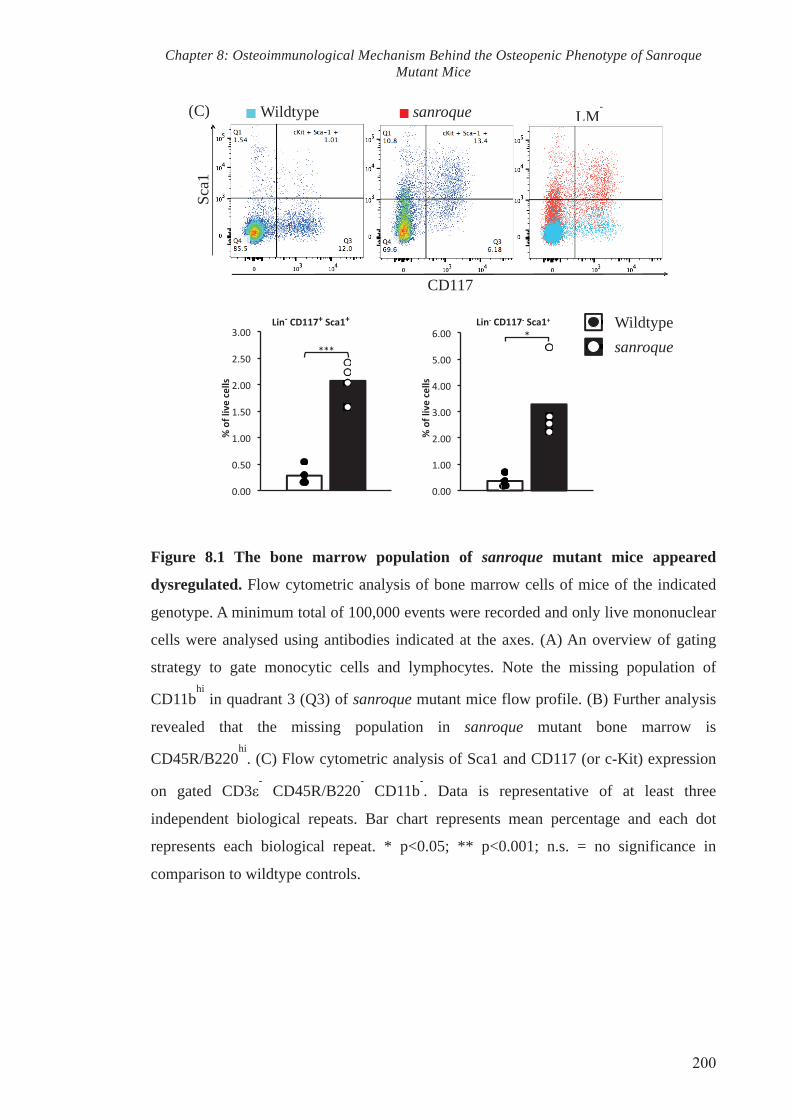
Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
200
Figure 8.1 The bone marrow population of sanroque mutant mice appeared
dysregulated. Flow cytometric analysis of bone marrow cells of mice of the indicated
genotype. A minimum total of 100,000 events were recorded and only live mononuclear
cells were analysed using antibodies indicated at the axes. (A) An overview of gating
strategy to gate monocytic cells and lymphocytes. Note the missing population of
CD11bhi
in quadrant 3 (Q3) of sanroque mutant mice flow profile. (B) Further analysis
revealed that the missing population in sanroque mutant bone marrow is
CD45R/B220hi
. (C) Flow cytometric analysis of Sca1 and CD117 (or c-Kit) expression
on gated CD3ε- CD45R/B220
- CD11b
-. Data is representative of at least three
independent biological repeats. Bar chart represents mean percentage and each dot
represents each biological repeat. * p<0.05; ** p<0.001; n.s. = no significance in
comparison to wildtype controls.
Wildtype
sanroque
(C) Wildtype sanroque LM-
Sca1
CD117
LM

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
201
Figure 8.2 Increased macrophage and reduced dendritic cell population in
sanroque bone marrow. Flow cytometric analysis of (A) macrophage and (B) dendritic
cell population of bone marrow cells. (C) Representative bar charts of indicated cell
population as percentage of live single cells. Data is representative of at three
independent biological repeats. * p<0.05; ** p<0.001; n.s. = no significance in
comparison to wildtype controls.
(A) Wildtype sanroque
CD
11b
CD115
Mϕ
CD11blo
CD115+
Wildtype sanroque yp q
CD
115
CD11c
Lin-
DCs
CD115-
CD11c+
(C)
Wildtype
sanroque
Mϕ DCs
(B)
Lin- CD117
+

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
202
Figure 8.3 Significant expansion of putative osteoclast progenitors in sanroque
bone marrow. (A) Flushed bone marrow cells were co-stained for RANK and CD115
(A) Wildtype sanroque Lin- CD117
+ CD11b
lo
CD115+
RANK+
CD115
Wildtypesanroque
(B) Wildtype sanroque Lin-CD117
+CD115
+
CD27
CD
11b
Wildtype
sanroque
yp q
CD27lo
CD27hi

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
203
to identify committed osteoclast precursors, which is increased in sanroque bone
marrow. (A) Pre-committed osteoclast precursors (Lin- CD117
+ CD115
+ CD11b
lo
CD27lo
), which have higher osteoclastogenic potential in comparison to OC/DC
common precursors (Lin- CD117
+ CD115
+ CD11b
lo CD27
hi) is also significantly higher
in sanroque bone marrow. Results are mean percentage of populations from three
independent biological repeats. * p<0.05; ** p<0.001; n.s. = no significance in
comparison to wildtype controls.

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
204
Figure 8.4 No difference in the percentages of CD4 and CD8 T cell population
between the bone marrow of sanroque mutant mice and wildtype. Flow cytometric
analysis of (A) CD4 and (B) CD8 T cells of wildtype and sanroque bone marrow. (C)
Bar charts represent mean percentage of populations from three independent biological
repeats. * p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype
controls.
(A) Wildtype sanroque Single live cells
CD
3ε
CD4
yp q g
CD3ε+
CD4+
(B) Wildtype sanroque Single live cells
CD3ε+
CD8+
CD
3ε
CD8
Wildtype
sanroque
(C)

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
205
(A) Wildtype
CD3εCD4CD8
sanroque
CD3εCD4CD8
CD70
Wildtype
sanroque
(B) Wildtype sanroque CD4+ CD4
CD70
CD70- CD70
+

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
206
Wildtype
sanroque
(C) Wildtype sanroque CD8+ yp q CD8
CD70- CD70
+
CD70
(D) Wildtype sanroque CD3ε- CD45R/B220
+
CD70
Wildtype
sanroque
CD70- CD70
+

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
207
Figure 8.5 Expression of CD70 in T, B and DCs. Representative of flow cytometric
analysis of CD70 expression in (A) all T cells, (B) CD4 T cells, (C) CD8 T cells, (D) B
cells, (E) DCs of wildtype and sanroque bone marrow. Results are representative of
three independent biological repeats and are presented as mean percentage. * p<0.05; **
p<0.001; n.s. = no significance in comparison to wildtype controls.
(E) Wildtype sanroque
CD70
CD11c+
CD70-
Wildtype
sanroque
CD70+

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
208
Figure 8.6 Increased follicular helper T cell in sanroque mutant mice. (A)
Representative flow cytometric analysis of CD4+ CD44+ CXCR5+ PD1+ TFH in
wildtype and sanroque spleens. (B) Average percentage of TFH in CD4 T cells. Results
are representative of three independent biological repeats and are presented as mean
percentage. * p<0.05; ** p<0.001; n.s. = no significance in comparison to wildtype
controls.
(A) Wildtype sanroque CD4+ CD44
+
PD-1
CXCR5
Wildtype
sanroque
(B)
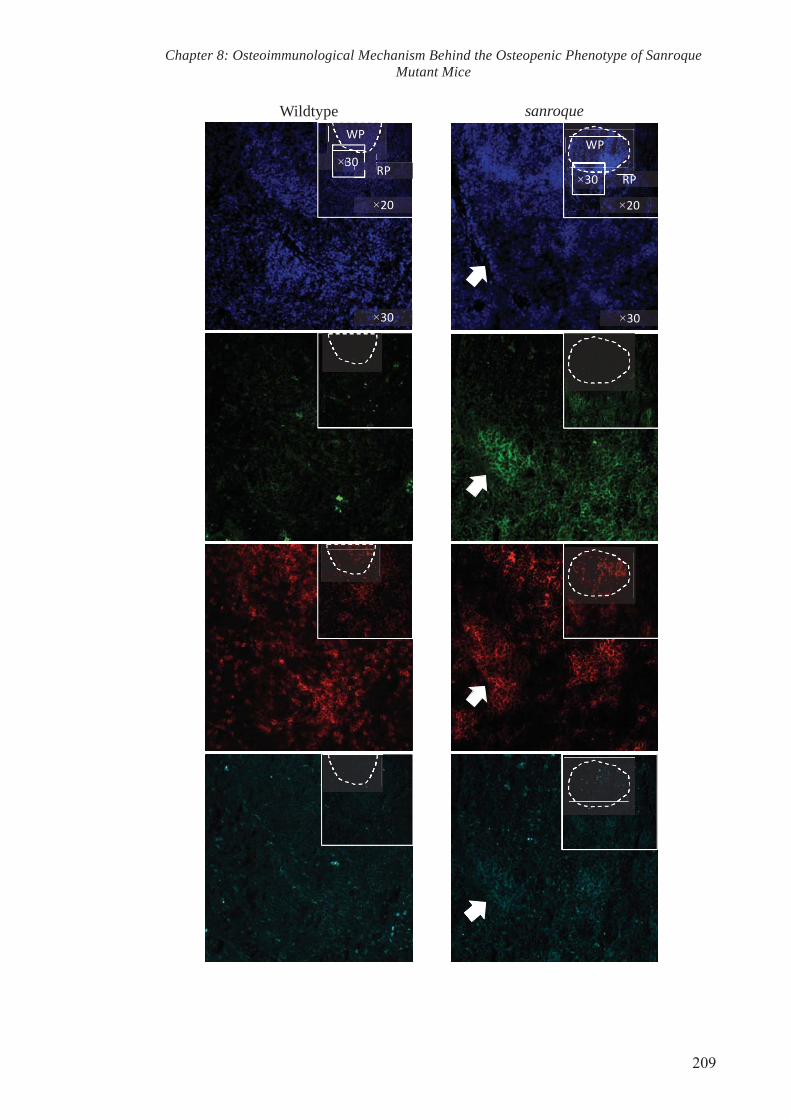
Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
209
Wildtype sanroque
×
×
×
p
×
×
×

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
210
Figure 8.7 Localisation of RANKL and TFH in spleen. Spleens were cryosectioned,
fixed and processed for immunofluorescence using specific antibodies to RANKL
(FITC), CD4 (DyLight-488) and CXCR5 (APC). Nucleuses were stained with DAPI
and white pulp (WP) is demarcated by the dotted white line. Areas of co-localisation
between CD4 and CXCR5 double positive cells with RANKL is denoted by yellow
colour in overlay with specific area indicated by the white arrow. Images are
representative of three independent biological repeats.
Wildtype sanroque p
DAPI RANKL (FITC) CD4 (DyLight488) CXCR5 (APC)

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
211
Figure 8.8 RANKL protein and gene expressions are elevated in sanroque spleens.
(A) Cell lysates were collected by homogenising spleen tissue and were subjected to
immunoblotting. Detection of RANKL and β-actin in the spleens of wildtype and
sanroque mutant mice. (B) Total RNA was harvested and subjected to RT-QPCR using
primers specific for OPG, RANKL and M-CSF. Gene expression analysis is expressed
relative to the house keeping gene Rn18S. Results are representative of two biological
repeats and are presented as mean ± SEM. * p<0.05; ** p<0.001; n.s. = no significance
in comparison to wildtype controls.
RANKL
β-actin
Wildtyp
e 1
sanroq
ue 1
Wildtyp
e 2
sanroq
ue 2
RANKL:β-actin 1.012 1.394 0.371 1.484
(B) (C)
Wildtype sanroque

Chapter 8: Osteoimmunological Mechanism Behind the Osteopenic Phenotype of Sanroque Mutant Mice
212
Figure 8.9 Increased common myeloid progenitors in sanroque spleen. (A)
Representative flow cytometric analysis of splenocytes of mice of the indicated
genotype. (B) Bar charts representing mean percentage of common myeloid progenitors
in total splenocytes. Data is representative of two independent biological repeats and are
presented as mean percentage of single live cells.
(A) Wildtype sanroque
CD
11b
CD45R/B220
CD3ε- CDCD33εε
Wildtype
sanroque
(B)

CHAPTER NINE
GENERAL OVERVIEW AND FUTURE DIRECTIONS

Chapter 9: General Overview and Future Directions
214
9. CHAPTER 9: GENERAL OVERVIEW AND FUTURE DIRECTIONS
9.1. Overview of thesis and general discussion
The homeostasis of the bone organ system is maintained by a process known as bone
remodelling. The bone remodelling process is driven by the coupled activity between
bone-resorbing osteoclasts and bone-forming osteoblasts. The cellular coupling between
osteoclastic and osteoblastic cells is heavily coordinated by systemic and local
regulatory systems to keep both remodelling and resorption processes synchronised
(Martin et al., 2006; Morgan et al., 2008). Many osteoimmunological studies on
inflammatory-related bone diseases have signified the dialogue between the skeletal and
the immune system. This is inevitable as spawned within the bone cavity are the
marrow stem cells. Whereas some mesenchymal stem cells differentiate into
osteoblastic and osteocytic cells, haematopoietic stem cells bifurcate into myeloid
progenitors, which give rise to macrophages, dendritic cells and osteoclasts, and
lymphoid progenitors that develop into lymphocytes and other immune cells (Arai et
al., 1999; Miyamoto et al., 2001; Pittenger et al., 1999). Thus, the development of bone
cells and immune cells occurs within the same microenvironment.
Furthermore, osteoclasts and immune cells share several regulatory molecules such as
cytokines, receptors, transcription factors and signalling molecules (Takayanagi, 2007).
Among these, RANKL demonstrates overlapping roles as the cytokine that is essential
for both osteoclast differentiation/function as well as immune development (Kong et al.,
1999a; Kong et al., 1999b). Under homeostatic conditions, RANKL is expressed by
osteoblasts and osteocytes to regulate osteoclast differentiation and activity (Lacey et
al., 1998; Nakashima et al., 2011). Conversely, immune cells also produce RANKL
during chronic immune activation or autoimmunity, thereby aggravating pathological
bone loss and remodelling (Kong et al., 1999a; Sato et al., 2006; Walsh et al., 2006).
Therefore, osteoimmunological interactions become a key player in the maintenance of
bone homeostasis and in the pathogenesis of systemic bone loss in autoimmune diseases
such as rheumatoid arthritis, spondylarthritis (Goldring, 2013), and systemic lupus
erythematosus (SLE) (Tang et al., 2013a). These disease conditions entail a risk of bone
fractures, causing significant discomfort and deformities. However, the exact
mechanism(s) underlying these pathological conditions remain poorly understood. Thus
understanding the molecular mechanisms involved in regulating the cross talk between
the bone organ and immune system could potentially provide an insight for the
development of novel therapeutic targets for these osteoimmune diseases.

