
HAL Id: tel-01666127https://tel.archives-ouvertes.fr/tel-01666127
Submitted on 18 Dec 2017
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
Synthèse et caractérisation de dérivés amphiphiles duxanthaneAudrey Roy
To cite this version:Audrey Roy. Synthèse et caractérisation de dérivés amphiphiles du xanthane. Autre. Université duHavre, 2015. Français. �NNT : 2015LEHA0028�. �tel-01666127�

ÉCOLE DOCTORALE NORMANDE DE CHIMIE
UNIVERSITÉ DU HAVREU.F.R DES SCIENCES & TECHNIQUES
N° attribué par la bibliothèque
THÈSE
En vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE DU HAVRESpécialité : Chimie et Physico-Chimie des Polymères
Présentée par
Audrey ROY
Le 18 Juin 2015
MEMBRES DU JURY
M. Jacques DESBRIERES Professeur, Université de Pau et des pays de L’Adour
Rapporteur
M. Dominique HOURDET Professeur, Université Pierre et Marie Curie (Paris 6)
Rapporteur
Mme Isabelle CAPRON Chargé de Recherche, INRA Nantes Examinateur
M. Luc PICTON Professeur, Université de Rouen Examinateur
M. Hervé ROLLAND Docteur-ingénieur, SEPPIC, Castres Examinateur
M. Michel GRISEL Professeur, Université du Havre Directeur de thèse
M. Frédéric RENOU Maître de Conférences, Université du Havre
Encadrant de thèse
Synthèse et caractérisation de dérivés amphiphiles du xanthane


REMERCIEMENTS
Ces travaux de thèse ont été réalisés à l'Unité de Recherche en Chimie Organique etMacromoléculaire (URCOM) de l'Université du Havre. Je remercie mon directeur de thèse, M. MichelGRISEL, professeur à l'Université du Havre, pour sa confiance pendant ces (presque) quatre années dethèse.
J'exprime ma profonde reconnaissance à mon encadrant de thèse, M. Frédéric RENOU, maître deconférences à l'Université du Havre, pour sa présence et son soutien sans faille durant ma thèse. Je leremercie pour sa disponibilité, ses conseils avisés, nos discussions scientifiques toujours bénéfiques, sapatience (si! si!) et sa pédagogie pour répondre à mes nombreuses questions de novice, notamment enrhéologie. Plus que tout, je le remercie pour m'avoir toujours encouragée à donner le meilleur de moi-même, et ce dans un climat de confiance et de respect mutuel.
Je remercie M. Dominique HOURDET, professeur à l'Université Pierre et Marie Curie, et M. JacquesDESBRIERES, professeur à l'Université de Pau et des Pays de l'Adour, pour avoir examiné ce travail en tantque rapporteur. Je les remercie pour leurs remarques et conseils concernant mon manuscrit. Je remercieégalement Mme Isabelle CAPRON, chargé de recherche à l'INRA de Nantes, M. Luc PICTON, professeur àl'Université de Rouen et M. Hervé ROLLAND, docteur-ingénieur chez SEPPIC, pour avoir accepté d'êtremembre de mon jury. Je remercie l'ensemble des mes membres de mon jury pour nos échanges fructueuxlors de ma soutenance.
J'exprime ma profonde gratitude à M. Sébastien COMESSE, maître de conférences à l'Université duHavre, pour son aide inestimable sur la partie modification chimique du xanthane. Je le remercie pour soninvestissement dans mon sujet de thèse, pourtant éloigné de ses axes de recherche (du côté obscur de laForce, comme diront certains…), et ses remarques et conseils toujours avisés. Je remercie du fond du cœurSébastien, sa compagne Alice et leur petite Lisa pour m'avoir intégrée dans leur famille lors de mon séjourau Havre. Merci pour votre amitié et votre soutien, notamment dans les moments difficiles.
Je remercie également M. Nicolas HUCHER, maître de conférences à l'Université du Havre, pour sonaide précieuse concernant la caractérisation de mes composés par RMN et fluorescence. Je le remercie dem’avoir formée à l'utilisation des spectromètres associés, ainsi que pour ses conseils.
Les analyses de dichroïsme circulaire ont été réalisées au laboratoire Polymères, Biopolymères etSurfaces de l'Université de Rouen. Je remercie Mme Isabelle DE, professeur à l'Université de Rouen, pourm'avoir formée à l'utilisation de l'appareil de dichroïsme circulaire.

Je remercie M. Luc PICTON et M. Zied SOUGUIR, chef de projet chez CELENYS, pour leurs conseilséclairés sur la modification chimique du xanthane.
J'exprime ma gratitude à l'ensemble des membres de l'URCOM pour leur accueil chaleureux. Jeremercie notamment Odile, Faustine et Natalie pour leur aide précieuse lors de mes enseignements. Unénorme merci à Vincent L., qui a toujours réparé, dans la bonne humeur, les nombreux appareils quitombaient mystérieusement en panne quand je les utilisais. Enfin, merci également à Sophie, notre superex-secrétaire, pour ses remarques et conseils avisés lors de l'organisation de la JEDNC 2014.
Je remercie mes collègues de bureau, doctorants, post-doctorants et stagiaires, pour la superambiance au cours de ma thèse. Merci à Adeline, Manon, Laura G., Catalin, Ronan, Leila, Igor, Olivier, Julien,Thomas, Philippe, Daria, Maxime mon stagiaire, Yanis, Baptiste, Claire, Mar, et surtout, surtout, Mathieu,Liliana, Aurélie, Pauline et Laura R. Merci à vous pour ces moments inoubliables passés ensemble.
Pour finir, je remercie ma mère, mon petit frère et ma famille pour leur soutien indéfectible et leuramour.
Je dédie ce manuscrit à mon père.


La thésarde et les neuf appareils
(Librement inspiré des Dix petits nègres d’Agatha CHRISTIE)
La thésarde utilisa le conductimètre un après-midi,
Ce dernier à jamais s’éteignit,
Ne resta plus que la thésarde et 8 appareils.
La thésarde une analyse ATG de routine lança,
La résistance du four ce jour-là grilla.
Ne resta plus que la thésarde et 7 appareils.
La thésarde soniquait des échantillons en biologie,
La sonde fut définitivement en panne un midi,
Ne resta plus que la thésarde et 6 appareils.
La thésarde faisait des mesures de polarimétrie,
Un tuyau d’eau tout un coup se fendit,
Ne resta plus que la thésarde et 5 appareils.
La thésarde lança une analyse RMN par une belle matinée,
Le système de contrôle de température refusa de démarrer,
Ne resta plus que la thésarde et 4 appareils.
La thésarde utilisait le rhéomètre récemment livré,
Un défaut de fabrication nécessita de le rapporter,
Ne resta plus que la thésarde et 3 appareils.
La thésarde programma une analyse rhéologique une journée,
Mais le bain thermostaté cessa de chauffer,
Ne resta plus que la thésarde et 2 appareils.

La thésarde avait lyophilisé des solutions pour la nuit,
Un des ballons implosa, et la pompe à vide périt,
Ne resta plus que la thésarde et 1 appareil.
La thésarde, en phase de rédaction, pesta,
Quand son clavier d’ordinateur brusquement lâcha,
Ne resta plus que la thésarde.
La thésarde, bien soulagée,
Soutenir, s’en est allée,
N’en resta plus… du tout.


SOMMAIRE Liste des abréviations 1 Liste des symboles 3
Introduction Générale 5
Chapitre 1 : Bibliographie 11
1.1. Introduction 13
1.2. Polymères amphiphiles 14
1.2.1. Définition et structure des polymères amphiphiles 14
1.2.2. Propriétés des polymères amphiphiles en solution 17
1.2.2.1. Propriétés viscosimétriques des polymères amphiphiles 17
1.2.2.1.1. Régime de concentration des polymères en solution 17
1.2.2.1.2. Comportement en écoulement des polymères amphiphiles 20
1.2.2.1.3. Paramètres influençant propriétés viscosimétriques des polymères amphiphiles 22
1.2.2.2. Propriétés viscoélastiques des polymères amphiphiles 26
1.2.2.2.1. Polymères non enchevêtrés : modèle de Rouse collant 28
1.2.2.2.2. Polymères enchevêtrés 31
1.3. 1.3. La gomme xanthane 39
1.3.1. Intérêt industriel du xanthane et procédé de production 40
1.3.2. Structure chimique et conformation du xanthane 42
1.3.2.1. Structure chimique du xanthane 42
1.3.2.2. Conformation du xanthane en solution : transition ordre-désordre 43
1.3.3. Propriétés du xanthane en solution 55
1.3.3.1. Domaines de concentration du xanthane en solution 55
1.3.3.2. Propriétés viscosimétriques du xanthane en solution 63
1.3.3.2.1. Viscosité intrinsèque du xanthane en solution 63
1.3.3.2.2. Courbes d’écoulement du xanthane en solution 68
1.3.3.3. Propriétés viscoélastiques du xanthane en solution 74
1.3.3.3.1. Influence de la température sur les propriétés viscoélastiques du xanthane 74
1.3.3.3.2. Influence de la concentration sur les propriétés viscoélastiques du xanthane 79
1.3.3.3.3. Superposition fréquence-température-concentration 82
1.3.3.3.4. Modèles d’organisation des chaînes en solution en régime semi-dilué enchevêtré
84
1.3.4. Modification chimique du xanthane 90
1.4. 1.4. Conclusion 94
1.5. Références bibliographiques 96

Chapitre 2 : Matériels et méthodes 109
2.1. Modification chimique du xanthane 111
2.1.1. Réactifs utilisés 111
2.1.2. Modification chimique du xanthane sous forme ordonnée 112
2.1.2.1. Processus de modification à température ambiante 113
2.1.2.2. Processus de modification à basse température (première étape) 113
2.1.3. Modification chimique du xanthane sous forme désordonnée 114
2.2. Résonance Magnétique Nucléaire (RMN) 115
2.2.1. Principes 115
2.2.1.1. Rappels sur le moment magnétique et la précession de Larmor 115
2.2.1.2. Phénomènes de résonance et de relaxation 116
2.2.1.3. Principe d’une analyse en RMN impulsionnelle 118
2.2.2. Appareil utilisé et préparation des échantillons 119
2.2.3. Méthode de détermination des degrés de substitution en acétate et pyruvate et de la
densité de greffage 120
2.2.3.1. Détermination des degrés de substitution en acétate et pyruvate 120
2.2.3.2. Détermination de la densité de greffage des xanthanes modifiés 122
2.3. Polarimétrie 123
2.3.1. Principes généraux 123
2.3.2. Présentation du polarimètre 125
2.3.3. Activité optique du xanthane 126
2.3.4. Appareil utilisé et préparation des échantillons 128
2.4. Fluorescence 128
2.4.1. Définitions et principes généraux 129
2.4.2. Principe d’une analyse de fluorescence 130
2.4.3. Caractéristiques de la sonde de fluorescence ANS 133
2.4.4. Appareil utilisé et préparation des solutions 134
2.5. Rhéologie 135
2.5.1. Définition et rappels 135
2.5.2. Mesures en écoulement 136
2.5.3. Mesures dynamiques en solution 137
2.5.4. Rhéologie interfaciale 140
2.5.4.1. Définition d’une interface 140
2.5.4.2. Principes de la rhéologie interfaciale de cisaillement 141
2.5.5. Rhéomètre utilisé et préparation des échantillons 144
2.5.5.1. Préparation des solutions 144
2.5.5.2. Analyses en solution 144
2.5.5.3. Analyses à l’interface eau/huile 145
Références bibliographiques 149

Chapitre 3 : Modification chimique du xanthane 153
3.1. Introduction 155
3.2. Couplage peptidique par des carbodiimides 156
3.3. Modification chimique du xanthane sous forme ordonnée 166
3.3.1. Processus de modification à température ambiante 166
3.3.1.1. Description du procédé 166
3.3.1.2. Mise en évidence de la conformation ordonnée du xanthane lors de sa modification
168
3.3.1.3. Analyse RMN 1H des échantillons et quantification de la densité de greffage 170
3.3.1.3.1. Description des spectres RMN et détermination des taux d’acétate et de pyruvate172
3.3.1.3.2. Densité de greffage des xanthanes modifiés suivant le protocole à température
ambiante 175
3.3.2. Processus de modification à basse température (première étape) 179
3.3.2.1. Influence de la température sur l’efficacité du couplage peptidique 179
3.3.2.2. Densité de greffage des xanthanes modifiés suivant le protocole à basse température
lors de la première étape 181
3.4. Modification chimique du xanthane sous forme désordonnée 187
3.4.1. Choix du solvant pour le protocole de modification 187
3.4.2. Mise en évidence de la conformation désordonnée du xanthane dans le DMSO 188
3.4.3. Protocole de modification du xanthane dans le DMSO 190
3.4.3.1. Choix des conditions expérimentales lors de la modification chimique 190
3.4.3.2. Choix des étapes de purification 192
3.4.3.3. Interprétation des spectres RMN 1H pour les xanthanes modifiés dans le DMSO 192
3.4.3.4. Densité de greffage estimée des xanthanes modifiés suivant le protocole dans le
DMSO 196
3.4.3.5. Comparaison de la densité de greffage entre les protocoles de modification sous forme
ordonnée et sous forme désordonnée 198
3.5. Conclusion 200
Références bibliographiques 202

Chapitre 4 : Propriétés en solution des xanthanes modifiés 207
4.1. Introduction 209
4.2. Comportement en solution des xanthanes modifiés sous forme ordonnée 210
4.2.1. Conformation en solution des chaînes modifiées sous forme ordonnée 210
4.2.2. Etude de l’organisation en solution des chaînes modifiées sous forme ordonnée par
fluorescence 213
4.2.3. Etude des propriétés rhéologiques dynamiques en solution des xanthanes modifiés sous
forme ordonnée 221
4.2.3.1. Courbes maîtresses fréquence-température 222
4.2.3.2. Loi d’Arrhénius 229
4.2.3.3. Viscosité dynamique 231
4.2.4. Modèle d’organisation des chaînes de xanthane modifié sous forme ordonnée en
solution 233
4.2.5. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous forme
ordonnée 236
4.2.5.1. Comportement du précurseur à l’interface eau/huile 238
4.2.5.2. Comportement des xanthanes modifiés sous forme ordonnée à l’interface eau/huile
246
4.2.5.3. Modèles d’organisation des chaînes de xanthane modifié sous forme ordonnée à
l’interface eau/huile 251
4.3. Comportement en solution des xanthanes modifiés sous forme désordonnée 256
4.3.1. Etude des propriétés rhéologiques en solution des xanthanes greffés sous forme
désordonnée 256
4.3.1.1. Comportement en solution du précurseur « désordonné » 257
4.3.1.2. Comportement en solution des xanthanes modifiés dans le DMSO 259
4.3.2. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous forme
désordonnée 267
4.3.2.1. Comportement à l’interface eau/huile du précurseur « désordonné » 268
4.3.2.2. Comportement à l’interface eau/huile des xanthanes modifiés dans le DMSO 270
4.4. Conclusion 275
Références bibliographiques 277
Conclusion Générale 281

Liste des abréviations
ANS : 8-(aniline)-1-naphthalènesulfonate
BT : Basse température
CAC : Concentration d’agrégation critique
Ccrit : Concentration critique
DCC : Dicyclohexylcarbodiimide
DSA : Degré de substitution en acétate
DSp : Degré de substitution en pyruvate
Ea : Energie d’activation
EDAC : 1-éthyl-3-(3-diméthylaminopropyl) carbodiimide
FD : Forme désordonnée
FO : Forme ordonnée
HOBt : Hydroxybenzotriazole
NHS : N-hydroxysuccinimide
RMN 1H : Résonance magnétique nucléaire du proton
TA : Température ambiante
TSP : 2.2.3.3-d(4)-3-(Triméthylsilyl)propanoate de sodium
X : Xanthane
- 1 -

- 2 -

Liste des symboles
: Pouvoir rotatoire (°)
aT : Facteur de décalage horizontal
bT : Facteur de décalage vertical
c* : Concentration critique de recouvrement (g/L)
c** : Concentration d’enchevêtrement (g/L)
f : Fréquence (Hz)
G’ : Module de conservation ou élastique (en solution, en Pa)
G’’ : Module de perte ou visqueux (en solution, en Pa)
Gs’ : Module interfacial de conservation (N/m)
Gs’’ : Module interfacial de perte (N/m)
: Déformation (%)
γ : Vitesse de cisaillement (s-1)
: Viscosité (Pa.s)
[η] : Viscosité intrinsèque (mL/g)
Imax : Intensité maximale d’émission
max : Longueur d’onde au maximum d’émission (nm)
: Contrainte de cisaillement (Pa)
: Ellipticité (mdeg)
Tm : Température de transition conformationnelle (°C)
: Temps de relaxation (s)
rep : Temps de reptation (s)
sticker : Durée d’interaction entre les groupements hydrophobes (s)
: Pulsation ou fréquence angulaire (rad/s)
%octyle : Densité de greffage en chaînes octyle
- 3 -

- 4 -

Introduction Générale
- 5 -

Introduction Générale
- 6 -

Introduction Générale
Les polymères amphiphiles sont des macromolécules constituées d’une partie hydrophile et
d’une partie hydrophobe, qui appartiennent à la classe des polymères associatifs. Ils possèdent la
capacité de pouvoir s’auto-organiser en solution aqueuse via des interactions intra et/ou
intermoléculaires attractives entre les chaînons hydrophobes. Dans le cadre de ces travaux, nous
nous intéressons aux polymères de structure peigne, c’est-à-dire constitués d’un squelette hydrophile
sur lequel ont été chimiquement greffés des groupements hydrophobes. Au-delà d’une concentration
critique, les entités hydrophobes s’associent de manière réversible au sein de microdomaines. Les
polymères amphiphiles se caractérisent donc par une organisation en solution de leurs chaînes très
différente de leurs homologues non modifiés. Cette particularité est à l’origine de leurs propriétés
viscosifiantes et épaississantes supérieures, ce qui explique leurs nombreuses applications
industrielles dans des domaines très variés : formulation de peinture, cosmétique, agroalimentaire,
récupération assistée du pétrole, etc. Par ailleurs, depuis les années 1980, le développement des
polysaccharides amphiphiles connaît un essor académique et industriel important. En effet, cette
catégorie de macromolécules possède des propriétés similaires à celles des polymères associatifs
synthétiques, tout en ayant l’avantage d’être biocompatibles, biodégradables et biosourcés.
Les propriétés des polymères amphiphiles sont contrôlables suivant un certain nombre de
paramètres bien identifiés dans la littérature : la nature des greffons hydrophobes, leur longueur et
leur répartition le long du squelette, ou encore la concentration en polymère, la température et la
force ionique du milieu. Toutefois, très peu d’études s’intéressent à l’influence de la conformation du
squelette hydrophile sur le comportement en solution de ces systèmes. Dans la grande majorité des
cas, le polysaccharide retenu pour être modifié chimiquement adopte une conformation de type
pelote en solution.
- -

Introduction Générale
Ainsi, l’objectif principal de ce travail de thèse consiste à caractériser l’impact de la conformation
du squelette hydrophile sur les propriétés d’un polysaccharide rendu amphiphile par greffage
covalent de résidus alkyle. Dans ce but, nous avons choisi le xanthane comme squelette. En effet, ce
polysaccharide présente la caractéristique d’exister sous deux formes différentes en solution, suivant
les conditions opératoires : une forme hélicoïdale rigide à basse température et haute force ionique,
et une conformation plus flexible à haute température et/ou très faible salinité. Cette transition
conformationnelle ordre-désordre est réversible notamment avec la température, et s’accompagne
principalement d’une modification prononcée des propriétés rhéologiques de ce polymère en
solution.
Nos travaux se sont articulés suivant deux grands axes. Le premier consiste en le développement
de protocoles opératoires permettant de greffer chimiquement des groupements hydrophobes sur le
xanthane, soit sous forme ordonnée, soit sous forme désordonnée. Le deuxième axe se focalise sur la
caractérisation des propriétés en solution de ces nouveaux polysaccharides, et en particulier leur
comportement rhéologique, afin de déterminer l’influence de la conformation adoptée par les
chaînes sur les propriétés en solution d’un polymère amphiphile.
Le premier chapitre est consacré à l’étude bibliographique, et se compose de deux parties. La
première partie décrit le comportement rhéologique en solution des polymères amphiphiles. Le lien
entre l’organisation de telles chaînes en solution et les propriétés observées est notamment abordé.
La deuxième partie présente le polysaccharide xanthane. Plus précisément, l’accent est mis sur les
différentes conformations adoptées par ce polymère en solution, suivant les conditions opératoires,
et les propriétés rhéologiques exhibées. Les différents exemples de modification chimique du
xanthane par greffage sont également indiqués dans cette section.
Le deuxième chapitre décrit succinctement les protocoles de modification chimique du xanthane
sous forme ordonnée ou sous forme désordonnée. Il recense également les différentes techniques de
- -

Introduction Générale
caractérisation utilisées lors de ces travaux, en décrivant à chaque fois le principe de la méthode, les
conditions expérimentales et le traitement des données effectué.
Le chapitre 3 présente plus en détail les protocoles de modification développés pour greffer des
groupements hydrophobes sur le xanthane soit sous forme ordonnée, soit sous forme désordonnée,
ainsi que les caractérisations des dérivés obtenus par RMN. Pour les deux protocoles, la modification
chimique a été effectuée via un couplage peptidique par des carbodiimides. Un rappel
bibliographique sur ce type de réaction est inclus en début de ce chapitre.
Le quatrième chapitre décrit les principaux résultats obtenus lors de la caractérisation des
propriétés en solution et à l’interface eau/huile des xanthanes modifiés, sous forme ordonnée ou
sous forme désordonnée. Dans les deux cas, on s’attache à démontrer la relation entre l’organisation
des chaînes amphiphiles en solution et les propriétés observées, afin de comprendre l’influence de la
conformation du squelette sur le comportement.
Enfin, nous finirons par les principales conclusions de ce travail et présenterons quelques
perspectives.
- -

Introduction Générale
- -

Chapitre 1 : Bibliographie
Chapitre 1 : Bibliographie
1.1. Introduction 13
1.2. Polymères amphiphiles 141.2.1. Définition et structure des polymères amphiphiles 14
1.2.2. Propriétés des polymères amphiphiles en solution 17
1.2.2.1. Propriétés viscosimétriques des polymères amphiphiles 171.2.2.1.1. Régime de concentration des polymères en solution 171.2.2.1.2. Comportement en écoulement des polymères amphiphiles 201.2.2.1.3. Paramètres influençant les propriétés viscosimétriques des polymères amphiphiles 22
1.2.2.2. Propriétés viscoélastiques des polymères amphiphiles 261.2.2.2.1. Polymères non enchevêtrés : modèle de Rouse collant 281.2.2.2.2. Polymères enchevêtrés 31
1.3. La gomme xanthane 391.3.1. Intérêt industriel du xanthane et procédé de production 40
1.3.2. Structure chimique et conformation du xanthane 42
1.3.2.1. Structure chimique du xanthane 421.3.2.2. Conformation du xanthane en solution : transition ordre-désordre 43
1.3.3. Propriétés du xanthane en solution 55
1.3.3.1. Domaines de concentration du xanthane en solution 551.3.3.2. Propriétés viscosimétriques du xanthane en solution 63
1.3.3.2.1. Viscosité intrinsèque du xanthane en solution 631.3.3.2.2. Courbes d’écoulement du xanthane en solution 68
1.3.3.3. Propriétés viscoélastiques du xanthane en solution 741.3.3.3.1. Influence de la température sur les propriétés viscoélastiques du xanthane 741.3.3.3.2. Influence de la concentration sur les propriétés viscoélastiques du xanthane 791.3.3.3.3. Superposition fréquence-température-concentration 821.3.3.3.4. Modèles d’organisation des chaînes en solution en régime semi-dilué enchevêtré 84
1.3.4. Modification chimique du xanthane 90
1.4. Conclusion 94
Références bibliographiques 96
- 11 -

Chapitre 1 : Bibliographie
- 12 -

Chapitre 1 : Bibliographie
1.1. Introduction
Les polymères amphiphiles sont des macromolécules comportant une partie hydrophile et une
partie hydrophobe. Ils se caractérisent par des propriétés rhéologiques et interfaciales bien
supérieures à celles de leur homologue purement hydrophile. L’origine de ces propriétés s’explique
par l’organisation particulière adoptée par les chaînes amphiphiles en solution, qui est le plus souvent
régie par les interactions entre les chaînons hydrophobes, comme illustré dans la figure 1-1 :
Figure 1-1 : Exemple d’association de polymère amphiphile en solution.
La description du comportement rhéologique en solution de ces polymères fait l’objet de la
première partie de ce chapitre. L’influence de la conformation des chaînes sur les propriétés exhibées
est notamment décrite, ainsi que le modèle rhéologique, dit « modèle de la reptation collante »,
développé pour expliquer le comportement de ces systèmes en solution.
Il faut savoir qu’il existe de très nombreux exemples de polymères amphiphiles décrits dans la
littérature. Parmi eux, on peut citer le cas des polysaccharides amphiphiles, qui connaissent un essor
important depuis les années 19801, du fait de leur abondance et de leur caractère biocompatible et
biodégradable. Néanmoins, peu d’études s’intéressent à l’impact de la conformation du squelette sur
les propriétés de ces polymères.
Afin de répondre à cette question, nous nous sommes intéressés au greffage de chaînons
hydrophobes sur le xanthane, un polysaccharide anionique hydrophile. En effet, ce dernier possède la
particularité de pouvoir exister sous deux conformations différentes2, 3 en solution, suivant les
conditions opératoires : une forme ordonnée hélicoïdale semi-rigide, à basse température et force
ionique élevée, et une conformation désordonnée de type pelote flexible à haute température et/ou
- 13 -

Chapitre 1 : Bibliographie
faible force ionique. De plus, l’adoption par les chaînes de l’une ou l’autre de ces conformations
impacte fortement les propriétés rhéologiques de ce polymère en solution. Ces caractéristiques du
xanthane sont décrites en détail dans la deuxième partie de ce chapitre.
1.2. Polymères amphiphiles
1.2.1. Définition et structure des polymères amphiphiles
Les polymères amphiphiles sont des macromolécules constituées d’une partie hydrophile et
d’une partie hydrophobe qui possèdent la capacité de s’organiser en solution aqueuse via des
interactions inter et/ou intramoléculaires entre les groupements hydrophobes. Ils appartiennent à la
classe des polymères associatifs, c’est-à-dire aux polymères ayant des groupements qui peuvent
former des microdomaines via des interactions physiques (liaisons hydrogène, hydrophobes,
ioniques, complexantes, etc.). Ces polymères présentent en solution une organisation de leurs
chaînes très différente de leurs homologues non associatifs. Ce phénomène est à l’origine de leurs
propriétés viscosifiantes et épaississantes majoritairement très supérieures aux polymères non
amphiphiles au-delà d’une concentration critique, appelée ccrit. C’est pourquoi ils sont le plus souvent
employés comme agents épaississants dans de nombreux domaines, comme la récupération assistée
du pétrole4, la cosmétique, l’agroalimentaire ou encore la formulation de peintures5, 6.
Les polymères amphiphiles sont le plus souvent des copolymères, obtenus soit par
polymérisation7-12 (radicalaire classique, par transfert d’électrons, en émulsion, etc.), soit par
modification chimique d’un squelette initial13-18. Du fait des très nombreuses méthodes d’obtention
de ce type de macromolécules, ils se caractérisent par une grande diversité architecturale. On peut
néanmoins les classer en deux catégories :
- Les copolymères à blocs, qui sont constitués d’un bloc hydrophile et d’un ou deux blocs
hydrophobes19, 20. Les polymères téléchéliques sont un des exemples les plus connus de ce
- 14 -

Chapitre 1 : Bibliographie
type de copolymère. Il s’agit de copolymères triblocs, composés d’une chaîne principale
hydrophile fonctionnalisée à ses deux extrémités par des groupements hydrophobes4, 19, 21.
- Les polymères de type peigne19, 21, qui comportent un squelette hydrophile porteur de
groupements hydrophobes latéraux, distribués tout le long de la chaîne. La répartition des
chaînons hydrophobes sur le squelette peut être plus ou moins contrôlée, suivant la méthode
de synthèse choisie.
Dans le cadre de cette étude, nous nous intéressons aux polymères amphiphiles de type peigne.
Afin de les décrire, nous utilisons les expressions suivantes dans ce manuscrit : les groupements
hydrophobes sont appelés chaînons latéraux, pendants, ou greffés ; le terme de densité de greffage
est employé pour désigner la quantité de chaînons hydrophobes dans la chaîne, quelle que soit la
méthode d’obtention du polymère ; les polymères de squelette identique aux macromolécules de
type peigne, mais sans groupements hydrophobes sont qualifiés de non modifiés/non greffés ou de
précurseur.
Dans le cas des polymères amphiphiles de type peigne, les greffons hydrophobes les plus
courants sont des chaînes carbonées ou fluorées22, 23, qui peuvent être linéaires12, 24-26, ramifiées25, 27, 28
ou aromatiques25, 29, 30. Des chaînons constitués de polyéther31 ou de poly(N-isopropylacrylamide)32, 33,
connus pour leur caractère thermo-associatif, sont également utilisés. Ce type de groupement ne sera
cependant pas étudié dans le cadre de cette thèse. Différentes répartitions des greffons sont
possibles le long du squelette hydrophile. En effet, ces derniers peuvent être disposés de manière
alternée, statistique, en blocs, ou encore être séparés de la chaîne principale par des espaceurs :
- 15 -

Chapitre 1 : Bibliographie
Figure 1-2 : Exemples d’architecture de polymères amphiphiles de type peigne34.
Le squelette hydrophile peut être un polymère neutre ou chargé35, 36. Il peut aussi être d’origine
synthétique ou naturelle. Les polysaccharides appartiennent à cette seconde catégorie. Il s’agit de
polymères constitués d’unités osidiques, reliées entre elles par des liaisons osidiques.
Historiquement, le premier polysaccharide amphiphile obtenu par modification chimique du
squelette est attribué à Landoll1, et consiste en une cellulose porteuse de chaînes époxyde alkylées.
Depuis ces travaux, la mise au point de dérivés amphiphiles à base de polysaccharides a connu un
essor important. En effet, les polysaccharides offrent l’avantage, par rapport aux polymères
synthétiques, d’être biocompatibles, biodégradables et issus de ressources naturelles et
renouvelables. En plus de la cellulose1, 18, 37-39, l’alginate13, 14, 35, 40, 41, le chitosane15, 26, 42, 43, le pullulane24,
44-48, le dextrane17, 49-51, le guar52 ou encore l’acide hyaluronique13, 43, 53, 54 ont fait l’objet de modification
chimique afin de leur conférer des propriétés amphiphiles. Les principales applications visées
concernent le domaine pharmaceutique et médical43, 55, avec par exemple la mise au point de
matrices pour l’encapsulation et la libération contrôlée de principes actifs41, 48, 53, 56, 57. Ces
polysaccharides peuvent également être utilisés en formulation, pour leurs propriétés émulsifiantes
et/ou interfaciales35, 38, 45, 51, 58, ou bien comme agent viscosifiant pour la récupération assistée du
pétrole4, 39.
- 16 -

Chapitre 1 : Bibliographie
1.2.2. Propriétés des polymères amphiphiles en solution
Les polymères amphiphiles possèdent la capacité de s’auto-organiser en solution du fait des
interactions hydrophobes entre leurs chaînons latéraux. Ces interactions peuvent être intra et/ou
intermoléculaires, et résultent en la formation de microdomaines constitués de groupements
appartenant à une ou plusieurs chaînes. La nature des interactions contrôle donc l’organisation et la
dynamique des chaînes, et par conséquent, les propriétés rhéologiques exhibées.
La description de ces propriétés fait l’objet de cette section. Plus particulièrement, on s’attache à
établir le lien entre les différentes organisations possibles en solution et le comportement observé.
De plus, il est possible de moduler la nature et la force des interactions hydrophobes et, par
conséquent, la conformation des chaînes en solution, suivant un certain nombre de paramètres
énoncés plus loin.
1.2.2.1. Propriétés viscosimétriques des polymères amphiphiles
1.2.2.1.1. Régime de concentration des polymères en solution
Le comportement en solution des polymères (associatifs ou non) est fortement régi par la
concentration et la masse molaire des macromolécules. Dans le cadre de ce travail, nous nous
intéressons aux polymères de masse molaire suffisamment élevée pour former des enchevêtrements
en solution. Pour cette catégorie de polymère, on peut définir trois régimes de concentration, comme
indiqué sur la figure 1-3 :
- 17 -

Chapitre 1 : Bibliographie
Figure 1-3 : Domaines de concentration de solutions de polymère (d’après Van Ruymbeke59).
En régime dilué, les chaînes macromoléculaires sont isolées et indépendantes les unes des
autres. Lorsque la concentration augmente, la distance entre chaînes diminue, jusqu’à ce que leurs
volumes hydrodynamiques s’interpénètrent. La concentration correspondant à ce phénomène est la
concentration critique de recouvrement c*. Elle marque le passage du régime dilué au régime semi-
dilué non enchevêtré, et s’accompagne d’une hausse de la viscosité59, 60.
Si l’on augmente encore la concentration en polymère, on atteint le régime semi-dilué
enchevêtré, régime dans lequel les chaînes commencent à s’enchevêtrer. La limite entre ce régime et
le précédent est définie par la concentration d’enchevêtrement c**. Cette dernière correspond à la
concentration à partir de laquelle les enchevêtrements deviennent élastiquement actifs, ce qui
confère à la solution des propriétés rhéologiques bien spécifiques (comportement rhéofluidifiant
marqué, propriétés viscoélastiques accrues).
Ces différents domaines se distinguent par des dépendances de la viscosité suivant la
concentration bien distinctes. Ce point sera développé en détail plus tard dans le manuscrit. De plus,
l’organisation adoptée par les chaînes en solution diffère selon le régime de concentration considéré
(cf. figure 1-4 A). Dans le cas des polymères sans interaction spécifique (autre que de volume exclu),
ces diverses organisations sont purement régies par des contraintes stériques/topologiques.
- 18 -