Chapter 9: General Overview and Future Directions
215
In an effort to identify novel factors that play significant roles in the maintenance and
regulation of the osteoimmune system, we systematically screened a chemically induced
(ENU) mutagenesis library for bone phenotypes. Using X-ray imaging, we discovered
the ENU-induced sanroque mutant mice line that exhibits reduced bone mass in the
hindlimbs, vertebrae and tail. Genetic mapping revealed that the mutant mouse line
carried a single point mutation in the Roquin (Rc3h1) gene, resulting in a methionine to
arginine substitution in the ROQ domain of the encoded protein. To the best of our
knowledge, this is the first study to document the novel role of Roquin in bone biology.
The structure of Roquin protein is characterised as a RING-type E3 ubiquitin ligase. An
E3 ubiquitin ligase, along with E1 ubiquitin activating enzyme and E2 ubiquitin
conjugating enzyme, is one of the enzymatic components that drive the ubiquitin
proteasome system (UPS). UPS is an ATP-dependent enzymatic cascade traditionally
described to be involved in the maintenance of protein homeostasis by marking the
protein of interest with a chain of ubiquitin (ubiquitination or ubiquitylation) so as to be
recognised by the proteasome for proteolysis. However, the role of UPS has extended
from just protein degradation, and is reported to be involved in transcription factor
activation, DNA repair, translational control, kinase activation, histone activity and
endocytosis (Pickart et al., 2004).
The sanroque mutant mouse line was first characterised by Vinuesa and colleagues in
2005 to display an autoimmune disease consistent to SLE, with splenomegaly and
lymphomegaly (Vinuesa et al., 2005a). Roquin is a repressor of inducible costimulator
(ICOS), a crucial co-stimulator receptor for TFH. As a result of the loss of function
mutation in Roquin, ICOS levels in sanroque mutant mice are elevated to promote
uncontrolled expansion of TFH, excessive production of IL-21 and high titres of
autoantibodies (Yu et al., 2007). Molecular studies revealed that Roquin represses
ICOS at the post-transcriptional level, whereby the ROQ protein domain binds directly
at the 3’ untranslated region (3’ UTR) of ICOS mRNA (Athanasopoulos et al., 2010;
Glasmacher et al., 2010). Roquin gene thus plays a regulatory role in immune
homeostasis in recognising self from non-self, thereby preventing self-reactive T cells
from providing assistance to B cell maturation and production of self-reactive
antibodies.
This study further extends the immunological role of Roquin to encompass its role in

Chapter 9: General Overview and Future Directions
216
the maintenance of bone homeostasis. MicroCT analysis revealed the reduction in mass
and deterioration in microarchitecture of the bone in sanroque mutant mice.
Histomorphometric analysis further confirmed the osteopenic bone phenotype and
provided some hints into a possible cellular mechanism that leads to the bone loss. The
increase in osteoclast number in vivo is evident in sanroque bone histology and this
correlated with the increased expression levels of osteoclast gene markers TRAP and
Cathepsin K. Unexpectedly, quantification of bone formation rate using double
fluorochrome labelling revealed impaired mineral apposition despite the slight increased
in osteoblast number and osteoid surface in vivo. These observations suggest that the
increased amount of osteoid surface in sanroque mice did not mineralise in a timely
manner, a hallmark of osteomalacia (Maricic, 2008). Consistently, Q-PCR analysis
revealed the significant reduction in osteocalcin expression, which plays a crucial role
during osteoblastic mineralisation (Neve et al., 2013), in the total bone of sanroque
mutant mice.
In an attempt to understand the cellular mechanism through which Roquin regulates
bone homeostasis, we performed a series of in vitro experiments to characterise
osteoblast and osteoclast activities in sanroque mutant mice. In vitro bone nodule
formation analysis was conducted by stimulating calvarial (COBs) and long bone
isolated osteoblasts (LBOBs) with osteogenic medium, which revealed that sanroque
mutant COBs and LBOBs responded to osteogenic induction differently. COBs isolated
from sanroque mutant mice failed to form bone nodules, whereas sanroque LBOBs
displayed a greater area of calcium deposition in comparison to wildtype LBOB culture.
The accelerated activity of sanroque LBOB correlates with the elevated expression of
alkaline phosphatase, osteocalcin, osteopontin and the transcription factor osterix.
However, the augmented calcium deposition by sanroque LBOB generated bone
nodules that appeared morphologically different from that of wildtype. This observation
warrants further investigation
The abnormality of sanroque mutant osteoblast activity is further shown by the
response of COBs and LBOBs toward BMP2, a key activator of bone formation (Rosen,
2009). BMP2 was not able to rescue the aberrant bone formation by sanroque COBs.
The insensitive response towards BMP2 is also observed in sanroque LBOBs. These
data suggest that either the receptor of BMP2 is dysfunctional in mutant osteoblasts, or

Chapter 9: General Overview and Future Directions
217
that the BMP2 signalling pathway may be altered due to the sanroque mutation. In
future studies, it would be useful to investigate the role of Roquin in BMP2 signalling.
The analysis of other key osteoblast gene markers such as the transcription factor Runx2
and collagen I would also provide valuable insight into the molecular mechanism
through which Roquin regulates osteoblastogenesis and mineralisation; respectively.
While Q-PCR analysis of the total bone revealed that RANKL:OPG is elevated in
sanroque mutant mice, co-culture experiments showed that sanroque mutant LBOBs
have reduced capability to support physiological osteoclastogenesis, implying that
mutant osteoblasts may not be the contributor of the elevated RANKL in vivo. This
experiment further indicated the atypical functions of mutant osteoblasts and further
highlighted the role of Roquin in osteoblast biology.
In vitro osteoclastogenesis assay revealed increased RANKL-induced osteoclast
formation in cell cultures derived from sanroque mutant BMMs. The accelerated
osteoclastogenesis observed in sanroque cell cultures was supported by the elevated
gene and protein expression of NFATc1, DC-Stamp, v-ATPase V0 subunit d2. BMMs
from sanroque mutant mice also appeared to be more sensitive towards RANKL
stimulation, as they were able to form osteoclasts at lower doses of RANKL. This
suggests that either the RANKL/RANK signalling pathways are elevated in sanroque
BMM, or that there may be an increase in osteoclast progenitors among BMM, which
are primed to form osteoclasts. In vitro bone resorption assays highlight the overzealous
activity of sanroque mutant osteoclasts, as each mutant multinucleated cell resorbed a
larger area of bone. The increased bone lytic activity is further supported by the
elevated cathepsin K secretion into the supernatant of sanroque mutant osteoclast
cultures when stimulated with bone powder. Consistent with the increased osteoclast
numbers observed in vivo, these in vitro observations showing the increase
osteoclastogenesis and lytic activity may partially attribute to the decreased bone mass
of sanroque mutant mice.
As aforementioned, it can be hypothesised that the increased osteoclastogenesis and
bone lytic activity in sanroque BMM and osteoclast cell culture could be due to the
increase in RANKL/RANK signalling pathway. The RANKL/RANK interaction
resulted in the posttranslational modification of the adapter protein TRAF6, reportedly
via ubiquitination, to activate six major signalling pathways: NFATc1, NF-κB,

Chapter 9: General Overview and Future Directions
218
Akt/PKB, JNK, ERK and p38, which then direct osteoclast-specific gene expression
leading to differentiation and function (Boyle et al., 2003; David et al., 2002; Feng,
2005; Gohda et al., 2005; Kobayashi et al., 2001; Lamothe et al., 2007; Li et al., 2002;
Takayanagi et al., 2000; Xing et al., 2002). Since Roquin was characterised as an E3
ubiquitin ligase, we conducted immunoprecipitation experiments that revealed no
difference in the ubiquitination level of TRAF6 between RANKL-stimulated wildtype
and sanroque BMM culture, suggesting that Roquin does not play a role in the
ubiquitination of TRAF6. Intriguingly, the levels of IκB-α phosphorylation in RANKL-
stimulated sanroque mutant BMMs remained higher, which equates to the inactivation
of NF-κB dimers to prevent the translocation to the nucleus and subsequent gene
transcription (Oeckinghaus et al., 2011). This suggests that Roquin may play a
regulatory role downstream of NF-κB canonical signalling, or that it may be involved in
other RANKL/RANK signalling cascades. Conversely, immunoblotting assay showed
that the downstream NFATc1 signalling is increased in RANKL-stimulated sanroque
mutant BMM, along with the phosphorylation of p38 and ERK1/2. However, other
RANKL/RANK signalling cascades including Akt/PKB and JNK have yet to be
examined. An in depth analysis of the complete RANKL/RANK signalling pathway
array is certainly required to identify the exact molecular pathway through which
Roquin affects osteoclastogenesis. Since the role of Roquin in regulating mRNA
homeostasis has been reported, it would also be worthwhile to do post-transcriptional
studies using tools such as microarrays and ribonomics.
Keeping in mind that sanroque mutant mice exhibit a Lupus-like autoimmune disease,
we explored the possible osteoimmunological interactions by performing co-culture
between BMM supplemented with mouse blood sera. Interestingly, medium
supplemented with sanroque mutant mouse sera elevates the proliferation of both
wildtype and sanroque BMMs. The levels of proliferation-inducing cytokines such as
GM-CSF and IL-6 in the blood serum should be measured as a part of future studies.
MTS assay further revealed that sanroque BMMs proliferate faster in the presence of
M-CSF. These data show that sanroque BMMs are probably more sensitive towards M-
CSF stimulation, or that they may have higher number of cells expressing M-CFS
receptor, c-fms, within the BMMs population of sanroque mice. Investigation of the M-
CSF downstream signalling pathway, such as ERK and AKT, in sanroque mutant
BMMs may provide the underlying molecular pathway in which Roquin is involved.

Chapter 9: General Overview and Future Directions
219
Nevertheless, a role for Roquin in osteoclasts has been established.
As previously hypothesised, increased putative osteoclast progenitors in sanroque
mutant mice may account for the increased osteoclast differentiation and activity in vivo
and in vitro. Subsequently, flow cytometry analysis revealed the significant expansion
of CD3e/CD45R/B220- CD11b+ cells, reported to be typical of cells undergoing
osteoclastogenesis (Jacquin et al., 2006), in the bone marrow of sanroque mutant mice.
Further characterisation of this population revealed increased populations of pre-
committed osteoclast precursors Lin- CD117+ CD115+ CD11blo CD27lo and the more
committed osteoclast precursors Lin- CD117+ CD11blo CD115 (or c-fms) and RANK
double positive cells (Xiao et al., 2013), suggesting that the bone marrow of sanroque
mutant mice are primed to differentiate into osteoclast progenitors.
The priming effect could first be initiated by the reduction of CD70, the ligand for
CD27 in the sanroque mutant mice. CD27/CD70 interaction is important in fine-tuning
the bifurcation of myeloid progenitors and inhibiting osteoclastogenesis to favour the
differentiation towards dendritic cells (Xiao et al., 2013). Additional flow cytometric
analysis revealed that CD4 T cells are the major CD70-bearing cells in the bone marrow
of wildtype mice, which is diminished in sanroque mutant mice. The reduced
expression of CD70 is compounded by the increased RANKL expression in sanroque
mouse, thus providing a more osteoclastogenic environment for the differentiation of
progenitor cells towards osteoclasts. OPG, the decoy receptor of RANKL, first
characterised to be expressed by osteoblasts to inhibit osteoclastogenesis, is also
reported be abundantly expressed by B cells, as knockout mice lacking B cells exhibit
osteoporosis (Li et al., 2007b). Sanroque mutant bone marrow showed a reduction in
CD45R/B220+ B cells, which may explain the increased RANKL:OPG ratio in
sanroque mutant mice.
Using Q-PCR analysis, elevated RANKL expression was observed in both the total
bone and the spleens of sanroque mutant mice. Western blotting further confirmed the
protein expression in the spleens of the sanroque mutant mice. For the first time, we
showed through confocal analysis that the expression of membrane-bound RANKL co-
localised with CD4 and CXCR5 cell surface markers, both of which are markers for
TFH, implicating TFH as a potential osteoclastogenic T cell subset that drives systemic
bone loss in pathological conditions.

Chapter 9: General Overview and Future Directions
220
9.2. Future Directions
The results from this study have underlined several areas that require further
investigation. Addressing these areas will provide deeper understanding into the
molecular mechanisms by which Roquin regulates osteoimmunological interactions
while further establishing the roles of Roquin in bone biology.
The role of Roquin in osteoimmunology and in the pathology of inflammatory
bone loss
Systemic bone loss is common amongst SLE patients; not only due to prolonged
administration of drugs, but the disease per se contributes to the deterioration of bone
microarchitecture in these patients (Tang et al., 2013a). Exacerbated localised and
systemic inflammation due to elevated pro-inflammatory cytokines results in
decoupling of bone remodelling to favour bone loss in lupus patients (Tang et al.,
2013b). The different cytokenic composition of sanroque blood sera is evident in the
co-culture with BMMs. However, a thorough analysis of serum soluble factors using
sensitive tools such as ELISA or multiplex measurements with a cytometric bead
approach is crucial in determining levels of pro-inflammatory cytokines as well as
RANKL and OPG.
The cross-regulation between the bone system and immune system means that there is
also a possibility whereby the autoimmunity is developed due to dysregulated bone
microenvironment. To determine whether the dysregulation in bone homeostasis
resulted in an autoimmune disease, a refined strategy using conditional gene
inactivation in osteoclasts or osteoblasts would be useful. Relying on DNA
recombinase, two recognition (loxP) sites are inserted to excise the flanked (floxed)
gene, i.e. Roquin, through Cre-mediated recombination expressed under a promoter
specific to the cell type, such as calcitonin receptor and osteopontin, which are specific
to osteoclasts and osteoblasts respectively (Friedel et al., 2011).
An alternative approach is to perform bone marrow transplantation, whereby wildtype
and sanroque mutant mice are irradiated and re-transplanted with sanroque and
wildtype bone marrow respectively. This method would be useful to investigate if the
existing osteopenic and autoimmune phenotypes of sanroque mutant mice could be
rescued by the bone marrow of wildtype mice. If the mutation in Roquin gene resulted
in the alteration of transplanted wildtype bone marrow, this may suggest that the

Chapter 9: General Overview and Future Directions
221
existing bone microenvironment may play a role in the pathogenesis of inflammatory-
related bone loss in sanroque mutant mice.
The collagen-induced arthritis (CIA) model has been extensively used to mimic human
rheumatoid arthritis (RA) in a quest to investigate potential mechanisms and to identify
potential targets for therapy. Performing CIA on wildtype and sanroque mutant mice
may provide insights to the osteoimmunological role of Roquin and perhaps further
highlight the osteoclastogenic potential of TFH in inflammatory bone loss.
Localisation of RANKL-bearing cell in sanroque mutant bone
Q-PCR analysis has revealed the significant increase in RANKL expression in the total
bone of sanroque mutant mice. In physiological conditions, RANKL is expressed by
osteoblasts to recruit osteoclast progenitors and induced osteoclastogenesis (Lacey et
al., 1998). However, in vitro osteoblast and BMM coculture revealed the impeded
ability of the mutant osteoblast to support osteoclastogenesis. Thus, the source of the
osteoclastogenic factor has yet to be determined. Imaging-flow cytometric analysis and
immunohistochemistry would be able to provide the identity of the RANKL-bearing
cell in the bone of sanroque mutant mice.
The effect of mutation in Roquin on stem cell niche of sanroque bone marrow
Preliminary flow cytometric analysis revealed atypical populations of the stem cell
niche in the mutant bone marrow. This indicates that the mutation in Roquin gene may
result in the alteration of haematopoiesis in sanroque mutant mice. This in turn may
contribute to the upregulation of common myeloid progenitors and putative osteoclast
progenitors observed in sanroque mice. In order to further investigate stem cell
populations within the niche, and determine exactly how the stem cell populations are
altered, flow cytometry with more specific antibodies against lineage negative bone
marrow cell populations should be performed. This includes the long-term
reconstituting HSCs (LT-HSC), short-term HSCs (ST-HSC) and multipotent
progenitors (MMP) which give rise to common lymphoid progenitors (CLP), B
cell/macrophage bipotent progenitors and common myeloid progenitors.
The role of Roquin in RANKL and BMP2 signalling pathways in sanroque
mutant mice
RANKL/RANK signalling cascades including Akt/PKB and JNK have yet to be

Chapter 9: General Overview and Future Directions
222
examined. In depth analysis of the complete RANKL/RANK signalling pathway array
behind the regulation of osteoclast formation and function by Roquin is certainly
required to identify the exact molecular pathway through which Roquin affects
osteoclastogenesis.
A recent study reported that BMP2 induced-bone marrow-derived mesenchymal stem
cells (BMMSCs) from SLE patients showed reduced capacity for osteogenic
differentiation due to repressed BMP/Smad pathway (Tang et al., 2013b). This suggests
that repressed BMP/Smad signalling pathway and impaired osteoblastic differentiation
may participate in the pathology of osteoporosis in SLE patients. Bearing in mind the
lupus like autoimmune disease, the sanroque mice emerge as a useful model to
investigate the molecular mechanisms involved in osteoblastogenesis in SLE patients.
Furthermore, our in vitro investigation indicated the atypical response of sanroque
mutant osteoblasts towards BMP2. Therefore, BMP2 signalling analysis would provide
insight into possible mechanisms behind the regulation of osteoblast differentiation and
activity by Roquin. These experiments are currently on going, with the results showing
altered phosphorylation of Smad 1, 5, 8 in sanroque mutant LBOBs.
Since the role of Roquin as an E3 ubiquitin ligase in regulating mRNA homeostasis has
been reported (Yu et al., 2007), it would also be worthwhile to do post-transcriptional
studies using tools such as microarrays and ribonomics.
Confirming TFH as a potential osteoclastogenic T cell subset
Several osteoimmunology studies have reported the expression of RANKL by T cells to
directly induce in vitro osteoclastogenesis (Horwood et al., 1999; Kong et al., 1999a;
Kong et al., 1999b; Kotake et al., 2001; Weitzmann et al., 2001; Wong et al., 1997b).
Using confocal analysis, we have shown that CD4 and CXCR5 double positive cells
express RANKL in sanroque mutant spleen, indicating that TFH could potentially be a
new member of the osteoclastogenic T cell subset along with TH17. However, such
observation has to be further validated by sorting TFH to analyse the expression of both
RANKL and OPG. Since the proliferation and differentiation of TFH is induced during
an immune response, vaccination maybe required to boost the rare population of TFH in
wildtype mice (Ma et al., 2014). In vitro co-culture between TFH and BMMs would be
able to further establish the potential osteoclastogenic role of the T cell subset. A few
vaccinated wildtype and sanroque mutant spleens may have to be pooled to perform the

Chapter 9: General Overview and Future Directions
223
in vitro co-culture experiment, which can be conducted as described (Kong et al.,
1999a), while TFH would have to be maintained throughout until the apparent
formation of osteoclasts (Nurieva et al., 2008).
Sample size planning
One of the limitations of this study is regarding the relatively small sample size. One of
the major obstacles in carrying out some experiments was due to insufficient supplies of
mutant mice from Australian National University (ANU) in Canberra, which was partly
owing to lack of breeding colonies. Many of the mice used in this experiment were
ordered as soon as we were informed by ANU on the availability of the breeding
colonies.
Due to logistical challenges, the earliest time point that we can acquire the animal is
usually when the mice turned 100 days old. In most experiments, three biological
repeats were used in this study and were sufficient to show significant difference
between wildtype and sanroque mutant mice. However, future research with larger
sample size should revisit the hypotheses of this study and to address areas mentioned
above, so as to provide robust statistical significance. Further bone characterisation of
the mice at different time points will also provide a clearer picture on the onset of
osteoporosis in sanroque mutant mice.
9.3. Conclusion
A hypothetical model illustrating the role of Roquin in osteoimmunological cellular
interaction in the pathogenesis of bone loss in sanroque mice is shown in Figure 9.1.
Collectively, this thesis has established the reduced bone mass of sanroque mutant
mice. The increased osteoclast number and activity, compounded by abnormality of
osteoblast function, favour bone loss in sanroque mutant mice. This is expedited by
expansion of putative osteoclast progenitors and the increased RANKL expression in
sanroque mutant mice. TFH may play a role in the pathogenesis of systemic bone loss
during chronic autoimmunity by expressing osteoclastogenic factors. The precise
molecular mechanism(s) through which Roquin regulates bone homeostasis remains to
be clarified. Nevertheless, our data demonstrates that the Roquin gene is an important
regulator of bone homeostasis via ENU mutagenesis and the sanroque mouse emerges
as a useful model to investigate the molecular genetics and mechanisms
osteoimmunology.
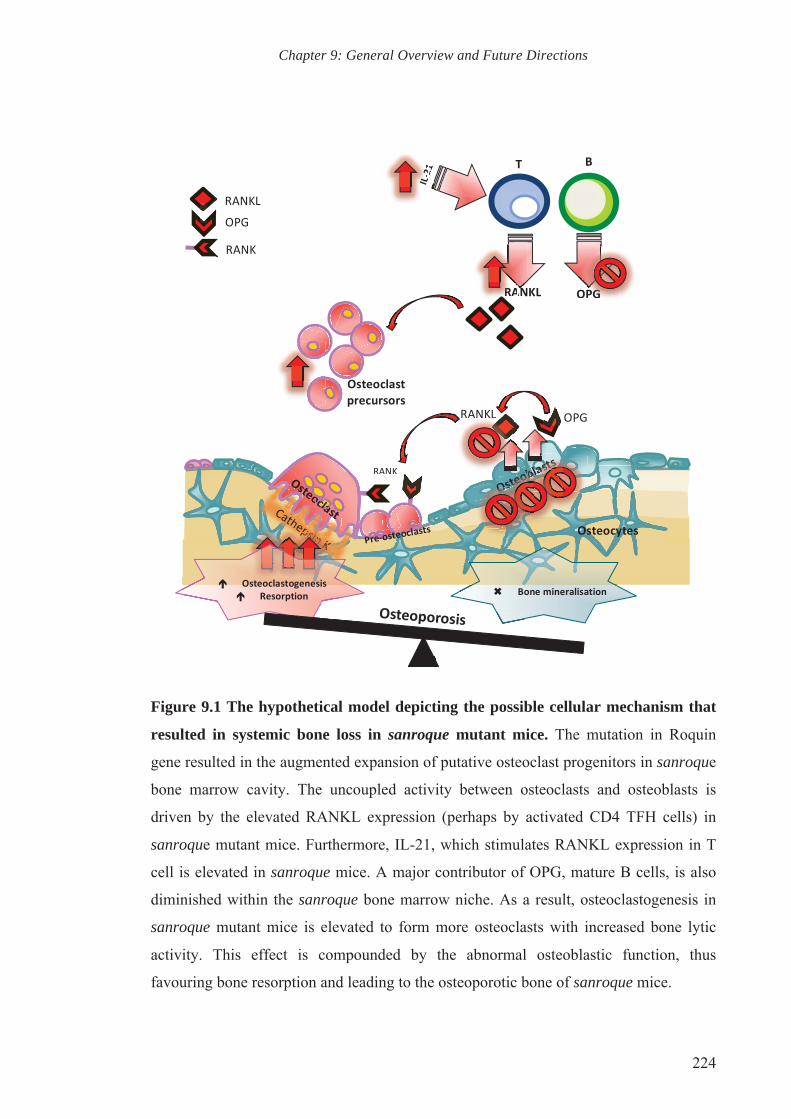
Chapter 9: General Overview and Future Directions
224
Figure 9.1 The hypothetical model depicting the possible cellular mechanism that
resulted in systemic bone loss in sanroque mutant mice. The mutation in Roquin
gene resulted in the augmented expansion of putative osteoclast progenitors in sanroque
bone marrow cavity. The uncoupled activity between osteoclasts and osteoblasts is
driven by the elevated RANKL expression (perhaps by activated CD4 TFH cells) in
sanroque mutant mice. Furthermore, IL-21, which stimulates RANKL expression in T
cell is elevated in sanroque mice. A major contributor of OPG, mature B cells, is also
diminished within the sanroque bone marrow niche. As a result, osteoclastogenesis in
sanroque mutant mice is elevated to form more osteoclasts with increased bone lytic
activity. This effect is compounded by the abnormal osteoblastic function, thus
favouring bone resorption and leading to the osteoporotic bone of sanroque mice.

CHAPTER TEN
REFERENCES

Chapter 10: References
226
10. CHAPTER 10: REFERENCES
Abdel Meguid MH, Hamad YH, Swilam RS, Barakat MS (2013). Relation of interleukin-6 in rheumatoid arthritis patients to systemic bone loss and structural bone damage. Rheumatol Int. 33(3): 697-703.
Abe K, Hirai M, Mizuno K, Higashi N, Sekimoto T, Miki T, et al. (2001). The YXXQ motif in gp 130 is crucial for STAT3 phosphorylation at Ser727 through an H7-sensitive kinase pathway. Oncogene 20(27): 3464-3474.
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997). beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J 16(13): 3797-3804.
Adachi T, Alam R (1998). The mechanism of IL-5 signal transduction. Am J Physiol 275(3 Pt 1): C623-633.
Adamopoulos IE, Danks L, Itonaga I, Locklin RM, Sabokbar A, Ferguson DJ, et al. (2006). Stimulation of osteoclast formation by inflammatory synovial fluid. Virchows Arch 449(1): 69-77.
Akhurst RJ, Hata A (2012). Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 11(10): 790-811.
Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. (2005). The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol 175(4): 2340-2348.
Akisaka T, Subita GP, Kawaguchi H, Shigenaga Y (1989). Different tartrate sensitivity and pH optimum for two isoenzymes of acid phosphatase in osteoclasts. An electron-microscopic enzyme-cytochemical study. Cell Tissue Res 255(1): 69-76.
Akiyama T, Bouillet P, Miyazaki T, Kadono Y, Chikuda H, Chung UI, et al. (2003). Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. EMBO J 22(24): 6653-6664.
Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L (2011). Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem 112(9): 2412-2423.
Alberts B (2008). Molecular biology of the cell. 5th edn. Garland Science: New York.
Alexander KA, Chang MK, Maylin ER, Kohler T, Muller R, Wu AC, et al. (2011). Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res 26(7): 1517-1532.

Chapter 10: References
227
Allen MR, Burr DB (2014). Bone Modeling and Remodeling. In: Burr DB, Allen MR (ed)^(eds). Basic and Applied Bone Biology, edn: Academic Press. p^pp 75-90.
Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, et al. (1998). Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 187(4): 461-468.
Andersen TL, Sondergaard TE, Skorzynska KE, Dagnaes-Hansen F, Plesner TL, Hauge EM, et al. (2009). A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol 174(1): 239-247.
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. (1997). A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390(6656): 175-179.
Andersson G, Ek-Rylander B, Hollberg K, Ljusberg-Sjolander J, Lang P, Norgard M, et al. (2003). TRACP as an osteopontin phosphatase. J Bone Miner Res 18(10): 1912-1915.
Ang E, Pavlos NJ, Rea SL, Qi M, Chai T, Walsh JP, et al. (2009). Proteasome inhibitors impair RANKL-induced NF-kappaB activity in osteoclast-like cells via disruption of p62, TRAF6, CYLD, and IkappaBalpha signaling cascades. J Cell Physiol 220(2): 450-459.
Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, et al. (1999). Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 190(12): 1741-1754.
Ardley HC, Robinson PA (2004). The role of ubiquitin-protein ligases in neurodegenerative disease. Neuro-degenerative diseases 1(2-3): 71-87.
Arron JR, Choi Y (2000a). Bone versus immune system. Nature 408(6812): 535-536.
Arron JR, Choi Y (2000b). Osteoimmunology: Bone versus immune system. Nature 408(6812): 535-536.
Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, et al. (2001). Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol 167(1): 1-5.

Chapter 10: References
228
Athanasopoulos V, Barker A, Yu D, Tan AH, Srivastava M, Contreras N, et al. (2010). The ROQUIN family of proteins localizes to stress granules via the ROQ domain and binds target mRNAs. FEBS J 277(9): 2109-2127.
Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, et al. (2002). A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med 196(7): 979-990.
Au-Yeung BB, Deindl S, Hsu LY, Palacios EH, Levin SE, Kuriyan J, et al. (2009). The structure, regulation, and function of ZAP-70. Immunol Rev 228(1): 41-57.
Avnet S, Cenni E, Perut F, Granchi D, Brandi ML, Giunti A, et al. (2007). Interferon-alpha inhibits in vitro osteoclast differentiation and renal cell carcinoma-induced angiogenesis. Int J Oncol 30(2): 469-476.
Awane M, Andres PG, Li DJ, Reinecker HC (1999). NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol 162(9): 5337-5344.
Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, et al. (2008). CTLA-4 directly inhibits osteoclast formation. Annals of the rheumatic diseases 67(11): 1603-1609.
Bab IA, Einhorn TA (1994). Polypeptide factors regulating osteogenesis and bone marrow repair. J Cell Biochem 55(3): 358-365.
Bachmann MF, Wong BR, Josien R, Steinman RM, Oxenius A, Choi Y (1999). TRANCE, a tumor necrosis factor family member critical for CD40 ligand-independent T helper cell activation. J Exp Med 189(7): 1025-1031.
Bakker AD, Klein-Nulend J (2012). Osteoblast isolation from murine calvaria and long bones. Methods Mol Biol 816: 19-29.
Balani D, Aeberli D, Hofstetter W, Seitz M (2013). Interleukin-17A stimulates granulocyte-macrophage colony-stimulating factor release by murine osteoblasts in the presence of 1,25-dihydroxyvitamin D(3) and inhibits murine osteoclast development in vitro. Arthritis Rheum 65(2): 436-446.
Baud CA (1968). Submicroscopic structure and functional aspects of the osteocyte. Clin Orthop Relat Res 56: 227-236.