Chapitre 1 : Bibliographie
Les polymères amphiphiles de masse molaire élevée présentent également trois domaines de
concentration61, 62 équivalents au régime dilué, semi-dilué non enchevêtré et semi-dilué enchevêtré
observés dans le cas des polymères non modifiés. Néanmoins, pour les polymères amphiphiles,
l’organisation des chaînes en solution est fortement impactée par les interactions hydrophobes. En
effet, ces dernières s’ajoutent, voire se substituent, aux contraintes topologiques. De plus, leur
caractère intra et/ou intermoléculaire est très dépendant de la concentration en polymère.
En règle générale, en régime dilué, les interactions sont essentiellement intramoléculaires17, 27, 28,
63, avec l’obtention d’agrégats constitués majoritairement d’une seule chaîne repliée sur elle-même.
Les chaînes adoptent, le plus souvent, des conformations plus compactes que leurs homologues non
modifiés. Par conséquent, la viscosité intrinsèque de solutions de polymères amphiphiles est le plus
souvent inférieure17, 27, 28, 63, et parfois égale62, à celle de polymères non modifiés correspondants.
Au fur et à mesure que la concentration augmente, les interactions intermoléculaires deviennent
privilégiées par rapport à celles intramoléculaires, du fait de la proximité de plus en plus importante
des chaînes. Le régime semi-dilué non enchevêtré est atteint lorsque les interactions
intermoléculaires sont en nombre suffisamment important pour créer des agrégats constitués de
plusieurs chaînes reliées entre elles par des microdomaines hydrophobes. La concentration associée à
ce phénomène est appelée concentration critique ccrit37, 46, 63-65 ou encore concentration d’agrégation
critique cac12, 28. Sa valeur est généralement inférieure à celle de la c* des polymères analogues non
amphiphiles. Cependant, pour certains systèmes, les deux concentrations sont identiques. C’est
pourquoi certains auteurs61, 62 parlent de concentration de recouvrement pour qualifier ccrit, ce qui
montre bien l’interdépendance entre les interactions intermoléculaires et la concentration.
Enfin, si la masse molaire est suffisamment grande, les polymères amphiphiles présentent
également un régime semi-dilué enchevêtré. Dans ce régime, les enchevêtrements topologiques
élastiquement actifs s’ajoutent aux microdomaines formés par les groupements hydrophobes61, 62.
- 19 -

Chapitre 1 : Bibliographie
Tout comme en régime non enchevêtré, la viscosité augmente fortement avec la concentration mais
avec une dépendance différente du précédent régime.
Les différentes organisations en solution suivant le régime de concentration des chaînes de
polymères, amphiphiles ou non, sont schématisées sur la figure 1-4 :
Figure 1-4 : Organisation des chaînes en solution suivant le régime de concentration pour un polymère non
amphiphile (partie a) et un polymère amphiphile (partie b, d’après Van Ruymbeke59).
En outre, à partir du régime semi-dilué non enchevêtré, la viscosité des polymères amphiphiles
est très supérieure à celle de leurs homologues non modifiés14, 28, 62, 66, 67. Cette caractéristique
s’explique par le fait qu’au-delà de la concentration critique, les groupements hydrophobes, en
interaction entre eux de manière intermoléculaire, deviennent des « points collants » qui ralentissent
les mouvements des chaînes68. Cette notion sera développée plus longuement lors de la description
des propriétés viscoélastiques des polymères amphiphiles.
1.2.2.1.2. Comportement en écoulement des polymères amphiphiles
En régime dilué, le comportement en écoulement des polymères amphiphiles est de type
newtonien25, 62 (viscosité indépendante de la vitesse de cisaillement), soit analogue aux
macromolécules non modifiées. En régime semi-dilué par contre, les interactions intermoléculaires
- 20 -

Chapitre 1 : Bibliographie
développées entre les greffons hydrophobes modifient les propriétés exhibées. Ce phénomène est
illustré sur la figure 1-5 :
Figure 1-5 : Influence des greffons hydrophobes sur le comportement en écoulement du chitosane à 2 g/L,
solvant : AcOH 0,3 M/AcONa 0,05 M (d’après Desbrieres et al.26).
Le chitosane présente un comportement newtonien sur toute la gamme de vitesses de
cisaillement. Le plateau newtonien est également observable pour l’échantillon CC10 (chitosane
substitué par une chaîne alkyle C10) mais sur une échelle de cisaillements beaucoup plus faible. Il est
suivi d’une zone de rhéofluidification à plus haut cisaillement. Ce comportement est provoqué par la
rupture des interactions hydrophobes sous l’effet du cisaillement et par l’alignement des chaînes dans
le sens de l’écoulement24, 25, 52, 69. Le caractère rhéofluidifiant est d’autant plus marqué que la longueur
des greffons hydrophobes est importante. Ainsi, dans le cas de CC12, la vitesse de cisaillement
critique, qui marque la limite entre le plateau newtonien et la zone de rhéofluidification, n’est pas
observable dans la gamme de cisaillements étudiée : le comportement du polysaccharide est
purement rhéofluidifiant. Le renforcement du comportement rhéofluidifiant peut également être
obtenu en augmentant par exemple la concentration en polymère12, 63, 70, la densité de greffage ou
encore la longueur des séquences hydrophobes au sein des chaînes11, 25, 28.
- 21 -

Chapitre 1 : Bibliographie
Dans certains cas, pour des concentrations en polymère proches de ccrit, on remarque une
augmentation de la viscosité16, 25, 52, 62 sous l’effet du cisaillement entre le plateau newtonien et le
domaine de rhéofluidification. Ce comportement, qualifié de rhéoépaississement34, serait dû à une
modification de l’équilibre entre les interactions hydrophobes intra et intermoléculaires16, 62. En effet,
l’étirement des chaînes par le cisaillement détruirait les interactions intramoléculaires tout en
favorisant celles intermoléculaires, d’où l’augmentation de viscosité34.
1.2.2.1.3. Paramètres influençant les propriétés viscosimétriques des polymères
amphiphiles
Il est possible de moduler le comportement viscosimétrique des polymères amphiphiles en
solution suivant la nature intra et/ou intermoléculaire des interactions hydrophobes, mais également
suivant la force de ces interactions. De ce fait, il existe plusieurs paramètres, autres que la
concentration en polymère, permettant de contrôler les interactions hydrophobes : la nature et le
taux de chaînons hydrophobes, la nature du squelette hydrophile, l’architecture du polymère, la force
ionique du milieu et la température. Ces points seront abordés dans les paragraphes suivants. L’effet
des tensioactifs sur le comportement des polymères amphiphiles ne sera, par contre, pas décrit.
La force et la nature des interactions sont contrôlables via la densité de groupements
hydrophobes au sein du polymère, leur nature (chaînons carbonés ou fluorés), leur structure (linéaire
ou branchée) ainsi que leur longueur. En effet, les interactions sont renforcées lorsque le groupement
est ramifié11, 25, lorsque sa longueur12, 65, 71, 72 et/ou ses proportions12, 24, 26, 36, 57, 62, 73 augmentent, ou
encore en utilisant des chaînes fluorocarbonées22, 23, 65, 69 au lieu de hydrocarbonées. L’ensemble de
ces modifications se traduit, au-delà de la concentration critique, par un comportement épaississant
accru14, 26, 65, 72.
- 22 -

Chapitre 1 : Bibliographie
Il est observé que la concentration critique ccrit diminue lorsque la longueur des chaînons
augmente12, 14, 26, 65, 71, 72, ou quand le groupement hydrophobe est ramifié au lieu de linéaire11, 25. Par
ailleurs, les polymères amphiphiles fluorés présentent aussi des valeurs de ccrit plus faibles que leurs
homologues hydrocarbonés22, 23, 65. De plus, en règle générale, la concentration critique décroît quand
la densité de greffage augmente12, 14, 24, 26, 41. L’hypothèse majoritairement admise pour ces
phénomènes est que les interactions intermoléculaires sont d’autant plus favorisées que
l’hydrophobie, la longueur et/ou la quantité de chaînons hydrophobes est importante.
Il est important de souligner que les polymères amphiphiles présentent un comportement
rhéologique particulier à partir d’une densité de greffage minimale de chaînons hydrophobes.
Cependant, cette densité ne doit pas dépasser une certaine valeur17, 65, 69, sous peine d’obtenir un
polymère avec des propriétés amoindries, voire insoluble dans l’eau. Ce constat est dû à la
compétition entre le caractère intra et intermoléculaire des interactions hydrophobes. En effet, à
partir d’une certaine densité de greffage, les interactions intramoléculaires deviennent de nouveau
majoritaires dans le milieu36, 46, 65, 69. Par ailleurs, une longueur minimale de chaînon hydrophobe,
généralement supérieure ou égale à 6 atomes de carbone, est aussi nécessaire afin d’obtenir des
propriétés associatives suffisantes15.
Bien que parfois mentionné46, l’impact de la rigidité du squelette hydrophile sur la nature intra
et/ou intermoléculaire des interactions hydrophobes n’a, à notre connaissance, été étudié que par
Inomata et al.74. Le polymère amphiphile examiné par ces auteurs est un polypeptide, le poly[N5-(2-
hydroxyethyl)-L-glutamine] comportant des groupements latéraux alkyle C12, C16 ou C18. Le squelette
hydrophile possède la particularité d’adopter deux conformations différentes en solution suivant le
solvant : une forme désordonnée de type pelote flexible dans l’eau, et une forme hélicoïdale dans
l’éthylène glycol. Les auteurs ont pu montrer, par fluorescence et rhéologie, que la conformation
rigide favorise les interactions de type intermoléculaire, quelle que soit la concentration en polymère.
- 23 -

Chapitre 1 : Bibliographie
Au contraire, pour le polypeptide sous forme pelote, la nature des interactions dépend de la
concentration : intramoléculaire à basse concentration, intermoléculaires au-dessus de la
concentration critique. Ces résultats mettent clairement en évidence l’influence de la rigidité de la
chaîne principale sur la nature intra/intermoléculaire des interactions hydrophobes.
Comme indiqué précédemment, les groupements hydrophobes peuvent adopter différentes
répartitions le long du squelette hydrophile. Cette répartition impacte directement les propriétés de
ces polymères en solution. En effet, il a été observé que les interactions hydrophobes sont renforcées
dans le cas de polymères ayant une structure de type blocs, par rapport à ceux ayant une répartition
statistique de leurs chaînons8, 10, 25, 75. Les polymères de type blocs se caractérisent ainsi par des
propriétés viscosifiantes plus importantes que leurs homologues statistiques. Par ailleurs, la présence
d’espaceurs76, 77 permet de rendre les groupements hydrophobes davantage disponibles pour former
des interactions intermoléculaires, ce qui se traduit par des systèmes possédant un pouvoir
épaississant accru.
La concentration en sel dans la solution affecte aussi la force des interactions entre les chaînons
hydrophobes. Globalement, dans le cas de polymères amphiphiles non chargés, l’augmentation de la
force ionique favorise les interactions hydrophobes du fait d’un phénomène de « salting–out »10, 12, 27,
69 : la présence de sel diminue la solubilité des composés hydrophobes au sein du milieu aqueux,
renforçant ainsi leurs propriétés associatives. L’influence de ce paramètre sur les propriétés dépend
de la nature des interactions hydrophobes, elle-même reliée à la concentration en polymère. Ainsi,
pour c < ccrit, l’ajout de sel renforce les interactions intramoléculaires. La chaîne amphiphile est, de ce
fait, d’autant plus collapsée que la force ionique est élevée, ce qui se traduit par une viscosité
intrinsèque de plus en plus faible27. Pour c > ccrit, le sel favorise les interactions intermoléculaires : la
viscosité augmente alors avec la salinité10, 12, 69. Néanmoins, pour chaque situation, une concentration
- 24 -

Chapitre 1 : Bibliographie
en sel trop élevée peut rendre le polymère complètement insoluble dans l’eau, ce qui entraîne des
phénomènes de séparation de phases27. Le cas des polyélectrolytes amphiphiles est plus complexe.
En effet, il existe une compétition entre les interactions hydrophobes attractives et les répulsions
électrostatiques au sein de ces systèmes16, 36, 65. Par conséquent, leur comportement est très
fortement dépendant de la concentration en sel. Ce point ne sera cependant pas discuté dans le
cadre de ce travail.
L’impact de la température sur les propriétés viscosimétriques des polymères amphiphiles est, à
ce jour, sujet à débat. En solution, ces macromolécules se caractérisent soit par une hausse 12, 17, 69, soit
par une diminution10, 17, 32, 66, 71, 78, 79 de leur viscosité quand la température augmente. Ce
comportement dépend également du domaine de concentration étudié et de la densité de greffage.
Deux hypothèses opposées sont proposées pour expliquer ces résultats contradictoires.
Pour certains auteurs12, 17, 69, 71, l’augmentation de la température favorise les interactions
hydrophobes. Ce phénomène s’expliquerait, soit par la nature entropique12 de ce type d’interaction,
soit par une modification de la qualité du solvant17, 69 (en l’occurrence l’eau) vis-à-vis de la chaîne
hydrophile sous l’effet de la chaleur. En effet, selon cette deuxième supposition, l’élévation de
température a pour conséquence d’affaiblir les liaisons hydrogène impliquées dans la solubilisation
du polymère. De ce fait, le solvant hydrate moins efficacement le polymère, ce qui entraîne
mécaniquement un renforcement des interactions hydrophobes. L’accroissement de la température
avantage donc ces interactions, mais de manière indirecte. L’évolution de la viscosité lors du
chauffage dépend alors du caractère intra ou intermoléculaire de ces interactions17, 71. Ce paramètre
est notamment régi par la concentration en macromolécules dans la solution. Ainsi, en milieu dilué,
les interactions sont majoritairement intramoléculaires : la hausse de température, en favorisant ces
interactions, entraîne un collapse plus important de la chaîne. Cette dernière présente donc une
viscosité intrinsèque d’autant plus faible que la température est élevée17, 71. En milieu semi-dilué, la
- 25 -

Chapitre 1 : Bibliographie
viscosité augmente avec la température si les interactions sont majoritairement intermoléculaires, et
baisse dans le cas contraire. Le comportement exhibé résulte alors de la densité de greffage du
polymère17, 71, puisque, comme mentionné précédemment, il existe une densité limite à partir de
laquelle les interactions intramoléculaires redeviennent majoritaires dans le milieu.
D’autres auteurs10, 28, 66, 79 considèrent au contraire que la température affecte défavorablement les
interactions hydrophobes. Biggs et al.10, par exemple, remarquent que des solutions de
polyacrylamides amphiphiles ont une viscosité de plus en plus faible lorsque la température
augmente. Ils attribuent ce phénomène à une rupture des liaisons hydrophobes sous l’effet de la
chaleur. Cette hypothèse s’appuie notamment sur leurs résultats de fluorescence, qui montrent une
réduction de la quantité de microdomaines hydrophobes avec la température. L’existence de tels
microdomaines en solution dépend du nombre de groupements hydrophobes en solution, mais
également du temps de présence des greffons dans ces microdomaines. Or, plusieurs études ont
montré que la dépendance en température de ce temps de vie est arrhénienne33, 71, 78. Par
conséquent, accroître la température accélère la vitesse de libération des groupements de leurs
microdomaines.
1.2.2.2. Propriétés viscoélastiques des polymères amphiphiles
Outre leur comportement très rhéofluidifiant et leur pouvoir épaississant accru, les polymères
amphiphiles se distinguent également de leur homologue non greffé par des propriétés
viscoélastiques différentes. Ce point est illustré sur la figure 1-6, qui représente l’évolution des
modules élastique G’ et visqueux G’’ en fonction de la fréquence, notée f, pour un alginate et ses
dérivés porteurs de chaînes de polycaprolactone à différentes densités de greffage57.
- 26 -

Chapitre 1 : Bibliographie
Figure 1-6 : Evolution de G’ (symboles pleins) et G’’ (symboles ouverts) en fonction de la fréquence, f, pour un
alginate et ses dérivés modifiés à différentes densités de greffage (d’après Colinet et al.57).
A basse fréquence, l’alginate présente un comportement de liquide visqueux, avec le module
élastique G’ inférieur au module visqueux G’’. A haute fréquence au contraire, l’inverse est observé :
le polysaccharide se comporte alors comme un solide. L’inversion de comportement survient à la
fréquence de relaxation dite fτ.
Le greffage de chaînons hydrophobes sur l’alginate entraîne une augmentation de plus en plus
marquée de la valeur des modules avec la densité de greffage, avec G’ supérieur à G’’. Ce phénomène
s’accompagne d’un décalage de fτ vers des valeurs de plus en plus basses, quand la densité de
greffage augmente. Pour l’alginate le plus modifié, on constate ainsi que G’ est supérieur à G’’ sur
toute la gamme de fréquences mesurées, et que les deux modules ont une faible dépendance en
fréquence. On a ici le comportement typique d’un gel.
Le comportement viscoélastique particulier des polymères amphiphiles s’explique par la
formation, à partir d’une certaine concentration, d’un réseau transitoire dans lequel les
microdomaines hydrophobes résultant des interactions intermoléculaires agissent comme des nœuds
- 27 -

Chapitre 1 : Bibliographie
de « réticulation » à durée de vie finie. Ce phénomène rend ainsi les chaînes de polymère
élastiquement actives. Il existe deux modèles permettant de décrire les propriétés rhéologiques de
ces systèmes au-delà de ccrit, suivant la présence ou non d’enchevêtrements. Pour remarque, ces
modèles s’appliquent pour tout polymère associatif, quel que soit le type d’interaction (liaisons
hydrogène, interaction hydrophobe, ionique, etc.).
1.2.2.2.1. Polymères non enchevêtrés : modèle de Rouse collant
Le modèle de Rouse collant80, 81 (ou « sticky Rouse model ») s’applique dans le cas des polymères
amphiphiles flexibles en régime semi-dilué non enchevêtré (c* < c < c**). D’après ce modèle, les
groupements hydrophobes sont considérés comme des « points collants », ou « stickers », qui sont
répartis de manière équidistante le long des chaînes et s’associent uniquement par paire (cf. figure 1-
7). Chaque sticker existe sous l’un ou l’autre des états suivants : « fermé » s’il est associé avec un
autre sticker (il forme alors un nœud de jonction), ou « ouvert » s’il est libre. Les portions de chaîne
entre chaque sticker sont qualifiées de « segments ». Ces segments sont considérés comme
élastiquement actifs lorsqu’ils sont délimités par deux stickers fermés, en interaction
intermoléculaire. Par conséquent, les stickers fermés de manière intramoléculaire ne participent ni à
la dynamique ni à l’élasticité du système. De même, les segments encadrés par des stickers ouverts
sont inactifs.
Figure 1-7 : Modèle de réseau transitoire pour des associations en paire (d’après Rubinstein et al.80 et Leibler et
al.81).
- 28 -

Chapitre 1 : Bibliographie
Les « nœuds », ainsi formés, ont une durée de vie finie qui dépend du temps d’association entre
stickers, τsticker, caractérisé par :
τ sticker=τ0 exp(Ea+EbkT ) Equation 1-1
Avec τ0 le temps de diffusion du sticker (de l’ordre de 10-9 s), Ea l’énergie d’activation nécessaire
pour former la jonction, Eb l’énergie de liaison des stickers et k la constante de Boltzmann.
La relaxation du système obéit à une variante du modèle de Rouse : les segments relaxent
lorsqu’ils diffusent d’une distance équivalente à leur taille. Cependant, la présence de stickers
intermoléculaires fermés empêche de tels mouvements. Par conséquent, la diffusion est possible
uniquement lorsque les stickers délimitant les segments sont à l’état ouvert. Elle est également
fortement ralentie par ces mêmes stickers, qui peuvent se réassocier au cours du mouvement, d’où le
terme de Rouse collant pour qualifier la dynamique de ces systèmes. Cette dernière dépend donc à la
fois du nombre de stickers à l’état fermé, et également de la durée d’association entre stickers.
On définit ainsi le temps de Rouse collant des chaînes associatives comme :
τ Rouse=τ sticker∗ ( f ∗ pinter )2
Equation 1-2
Avec f le nombre de stickers par chaîne et p inter la fraction de stickers fermés en interaction
intermoléculaire.
Le module élastique au temps de Rouse collant, G’, reste identique à celui des polymères non
associatifs, soit, pour une chaîne constituée de N monomères de taille b et de fraction volumique φ :
G'=kT∗ϕ
N∗ b3Equation 1-3
- 29 -

Chapitre 1 : Bibliographie
La viscosité est donnée par la relation suivante :
η=G'∗τ Rouse=G'∗ τ sticker∗ ( f ∗ pinter )
2Equation 1-4
En régime dilué, les interactions entre stickers sont majoritairement intramoléculaires : la
viscosité est faible, car pinter est proche de 0. La forte hausse de viscosité lors du passage du régime
dilué au régime semi-dilué non enchevêtré s’explique par l’augmentation importante du nombre
d’interactions intermoléculaires lorsque ccrit est atteint.
Si la concentration croît encore, une renormalisation du temps de vie des nœuds est nécessaire.
En effet, à concentration élevée, le nombre de stickers ouverts est faible. Par conséquent, un sticker
s’associe et se dissocie plusieurs fois avec son partenaire avant de pouvoir entrer en interaction avec
un nouveau sticker disponible. Ce phénomène se traduit par une augmentation de τ sticker avec la
concentration, ce qui explique la forte dépendance de la viscosité avec la concentration dans ce
régime.
Ce modèle a été développé selon l’hypothèse où les associations se font toujours par paire, ce qui
coïncide rarement avec les situations rencontrées expérimentalement. Néanmoins, les prédictions du
modèle s’appliquent également pour des polymères associatifs dont les nœuds comportent plus de
deux stickers. Dans ce cas, τsticker est défini comme la durée nécessaire pour que le sticker se détache
du microdomaine résultant des associations entre plusieurs stickers.
Les propriétés viscoélastiques des polymères amphiphiles en régime linéaire peuvent être
modélisées par un élément de Maxwell, comme illustré sur la figure 1-8 :
- 30 -

Chapitre 1 : Bibliographie
Figure 1-8 : Propriétés viscoélastiques d’un polymère téléchélique82.
Le système est décrit par un unique temps de relaxation τ. A haute fréquence, le système se
comporte comme un gel chimique. Au contraire, à basse fréquence le polymère amphiphile présente
les caractéristiques d’un liquide visqueux. Ces résultats sont en accord avec le modèle de Rouse
collant. L’unique temps de relaxation correspond au temps de vie d’un chaînon hydrophobe dans un
nœud, τsticker. Pour t < τsticker (hautes fréquences), les stickers ne peuvent pas sortir des microdomaines
hydrophobes, d’où les propriétés de type gel observées. En revanche, pour t > τ sticker (basses
fréquences), les stickers ont suffisamment de temps pour se libérer de leur nœud. Par conséquent, la
majorité des stickers sont ouverts : le système peut alors couler. Le modèle de Rouse collant est
particulièrement adapté pour décrire le comportement viscoélastique des polymères téléchéliques19,
21, 82, 83.
1.2.2.2.2. Polymères enchevêtrés
Le modèle de Rouse collant n’est cependant pas applicable pour les polymères amphiphiles en
régime enchevêtré. En effet, dans ce régime de concentration, les enchevêtrements topologiques, qui
s’ajoutent aux microdomaines hydrophobes, contribuent également à la dynamique du système. Ce
- 31 -

Chapitre 1 : Bibliographie
constat a donné lieu au développement du modèle de la reptation collante. Ce dernier combine à la
fois la théorie de la reptation, utilisée pour expliquer le comportement rhéologique de polymères
enchevêtrés non associatifs, et celle de Rouse collante.
a. Polymères non associatifs en régime enchevêtré : modèle de la reptation
Pour rappel, en régime enchevêtré, une chaîne est gênée dans ses mouvements par ses voisines,
du fait des enchevêtrements topologiques. Afin de rendre compte de cette situation, Doi et
Edwards84, ainsi que de Gennes85, ont développé le modèle de la reptation. D’après ce dernier, la
chaîne étudiée est confinée dans un tube délimité par les enchevêtrements de la macromolécule
avec ses voisines, comme illustré sur la figure 1-9 :
Figure 1-9 : Chaîne confinée dans son tube de reptation de diamètre a (d’après Van Ruymbeke59).
La chaîne est divisée en segments de longueur l qui sont circonscrits par les enchevêtrements.
L’axe passant par le centre du tube est appelé « chaîne primitive ». Les mouvements de la chaîne
suivant cet axe sont libres, et suivent une dynamique de type Rouse. Les déplacements latéraux, par
contre, sont fortement limités par les parois du tube. Par conséquent, la chaîne ne peut diffuser dans
le tube que par un mouvement de reptation, semblable à celui d’un serpent.
- 32 -
Segment de longueur lentre 2 enchevêtrements Chaîne confinée dans le tube
Enchevêtrement

Chapitre 1 : Bibliographie
La relaxation de la chaîne s’effectue en plusieurs étapes, chacune étant associée à un temps
caractéristique (cf. figure 1-10).
Figure 1-10 : Relaxation des polymères en régime enchevêtré (d’après Leibler et al.81).
τe correspond au temps de relaxation des segments de chaîne entre enchevêtrements
(dynamique de Rouse). Le temps de reptation, τrep, est le temps nécessaire pour que la chaîne sorte
de son tube.
Pour t < τe, la relaxation, qui suit une dynamique de type Rouse, se produit uniquement au sein
des segments. La position de ces derniers fluctue librement au sein du tube, mais sans toucher les
parois. Par conséquent, les segments ne ressentent pas la présence des enchevêtrements : la chaîne
ne « voit » pas le tube.
A partir de t > τe, les fluctuations sont suffisamment importantes pour que la chaîne perçoive les
enchevêtrements. Ces derniers agissent comme des nœuds de réticulation, ce qui se traduit, au
niveau des propriétés rhéologiques, par l’apparition d’un plateau caoutchoutique entre τ e et τrep, de
module :
- 33 -

Chapitre 1 : Bibliographie
G0≈ cRT( 1N e ) Equation 1-5
Avec c la concentration en monomère et Ne le nombre de monomères entre enchevêtrement.
Puis, lorsque la fréquence diminue, le régime de reptation est atteint : les mouvements de
relaxation latéraux sont limités aux parois du tube, seuls les déplacements le long du tube sont
autorisés. La chaîne commence à diffuser petit à petit hors de son tube par reptation.
La relaxation est complète lorsque la chaîne est entièrement sortie du tube, soit à t > τ rep. Au delà
de τrep, les chaînes coulent librement les unes par rapport aux autres, et le système se comporte
comme un liquide visqueux. Le temps de reptation, ou temps de relaxation terminal, est donné par :
τ rep=τ e( NN e)3,5
Equation 1-6
Avec τR le temps de Rouse d’une chaîne constituée de N monomères. Il ressort que τ rep dépend
très fortement de la densité d’enchevêtrement.
b. Polymères associatifs en régime enchevêtré : modèle de la reptation collante
Le modèle de la reptation collante80, 81 ajoute la notion de tube de reptation à la théorie de Rouse
collante afin de modéliser l’effet des enchevêtrements sur la dynamique des polymères amphiphiles.
En d’autres termes, le système peut être visualisé comme une chaîne obéissant à une dynamique de
type Rouse collante, mais confinée dans un tube délimité par les enchevêtrements. De plus, on
considère que le nombre d’enchevêtrements est très supérieur à celui des stickers. La relaxation de la
chaîne s’effectue suivant une succession d’étapes élémentaires :
- 34 -

Chapitre 1 : Bibliographie
Figure 1-11: Modèle de la reptation collante81.
Soit une chaîne P confinée dans son tube de reptation et présentant 3 stickers (positions c, i et d).
A un temps t (partie a), les chaînes P et P1 sont en interaction via les deux stickers fermés en position
i. Ces stickers empêchent les segments de chaîne compris entre c et d de diffuser par reptation. Le
mouvement devient possible uniquement lorsque les stickers i s’ouvrent et ne se réassocient pas. La
chaîne peut alors se mouvoir au sein du tube, jusqu’à ce que le sticker ainsi libéré entre en interaction
avec un autre sticker (ici f de la chaîne 2, partie b). Ces étapes élémentaires se répètent jusqu’à ce
que la chaîne sorte complètement de son tube. La reptation de la chaîne hors de son tube est donc
possible même si tous les stickers ne sont pas conjointement à l’état ouvert.
- 35 -
Sticker fermé
Sticker ouvert

Chapitre 1 : Bibliographie
La figure 1-12 schématise la réponse viscoélastique de tels polymères en accord avec le modèle
de la reptation collante, tout en la comparant à celle des polymères non associatifs :
Figure 1-12 : Réponse viscoélastique des polymères associatifs (en rouge) et non associatifs (en bleu, d’après
Leibler et al.81).
Le processus de relaxation des polymères associatifs présente quatre régimes, définis par trois
temps caractéristiques : le temps de type Rouse des chaînes entre enchevêtrements, τe, la durée des
interactions entre stickers, τsticker, et le temps de reptation τrep pour le polymère associatif (τ0rep dans le
cas du polymère non modifié).
Pour t < τe, seuls les segments entre les enchevêtrements relaxent suivant une dynamique de
type Rouse, sans influence des stickers. Dans ce régime, la relaxation des chaînes associatives est
identique à celle du polymère non modifié.
Pour τe < t < τsticker, les stickers n’ont pas le temps de se libérer des microdomaines hydrophobes.
Ces derniers agissent donc comme des nœuds de réticulation physique. De plus, à partir de t > τe, la
chaîne devient également sensible à la présence des enchevêtrements topologiques. Par conséquent,
la relaxation du système dans ce régime se caractérise par un plateau caoutchoutique, analogue à
- 36 -

Chapitre 1 : Bibliographie
celui observé pour les polymères non modifiés, mais de module plus élevé du fait de la contribution
supplémentaire des microdomaines à celle des enchevêtrements :
G1≈cRT ∗( ρN s
+1Ne ) Equation 1-7
Avec ρ la masse volumique et Ns nombre de monomères entre sticker.
Pour τsticker < t < τrep, la contrainte contenue dans les microdomaines hydrophobes est relaxée.
Seuls les enchevêtrements continuent d’agir comme des nœuds élastiquement actifs. Le système se
comporte toujours comme un gel (présence d’un deuxième plateau caoutchoutique), mais de module
G0 identique à celui des polymères non greffés (cf. équation 1-5). Le temps de reptation est défini
par :
τ rep=τ Rouse( NN e )1,5
≈( NN e)1,5 2 f 2τ sticker1−9/ pinter+12/ pinter
2 Equation 1-8
Avec f le nombre de stickers par chaîne et p inter la fraction de stickers en interaction
intermoléculaire. Le temps de reptation des polymères associatifs dépend donc des enchevêtrements
topologiques, mais également des stickers, même s’ils ne forment plus de nœuds élastiquement
actifs. Ces derniers agissent comme des points collants qui ralentissent la reptation de la chaîne hors
de son tube, selon le mécanisme présenté plus haut.
Enfin, pour t > τrep, les chaînes libérées de tout nœud, coulent les unes par rapport aux autres. On
retrouve donc le comportement d’un liquide visqueux.
La viscosité des polymères associatifs est définie par :
η≈G0 τ rep≈cRTN e (
NN e )
1,5 2 f 2 τ sticker1−9/ p inter+12/ pinter
2 Equation 1-9
- 37 -

Chapitre 1 : Bibliographie
La viscosité des polymères amphiphiles est donc d’autant plus importante que la quantité de
stickers au sein des chaînes et/ou la durée de vie des interactions hydrophobes sont élevées.
Pour résumer
D’après le modèle de la reptation collante, la relaxation des polymères associatifs/amphiphiles se
distingue de celle des polymères non modifiés par la présence de deux plateaux caoutchoutiques,
auxquels sont associés deux temps caractéristiques. Le premier temps, τsticker, à haute fréquence, est
indépendant de tout paramètre moléculaire (longueur des chaînes, nombre de stickers, etc.). Le
deuxième temps, τrep, à basse fréquence, varie suivant la longueur des chaînes, le nombre de stickers,
et également τsticker. Le modèle prédit également que les polymères amphiphiles possèdent des
propriétés épaississantes renforcées par rapport à leurs homologues non modifiés. De plus, les
systèmes amphiphiles ont un temps de relaxation terminal beaucoup plus élevé que les chaînes non
modifiées, ce qui se traduit par un comportement de type gel dans des gammes de fréquences pour
lesquelles leur homologue est un liquide visqueux. L’ensemble de ces prévisions se trouvent
confirmées par les résultats expérimentaux décrits dans la littérature28, 37, 52, 57, 62.
Pour autant, les données bibliographiques ne semblent pas en accord avec la prédiction du
modèle concernant l’existence de ces deux plateaux. En effet, la quasi-totalité des polymères
associatifs n’exhibent qu’un seul plateau avant la zone terminale d’écoulement. Néanmoins, Müller et
al.86 ont pu observer, par des analyses viscoélastiques et diélectriques, les deux plateaux
caoutchoutiques dans le cas de dérivés de polybutadiène s’auto-associant via des liaisons hydrogène.
Ce résultat renforce la pertinence de la théorie de la reptation collante pour expliquer le
comportement des polymères associatifs, et donc amphiphiles. L’absence du premier plateau pour la
majorité des polymères amphiphiles s’explique par le temps très court d’association entre stickers, ce
qui rend difficile l’observation expérimentale de ce plateau.
- 38 -

Chapitre 1 : Bibliographie
Par ailleurs, d’après la théorie de la reptation collante, les propriétés rhéologiques des polymères
amphiphiles sont d’autant plus améliorées que le nombre de stickers par chaîne ou la durée
d’association entre stickers, τsticker, sont élevés. Une fois encore, les résultats décrits dans la littérature
vont dans ce sens : les polymères amphiphiles ont un temps de reptation qui augmente avec la
densité de greffage57, 67 ou la longueur des chaînons hydrophobes57. Ce phénomène est, du reste,
illustré sur la figure 1-6.
Par la suite, les analyses viscoélastiques décrites dans ce manuscrit s’effectuant en régime
enchevêtré, seule la théorie de la reptation collante sera considérée. Ces modèles, qui élucident
l’origine des propriétés rhéologiques accrues des polymères amphiphiles par rapport à leurs
homologues non modifiés, montrent que leur dynamique est contrôlée par le nombre et la durée de
vie des interactions entre chaînons hydrophobes.
1.3. La gomme xanthane
Dans le cadre de ces travaux, nous nous intéressons à greffer chimiquement des chaînons
hydrophobes sur le xanthane. Cette partie a pour objectif de décrire la structure et le comportement
en solution de ce polysaccharide. En particulier, le xanthane est connu pour adopter deux
conformations différentes2, 3 en solution, suivant les conditions opératoires : une forme ordonnée
hélicoïdale semi-rigide, et une forme désordonnée de type pelote flexible, qui ont des propriétés
rhéologiques bien distinctes. Cette section s’attache notamment à mettre en relief le lien entre les
différentes conformations pouvant être adoptées par les chaînes et les propriétés viscosimétriques et
rhéologiques du polymère. Enfin, les différents exemples de modification du xanthane par greffage
sont également présentés dans cette partie.
- 39 -

Chapitre 1 : Bibliographie
1.3.1. Intérêt industriel du xanthane et procédé de production
Le xanthane est un hétéropolysaccharide microbien obtenu par fermentation de sucre en
présence de la bactérie Xanthomonas campestris. Cette gomme fut découverte à la fin des années
1950 par les scientifiques du département américain de l’Agriculture, lors d’un criblage visant à
identifier des micro-organismes capables de produire des polymères hydrosolubles d’intérêt
commercial87, 88.
Très rapidement, le xanthane fut l’objet de nombreuses recherches académiques et industrielles,
du fait de ses propriétés rhéologiques particulières. En effet, ce polysaccharide possède un pouvoir
épaississant important, et ce, dès de faibles concentrations en polymère, ainsi qu’un caractère
fortement rhéofluidifiant. De plus, à forte concentration en sel, les solutions aqueuses de xanthane
conservent une viscosité importante jusqu’à des températures élevées (70-80°C). Enfin, ce polymère
est connu pour interagir avec d’autres polysaccharides, tels les galactomannanes89-92 (guar, caroube,
etc.). Les mélanges ainsi obtenus possèdent une viscosité bien supérieure à celles des solutions de
polysaccharides seuls. Ce phénomène est qualifié de synergie (positive) des systèmes
galactomannane/xanthane. Ainsi, le xanthane est surtout utilisé industriellement comme agent de
contrôle de viscosité des systèmes aqueux et/ou comme stabilisant dans les émulsions et
suspensions93-95.
Le premier procédé industriel de production du xanthane fut élaboré dès 1960 par l’entreprise
Kelco®, et ce polysaccharide devint disponible sur le marché à partir de 196488, 94. De nos jours, le
xanthane est le polysaccharide d’origine microbienne le plus vendu au monde. En 2012, la production
mondiale de gomme xanthane est estimée à plus de 110 000 tonnes ; les principaux producteurs sont
la Chine (près des deux-tiers de la production), les Etats-Unis, la France, l’Autriche et le Japon96.
Le principal domaine d’application du xanthane est l’agroalimentaire93, 95. Cette gomme est
autorisée dans l’industrie alimentaire aux Etats-Unis depuis 196995, en France depuis 1974 et en
- 40 -

Chapitre 1 : Bibliographie
Europe depuis 1982 (additif E415)88. Il est présent notamment dans les assaisonnements de salade,
les boissons, les plats cuisinés et surgelés en tant qu’épaississant, agent stabilisant ou encore pour
améliorer la texture et l’apparence du produit final. Le xanthane est également employé dans de
nombreux autres domaines, tels que l’industrie pétrolière (pour la récupération assistée du pétrole),
les produits cosmétiques et pharmaceutiques, les revêtements de surface (peinture, vernis,
impression sur papier ou textile) ou encore les explosifs87, 93-95.
Le procédé industriel d’obtention de la gomme est un processus le plus souvent discontinu (dit en
batch), qui comporte un certain nombre d’étapes présentées dans la figure 1-13 :
Figure 1-13: Procédé de production de la gomme xanthane93, 94.
La première étape de ce procédé consiste en la synthèse de la gomme par fermentation aérobie
en milieu stérile et en présence de la bactérie Xanthomonas campestris. Le milieu de culture contient
divers nutriments nécessaires à la bactérie : des sels de magnésium, de potassium ou de calcium, des
sources d’azote et de carbone (glucose, fructose ou amidon). La réaction se déroule à température et
- 41 -

Chapitre 1 : Bibliographie
pH contrôlés (respectivement entre 28 et 32°C et entre 6,5 et 7,5) jusqu’à consommation totale du
carbone.
En fin de fermentation, le moût réactionnel contient, outre le xanthane, des cellules
bactériennes, des protéines et des nutriments résiduels. Plusieurs étapes de purification sont alors
nécessaires pour éliminer ces impuretés. En premier lieu, le moût est stérilisé thermiquement (entre
80 et 130°C) pendant une dizaine de minutes afin de tuer les bactéries Xanthomonas. Après
refroidissement du milieu, le xanthane est alors précipité dans de l’isopropanol. Les fibres sont
ensuite récupérées par filtration ou centrifugation, séchées, broyées et tamisées, et la poudre finale
obtenue conditionnée. L’isopropanol usagé, provenant des étapes de précipitation et de séchage, est
purifié par distillation et réinjecté dans le processus de production. Par ailleurs, suivant les
applications visées, des traitements supplémentaires peuvent être effectués sur la gomme lors de la
fermentation, du processus de purification et/ou après séchage. Le moût peut, par exemple, être
chauffé en milieu basique afin d’ôter les fonctions acétate présentes dans le polymère, ou bien
soumis à des étapes de purification plus poussées, dans le but d’éliminer toute trace de débris
cellulaires93.
1.3.2. Structure chimique et conformation du xanthane
1.3.2.1. Structure chimique du xanthane
Le xanthane est un hétéropolysaccharide anionique constitué d’un squelette cellulosique linéaire
de β-D-glucoses dont une unité sur deux est substituée en C3 par une chaîne latérale trisaccharidique.
Cette dernière comporte un α-D-mannose, un acide β-D-glucuronique et un β-D-mannose terminal 97,
98 :
- 42 -

Chapitre 1 : Bibliographie
Figure 1-14 : Structure chimique du xanthane.
Le xanthane comporte également des groupements acétate et pyruvate, situés respectivement
sur le carbone 6 du mannose interne et sur les carbones 4 et 6 du mannose terminal. La proportion
de ces groupements varie d’une gomme xanthane à une autre selon la nature de la souche
bactérienne utilisée99, 100, les conditions de fermentation94, 101-103 ou encore les traitements post-
fermentation93.
Les propriétés anioniques du xanthane s’expliquent par la présence, le long de la chaîne latérale,
de une à deux fonctions acide carboxylique par unité de répétition, suivant le degré de substitution
en pyruvate. Dans la majorité des échantillons de xanthane commerciaux, ces fonctions sont sous
forme carboxylate, et saponifiées par des ions Na+, K+ ou encore Ca2+. Le pKa de ces fonctions acides
est compris entre 2,6 et 4, suivant la température et la force ionique du milieu104, 105.
1.3.2.2. Conformation du xanthane en solution : transition ordre-désordre
En solution, le xanthane est caractérisé par une transition conformationnelle ordre-désordre2, 3,
au cours de laquelle les chaînes passent d’une conformation ordonnée semi-rigide, à une
conformation désordonnée flexible. Cette transition est définie par une température de transition,
Tm, et s’accompagne d’une modification des propriétés rhéologiques du xanthane. De nombreuses
techniques permettent de mettre en évidence cette transition et de caractériser les différentes
conformations obtenues, telles que le dichroïsme circulaire2, 3, 106, 107, la polarimétrie3, 108-113, l’analyse
- 43 -

Chapitre 1 : Bibliographie
calorimétrique différentielle114-116, ou encore la RMN3, 113, 117, la diffusion de la lumière108, 118-121, la
microscopie122-125 et la rhéologie116, 126-130.
Suivant la température et la force ionique du milieu, le polymère peut se présenter sous trois
conformations différentes109. En effet, à basse température et force ionique élevée, le xanthane existe
sous une forme ordonnée dite native. Cet état correspond à celui adopté par les chaînes dans le
moût réactionnel n’ayant subi aucun traitement thermique. Si le milieu est chauffé à une
température supérieure à Tm et/ou dilué en présence d’une très faible quantité de sel ([NaCl] < 10 -4
M), le xanthane adopte alors une conformation désordonnée, qualifiée de dénaturée. La transition
de l’état natif à l’état dénaturé est un processus irréversible. L’ajout de sel à la solution ou le
refroidissement de cette dernière à une température inférieure à Tm permet aux chaînes d’adopter
de nouveau une conformation ordonnée, mais différente de celle de l’état natif. On parle alors de la
forme renaturée du xanthane. Contrairement à la transition de la forme native vers la forme
renaturée, le passage entre les états dénaturés et renaturés du xanthane est réversible.
Il faut savoir que la conformation du xanthane est toujours un sujet de débat, en particulier en ce
qui concerne les formes natives et renaturées de la gomme.
A l’état solide, des analyses de diffraction des rayons X montrent que le xanthane adopte une
conformation hélicoïdale131, 132. Ainsi, Moorhouse et al.131 établissent que la structure secondaire de ce
polysaccharide consiste en une hélice droite quintuple 51 de pas 4.7 nm, avec les chaînes latérales
repliées et alignées le long du squelette cellulosique (cf. figure 1-15).
- 44 -

Chapitre 1 : Bibliographie
Figure 1-15 : Hélice 51 du xanthane vue a : perpendiculaire à l’axe de l’hélice. b : parallèle à l’axe de l’hélice131.
Cette structure est stabilisée par des liaisons hydrogène intramoléculaires entre les chaînes
latérales et principales. Pour ces auteurs, le xanthane adopterait la même conformation ordonnée en
solution, soit celle d’une simple hélice. Okuyama et al.132, au contraire, considèrent que le xanthane à
l’état solide se présente sous forme d’une double hélice composée de deux chaînes antiparallèles et
stabilisées par des liaisons hydrogène intermoléculaires (cf. figure 1-16) :
- 45 -

Chapitre 1 : Bibliographie
Figure 1-16 : Double hélice 51 de xanthane a : parallèle . b : antiparallèle (vue perpendiculaire par rapport à
l’axe de l’hélice)132.
Pour ces auteurs, les chaînes adoptent également cette configuration en milieu aqueux. Dans les
deux cas, les données de rayon X sont en accord avec les deux modèles proposés, ce qui ne permet
pas de conclure sur la nature exacte de la structure secondaire du xanthane.
En solution, les deux conformations ordonnées du xanthane (simple et double hélice) ont été
observées, sans qu’il soit possible d’attribuer de façon définitive l’une ou l’autre de ces structures à
l’état natif ou renaturé du xanthane. L’obtention d’une simple ou double hélice dépend de nombreux
paramètres, dont la température, la concentration en polymère, mais également les traitements de
- 46 -

Chapitre 1 : Bibliographie
purification subis par l’échantillon après le processus de fermentation (en particulier les éventuelles
étapes de pasteurisation et/ou de dilution). A ce jour, plusieurs scénarii existent dans la littérature
afin d’expliquer la transition ordre-désordre du xanthane et les conformations natives, dénaturées et
renaturées associées.
Certains auteurs considèrent que la forme ordonnée du xanthane est celle d’une hélice
constituée d’un seul brin2, 3, 111, 133. La transition ordre-désordre est alors un processus intramoléculaire
qui s’explique par une rupture des liaisons hydrogène stabilisant l’hélice et un « déploiement » des
chaînes latérales qui s’éloignent du squelette cellulosique2. La conformation désordonnée obtenue
est celle d’une pelote flexible, comme indiquée sur la figure 1-17 :
Figure 1-17 : Transition ordre-désordre du xanthane selon un processus intramoléculaire (d’après Norton et
al.111).
Ce modèle s’appuie notamment sur des expériences de dénaturation du xanthane qui montrent
que la masse molaire du polymère ne varie pas lors de la transition ordre-désordre108, 130, 134. De plus, il
a été observé, par polarimétrie et dichroïsme circulaire, que la transition conformationnelle est
indépendante de la concentration en polymère2, 3. Par ailleurs, Norton et al.111 ont étudié la vitesse de
dénaturation de ce polysaccharide, et ont mis en évidence que la cinétique de transition est d’ordre
1, en accord avec un processus intramoléculaire. Ils en déduisent également que des zones
ordonnées et désordonnées peuvent coexister au sein de la même chaîne de polymère. Jones et al.133
- 47 -
Forme ordonnée Forme désordonnée
Dénaturation
Renaturation
Chaîne latérale
Chaîne principale
Zone ordonnée
Zone désordonnée

Chapitre 1 : Bibliographie
concluent également, par analyse de la conductivité de solutions aqueuses de xanthane, que ce
dernier adopte une conformation ordonnée hélicoïdale simple brin. Il est important de souligner,
néanmoins, que les études menées par Morris et al.3, Norton et al.111 et Jones et al.133 ont été
réalisées sur des échantillons de polymère ayant préalablement subi un traitement thermique. De ce
fait, il est probable que la forme ordonnée caractérisée par ces auteurs soit celle du xanthane à l’état
renaturé, et non pas natif, ce qui ne permet pas de conclure quant à la conformation adoptée par les
chaînes sous forme native.
Milas et al. se sont intéressés aux conformations ordonnées d’un xanthane issu d’un moût de
fermentation non pasteurisé, dont une fraction a été dénaturée thermiquement134 ou par dilution en
l’absence de sel130. Leurs clichés de microscopie électronique124 montrent que les deux formes
ordonnées consistent en une hélice comprenant une seule chaîne, et de même masse molaire130, 134,
135, en accord avec le modèle de la simple hélice subissant une transition conformationnelle
intramoléculaire. Cependant, leurs résultats indiquent également que la forme renaturée du
xanthane est plus visqueuse130, 134 et plus rigide124, 134 que la forme native. Les deux conformations ne
sont donc pas identiques. Ils en concluent que le xanthane à l’état natif est une simple hélice un brin 2,
124, alors que la forme renaturée consiste en une double hélice formée d’un seul brin replié sur lui-
même134, 135, d’où sa rigidité plus importante.
Figure 1-18 : Forme native, dénaturée et renaturée du xanthane d’après Milas et al.2, 124, 134, 135.
- 48 -

Chapitre 1 : Bibliographie
D’autres auteurs considèrent que la conformation native du xanthane est celle d’une double
hélice constituée de deux chaînes. La transition ordre-désordre est alors intermoléculaire et consiste
en la dissociation de cette double hélice en deux brins simples, de type pelote136-138. Deux modèles
d’hélice sont envisagés : soit une double hélice constituée de deux hélices simples qui interagissent
latéralement l’une avec l’autre114, 125 (figure 1-19 (a)), soit une double hélice constituée de deux brins
appariés, similaire à la structure de l’ADN, et qui « fond » par ses extrémités lors de la dénaturation110
(figure 1-19 (b)). Actuellement, ce second modèle, dit modèle de Liu et al.139, est le plus
communément admis dans la littérature.
Figures 1-19 : Modèle de la double hélice a : deux simples hélices en interaction latérale87. b : une double hélice
de deux brins appariés140.
De nombreux auteurs supportent le modèle de la double hélice. Stokke et Christensen140 ont, par
exemple, étudié la dépolymérisation du xanthane en milieu acide118, 141 ou en présence d’eau
oxygénée et de fer142. Ils observent ainsi que la dégradation du xanthane comporte deux étapes : une
première phase de dépolymérisation « lente », avec une faible variation de la masse molaire au cours
du temps, suivie d’une deuxième phase « rapide », qui se caractérise par une diminution importante
de la masse molaire. Ces résultats peuvent s’expliquer par le modèle de la double hélice : au début de
la dégradation, les interactions intermoléculaires entre les deux brins de la double hélice empêchent
les fragments dépolymérisés de se libérer de la structure. Par conséquent, la masse molaire évolue
peu au cours du temps. Au fur et à mesure de la dépolymérisation, les liaisons glycosidiques sont
- 49 -
Zone de jonction ordonnée
Zones de jonction désordonnées
(b)

Chapitre 1 : Bibliographie
brisées, jusqu’à un seuil critique à partir duquel la conformation en double hélice ne peut plus être
maintenue. A partir de ce point, les fragments dépolymérisés sont relâchés, ce qui se traduit par une
chute de la masse molaire. Il a aussi été observé, par microscopie électronique 118, 125, que le xanthane
natif adopte une structure hélicoïdale de deux brins qui se dissocie plus ou moins totalement en
simples chaînes. Plusieurs études des conformations du xanthane, basées sur la diffusion de la
lumière, semblent pareillement confirmer la structure en double hélice de ce polysaccharide à l’état
natif. Muller et al.138 et Capron et al.126 ont notamment utilisé cette technique pour déterminer les
masses molaires d’un xanthane natif et renaturé après traitement thermique. Ils ont ainsi remarqué
que la forme native du xanthane se caractérise par une masse molaire deux fois plus élevée que celle
de la forme renaturée. Un résultat similaire, avec la même technique, a été obtenu par Lecourtier et
al.109 pour des échantillons dénaturés par dilution en absence de sel. Cette diminution de masse
molaire s’accompagne également d’une chute de la viscosité109, 137. Liu et al.110 ont également observé,
par diffusion statique de la lumière, que la masse molaire du xanthane natif est divisée par deux
lorsque ce dernier est dissous dans le cadoxen (mélange de cadmiun, d’hydroxyde de sodium et
d’éthylène diamine), solvant dans lequel le xanthane est sous forme désordonnée. Ce résultat a été
confirmé par Sato et al.143 et Hatakenaka et al.144 via des expériences de sédimentation et de viscosité.
Toutefois, le xanthane est un polysaccharide qui a une forte tendance à l’agrégation145-148, et ce,
dès de faibles concentrations en polymère147, 148. Du fait de cette caractéristique, des masses molaires
très variables, comprises entre 2 et 50*106 g/mol pour ce polymère, ont été reportées dans la
littérature109, 147, 149, 150. Bien que les valeurs de masse molaire les plus élevées s’expliquent sûrement
par la présence d’agrégats, il est difficile d’affirmer si les masses molaires plus faibles, de l’ordre de
106 g/mol, correspondent réellement aux chaînes isolées. Par conséquent, il se peut que la
diminution de masse molaire observée lors de la dénaturation (thermique ou par dissolution dans le
cadoxen) d’un xanthane natif ne provienne pas de la dissociation d’une double hélice, mais
essentiellement d’agrégats111, 128. Une autre explication possible serait que de tels traitements
- 50 -

Chapitre 1 : Bibliographie
provoquent une dégradation du polymère128, 144, 151. Plus généralement, les données issues
d’expériences de dénaturation du xanthane natif, en particulier par voie thermique, sont souvent
contradictoires, ce qui explique en partie la controverse sur les conformations adoptées par le
xanthane en solution aqueuse. Milas et al. 134, 152, par exemple, n’observent pas de diminution de
masse molaire pour un xanthane natif ayant subi un traitement thermique à 80°C en présence de
0,01 M NaCl. Pour ces auteurs, ce résultat confirme la conformation simple hélice du polymère. Au
contraire, Muller et al.138 ont constaté, par mesure de masse molaire, que la dissociation de la double
hélice native dans 0,1 M de NaCl est possible si l’échantillon est chauffé au minimum 15 minutes à
110°C. Kawakami et al.129 n’obtiennent pas ce résultat pour un échantillon commercial dénaturé dans
de l’eau ultrapure à 95°C pour des durées comprises entre 2 et 9 heures, bien qu’ils obtiennent une
masse molaire deux fois plus faible pour le même polymère dans le cadoxen. Il apparait donc que la
dénaturation du xanthane natif est un processus complexe, qui est fortement influencé par les
conditions opératoires utilisées, telles que la température, la force ionique, la durée de chauffage ou
encore l’histoire thermique du polysaccharide et sa structure chimique.
De plus, la concentration en polymère lors de la dénaturation du xanthane semble également
jouer un rôle important sur sa conformation finale en solution. Capron et al.126 ont ainsi noté que,
pour des concentrations en polysaccharide supérieures à 10 g/L dans 0,1 M NaCl, la dénaturation
thermique d’un xanthane natif s’accompagne d’une augmentation de la masse molaire, et non pas
d’une diminution comme observée à de plus faibles concentrations. Le xanthane renaturé obtenu se
caractérise également par une viscosité plus élevée que celle de la forme native, et par des propriétés
viscoélastiques renforcées126, 187. Capron et al. en déduisent qu’en milieu concentré, la double hélice
native ne peut se dissocier que partiellement, du fait des contraintes stériques générées par les
chaînes voisines. Matsuda et al.106 ont également observé cette augmentation de masse molaire avec
la concentration lors de la transition conformationnelle, et arrivent aux mêmes conclusions que
Capron. La viscosité plus élevée de la forme renaturée par rapport à la forme native avec la
- 51 -
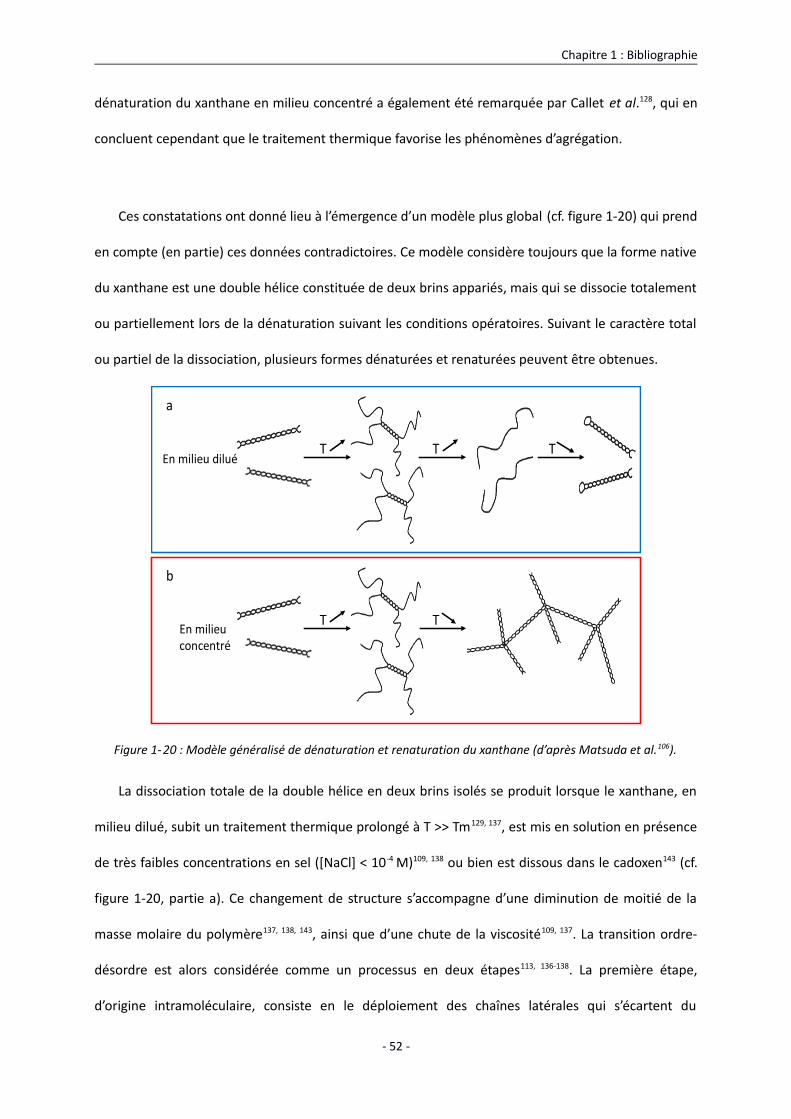
Chapitre 1 : Bibliographie
dénaturation du xanthane en milieu concentré a également été remarquée par Callet et al.128, qui en
concluent cependant que le traitement thermique favorise les phénomènes d’agrégation.
Ces constatations ont donné lieu à l’émergence d’un modèle plus global (cf. figure 1-20) qui prend
en compte (en partie) ces données contradictoires. Ce modèle considère toujours que la forme native
du xanthane est une double hélice constituée de deux brins appariés, mais qui se dissocie totalement
ou partiellement lors de la dénaturation suivant les conditions opératoires. Suivant le caractère total
ou partiel de la dissociation, plusieurs formes dénaturées et renaturées peuvent être obtenues.
Figure 1-20 : Modèle généralisé de dénaturation et renaturation du xanthane (d’après Matsuda et al.106).
La dissociation totale de la double hélice en deux brins isolés se produit lorsque le xanthane, en
milieu dilué, subit un traitement thermique prolongé à T >> Tm129, 137, est mis en solution en présence
de très faibles concentrations en sel ([NaCl] < 10-4 M)109, 138 ou bien est dissous dans le cadoxen143 (cf.
figure 1-20, partie a). Ce changement de structure s’accompagne d’une diminution de moitié de la
masse molaire du polymère137, 138, 143, ainsi que d’une chute de la viscosité109, 137. La transition ordre-
désordre est alors considérée comme un processus en deux étapes113, 136-138. La première étape,
d’origine intramoléculaire, consiste en le déploiement des chaînes latérales qui s’écartent du
- 52 -
En milieu diluéT T T
En milieu concentré
T T
a
b

Chapitre 1 : Bibliographie
squelette cellulosique. La forme dénaturée obtenue peut être visualisée comme une double hélice
« étendue », moins compacte que la forme native. La deuxième phase de la transition correspond à la
dissociation progressive de la double hélice en deux brins isolés. Le modèle couramment admis 126, 144
pour expliquer cette dissociation est celui de Liu et al.110, 139, consistant en une double hélice qui
« fond » par ses extrémités. Cette étape est beaucoup plus lente que la première, et nécessite des
conditions de dénaturation plus extrêmes (températures élevées, temps de dénaturation
suffisamment longs123, 129, 138, force ionique faible). L’état dénaturé final est alors celui d’une simple
chaîne en pelote statistique. Lors de la renaturation, les chaînes isolées se replient sur elles-mêmes
pour former une double hélice constituée d’un seul brin122, 153, plus rigide que la forme native137. De
telles structures ont notamment pu être observées par microscopie électronique122, 123.
Lorsque les conditions de dénaturation ne sont pas suffisantes pour assurer une dissociation
totale de la double hélice, des conformations dénaturées et renaturées intermédiaires entre la
double hélice native et le simple brin dénaturé sont alors observées114, 125. La transition ordre-
désordre obéit au même processus en deux étapes que celui présenté auparavant, mais qui s’arrête
au moment de la dissociation partielle de la double hélice136 (cf. figure 1-20, partie b). La forme
désordonnée consiste alors en une succession de zones désordonnées de type pelote maintenues
ensemble par des « nœuds » de doubles hélices110, 126, les proportions de chaque zone dépendant des
conditions de dénaturation. Lors de la renaturation, une partie des zones désordonnées peut
reformer des doubles hélices, composées soit des chaînes « initiales », soit de deux chaînes issues de
deux hélices différentes106, 108. La renaturation aboutit alors à la formation d’agrégats, voire même
d’un réseau, ce qui peut expliquer le comportement de type gel faible observé dans certains cas pour
la forme renaturée126, 127, 187. Par conséquent, la conformation renaturée est également une double
hélice deux brins, mais moins ordonnée et moins compacte que la forme native 129, 137. De ce fait, sa
masse molaire est identique à celle de la forme native mais sa viscosité est plus élevée109, 128, 138.
- 53 -

Chapitre 1 : Bibliographie
Il faut savoir que la température de transition conformationnelle, Tm, est influencée par la force
ionique du milieu, le pH ou encore la teneur en acétate et pyruvate au sein du polysaccharide, ce qui
complique la comparaison des données de la littérature et contribue à la controverse sur la structure
secondaire du xanthane. Il a été ainsi observé que la concentration en sel influe sur la valeur de la
température de transition conformationnelle3, 100, 111, 113, 154 : la Tm est proportionnelle au logarithme de
la force ionique du milieu2, 108. La présence de sel renforce donc la stabilité thermique de la forme
ordonnée. En effet, l’ajout de cations permet d’écranter les charges négatives portées par les chaînes
latérales du polysaccharide, éliminant ainsi les répulsions électrostatiques entre ces groupes et
stabilisant la forme hélicoïdale. Pour les mêmes raisons, la Tm change aussi avec le pH et augmente
très fortement lorsque le pH diminue112, 115, 141.
Par ailleurs, la Tm varie également avec les proportions en acétate et pyruvate au sein du
polymère. Les groupements pyruvate, chargés négativement, déstabilisent la conformation ordonnée
du xanthane. De ce fait, la Tm est d’autant plus basse que la teneur en pyruvate est importante 112, 155.
Au contraire, la Tm augmente avec la teneur en fonctions acétate : ces dernières stabilisent la forme
hélicoïdale100, 112. Cet effet stabilisant pourrait s’expliquer par des interactions apolaires entre les
groupements méthyle des acétates au sein de l’hélice155 ou encore par l’existence de liaisons
hydrogène entre ces groupements et le squelette cellulosique du xanthane du fait de la présence
d’atomes d’oxygène107.
- 54 -

Chapitre 1 : Bibliographie
Pour résumer :
La transition ordre-désordre du xanthane est un processus dépendant très fortement des
conditions opératoires utilisées, telles que la température, la durée de dénaturation, la force ionique
ou encore la concentration en polymère, mais également de paramètres intrinsèques au
polysaccharide (taux d’acétate et de pyruvate). Suivant ces éléments, le xanthane adopte trois
conformations en solution : les conformations natives et renaturées, rigides et hélicoïdales, et une
conformation désordonnée flexible de type pelote. A ce jour, il n’est pas possible d’affirmer avec
certitude si les formes natives et renaturées consistent en une simple ou double hélice, au regard des
nombreuses données contradictoires de la littérature. Néanmoins, ces contradictions peuvent
s’expliquer en partie par le fait que les différentes études n’ont pas été réalisées dans des conditions
identiques. En particulier, tous les facteurs qui influencent le processus de dénaturation n’ont pas
toujours été pris en considération lors de ces expériences. Actuellement cependant, un consensus
semble s’établir dans la littérature autour de la conformation native du xanthane : le modèle
majoritairement soutenu est celui de la double hélice deux brins, qui se dissocie totalement ou
partiellement en simples brins suivant les conditions opératoires. Cette transition s’accompagne
d’une modification des propriétés, notamment rhéologiques, du xanthane en solution.
1.3.3. Propriétés du xanthane en solution
1.3.3.1. Domaines de concentration du xanthane en solution
Comme décrit précédemment, les polymères de haute masse molaire présentent trois régimes
de concentration en solution : le régime dilué (pour c < c*), le régime semi-dilué non enchevêtré (c* <
c < c**) et le régime semi-dilué enchevêtré (c > c**). Les régimes de concentration d’un polymère en
solution sont le plus souvent déterminés par viscosimétrie, en traçant l’évolution du logarithme de la
- 55 -
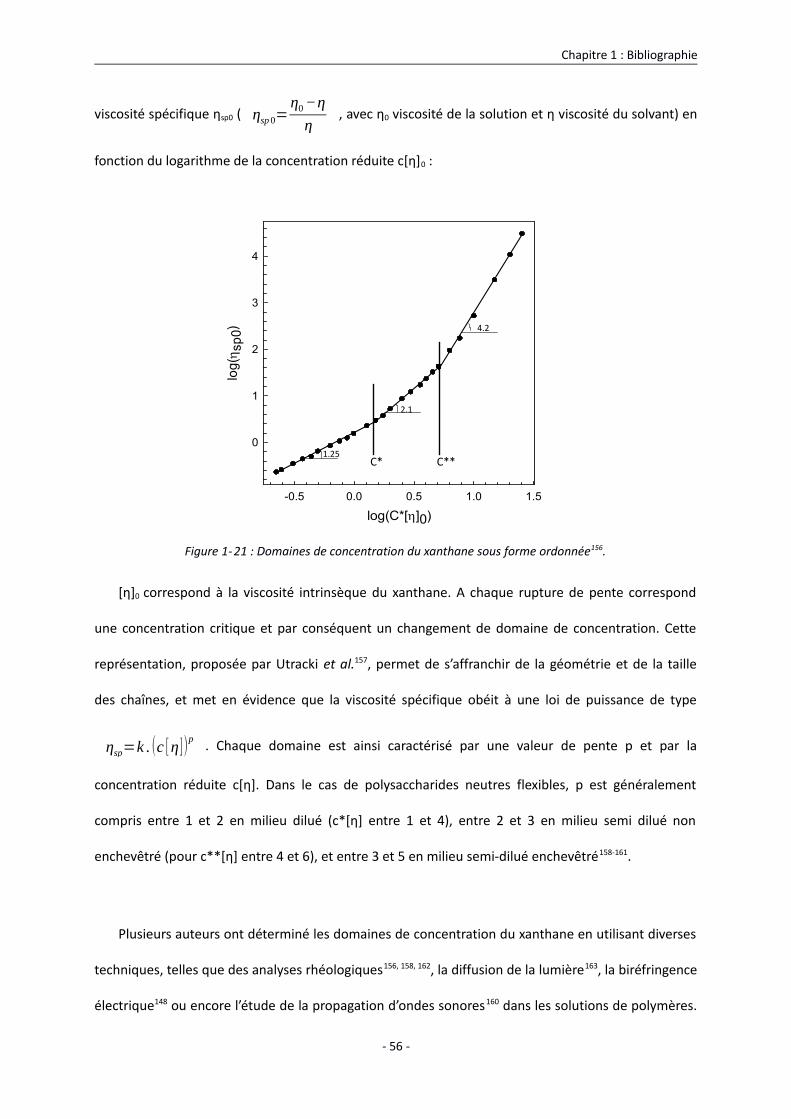
Chapitre 1 : Bibliographie
viscosité spécifique ηsp0 ( ηsp 0=η0−η
η, avec η0 viscosité de la solution et η viscosité du solvant) en
fonction du logarithme de la concentration réduite c[η]0 :
Figure 1-21 : Domaines de concentration du xanthane sous forme ordonnée156.
[η]0 correspond à la viscosité intrinsèque du xanthane. A chaque rupture de pente correspond
une concentration critique et par conséquent un changement de domaine de concentration. Cette
représentation, proposée par Utracki et al.157, permet de s’affranchir de la géométrie et de la taille
des chaînes, et met en évidence que la viscosité spécifique obéit à une loi de puissance de type
ηsp=k . (c [η ])p . Chaque domaine est ainsi caractérisé par une valeur de pente p et par la
concentration réduite c[η]. Dans le cas de polysaccharides neutres flexibles, p est généralement
compris entre 1 et 2 en milieu dilué (c*[η] entre 1 et 4), entre 2 et 3 en milieu semi dilué non
enchevêtré (pour c**[η] entre 4 et 6), et entre 3 et 5 en milieu semi-dilué enchevêtré158-161.
Plusieurs auteurs ont déterminé les domaines de concentration du xanthane en utilisant diverses
techniques, telles que des analyses rhéologiques156, 158, 162, la diffusion de la lumière163, la biréfringence
électrique148 ou encore l’étude de la propagation d’ondes sonores160 dans les solutions de polymères.
- 56 -
log(C*[h]0)-0.5 0.0 0.5 1.0 1.5
log(h sp0)
0
1
2
3
4
C* C**1.25
2.1
4.2