Chapter 10: References
229
Bendre MS, Gaddy-Kurten D, Mon-Foote T, Akel NS, Skinner RA, Nicholas RW, et al. (2002). Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res 62(19): 5571-5579.
Bendre MS, Margulies AG, Walser B, Akel NS, Bhattacharrya S, Skinner RA, et al. (2005). Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res 65(23): 11001-11009.
Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ (2003). Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 33(1): 28-37.
Bertolini DR, Nedwin GE, Bringman TS, Smith DD, Mundy GR (1986). Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 319(6053): 516-518.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441(7090): 235-238.
Bhargava U, Bar-Lev M, Bellows CG, Aubin JE (1988). Ultrastructural analysis of bone nodules formed in vitro by isolated fetal rat calvaria cells. Bone 9(3): 155-163.
Blair HC, Zaidi M, Huang CL, Sun L (2008). The developmental basis of skeletal cell differentiation and the molecular basis of major skeletal defects. Biol Rev Camb Philos Soc 83(4): 401-415.
Boe A, Baiocchi M, Carbonatto M, Papoian R, Serlupi-Crescenzi O (1999). Interleukin 6 knock-out mice are resistant to antigen-induced experimental arthritis. Cytokine 11(12): 1057-1064.
Borzi RM, Mazzetti I, Macor S, Silvestri T, Bassi A, Cattini L, et al. (1999). Flow cytometric analysis of intracellular chemokines in chondrocytes in vivo: constitutive expression and enhancement in osteoarthritis and rheumatoid arthritis. FEBS lett 455(3): 238-242.
Boskey AL, Gadaleta S, Gundberg C, Doty SB, Ducy P, Karsenty G (1998). Fourier transform infrared microspectroscopic analysis of bones of osteocalcin-deficient mice provides insight into the function of osteocalcin. Bone 23(3): 187-196.

Chapter 10: References
230
Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. (2006). ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol 177(7): 4927-4932.
Bossard MJ, Tomaszek TA, Thompson SK, Amegadzie BY, Hanning CR, Jones C, et al. (1996). Proteolytic activity of human osteoclast cathepsin K. Expression, purification, activation, and substrate identification. J Biol Chem 271(21): 12517-12524.
Bougeret C, Mansur IG, Dastot H, Schmid M, Mahouy G, Bensussan A, et al. (1992). Increased surface expression of a newly identified 150-kDa dimer early after human T lymphocyte activation. J Immunol 148(2): 318-323.
Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25(7): 1468-1486.
Boyce BF, Xing L (2008). Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473(2): 139-146.
Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. (2002). High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med 346(20): 1513-1521.
Boyle WJ, Simonet WS, Lacey DL (2003). Osteoclast differentiation and activation. Nature 423(6937): 337-342.
Bozec A, Zaiss MM, Kagwiria R, Voll R, Rauh M, Chen Z, et al. (2014). T cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med 6(235): 235ra260.
Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
Brandes ME, Allen JB, Ogawa Y, Wahl SM (1991). Transforming growth factor beta 1 suppresses acute and chronic arthritis in experimental animals. J Clin Invest 87(3): 1108-1113.
Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. (2000). Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med 192(11): 1545-1552.

Chapter 10: References
231
Brennan FM, Chantry D, Turner M, Foxwell B, Maini R, Feldmann M (1990). Detection of transforming growth factor-beta in rheumatoid arthritis synovial tissue: lack of effect on spontaneous cytokine production in joint cell cultures. Clin Exp Immunol 81(2): 278-285.
Bromley M, Woolley DE (1984). Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum 27(9): 968-975.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. (1998). osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12(9): 1260-1268.
Burchill MA, Nardelli DT, England DM, DeCoster DJ, Christopherson JA, Callister SM, et al. (2003). Inhibition of interleukin-17 prevents the development of arthritis in vaccinated mice challenged with Borrelia burgdorferi. Infect Immun 71(6): 3437-3442.
Cain CJ, Rueda R, McLelland B, Collette NM, Loots GG, Manilay JO (2012). Absence of sclerostin adversely affects B-cell survival. J Bone Miner Res 27(7): 1451-1461.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425(6960): 841-846.
Cao L, Moriishi T, Miyazaki T, Iimura T, Hamagaki M, Nakane A, et al. (2011). Comparative morphology of the osteocyte lacunocanalicular system in various vertebrates. J Bone Miner Metab 29(6): 662-670.
Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB (2009). Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res 24(4): 597-605.
Carlsten H (2005). Immune responses and bone loss: the estrogen connection. Immunol Rev 208: 194-206.
Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. (1994). Activation of human dendritic cells through CD40 cross-linking. J Exp Med 180(4): 1263-1272.
Centrella M, Horowitz MC, Wozney JM, McCarthy TL (1994). Transforming growth factor-beta gene family members and bone. Endocr Rev 15(1): 27-39.

Chapter 10: References
232
Centrella M, McCarthy TL, Canalis E (1988). Tumor necrosis factor-alpha inhibits collagen synthesis and alkaline phosphatase activity independently of its effect on deoxyribonucleic acid synthesis in osteoblast-enriched bone cell cultures. Endocrinology 123(3): 1442-1448.
Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, et al. (2008). Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol 181(2): 1232-1244.
Chen G, Deng C, Li YP (2012). TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8(2): 272-288.
Chen ZJ (2005). Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 7(8): 758-765.
Cheng B, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX (2001). Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells. J Bone Miner Res 16(2): 249-259.
Cheng J, Montecalvo A, Kane LP (2011). Regulation of NF-kappaB induction by TCR/CD28. Immunol Res 50(2-3): 113-117.
Cheon H, Yu SJ, Yoo DH, Chae IJ, Song GG, Sohn J (2002). Increased expression of pro-inflammatory cytokines and metalloproteinase-1 by TGF-beta1 in synovial fibroblasts from rheumatoid arthritis and normal individuals. Clin Exp Immunol 127(3): 547-552.
Chim SM, Tickner J, Chow ST, Kuek V, Guo B, Zhang G, et al. (2013). Angiogenic factors in bone local environment. Cytokine Growth Factor Rev 24(3): 297-310.
Cho SW, Soki FN, Koh AJ, Eber MR, Entezami P, Park SI, et al. (2014). Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc Natl Acad Sci U S A 111(4): 1545-1550.
Chow JW, Wilson AJ, Chambers TJ, Fox SW (1998). Mechanical loading stimulates bone formation by reactivation of bone lining cells in 13-week-old rats. J Bone Miner Res 13(11): 1760-1767.
Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. (2004). T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol 173(1): 68-78.

Chapter 10: References
233
Chung DJ, Castro CH, Watkins M, Stains JP, Chung MY, Szejnfeld VL, et al. (2006). Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J Cell Sci 119(Pt 20): 4187-4198.
Chung UI, Kawaguchi H, Takato T, Nakamura K (2004). Distinct osteogenic mechanisms of bones of distinct origins. J Orthop Sci 9(4): 410-414.
Clark WD, Smith EL, Linn KA, Paul-Murphy JR, Muir P, Cook ME (2005). Osteocyte apoptosis and osteoclast presence in chicken radii 0-4 days following osteotomy. Calcif Tissue Int 77(5): 327-336.
Clarke B (2008). Normal bone anatomy and physiology. Clin J Am Soc Nephrol : 3 Suppl 3: S131-139.
Cohen P (2014). Immune diseases caused by mutations in kinases and components of the ubiquitin system. Nat Immunol 15(6): 521-529.
Cohen SL, Moore AM, Ward WE (2004). Interleukin-10 knockout mouse: a model for studying bone metabolism during intestinal inflammation. Inflamm Bowel Dis 10(5): 557-563.
Coquet JM, Middendorp S, van der Horst G, Kind J, Veraar EA, Xiao Y, et al. (2013). The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity 38(1): 53-65.
Corisdeo S, Gyda M, Zaidi M, Moonga BS, Troen BR (2001). New insights into the regulation of cathepsin K gene expression by osteoprotegerin ligand. Biochem Biophys Res Commun 285(2): 335-339.
Corsi A, Collins MT, Riminucci M, Howell PG, Boyde A, Robey PG, et al. (2003). Osteomalacic and hyperparathyroid changes in fibrous dysplasia of bone: core biopsy studies and clinical correlations. J Bone Miner Res 18(7): 1235-1246.
D.Baeten, Emery P, Sieper J, Braun J, Heijde Dvd, McInnes I, et al. (2011). The Anti-IL17A Monoclonal Antibody Secukinumab (AIN457) Showed Good Safety and Efficacy in the Treatment of Active Ankylosing Spondylitis. In: EULAR 2011 (Abstract 0174). 25-28 May 2011, London, UK: The Annual European Congress of Rheumatology.
Danks L, Sabokbar A, Gundle R, Athanasou NA (2002). Synovial macrophage-osteoclast differentiation in inflammatory arthritis. Ann Rheum Dis 61(10): 916-921.

Chapter 10: References
234
David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF (2002). JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci 115(Pt 22): 4317-4325.
DeKoter RP, Walsh JC, Singh H (1998). PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J 17(15): 4456-4468.
Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, et al. (2001). Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res 16(10): 1846-1853.
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. (2000). Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103(2): 351-361.
Deonarain R, Alcami A, Alexiou M, Dallman MJ, Gewert DR, Porter AC (2000). Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol 74(7): 3404-3409.
Derynck R, Zhang YE (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425(6958): 577-584.
Desgeorges A, Gabay C, Silacci P, Novick D, Roux-Lombard P, Grau G, et al. (1997). Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J Rheumatol 24(8): 1510-1516.
Di Giovine FS, Nuki G, Duff GW (1988). Tumour necrosis factor in synovial exudates. Ann Rheum Dis 47(9): 768-772.
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. (2007). Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13(2): 156-163.
Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ (1995). Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 121(6): 1845-1854.
Dierkes C, Kreisel M, Schulz A, Steinmeyer J, Wolff JC, Fink L (2009). Catabolic properties of microdissected human endosteal bone lining cells. Calcif Tissue Int 84(2): 146-155.

Chapter 10: References
235
Dobnig H, Turner RT (1995). Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 136(8): 3632-3638.
Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. (2001a). ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 409(6816): 97-101.
Dong C, Temann UA, Flavell RA (2001b). Cutting edge: critical role of inducible costimulator in germinal center reactions. J Immunol 166(6): 3659-3662.
Doty SB (1981). Morphological evidence of gap junctions between bone cells. Calcif Tissue Int 33(5): 509-512.
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. (1999). RANK is essential for osteoclast and lymph node development. Genes Dev 13(18): 2412-2424.
Dresner-Pollak R, Gelb N, Rachmilewitz D, Karmeli F, Weinreb M (2004). Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology 127(3): 792-801.
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997). Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89(5): 747-754.
Dudley HR, Spiro D (1961). The Fine Structure of Bone Cells. J Biophys Biochem Cytol 11(3): 627-649.
Duplomb L, Baud'huin M, Charrier C, Berreur M, Trichet V, Blanchard F, et al. (2008). Interleukin-6 inhibits receptor activator of nuclear factor kappaB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of Serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology 149(7): 3688-3697.
Duplomb L, Dagouassat M, Jourdon P, Heymann D (2007). Concise review: embryonic stem cells: a new tool to study osteoblast and osteoclast differentiation. Stem Cells 25(3): 544-552.
Edwards JR, Mundy GR (2011). Advances in osteoclast biology: old findings and new insights from mouse models. Nat Rev Rheumatol 7(4): 235-243.

Chapter 10: References
236
Ek-Rylander B, Flores M, Wendel M, Heinegard D, Andersson G (1994). Dephosphorylation of osteopontin and bone sialoprotein by osteoclastic tartrate-resistant acid phosphatase. Modulation of osteoclast adhesion in vitro. J Biol Chem 269(21): 14853-14856.
Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. (1994). Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet 344(8930): 1105-1110.
Eriksen EF (1986). Normal and pathological remodeling of human trabecular bone: three dimensional reconstruction of the remodeling sequence in normals and in metabolic bone disease. Endocr Rev 7(4): 379-408.
Erlebacher A, Derynck R (1996). Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol 132(1-2): 195-210.
Everts V, Beertsen W (2005). External Lysosomes: The Osteoclast and Its Unique Capacities to Degrade Mineralised Tissues. In: Saftig P. Lysosomes, edn. Georgetown, Tex. New York, N.Y.: Landes Bioscience/Eurekah.com ; Springer Science+Business Media. p^pp 197 p.
Everts V, Delaisse JM, Korper W, Jansen DC, Tigchelaar-Gutter W, Saftig P, et al. (2002). The bone lining cell: its role in cleaning Howship's lacunae and initiating bone formation. J Bone Miner Res 17(1): 77-90.
Fakhry M, Hamade E, Badran B, Buchet R, Magne D (2013). Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J Stem Cells 5(4): 136-148.
Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG (2009). Follicular helper T cells: lineage and location. Immunity 30(3): 324-335.
Feng X (2009). Chemical and Biochemical Basis of Cell-Bone Matrix Interaction in Health and Disease. Curr Chem Biol 3(2): 189-196.
Feng X (2005). RANKing intracellular signaling in osteoclasts. IUBMB life 57(6): 389-395.
Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, et al. (2006). A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 38(1): 41-47.

Chapter 10: References
237
Filvaroff E, Erlebacher A, Ye J, Gitelman SE, Lotz J, Heillman M, et al. (1999). Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 126(19): 4267-4279.
Fleige S, Pfaffl MW (2006). RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med 27(2-3): 126-139.
Fontana A, Hengartner H, Weber E, Fehr K, Grob PJ, Cohen G (1982). Interleukin 1 activity in the synovial fluid of patients with rheumatoid arthritis. Rheumatol Int 2(2): 49-53.
Fox SW, Fuller K, Bayley KE, Lean JM, Chambers TJ (2000). TGF-beta 1 and IFN-gamma direct macrophage activation by TNF-alpha to osteoclastic or cytocidal phenotype. J Immunol 165(9): 4957-4963.
Francois RJ, Neure L, Sieper J, Braun J (2006). Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann Rheum Dis 65(6): 713-720.
Franz-Odendaal TA, Hall BK, Witten PE (2006). Buried alive: how osteoblasts become osteocytes. Dev Dyn 235(1): 176-190.
Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. (1997). Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev 11(24): 3482-3496.
Frey O, Petrow PK, Gajda M, Siegmund K, Huehn J, Scheffold A, et al. (2005). The role of regulatory T cells in antigen-induced arthritis: aggravation of arthritis after depletion and amelioration after transfer of CD4+CD25+ T cells. Arthritis Res Ther 7(2): R291-301.
Friedel RH, Wurst W, Wefers B, Kuhn R (2011). Generating conditional knockout mice. Methods Mol Biol 693: 205-231.
Frost HM (1973). Bone remodeling and its relationship to metabolic bone diseases. edn. Thomas: Springfield, Ill.,.
Fujii M, Takeda K, Imamura T, Aoki H, Sampath TK, Enomoto S, et al. (1999). Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell 10(11): 3801-3813.