Chapitre 1 : Bibliographie
Dans la majorité des cas, ces études ont été réalisées en présence de sel, soit dans des conditions
pour lesquelles le xanthane est sous forme ordonnée. Le tableau 1-1 récapitule les différentes valeurs
des concentrations critiques décrites dans la littérature :
Auteurs Solvant c* (g/L) c** (g/L) Techniques
Capron164 NaCl 0,1M 0,21 - Rhéologie
Cuvelier et al.156 NaCl 0,1M 0,28 1,1 Rhéologie
Launay et al.165 NaCl 0,1M 0,24 0,92 Rhéologie
Meyer et al.148 NaCl 0,05M - 2,0 Biréfringence
Dichroïsme circulaire
Milas et al.162 NaCl 0,1M 0,13 0,78 Rhéologie
Peirera et al.160 KCl 0,1M 0,25 - Ondes sonores
Southwick et al.163 NaN3 0,03M 0,15 0,70 Diffusion de la lumière
Wyatt et al.166 NaCl 0,05M 0,20 0,80 Rhéologie
Tableau 1-1: Valeurs des concentrations critiques du xanthane, forme ordonnée.
On constate que les données de c* et de c** divergent quelque peu d’un auteur à l’autre.
Plusieurs paramètres sont à l’origine de ces différences. En premier lieu, il est important de souligner
que la notion de concentration critique est davantage un concept qu’une description réaliste des
phénomènes observés. En effet, la transition entre les différents régimes de concentration s’effectue
progressivement. De ce fait, il peut être difficile, dans certains cas, d’identifier précisément c* et c**.
De plus, la détermination de ces concentrations peut dépendre également de la technique d’analyse
utilisée. Par ailleurs, le xanthane est connu pour avoir une forte tendance à l’agrégation, et ce, même
à très faible concentration 145-148, ce qui peut compliquer d’autant plus la quantification de ses
domaines de concentration. La nature de l’échantillon de xanthane pourrait aussi avoir une influence
sur c* et c**, et plus particulièrement la teneur en pyruvate155, 167, la polydispersité, généralement
très élevée (indice de polydispersité compris entre 4 et 6) et/ou surtout la masse molaire du
polysaccharide168. Cependant, les données de la littérature sur ce point sont contradictoires 169-171 et ne
permettent pas de statuer quant à l’influence exacte de ce paramètre sur les valeurs des
concentrations critiques.
- 57 -

Chapitre 1 : Bibliographie
Pour autant, on peut noter que les valeurs des concentrations critiques du xanthane sous forme
ordonnée sont du même ordre de grandeur. On peut approximer, au regard des résultats décrits dans
la littérature, que c* ≈ 0,20 g/L et c** ≈ 1,0 g/L.
De plus, comme mentionné précédemment, chaque régime de concentration est caractérisé par
une pente p et par la concentration réduite c[η]. En régime dilué, p est compris entre 1 et 1,54. La
transition de ce régime au régime semi-dilué non enchevêtré est généralement atteinte pour c*[η]
≈1. En régime semi-dilué non enchevêtré, p est compris entre 2 et 2,6. Le régime semi-dilué
enchevêtré est obtenu pour c**[η] ≈ 5-8, et est défini par une valeur de p comprise entre 3,5 et 4,7.
Bien que le xanthane sous forme ordonnée soit rigide, les données de p et de c*[η] pour chaque
domaine de concentration sont en accord avec celles déterminées pour des polysaccharides neutres
flexibles158-161.
La valeur de la pente dans le domaine semi-dilué enchevêtré donne des informations sur la
nature des interactions entre les chaînes. Ainsi, il a été observé que les polymères linéaires neutres et
flexibles qui interagissent en solution uniquement via des enchevêtrements physiques ont une pente
comprise entre 3 et 4. D’après de Gennes172, la viscosité de tels systèmes obéit à une loi d’échelle de
la forme : η≈M3 .c3,75 pour c > c**. Au contraire, certains polysaccharides, comme les gommes
guar ou caroube, présentent des pentes dans le domaine semi-dilué enchevêtré supérieures à 4161, 165.
Ce résultat serait dû à l’existence d’interactions spécifiques attractives entre les chaînes en solution,
qui s’ajouteraient ou se substitueraient aux enchevêtrements physiques (on parle alors de chaînes
« hyperenchevêtrées »)159. Dans le cas du xanthane sous forme ordonnée, la nature des interactions
en solution en régime enchevêtré n’est pas la même suivant que le xanthane soit sous forme native
ou renaturée. Arendt et al.173 et Milas et al.162 ont ainsi étudié les régimes de concentration d’un
xanthane natif, provenant d’un moût réactionnel non pasteurisé. En régime semi-dilué enchevêtré, ils
ont déterminé respectivement une pente p de 3,5 et 3,7. Milas et al.162 en concluent que les
- 58 -

Chapitre 1 : Bibliographie
propriétés rhéologiques du xanthane natif sont uniquement gouvernées par des enchevêtrements
physiques. Au contraire, les mêmes auteurs trouvent un p de 4,24 pour un xanthane commercial 170.
Wyatt et al.166 obtiennent un résultat similaire pour le même type d’échantillon. Dans les deux cas, il
n’est pas indiqué si les poudres commerciales ont subi des traitements thermiques lors des étapes de
purification. Toutefois, Cuvelier et al.156 et Capron et al.164, qui ont étudié des xanthanes commerciaux
à l’état renaturé, ont trouvé des valeurs de p proches de celles de Milas et Wyatt, ce qui laisse
supposer que les échantillons de ces auteurs sont renaturés. Il apparait donc que les deux formes
ordonnées du xanthane n’ont pas les mêmes propriétés en solution. En particulier, l’organisation des
chaînes renaturées en régime semi-dilué enchevêtré ne peut pas être complètement décrite par un
simple mécanisme d’enchevêtrement, mais doit prendre en compte l’existence d’interactions
spécifiques attractives entre les chaînes156.
En contrepartie, il existe très peu d’exemples dans la littérature concernant les domaines de
concentration du xanthane en l’absence de sel, c’est-à-dire quand ce polymère est sous forme
désordonnée. A notre connaissance, une seule étude, menée par Wyatt et al.166, présente les
domaines de concentration de ce polysaccharide dans de telles conditions (cf. figure 1-22).
- 59 -

Chapitre 1 : Bibliographie
Figure 1-22 : Domaines de concentration du xanthane sous forme désordonnée (d’après Wyatt et al.166).
Comme pour la forme ordonnée, on observe les trois domaines de concentration suivants : le
régime dilué, caractérisé par une pente p de 1,6 (pour c < c*, c* = 0,07 g/L), le régime semi-dilué non
enchevêtré, avec p = 0,5 (pour c* < c < c**, avec c** concentration d’enchevêtrement, égale à 0,4
g/L), et le régime semi-dilué enchevêtré (c > c**), avec une dépendance en concentration de 1,5. De
plus, on a l’apparition d’un quatrième régime de concentration (dans lequel η ~ c3,75.), à une
concentration critique cd de 2 g/L.
Le tableau 1-2 compare les valeurs des concentrations critiques pour le xanthane sous forme
ordonnée et désordonnée :
Xanthane C* (g/L) C** (g/L) Ce (g/L)
Forme ordonnée 0,2 1 -
Forme désordonnée 0,07 0,4 2
Tableau 1-2 : Concentrations critiques du xanthane sous forme ordonnée et désordonnée.
Le comportement du xanthane en l’absence de sel est très différent de celui sous forme
ordonnée. En premier lieu, les concentrations critiques c* et c** sont plus faibles que celles de son
analogue ordonné. Par ailleurs, les dépendances en concentration ne sont pas les mêmes entre les
- 60 -

Chapitre 1 : Bibliographie
deux formes. Enfin, on observe la présence de ce quatrième domaine de concentration, non observé
lorsqu’on ajoute du sel. D’après les auteurs, ces différences s’expliqueraient par le caractère
polyélectrolyte du xanthane. En effet, en l’absence de sel, les charges négatives portées par les
chaînes latérales ne sont pas écrantées. Par conséquent, les chaînes, à l’état désordonné, sont étirées
sous l’effet des répulsions électrostatiques. Leur volume hydrodynamique est plus important que
leurs homologues neutralisés, ce qui explique pourquoi les chaînes chargées peuvent interagir entre
elles à des concentrations plus faibles.
Plus spécifiquement, le comportement du xanthane écranté par du sel peut être prédit par la
théorie de Dobrynin et al.174 à propos des polyélectrolytes flexibles. Selon ce modèle, les
polyélectrolytes peuvent être décrits comme une succession de blobs électrostatiques, de longueur
De, qui se repoussent les uns les autres sous l’effet des répulsions. Pour une longueur inférieure à la
longueur de corrélation ξ, les chaînes peuvent être visualisées comme des bâtonnets rigides de blobs
électrostatiques. Au-dessus de ξ en revanche, les chaînes deviennent flexibles et sont modélisées par
une marche aléatoire de blobs de corrélation (chaque blob de corrélation contient plusieurs blobs
électrostatiques), comme indiqué sur la figure 1-23 :
Figure 1-23 : Représentation schématique d’un polyélectrolyte en régime semi-dilué174.
- 61 -
Blob électrostatique Blob
de corrélation

Chapitre 1 : Bibliographie
Selon cette théorie, les polyélectrolytes présentent quatre domaines de concentration, délimités
par trois concentrations critiques : la concentration de recouvrement c* des blobs de corrélation, la
concentration d’enchevêtrement c** et la concentration cd de recouvrement des blobs
électrostatiques. La dépendance en concentration de chaque domaine est la suivante :
- Régime semi-dilué non enchevêtré (c* < c < c**) : η ~ c0,5.
- Régime semi-dilué enchevêtré (c** < c < cd) : η ~ c1,.5.
- Régime « concentré » (c > cd) : η ~ c3,75. Dans ce régime, le comportement du polyélectrolyte
devient identique à celui d’un polymère neutre flexible en bon solvant qui présente
uniquement des enchevêtrements physiques.
Le comportement du xanthane chargé et dénaturé obéit à cette théorie. La conformation
adoptée par ce polysaccharide impacte donc fortement ses propriétés en solution. En régime
« concentré », on remarque que la viscosité évolue avec la concentration selon la loi de puissance η ~
c3,75. Cette loi est identique à celle prédite par de Gennes concernant les polymères neutres flexibles
comportant uniquement des enchevêtrements physiques. Il semblerait donc que sous forme
dénaturée, le xanthane ne présente pas d’interactions spécifiques attractives entre les chaînes,
contrairement à ce qui a pu être observé à l’état renaturé.
- 62 -

Chapitre 1 : Bibliographie
Pour résumer :
La conformation du xanthane influence ses propriétés en solution, notamment les valeurs des
concentrations critiques c* et c** et les lois d’échelle associées à chaque domaine de concentration.
Il a été mentionné précédemment que le xanthane présente des propriétés rhéologiques
intéressantes en régime semi-dilué enchevêtré, du fait de la formation d’un réseau tridimensionnel.
Les données de la littérature, décrites ci-dessus, semblent indiquer que la nature des interactions
entre chaînes dans ce régime n’est pas la même suivant que le xanthane soit sous forme native,
dénaturée ou renaturée. En particulier, les propriétés de la forme renaturée seraient régies par des
interactions spécifiques entre chaînes différentes des simples enchevêtrements physiques a priori
observés pour les états natifs et dénaturés. Ces points sont décrits plus en détail dans les sections
suivantes du manuscrit.
1.3.3.2. Propriétés viscosimétriques du xanthane en solution
1.3.3.2.1. Viscosité intrinsèque du xanthane en solution
Comme indiqué précédemment, les propriétés du xanthane en solution dépendent du régime de
concentration. En régime dilué, les propriétés viscosimétriques de la gomme sont notamment
caractérisées par la viscosité intrinsèque [η]. Ce paramètre, qui rend compte du volume
hydrodynamique occupé par les chaînes en solution, dépend notamment de la masse molaire et de la
conformation (rigidité) du polymère étudié.
- 63 -

Chapitre 1 : Bibliographie
Le tableau 1-3 indique quelques valeurs de viscosité intrinsèque reportées dans la littérature pour
du xanthane en solution aqueuse :
Auteurs Etat Solvant Mw (106 g/mol) [η] mL/g
Capron et al.137 Natif
Natif
NaCl 0,1M
NaCl 0,01M
5,3
5,2
7 150
7 400
Khouryieh et al.175 -
-
-
Eau
NaCl 0,002M
NaCl 0,04M
2,65
-
-
15 570
9 290
7 320
Lecourtier et al.109 Natif
Renaturé
NaCl 0,1M
NaCl 0,1M
4,8
2,1
3 400
18 000
Lund et al.176 Natif
Natif
NaCl 0,1M
NaCl 1M
5,2
5,4
5 500
5 350
Milas et al.130 Natif
Renaturé
NaCl 0,01M
NaCl 0,01M
5,25
5,25
10 300
13 200
Muller et al.138 Natif
Renaturé 1
Renaturé 2
NaCl 0,1M
NaCl 0,1M
NaCl 0,1M
4,8
4,0
2,1
3 400
8 500
6 100
Tableau 1-3 : Viscosité intrinsèque du xanthane.
On constate que la viscosité intrinsèque est très variable d’un échantillon à l’autre. Plusieurs
paramètres peuvent être à l’origine de ces différences : la concentration en sel au sein de la solution,
la masse molaire du xanthane ou encore sa forme (native, dénaturée ou renaturée).
Les données de Khouryieh et al.175 montrent que la viscosité intrinsèque du xanthane diminue
quand la concentration en sel augmente. Ce constat a également été effectué par Milas et al.130 et
Lecourtier et al.109. Il s’agit là d’un résultat classique pour un polyélectrolyte : en présence de sel,
l’écrantage des charges portées par le polymère entraîne un effondrement de la chaîne. Cette
dernière occupe un volume moins important en solution, d’où une viscosité d’autant plus diminuée
que la concentration en sel est élevée. Néanmoins, et contrairement aux polyélectrolytes flexibles, la
viscosité du xanthane devient indépendante de la force ionique à partir d’une très faible
- 64 -

Chapitre 1 : Bibliographie
concentration en sel, de l’ordre de 0,005 M109, 166 . Au-delà de cette salinité, la viscosité n’évolue
quasiment pas, comme l’indique les résultats de Capron et al.137 et Lund et al.176. De plus, elle reste
très élevée pour un polyélectrolyte dans de telles conditions. Ce phénomène s’explique par la
stabilisation, en présence de sel, de la forme ordonnée du xanthane. Cette dernière, très rigide,
possède un volume hydrodynamique conséquent, ce qui se traduit par une viscosité importante.
Cette insensibilité à la force ionique, couplée avec une viscosité intrinsèque pouvant être 10 fois
supérieure à celle des carboxyméthylcelluloses ou des galactommannanes93 de masse molaire
équivalente, expliquent notamment l’intérêt industriel du xanthane en tant qu’épaississant ou agent
de suspension.
Il est connu que la masse molaire d’un polymère influence très fortement sa viscosité : cette
dernière diminue avec Mw. Sur ce point, les expériences menées par Liu et al.110, consistant
notamment à mesurer la viscosité intrinsèque d’échantillons plus ou moins soniqués (donc de masse
molaire différente), mettent en avant le fort impact de Mw sur [η]. De ce fait, les variations de [η]
observées dans la littérature peuvent s’expliquer par les différences de masse molaire des polymères
étudiés.
La viscosité du xanthane dépend également de la forme, native, dénaturée ou renaturée,
adoptée par les chaînes en solution. En particulier, les états natifs et renaturés se caractérisent par
des viscosités intrinsèques bien distinctes, comme illustré dans le tableau 1-3. Globalement, la
viscosité de la forme renaturée est plus importante que celle de la forme native. Ce phénomène
s’expliquerait par le fait que les chaînes renaturées adoptent une conformation hélicoïdale moins
ordonnée que l’état natif (donc moins compacte). Par conséquent, les chaînes occupent un volume
plus important. Un autre paramètre influant [η] serait la nature de la forme renaturée. En effet,
comme expliqué précédemment, la dénaturation de la double hélice native de xanthane peut aboutir
- 65 -

Chapitre 1 : Bibliographie
à une forme renaturée hélicoïdale constituée d’un ou de deux brins, moins ordonnée que l’état natif,
suivant les conditions de dénaturation. Or, il a été remarqué que la viscosité finale de la forme
renaturée dépend des conditions de dénaturation appliquées. Par exemple, Milas et al.162 ont noté
que la viscosité de l’état renaturé, obtenu par dénaturation à 80°C pendant une minute, est
supérieure à celle de la forme native. Cette viscosité reste cependant du même ordre de grandeur. Au
contraire, Muller et al.138 ont soumis un xanthane natif à une dénaturation plus drastique (110°C
pendant 10 minutes, échantillon renaturé 1) : l’état renaturé présente une viscosité plus de deux fois
supérieure à celle de la conformation native. Cependant, si les conditions de dénaturation sont trop
poussées (température très élevée et/ou pendant une durée importante), la viscosité intrinsèque de
la forme renaturée diminue (échantillon 2). Ces auteurs en déduisent que l’échantillon renaturé 1 a
subi une dénaturation incomplète, et est par conséquent sous forme d’une double hélice moins
ordonnée que celle native. Cette conformation occupe un volume hydrodynamique beaucoup plus
important que la forme native en solution, d’où une viscosité accrue. L’échantillon renaturé 2
correspondrait à une forme renaturée issue d’une dénaturation totale, composée d’un seul brin, et
de volume hydrodynamique plus faible. Des résultats et conclusions similaires ont été obtenus par
Lecourtier et al.109 et Capron et al.137. Il est donc possible de contrôler la viscosité du xanthane
renaturé suivant les conditions de dénaturation utilisées. Si ces dernières sont trop faibles, le
xanthane renaturé adopte une conformation très proche de celle du xanthane natif, ce qui se traduit
par une viscosité similaire et pourrait expliquer les résultats de Milas et al.130, 162.
Tout comme pour la forme renaturée, la conformation exacte de l’état dénaturé influence aussi la
viscosité intrinsèque du xanthane. En effet, selon le modèle de la double hélice, la forme dénaturée
peut se présenter comme une pelote constituée d’une seule chaîne (dénaturation totale) ou bien
comme une hélice partiellement dissociée, comportant des zones ordonnées en double hélice et des
zones désordonnées en pelote (dénaturation incomplète). L’importance de ce paramètre sur la
viscosité intrinsèque du xanthane a été notamment mise en évidence par Lecourtier et al.109. Leurs
- 66 -

Chapitre 1 : Bibliographie
travaux portent sur l’évolution de la viscosité intrinsèque d’un xanthane natif dissous dans des
solutions à différentes forces ioniques :
Figure 1-24: Influence de la salinité sur la viscosité intrinsèque du xanthane109.
Pour des concentrations en sel entre 10-1 et 10-3 M, la viscosité intrinsèque reste quasiment
constante, ce qui s’explique par la forme native ordonnée du xanthane. A partir de 10 -3 M, on observe
une augmentation significative de la viscosité, la force ionique n’étant plus suffisante pour maintenir
la conformation native. La double hélice commence alors à se dissocier. Cette dissociation, partielle,
est de plus en plus importante au fur et à mesure que la salinité diminue, ce qui se traduit par un
volume hydrodynamique et, par conséquent, une viscosité, de plus en plus élevés. Aux alentours
d’une concentration en NaCl de 10-4 M, la double hélice est presque complètement dissociée : l’état
dénaturé est alors majoritairement constitué de zones désordonnées comportant encore quelques
nœuds de jonction en double hélice. La viscosité intrinsèque atteinte est alors maximale (de l’ordre
de 60 000 mL/g). En-deçà de cette concentration en sel, la viscosité intrinsèque chute brutalement :
la double hélice s’est complètement dissociée, la forme dénaturée consiste en une pelote constituée
d’une seule chaîne de polymère, qui occupe un volume beaucoup plus faible ([η] = 18 000 mL/g, en
- 67 -
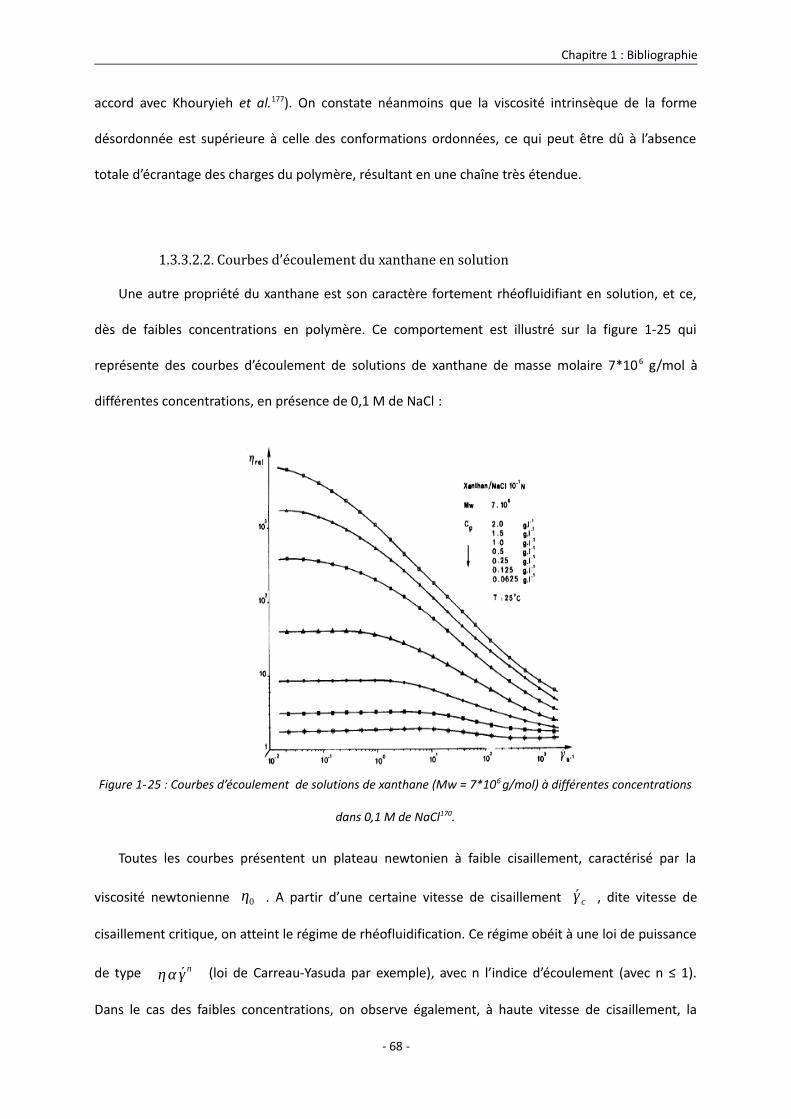
Chapitre 1 : Bibliographie
accord avec Khouryieh et al.177). On constate néanmoins que la viscosité intrinsèque de la forme
désordonnée est supérieure à celle des conformations ordonnées, ce qui peut être dû à l’absence
totale d’écrantage des charges du polymère, résultant en une chaîne très étendue.
1.3.3.2.2. Courbes d’écoulement du xanthane en solution
Une autre propriété du xanthane est son caractère fortement rhéofluidifiant en solution, et ce,
dès de faibles concentrations en polymère. Ce comportement est illustré sur la figure 1-25 qui
représente des courbes d’écoulement de solutions de xanthane de masse molaire 7*106 g/mol à
différentes concentrations, en présence de 0,1 M de NaCl :
Figure 1-25 : Courbes d’écoulement de solutions de xanthane (Mw = 7*106 g/mol) à différentes concentrations
dans 0,1 M de NaCl170.
Toutes les courbes présentent un plateau newtonien à faible cisaillement, caractérisé par la
viscosité newtonienne η0 . A partir d’une certaine vitesse de cisaillement γ c , dite vitesse de
cisaillement critique, on atteint le régime de rhéofluidification. Ce régime obéit à une loi de puissance
de type ηα γ n (loi de Carreau-Yasuda par exemple), avec n l’indice d’écoulement (avec n ≤ 1).
Dans le cas des faibles concentrations, on observe également, à haute vitesse de cisaillement, la
- 68 -

Chapitre 1 : Bibliographie
présence du second plateau newtonien, de viscosité η∞ , dont la valeur est proche de celle du
solvant utilisé.
On constate que le comportement du xanthane dépend très fortement de sa concentration en
solution. Dans tous les cas, le xanthane présente un caractère rhéofluidifiant, même en régime dilué.
Lorsque la concentration augmente, le deuxième plateau newtonien disparaît pour c > c*, le premier
plateau est de moins en moins visible et γ c diminue. Par ailleurs, le comportement rhéofluidifiant
est de plus en plus marqué. Cette propriété est caractérisée par l’indice d’écoulement, n, dont la
valeur absolue augmente et tend vers 1 lorsque le polymère est de plus en plus rhéofluidifiant. Wyatt
et al.166 obtiennent ainsi des indices n qui passent de -0,08, soit très peu rhéofluidifiant, à -0,62, soit
rhéofluidifiant, lorsque la concentration en xanthane, en présence de 50 mM de NaCl, augmente de
0,1 à 2 g/L. Cette augmentation de la valeur absolue de n avec la concentration est également
observée par Carnali et al.178. Ces derniers trouvent des valeurs de n allant de -0,65 à -0,86 pour des
concentrations en polymère variant de 5 à 30 g/L en présence de 0,02 M de KCl. Cette évolution de n
traduit bien le caractère de plus en plus rhéofluidifiant de ce polymère quand la concentration
augmente. Plus particulièrement, le comportement rhéofluidifiant devient très marqué (n > 0,6)
lorsque le xanthane est en régime semi-dilué enchevêtré (c > c**, c** ≈ 1 g/L)166, 170.
Le comportement rhéofluidifiant est souvent observé pour les solutions de polymères en régime
semi-dilué. Il est dû, le plus souvent, au désenchevêtrement des chaînes sous l’effet du cisaillement.
Dans le cas du xanthane, cependant, le caractère rhéofluidifiant est présent dès le régime dilué 166, 170.
Ce résultat semble indiquer que le comportement de ce polymère en solution n’est pas
complètement, voire pas du tout, régi par de simples enchevêtrements physiques. Par contre, la
rigidité importante du xanthane116 peut être à l’origine de cette forte tendance à la rhéofluidification :
- 69 -

Chapitre 1 : Bibliographie
sous l’effet du cisaillement, les chaînes semi-rigides s’alignent dans le sens de l’écoulement, ce qui
facilite celui-ci.
La nature des mécanismes responsables des propriétés en écoulement du xanthane fait l’objet
d’interrogations dans la littérature. Le comportement en solution de ce polysaccharide obéit-il au
modèle des simples enchevêtrements physiques ou bien d’autres interactions, plus spécifiques,
gouvernent-elles les propriétés ? Certains auteurs ont tenté de répondre à cette question en utilisant
le principe de superposition de Cox-Merz. Ce principe empirique énonce que les courbes, viscosité de
cisaillement η en fonction de la vitesse de cisaillement γ , et viscosité complexe η* en fonction de
la pulsation ω, se superposent lorsque le polymère n’est soumis qu’à des enchevêtrements. Au
contraire, si l’organisation des chaînes en solution résulte d’interactions particulières, la superposition
des courbes n’est alors plus possible. Dans le cas du xanthane, des résultats contradictoires sont
décrits dans la littérature pour des solutions de xanthane en régime semi-dilué enchevêtré. Wyatt et
al.166 constatent que la loi de Cox-Merz est respectée sur toute la gamme de fréquences/cisaillements
étudiée pour des solutions de xanthane en l’absence de sel. Carnali et al.178 obtiennent également
une bonne superposition de leurs données concernant des solutions de xanthane sous forme
ordonnée. Ces résultats semblent indiquer que les chaînes de xanthane forment un réseau
enchevêtré en solution, sans interaction spécifique. A l’inverse, Ross-Murphy et al.179, Frangou et al.180
et Santore et al.181 remarquent que la loi de Cox-Merz n’est pas applicable, quel que soit l’échantillon
de xanthane étudié (natif ou renaturé). Ils en déduisent que le comportement en écoulement du
xanthane s’explique par l’existence d’interactions non covalentes en solution, et non pas par les seuls
enchevêtrements physiques. Ces interactions sont faibles, et de ce fait facilement détruites sous
l’effet du cisaillement, d’où le caractère fortement rhéofluidifiant du xanthane. D’autres auteurs 116, 162,
182 constatent cependant que la loi de Cox-Merz peut être appliquée aux faibles vitesses de
cisaillements/fréquences. Choppe et al.182 ont ainsi montré que cette loi est respectée pour le plateau
newtonien et jusqu’à quatre décades de vitesses de cisaillement au-delà de γ . A haut cisaillement
par contre, la viscosité complexe devient supérieure à celle d’écoulement. Les interprétations
- 70 -

Chapitre 1 : Bibliographie
concernant ces données sont néanmoins contraires. Pour Milas et al.162, un tel résultat signifie que le
comportement en écoulement du xanthane est uniquement régi par de simples enchevêtrements.
Choppe et al.182, par contre, considèrent que les chaînes en solution forment un réseau transitoire
gouverné par des interactions particulières. Quant à Pelletier et al.116, ils constatent que le non-
respect de la loi de Cox-Merz à haute vitesse de cisaillement est en accord avec le modèle développé
par Kuzuu et Doi183 sur les propriétés en écoulement de solutions concentrées de polymères rigides.
C’est pourquoi ils en concluent que le comportement rhéofluidifiant du xanthane est dû à sa forte
rigidité.
D’autres paramètres influencent aussi le comportement en écoulement du xanthane : la
température, la force ionique du milieu ou encore la forme native ou renaturée des chaînes.
L’influence de la force ionique sur les propriétés en écoulement du xanthane varie suivant le
régime de concentration du polymère184, 185. En effet, en milieu dilué et semi-dilué non enchevêtré, la
viscosité diminue lorsque la concentration en sel augmente. De plus, la vitesse de cisaillement
critique γ c devient plus élevée avec la concentration en sel. Wyatt et al.184 constatent ainsi qu’en
régime dilué, le xanthane a un comportement newtonien en présence de sel, alors qu’il est
rhéofluidifiant à force ionique nulle. Ce résultat s’explique par l’écrantage des charges négatives
portées par le polymère. Ce phénomène entraîne une contraction de son volume en solution, comme
vu précédemment et, de ce fait, une viscosité plus faible et des propriétés rhéofluidifiantes moins
marquées pour une même concentration en macromolécule. Pour autant, cette dépendance à la
concentration en sel est limitée : le comportement en écoulement n’évolue plus à partir d’une force
ionique limite, de l’ordre de 0,01 M184, 185qui correspond à la quantité de sel nécessaire pour stabiliser
la forme ordonnée109 via un écrantage total des charges. En régime semi-dilué enchevêtré par contre,
la viscosité augmente avec la force ionique, alors que γ c diminue186. Les auteurs en déduisent que
l’ajout de sel, en écrantant les charges portées par le xanthane, favorise la formation d’interactions
- 71 -

Chapitre 1 : Bibliographie
non covalentes spécifiques entre les chaînes, ce qui renforce le réseau intermoléculaire en régime
enchevêtré et résulte en des propriétés rhéofluidifiantes renforcées.
Enfin, la forme native, dénaturée ou renaturée du xanthane impacte directement ses propriétés
en écoulement. Les données de la littérature sont contradictoires. Oviatt et al.187 constatent ainsi que
le comportement en écoulement d’un xanthane commercial avant et après traitement thermique
varie grandement. En particulier, les échantillons renaturés se caractérisent par une viscosité plus
importante, et un comportement rhéofluidifiant accentué par rapport au xanthane non traité
thermiquement. Pour ces auteurs, ces différences s’expliquent par la dénaturation partielle de la
double hélice native lors du chauffage. Elle aboutit à la formation d’un réseau transitoire comportant
davantage de nœuds de jonctions physiques que le réseau « natif », aboutissant à un renforcement
des propriétés rhéofluidifiantes. Milas et al.162, ou encore Santore et al.181, n’observent, au contraire,
aucune différence de comportement entre le xanthane natif, issu d’un moût de fermentation, et sa
conformation renaturée. Ces contradictions peuvent s’expliquer par la nature des xanthanes étudiés
(commercial dans le cas de Oviatt, issu d’un moût pour Milas et Santore) ou encore par les
divergences dans les traitements thermiques subis par les polymères.
- 72 -

Chapitre 1 : Bibliographie
- 73 -
Pour résumer :
Le xanthane est un polysaccharide caractérisé par une viscosité intrinsèque très élevée et un
comportement fortement rhéofluidifiant en régime enchevêtré, ce qui explique ses nombreuses
applications industrielles en tant qu’agent épaississant ou encore agent de texture. Par ailleurs, les
propriétés viscosimétriques de ce polymère dépendent essentiellement de la conformation native,
dénaturée ou renaturée, des chaînes en solution, ainsi que des domaines de concentration et de la
force ionique.
Globalement, la forme dénaturée se caractérise par une viscosité intrinsèque plus importante
que celle des états ordonnés, ainsi que par des propriétés rhéofluidifiantes renforcées en régime
dilué et semi-dilué non enchevêtré. Ces spécificités s’expliquent par le caractère polyélectrolyte du
xanthane en l’absence de sel.
Les formes ordonnées, quant à elles, présentent un comportement rhéofluidifiant très
marqué, qui est accru lorsque la concentration en sel augmente. Bien que les données à ce sujet
soient ambiguës, il semblerait que les propriétés des états natifs et renaturés diffèrent, et ce
d’autant plus que les conditions de dénaturation sont poussées. La forme renaturée se caractérise
ainsi par une viscosité et un caractère rhéofluidifiant plus importants que la forme native.
Enfin, les analyses de viscosimétrie ont mis en évidence qu’en milieu semi-dilué enchevêtré (c
> c*), la conformation ordonnée forme un réseau intermoléculaire majoritairement gouverné par
des interactions non covalentes spécifiques, et non par de simples enchevêtrements physiques,
contrairement à la forme dénaturée. Les données contradictoires de la littérature ne permettent
cependant pas d’affirmer si un tel réseau est présent à la fois pour les formes natives et
renaturées. Néanmoins, l’existence de ces interactions spécifiques semble être à l’origine des
propriétés en écoulement particulières du xanthane, et également de son comportement
viscoélastique.