Chapter 10: References
238
Fujioka K, Kishida T, Ejima A, Yamamoto K, Fujii W, Murakami K, et al. (2014). Inhibition of osteoclastogenesis by osteoblast-like cells genetically engineered to produce interleukin-10. Biochem Biophys Res Commun 456(3): 785-91.
Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, et al. (1996). Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity 5(5): 449-460.
Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ (1993). Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med 178(5): 1733-1744.
Fuller K, Wong B, Fox S, Choi Y, Chambers TJ (1998). TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med 188(5): 997-1001.
Fullilove S, Jellis J, Hughes SP, Remick DG, Friedland JS (2000). Local and systemic concentrations of tumour necrosis factor-alpha, interleukin-6 and interleukin-8 in bacterial osteomyelitis. Trans R Soc Trop Med Hyg 94(2): 221-224.
Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. (2007). IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest 117(1): 122-132.
Geiser AG, Zeng QQ, Sato M, Helvering LM, Hirano T, Turner CH (1998). Decreased bone mass and bone elasticity in mice lacking the transforming growth factor-beta1 gene. Bone 23(2): 87-93.
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996). Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 273(5279): 1236-1238.
Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, et al. (2010). LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 62(4): 929-939.
Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. (2000). Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology 141(11): 3956-3964.

Chapter 10: References
239
Gilbert SF (2000a). Osteogenesis: The development of Bones. In: (ed)^(eds). Development Biology, 6th edn. Sunderland, Mass.: Sinauer Associates. p^pp xviii, 749 p.
Gilbert SF (2000b). The paraxial and intermediate mesoderm. In: (ed)^(eds). Developmental Biology, 6th edn. Sunderland, Mass.: Sinauer Associates. p^pp 465-490.
Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. (2010). Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol 11(8): 725-733.
Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J (2005). RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J 24(4): 790-799.
Goldring SR (2013). Osteoimmunology and bone homeostasis: relevance to spondyloarthritis. Curr Rheumatol Rep 15(7): 342.
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. (2001). LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107(4): 513-523.
Gordon CM (2006). Bone loss in children with Crohn disease: Evidence of "osteoimmune" alterations. Journal Pediatr 148(4): 429-432.
Gough AK, Lilley J, Eyre S, Holder RL, Emery P (1994). Generalised bone loss in patients with early rheumatoid arthritis. Lancet 344(8914): 23-27.
Gowen M, Mundy GR (1986). Actions of recombinant interleukin 1, interleukin 2, and interferon-gamma on bone resorption in vitro. J Immunol 136(7): 2478-2482.
Gratacos J, Collado A, Filella X, Sanmarti R, Canete J, Llena J, et al. (1994). Serum cytokines (IL-6, TNF-alpha, IL-1 beta and IFN-gamma) in ankylosing spondylitis: a close correlation between serum IL-6 and disease activity and severity. Br J Rheumatol 33(10): 927-931.
Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, et al. (2000). Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 43(2): 250-258.

Chapter 10: References
240
Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, et al. (1994). c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 266(5184): 443-448.
Grimston SK, Brodt MD, Silva MJ, Civitelli R (2008). Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1). J Bone Miner Res 23(6): 879-886.
Gu G, Mulari M, Peng Z, Hentunen TA, Vaananen HK (2005). Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Biochem Biophys Res Commun 335(4): 1095-1101.
Guihard P, Danger Y, Brounais B, David E, Brion R, Delecrin J, et al. (2012). Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 30(4): 762-772.
Gunness-Hey M, Hock JM (1984). Increased trabecular bone mass in rats treated with human synthetic parathyroid hormone. Metab Bone Dis Relat Res 5(4): 177-181.
Hamann C, Picke AK, Campbell GM, Balyura M, Rauner M, Bernhardt R, et al. (2014). Effects of parathyroid hormone on bone mass, bone strength, and bone regeneration in male rats with type 2 diabetes mellitus. Endocrinology 155(4): 1197-1206.
Harada A, Sekido N, Akahoshi T, Wada T, Mukaida N, Matsushima K (1994). Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol 56(5): 559-564.
Harada S, Rodan GA (2003). Control of osteoblast function and regulation of bone mass. Nature 423(6937): 349-355.
Hashizume M, Hayakawa N, Mihara M (2008). IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology 47(11): 1635-1640.
Hashizume M, Mihara M (2010). High molecular weight hyaluronic acid inhibits IL-6-induced MMP production from human chondrocytes by up-regulating the ERK inhibitor, MKP-1. Biochem Biophys Res Commun 403(2): 184-189.
Hattner R, Epker BN, Frost HM (1965). Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 206(983): 489-490.

Chapter 10: References
241
Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F (2001). Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res 16(9): 1575-1582.
Hayman AR, Jones SJ, Boyde A, Foster D, Colledge WH, Carlton MB, et al. (1996). Mice lacking tartrate-resistant acid phosphatase (Acp 5) have disrupted endochondral ossification and mild osteopetrosis. Development 122(10): 3151-3162.
He Y, Staser K, Rhodes SD, Liu Y, Wu X, Park SJ, et al. (2011). Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One 6(9): e24780.
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334 ( Pt 2): 297-314.
Henriksen K, Neutzsky-Wulff AV, Bonewald LF, Karsdal MA (2009). Local communication on and within bone controls bone remodeling. Bone 44(6): 1026-1033.
Hering S, Isken F, Janott J, Jost C, Pommer A, Muhr G, et al. (2001). Analysis of TGFbeta3 gene expression and protein levels in human bone and serum. Exp Clin Endocrinol Diabetes 109(2): 107-115.
Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T (1990). Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63(6): 1149-1157.
Hildebrand T, Ruegsegger P (1997). Quantification of Bone Microarchitecture with the Structure Model Index. Comput Methods Biomech Biomed Engin 1(1): 15-23.
Hill PA (1998). Bone remodelling. Br J Orthod 25(2): 101-107.
Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, et al. (1988). Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 18(11): 1797-1801.
Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA (2004). The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem 279(14): 13984-13992.
Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S (1999). Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate

Chapter 10: References
242
osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 25(3): 255-259.
Hong MH, Williams H, Jin CH, Pike JW (2000). The inhibitory effect of interleukin-10 on mouse osteoclast formation involves novel tyrosine-phosphorylated proteins. J Bone Miner Res 15(5): 911-918.
Hopkins SJ, Humphreys M, Jayson MI (1988). Cytokines in synovial fluid. I. The presence of biologically active and immunoreactive IL-1. Clin Exp Immunol 72(3): 422-427.
Horowitz M, Jilka RL (1992). Colony stimulating factors in bone remodeling. In: Cytokines and Bone Metabolism, Gowen M (ed). Boca Raton, FL: CRC Press.
Horowitz MC, Bothwell AL, Hesslein DG, Pflugh DL, Schatz DG (2005). B cells and osteoblast and osteoclast development. Immunol Rev 208: 141-153.
Horowitz MC, Xi Y, Pflugh DL, Hesslein DG, Schatz DG, Lorenzo JA, et al. (2004). Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J Immunol 173(11): 6583-6591.
Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT (1999). Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun 265(1): 144-150.
Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, et al. (2009). IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 16(10): 1332-1343.
Huang H, Li Y, Qi X (2013). Cytokine signaling in the differentiation of innate effector cells. Jak-Stat 2(1): e23531.
Huang W, O'Keefe RJ, Schwarz EM (2003). Exposure to receptor-activator of NFkappaB ligand renders pre-osteoclasts resistant to IFN-gamma by inducing terminal differentiation. Arthritis Res Ther 5(1): R49-59.
Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R (1996). Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev 59(1): 3-10.

Chapter 10: References
243
Hume DA, Loutit JF, Gordon S (1984). The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80: macrophages of bone and associated connective tissue. J Cell Sci 66: 189-194.
Iannone F, De Bari C, Dell'Accio F, Covelli M, Cantatore FP, Patella V, et al. (2001). Interleukin-10 and interleukin-10 receptor in human osteoarthritic and healthy chondrocytes. Clin Exp Rheumatol. 19(2): 139-145.
Igarashi H, Gregory SC, Yokota T, Sakaguchi N, Kincade PW (2002). Transcription from the RAG1 locus marks the earliest lymphocyte progenitors in bone marrow. Immunity 17(2): 117-130.
Iolascon G, Resmini G, Tarantino U (2013). Mechanobiology of bone. Aging Clin Exp Res 25 Suppl 1: S3-7.
Irie N, Takada Y, Watanabe Y, Matsuzaki Y, Naruse C, Asano M, et al. (2009). Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem 284(21): 14637-14644.
Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, et al. (2009). Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 458(7237): 524-528.
Ishimi Y, Miyaura C, Jin CH, Akatsu T, Abe E, Nakamura Y, et al. (1990). IL-6 is produced by osteoblasts and induces bone resorption. J Immunol 145(10): 3297-3303.
Ivanov, II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6): 1121-1133.
Ivashkiv LB, Donlin LT (2014). Regulation of type I interferon responses. Nat Rev Immunol 14(1): 36-49.
Jacome-Galarza CE, Lee SK, Lorenzo JA, Aguila HL (2013). Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res 28(5): 1203-1213.
Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL (2006). Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res 21(1): 67-77.

Chapter 10: References
244
Ji JD, Park-Min KH, Shen Z, Fajardo RJ, Goldring SR, McHugh KP, et al. (2009). Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J Immunol 183(11): 7223-7233.
Jiang H, Harris MB, Rothman P (2000). IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol 105(6 Pt 1): 1063-1070.
Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC (1998). Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res 13(5): 793-802.
Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC (1999). Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest 104(4): 439-446.
Jing D, Baik AD, Lu XL, Zhou B, Lai X, Wang L, et al. (2014). In situ intracellular calcium oscillations in osteocytes in intact mouse long bones under dynamic mechanical loading. FASEB J 28(4):1582-92.
Jones SJ, Gray C, Sakamaki H, Arora M, Boyde A, Gourdie R, et al. (1993). The incidence and size of gap junctions between the bone cells in rat calvaria. Anat Embryol (Berl) 187(4): 343-352.
Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. (2003). Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity 18(6): 751-762.
Jung Y, Wang J, Schneider A, Sun YX, Koh-Paige AJ, Osman NI, et al. (2006). Regulation of SDF-1 (CXCL12) production by osteoblasts; a possible mechanism for stem cell homing. Bone 38(4): 497-508.
Jung YK, Kim GW, Park HR, Lee EJ, Choi JY, Beier F, et al. (2013). Role of interleukin-10 in endochondral bone formation in mice: anabolic effect via the bone morphogenetic protein/Smad pathway. Arthritis Rheum 65(12): 3153-3164.
Kamalakar A, Bendre MS, Washam CL, Fowler TW, Carver A, Dilley JD, et al. (2014). Circulating interleukin-8 levels explain breast cancer osteolysis in mice and humans. Bone 61: 176-185.
Kamioka H, Ishihara Y, Ris H, Murshid SA, Sugawara Y, Takano-Yamamoto T, et al. (2007). Primary cultures of chick osteocytes retain functional gap junctions between osteocytes and between osteocytes and osteoblasts. Microsc Microanal 13(2): 108-117.

Chapter 10: References
245
Kaneda T, Nojima T, Nakagawa M, Ogasawara A, Kaneko H, Sato T, et al. (2000). Endogenous production of TGF-beta is essential for osteoclastogenesis induced by a combination of receptor activator of NF-kappa B ligand and macrophage-colony-stimulating factor. J Immunol 165(8): 4254-4263.
Kaneko S, Satoh T, Chiba J, Ju C, Inoue K, Kagawa J (2000). Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell Mol Ther 6(2): 71-79.
Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C (2003). IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol 24(1): 25-29.
Kar R, Riquelme MA, Werner S, Jiang JX (2013). Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J Bone Miner Res 28(7): 1611-1621.
Kariuki SN, Niewold TB (2010). Genetic regulation of serum cytokines in systemic lupus erythematosus. Transl Res 155(3): 109-117.
Karlson EW, Chibnik LB, Tworoger SS, Lee IM, Buring JE, Shadick NA, et al. (2009). Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arthritis Rheum 60(3): 641-652.
Karsdal MA, Martin TJ, Bollerslev J, Christiansen C, Henriksen K (2007). Are nonresorbing osteoclasts sources of bone anabolic activity? J Bone Miner Res 22(4): 487-494.
Katavic V, Lukic IK, Kovacic N, Grcevic D, Lorenzo JA, Marusic A (2003). Increased bone mass is a part of the generalized lymphoproliferative disorder phenotype in the mouse. J Immunol 170(3): 1540-1547.
Kawakami A, Eguchi K, Matsuoka N, Tsuboi M, Koji T, Urayama S, et al. (1997). Fas and Fas ligand interaction is necessary for human osteoblast apoptosis. J Bone Miner Res 12(10): 1637-1646.
Kayal RA (2013). The role of osteoimmunology in periodontal disease. Biomed Res Int 2013: 639368.
Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD (2003). Interleukin-4 and interleukin-13 signaling connections maps. Science 300(5625): 1527-1528.

Chapter 10: References
246
Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB (2012). Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 50(5): 1115-1122.
Key LL, Jr., Rodriguiz RM, Willi SM, Wright NM, Hatcher HC, Eyre DR, et al. (1995). Long-term treatment of osteopetrosis with recombinant human interferon gamma. N Engl J Med 332(24): 1594-1599.
Kikuta J, Kawamura S, Okiji F, Shirazaki M, Sakai S, Saito H, et al. (2013). Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc Natl Acad Sci U S A 110(17): 7009-7013.
Kim D, Mebius RE, MacMicking JD, Jung S, Cupedo T, Castellanos Y, et al. (2000). Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J Exp Med 192(10): 1467-1478.
Kim HS, Kim DK, Kim AR, Mun SH, Lee SK, Kim JH, et al. (2012). Fyn positively regulates the activation of DAP12 and FcRgamma-mediated costimulatory signals by RANKL during osteoclastogenesis. Cell Signal 24(6): 1306-1314.
Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, et al. (2009). The mechanism of osteoclast differentiation induced by IL-1. J Immunol 183(3): 1862-1870.
Kim K, Kim JH, Lee J, Jin HM, Kook H, Kim KK, et al. (2007a). MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 109(8): 3253-3259.
Kim K, Lee SH, Ha Kim J, Choi Y, Kim N (2008). NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol Endocrinol 22(1): 176-185.
Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, et al. (2005). Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med 202(5): 589-595.
Kim SY, Han SW, Kim GW, Lee JM, Kang YM (2004). TGF-beta1 polymorphism determines the progression of joint damage in rheumatoid arthritis. Scand J Rheumatol 33(6): 389-394.
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB (2007b). Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun 357(4): 1046-1052.

Chapter 10: References
247
Kitami S, Tanaka H, Kawato T, Tanabe N, Katono-Tani T, Zhang F, et al. (2010). IL-17A suppresses the expression of bone resorption-related proteinases and osteoclast differentiation via IL-17RA or IL-17RC receptors in RAW264.7 cells. Biochimie 92(4): 398-404.
Kitazawa R, Kimble RB, Vannice JL, Kung VT, Pacifici R (1994). Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J Clin Invest 94(6): 2397-2406.
Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. (2001). Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 20(6): 1271-1280.
Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, et al. (1994). Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J Clin Invest 93(3): 921-928.
Koch AE, Kunkel SL, Shah MR, Fu R, Mazarakis DD, Haines GK, et al. (1995a). Macrophage inflammatory protein-1 beta: a C-C chemokine in osteoarthritis. Clin Immunol Immunopathol 77(3): 307-314.
Koch AE, Kunkel SL, Shah MR, Hosaka S, Halloran MM, Haines GK, et al. (1995b). Growth-related gene product alpha. A chemotactic cytokine for neutrophils in rheumatoid arthritis. J Immunol 155(7): 3660-3666.
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, et al. (2005). Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol 167(1): 141-149.
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. (2014). Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20(1): 62-68.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. (1999a). Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 402(6759): 304-309.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. (1999b). OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397(6717): 315-323.