Chapitre 1 : Bibliographie
1.3.3.3. Propriétés viscoélastiques du xanthane en solution
Plusieurs auteurs se sont intéressés à la caractérisation des propriétés viscoélastiques du
xanthane en solution. Dans la plupart des cas, leurs études se sont focalisées sur l’évolution des
propriétés suivant la température92, 116, 127, 182, 185 ou la concentration en polymère156, 162, 166, 178, 182, 185, 187,
188.
1.3.3.3.1. Influence de la température sur les propriétés viscoélastiques du xanthane
La figure 1-26 représente l’évolution des modules élastique G’ et visqueux G’’ en fonction de la
fréquence, f, à différentes températures (entre 5 et 90°C) pour une solution de xanthane à 10 g/L
sans ajout de sel.
Figure 1-26 : Evolution des modules élastique G’ (symboles noirs) et visqueux G’’ (symboles blancs) en fonction
de la fréquence à différentes températures, solution de xanthane à 10 g/L sans ajout de sel.182.
A basse température, le module élastique G’ est supérieur au module visqueux G’’ sur toute la
gamme de fréquences, ce qui est caractéristique d’un comportement de type solide. Lorsque la
température augmente, les valeurs des deux modules diminuent et leur dépendance en fréquence
augmente. On constate également l’apparition de la fréquence de relaxation, fτ, fréquence en-
- 74 -

Chapitre 1 : Bibliographie
dessous de laquelle le xanthane se comporte comme un liquide viscoélastique (avec G’ < G’’). De
plus, la fréquence de relaxation augmente avec la température, et sort de la gamme mesurable à
70°C. A partir de cette température, le comportement observé est celui d’un liquide de Maxwell, avec
G’ inférieur à G’’ sur toute la gamme de fréquences, et des dépendances en fréquence proches de 2
et 1, respectivement.
A partir de ces données, il est possible d’appliquer le principe d’équivalence temps-température
afin de construire une courbe maîtresse fréquence-température. Ce principe permet de savoir si les
processus de relaxation d’une solution de polymère sont identiques d’une température à une autre.
Une telle construction consiste, dans un premier temps, à fixer une température de référence Tref. Les
valeurs de fréquences et de modules obtenues aux autres températures sont alors multipliées
respectivement par un facteur aT (décalage horizontal) et bT (décalage vertical), de manière à
superposer les courbes associées à celles de référence. La courbe maîtresse ainsi construite permet
de modéliser le comportement du polymère étudié sur une gamme de fréquences plus étendue que
celle accessible expérimentalement. aT et bT représentent respectivement la variation du temps de
relaxation et celle de l’amplitude des modules avec la température. Ce type de construction a été
réalisée par Choppe et al.182, Rochefort et al.185, Pelletier et al.116, ou encore Renou et al.92 pour le
xanthane. La courbe maîtresse résultante de Choppe et al.182 est représentée sur la figure 1-27 :
- 75 -

Chapitre 1 : Bibliographie
Figure 1-27 : Courbe maîtresse fréquence-température pour une solution de xanthane à 10 g/L sans ajout de sel
(Tref = 20°C)182.
A haute fréquence et basse température, le xanthane présente un comportement de type gel
faible, avec G’ supérieur à G’’ et une faible dépendance en fréquence des deux modules (G’ ∝
f0,16). A basse fréquence et haute température, le comportement est celui d’un liquide viscoélastique,
avec G’ inférieur à G’’. De plus, la dépendance en fréquence de ces deux modules est caractérisée par
les lois de puissance suivante : G’ ∝ f2 et G’’ ∝ f1, ce qui est en accord avec celle d’un liquide
de Maxwell. Pelletier et al.116 obtiennent une courbe maîtresse similaire pour une solution de
xanthane à 10 g/L en présence de 8 mM de NaCl, chauffée entre 25 et 80°C. Ces derniers
déterminent notamment qu’à haute fréquence, la dépendance en fréquence des deux modules, très
faible, obéit aux lois de puissance suivantes : G’ ∝ ω0,3 et G’’ ∝ ω0,1. Au contraire, à basse
fréquence, la dépendance en fréquence des deux modules est plus marquée (G’ ∝ ω1,33 et G’’
∝ ω0,75), mais ne correspond pas celle attendue dans le cas d’un liquide de Maxwell, car les
mesures ne sont pas effectuées à des températures suffisamment élevées et/ou des concentrations
suffisamment faibles.
- 76 -

Chapitre 1 : Bibliographie
Rochefort et al.185 ont aussi étudié les propriétés viscoélastiques du xanthane à 5 g/L avec et sans
sel, de 25 à 80°C. En présence de sel, une courbe maîtresse, d’allure semblable à celle de Choppe et
al.182 et de Pelletier et al.116, est décrite. Au contraire, en l’absence de sel, il n’est pas possible de
construire une seule et unique courbe maîtresse. Les courbes ne se superposent pas à partir de 60°C.
Les auteurs attribuent ce phénomène à la transition ordre-désordre du xanthane, qui, dans leur
exemple, survient entre 40 et 60°C. Cette transition entraînerait une modification de la structure de la
solution, et donc des phénomènes de relaxation associés, ce qui expliquerait l’impossibilité
d’appliquer le principe d’équivalence temps-température. En présence de sel par contre, la
température de transition conformationnelle est décalée vers 80°C : la construction de la courbe
maîtresse est alors possible.
Les résultats de Rochefort et al.185, en l’absence de sel, sont cependant en contradiction avec ceux
de Pelletier et al.116, Choppe et al.182, et Renou et al.92 .En effet, ces trois auteurs obtiennent une
unique courbe maîtresse dans des gammes de températures dans lesquelles intervient le passage de
la forme ordonnée à désordonnée. La capacité à réaliser une telle construction indique que les deux
conformations sont régies par les mêmes mécanismes de relaxation. Le non respect du principe de
superposition dans le cas de Rochefort et al.185 peut s’expliquer par leur choix de faire varier bT selon
la température absolue, en accord avec le modèle de l’élasticité caoutchoutique. Or, il est peu
probable que le xanthane obéisse à ce modèle. Par ailleurs, Pelletier et al.116, par exemple, arrivent à
construire leur courbe maîtresse en fixant bT égal à 1, bien que la superposition des G’’ ne soit pas
optimale. Les meilleures superpositions sont obtenues dans le cas de Choppe et al.182 et Renou et
al.92, qui font varier librement bT. Choppe et al.182 remarquent ainsi que la variation de bT avec la
température est sigmoïdale: bT diminue linéairement jusqu’à 40°C environ, soit aux alentours de la
Tm, puis chute plus brutalement ensuite. D’après ce constat, l’élasticité du xanthane ne peut pas être
modélisée par les théories classiques.
- 77 -
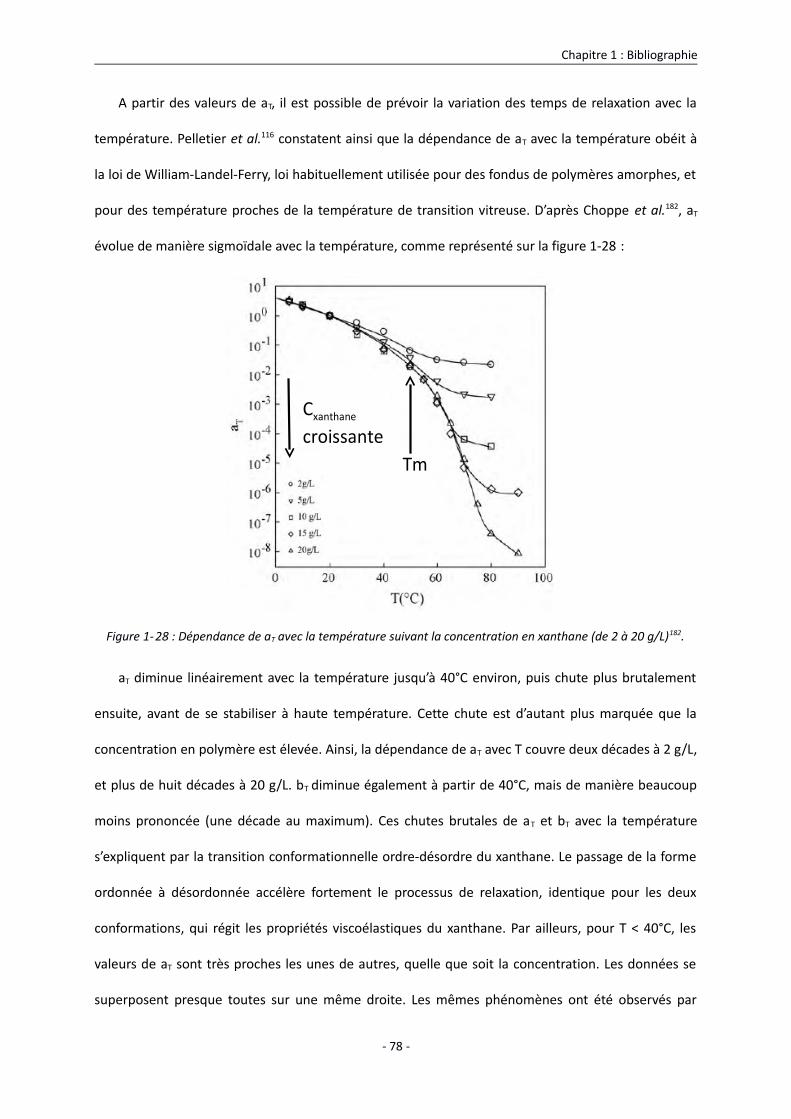
Chapitre 1 : Bibliographie
A partir des valeurs de aT, il est possible de prévoir la variation des temps de relaxation avec la
température. Pelletier et al.116 constatent ainsi que la dépendance de aT avec la température obéit à
la loi de William-Landel-Ferry, loi habituellement utilisée pour des fondus de polymères amorphes, et
pour des température proches de la température de transition vitreuse. D’après Choppe et al.182, aT
évolue de manière sigmoïdale avec la température, comme représenté sur la figure 1-28 :
Figure 1-28 : Dépendance de aT avec la température suivant la concentration en xanthane (de 2 à 20 g/L)182.
aT diminue linéairement avec la température jusqu’à 40°C environ, puis chute plus brutalement
ensuite, avant de se stabiliser à haute température. Cette chute est d’autant plus marquée que la
concentration en polymère est élevée. Ainsi, la dépendance de aT avec T couvre deux décades à 2 g/L,
et plus de huit décades à 20 g/L. bT diminue également à partir de 40°C, mais de manière beaucoup
moins prononcée (une décade au maximum). Ces chutes brutales de aT et bT avec la température
s’expliquent par la transition conformationnelle ordre-désordre du xanthane. Le passage de la forme
ordonnée à désordonnée accélère fortement le processus de relaxation, identique pour les deux
conformations, qui régit les propriétés viscoélastiques du xanthane. Par ailleurs, pour T < 40°C, les
valeurs de aT sont très proches les unes de autres, quelle que soit la concentration. Les données se
superposent presque toutes sur une même droite. Les mêmes phénomènes ont été observés par
- 78 -
Tm
Cxanthane
croissante

Chapitre 1 : Bibliographie
Renou et al.92. Pour T < Tm, il est possible d’appliquer la loi d’Arrhenius et de déterminer une énergie
d’activation Ea pour le système. Choppe et al.182 trouvent ainsi une énergie d’activation Ea de 79
kJ/mol et Renou et al.92 obtiennent Ea = 73 kJ/mol. Pour T > Tm en revanche, il n’est pas possible
d’appliquer la loi d’Arrhenius.
1.3.3.3.2. Influence de la concentration sur les propriétés viscoélastiques du xanthane
Les propriétés viscoélastiques sont aussi fortement influencées par la concentration en polymère,
comme l’illustre la figure 1-29 :
Figure 1-29 : Influence de la concentration sur les propriétés viscoélastiques du xanthane156.
En régime dilué et semi-dilué non enchevêtré, la solution présente un comportement de liquide
viscoélastique sur quasiment toute la gamme de fréquences166. En régime semi-dilué, il est possible
d’observer, à haute fréquence, la fréquence de relaxation ωτ. Cuvelier et al.156 ont remarqué que dans
ces régimes de concentration, ωτ augmente avec la concentration. A l’inverse, en régime concentré, le
comportement bascule de liquide viscoélastique à celui de gel faible. Notamment, ω τ diminue lorsque
la concentration augmente.
- 79 -

Chapitre 1 : Bibliographie
Avec ces données, on peut construire des courbes maîtresses fréquence-concentration. Dans ce
cas, les facteurs de décalage de fréquence et de modules sont appelés respectivement ac et bc. Ce
type de construction a été réalisée par Oviatt et al.187, Milas et al.162, Cuvelier et al.156 ou encore Tam
et al.188.
Comme mentionné plus haut, Cuvelier et al.156 observent une dépendance en concentration de ωτ
différente suivant les régimes de concentration. C’est pourquoi ces auteurs ont construit deux
courbes maîtresses fréquence-concentration distinctes : une correspondant au régime dilué et semi-
dilué non enchevêtré (figure 1-30 a), et l’autre au régime concentré (figure 1-30 b).
Figures 1-30 a : Courbes maîtresses fréquence-concentration du xanthane en régime dilué et semi-dilué non
enchevêtré. b : en régime concentré156.
- 80 -

Chapitre 1 : Bibliographie
Pour c < c**, G’ est inférieur à G’’ sur presque toute la gamme de fréquences. Un résultat
similaire a été obtenu par Carrière et al.189, Wyatt et al.166 ou encore Tam et al.188. De plus, dans ces
régimes de concentration, G’ tend à être proportionnel à ω2 et G’’ à ω 162, 188, ce qui est caractéristique
d’un liquide de Maxwell. Dans ces régimes de concentration, Cuvelier et al.156 constatent que les
facteurs ac et bc obéissent respectivement aux lois de puissance suivantes : ac ∝ c-1 et bc ∝ c-
2,9.
Pour c > c**, le comportement passe de liquide à basse fréquence à celui d’un gel faible à haute
fréquence. Ce phénomène est également observé dans ce régime de concentration par Milas et al.162
et Oviatt et al.187. De plus, la dépendance en concentration des facteurs a c et bc est différente de celle
déterminée en régime semi-dilué non enchevêtré. En effet, en régime concentré, a c est proportionnel
à c7,4, alors que bc reste presque constant et égal à 1.
Pour les auteurs, ces différences dans les lois de puissance signifient que les chaînes en solution
ne sont pas soumises aux mêmes mécanismes de relaxation lorsqu’on passe du régime semi-dilué
non enchevêtré à celui concentré. Pour c* < c < c**, les chaînes se recouvrent progressivement,
résultant en la formation d’un réseau intermoléculaire. Les modules évoluent donc avec la
concentration. A partir de c = c**, par contre, les modules deviennent indépendants de la
concentration (bc ≈ 1). Ce résultat indique que l’ajout de nouvelles chaînes ne crée pas de nouvelles
jonctions, contrairement aux observations pour des polymères obéissant au mécanisme de simples
enchevêtrements physiques. Au contraire, les chaînes ajoutées viennent renforcer les nœuds de
jonction déjà existants, ce qui augmente considérablement leur temps de relaxation et explique la
forte dépendance de ac avec la concentration dans ce régime. Les auteurs en concluent que
l’organisation des chaînes de xanthane en régime concentré ne peut pas être décrite par des
enchevêtrements physiques, mais par des interactions non covalentes spécifiques.
Milas et al.162 se sont également intéressés aux propriétés viscoélastiques du xanthane sous
forme ordonnée dans les trois régimes de concentration. Dans leur cas cependant, ils obtiennent une
- 81 -

Chapitre 1 : Bibliographie
seule et unique courbe maîtresse de 0,2 à 3 g/L. Cette dernière présente un comportement de liquide
visqueux à basse fréquence, et de solide à haute fréquence. De plus, cette courbe maîtresse est
obtenue en fixant bc ∝ c-1, en accord avec le modèle de Rouse pour des systèmes à
enchevêtrements physiques. La même dépendance en concentration de bc est utilisée par Oviatt et
al.187 pour construire des courbes maîtresses fréquence-concentration de 10 à 30 g/L en xanthane en
présence de 0,1 M de NaCl. Milas et al.162 en déduisent que les mécanismes de relaxation du
xanthane restent identiques d’un régime de concentration à l’autre, et s’expliquent par de simples
enchevêtrements, à l’opposé de l’opinion émise par Cuvelier et al.156.
Cette contradiction dans les données peut s’expliquer par la nature différente des échantillons de
xanthane analysés. En effet, Cuvelier et al.156 ont caractérisé les propriétés viscoélastiques d’un
xanthane commercial, auquel ils ont appliqué un cycle de dénaturation-renaturation thermique
poussé (1h à 80°C). Au contraire, l’échantillon de Milas est issu d’un moût de fermentation non traité,
soumis à une dénaturation thermique très brève (1 minute à 80°C). Par conséquent, il est fort
probable que les échantillons de Milas et de Cuvelier aient des conformations très différentes. De
plus, à notre connaissance, Milas et Cuvelier sont les seuls auteurs ayant construit des courbes
maîtresses fréquence-concentration qui balayent tous les régimes de concentration. En règle
générale, les courbes maîtresses décrites dans la littérature concernent soit les milieux dilué et semi-
dilué non enchevêtré188, soit le milieu concentré187. Il est, par conséquent, difficile de conclure sur la
pertinence des conclusions de Milas et de Cuvelier sur l’influence des régimes de concentration sur
les propriétés viscoélastiques du xanthane, en l’absence d’autres points de comparaison.
1.3.3.3.3. Superposition fréquence-température-concentration
A partir des courbes maîtresses fréquence-température à différentes concentrations, il est
possible de construire une courbe maîtresse fréquence-température-concentration92, 182, en
appliquant les facteurs de décalage ac et bc.
- 82 -

Chapitre 1 : Bibliographie
Figure 1-31 : Courbe maîtresse fréquence-température-concentration, avec G’ symboles pleins et G’’ symboles
ouverts. Cref = 10 g/L et Tref = 20°C92.
A hautes fréquences, on retrouve le comportement de type gel faible, et celui de liquide visqueux
à basses fréquences, avec G’ ∝ ω2 et G’’ ∝ ω. Ce type de construction indique que les
processus de relaxation au sein du xanthane sont identiques, quelles que soient la concentration et la
température. D’après Choppe et al.182, la dépendance en concentration de ac suit une loi
exponentielle sur près de huit décades, alors que bc reste proche de 1. La même variation de ac est
constatée par Renou et al.92 Cependant, ces derniers trouvent que bc varie aussi exponentiellement
avec la concentration, mais sur moins d’une décade. D’après ces résultats, G’ et G’’ ne sont pas
proportionnels à c1, phénomène habituellement observé pour des polymères soumis à des
enchevêtrements. Ceci implique que la relaxation est fortement dépendante de la concentration,
alors que les modules sont quasiment insensibles. Ce dernier point corrobore les observations de
Milas et al.162. Par conséquent, les auteurs en déduisent que la dynamique de relaxation du xanthane
est contrôlée par des interactions spécifiques entre chaînes.
- 83 -

Chapitre 1 : Bibliographie
1.3.3.3.4. Modèles d’organisation des chaînes en solution en régime semi-dilué
enchevêtré
Le comportement viscoélastique du xanthane semble fortement dépendre de sa conformation en
solution, bien que les données de la littérature, soient, une fois encore, ambiguës. Certains auteurs
soutiennent que les chaînes ordonnées en solution sont soumises uniquement à des
enchevêtrements, d’autres qu’il existe des interactions spécifiques à l’origine des propriétés
particulières de ce polysaccharide. Sur ce sujet, plusieurs modèles d’organisation des chaînes en
solution ont été proposés afin d’expliquer ces contradictions de la littérature.
Pour Milas et al.162, le xanthane sous forme ordonnée en solution se comporte comme un
polymère soumis uniquement à des contraintes topologiques. Leurs conclusions se basent sur le fait
que leur échantillon obéisse à la loi de Cox-Merz, que la valeur de la pente en régime semi-dilué
enchevêtré respecte la loi de de Gennes, et qu’il soit possible de construire une courbe maîtresse
fréquence-concentration en appliquant le modèle de Rouse (avec bc ∝ c-1). De plus, la courbe
maîtresse obtenue couvre toute la gamme de concentrations allant du régime dilué au régime
enchevêtré. Par ailleurs, ces auteurs ont étudié les propriétés viscoélastiques d’un xanthane natif, issu
d’un moût de fermentation non pasteurisé, mais également renaturé suite à un traitement thermique
à 80°C. Ils constatent que la forme renaturée présente les mêmes caractéristiques rhéologiques que
celle native : sa dynamique est régie par des enchevêtrements physiques. Néanmoins, on peut
souligner que leurs conditions de dénaturation se caractérisent par un temps de chauffage très court
(une minute). Par conséquent, l’état renaturé est probablement très similaire à celui natif, d’où les
similitudes de comportement.
D’autres auteurs ont mis en évidence l’existence d’interactions spécifiques au sein des solutions
de xanthane sous forme ordonnée, en milieu enchevêtré, qui seraient responsables de ses propriétés
rhéologiques particulières. Au regard des données de la littérature, il n’est cependant pas possible
d’affirmer si de telles interactions sont présentes à l’état natif et/ou renaturé. Cette hypothèse se
- 84 -

Chapitre 1 : Bibliographie
fonde sur plusieurs critères : les valeurs des pentes du régime enchevêtré supérieures à la loi de de
Gennes, le non respect de la loi de Cox-Merz, et également la construction de courbes maîtresses
fréquence-température ou fréquence-concentration qui ne se conforment pas au modèle des
polymères à simples enchevêtrements. Plusieurs types d’interactions sont envisagés dans la
littérature : interactions ioniques179, 180, hydrophobes178, 181, ou encore liaisons hydrogène156, 179, 180, 182, 184,
185. Ces dernières ont été mises en évidence par Ross-Murphy et al.179 et Frangou et al.180. Ils ont
étudié le comportement viscoélastique d’un xanthane renaturé, dont une fraction a été traitée
thermiquement en présence d’urée (jusqu’à une concentration de 4 M), un agent dénaturant capable
de détruire les interactions hydrogène. D’après leurs analyses de polarimétrie, l’urée n’empêche pas
la renaturation du xanthane en conformation ordonnée. Néanmoins, la concentration d’urée impacte
fortement les propriétés rhéologiques du polymère. En effet, les solutions de xanthane ordonné
passent d’un comportement rhéofluidifiant à newtonien, avec des modules plus faibles, lorsque la
concentration en urée augmente. De plus, la loi de Cox-Merz devient applicable pour ces systèmes,
contrairement à l’échantillon témoin. Par ailleurs, les propriétés viscoélastiques de type gel,
observables pour le témoin, disparaissent au profit de celles d’un liquide viscoélastique pour une
concentration en urée croissante. D’après ces résultats, les liaisons hydrogène contrôlent donc
majoritairement le comportement rhéologique du xanthane.
L’organisation des chaînes en solution en régime enchevêtré consisterait alors en un réseau
transitoire, constitué des hélices semi-rigides de xanthane arrangées parallèlement les unes aux
autres et en interactions entre elles via des liaisons hydrogène156, 179, 182, 185. Les liaisons hydrogène
peuvent être considérées comme des nœuds de jonction ayant des temps de vie finis. Du fait de leur
énergie moyenne, elles se rompent facilement sous l’effet de la chaleur ou du cisaillement. Cette
caractéristique expliquerait le passage du comportement de type gel faible à liquide viscoélastique
avec l’augmentation de la température, ou encore le caractère rhéofluidifiant du xanthane. De plus,
l’alignement parallèle des hélices de xanthane a sans doute pour conséquence de créer un nombre
fini de nœuds élastiquement actifs. En effet, les chaînes en conformation ordonnée sont rigides : la
- 85 -

Chapitre 1 : Bibliographie
disposition spatiale de tels objets en solution est fortement régie par les contraintes stériques. Les
hélices ne peuvent pas se disposer de manière aléatoire dans l’espace, mais sont sûrement
contraintes à suivre des modèles d’organisation bien définis. Des analyses de diffusion de la lumière,
réalisées par Milas et Rinaudo135 en régime enchevêtré, ont montré que, localement, le réseau
adopte une organisation hexagonale similaire à celles de bâtonnets rigides, ce qui confirmerait le
modèle proposé. Par conséquent, à partir d’une certaine concentration, le réseau est entièrement
formé. Cette concentration critique est probablement la concentration d’enchevêtrement c**, d’où la
différence de dépendance en concentration de bc observée par Cuvelier et al.156 lors du passage du
régime semi-dilué non enchevêtré à celui enchevêtré. L’ajout de nouvelles chaînes ne fait que
renforcer les nœuds déjà existants, et en particulier leur temps de relaxation, d’où la forte
dépendance de ac. La faible variation de bc s’explique par le fait que les chaînes introduites ne créent
pas de nouveaux nœuds (et ne participent donc pas à l’élasticité du réseau), contrairement aux
systèmes à enchevêtrements. C’est pourquoi bc, en régime enchevêtré, n’est pas proportionnel à c -1
(modèle de l’élasticité caoutchoutique).
Le modèle du réseau transitoire permet aussi d’expliquer l’effet de la force ionique sur les
propriétés rhéologiques. En effet, plusieurs études116, 184, 185 montrent que l’augmentation de la
concentration en sel entraîne une amélioration des propriétés de gel faible : la fréquence de
relaxation diminue, alors que les modules augmentent légèrement. L’ajout de sel permet d’écranter
les charges négatives portées par le xanthane, qui ont tendance à déstabiliser le réseau transitoire du
fait des répulsions électrostatiques. L’accroissement de la force ionique renforce à la fois la
conformation ordonnée des chaînes, via l’augmentation de Tm associée, et les liaisons hydrogène
entre les hélices, d’où le décalage de la fréquence de relaxation vers des valeurs plus faibles quand la
concentration en sel augmente. Cet effet de la concentration en sel sur la dynamique de relaxation du
xanthane a notamment été décrit par Choppe et al.182, qui ont pu construire des courbes maîtresses
fréquence-température-force ionique. Ces derniers ont également démontré que les modules varient
relativement peu avec la salinité. Ainsi, la présence de sel n’augmente pas, ou très peu, le nombre de
- 86 -

Chapitre 1 : Bibliographie
nœuds élastiquement actifs. Ici aussi, ce phénomène a sans doute pour cause la forte rigidité des
chaînes ordonnées de xanthane, qui limite le nombre de nœuds de jonction participant au réseau.
De nombreuses études ont également montré que les conditions de dénaturation influencent
fortement les propriétés rhéologiques du xanthane. En effet, la forme renaturée se caractérise le plus
souvent par une viscosité plus importante que la conformation native et des propriétés
rhéofluidifiantes accrues. Plusieurs auteurs126, 127, 187 ont aussi prouvé que le traitement thermique a
pour conséquence de renforcer le comportement de type gel du xanthane en régime concentré.
Oviatt et al.187 parviennent ainsi à construire une courbe maîtresse fréquence-concentration pour des
échantillons de faible masse molaire (Mw < 5*105 g/mol), autoclavés à 120°C pendant 20 minutes,
alors que la même construction est impossible pour les témoins (non chauffés). Ils en concluent que
le cycle de dénaturation-renaturation modifie l’organisation des chaînes en solution, du fait de la
transition ordre-désordre du xanthane. Plus spécifiquement, les conditions thermiques utilisées
entraînent une dénaturation partielle de la conformation double hélice. Lors de la renaturation, les
brins libres se réassocient en double hélice avec leurs plus « proches voisins », qui peuvent être issus
de la même hélice initiale ou d’une hélice environnante (cf. figure 1-20). Ce phénomène aboutit à la
formation d’un réseau en solution où les nœuds de jonctions sont des doubles hélices. Capron et
al.126 et Lee et al.127 attribuent également l’amélioration des propriétés viscoélastiques du xanthane
sous l’effet d’un traitement thermique à la dénaturation partielle des chaînes. Capron et al.126,
néanmoins, considèrent que les nœuds de jonctions consistent en des interactions intermoléculaires
entre les chaînes latérales du xanthane, libérées de leur association avec le squelette lors de la
dénaturation, et non pas en des doubles hélices. Leur hypothèse se base notamment sur le fait que le
réseau formé est facilement détruit sous l’effet du cisaillement. Les interactions stabilisant le réseau
sont donc de faible énergie, ce qui parait difficile à concevoir si le réseau résulte de l’appariement
intermoléculaire de deux brins pour former des doubles hélices. Une autre possibilité est que les
- 87 -

Chapitre 1 : Bibliographie
nœuds de jonction sont bien des doubles hélices, mais constituées d’hélices simples interagissant
latéralement l’une avec l’autre (cf. figures 1-19).
Il faut savoir que très peu d’études viscosimétriques et rhéologiques ont été effectuées sur le
xanthane en conformation désordonnée. Les analyses viscosimétriques de Wyatt et al.184 semblent
indiquer que le réseau formé sous cette conformation est régi uniquement par des contraintes
topologiques. Rochefort et al.185 ont soumis une solution de xanthane à 5 g/L avec et sans sel, à un
fort cisaillement, puis ont étudié la cinétique de retour à l’état d’équilibre. Dans le cas de la solution
contenant du sel, les propriétés viscoélastiques sont identiques avant et après l’application du
cisaillement. Les auteurs en déduisent que, sous forme ordonnée, les chaînes hélicoïdales sont
capables de former un réseau intermoléculaire qui s’auto-répare très rapidement après sollicitation.
Au contraire, lorsque la même analyse est effectuée sur la solution sans sel, ils observent que la
valeur des modules chute de moitié : la structure de la solution est complètement détruite sous
l’effet du cisaillement. Néanmoins, au cours du temps, les modules augmentent progressivement,
sans toutefois revenir à leur valeur initiale. Pour les auteurs, ce résultat signifie que les chaînes sous
forme désordonnée sont capables de reformer un réseau via des interactions intermoléculaires, mais
que la cinétique de réassociation est très lente et incomplète, contrairement au cas de la forme
ordonnée. En particulier, la présence des chaînes latérales chargées en l’absence de sel gênerait
fortement la reformation du réseau du fait des contraintes électrostatiques. L’existence d’interactions
intermoléculaires pour la forme désordonnée semble également confirmée par le fait que Pelletier et
al.116, Choppe et al.182 et Renou et al.92 parviennent à construire des courbes maîtresses fréquence-
température sur une gamme de températures englobant la transition conformationnelle, ce qui laisse
supposer que les mouvements de relaxation des deux formes sont identiques et gérés par les mêmes
processus. Il faut toutefois noter que les expériences menées par ces auteurs et Rochefort et al.185
sont réalisées pour des concentrations en xanthane élevées (entre 5 et 10 g/L). Il est donc possible
que les conformations désordonnées étudiées correspondent à une dénaturation partielle de la
- 88 -

Chapitre 1 : Bibliographie
double hélice, et que les propriétés de l’état complètement dénaturé soit très différentes. A ce jour,
les données de la littérature ne permettent pas de conclure sur ce point.
Pour résumer :
La température a un fort impact sur les propriétés viscoélastiques du xanthane en solution. Il a
ainsi été observé que le comportement rhéologique de ce polysaccharide évolue de gel faible à
liquide viscoélastique lorsque la température augmente. De plus, le principe de superposition temps-
température peut être appliqué sur les données expérimentales, ce qui permet de construire des
courbes maîtresses fréquence-température sur une gamme de fréquences plus étendue que celle
accessible lors de l’analyse. Une telle construction indique que les processus de relaxation au sein des
chaînes sont identiques d’une température à une autre. Plus précisément, il est possible, dans la
majorité des cas92, 116, 182, de construire une unique courbe maîtresse dans les gammes de
températures où se produit la transition ordre-désordre du xanthane. Ce résultat montre que les
deux conformations sont contrôlées par les mêmes mécanismes de relaxation, et que ces derniers ne
sont pas modifiés par le passage de la forme ordonnée à désordonnée. La transition
conformationnelle contribue néanmoins à fortement accélérer les processus de relaxation. Par
ailleurs, pour T < Tm, l’évolution de aT avec la température obéit à une loi arrhénienne, caractérisée
par une unique énergie d’activation Ea de l’ordre de 70-80 kJ/mol92, 182.
Les propriétés rhéologiques du xanthane dépendent également de la concentration en polymère
dans le milieu. En régime dilué et semi-dilué, le comportement observé est celui d’un liquide
viscoélastique, quelle que soit la concentration. En milieu concentré par contre, le comportement
évolue de liquide viscoélastique à gel faible lorsque la concentration augmente. Plusieurs auteurs 156,
162, 187, 188 ont pu construire des courbes maîtresses fréquence-concentration à partir des données
expérimentales. On observe néanmoins des divergences dans les constructions de ces courbes. Milas
et al.162 parviennent, par exemple, à construire une unique courbe maîtresse fréquence-
- 89 -

Chapitre 1 : Bibliographie
concentration pour les trois régimes de concentration, alors que Cuvelier et al.156 obtiennent deux
courbes maîtresses distinctes en régime semi-dilué et en régime concentré.
Ces résultats contradictoires en régime concentré posent la question de l’organisation adoptée
par le xanthane sous forme ordonnée en solution. Selon Milas et al.162, cette organisation est
contrôlée par de simples enchevêtrements topologiques. D’autres auteurs, en revanche, considère
que l’organisation des chaînes est régie par l’existence d’interactions spécifiques non covalentes156, 178,
179, 182, 184, 185, 190. Le modèle le plus communément admis considère que les chaînes sous forme
ordonnée forment un réseau transitoire constitué d’hélices semi-rigides arrangées parallèlement les
unes aux autres et en interaction entre elles via des liaisons hydrogène156, 179, 182, 185. Ces dernières
peuvent être considérées comme des nœuds de jonction de temps de vie fini.
1.3.4. Modification chimique du xanthane
Le xanthane a fait l’objet de plusieurs études visant à améliorer ses propriétés en solution ou à lui
en conférer de nouvelles afin d’élargir sa gamme d’application. Les stratégies employées pour
atteindre ces objectifs consistent à mélanger le xanthane avec d’autres composés, notamment des
polymères, ou bien à modifier chimiquement les chaînes de ce polysaccharide. Ces modifications
peuvent consister à faire varier les taux d’acétate et/ou de pyruvate112, 155, 169, 191, 192, ôter des sous-
unités de la chaîne latérale194, ou encore greffer des composés sur les fonctions hydroxyle194-210 ou
acide carboxylique211-213 du polymère. Dans le cadre de notre étude, nous nous intéressons à cette
dernière catégorie de modification. En règle générale, les greffons introduits ont pour rôle soit
d’apporter directement de nouvelles propriétés au polysaccharide, du fait de leur nature (greffon
hydrophobe, ou ayant une activité biologique spécifique par exemple), soit de permettre la
réticulation chimique du xanthane afin de créer un hydrogel.
- 90 -

Chapitre 1 : Bibliographie
A ce jour, la plupart des travaux réalisés sur la modification chimique du xanthane ciblent comme
site de greffage les fonctions hydroxyle de ce polymère194-210. Les objectifs visés sont majoritairement
la mise au point de dérivés pouvant piéger des polluants organiques195 ou des métaux196, 197, ou bien
l’élaboration d’un polymère à application médicale et pharmaceutique200, 202-204, 209, 210, 214. Parmi ces
études, on peut citer celle de Dumitriu et al.202 et de Mendes et al.203. Les premiers s’intéressent à la
mise au point d’une matrice de rétention de molécules thérapeutiques. Dans leur cas, ils greffent le
principe actif, à savoir l’acide nalidixique (agent antiseptique et désinfectant) sur le xanthane via une
réaction d’estérification dans le DMSO. Cet acide peut être par la suite facilement libéré par hydrolyse
acide, dans des conditions de pH compatibles avec celles de l’estomac. Mendes et al.203 parviennent à
synthétiser un xanthane amphiphile via le greffage de l’acide palmitique, un acide gras comportant 16
carbones, sur les fonctions alcool du xanthane. En solution, dans des conditions de pH et de force
ionique physiologique, ce polysaccharide possède la particularité de s’auto-assembler et de former
des microcapsules capables d’emprisonner des cellules de cartilage, tout en assurant leur survie et
leur prolifération.
Certaines modifications effectuées sur les fonctions alcool du xanthane ont pour but d’introduire
des groupements pour réticuler chimiquement le polysaccharide, et créer ainsi un hydrogel204-210, 215.
Un des premiers exemples d’hydrogels est celui synthétisé par Dumitriu et al.204 : les chaînes de
xanthane sont réticulées chimiquement via de l’épichlorhydrine, un agent de réticulation également
employé par Alupei et al.205. Ce composé présente néanmoins l’inconvénient d’être toxique, ce qui
limite grandement les domaines d’utilisation possibles de ces gels. Pour lever cette difficulté,
plusieurs solutions, utilisant des produits non dangereux pour l’homme et/ou l’environnement, ont
été proposées dans la littérature. Certains auteurs207, 208, 215 choisissent ainsi de greffer des dérivés
acrylates sur le xanthane, afin d’introduire des insaturations pouvant être réticulées par voie
radicalaire. D’autres209, 210 emploient le trimétaphosphate de sodium (STMP), un agent de réticulation
non toxique. La plupart des gels ainsi synthétisés ont des applications potentielles comme matrice de
rétention de principes actifs207-209, 215.
- 91 -

Chapitre 1 : Bibliographie
En contrepartie, très peu d’études se sont focalisées sur la modification chimique du xanthane au
niveau de ses fonctions acide carboxylique. A notre connaissance, Arimura et al.211, Bejenariu et al.212
et Mendes et al.213 sont les seuls auteurs à avoir exploité ces sites de greffage pour modifier le
xanthane. Ainsi, Arimura et al.211 ont greffé de l’amylose sur le xanthane via une synthèse
chemoenzymatique en deux étapes. La première étape consiste en le greffage d’un
maltoolisaccharide sur les acides carboxyliques du xanthane via une réaction de couplage peptidique
avec des carbodiimides. Lors de la deuxième étape, l’amylose est polymérisé enzymatiquement sur le
polysaccharide, le maltoolisaccharide servant de site de fixation. Le polymère obtenu présente la
capacité de former un gel physique dans un liquide ionique et dans l’eau. Bejenariu et al.212 créent un
hydrogel de xanthane via une réaction de couplage peptidique par les carbodiimides entre les
fonctions acide carboxylique du polymère et l’acide adipique dihydrazide comme agent de
réticulation. Dans leurs conditions de synthèse (milieu acide, 90°C, 10 -3 M de LiNO3, concentration en
polysaccharide de 25 g/L), les chaînes de xanthane sont sous forme ordonnée. Dans ce cas
également, l’hydrogel synthétisé interagit avec des espèces chargées positivement. Mendes et al.213,
quant à eux, ont greffé un phospholipide comportant deux chaînes hydrophobes de 17 carbones sur
les acides carboxyliques du xanthane par du couplage peptidique en présence de carbodiimide. Leurs
analyses de dichroïsme circulaire indiquent que les chaînes de xanthane restent sous forme
hélicoïdale même après modification. Tout comme pour le xanthane amphiphile modifié au niveau
des fonctions alcool, ce polymère est également capable, dans des conditions physiologiques, de
former des microcapsules pouvant contenir et maintenir vivantes des cellules de cartilage.
- 92 -

Chapitre 1 : Bibliographie
- 93 -
Pour résumer :
Il existe dans la littérature plusieurs exemples de modifications chimiques du xanthane via le
greffage d’espèces chimiques sur son squelette. La plupart des modifications sont réalisées pour des
objectifs d’applications médicales et pharmaceutiques. L’obtention d’hydrogels chimiques de
xanthane s’inscrit, par exemple, dans cette perspective. Bien qu’intéressant, ce type de système
n’entre cependant pas dans le cadre de nos travaux.
On note que la majorité des greffages s’effectue sur les fonctions alcool du polymère, et non pas
sur ses fonctions acide carboxylique. Or, ces dernières, beaucoup moins nombreuses, permettent un
meilleur contrôle de la régiosélectivité des réactions et, par conséquent, des propriétés apportées.
De plus, il a été indiqué précédemment dans le manuscrit que la conformation adoptée par les
chaînes de xanthane en solution affecte fortement son comportement. Il est donc probable que les
propriétés des xanthanes modifiés soient également influencées par ce paramètre. Pour autant, à
l’exception de Mendes et al.213 et Bejenariu et al.212, aucun auteur ne mentionne la conformation des
chaînes de xanthane modifié en solution. Plus précisément, il n’existe aucune étude s’intéressant à
l’impact de la conformation du xanthane greffé sur ses propriétés, en particulier rhéologiques.
Par ailleurs, la plupart des chaînes greffées sur le xanthane, hors le cas des hydrogels, sont soit
hydrophiles, soit constituées de courts segments hydrophobes. Mendes et al.203, 213 sont les seuls
auteurs, à notre connaissance, à avoir greffé des chaînes hydrophobes suffisamment longues sur le
xanthane pour que ce dernier présente un comportement amphiphile. Néanmoins, les propriétés
rhéologiques de ces xanthanes n’ont pas été étudiées.
Au regard de ces conclusions, il apparaît donc que la caractérisation des propriétés rhéologiques
de dérivés amphiphiles du xanthane, et l’influence de leur conformation sur le comportement en
solution, n’a, à ce jour, jamais été effectuée.

Chapitre 1 : Bibliographie
1.4. Conclusion
Les polymères amphiphiles flexibles se caractérisent par des propriétés rhéologiques en solution
bien supérieures à leur analogue non modifié. Cette particularité s’explique par l’existence
d’interactions intra et/ou intermoléculaires entre les greffons hydrophobes, qui régissent
l’organisation des chaînes en solution. Il existe dans la littérature de très nombreux exemples de
polymères amphiphiles d’origine synthétique ou naturelle. En particulier, les polysaccharides
amphiphiles, qui appartiennent à cette deuxième catégorie, connaissent un essor important depuis
les travaux de Landoll1, car ils sont biocompatibles, biodégradables et issus de ressources abondantes
et renouvelables. Toutefois, on peut remarquer que la grande majorité des polysaccharides choisis
dans la littérature pour être modifiés de manière hydrophobe adoptent des conformations de type
pelote flexible en solution. Très peu d’auteurs s’intéressent à la modification d’un polymère adoptant
une conformation autre en solution, voire pouvant changer de conformation suivant les conditions
opératoires.
Le xanthane est un polysaccharide hydrophile caractérisé par une transition conformationnelle
ordre-désordre en solution selon la température et la force ionique du milieu. Les chaînes passent
d’une forme hélicoïdale rigide à basse température et haute force ionique, à une conformation plus
flexible à haute température et/ou très faible salinité. Cette transition s’accompagne notamment
d’une modification prononcée des propriétés rhéologiques de ce polymère en solution. Il existe
plusieurs exemples dans la littérature de modifications chimiques du xanthane via le greffage
d’espèces chimiques sur son squelette. Dans la majorité des cas cependant, la modification est
réalisée au niveau des fonctions alcool du polymère. Par comparaison, les groupements acide
carboxylique sont beaucoup moins sujets à greffage, bien qu’ils offrent l’avantage d’être moins
nombreux et d’ainsi assurer un meilleur contrôle de la régiosélectivité des réactions. En outre, il
existe très peu d’exemples de xanthane chimiquement modifié afin de lui conférer des propriétés
amphiphiles203, 213. De plus, le comportement rhéologique de ces systèmes n’a pas été étudié. Par
- 94 -

Chapitre 1 : Bibliographie
ailleurs, aucune des études décrites dans la littérature ne s’intéresse à l’influence de la conformation
du xanthane greffé sur ses propriétés physico-chimiques en solution. Ainsi, l’objectif de ces travaux
est de greffer chimiquement des groupements hydrophobes sur le xanthane afin de le rendre
amphiphile, puis d’étudier l’impact de la conformation adoptée par les chaînes sur le comportement
rhéologique du polysaccharide en solution. Ces travaux ont nécessité, dans un premier temps, de
mettre au point deux protocoles pour greffer à façon des chaînons hydrophobes sur les fonctions
acide carboxylique du xanthane, sous forme ordonnée et sous forme désordonnée. Cette partie de
nos travaux est décrite dans le chapitre 3 de ce manuscrit. Les propriétés physico-chimiques en
solution des échantillons obtenus ont ensuite été caractérisées par fluorescence et rhéologie. En
particulier, nous avons cherché à relier la conformation adoptée par les chaînes au comportement
observé. Nous avons également étudié l’influence de la densité de greffage, que ce soit à l’état
ordonné ou désordonné, sur les propriétés des xanthanes amphiphiles. Les résultats obtenus sont
présentés dans le chapitre 4 de ce manuscrit.
- 95 -

Chapitre 1 : Bibliographie
Références bibliographiques
1. Landoll, L. M., Nonionic polymer surfactants. Journal of Polymer Science: Polymer ChemistryEdition 1982, 20, 443-455.
2. Milas, M.; Rinaudo, M., Conformational investigation on the bacterial polysaccharide xanthan.Carbohydrate Research 1979, 76, 189-196.
3. Morris, E. R.; Rees, D. A.; Young, G.; Walkinshaw, M. D.; Darke, A., Order-disorder transition for abacterial polysaccharide in solution. A role for polysaccharide conformation in recognition betweenXanthomonas pathogen and its plant host. Journal of Molecular Biology 1977, 110, 1-16.
4. Wever, D. A. Z.; Picchioni, F.; Broekhuis, A. A., Polymers for enhanced oil recovery: A paradigm forstructure-property relationship in aqueous solution. Progress in Polymer Science 2011, 36, 1558-1628.
5. Reuvers, A. J., Control of rheology of water-borne paints using associative thickeners. Progress inOrganic Coatings 1999, 35, 171-181.
6. Anwari, F. M.; Schwab, F. G., Optimizing latex paint rheology with associative thickeners. InPolymers in Aqueous Media, American Chemical Society: 1989; Vol. 223, pp 527-542.
7. Branham, K. D.; Davis, D. L.; Middleton, J. C.; McCormick, C. L., Water-soluble polymers: 59.Investigation of the effects of polymer microstructure on the associative behaviour of amphiphilicterpolymers of acrylamide, acrylic acid and N-[(4-decyl)phenyl]acrylamide. Polymer 1994, 35, 4429-4436.
8. Lacik, I.; Selb, J.; Candau, F., Compositional heterogeneity effects in hydrophobically associatingwater-soluble polymers prepared by micellar copolymerization. Polymer 1995, 36, 3197-3211.
9. Li, M.; Li, G. L.; Zhang, Z.; Li, J.; Neoh, K.-G.; Kang, E.-T., Self-assembly of pH-responsive andfluorescent comb-like amphiphilic copolymers in aqueous media. Polymer 2010, 51, 3377-3386.
10. Biggs, S.; Selb, J.; Candau, F., Copolymers of acrylamideN-alkylacrylamide in aqueous solution: theeffects of hydrolysis on hydrophobic interactions. Polymer 1993, 34, 580-591.
11. Candau, F.; Selb, J., Hydrophobically-modified polyacrylamides prepared by micellarpolymerization. Advances in Colloid and Interface Science 1999, 79, 149-172.
12. McCormick, C. L.; Nonaka, T.; Johnson, C. B., Water-soluble copolymers: 27. Synthesis andaqueous solution behaviour of associative acrylamideN-alkylacrylamide copolymers. Polymer 1988,29, 731-739.
13. Pelletier, S.; Hubert, P.; Lapicque, F.; Payan, E.; Dellacherie, E., Amphiphilic derivatives of sodiumalginate and hyaluronate: synthesis and physico-chemical properties of aqueous dilute solutions.Carbohydrate Polymers 2000, 43, 343-349.
14. Colinet, I.; Dulong, V.; Hamaide, T.; Le Cerf, D.; Picton, L., New amphiphilic modifiedpolysaccharides with original solution behaviour in salt media. Carbohydrate Polymers 2009, 75, 454-462.
15. Desbrières, J.; Martinez, C.; Rinaudo, M., Hydrophobic derivatives of chitosan: Characterizationand rheological behaviour. International Journal of Biological Macromolecules 1996, 19, 21-28.
16. Sinquin, A.; Hubert, P.; Dellacherie, E., Amphiphilic derivatives of alginate: Evidence for intra- andintermolecular hydrophobic associations in aqueous solution. Langmuir 1993, 9, 3334-3337.
- 96 -

Chapitre 1 : Bibliographie
17. Durand, A., Aqueous solutions of amphiphilic polysaccharides: Concentration and temperatureeffect on viscosity. European Polymer Journal 2007, 43, 1744-1753.
18. Zhang, L. M., Cellulosic associative thickeners. Carbohydrate Polymers 2001, 45, 1-10.
19. Chassenieux, C.; Nicolai, T.; Benyahia, L., Rheology of associative polymer solutions. CurrentOpinion in Colloid & Interface Science 2011, 16, 18-26.
20. Alexandridis, P., Amphiphilic copolymers and their applications. Current Opinion in Colloid &Interface Science 1996, 1, 490-501.
21. Winnik, M. A.; Yekta, A., Associative polymers in aqueous solution. Current Opinion in Colloid &Interface Science 1997, 2, 424-436.
22. Berret, J.-F.; Calvet, D.; Collet, A.; Viguier, M., Fluorocarbon associative polymers. Current Opinionin Colloid & Interface Science 2003, 8, 296-306.
23. Petit-Agnely, F.; Iliopoulos, I., Aggregation mechanism of amphiphilic associating polymersstudied by 19F and 13C nuclear magnetic resonance. The Journal of Physical Chemistry B 1999, 103,4803-4808.
24. Bataille, I.; Huguet, J.; Muller, G.; Mocanu, G.; Carpov, A., Associative behaviour ofhydrophobically modified carboxymethylpullulan derivatives. International Journal of BiologicalMacromolecules 1997, 20, 179-191.
25. Volpert, E.; Selb, J.; Candau, F., Influence of the hydrophobe structure on composition,microstructure, and rheology in associating polyacrylamides prepared by micellar copolymerization.Macromolecules 1996, 29, 1452-1463.
26. Desbrieres, J., Autoassociative natural polymer derivatives: The alkylchitosans. Rheologicalbehaviour and temperature stability. Polymer 2004, 45, 3285-3295.
27. Argillier, J. F.; Audibert, A.; Lecourtier, J.; Moan, M.; Rousseau, L., Solution and adsorptionproperties of hydrophobically associating water-soluble polyacrylamides. Colloids and Surfaces A:Physicochemical and Engineering Aspects 1996, 113, 247-257.
28. Volpert, E.; Selb, J.; Candau, F., Associating behaviour of polyacrylamides hydrophobicallymodified with dihexylacrylamide. Polymer 1998, 39, 1025-1033.
29. McCormick, C. L.; Middleton, J. C.; Grady, C. E., Water soluble copolymers: 38. Synthesis andcharacterization of electrolyte responsive terpolymers of acrylamide, N-(4-butyl)phenylacrylamide,and sodium acrylate, sodium-2-acrylamido-2-methylpropanesulphonate or sodium-3-acrylamido-3-methylbutanoate. Polymer 1992, 33, 4184-4190.
30. Maiti, S.; Jayachandran, K. N.; Chatterji, P. R., Probing the association behavior ofpoly(ethyleneglycol) based amphiphilic comb-like polymer. Polymer 2001, 42, 7801-7808.
31. Dulong, V.; Mocanu, G.; Picton, L.; Le Cerf, D., Amphiphilic and thermosensitive copolymers basedon pullulan and Jeffamine®: Synthesis, characterization and physicochemical properties.Carbohydrate Polymers 2012, 87, 1522-1531.
32. Bokias, G.; Hourdet, D.; Iliopoulos, I.; Staikos, G.; Audebert, R., Hydrophobic interactions ofpoly(N-isopropylacrylamide) with hydrophobically modified poly(sodium acrylate) in aqueoussolution. Macromolecules 1997, 30, 8293-8297.
33. Hourdet, D.; Gadgil, J.; Podhajecka, K.; Badiger, M. V.; Brûlet, A.; Wadgaonkar, P. P.,Thermoreversible behavior of associating polymer solutions: Thermothinning versusthermothickening. Macromolecules 2005, 38, 8512-8521.
34. Wang, J. Etude rhéologique du comportement rhéoépaississant d'un polymère amphiphile thèse,Université du Maine, Le Mans, 2008.
- 97 -

Chapitre 1 : Bibliographie
35. Babak, V.; Skotnikova, E.; Pelletier, S.; Hubert, P.; Dellacherie, E., Hydrophobically associatingalginate derivatives: surface tension properties of their mixed aqueous solutions with oppositelycharged surfactants. Journal of Colloid and Interface Science 2000, 225, 505-510.
36. Wang, K. T.; Iliopoulos, I.; Audebert, R., Viscometric behaviour of hydrophobically modifiedpoly(sodium acrylate). Polymer Bulletin 1988, 20, 577-582.
37. Charpentier-Valenza, D.; Merle, L.; Mocanu, G.; Picton, L.; Muller, G., Rheological properties ofhydrophobically modified carboxymethyl celluloses varying by their hydrophili-lipophilic balance.Carbohydrate Polymers 2005, 60, 87-94.
38. Akiyama, E.; Kashimoto, A.; Fukuda, K.; Hotta, H.; Suzuki, T.; Kitsuki, T., Thickening properties andemulsification mechanisms of new derivatives of polysaccharides in aqueous solution. Journal ofColloid and Interface Science 2005, 282, 448-457.
39. Kjøniksen, A.-L.; Beheshti, N.; Kotlar, H. K.; Zhu, K.; Nyström, B., Modified polysaccharides for usein enhanced oil recovery applications. European Polymer Journal 2008, 44, 959-967.
40. Yang, J.-S.; Xie, Y.-J.; He, W., Research progress on chemical modification of alginate: A review.Carbohydrate Polymers 2011, 84, 33-39.
41. Leonard, M.; Rastello de Boisseson, M.; Hubert, P.; Dalençon, F.; Dellacherie, E., Hydrophobicallymodified alginate hydrogels as protein carriers with specific controlled release properties. Journal ofControlled Release 2004, 98, 395-405.
42. Nyström, B.; Kjøniksen, A.-L.; Iversen, C., Characterization of association phenomena in aqueoussystems of chitosan of different hydrophobicity. Advances in Colloid and Interface Science 1999, 79,81-103.
43. Rinaudo, M., New amphiphilic grafted copolymers based on polysaccharides. CarbohydratePolymers 2011, 83, 1338-1344.
44. Duval, C.; Huguet, J.; Muller, G., Self-assembly and hydrophobic clusters of amphiphilicpolysaccharides. Colloids and Surfaces A: Physicochemical Engineering Aspects 2003, 220, 105-115.
45. Henni, W.; Deyme, M.; Stchakovsky, M.; LeCerf, D.; Picton, L.; Rosilio, V. r., Aggregation ofhydrophobically modified polysaccharides in solution and at the air-water interface. Journal of Colloidand Interface Science 2005, 281, 316-324.
46. Simon, S.; Dugast, J. Y.; Le Cerf, D.; Picton, L.; Muller, G., Amphiphilic polysaccharidescharacterization in dilute regime. competition between intramolecular and intermolecularinteractions. Polymer 2003, 44, 7917-7924.
47. Souguir, Z.; Roudesli, S.; Picton, L.; Le Cerf, D.; About-Jaudet, E., Novel cationic and amphiphilicpullulan derivatives I: synthesis and characterization. European Polymer Journal 2007, 43, 4940-4950.
48. Akiyoshi, K.; Kobayashi, S.; Shichibe, S. M., D.; Baudys, M.; Kim, S. W.; Sunamoto, J., Self-assembled hydrogel nanoparticles of cholesterol-bearing pullulan as a carrier of protein drugs:complexation and stabilization of insulin. Journal of Controlled Release 1998, 54, 313-320.
49. Rotureau, E.; Dellacherie, E.; Durand, A., Viscosity of aqueous solutions of polysaccharides andhydrophobically modified polysaccharides: Application of Fedors equation. European Polymer Journal2006, 42, 1086-1092.
50. Vieira, N. A. B.; Moscardini, M. S.; Tiera, V. A. d. O.; Tiera, M. J., Aggregation behavior ofhydrophobically modified dextran in aqueous solution: a fluorescence probe study. CarbohydratePolymers 2003, 53, 137-143.
51. Rouzes, C.; Durand, A.; Leonard, M.; Dellacherie, E., Surface activity and emulsification propertiesof hydrophobically modified dextrans. Journal of Colloid and Interface Science 2002, 253, 217-223.
- 98 -

Chapitre 1 : Bibliographie
52. Aubry, T.; Moan, M., Rheological behavior of a hydrophobically associating water soluble polymer.Journal of Rheology 1994, 38, 1681-1692.
53. Mayol, L.; Biondi, M.; Russo, L.; Malle, B. M.; Schwach-Abdellaoui, K.; Borzacchiello, A.,Amphiphilic hyaluronic acid derivatives toward the design of micelles for the sustained delivery ofhydrophobic drugs. Carbohydrate Polymers 2014, 102, 110-116.
54. Creuzet, C.; Kadi, S.; Rinaudo, M.; Auzély-Velty, R., New associative systems based on alkylatedhyaluronic acid. Synthesis and aqueous solution properties. Polymer 2006, 47, 2706-2713.
55. Pelletier, E.; Hubert, P.; Payan, E.; Marchal, P.; Choplin, L.; Dellacherie, E., Amphiphilic derivativesof sodium alginate and hyaluronate for cartilage repair: rheological properties. Journal of BiomedicalMaterial Research 2001, 54, 102-108.
56. Hassani, L. N.; Hendra, F. d. r.; Bouchemal, K., Auto-associative amphiphilic polysaccharides asdrug delivery systems. Drug Discovery Today 2012, 17, 608-614.
57. Colinet, I.; Dulong, V.; Hamaide, T.; Le Cerf, D.; Picton, L., Unusual rheological properties of newassociative polysaccharide in salt media. Carbohydrate Polymers 2009, 77, 743-749.
58. Rotureau, E.; Leonard, M.; Dellacherie, E.; Durand, A., Amphiphilic derivatives of dextran:Adsorption at air/water and oil/water interfaces. Journal of Colloid and Interface Science 2004, 279,68-77.
59. Supports de présentation de Van Ruymbeke, E., Molecular View on Viscoelasticity. University ofWestern Macedonia, 2009.
60. Perrin, P.; Hourdet, D., Polymères en solution. Techniques de l'ingénieur: traité Plastiques etComposites 1997, A3050, 1-19.
61. Cram, S. L.; Brown, H. R.; Spinks, G. M.; Hourdet, D.; Creton, C., Hydrophobically modifieddimethylacrylamide synthesis and rheological behavior. Macromolecules 2005, 38, 2981-2989.
62. Regalado, E. J.; Selb, J.; Candau, F., Viscoelastic behavior of semidilute solutions of multistickerpolymer chains. Macromolecules 1999, 32, 8580-8588.
63. Candau, F.; Regalado, E. J.; Selb, J., Scaling behavior of the zero shear viscosity of hydrophobicallymodified poly(acrylamide)s. Macromolecules 1998, 31, 5550-5552.
64. Tanaka, R.; Meadows, J.; Phillips, G. O.; Williams, P. A., Viscometric and spectroscopic studies onthe solution behaviour of hydrophobically modified cellulosic polymers. Carbohydrate Polymers 1990,12, 443-459.
65. Petit, F.; Iliopoulos, I.; Audebert, R.; Szönyi, S., Associating polyelectrolytes with perfluoroalkylside chains: Aggregation in aqueous solution, association with surfactants, and comparison withhydrogenated analogues. Langmuir 1997, 13, 4229-4233.
66. Kujawa, P.; Audibert-Hayet, A.; Selb, J.; Candau, F., Rheological properties of multistickerassociative polyelectrolytes in semidilute aqueous solutions. Journal of Polymer Science Part B:Polymer Physics 2004, 42, 1640-1655.
67. Colinet, I.; Dulong, V.; Mocanu, G.; Picton, L.; Le Cerf, D., New amphiphilic and pH-sensitivehydrogel for controlled release of a model poorly water-soluble drug. European Journal ofPharmaceutics and Biopharmaceutics 2009, 73, 345-350.
68. Rubinstein, M.; Semenov, A. N., Thermoreversible gelation in solutions of associating polymers. 2.Linear dynamics. Macromolecules 1998, 31, 1386-1397.
69. Xie, X.; Hogen-Esch, T. E., Copolymers of N,N-dimethylacrylamide and 2-(N-ethylperfluorooctanesulfonamido)ethyl acrylate in aqueous media and in bulk. Synthesis andproperties. Macromolecules 1996, 29, 1734-1745.
- 99 -

Chapitre 1 : Bibliographie
70. Sinquin, A.; Hubert, P.; Marchal, P.; Choplin, L.; Dellacherie, E., Rheological properties of semi-dilute aqueous solutions of hydrophobically modified propylene glycol alginate derivatives. Colloidsand Surfaces A: Physicochemical Engineering Aspects 1996, 112, 193-200.
71. Durand, A.; Dellacherie, E., Aqueous solutions of native and hydrophobically modifiedpolysaccharides: Temperature effect. Biomacromolecules 2006, 7, 958-964.
72. Wang, T. K.; Iliopoulos, I.; Audebert, R., Aqueous-solution behavior of hydrophobically modifiedpoly(acrylic acid). In Water-Soluble Polymers, American Chemical Society: 1991; Vol. 467, pp 218-231.
73. McCormick, C. L.; Morgan, S. E.; Hester, R., D. , Roles of molecular structure and solvation on dragreduction in aqueous solutions. In Water-Soluble Polymers, American Chemical Society: 1991; Vol.467, pp 320-337.
74. Inomata, K.; Takai, T.; Ohno, N.; Yamaji, Y.; Yamada, E.; Sugimoto, H.; Nakanishi, E., Helix-coiltransition and association behavior of water-soluble polypeptides having hydrophobic alkyl sidechains. In Gels: Structures, Properties, and Functions, Springer Berlin Heidelberg: 2009; Vol. 136, pp15-22.
75. Hill, A.; Candau, F.; Selb, J., Aqueous solution properties of hydrophobically associatingcopolymers. In Trends in Colloid and Interface Science V, Steinkopff: 1991; Vol. 84, pp 61-65.
76. Noda, T.; Hashidzume, A.; Morishima, Y., Effects of spacer length on the side-chain micellization inrandom copolymers of sodium 2-(acrylamido)-2-methylpropanesulfonate and methacrylatessubstituted with ethylene oxide-based surfactant moieties. Macromolecules 2001, 34, 1308-1317.
77. Tam, K. C.; Farmer, M. L.; Jenkins, R. D.; Bassett, D. R., Rheological properties of hydrophobicallymodified alkali-soluble polymers—effects of ethylene–oxide chain length. Journal of Polymer SciencePart B: Polymer Physics 1998, 36, 2275-2290.
78. Caputo, M.-R.; Selb, J.; Candau, F., Effect of temperature on the viscoelastic behaviour ofentangled solutions of multisticker associating polyacrylamides. Polymer 2004, 45, 231-240.
79. Kujawa, P.; Audibert-Hayet, A.; Selb, J.; Candau, F., Effect of ionic strength on the rheologicalproperties of multisticker associative polyelectrolytes. Macromolecules 2006, 39, 384-392.
80. Rubinstein, M.; Semenov, A. N., Dynamics of entangled solutions of associating polymers.Macromolecules 2001, 34, 1058-1068.
81. Leibler, L.; Rubinstein, M.; Colby, R. H., Dynamics of reversible networks. Macromolecules 1991,24, 4701-4707.
82. Annable, T.; Buscall, R.; Ettelaie, R.; Whittlestone, D., The rheology of solutions of associatingpolymers - comparison of experimental behavior with transient network theory. Journal of Rheology1993, 37, 695-726.
83. Tam, K. C.; Jenkins, R. D.; Winnik, M. A.; Bassett, D. R., A structural model of hydrophobicallymodified urethane-ethoxylate (HEUR) associative polymers in shear flows. Macromolecules 1998, 31,4149-4159.
84. Doi, M.; Edwards, S. F., The Theory of Polymer Dynamics. Clarendon Press: Oxford, 1986.
85. de Gennes, P. G., Reptation of a polymer chain in the presence of fixed obstacles. The Journal ofChemical Physics 1971, 55, 572-579.
86. Müller, M.; Seidel, U.; Stadler, R., Influence of hydrogen bonding on the viscoelastic properties ofthermoreversible networks: analysis of the local complex dynamics. Polymer 1995, 36, 3143-3150.
87. Morris, V. J., Bacterial polysaccharides. In Food Polysaccharides and their applications, secondedition, Stephen, A. M.; Phillips, G. O., Eds. Taylor & Francis Group, LLC: 2006; pp 413-443.
- 100 -

Chapitre 1 : Bibliographie
88. Born, K.; Langendorff, V.; Boulenguer, P., Xanthan. In Polysaccharides I: Polysaccharides fromProkaryotes, Vandamme, E. J.; de Baets, S.; Steinbuchel, A., Eds. WILEY-VCH: 2002; Vol. 5, pp 259-269.
89. Bresolin, T.; Sander, P.; Reicher, F.; Sierakowski, M.; Rinaudo, M.; Ganterb, J., Viscometric studieson xanthan and galactomannan systems. Carbohydrate Polymers 1997, 33, 131-138.
90. Secouard, S.; Grisel, M.; Malhiac, C., Flavour release study as a way to explain xanthan–galactomannan interactions. Food Hydrocolloids 2007, 21, 1237-1244.
91. Desplanques, S.; Grisel, M.; Malhiac, C.; Renou, F., Stabilizing effect of acacia gum on the xanthanhelical conformation in aqueous solution. Food Hydrocolloids 2014, 35, 181-188.
92. Renou, F.; Petibon, O.; Malhiac, C.; Grisel, M., Effect of xanthan structure on its interaction withlocust bean gum: Toward prediction of rheological properties. Food Hydrocolloids 2013, 32, 331-340.
93. Hublik, G., Xanthan. Polymer Science A: Comprehensive Review 2012, 10, 221-229.
94. Garcia-Ochoa, F.; Santos, V. E.; Casas, J. A.; Gomez, E., Xanthan gum: production, recovery, andproperties. Biotechnology Advances 2000, 18, 549-579.
95. Katzbauer, B., Properties and applications of xanthan gum. Polymer Degradation and Stability1998, 59, 81-84.
96. Miles, R. Xanthan gum: another possibly sticky situation?≤ htttp://spendmatters.com/2012/09/24/xanthan-gum-another-possible-sticky-situation/ ≥, dernièreconsultation le 20/01/2015.
97. Jansson, P.; Kenne, L.; Lindberg, B., Structure of the extracellular polysaccharide fromXanthomonas campestri. Carbohydrate Research 1975, 45, 275-282.
98. Melton, L. D.; Mindt, L.; Rees, D. A., Covalent structure of the extracellular polysaccharide fromXanthomonas campestris: evidence from partial hydrolysis studies. Carbohydrate Research 1976, 46,245-257.
99. Torres, L.; Brito, E.; Galindo, E.; Choplin, L., Viscous behavior of xanthan aqueous solutions from avariant strain of Xanthomonas campestri. Journal of Fermentation and Bioengineering 1993, 75, 58-64.
100. Shatwell, K.; Sutherland, I. W.; Dea, I. C. M.; Ross-Murphy, S. B., The influence of acetyl andpyruvate substituents on the helix coil transition behaviour of xanthan. Carbohydrate Research 1990,206, 87-103.
101. Papagianni, M.; Psomas, S. K.; Batsilas, L.; Paras, S. V.; Kyriakidis, D. A.; Liakopoulou-Kyriakides, M., Xanthan production by Xanthomonas Campestri in batch cultures. ProcessBiochemistry 2001, 37, 73-80.
102. Casas, J. A.; Santos, V. E.; Garcia-Ochoa, F., Xanthan gum production under severaloperational conditions: molecular structure and rheological properties. Enzyme and MicrobialTechnology 2000, 26, 282-291.
103. Borges, C. D.; de Paula, R. C. M.; Feitosa, J. P. A.; Vendruscolo, C. T., The influence of thermaltreatment and operational conditions on xanthan produced by X. arboricola pv pruni strain 106.Carbohydrate Polymers 2009, 75, 262-268.
104. Milas, M.; Rinaudo, M., Investigation on conformational properties of xanthan in aqueoussolutions. In Solution Properties of Polysaccharides, American Chemical Society: 1981; Vol. 150, pp25-30.
105. Holzwarth, G., Conformation of the extracellular polysaccharide of Xanthomonas campestris.Biochemistry 1976, 15, 4333-4339.
- 101 -

Chapitre 1 : Bibliographie
106. Matsuda, Y.; Biyajima, Y.; Sato, T., Thermal denaturation, renaturation, and aggregation of adouble-helical polysaccharide xanthan in aqueous solution. Polymer Journal 2009, 41, 526-532.
107. Dentini, M.; Crescenzi, V.; Blasi, D., Conformational properties of xanthan derivatives in diluteaqueous solution. International Journal of Biological Macromolecules 1984, 6, 93.
108. Hacche, L. S. W., G. E.; Brant, D. A., Light-scattering investigation of the temperature-drivenconformation change in xanthan. Macromolecules 1987, 20, 2179-2187.
109. Lecourtier, J.; Chauveteau, G.; Muller, G., Salt-induced extension and dissociation of a nativedouble-stranded xanthan. International Journal of Biological Macromolecules 1986, 8, 306-310.
110. Liu, W.; Sato, T.; Norisuye, T.; Fujita, H., Thermally induced conformational change of xanthanin 0.01M aqueous sodium chloride. Carbohydrate Research 1987, 160, 267-281.
111. Norton, I. T.; Goodall, D. M.; Frangou, S. A.; Morris, E. R.; Rees, D. A., Mechanism anddynamics of conformational ordering in xanthan polysaccharide. Journal of Molecular Biology 1984,175, 371-394.
112. Holzwarth, G.; Ogletree, J., Pyruvate-free xanthan. Carbohydrate Research 1979, 76, 277-280.
113. Foss, S., Thermal stability and chain conformational studies of xanthan at different ionicstrengths. Carbohydrate Polymers 1987, 7, 421-433.
114. Paoletti, S.; Cesaro, A.; Delben, F., Thermally induced conformational transition of xanthanpolyelectrolyte. Carbohydrate Research 1983, 123, 173-178.
115. Agoub, A. A.; Smith, A. M.; Giannouli, P.; Richardson, R. K.; Morris, E. R., "Melt-in-the-mouth"gels from mixtures of xanthan and konjac glucomannan under acidic conditions: A rheological andcalorimetric study of the mechanism of synergistic gelation. Carbohydrate Polymers 2007, 69, 713-724.
116. Pelletier, E.; Viebke, C.; Meadows, J.; Williams, P. A., A rheological study of the order-disorderconformation transition of xanthan gum. Biopolymers 2001, 59, 339-346.
117. Gamini, A.; de Bleijser, J.; Leyte, J. C., Physico-chemical properties of aqueous solutions ofxanthan: An n.m.r. study. Carbohydrate Research 1991, 220, 33-47.
118. Christensen, B. E.; Smidsrød, O.; Elgsaeter, A.; Stokke, B. T., Depolymerization of double-stranded xanthan by acid hydrolysis: characterization of partially degraded double strands and single-stranded oligomers released from the ordered structures. Macromolecules 1993, 26, 6111-6120.
119. Coviello, T.; Kajiwara, K.; Burchard, W.; Dentini, M.; Crescenzi, V., Solution properties ofxanthan. 1. Dynamic and static light scattering from native and modified xanthans in dilute solutions.Macromolecules 1986, 19, 2826-2831.
120. Muller, G.; Lecourtier, J.; Allain, C., Conformation of the xanthan molecule in an orderedstructure. Makromolecular Chemistry, Rapid Communication 1984, 5, 203-208.
121. Paradossi, G.; Brant, D. A., Light scattering study of a series of xanthan fractions in aqueoussolution. Macromolecules 1982, 15, 874-879.
122. Camesano, T. A.; Wilkinson, K. J., Single molecule study of xanthan conformation using atomicforce microscopy. Biomacromolecules 2001, 2, 1184-1191.
123. Capron, I.; Alexandre, S.; Muller, G., An atomic force microscopy study of the molecularorganisation of xanthan. Polymer 1998, 39, 5725-5730.
124. Milas, M.; Rinaudo, M.; Tinland, B.; de Murcia, G., Evidence for a single stranded xanthanchain by electron microscopy. Polymer Bulletin 1988, 19, 567-572.
- 102 -