Chapter 10: References
248
Konig A, Krenn V, Toksoy A, Gerhard N, Gillitzer R (2000). Mig, GRO alpha and RANTES messenger RNA expression in lining layer, infiltrates and different leucocyte populations of synovial tissue from patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Virchows Archiv 436(5): 449-458.
Kopesky P, Tiedemann K, Alkekhia D, Zechner C, Millard B, Schoeberl B, et al. (2014). Autocrine signaling is a key regulatory element during osteoclastogenesis. Biol Open 3(8): 767-76.
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009). IL-17 and Th17 Cells. Annu Rev Immunol 27: 485-517.
Kotake S, Nanke Y, Mogi M, Kawamoto M, Furuya T, Yago T, et al. (2005). IFN-gamma-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur J Immunol 35(11): 3353-3363.
Kotake S, Udagawa N, Hakoda M, Mogi M, Yano K, Tsuda E, et al. (2001). Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum 44(5): 1003-1012.
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. (1999). IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9): 1345-1352.
Kristensen HB, Andersen TL, Marcussen N, Rolighed L, Delaisse JM (2013). Increased presence of capillaries next to remodeling sites in adult human cancellous bone. J Bone Miner Res 28(3): 574-585.
Kukita T, Wada N, Kukita A, Kakimoto T, Sandra F, Toh K, et al. (2004). RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med 200(7): 941-946.
Kular J, Tickner J, Chim SM, Xu J (2012). An overview of the regulation of bone remodelling at the cellular level. Clin Biochem 45(12): 863-873.
Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. (1993). Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A 90(2): 770-774.
Kurt-Jones EA, Mandell L, Whitney C, Padgett A, Gosselin K, Newburger PE, et al. (2002). Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood 100(5): 1860-1868.

Chapter 10: References
249
Kwok SK, Cho ML, Park MK, Oh HJ, Park JS, Her YM, et al. (2012). Interleukin-21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen-induced arthritis. Arthritis Rheum 64(3): 740-751.
Kwon RY, Temiyasathit S, Tummala P, Quah CC, Jacobs CR (2010). Primary cilium-dependent mechanosensing is mediated by adenylyl cyclase 6 and cyclic AMP in bone cells. FASEB J 24(8): 2859-2868.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. (1998). Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93(2): 165-176.
Lafage-Proust MH, Prisby R, Roche B, Vico L (2010). Bone vascularization and remodeling. Joint Bone Spine 77(6): 521-524.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000). TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106(12): 1481-1488.
Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG (2007). TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun 359(4): 1044-1049.
Leaffer D, Sweeney M, Kellerman LA, Avnur Z, Krstenansky JL, Vickery BH, et al. (1995). Modulation of osteogenic cell ultrastructure by RS-23581, an analog of human parathyroid hormone (PTH)-related peptide-(1-34), and bovine PTH-(1-34). Endocrinology 136(8): 3624-3631.
Lee CA, Einhorn TA (2001). The Bone Organ System: Form and Function. In: Marcus R, Feldman D, Kelsey JL (ed)^(eds). Osteoporosis, 2nd edn. San Diego, CA: Academic Press.
Lee KL, Hoey DA, Spasic M, Tang T, Hammond HK, Jacobs CR (2014). Adenylyl cyclase 6 mediates loading-induced bone adaptation in vivo. FASEB J 28(3): 1157-1165.
Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, et al. (2006). v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med 12(12): 1403-1409.
Lee Y (2013). The role of interleukin-17 in bone metabolism and inflammatory skeletal diseases. BMB Rep 46(10): 479-483.

Chapter 10: References
250
Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, et al. (2010). Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum 62(10): 2876-2885.
Leisen JC, Duncan H, Riddle JM, Pitchford WC (1988). The erosive front: a topographic study of the junction between the pannus and the subchondral plate in the macerated rheumatoid metacarpal head. J Rheumatol 15(1): 17-22.
Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q (2010). Cross talk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 116(2): 210-217.
Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC (2007a). Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J Biol Chem 282(45): 33098-33106.
Li X, Udagawa N, Itoh K, Suda K, Murase Y, Nishihara T, et al. (2002). p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 143(8): 3105-3113.
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. (2007b). B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109(9): 3839-3848.
Lian JB, Stein GS (2001). Osteoblast Biology. In: Marcus R, Feldman D, Kelsey J (ed)^(eds). Osteoporosis, edn, Vol. 1. Burlington, Mass: Elsevier Acadamic Press.
Ling Y, Rios HF, Myers ER, Lu Y, Feng JQ, Boskey AL (2005). DMP1 depletion decreases bone mineralization in vivo: an FTIR imaging analysis. J Bone Miner Res 20(12): 2169-2177.
Lisignoli G, Toneguzzi S, Grassi F, Piacentini A, Tschon M, Cristino S, et al. (2002). Different chemokines are expressed in human arthritic bone biopsies: IFN-gamma and IL-6 differently modulate IL-8, MCP-1 and rantes production by arthritic osteoblasts. Cytokine 20(5): 231-238.
Lisignoli G, Toneguzzi S, Pozzi C, Piacentini A, Grassi F, Ferruzzi A, et al. (1999). Chemokine expression by subchondral bone marrow stromal cells isolated from osteoarthritis (OA) and rheumatoid arthritis (RA) patients. Clin Exp Immunol 116(2): 371-378.

Chapter 10: References
251
Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, et al. (2002). A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet 70(1): 11-19.
Liu XH, Kirschenbaum A, Yao S, Levine AC (2005). Cross-talk between the interleukin-6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-{kappa}B (RANK) ligand/RANK system. Endocrinology 146(4): 1991-1998.
Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4): 402-408.
Lloyd SA, Lewis GS, Zhang Y, Paul EM, Donahue HJ (2012). Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res 27(11): 2359-2372.
Loiselle AE, Paul EM, Lewis GS, Donahue HJ (2013). Osteoblast and osteocyte-specific loss of Connexin43 results in delayed bone formation and healing during murine fracture healing. J Orthop Res 31(1): 147-154.
Lourenco EV, La Cava A (2009). Cytokines in systemic lupus erythematosus. Curr Mol Med 9(3): 242-254.
Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB (2002). Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res 51(2): 102-104.
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. (2004). Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50(2): 650-659.
Luchin A, Purdom G, Murphy K, Clark MY, Angel N, Cassady AI, et al. (2000). The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J Bone Miner Res 15(3): 451-460.
Lucht U (1972). Osteoclasts and their relationship to bone as studied by electron microscopy. Z Zellforsch Mikrosk Anat 135(2): 211-228.
Ma CS, Deenick EK (2014). Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol 92(1): 64-71.

Chapter 10: References
252
Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, et al. (2012). Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol 2012: 827480.
Macias MP, Fitzpatrick LA, Brenneise I, McGarry MP, Lee JJ, Lee NA (2001). Expression of IL-5 alters bone metabolism and induces ossification of the spleen in transgenic mice. J Clin Invest 107(8): 949-959.
Maecker HT, Trotter J (2006). Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry A 69(9): 1037-1042.
Mak TW, Shahinian A, Yoshinaga SK, Wakeham A, Boucher LM, Pintilie M, et al. (2003). Costimulation through the inducible costimulator ligand is essential for both T helper and B cell functions in T cell-dependent B cell responses. Nat Immunol. 4(8): 765-772.
Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, et al. (2007). Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc Natl Acad Sci U S A 104(33): 13325-13330.
Mangashetti LS, Khapli SM, Wani MR (2005). IL-4 inhibits bone-resorbing activity of mature osteoclasts by affecting NF-kappa B and Ca2+ signaling. J Immunol 175(2): 917-925.
Mann GN, Jacobs TW, Buchinsky FJ, Armstrong EC, Li M, Ke HZ, et al. (1994). Interferon-gamma causes loss of bone volume in vivo and fails to ameliorate cyclosporin A-induced osteopenia. Endocrinology 135(3): 1077-1083.
Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, et al. (1997). High susceptibility to collagen-induced arthritis in mice lacking IFN-gamma receptors. J Immunol 158(11): 5501-5506.
Mansour A, Anginot A, Mancini SJ, Schiff C, Carle GF, Wakkach A, et al. (2011). Osteoclast activity modulates B-cell development in the bone marrow. Cell Res 21(7): 1102-1115.
Maricic M (2008). Osteomalacia. Curr Osteoporos Rep 6(4): 130-133.
Martin TJ, Quinn JM, Gillespie MT, Ng KW, Karsdal MA, Sims NA (2006). Mechanisms involved in skeletal anabolic therapies. Ann N Y Acad Sci 1068: 458-470.

Chapter 10: References
253
Martin TJ, Sims NA (2005). Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 11(2): 76-81.
Matsuno H, Yudoh K, Katayama R, Nakazawa F, Uzuki M, Sawai T, et al. (2002). The role of TNF-alpha in the pathogenesis of inflammation and joint destruction in rheumatoid arthritis (RA): a study using a human RA/SCID mouse chimera. Rheumatology 41(3): 329-337.
Matsuo K, Irie N (2008). Osteoclast-osteoblast communication. Arch Biochem Biophys 473(2): 201-209.
Matsuo K, Otaki N (2012). Bone cell interactions through Eph/ephrin: bone modeling, remodeling and associated diseases. Cell Adh Migr 6(2): 148-156.
McClugage SG, Jr., McCuskey RS (1973). Relationship of the microvascular system to bone resorption and growth in situ. Microvasc Res 6(1): 132-134.
McInnes IB, Sieper J, Braun J, Emery P, van der Heijde D, Isaacs JD, et al. (2014). Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 73(2): 349-356.
Meghji S (1992). Bone remodelling. Br Dent J 172(6): 235-242.
Menton DN, Simmons DJ, Chang SL, Orr BY (1984). From bone lining cell to osteocyte--an SEM study. The Anatomical record 209(1): 29-39.
Meury T, Akhouayri O, Jafarov T, Mandic V, St-Arnaud R (2010). Nuclear alpha NAC influences bone matrix mineralization and osteoblast maturation in vivo. Mol Cell Biol 30(1): 43-53.
Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. (2009). Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15(7): 774-780.
Millan FA, Denhez F, Kondaiah P, Akhurst RJ (1991). Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development 111(1): 131-143.
Miller SC, Jee WS (1987). The bone lining cell: a distinct phenotype? Calcif Tissue Int 41(1): 1-5.

Chapter 10: References
254
Miyamoto T, Ohneda O, Arai F, Iwamoto K, Okada S, Takagi K, et al. (2001). Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood 98(8): 2544-2554.
Miyamoto T, Suda T (2003). Differentiation and function of osteoclasts. Keio J Med 52(1): 1-7.
Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, et al. (2000). Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol 148(2): 333-342.
Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, et al. (1998). Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun 247(3): 610-615.
Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, et al. (2004). The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci U S A 101(16): 6158-6163.
Mohamed SG, Sugiyama E, Shinoda K, Taki H, Hounoki H, Abdel-Aziz HO, et al. (2007). Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone 41(4): 592-602.
Morgan EF, Barnes GL, Einhorn TA (2008). The Bone Organ System: Form and Function. In: Marcus R, Feldman D, Nelson DA, Rosen CJ (ed)^(eds). Osteoporosis, 3rd edn, Vol. 1. Burlington, Mass.: Academic Press/Elsevier. p^pp 2 v. (xxiv, 1941 p., [1918] p. of plates).
Morgan ME, Flierman R, van Duivenvoorde LM, Witteveen HJ, van Ewijk W, van Laar JM, et al. (2005). Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum 52(7): 2212-2221.
Morko J, Kiviranta R, Mulari MT, Ivaska KK, Vaananen HK, Vuorio E, et al. (2009). Overexpression of cathepsin K accelerates the resorption cycle and osteoblast differentiation in vitro. Bone 44(4): 717-728.
Morony S, Capparelli C, Lee R, Shimamoto G, Boone T, Lacey DL, et al. (1999). A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resorption induced by IL-1beta, TNF-alpha, PTH, PTHrP, and 1, 25(OH)2D3. J Bone Miner Res 14(9): 1478-1485.

Chapter 10: References
255
Moseley TA, Haudenschild DR, Rose L, Reddi AH (2003). Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev 14(2): 155-174.
Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, et al. (2013). M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 497(7448): 239-243.
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835): 256-260.
Mukai T, Ishida S, Ishikawa R, Yoshitaka T, Kittaka M, Gallant R, et al. (2014). SH3BP2 Cherubism Mutation Potentiates TNF-alpha-Induced Osteoclastogenesis Via NFATc1 and TNF-alpha-Mediated Inflammatory Bone Loss. J Bone Miner Res 29(12): 2618-35.
Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. (1996). Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382(6592): 635-638.
Nakae S, Nambu A, Sudo K, Iwakura Y (2003). Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171(11): 6173-6177.
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. (2011). Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17(10): 1231-1234.
Nardelli DT, Burchill MA, England DM, Torrealba J, Callister SM, Schell RF (2004). Association of CD4+ CD25+ T cells with prevention of severe destructive arthritis in Borrelia burgdorferi-vaccinated and challenged gamma interferon-deficient mice treated with anti-interleukin-17 antibody. Clin Diagn Lab Immunol 11(6): 1075-1084.
Negishi-Koga T, Shinohara M, Komatsu N, Bito H, Kodama T, Friedel RH, et al. (2011). Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med 17(11): 1473-1480.
Neidhart M, Baraliakos X, Seemayer C, Zelder C, Gay RE, Michel BA, et al. (2009). Expression of cathepsin K and matrix metalloproteinase 1 indicate persistent osteodestructive activity in long-standing ankylosing spondylitis. Ann Rheum Dis 68(8): 1334-1339.

Chapter 10: References
256
Nelms KA, Goodnow CC (2001). Genome-wide ENU mutagenesis to reveal immune regulators. Immunity 15(3): 409-418.
Ness TJ, Gebhart GF (1991). Interactions between visceral and cutaneous nociception in the rat. II. Noxious visceral stimuli inhibit cutaneous nociceptive neurons and reflexes. J Neurophysiol 66(1): 29-39.
Neve A, Corrado A, Cantatore FP (2013). Osteocalcin: skeletal and extra-skeletal effects. J Cell Physiol 228(6): 1149-1153.
Neve A, Corrado A, Cantatore FP (2012). Osteocytes: central conductors of bone biology in normal and pathological conditions. Acta Physiol 204(3): 317-330.
Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. (2007). IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol 19(6): 695-702.
Noble BS, Stevens H, Loveridge N, Reeve J (1997). Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone 20(3): 273-282.
Nolte MA, Arens R, van Os R, van Oosterwijk M, Hooibrink B, van Lier RA, et al. (2005). Immune activation modulates hematopoiesis through interactions between CD27 and CD70. Nat Immunol 6(4): 412-418.
Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA (2009). Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev 229(1): 216-231.
Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, et al. (2009). Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol 182(1): 613-622.
Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. (2008). Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 29(1): 138-149.
Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. (2009). Bcl6 mediates the development of T follicular helper cells. Science 325(5943): 1001-1005.
O'Brien CA (2009). Control of RANKL gene expression. Bone 46(4): 911-9.