Chapitre 1 : Bibliographie
125. Stokke, B. T.; Elgsaeter, A.; Smidsrod, O., Electron microscopic study of single-and double-stranded xanthan. International Journal of Biological Macromolecules 1986, 8, 217-225.
126. Capron, I.; Brigand, G.; Muller, G., Thermal denaturation and renaturation of a fermentationbroth of xanthan: rheological consequences. International Journal of Biological Macromolecules1998, 23, 215-225.
127. Lee, H.-C.; Brant, D. A., Rheology of concentrated isotropic and anisotropic xanthan solutions:3. Temperature dependence. Biomacromolecules 2002, 3, 742-753.
128. Callet, F.; Milas, M.; Rinaudo, M., On the role of thermal treatments on the properties ofxanthan solutions. Carbohydrate Polymers 1989, 11, 127-137.
129. Kawakami, K.; Okabe, Y.; Norisuye, T., Dissociation of dimerized xanthan in aqueous solution.Carbohydrate Polymers 1990, 14, 189-203.
130. Milas, M.; Rinaudo, M., Properties of xanthan gum in aqueous solutions: Role of theconformational transition. Carbohydrate Research 1986, 158, 191-204.
131. Moorhouse, R.; Walkinshaw M, D.; Arnott, S., Xanthan Gum: Molecular conformation andinteractions. In Extracellular Microbial Polysaccharides, American Chemical Society: 1977; Vol. 45, pp90-102.
132. Okuyama, K.; Arnott, S.; Moorhouse, R.; Walkinshaw M, D.; Atkins E. D, T.; Wolf-Ullish, C. H.,Fiber diffraction studies of bacterial polysaccharides. In Fiber Diffraction Methods, American ChemicalSociety: 1980; Vol. 141, pp 411-427.
133. Jones, S. A.; Goodall, D. M.; Cutler, A. N.; Norton, I. T., Application of conductivity studies andpolyelectrolyte theory to the conformation and order-disorder transition of xanthan polysaccharide.European Biophysics Journal 1987, 15, 185-191.
134. Milas, M.; Reed, W. F.; Printz, S., Conformations and flexibility of native and renaturedxanthan in aqueous solutions. International Journal of Biological Macromolecules 1996, 18, 211-221.
135. Milas, M.; Rinaudo, M.; Duplessix, R.; Borsali, R.; Lindner, P., Small angle neutron scatteringfrom polyelectrolyte solutions: From disordered to ordered xanthan chain conformation.Macromolecules 1995, 28, 3119-3124.
136. Bezemer, L.; Ubbink, J.; de Kooker, J.; Kuil, M.; Leyte, J., On the conformational transition ofnative xanthan. Macromolecules 1993, 26, 6436-6446.
137. Capron, I.; Brigand, G.; Muller, G., About the native and renatured conformation of xanthanexopolysaccharide. Polymer 1997, 38, 5289-5295.
138. Muller, G.; Lecourtier, J., Temperature-induced extension and dissociation of native xanthan.Carbohydrate Polymers 1988, 9, 213-225.
139. Liu, W.; Norisuye, T., Thermally induced conformation change of xanthan: interpretation ofviscosity behavior in 0.01M aqueous sodium chloride. International Journal of BiologicalMacromolecules 1988, 10, 44-50.
140. Stokke, B. T.; Christensen, B. E., Release of disordered xanthan oligomers upon partial acidhydrolysis of double-stranded xanthan. Food Hydrocolloids 1996, 10, 83-89.
141. Christensen, B.; Smidsrod, O., Hydrolysis of xanthan in dilute acid: Effects on chemicalcomposition, conformation, and intrinsic viscosity. Carbohydrate Research 1991, 214, 55-69.
142. Christensen, B. E.; Myhr, M. H.; Smidsrød, O., Degradation of double-stranded xanthan byhydrogen peroxide in the presence of ferrous ions: comparison to acid hydrolysis. CarbohydrateResearch 1996, 280, 85-99.
- 103 -

Chapitre 1 : Bibliographie
143. Sato, T.; Norisuye, T.; Fujita, H., Double-stranded helix of xanthan: dimensional andhydrodynamic properties in 0.1M aqueous sodium chloride. Macromolecules 1984, 17, 2696-2700.
144. Hatakenaka, K.; Liu, W.; Norisuye, T., Stability of xanthan in aqueous sodium chloride atelevated temperature. International Journal of Biological Macromolecules 1987, 9, 346-348.
145. Berth, G.; Dautzenberg, H.; Christensen, B.; Harding, S.; Rother, G.; Smidsrød, O., Static lightscattering studies on xanthan in aqueous solutions. Macromolecules 1996, 29, 3491-3498.
146. Esquenet, C.; Buhler, E., Aggregation behavior in semidilute rigid and semirigidpolysaccharide solutions. Macromolecules 2002, 35, 3708-3716.
147. Southwick, J.; Lee, H.; Jamieson, A.; Blackwell, J., Self-association of xanthan in aqueous-solvent systems. Carbohydrate Research 1980, 84, 287-295.
148. Meyer, E.; Fuller, G.; Clark, R.; Kuliche, W., Investigation of xanthan gum solution behaviorunder shear flow using rheooptical techniques. Macromolecules 1993, 26, 504-511.
149. Holzwarth, G., Molecular weight of xanthan polysaccharide. Carbohydrate Research 1978, 66,173-186.
150. Dintzis, F. R.; Babcock, G. E.; Tobin, R., Studies on dilute solutions and dispersion of thepolysaccharide from Xanthomonas campestris NRRL B-1459. Carbohydrate Research 1970, 13, 257-267.
151. Milas, M.; Tinland, B., Behaviour of xanthan in cadoxen. Carbohydrate Polymers 1990, 13, 47-56.
152. Milas, M.; Rinaudo, M., On the existence of two different secondary structures for thexanthan in aqueous solutions. Polymer Bulletin 1984, 12, 507-514.
153. Bordi, F.; Cametti, C.; Paradossi, G., Conformational changes of xanthan in salt free aqueoussolutions: a low frequency electrical conductivity study. Journal of Physical Chemistry 1996, 100,7148-7154.
154. Muller, G.; Aurhourrache, M.; Lecourtier, J.; Chauveteau, G., Salt dependence of theconformation of a single-stranded xanthan. International Journal of Biological Macromolecules 1986,8, 167-172.
155. Smith, I. H.; Symes, K. C.; Lawson, C. J.; Morris, E. R., Influence of the pyruvate content ofxanthan on macromolecular association in solution. International Journal of BiologicalMacromolecules 1981, 3, 129-134.
156. Cuvelier, G.; Launay, B., Concentration regimes in xanthan gum solutions deduced from flowand viscoelastic properties. Carbohydrate Polymers 1986, 6, 321-333.
157. Utracki, L.; Simha, R., Corresponding state relations for the viscosity of moderatelyconcentrated polymer solutions. Journal of Polymer Science Part A: General Papers 1963, 1, 1089-1098.
158. Launay, B.; Cuvelier, G.; Martinez-Reyes, S., Viscosity of locust bean, guar and xanthan gumsolutions in the Newtonian domain: a critical examination of the log ([eta]sp)o-log c[[eta]]o mastercurves. Carbohydrate Polymers 1997, 34, 385-395.
159. Morris, E. R.; Cutler, A. N.; Ross-Murphy, S. B.; Rees, D. A.; Price, J., Concentration and shearrate dependence of viscosity in random coil polysaccharide solutions. Carbohydrate Polymers 1981, 1,5-21.
160. Pereira, M. C.; Wyn-Jones, E.; Morris, E. R.; Ross-Murphy, S. B., Characterisation of interchainassociation in polysaccharide solutions by ultrasonic relaxation and velocity. Carbohydrate Polymers1982, 2, 103-113.
- 104 -

Chapitre 1 : Bibliographie
161. Robinson, G.; Ross-Murphy, S. B.; Morris, E. R., Viscosity-molecular weight relationships,intrinsic chain flexibility, and dynamic solution properties of guar galactomannan. CarbohydrateResearch 1982, 107, 17-32.
162. Milas, M.; Rinaudo, M.; Knipper, M.; Schuppiser, J. C., Flow and viscoelasticproperties ofxanthan gum solutions. Macromolecules 1990, 23, 2507-2511.
163. Southwick, J.; Brant, D. A., Quasielastic light-scattering studies of xanthan in solution. ACSSymposium Series 1981.
164. Capron, I. Traitement thermique des moûts de fermentation et organisation moléculaire duxanthane, conséquences rhéologiques. thèse, Université de Rouen, Rouen, 1996.
165. Launay, B.; Doublier, J. L.; Cuvelier, G., Flow properties of aqueous solutions and dispersionsof polysaccharides. In Functional Properties of Food Macromolecules, 2nd edition, Hill, S. E.; Mitchell,J. R.; Ledward, D. A., Eds. Springer: 1998; pp 1-78.
166. Wyatt, N. B.; Liberatore, M. W., Rheological and viscosity scaling of the polyelectrolytexanthan gum. Journal of Applied Polymer Science 2009, 114, 4076-4084.
167. Cheetham, N. W. H.; Nik Norma, N. M., The effect of pyruvate on viscosity properties ofxanthan. Carbohydrate Polymers 1989, 10, 55-60.
168. Takada, Y.; Sato, T.; Teramoto, A., Dynamics of stiff-chain polymers in isotropic solution. 2.Viscosity of aqueous solutions of xanthan, a rigid double-helical polysaccharide. Macromolecules1991, 24, 6215-6219.
169. Bradshaw; Nisbet; Kerr; Sutherland, Modified xanthan: its preparation and viscosity.Carbohydrate Polymers 1983, 3, 23-38.
170. Milas, M.; Rinaudo, M.; Tinland, B., The viscosity dependence on concentration, molecularweight and shear rate of xanthan solutions. Polymer Bulletin 1985, 14, 157-164.
171. Callet, F.; Milas, M.; Rinaudo, M., Influence of acetyl and pyruvate contents on rheologicalproperties of xanthan in dilute solution. International Journal of Biological Macromolecules 1987, 9,291-293.
172. de Gennes, P. G., Brownian motions of flexible polymer chains. Nature 1979, 282, 367-370.
173. Arendt, O.; Kuliche, W., Determination of the viscoelastic properties of a homologous seriesof the fermentation polymer xanthan gum. Die Angewandte Makromolekulare Chemie 1998, 259, 61-67.
174. Dobrynin, A. V.; Rubinstein, M., Theory of polyelectrolytes in solutions and at surfaces.Progress in Polymer Science 2005, 30, 1049-1118.
175. Khouryieh, H. A.; Herald, T. J.; Aramouni, F.; Alavi, S., Intrinsic viscosity and viscoelasticproperties of xanthan/guar mixtures in dilute solutions: Effect of salt concentration on the polymerinteractions. Food Research International 2007, 40, 883-893.
176. Lund, T.; Lecourtier, J.; Muller, G., Properties of xanthan solutions after long-term heattreatment at 90°C. Polymer Degradation and Stability 1990, 27, 211-225.
177. Khouryieh, H. A.; Herald, T. J.; Aramouni, F.; Alavi, S., Influence of mixing temperature onxanthan conformation and interaction of xanthan-guar gum in dilute aqueous solutions. FoodResearch International 2006, 39, 964-973.
178. Carnali, J. O., A dispersed anisotropic phase as the origin of the weak-gel properties ofaqueous xanthan gum. Journal of Applied Polymer Science 1991, 43, 929-941.
179. Ross-Murphy, S. B.; Morris, V. J.; Morris, E. R., Molecular viscoelasticity of xanthanpolysaccharide. Faraday Symposia of the Chemical Society 1983, 18, 115-129.
- 105 -

Chapitre 1 : Bibliographie
180. Frangou, S. A.; Morris, E. R.; Rees, D. A.; Richardson, R. K.; Ross-Murphy, S. B., Molecularorigin of xanthan solution rheology: Effect of urea on chain conformation and interactions. Journal ofPolymer Science: Polymer Letters Edition 1982, 20, (10), 531-538.
181. Santore, M. M.; Prud'homme, R. K., Rheology of a xanthan broth at low stresses and strains.Carbohydrate Polymers 1990, 12, 329-335.
182. Choppe, E.; Puaud, F.; Nicolai, T.; Benyahia, L., Rheology of xanthan as a function oftemperature, concentration and ionic strength. Carbohydrate Polymers 2010, 82, 1228-1235.
183. Kuzuu, N. Y.; Doi, M., Nonlinear viscoelasticity of concentrated solution of rod-like polymers.Polymer Journal 1980, 12, 883-890.
184. Wyatt, N. B.; Gunther, C. M.; Liberatore, M. W., Increasing viscosity in entangledpolyelectrolyte solutions by the addition of salt. Polymer 2011, 52, 2437-2444.
185. Rochefort, W. E.; Middleman, S., Rheology of xanthan gum: salt, temperature, and straineffects in oscillatory and steady shear experiments. Journal of Rheology 1987, 31, 337-369.
186. Whitcomb, P. J.; Macosko, C. W., Rheology of xanthan gum. Journal of Rheology 1978, 22,493-505.
187. Oviatt, H. W.; Brant, D. A., Viscoelastic behavior of thermally treated aqueous xanthansolutions in the semidilute concentration regime. Macromolecules 1994, 27, 2402-2408.
188. Tam, K. C.; Tiu, C., Steady and dynamic shear properties of aqueous polymer solutions.Journal of Rheology 1989, 33, 257-280.
189. Carriere, C. J.; Amis, E. J.; Schrag, J. L.; Ferry, J. D., Dilute - solution dynamic viscoelasticproperties of xanthan polysaccharide. Journal of Rheology 1993, 37, 469-478.
190. Frangou, S. A.; Morris, E. R.; Rees, D. A.; Richardson, R. K.; Ross-Murphy, S. B., Molecularorigin of xanthan solution rheology: Effect of urea on chain conformation and interactions. Journal ofPolymer Science: Polymer Letters Edition 1982, 20, 531-538.
191. Tako, M.; Nakamura, S., Rheological Properties of deacetylated xanthan in aqueous media.Agricultural and Biological Chemistry 1984, 48, 2987-2993.
192. Tako, M.; Nakamura, S., Rheological properties of depyruvated xanthan in aqueous solution.Agricultural and Biological Chemistry 1988, 52, 1585-1586.
193. Millane, R. P.; Narasaiah, T. V., X-ray fiber diffraction studies of a variant of xanthan gum inwhich the sidechain terminal mannose unit is absent. Carbohydrate Polymers 1990, 12, 315-321.
194. Su, L.; Ji, W. K.; Lan, W. Z.; Dong, X. Q., Chemical modification of xanthan gum to increasedissolution rate. Carbohydrate Polymers 2003, 53, 497-499.
195. Ghorai, S.; Sarkar, A. K.; Panda, A. B.; Pal, S., Effective removal of Congo red dye from aqueoussolution using modified xanthan gum/silica hybrid nanocomposite as adsorbent. BioresourceTechnology 2013, 144, 485-491.
196. Ghorai, S.; Sinhamahpatra, A.; Sarkar, A.; Panda, A. B.; Pal, S., Novel biodegradablenanocomposite based on XG-g-PAM/SiO2: Application of an efficient adsorbent for Pb2+ ions fromaqueous solution. Bioresource Technology 2012, 119, 181-190.
197. Sand, A.; Yadav, M.; Behari, K., Graft copolymerization of 2-acrylamidoglycolic acid on toxanthan gum and study of its physicochemical properties. Carbohydrate Polymers 2010, 81, 626-632.
198. Behari, K.; Pandey, P. K.; Kumar, R.; Taunk, K., Graft copolymerization of acrylamide ontoxanthan gum. Carbohydrate Polymers 2001, 46, 185-189.
- 106 -

Chapitre 1 : Bibliographie
199. Kumar, I.; Mir, N. A.; Rode, C. V.; Wakhloo, B. P., Intramolecular Huisgen [3+2] cycloaddition inwater: synthesis of fused pyrrolidine–triazoles. Tetrahedron: Asymetry 2012, 23, 225-229.
200. Mundargi, R. C.; Patil, S. A.; Aminabhavi, T. M., Evaluation of acrylamide-grafted-xanthan gumcopolymer matrix tablets for oral controlled delivery of antihypertensive drugs. CarbohydratePolymers 2007, 69, 130-141.
201. Ahuja, M.; Kumar, A.; Singh, K., Synthesis, characterization and in vitro release behavior ofcarboxymethyl xanthan. International Journal of Biological Macromolecules 2012, 51, 1086-1090.
202. Dumitiru, S.; Popa, M.; Dumitriu, M.; Pandele, T., Synthesis and characterization of nalidixicacid conjugate. Journal of Bioactive and Compatible Polymers 1990, 5, 310-315.
203. Mendes, A. C.; Baran, E. T.; Numes, C.; Coimbra, M.; Azevedo, H.; Reis, R. L., Palmitoylation ofxanthan polysaccharide for self-assembly microcapsule formation and encapsulation of cells inphysiological conditions. Soft Matter 2011, 7, 9647-9658.
204. Dumitriu, S.; Dumitriu, M.; Teaca, G., Bioactive polymers 65 - studies of cross-linked xanthanhydrogels as supports in drug retardation. Clinical Materials 1990, 6, 265-276.
205. Alupei, I. C.; Popa, M.; Hamcerencu, M.; Abadie, M. J. M., Superabsorbant hydrogels based onxanthan and poly(vinyl alcohol): 1. The study of the swelling properties. European Polymer Journal2002, 38, 2313-2320.
206. Jampala, S. N.; Manolache, S.; Gunasekaran, S.; Denes, F. S., Plasma-enhanced modification ofxanthan gum and its effect on rheological properties. Journal of Agricultural and Food Chemistry2005, 53, 3618-3625.
207. Hamcerencu, M.; Desbrieres, J.; Popa, M.; Khoukh, A.; Riess, G., New unsaturated derivativesof xanthan gum: Synthesis and characterization. Polymer 2007, 48, 1921-1929.
208. Gils, P. S.; Ray, D.; Sahoo, P. K., Characteristics of xanthan gum-based biodegradablesuperporous hydrogel. International Journal of Biological Macromolecules 2009, 45, 364-371.
209. Shalviri, A.; Liu, Q.; Abdekhodaie, M. J.; Wu, X. Y., Novel modified starch - xanthan gumhydrogels for controlled drug delivery: Synthesis and characterization. Carbohydrate Polymers 2010,79, 898-907.
210. Bejenariu, A.; Popa, M.; Dulong, V.; Picton, L.; Le Cerf, D., Trisodium trimetaphosphatecrosslinked xanthan networks: synthesis, swelling, drug loading and releasing behaviour. PolymerBulletin 2009, 62, 525-539.
211. Arimura, T.; Omagari, Y.; Yamamoto, K.; Kadokawa, J.-i., Chemoenzymatic synthesis andhydrogelation of amylose-grafted xanthan gums. International Journal of Biological Macromolecules2011, 49, 498-503.
212. Bejenariu; Popa, M.; Le Cerf, D.; Picton, L., Stiffness xanthan hydrogels: synthesis, swellingcharacteristics and controlled release properties. Polymer Bulletin 2008, 61, 631-641.
213. Mendes, A. C.; Baran, E. T.; Reis, R. L.; Azevedo, H. S., Fabrication of phospholipid - xanthanmicrocapsules by combining microfluidics with self-assembly. Acta Biomaterialia 2013, 9, 6675-6685.
214. Kumar, A.; Singh, K.; Ahuja, M., Xanthan-g-poly(acrylamide): Microwave-assisted synthesis,characterization and in vitro release behavior. Carbohydrate Polymers 2009, 76, 261-267.
215. Hamcerencu, M.; Desbrieres, J.; Popa, M.; Riess, G. r., Stimuli-sensitive xanthan derivatives/N-isopropylacrylamide hydrogels: Influence of cross-linking agent on interpenetrating polymer networkproperties. Biomacromolecules 2009, 10, 1911-1922.
- 107 -

Chapitre 1 : Bibliographie
- 108 -

Chapitre 2 : Matériels et Méthodes
Chapitre 2 : Matériels et méthodes
2.1. Modification chimique du xanthane 1112.1.1. Réactifs utilisés 1112.1.2. Modification chimique du xanthane sous forme ordonnée 112
2.1.2.1. Processus de modification à température ambiante 1132.1.2.2. Processus de modification à basse température (première étape) 113
2.1.3. Modification chimique du xanthane sous forme désordonnée 114
2.2. Résonance Magnétique Nucléaire (RMN) 1152.2.1. Principes 115
2.2.1.1. Rappels sur le moment magnétique et la précession de Larmor 1152.2.1.2. Phénomènes de résonance et de relaxation 1162.2.1.3. Principe d’une analyse en RMN impulsionnelle 118
2.2.2. Appareil utilisé et préparation des échantillons 1192.2.3. Méthode de détermination des degrés de substitution en acétate et pyruvate et de la densité de greffage 120
2.2.3.1. Détermination des degrés de substitution en acétate et pyruvate 1202.2.3.2. Détermination de la densité de greffage des xanthanes modifiés 122
2.3. Polarimétrie 1232.3.1. Principes généraux 1232.3.2. Présentation du polarimètre 1252.3.3. Activité optique du xanthane 1262.3.4. Appareil utilisé et préparation des échantillons 128
2.4. Fluorescence 1282.4.1. Définitions et principes généraux 1292.4.2. Principe d’une analyse de fluorescence 1302.4.3. Caractéristiques de la sonde de fluorescence ANS 1332.4.4. Appareil utilisé et préparation des solutions 134
2.5. Rhéologie 1352.5.1. Définition et rappels 1352.5.2. Mesures en écoulement 1362.5.3. Mesures dynamiques en solution 1372.5.4. Rhéologie interfaciale 140
2.5.4.1. Définition d’une interface 1402.5.4.2. Principes de la rhéologie interfaciale de cisaillement 141
2.5.5. Rhéomètre utilisé et préparation des échantillons 1442.5.5.1. Préparation des solutions 1442.5.5.2. Analyses en solution 1442.5.5.3. Analyses à l’interface eau/huile 145
Références bibliographiques 149
- 109 -

Chapitre 2 : Matériels et Méthodes
- 110 -

Chapitre 2 : Matériels et Méthodes
Ce chapitre décrit les protocoles de modification développés afin de greffer des chaînons alkyle
hydrophobe sur le xanthane, sous forme ordonnée ou désordonnée. Nous détaillons également ici les
principales techniques de caractérisation physico-chimique utilisées dans cette étude : la RMN du
proton, la polarimétrie, la fluorescence, la rhéologie en solution et interfaciale.
2.1. Modification chimique du xanthane
Cette partie présente succinctement les protocoles de modification développés pour greffer des
chaînons hydrophobes sur le xanthane. Une description plus complète de ces protocoles est
effectuée dans le chapitre 3.
2.1.1. Réactifs utilisés
Le xanthane employé dans cette étude est fourni par Danisco. Cet échantillon, noté XDan1, se
caractérise par un très faible taux en protéine, inférieur à 1 % (déterminé par mesure UV selon la
procédure de Bradford1, protéine utilisée pour la calibration : albumine de sérum bovin2). Les degrés
de substitution en acétate (DSA) et pyruvate (DSP) de ce polysaccharide ont été obtenus par RMN 1H
et sont respectivement de 0,87 et 0,49 (cf. partie 2.2.3). Le taux d’humidité, déterminé par
thermogravimétrie, est de 10,8 %.
Le processus de modification chimique du xanthane, sous forme ordonnée ou désordonnée,
nécessite que ce dernier soit préalablement acidifié. Dans ce but, une solution aqueuse de xanthane
à 5 g/L est préparée par solubilisation de 2,24 g de xanthane dans 400 mL d’eau ultrapure par
agitation mécanique pendant 6 h à température ambiante. Le milieu est ensuite dialysé pendant 24 h
contre de l’eau ultrapure ajustée à pH = 3 avec une solution aqueuse de HCl à 1 M. La solution finale
est qualifiée de mère. Elle est conservée une semaine au maximum à 4°C pour les protocoles de
modification du xanthane sous forme ordonnée, ou lyophilisée pendant 24 h afin être stockée sous
- 111 -

Chapitre 2 : Matériels et Méthodes
forme solide avant d’être resolubilisée dans le solvant adéquat pour la modification du polymère à
l’état désordonné.
Les réactifs employés lors des protocoles de modification du xanthane sont les trois agents de
couplage suivants : 1-éthyl-3-(3-diméthylaminopropyl) carbodiimide (EDAC, Alfa Aesar, 98 %), N-
hydroxysuccinimide (NHS, Acros Organics, 98 %) et hydroxybenzotriazole (HOBt, Acros Organics, 98
%), ainsi que la n-octylamine (Sigma Aldrich, 97 %). Ces produits commerciaux n’ont pas fait l’objet de
purification préliminaire avant utilisation.
Les solvants de réaction sont l’eau ultrapure (grade 1, résistivité de 18,2 MΩ.cm à 25°C) ou le
diméthylsulfoxyde (DMSO, Carlo Erba, 99,8 %). Ce dernier a été employé sans purification préalable.
Les membranes de dialyse CelluSep T2, en cellulose régénérée, ont un diamètre de 25,5 mm et
un cut-off de 6 000 à 8 000 g/mol.
2.1.2. Modification chimique du xanthane sous forme ordonnée
La modification chimique du xanthane sous forme ordonnée s’effectue dans l’eau, en deux
étapes, grâce à une réaction de couplage peptidique entre les fonctions acide carboxylique du
polymère et les groupements amine porteurs des chaînons hydrophobes. La quantité (en mole) des
différents réactifs à introduire dans le milieu est calculée en fonction du nombre de fonctions acide
carboxylique portées par le polysaccharide. Cette quantité est la même pour tous les réactifs autre
que le xanthane. Par exemple, une réaction pour laquelle noctylaminenCOOH
=3 (avec noctylamine nombre de
moles d’octylamine et nCOOH nombre de moles de fonctions acide carboxylique) signifie que le nombre
de moles de chaque réactif est 3 fois supérieur à celui des acides carboxyliques.
- 112 -

Chapitre 2 : Matériels et Méthodes
On distingue deux protocoles de modification sous forme ordonnée : le processus à température
ambiante, qui permet d’obtenir des xanthanes modifiés avec des densités de greffage inférieures à 25
%, et le processus à basse température lors de la première étape pour des densités de greffage
pouvant atteindre près de 40 %.
2.1.2.1. Processus de modification à température ambiante
Le milieu réactionnel est une solution de xanthane acidifié à 1 g/L obtenue par dilution de la
solution mère dans de l’eau ultrapure, puis agitée à température ambiante jusqu’à homogénéisation
totale de la solution. La première étape du procédé consiste en l’ajout, en goutte à goutte rapide, de
l’EDAC puis du NHS tous deux dissous dans de l’eau ultrapure, dans le milieu. Le pH de la solution est
alors ajusté à 4,5 avec une solution aqueuse de HCl 1 M et/ou de NaOH 1 M, et le milieu est laissé
sous agitation à température ambiante pendant 2 h. Lors de la deuxième étape, une solution aqueuse
d’octylamine est introduite en goutte à goutte rapide dans le milieu, puis le pH est ajusté à 10 par
NaOH 1 M. La réaction est finalement laissée sous agitation à température ambiante pendant une
nuit.
Le milieu réactionnel est purifié par dialyses successives contre eau acidifiée à pH = 3 par HCl 1 M
(24 h), puis contre eau salée (0,1 M de NaCl, 24 h), eau distillée (7 jours) et enfin eau ultrapure (24 h).
La solution est ensuite concentrée sous pression réduite à 40°C puis le polymère en solution est
lyophilisé pendant 24 h pour obtenir le produit sous la forme d’un solide blanc.
2.1.2.2. Processus de modification à basse température (première étape)
Ce protocole est une variante du procédé 1, dans lequel un contrôle de la température est
appliqué lors de la première étape de la réaction. Avant l’ajout des agents de couplage, le milieu est
refroidi à une température inférieure à 5°C via un bain de glace. Cette température est maintenue
- 113 -

Chapitre 2 : Matériels et Méthodes
pendant toute cette phase de la réaction. Le bain de glace est ôté lors du début de la deuxième
étape, avant l’ajout de l’octylamine. Le reste du protocole (synthèse, purification et récupération du
polymère) est identique à celui décrit dans le paragraphe précédent.
2.1.3. Modification chimique du xanthane sous forme désordonnée
Le xanthane modifié sous forme désordonnée est également obtenu par une réaction de
couplage peptidique entre les fonctions acide carboxylique du polysaccharide et les groupements
amine. Cependant, dans ce cas, le co-agent de couplage employé est le HOBt, et la réaction se
déroule dans le DMSO. La quantité des différents réactifs (autres que le xanthane) est identique et
calculée par rapport au nombre de fonctions acide carboxylique.
Le milieu réactionnel est obtenu par solubilisation, à 80°C et sous agitation, d’un xanthane
acidifié et lyophilisé (provenant de la solution mère décrite précédemment) dans le DMSO. Une fois
homogène, le milieu est laissé au repos jusqu’à ce que sa température revienne à l’ambiante. Des
solutions de HOBt, puis d’EDAC, dans le DMSO, sont alors introduites en goutte à goutte rapide dans
le milieu. Au bout de deux heures, une solution d’octylamine dans le DMSO est aussi ajoutée de la
même manière dans le milieu. Ce dernier est laissé sous agitation à température ambiante pendant la
nuit.
La purification du milieu réactionnel est réalisée par dialyses successives contre DMSO (24 h), eau
acide à pH = 3/isopropanol 70/30 en volume (24 h), eau salée/isopropanol 70/30 en volume (NaCl 0,1
M, 24 h), DMSO (24 h), eau distillée (7 jours) et ultrapure (24 h). La solution est ensuite concentrée
sous pression réduite à 40°C puis le polymère en solution est lyophilisé pendant 24 h pour obtenir le
produit sous la forme d’un solide blanc.
- 114 -

Chapitre 2 : Matériels et Méthodes
2.2. Résonance Magnétique Nucléaire (RMN)
La Résonance Magnétique Nucléaire (RMN) est une technique spectroscopique non destructive
qui permet de déterminer la structure des molécules en milieu liquide ou solide. Appliquée aux
polymères modifiés, elle permet également, sous certaines conditions, de quantifier la densité de
greffage de ces composés.
2.2.1. Principe s
2.2.1.1. Rappels sur le moment magnétique et la précession de Larmor3
La résonance magnétique nucléaire est un phénomène observable uniquement pour des noyaux
de nombre quantique de spin, ou spin (noté S), non nul. Un noyau peut être modélisé comme une
sphère chargée positivement en rotation sur elle-même. Ce mouvement entraîne l’apparition d’un
champ magnétique local, appelé moment magnétique nucléaire μ . Sous l’effet d’un champ
magnétique externe B0 , ce moment magnétique peut adopter différentes orientations par
rapport à la direction du champ externe appliqué. Le nombre d’orientations possibles de μ est
donné par le spin S. Ainsi, dans le cas de l’hydrogène 1H, de spin S = + ½, on a deux orientations
possibles :
- Une orientation parallèle à B0 , dit état α (de nombre quantique magnétique m = + ½).
- Une orientation antiparallèle à B0 , dit état β (m = - ½).
Chacun de ces états est caractérisé par une énergie, donnée par la relation :
E=−m∗ h∗ ν0 Equation 2-1
Avec h la constante de Planck et ν0 la fréquence de précession de Larmor. En effet, chaque
moment magnétique tourne autour de l’axe du champ externe, selon un mouvement dit de
- 115 -

Chapitre 2 : Matériels et Méthodes
précession. La fréquence de Larmor détermine la vitesse de ce mouvement. Cette fréquence dépend
notamment du noyau étudié et de son environnement électronique.
En l’absence d’un champ magnétique externe, il n’y a pas d’orientation spécifique de μ : les
états sont dits dégénérés (de même énergie). L’application du champ B0 permet de lever la
dégénérescence, comme illustré sur la figure 2-1 :
Figure 2-1 : Levée de dégénérescence des spins de 1H sous l’effet d’un champ magnétique externe (d’après
Akoka3).
L’absorption de photons de fréquence ν0 permet de passer de l’état α (état fondamental) à l’état β
(état excité) : il s’agit du phénomène de résonance.
2.2.1.2. Phénomènes de résonance et de relaxation3, 4
Une molécule comporte plusieurs atomes. Chaque atome de spin non nul possède un moment
magnétique lorsqu’il est soumis à un champ extérieur. On définit l’aimantation, notée M , comme
la somme vectorielle des moments magnétiques de chaque atome constituant l’échantillon étudié. Il
s’agit du paramètre mesuré lors d’une analyse RMN.
- 116 -