Chapter 10: References
257
O'Neill HC, Griffiths KL, Periasamy P, Hinton RA, Hey YY, Petvises S, et al. (2011). Spleen as a site for hematopoiesis of a distinct antigen presenting cell type. Stem Cells Int 2011: 954275.
Oeckinghaus A, Hayden MS, Ghosh S (2011). Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12(8): 695-708.
Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, et al. (1998). Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci U S A 95(14): 8222-8226.
Onyia JE, Bidwell J, Herring J, Hulman J, Hock JM (1995). In vivo, human parathyroid hormone fragment (hPTH 1-34) transiently stimulates immediate early response gene expression, but not proliferation, in trabecular bone cells of young rats. Bone 17(5): 479-484.
Orimo H (2010). The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch 77(1): 4-12.
Ortega N, Behonick DJ, Werb Z (2004). Matrix remodeling during endochondral ossification. Trends Cell Biol 14(2): 86-93.
Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ (2000). Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci U S A 97(21): 11439-11444.
Palmqvist P, Persson E, Conaway HH, Lerner UH (2002). IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol 169(6): 3353-3362.
Palumbo C (1986). A three-dimensional ultrastructural study of osteoid-osteocytes in the tibia of chick embryos. Cell Tissue Res 246(1): 125-131.
Palumbo C, Palazzini S, Marotti G (1990). Morphological study of intercellular junctions during osteocyte differentiation. Bone 11(6): 401-406.
Pappalardo A, Thompson K (2013). Activated gammadelta T cells inhibit osteoclast differentiation and resorptive activity in vitro. Clin Exp Immunol 174(2): 281-291.
Parfitt AM (2001). The bone remodeling compartment: a circulatory function for bone lining cells. J Bone Miner Res 16(9): 1583-1585.

Chapter 10: References
258
Parfitt AM (1990). Bone-forming cells in clinical conditons. In: Hall BK (ed)^(eds). Bone, edn, Vol. 1: the osteoblast and osteocyte. Boca Raton, FL.: Telford Press and CRC Press. p^pp 351-429.
Parfitt AM, Mundy GR, Roodman GD, Hughes DE, Boyce BF (1996). A new model for the regulation of bone resorption, with particular reference to the effects of bisphosphonates. J Bone Miner Res 11(2): 150-159.
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. (2004). Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279(9): 7370-7377.
Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. (2000). Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 408(6808): 57-63.
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ (2008). Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A 105(52): 20764-20769.
Pelton RW, Saxena B, Jones M, Moses HL, Gold LI (1991). Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 115(4): 1091-1105.
Perlot T, Penninger JM (2012). Development and function of murine B cells lacking RANK. J Immunol 188(3): 1201-1205.
Perng DW, Yang DM, Hsiao YH, Lo T, Lee OK, Wu MT, et al. (2012). miRNA-146a expression positively regulates tumor necrosis factor-alpha-induced interleukin-8 production in mesenchymal stem cells and differentiated lung epithelial-like cells. Tissue Eng Part A 18(21-22): 2259-2267.
Peterlik M, Hoffmann O, Swetly P, Klaushofer K, Koller K (1985). Recombinant gamma-interferon inhibits prostaglandin-mediated and parathyroid hormone-induced bone resorption in cultured neonatal mouse calvaria. FEBS Lett 185(2): 287-290.
Pettit AR, Chang MK, Hume DA, Raggatt LJ (2008). Osteal macrophages: a new twist on coupling during bone dynamics. Bone 43(6): 976-982.

Chapter 10: References
259
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, et al. (2001). TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol 159(5): 1689-1699.
Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM (2006). RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology 45(9): 1068-1076.
Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9): e45.
Piccinini AM, Midwood KS (2010). DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010: pii: 672395.
Pickart CM, Eddins MJ (2004). Ubiquitin: structures, functions, mechanisms. BBA Clin 1695(1-3): 55-72.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. (1999). Multilineage potential of adult human mesenchymal stem cells. Science 284(5411): 143-147.
Plotkin LI, Manolagas SC, Bellido T (2002). Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem 277(10): 8648-8657.
Pohlers D, Beyer A, Koczan D, Wilhelm T, Thiesen HJ, Kinne RW (2007). Constitutive upregulation of the transforming growth factor-beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther 9(3): R59.
Pratama A, Ramiscal RR, Silva DG, Das SK, Athanasopoulos V, Fitch J, et al. (2013). Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity 38(4): 669-680.
Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. (1995). Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 11(4): 409-414.
Qi B, Cong Q, Li P, Ma G, Guo X, Yeh J, et al. (2014). Ablation of Tak1 in osteoclast progenitor leads to defects in skeletal growth and bone remodeling in mice. Sci Rep 4: 7158.

Chapter 10: References
260
Quinn JM, Itoh K, Udagawa N, Hausler K, Yasuda H, Shima N, et al. (2001). Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res 16(10): 1787-1794.
Raggatt LJ, Partridge NC (2010). Cellular and molecular mechanisms of bone remodeling. J Biol Chem 285(33): 25103-25108.
Ramsdell F, Seaman MS, Miller RE, Tough TW, Alderson MR, Lynch DH (1994). gld/gld mice are unable to express a functional ligand for Fas. European journal of immunology 24(4): 928-933.
Rasheed AU, Rahn HP, Sallusto F, Lipp M, Muller G (2006). Follicular B helper T cell activity is confined to CXCR5(hi)ICOS(hi) CD4 T cells and is independent of CD57 expression. Eur J Immunol 36(7): 1892-1903.
Rasmussen H, Bordier P (1973). The physiological and cellular basis of metabolic bone disease. edn. Williams & Wilkins Co.: Baltimore,.
Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA (2008). The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev 22(8): 992-997.
Ratkay LG, Zhang L, Tonzetich J, Waterfield JD (1993). Complete Freund's adjuvant induces an earlier and more severe arthritis in MRL-lpr mice. J Immunol 151(9): 5081-5087.
Reddy SV, Roodman GD (1998). Control of osteoclast differentiation. Crit Rev Eukaryot Gene Expr 8(1): 1-17.
Redlich K, Gortz B, Hayer S, Zwerina J, Doerr N, Kostenuik P, et al. (2004). Repair of local bone erosions and reversal of systemic bone loss upon therapy with anti-tumor necrosis factor in combination with osteoprotegerin or parathyroid hormone in tumor necrosis factor-mediated arthritis. Am J Pathol 164(2): 543-555.
Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, et al. (2002). Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest 110(10): 1419-1427.
Rifkin BR, Heijl L (1979). The occurrence of mononuclear cells at sites of osteoclastic bone resorption in experimental periodontitis. J Periodontol 50(12): 636-640.

Chapter 10: References
261
Ritchlin C, Haas-Smith SA (2001). Expression of interleukin 10 mRNA and protein by synovial fibroblastoid cells. J Rheumatol 28(4): 698-705.
Rodriguiz RM, Key LL, Jr., Ries WL (1993). Combination macrophage-colony stimulating factor and interferon-gamma administration ameliorates the osteopetrotic condition in microphthalmic (mi/mi) mice. Pediatr Res 33(4 Pt 1): 384-389.
Romas E, Sims NA, Hards DK, Lindsay M, Quinn JW, Ryan PF, et al. (2002). Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. Am J Pathol 161(4): 1419-1427.
Rosen V (2009). BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev 20(5-6): 475-480.
Rothe L, Collin-Osdoby P, Chen Y, Sunyer T, Chaudhary L, Tsay A, et al. (1998). Human osteoclasts and osteoclast-like cells synthesize and release high basal and inflammatory stimulated levels of the potent chemokine interleukin-8. Endocrinology 139(10): 4353-4363.
Roths JB, Murphy ED, Eicher EM (1984). A new mutation, gld, that produces lymphoproliferation and autoimmunity in C3H/HeJ mice. J Exp Med 159(1): 1-20.
Rubin E, Farber JL (1999). Pathology. 3rd edn. Lippincott-Raven: Philadelphia.
Runyan RB, Potts JD, Weeks DL (1992). TGF-beta 3-mediated tissue interaction during embryonic heart development. Mol Reprod Dev 32(2): 152-159.
Rybak A, Fuchs H, Hadian K, Smirnova L, Wulczyn EA, Michel G, et al. (2009). The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol 11(12): 1411-1420.
Sack U, Kinne RW, Marx T, Heppt P, Bender S, Emmrich F (1993). Interleukin-6 in synovial fluid is closely associated with chronic synovitis in rheumatoid arthritis. Rheumatol Int 13(2): 45-51.
Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R (2009). T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev 228(1): 9-22.
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. (1997). TGFbeta2 knockout mice have multiple developmental defects that are

Chapter 10: References
262
non-overlapping with other TGFbeta knockout phenotypes. Development 124(13): 2659-2670.
Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, et al. (2009). MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 138(2): 300-313.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. (2006). Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203(12): 2673-2682.
Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B (2000). CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med 192(11): 1553-1562.
Schmid P, Cox D, Bilbe G, Maier R, McMaster GK (1991). Differential expression of TGF beta 1, beta 2 and beta 3 genes during mouse embryogenesis. Development 111(1): 117-130.
Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, et al. (2013). Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta-analysis informing a consensus statement. Ann Rheum Dis 72(4): 583-589.
Schwandner R, Yamaguchi K, Cao Z (2000). Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med 191(7): 1233-1240.
Serwold T, Ehrlich LI, Weissman IL (2009). Reductive isolation from bone marrow and blood implicates common lymphoid progenitors as the major source of thymopoiesis. Blood 113(4): 807-815.
Severe N, Dieudonne FX, Marie PJ (2013). E3 ubiquitin ligase-mediated regulation of bone formation and tumorigenesis. Cell Death Dis 4: e463.
Shalom-Barak T, Quach J, Lotz M (1998). Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem 273(42): 27467-27473.
Shapiro F (2008). Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater 15: 53-76.

Chapter 10: References
263
Shinar DM, Endo N, Halperin D, Rodan GA, Weinreb M (1993). Differential expression of insulin-like growth factor-I (IGF-I) and IGF-II messenger ribonucleic acid in growing rat bone. Endocrinology 132(3): 1158-1167.
Shu L, Zhang H, Boyce BF, Xing L (2013). Ubiquitin E3 ligase Wwp1 negatively regulates osteoblast function by inhibiting osteoblast differentiation and migration. J Bone Miner Res 28(9): 1925-1935.
Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. (1992). Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 359(6397): 693-699.
Silfversward CJ, Larsson S, Ohlsson C, Frost A, Nilsson O (2007). Reduced cortical bone mass in mice with inactivation of interleukin-4 and interleukin-13. J Orthop Res 25(6): 725-731.
Silfversward CJ, Sisask G, Larsson S, Ohlsson C, Frost A, Ljunggren O, et al. (2008). Bone formation in interleukin-4 and interleukin-13 depleted mice. Acta Orthop 79(3): 410-420.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. (1997). Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89(2): 309-319.
Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. (2010). Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum 62(1): 234-244.
Sims NA, Jenkins BJ, Quinn JM, Nakamura A, Glatt M, Gillespie MT, et al. (2004). Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest 113(3): 379-389.
Sims NA, Quinn JM (2014). Osteoimmunology: oncostatin M as a pleiotropic regulator of bone formation and resorption in health and disease. Bonekey Rep 3: 527.
Smith-Garvin JE, Koretzky GA, Jordan MS (2009). T cell activation. Annu Rev Immunol 27: 591-619.
So H, Rho J, Jeong D, Park R, Fisher DE, Ostrowski MC, et al. (2003). Microphthalmia transcription factor and PU.1 synergistically induce the leukocyte receptor osteoclast-associated receptor gene expression. J Biol Chem 278(26): 24209-24216.

Chapter 10: References
264
Solmesky LJ, Abekasis M, Bulvik S, Weil M (2009). Bone morphogenetic protein signaling is involved in human mesenchymal stem cell survival in serum-free medium. Stem Cells Dev 18(9): 1283-1292.
Spector TD, Hall GM, McCloskey EV, Kanis JA (1993). Risk of vertebral fracture in women with rheumatoid arthritis. Bmj 306(6877): 558.
Spolski R, Leonard WJ (2014). Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov 13(5): 379-395.
St-Jacques B, Hammerschmidt M, McMahon AP (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13(16): 2072-2086.
Steele DG, Bramblett CA (1988). The anatomy and biology of the human skeleton. 1st edn. Texas A&M University Press: College Station.
Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K, et al. (2008). APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin ligase. Mol cell 32(5): 652-661.
Sun SC (2011). Non-canonical NF-kappaB signaling pathway. Cell Res 21(1): 71-85.
Sundaram K, Nishimura R, Senn J, Youssef RF, London SD, Reddy SV (2007). RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res 313(1): 168-178.
Taichman RS, Emerson SG (1994). Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med 179(5): 1677-1682.
Taichman RS, Reilly MJ, Emerson SG (1996). Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 87(2): 518-524.
Takahashi N, Udagawa N, Akatsu T, Tanaka H, Isogai Y, Suda T (1991). Deficiency of osteoclasts in osteopetrotic mice is due to a defect in the local microenvironment provided by osteoblastic cells. Endocrinology 128(4): 1792-1796.
Takahashi T, Kurihara N, Takahashi K, Kumegawa M (1986). An ultrastructural study of phagocytosis in bone by osteoblastic cells from fetal mouse calvaria in vitro. Arch Oral Biol 31(10): 703-706.

Chapter 10: References
265
Takai H, Kanematsu M, Yano K, Tsuda E, Higashio K, Ikeda K, et al. (1998). Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J Biol Chem 273(42): 27091-27096.
Takatsu K, Tominaga A, Harada N, Mita S, Matsumoto M, Takahashi T, et al. (1988). T cell-replacing factor (TRF)/interleukin 5 (IL-5): molecular and functional properties. Immunol Rev 102: 107-135.
Takayanagi H (2005). Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med 83(3): 170-179.
Takayanagi H (2007). Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 7(4): 292-304.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. (2002a). Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3(6): 889-901.
Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, et al. (2002b). RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature 416(6882): 744-749.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. (2000). T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408(6812): 600-605.
Takeshita S, Kaji K, Kudo A (2000). Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res 15(8): 1477-1488.
Talmage RV (1969). Calcium homeostasis--calcium transport--parathyroid action. The effects of parathyroid hormone on the movement of calcium between bone and fluid. Clin Orthop Relat Res 67: 211-223.
Talmage RV (1970). Morphological and physiological considerations in a new concept of calcium transport in bone. Am J Anat 129(4): 467-476.
Talmage RV, Cooper CW, Park HZ (1970). Regulation of calcium transport in bone by parathyroid hormone. Vitam Horm 28: 103-140.

Chapter 10: References
266
Tanaka K, Hashizume M, Mihara M, Yoshida H, Suzuki M, Matsumoto Y (2014). Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clin Exp Immunol 175(2): 172-180.
Tanaka-Kamioka K, Kamioka H, Ris H, Lim SS (1998). Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner Res 13(10): 1555-1568.
Tang XL, Griffith JF, Qin L, Hung VW, Kwok AW, Zhu TY, et al. (2013a). SLE disease per se contributes to deterioration in bone mineral density, microstructure and bone strength. Lupus 22(11): 1162-1168.
Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. (2009). TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15(7): 757-765.
Tang Y, Xie H, Chen J, Geng L, Chen H, Li X, et al. (2013b). Activated NF-kappaB in bone marrow mesenchymal stem cells from systemic lupus erythematosus patients inhibits osteogenic differentiation through downregulating Smad signaling. Stem Cells Dev 22(4): 668-678.
Tasat DR, Orona NS, Mandalunis PM, Cabrini RL, Ubios AM (2007). Ultrastructural and metabolic changes in osteoblasts exposed to uranyl nitrate. Arch Toxicol 81(5): 319-326.
Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. (2007). Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab 5(6): 464-475.
Taylor AF, Saunders MM, Shingle DL, Cimbala JM, Zhou Z, Donahue HJ (2007). Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am J Physiol Cell Physiol 292(1): C545-552.
Teitelbaum SL (2000). Bone resorption by osteoclasts. Science 289(5484): 1504-1508.
Temiyasathit S, Tang WJ, Leucht P, Anderson CT, Monica SD, Castillo AB, et al. (2012). Mechanosensing by the primary cilium: deletion of Kif3A reduces bone formation due to loading. PLoS One 7(3): e33368.
Terrier B, Costedoat-Chalumeau N, Garrido M, Geri G, Rosenzwajg M, Musset L, et al. (2012). Interleukin 21 correlates with T cell and B cell subset alterations in systemic lupus erythematosus. J Rheumatol 39(9): 1819-1828.

Chapter 10: References
267
Teti A (2011). Bone development: overview of bone cells and signaling. Curr Osteoporos Rep 9(4): 264-273.
Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, et al. (2008). Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med 205(6): 1395-1408.
Thomson BM, Mundy GR, Chambers TJ (1987). Tumor necrosis factors alpha and beta induce osteoblastic cells to stimulate osteoclastic bone resorption. J Immunol 138(3): 775-779.
Tondravi MM, McKercher SR, Anderson K, Erdmann JM, Quiroz M, Maki R, et al. (1997). Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature 386(6620): 81-84.
Tranquilli Leali P, Doria C, Zachos A, Ruggiu A, Milia F, Barca F (2009). Bone fragility: current reviews and clinical features. Clin Cases Miner Bone Metab 6(2): 109-113.
Troughton PR, Platt R, Bird H, el-Manzalawi E, Bassiouni M, Wright V (1996). Synovial fluid interleukin-8 and neutrophil function in rheumatoid arthritis and seronegative polyarthritis. Br J Rheumatol 35(12): 1244-1251.
Tu-Rapp H, Hammermuller A, Mix E, Kreutzer HJ, Goerlich R, Kohler H, et al. (2004). A proinflammatory role for Fas in joints of mice with collagen-induced arthritis. Arthritis Res Ther 6(5): R404-414.
Turner CH, Owan I, Alvey T, Hulman J, Hock JM (1998). Recruitment and proliferative responses of osteoblasts after mechanical loading in vivo determined using sustained-release bromodeoxyuridine. Bone 22(5): 463-469.
Turner RT, Iwaniec UT, Wong CP, Lindenmaier LB, Wagner LA, Branscum AJ, et al. (2013). Acute exposure to high dose gamma-radiation results in transient activation of bone lining cells. Bone 57(1): 164-173.
Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, et al. (1999). Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone 25(5): 517-523.
Urist MR (1965). Bone: formation by autoinduction. Science 150(3698): 893-899.

Chapter 10: References
268
Uzbekov RE, Maurel DB, Aveline PC, Pallu S, Benhamou CL, Rochefort GY (2012). Centrosome fine ultrastructure of the osteocyte mechanosensitive primary cilium. Microsc Microanal 18(6): 1430-1441.
Uzer G, Pongkitwitoon S, Ian C, Thompson WR, Rubin J, Chan ME, et al. (2014). Gap junctional communication in osteocytes is amplified by low intensity vibrations in vitro. PLoS One 9(3): e90840.
Vaananen HK, Zhao H, Mulari M, Halleen JM (2000). The cell biology of osteoclast function. J Cell Sci 113 ( Pt 3): 377-381.
Van bezooijen RL, Farih-Sips HC, Papapoulos SE, Lowik CW (1999). Interleukin-17: A new bone acting cytokine in vitro. J Bone Miner Res 14(9): 1513-1521.
van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. (2011). Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum 63(1): 73-83.
Verborgt O, Gibson GJ, Schaffler MB (2000). Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res 15(1): 60-67.
Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P (1997). Accelerated collagen-induced arthritis in IFN-gamma receptor-deficient mice. J Immunol 158(11): 5507-5513.
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. (2005a). A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435(7041): 452-458.
Vinuesa CG, Tangye SG, Moser B, Mackay CR (2005b). Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 5(11): 853-865.
Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL (2004). Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103(9): 3258-3264.
Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C (2008). A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity 29(1): 127-137.

Chapter 10: References
269
Wada T, Nakashima T, Hiroshi N, Penninger JM (2006). RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 12(1): 17-25.
Wajant H, Pfizenmaier K, Scheurich P (2003). Tumor necrosis factor signaling. Cell Death Differ 10(1): 45-65.
Walker EC, McGregor NE, Poulton IJ, Pompolo S, Allan EH, Quinn JM, et al. (2008). Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res 23(12): 2025-2032.
Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, et al. (2006). Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol 24: 33-63.
Walunas TL, Bakker CY, Bluestone JA (1996). CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med 183(6): 2541-2550.
Wang X, Dong C (2013). The CD70-CD27 axis, a new brake in the T helper 17 cell response. Immunity 38(1): 1-3.
Waring P, Mullbacher A (1999). Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol 77(4): 312-317.
Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, et al. (1992). The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol 148(4): 1274-1279.
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL (2005). IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest 115(2): 282-290.
Wein MN, Jones DC, Glimcher LH (2005). Turning down the system: counter-regulatory mechanisms in bone and adaptive immunity. Immunol Rev 208: 66-79.
Weitzmann MN (2013). The Role of Inflammatory Cytokines, the RANKL/OPG Axis, and the Immunoskeletal Interface in Physiological Bone Turnover and Osteoporosis. Scientifica 2013: 125705.
Weitzmann MN, Cenci S, Haug J, Brown C, DiPersio J, Pacifici R (2000). B lymphocytes inhibit human osteoclastogenesis by secretion of TGFbeta. J Cell Biochem 78(2): 318-324.

Chapter 10: References
270
Weitzmann MN, Cenci S, Rifas L, Haug J, Dipersio J, Pacifici R (2001). T cell activation induces human osteoclast formation via receptor activator of nuclear factor kappaB ligand-dependent and -independent mechanisms. J Bone Miner Res 16(2): 328-337.
Wiesmann A, Phillips RL, Mojica M, Pierce LJ, Searles AE, Spangrude GJ, et al. (2000). Expression of CD27 on murine hematopoietic stem and progenitor cells. Immunity 12(2): 193-199.
Wilfinger WW, Mackey K, Chomczynski P (1997). Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. BioTechniques 22(3): 474-476, 478-481.
Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, et al. (1997a). TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med 186(12): 2075-2080.
Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. (1997b). TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem 272(40): 25190-25194.
Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, et al. (1988). Novel regulators of bone formation: molecular clones and activities. Science 242(4885): 1528-1534.
Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. (2009). BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10(11): R130.
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. (2008a). Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A 105(44): 16976-16981.
Wu X, McKenna MA, Feng X, Nagy TR, McDonald JM (2003). Osteoclast apoptosis: the role of Fas in vivo and in vitro. Endocrinology 144(12): 5545-5555.
Wu Y, Humphrey MB, Nakamura MC (2008b). Osteoclasts - the innate immune cells of the bone. Autoimmunity 41(3): 183-194.
Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, et al. (2012). Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med 18(7): 1095-1101.

Chapter 10: References
271
Xiao Y, Song JY, de Vries TJ, Fatmawati C, Parreira DB, Langenbach GE, et al. (2013). Osteoclast precursors in murine bone marrow express CD27 and are impeded in osteoclast development by CD70 on activated immune cells. Proc Natl Acad Sci U S A 110(30): 12385-12390.
Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, et al. (2006). Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J Biol Chem 281(41): 30884-30895.
Xing L, Bushnell TP, Carlson L, Tai Z, Tondravi M, Siebenlist U, et al. (2002). NF-kappaB p50 and p52 expression is not required for RANK-expressing osteoclast progenitor formation but is essential for RANK- and cytokine-mediated osteoclastogenesis. J Bone Miner Res 17(7): 1200-1210.
Xu J, Tan JW, Huang L, Gao XH, Laird R, Liu D, et al. (2000). Cloning, sequencing, and functional characterization of the rat homologue of receptor activator of NF-kappaB ligand. J Bone Miner Res 15(11): 2178-2186.
Xu J, Wu HF, Ang ES, Yip K, Woloszyn M, Zheng MH, et al. (2009a). NF-kappaB modulators in osteolytic bone diseases. Cytokine Growth Factor Rev 20(1): 7-17.
Xu L, Feng X, Tan W, Gu W, Guo D, Zhang M, et al. (2013). IL-29 enhances Toll-like receptor-mediated IL-6 and IL-8 production by the synovial fibroblasts from rheumatoid arthritis patients. Arthritis Res Ther 15(5): R170.
Xu LX, Kukita T, Kukita A, Otsuka T, Niho Y, Iijima T (1995). Interleukin-10 selectively inhibits osteoclastogenesis by inhibiting differentiation of osteoclast progenitors into preosteoclast-like cells in rat bone marrow culture system. J Cell Physiol 165(3): 624-629.
Xu Z, Hurchla MA, Deng H, Uluckan O, Bu F, Berdy A, et al. (2009b). Interferon-gamma targets cancer cells and osteoclasts to prevent tumor-associated bone loss and bone metastases. J Biol Chem 284(7): 4658-4666.
Xynos ID, Hukkanen MV, Batten JJ, Buttery LD, Hench LL, Polak JM (2000). Bioglass 45S5 stimulates osteoblast turnover and enhances bone formation In vitro: implications and applications for bone tissue engineering. Calcif Tissue Int 67(4): 321-329.
Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, et al. (2005). DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med 202(3): 345-351.

Chapter 10: References
272
Yagi M, Ninomiya K, Fujita N, Suzuki T, Iwasaki R, Morita K, et al. (2007). Induction of DC-STAMP by alternative activation and downstream signaling mechanisms. J Bone Miner Res 22(7): 992-1001.
Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, et al. (2009). IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem 108(4): 947-955.
Yamaguchi A, Komori T, Suda T (2000). Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr Rev 21(4): 393-411.
Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, et al. (1999). Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126(8): 1571-1580.
Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, et al. (2006). Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem 281(17): 11846-11855.
Yasui T, Kadono Y, Nakamura M, Oshima Y, Matsumoto T, Masuda H, et al. (2011). Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and Traf6. J Bone Miner Res 26(7): 1447-1456.
Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K (1992). Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J Biol Chem 267(31): 22506-22511.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC bioinformatics 13: 134.
Yokomizo T, Dzierzak E (2008). Fine-tuning of hematopoietic stem cell homeostasis: novel role for ubiquitin ligase. Genes Dev 22(8): 960-963.
Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, et al. (1997). Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol 17(7): 4015-4023.

Chapter 10: References
273
Yu D, Tan AH, Hu X, Athanasopoulos V, Simpson N, Silva DG, et al. (2007). Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature 450(7167): 299-303.
Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM, et al. (1998). OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol 161(11): 6113-6121.
Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, Moon JJ, et al. (2001). Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol 166(3): 1482-1491.
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, et al. (2007). Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum 56(12): 4104-4112.
Zaiss MM, Sarter K, Hess A, Engelke K, Bohm C, Nimmerjahn F, et al. (2010). Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum 62(8): 2328-2338.
Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ (2007). The molecular basis of IL-21-mediated proliferation. Blood 109(10): 4135-4142.
Zhang DE, Hetherington CJ, Chen HM, Tenen DG (1994). The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol 14(1): 373-381.
Zhang F, Tanaka H, Kawato T, Kitami S, Nakai K, Motohashi M, et al. (2011a). Interleukin-17A induces cathepsin K and MMP-9 expression in osteoclasts via celecoxib-blocked prostaglandin E2 in osteoblasts. Biochimie 93(2): 296-305.
Zhang H, Wu C, Matesic LE, Li X, Wang Z, Boyce BF, et al. (2013a). Ubiquitin E3 ligase Itch negatively regulates osteoclast formation by promoting deubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem 288(31): 22359-22368.
Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. (2008). Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev 226: 29-40.

Chapter 10: References
274
Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R (2001). Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci U S A 98(3): 974-979.
Zhang Y, Liu B, Ma Y, Jin B (2013b). Sema 4D/CD100-plexin B is a multifunctional counter-receptor. Cell Mol Immunol 10(2): 97-98.
Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, et al. (2011b). Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One 6(8): e23516.
Zhang YE (2009). Non-Smad pathways in TGF-beta signaling. Cell Res 19(1): 128-139.
Zhang YH, Lin JX, Vilcek J (1990). Interleukin-6 induction by tumor necrosis factor and interleukin-1 in human fibroblasts involves activation of a nuclear factor binding to a kappa B-like sequence. Mol Cell Biol 10(7): 3818-3823.
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, et al. (2006). Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4(2): 111-121.
Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D (2003). E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J Biol Chem 278(30): 27939-27944.
Zheng ZH, Li XY, Ding J, Jia JF, Zhu P (2008). Allogeneic mesenchymal stem cell and mesenchymal stem cell-differentiated chondrocyte suppress the responses of type II collagen-reactive T cells in rheumatoid arthritis. Rheumatology 47(1): 22-30.
Zhou P, Kitaura H, Teitelbaum SL, Krystal G, Ross FP, Takeshita S (2006). SHIP1 negatively regulates proliferation of osteoclast precursors via Akt-dependent alterations in D-type cyclins and p27. J Immunol 177(12): 8777-8784.
Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH (1999). A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400(6745): 687-693.
Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. (2007). Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 109(9): 3706-3712.

Chapter 10: References
275
Zittermann A (2001). Effects of vitamin K on calcium and bone metabolism. Curr Opin Clin Nutr Metab Care 4(6): 483-487.
Zola H, Flego L (1992). Expression of interleukin-6 receptor on blood lymphocytes without in vitro activation. Immunology 76(2): 338-340.

APPENDIX

Appendix
277
Supplementary Figure 1 Female mutant sanroque mice display reduced bone
mass. MicroCT morphometric analysis of tibias from 83 ± 13 days-old female wildtype
and sanroque mice. Data is presented as mean ± SEM. * p<0.005; ** p<0.001; n.s. = no
significance in comparison to wildtype littermates.
sanroque
Wildtype

Appendix
278
Supplementary Figure 2 Left shift is observed in peripheral blood from sanroque
mutant mice. Geimsa-stained peripheral blood smears from wildtype and sanroque
mutant mice revealed increase number of immature leukocyte in sanroque mice
(personal communication with W/Prof. Wendy Erber of UWA School of Pathology and
Laboratory Medicine).
Wildtype sanroque
×20 ×20
×40 ×40

- END -
Recommended