Chapitre 2 : Matériels et Méthodes
Soit un échantillon caractérisé en RMN 1H. En l’absence de tout champ magnétique externe, les
moments magnétiques sont orientés de manière aléatoire (cf. figure 2-2 a) : l’aimantation est nulle.
Sous l’effet du champ B0 , ces moments s’alignent parallèlement (état α, m = + ½) ou
antiparallèlement (état β, m = - ½) par rapport au champ (cf. figure 2-2 b). Le nombre de noyaux à
l’état α, (état le plus stable énergétiquement) est supérieur à celui des noyaux à l’état β. Par
conséquent, l’aimantation est non-nulle et dirigée dans la direction du champ B0 (cf. figure 2-2 c).
Par convention, on considère que le champ B0 est dirigé suivant l’axe z, dit axe longitudinal.
Figure 2-2 : Orientation des moments magnétiques nucléaires (S = ½) a : en l’absence de champ externe. b : en
présence d’un champ magnétique externe B0 . c : aimantation résultant de l’orientation des moments
magnétiques dans le champ B0 (d’après Akoka3).
L’aimantation n’est pas observable tant qu’elle est orientée dans le même sens que B0 (cf.
figure 2-3 A). Afin de pouvoir la mesurer, elle est basculée de 90° via une impulsion radiofréquence
dont la composante magnétique est un champ B1 .perpendiculaire à B0 (cf. figure 2-3 B). Par
convention, le plan dans lequel est appliqué B1 est le plan (xy), appelé plan transversal.
- 117 -
m = -1/2
m = +1/2

Chapitre 2 : Matériels et Méthodes
L’impulsion appliquée a une fréquence proche des fréquences de précession des noyaux de
l’échantillon. Elle entraîne donc l’adsorption de photons d’énergie égale à ΔE : les atomes de
l’échantillon entrent en résonance. Lorsque le rayonnement cesse, le système retourne à l’équilibre
en émettant des photons d’énergie ΔE : c’est le phénomène de relaxation. L’aimantation revient
progressivement à sa position d’équilibre, parallèle à B0 , via un mouvement de précession autour
de l’axe longitudinal (cf. figure 2-3 C).
Figure 2-3 : Représentation schématique de l’application d’une radiofréquence B1 sur la magnétisation d’un
échantillon (d’après Maes4).
2.2.1.3. Principe d’une analyse en RMN impulsionnelle
L’objectif de l’analyse RMN est d’accéder à l’aimantation de l’échantillon, afin de remonter à sa
structure. Dans ce but, une expérience RMN impulsionnelle de base comporte les deux phases
suivantes4, 5 :
- Une phase d’excitation, au cours de laquelle une impulsion radiofréquence dont la fréquence
est proche de celles de résonance des noyaux de l’échantillon, est appliquée sur l’échantillon.
Son amplitude et sa durée sont calibrées pour produire un basculement d’aimantation dans
le plan transversal (XY). Cette étape dure généralement quelques microsecondes.
- Une phase de détection pendant laquelle l’aimantation basculée est mesurée.
- 118 -

Chapitre 2 : Matériels et Méthodes
Comme mentionné précédemment, l’aimantation basculée adopte un mouvement de précession
par rapport à l’axe longitudinal, de fréquence ν0. En plaçant une bobine dans un des axes
perpendiculaires au plan transversal, il est possible de mesurer la valeur de M au cours du temps. En
effet, la rotation de M lors de la relaxation génère un flux magnétique périodique à travers la
bobine, ce qui se traduit par un signal électrique de fréquence ν 0 et d’amplitude M. Ce signal est
qualifié de « signal de précession libre » ou FID pour « Free Induction Decay ». En règle générale, il
est constitué de plusieurs fréquences différentes détectées simultanément. Le FID donne, après
traitement mathématique par transformée de Fourier, un spectre RMN résolu en fréquence, dans
lequel chaque pic, ou groupe de pics magnétiquement équivalents, correspond à un atome avec un
environnement chimique particulier.
Il est donc possible, à partir du spectre, d’identifier la connectivité des différents atomes
constitutifs de la molécule, et par conséquent de définir sa structure. De plus, en solution, suivant les
conditions d’analyse, la RMN permet de quantifier le nombre d’atomes magnétiquement équivalents,
via l’intégration des aires situées sous les pics du spectre. Par cette technique, on peut notamment
quantifier la densité de greffage des xanthanes amphiphiles obtenus par modification chimique, ainsi
que les degrés de substitution en acétate et pyruvate.
2.2.2. Appareil utilisé et préparation des échantillons
Les spectres RMN 1H des échantillons ont été enregistrés à 80°C (353,15°K) sur un appareil Brüker
Avance 300 MHz équipé d’une sonde SEI inverse (1H/13C) à haute sensibilité en proton, avec un délai
inter-impulsion d1 de 6 s (soit 5 fois supérieur à T1, temps de relaxation longitudinal ou encore temps
de relaxation spin-réseau, ce qui permet une exploitation quantitative des spectres) et un nombre
d’itérations de 256, permettant d’obtenir un spectre RMN dont le rapport signal/bruit est également
compatible avec une exploitation quantitative. Les déplacements chimiques (δ) sont indiqués en
parties par million (ppm) par rapport au 2.2.3.3-d(4)-3-(Triméthylsilyl)propanoate de sodium (TSP),
- 119 -

Chapitre 2 : Matériels et Méthodes
utilisé comme référence interne (δ = 0 ppm). Le logiciel MestReNova est utilisé pour le traitement des
données. Pour toutes les analyses, des solutions de xanthane (modifié ou non) à 5 g/L dans du D 2O
ont été préparées suivant le protocole suivant.
Il a été observé expérimentalement que la forte viscosité des solutions de xanthane modifié
affecte la relaxation des protons, et rend notamment impossible toute interprétation quantitative des
spectres. C’est pourquoi la viscosité des solutions est diminuée avant analyse par dépolymérisation
par le peroxyde d’hydrogène en milieu alcalin, en accord avec le processus développé par Wu et al6.
NaOH (7,4 μL, 1 M) et H2O2 (25 μL) sont ajoutés à 1,5 mL d’une solution de xanthane à 6 g/L dans du
D2O. La solution est ensuite chauffée sous agitation à 80°C pendant 1 h, puis refroidie dans un bain de
glace pendant 30 minutes afin d’arrêter la dépolymérisation. Une fraction de la solution
dépolymérisée est alors lyophilisée pendant 24 h afin d’obtenir 5 mg de polymère. La poudre est
finalement mise à dissoudre à température ambiante pendant une nuit dans 1 mL de D2O contenant
5*10-3 M de TSP. On conserve ainsi tous les oligomères, monomères et fragments issus de la
dépolymérisation.
2.2.3. Méthode de détermination des degrés de substitution en acétate et pyruvate
et de la densité de greffage
2.2.3.1. Détermination des degrés de substitution en acétate et pyruvate
Le xanthane comporte des groupements acétate et pyruvate sur ses chaînes latérales, en
proportions variables suivant les conditions de fermentation. Ces fonctions comportent chacune trois
protons équivalents. Elles sont observables sur le spectre RMN sous forme de deux singulets bien
distincts : l’un à 1,36 ppm et l’autre à 2,06 ppm, correspondant respectivement aux protons de
l’acétate et du pyruvate7, 8. Le TSP, référence interne, apparaît sous forme d’un singulet normalisé à 0
ppm, qui intègre pour neuf protons. En connaissant la quantité de TSP introduite dans le tube, il est
possible de déterminer la proportion d’acétate et de pyruvate dans le xanthane étudié, via les
- 120 -

Chapitre 2 : Matériels et Méthodes
intégrations des pics associés à ces groupements. Ces intégrations sont déterminées en utilisant
l’algorithme GSD (Global Spectral Deconvolution) fourni par le logiciel MestreNova.
La méthode de quantification des degrés de substitution en acétate et en pyruvate utilisée dans
le cadre de ces travaux a été développée par Esquenet et al9.
Soit A le nombre de moles de groupements acétate par gramme de polymère et P le nombre de
moles de groupements pyruvate par gramme de polymère :
A=I acé tate∗ [TSP ]
I TSP/3∗ [Xanthane ] et P=
I pyruvate∗ [TSP ]
ITSP /3∗ [Xanthane ]Equation 2-2
Avec Iacétate, Ipyruvate et ITSP les intégrations des pics de l’acétate, du pyruvate et du TSP, [TSP] la
concentration de la référence (en mol/L) et [Xanthane] la concentration en g/L.
On définit DSA et DSP, respectivement le degré de substitution en acétate et pyruvate, comme
étant le nombre de groupements acétate/pyruvate par unité de répétition (en pourcentage) :
DSA=A∗M xanthane
1−P∗M pyruvate− A∗M acetate
Equation 2-3
Et DSP=P∗M xanthane
1−P∗M pyruvate− A∗M acetate
Equation 2-4
Avec Mxanthane, Macétate et Mpyruvate les masses molaires respectivement de l’unité de répétition du
xanthane sans acétate ni pyruvate, du groupement acétate et du pyruvate. Dans le cadre de notre
étude, le xanthane employé se caractérise par DSA = 87 % et DSP = 49 %.
Par conséquent, la masse molaire de l’unité de répétition du xanthane non modifié, MUR, est :
MUR = Mxanthane + Macétate * DSA + Mpyruvate * DSP Equation 2-5
- 121 -

Chapitre 2 : Matériels et Méthodes
2.2.3.2. Détermination de la densité de greffage des xanthanes modifiés
La densité de greffage des xanthanes amphiphiles, définie comme étant le nombre d’amine
greffée par unité de répétition, est calculée suivant la formule suivante :
octyle=noctylaminenUR'
Equation 2-6
Avec noctylamine le nombre de moles d’octylamine greffée sur les fonctions acide carboxylique du
xanthane, et nUR’ le nombre de moles de l’unité de répétition du xanthane modifié. Comme le
xanthane utilisé possède 1 acide glucuronique et 0,49 groupement pyruvate par unité de répétition,
la densité de greffage est comprise entre 0 et 149 %.
Le nombre de moles d’octylamine greffée est déterminé à partir des intégrations du pic des
protons du méthyle terminal de l’amine (0,88 ppm) et du pic de la référence TSP :
noctyle=3∗ I octylamine∗ nTSP
ITSPEquation 2-7
Avec nTSP le nombre de moles de TSP dans le tube.
Le nombre de moles de l’unité de répétition du xanthane modifié, nUR’, est obtenu par l’équation
suivante :
nUR'=mxanthane
MUR'
Equation 2-8
Avec mxanthane la masse de xanthane (en gramme) dans le tube et MUR’ la masse molaire de l’unité
de répétition du xanthane modifié. Cette dernière correspond à :
MUR' = MUR + %octyle*Moctyle - %octyle*MH2O Equation 2-9
- 122 -

Chapitre 2 : Matériels et Méthodes
Pour chaque chaîne d’octylamine greffée sur le xanthane, une molécule d’eau est éliminée. De
plus, MUR’ évolue avec la densité de greffage.
La détermination de la densité de greffage, %octyle, nécessite de connaître MUR’ (équation 2-6 et
équation 2-8). Or, MUR’ ne peut être calculée si l’on a déjà %octyle (équation 2-9). Pour résoudre ce
système, nous utilisons la méthode itérative suivante :
- Nous fixons une valeur pour %octyle.
- Cette valeur est injectée dans l’équation 2-9 pour avoir une première estimation de MUR’.
- L’équation 2-8 est ensuite résolue pour obtenir nUR’.
- Puis la valeur de nUR’ est utilisée dans l’équation 2-6 pour déterminer %octyle.
Ce cycle d’itération est répété jusqu’à obtenir une valeur constante de %octyle. Dans notre cas, la
densité de greffage n’évolue plus à partir de 3 itérations.
2.3. Polarimétrie
2.3.1. Principes généraux
Un rayonnement lumineux peut être représenté par une onde électromagnétique. Cette dernière
est constituée d’un champ magnétique M et d’un champ électrique E tous deux
perpendiculaires à la direction de propagation du faisceau10, 11. Ces deux champs oscillent
simultanément dans des directions différentes.
La polarisation s’intéresse à l’orientation du champ électrique associé à une onde. Ainsi, une
onde peut être polarisée linéairement (la direction de E est constante au cours du temps),
circulairement (le champ électrique tourne autour de l’axe de propagation avec une fréquence
- 123 -

Chapitre 2 : Matériels et Méthodes
identique à celle de l’onde), elliptiquement (combinaison de la polarisation linéaire et de la circulaire)
ou non polarisée (la direction du champ électrique varie aléatoirement au cours du temps).
De plus, une onde plane polarisée linéairement peut être décomposée en deux ondes polarisées
circulairement, mais de sens de rotation opposé10, 12. Ce phénomène est illustré sur la figure 2-4:
Figure 2-4 : Décomposition d’une onde polarisée linéairement en ses deux composantes polarisées
circulairement, mais de sens opposé (d’après Robert et al.12).
L’intersection de l’onde polarisée linéairement avec un plan perpendiculaire au sens de
propagation donne une droite (cf. figure 2-4 A). L’intersection des ondes polarisées circulairement
avec le même plan donne deux cercles : un tournant vers la droite (cf. figure 2-4 B), l’autre vers la
gauche (cf. figure 2-4 C), avec des vitesses identiques mais de signe contraire. Le vecteur E est
alors la somme vectorielle des deux vecteurs, d’amplitude identique, associés à chacune des ondes
circulaires :
E=ED+ EG Equation 2-10
Chacune des ondes polarisées circulaires est caractérisée par un indice de réfraction 12, ηD ou ηG.
Ces indices mesurent les variations de vitesse de propagation de ces ondes lorsqu’elles traversent un
milieu. Dans certains milieux, les deux indices sont identiques : l’onde lumineuse conserve sa
polarisation linéaire. Dans d’autres milieux par contre, l’interaction entre l’onde et la matière entraîne
un ralentissement de la propagation d’une des ondes circulaires par rapport à l’autre : les deux
- 124 -

Chapitre 2 : Matériels et Méthodes
indices sont alors différents. Le vecteur résultant E est alors pivoté d’un angle α par rapport à
l’axe de propagation de l’onde polarisée linéairement, comme indiqué sur les figures 2-5 :
Figures 2-5 : Effet du milieu sur l’orientation d’une onde plane polarisée linéairement A : quand ηR = ηG. B :
quand ηR ≠ ηG (d’après Robert et al.12).
Un milieu capable de provoquer cette rotation du vecteur est dit optiquement actif : il s’agit du
phénomène de biréfringence circulaire. L’angle α est appelé le pouvoir rotatoire, ou encore l’activité
optique, du milieu considéré. Lorsque la rotation s’effectue vers la droite, le milieu est qualifié de
dextrogyre. Lorsque la rotation a lieu vers la gauche, le milieu est qualifié de lévogyre. Globalement,
la biréfringence circulaire est observée dans des milieux contenant des composés chiraux.
2.3.2. Présentation du polarimètre
Un polarimètre est un instrument analytique qui permet de mesurer l’angle de rotation du plan
de polarisation d’un faisceau lumineux après avoir traversé un échantillon10, 12. La figure 2-6 donne
une représentation schématique de ce type d’appareil fonctionnant selon le principe optique nul :
- 125 -

Chapitre 2 : Matériels et Méthodes
Figure 2-6 : Schéma d’un polarimètre (d’après Robert et al.12).
L’appareil comporte deux lames polaroïdes (le polarisateur et l‘analyseur) qui encadrent la cellule
de mesure. Ces deux lames orientent le faisceau dans une seule direction. Le polarisateur est fixe par
rapport à l’axe de propagation du faisceau lumineux. L’analyseur, par contre, tourne autour de l’axe
selon un angle θ. En l’absence de milieu optiquement actif, l’intensité de la lumière envoyée sur les
deux polaroïdes est maximale si les lames sont orientées dans le même sens, et nulle si l’analyseur
est basculé de 90° par rapport au polarisateur (on parle alors d’extinction).
Avant toute mesure, les deux polaroïdes sont en position d’extinction l’un par rapport à l’autre :
aucune lumière n’est mesurée au niveau du détecteur. L’introduction d’un composé optiquement
actif entre les deux lames change l’orientation du plan de polarisation du faisceau à l’entrée de
l’analyseur, ce qui entraîne le passage d’un rayon lumineux vers le détecteur. L’analyseur est alors
pivoté d’un angle θ afin de rétablir la position d’extinction. Cet angle θ correspond à α, c’est-à-dire au
pouvoir rotatoire de l’échantillon placé dans la cuve de mesure.
2.3.3. Activité optique du xanthane
Le xanthane possède plusieurs carbones asymétriques. Par conséquent, il possède une activité
optique. Plus spécifiquement, pour ce polysaccharide, il a été observé que la rotation du plan de
polarisation est différente suivant que les chaînes soient sous forme ordonnée ou désordonnée 13-17.
Ce phénomène est illustré sur la figure 2-7:
- 126 -

Chapitre 2 : Matériels et Méthodes
Figure 2-7 : Evolution de α avec la température pour une solution de Xdan1 à 1 g/L dans NaCl 10-2 M.
A basse température, α est quasiment constant et égal à -0,030. A partir de T > 60°C, on
remarque que le pouvoir rotatoire augmente fortement, puis se stabilise à environ -0,010 pour T ≥
80°C. Globalement, l’angle de rotation évolue de manière sigmoïdale avec la température, ce qui est
caractéristique de la transition conformationnelle du xanthane14, 16 : forme ordonnée hélicoïdale à
basse température et forme désordonnée plus flexible à haute température. En appliquant la
méthode des tangentes, il est possible de déterminer la température de transition ordre-désordre du
polymère, Tm. Dans le cas de notre exemple, Tm est égale à 67°C pour du Xdan1 dissous à 1 g/L dans
une solution aqueuse de 0,01 M de NaCl. Dans la suite du manuscrit, nous utilisons ou normalisé pour
représenter l’évolution du pouvoir rotatoire d’un échantillon de xanthane en fonction de la
température. normalisé est obtenu à partir de la formule suivante :
ɑnormalis é=ɑ−ɑmaxɑmin−ɑmax
, avec 0 ≤ normalisé ≤ 1 Equation 2-11
- 127 -

Chapitre 2 : Matériels et Méthodes
2.3.4. Appareil utilisé et préparation des échantillons
Le polarimètre utilisé est un Perkin-Elmer 241, qui fonctionne suivant le principe optique nul. Il
est équipé de deux sources lumineuses (mercure et sodium) et d’un monochromateur pour fixer la
longueur d’onde d’analyse. Les mesures optiques ont été réalisées à 436 nm 2, 18 (lampe à mercure),
dans une cellule de verre thermostatée (longueur du trajet optique 1 dm). Les angles de rotation sont
mesurés tous les 5°C entre 25 et 90°C, après 30 minutes d’équilibration thermique.
La caractérisation de la température de transition conformationnelle des échantillons de
xanthane modifié a été effectuée sur des solutions de polysaccharide à 1 g/L dans de l’eau ultrapure.
Ces dernières ont été obtenues par dissolution de la masse requise du polymère dans l’eau à
température ambiante pendant une nuit. Le pH et la conductivité de la solution ont, par la suite, été
respectivement ajustés à 4,3 avec 0,01 M de NaCl et/ou de HCl, et 1 mS/cm avec NaCl(s). Les
solutions ont enfin été centrifugées à une force centrifuge relative de 362 g pendant 3 minutes afin
d’enlever toute bulle d’air.
2.4. Fluorescence
La technique de fluorescence est couramment utilisée lors de l’étude de composés amphiphiles.
En effet, cette méthode analytique non destructive permet de mettre en évidence la présence de
domaines hydrophobes au sein des systèmes étudiés, voire de les quantifier. Elle est, par exemple,
employée en biologie afin de caractériser l’organisation des protéines ou des membranes dans les
systèmes vivants19-21, ou encore en physico-chimie, afin de déterminer la concentration
micellaire/d’agrégation critique de tensioactifs22-24 ou de polymères amphiphiles25-29 en solution.
- 128 -

Chapitre 2 : Matériels et Méthodes
2.4.1. Définitions et principes généraux
Toute molécule est capable d’absorber de l’énergie sous forme de photon pour passer d’un état
fondamental à un état excité. Le retour à l’état fondamental s’effectue suivant divers processus30,
comme illustré sur la figure 2-8 :
Figure 2-8 : Diagramme de Perrin-Jablonski : processus lumineux possibles après excitation d’une molécule
(d’après Valeur30).
La molécule peut exister sous différents états électroniques (traits épais), singulets (S) ou triplets
(T), auxquels sont associés des niveaux vibrationnels (traits fins). Lors de l’excitation par absorption
de photon, la molécule passe de son état fondamental S0 à un état excité S1 ou Sn. Le retour à l’état
d’équilibre s’effectue principalement par désexcitation non-radiative, essentiellement via des chocs
moléculaires. La conversion interne CI (transition entre deux états électroniques de même
multiplicité de spin) ou le croisement intersystème CIS (transition entre deux niveaux vibrationnels
appartenant à des états électroniques de multiplicités différentes) sont des processus de
désexcitation non-radiatifs. Certaines molécules cependant sont susceptibles de rendre plus ou moins
- 129 -

Chapitre 2 : Matériels et Méthodes
favorables les désexcitations radiatives par fluorescence ou phosphorescence. Dans le cadre de cette
étude, nous n’aborderons que le premier cas de figure de ces phénomènes de luminescence.
Le phénomène de fluorescence correspond à l’émission spontanée de photon lors de la relaxation
S1 → S0 de la molécule. Ce phénomène est très rapide (de l’ordre de 10 -7 à 10-10s). Il s’accompagne
généralement d’une légère perte d’énergie, dissipée par relaxation vibrationnelle mais surtout
amplifiée par les effets de relaxation de solvatation (en effet, le moment dipolaire des molécules
fluorescentes à l’état excité est souvent très différent de celui à l’état fondamental). C’est pourquoi le
spectre de fluorescence est situé, dans la plupart des cas, à des longueurs d’onde plus grandes (soit
des énergies plus basses) que celui d’absorption.
2.4.2. Principe d’une analyse de fluorescence
Expérimentalement, effectuer une analyse de fluorescence consiste à envoyer un faisceau
lumineux sur un échantillon à une ou plusieurs longueurs d’onde d’excitation, λE, puis à enregistrer
l’émission de lumière I(λE, λF) en fonction de la longueur d’onde, λF, lorsque l’échantillon revient à l’état
fondamental30 (cf. figure 2-9) :
Figure 2-9 : Schéma d’une analyse de fluorescence (d’après Valeur30).
ka est la constante de vitesse d’absorption de photons par les espèces fluorophores M (de l’ordre
de 1015 s-1), kSr est la constante de vitesse associée aux processus de désexcitation radiatifs (avec
- 130 -

Chapitre 2 : Matériels et Méthodes
émission de fluorescence) et kSnr celle des processus non radiatifs (toutes les deux de l’ordre de 10 7 -
1010 s-1).
La source lumineuse émet un flux constant de photons lors de l’analyse. Soit N0 le nombre de
photons incidents pénétrant l’échantillon (en mol/L). Une fraction α de ces photons est adsorbée par
les fluorophores. Par conséquent, la quantité de photons adsorbés par unité de volume et de temps
est :
ka∗α∗N0=α∗ I 0 Equation 2-12
Avec I0 l’intensité de la lumière incidente (en moles de photons par litre par seconde).
L’illumination étant continue au cours de la mesure, la concentration en fluorophores excités,
[M*], est constante au cours du temps : on est en régime stationnaire. De ce fait, la vitesse de
variation de [M*] en fonction du temps vaut :
d [M✳ ]dt
=ka∗α∗ N0−(k Sr+kSnr )∗ [M✳ ]=α∗ I 0−(k Sr+k Snr)∗ [M ✳ ]=0 Equation 2-13
Donc :
[M ✳ ]=α∗ I 0kSr+k Snr
=0 Equation 2-14
L’intensité de fluorescence stationnaire, IF (λE, λF), grandeur mesurée lors de l’analyse, est définie
comme le nombre total de photons de fluorescence émis par les fluorophores par unité de temps et
de volume :
I F ( λE , λ F)=k Sr∗ [M ✳ ]=α ∗ I 0∗ kSrkSr+kSnr
=α∗ I 0∗φF
Equation 2-15
- 131 -

Chapitre 2 : Matériels et Méthodes
ФF représente le rendement quantique de fluorescence, soit la fraction de molécules M excitées
qui retournent à l’état fondamental S0 avec émission de photons de fluorescence, ou, en d’autres
termes, le rapport du nombre de photons émis sur le nombre de photons adsorbés. Cette grandeur
dépend de la nature du composé fluorophore étudié et du milieu.
L’intensité de fluorescence stationnaire peut également s’exprimer sous la forme :
IF ( λE , λ F)=k∗F λ( λF )∗ IA (λE) Equation 2-16
Fλ(λF) est le spectre de fluorescence ou d’émission du fluorophore étudié. Il reflète la distribution
de la probabilité des différentes transitions à partir du plus bas niveau vibrationnel de S 1 vers les
divers niveaux vibrationnels de S0. IA(λE) correspond à l’intensité adsorbée par les molécules
fluorophores. Quant à k, il s’agit d’un facteur instrumental qui dépend du spectrofluorimètre
employé. Par conséquent, il n’est pas possible de comparer les valeurs numériques de I F(λF) d’un
même échantillon analysé sur deux appareils différents. C’est pourquoi l’intensité est exprimée en
unités arbitraires.
Une analyse de fluorescence requiert, bien entendu, que le système étudié fluoresce. Or, toutes
les molécules ne possèdent pas cette propriété. En effet, la fluorescence d’un composé nécessite que
ce dernier possède une ou plusieurs doublets électroniques de type π. Les molécules utilisées en
fluorescence, qualifiées de sonde de fluorescence31, sont donc généralement constituées d’un ou
plusieurs cycles aromatiques sièges d’une délocalisation électronique. Les sondes peuvent être
intrinsèques (le système considéré fluoresce naturellement) ou extrinsèques (ajoutées au système en
solution). Dans la deuxième situation, elles peuvent être soit liées de manière covalente au polymère
étudié, soit associées à ce dernier via des interactions intermoléculaires (hydrogènes, ioniques,
hydrophobes, etc.). De plus, la plupart des sondes de fluorescence sont sensibles à une propriété
particulière de leur microenvironnement : leur spectre d’émission varie suivant cette propriété. Il
- 132 -

Chapitre 2 : Matériels et Méthodes
existe ainsi des sondes sensibles au pH, à la viscosité, à la présence d’ions, à la polarité ou encore au
caractère hydrophile/hydrophobe du milieu étudié, ce qui permet d’obtenir une grande richesse
d’informations sur le système étudié en fonction de la sonde employée. Dans le cadre de ce travail,
nous utilisons une sonde fluorescente à la fois sensible à la polarité et à la présence d’eau, le 8-
(aniline)-1-naphthalènesulfonate (ANS).
2.4.3. Caractéristiques de la sonde de fluorescence ANS
Les sondes dites de polarité se caractérisent par un spectre d’absorption et d’émission dont les
longueurs d’onde de maximum d’absorption/d’émission changent fortement suivant la polarité du
solvant31. Ces composés sont aussi appelés solvatochromiques. Dans le cas de l’ANS, le spectre
d’émission se décale vers le bleu quand la polarité diminue21, 24, 28, 32, comme illustré sur la figure 2-10 :
Figure 2-10 : Spectre d’émission de l’ANS dans a : l’eau. b : 1,7*10-2 M. c : 2,5*10-1 M. d : 3,6*10-1 M. e : 4,9*10-1
M de dextrane amphiphile28.
Le milieu contient des macromolécules de dextrane modifié hydrophobiquement en
concentration croissante. On observe nettement le décalage de la longueur d’onde au maximum
- 133 -

Chapitre 2 : Matériels et Méthodes
d’émission, λmax, lorsque la concentration en polymère augmente (soit quand le milieu devient de plus
en plus apolaire). La valeur de la longueur d’onde au maximum d’émission donne ainsi des
informations sur la polarité du système étudié.
Par ailleurs, l’ANS possède la particularité d’être sensible à la présence d’eau dans le milieu 21, 28, 29,
31, 33. Ainsi, en milieu non aqueux, l’émission de fluorescence IF(λE, λF) est très importante. Au contraire,
en présence d’eau, l’intensité mesurée est très faible. L’eau est donc une molécule d’extinction de
fluorescence de l’ANS. Cette sonde permet, par conséquent, de détecter la présence de zones
hydrophobes au sein de systèmes aqueux étudiés. Elle est notamment utilisée pour cette propriété
dans la caractérisation structurale de composés biologiques20, 21, 34 ou de polymères amphiphiles28, 29.
2.4.4. Appareil utilisé et préparation des solutions
Les analyses de fluorescence sont réalisées sur un spectrofluorimètre Varian Cary Eclipse avec
l’ANS comme sonde. La longueur d’onde d’excitation est de 350 nm, et le spectre d’émission est
enregistré de 380 à 690 nm. L’intensité d’excitation est réglée de façon à minimiser les phénomènes
de photoblanchiment pendant l’expérience. Le photomultiplicateur a été réglé de façon à obtenir le
signal le plus intense sans saturation. Les solutions de xanthane à caractériser sont obtenues par
dissolution, sous agitation à température ambiante pendant une nuit, de la masse requise de
polymère dans un milieu tamponné (pH = 4,5) de 20 mM d’acétate de sodium contenant 2*10 -5 M
d’ANS. Ainsi, l’intensité d’émission de fluorescence qui sera mesurée ne dépendra pas de la
concentration en sonde mais, principalement, de l’environnement immédiat de celle-ci.
- 134 -

Chapitre 2 : Matériels et Méthodes
2.5. Rhéologie
2.5.1. Définition et rappels
La rhéologie est la science qui s’intéresse à la déformation et à l’écoulement de la matière sous
l’effet d’une contrainte appliquée35. Elle permet d’accéder à un certain nombre de grandeurs
physiques du matériau étudié, comme la viscosité de solutions ou de fondus via des mesures en
écoulement, ou aux modules viscoélastiques dans le cas de mesures dynamiques.
Pour rappel, les matériaux peuvent être classés en trois catégories36 :
- Le solide élastique, qui se déforme instantanément et réversiblement lorsqu’il est soumis à
une contrainte. Le modèle analogique associé à ce comportement est celui du ressort de
Hooke. Le solide élastique possède la caractéristique de restituer entièrement l’énergie
emmagasinée lors de la déformation quand cette dernière est relâchée.
- Le liquide visqueux ou newtonien, qui coule uniquement, et de manière irréversible,
lorsqu’une contrainte est appliquée. Ce comportement est modélisé par un amortisseur
infini, ou encore un piston. Le liquide visqueux dissipe entièrement l’énergie fournie pour le
déformer.
- Le matériau viscoélastique, qui a un comportement intermédiaire entre le solide élastique et
le liquide visqueux. Les modèles analogiques associés à ce système sont le modèle de
Maxwell (ressort et amortisseur montés en série) ou le modèle de Kelvin-Voigt (ressort et
amortisseur montés en parallèle), voire des associations en série ou en parallèle de ces
différents éléments (modèles dits généralisés).
La grande majorité des matériaux, notamment polymères, ont un comportement viscoélastique.
Les modèles du solide élastique et du liquide visqueux peuvent être considérés comme des cas
idéaux.
- 135 -

Chapitre 2 : Matériels et Méthodes
2.5.2. Mesures en écoulement
Un matériau soumis à une contrainte peut se déformer. Sa déformation dépend notamment de
l’intensité de la contrainte appliquée et de sa répartition sur la surface du matériau. Dans le cas d’un
écoulement laminaire, le matériau peut être modélisé comme une superposition de couches minces
qui glissent les unes par rapport aux autres, sans transfert de matière37 :
Figure 2-11 : Représentation de l’écoulement laminaire d’un matériau.
Le mouvement des couches entraîne l’apparition d’une force de cisaillement, F, parallèle à la
surface de la couche. La contrainte de cisaillement, notée σ, est définie comme étant la force de
cisaillement F normalisée par l’aire de la surface, S, sur laquelle agit F :
σ=FS
Equation 2-17
Avec F en N, S en m2 et, par conséquent, σ en N/m2, soit en Pa.
Le déplacement relatif des couches les unes par rapport aux autres correspond à la déformation
du matériau, ɣ, et la vitesse (ou taux) de cisaillement, notée ɣ , à la dérivée de la déformation par
rapport au temps (en s-1).
ɣ=dxdyet ɣ=
∂ɣ∂ t
Equation 2-18
Ces grandeurs permettent d’obtenir la viscosité du fluide, η (en Pa.s), via la relation de Newton :
- 136 -

Chapitre 2 : Matériels et Méthodes
η=σɣ
Equation 2-19
Afin de pouvoir exploiter les résultats, les mesures doivent être réalisées dans le régime
stationnaire d’écoulement, régime dans lequel la viscosité est indépendante du temps. De plus, il est
également préférable, afin d’interpréter au mieux les données, de travailler dans le domaine linéaire.
En effet, dans ce domaine, la réponse du matériau est indépendante de la contrainte ou de la
déformation imposée (la viscosité est donc constante).
Pour un système purement élastique, la contrainte peut s’exprimer en fonction de la déformation
suivant la formule suivante :
G=σɣ
Equation 2-20
Avec G le module de cisaillement du matériau étudié (en Pa).
2.5.3. Mesures dynamiques en solution
Le comportement rhéologique d’un échantillon peut également être déterminé via l’application
d’une sollicitation sinusoïdale, à des fréquences et déformations variables36, 38. Soit un échantillon
sollicité via une déformation sinusoïdale qui évolue au cours du temps, ɣ(t) :
ɣ ( t )=ɣ o sin (ωt ) Equation 2-21
Avec ω la pulsation ou fréquence angulaire (en rad/s) et ω/2π la fréquence des oscillations.
Dans le domaine de réponse linéaire, la contrainte résultante est également une fonction
sinusoïdale en fonction du temps. Cette dernière, notée σ(t), est de même fréquence, mais présente
un déphasage δ (aussi appelé angle de perte) par rapport à la déformation :
σ ( t )=σO sin (ωt+δ ) Equation 2-22
- 137 -

Chapitre 2 : Matériels et Méthodes
L’angle de perte δ prend différentes valeurs suivant le caractère viscoélastique du matériau
étudié :
- δ = 0 dans le cas d’un solide élastique idéal.
- δ = π/2 dans le cas d’un liquide visqueux idéal.
- 0 ≤ δ ≤ π/2 pour un matériau viscoélastique.
Après développement du sinus, σ(t) s’écrit de la manière suivante :
σ ( t )=σ osin (ωt )cos (δ )+σ ocos (ωt )sin (δ ) Equation 2-23
Cette équation fait apparaître deux termes : l’un en phase avec la sollicitation et l’autre en
déphasage de π/2 avec cette sollicitation. Le premier terme est relié au module de conservation G’
(ou module élastique) et le second au module de perte G’’ (ou module visqueux), tous deux exprimés
en N.m-2 ou en Pa. Par identification, on obtient :
G'=σoɣ ocos (δ )et G' '
=σoɣosin (δ ) Equation 2-24
Il est également possible d’exprimer ces différentes grandeurs sous forme de nombres
complexes. On a ainsi :
ɣ ( t )=ɣ0 eiω t Equation 2-25
Et
σ ( t )=σ 0ei (ωt+δ )=G∗ ɣ0 Equation 2-26
Avec G le module complexe de cisaillement, qui peut se décomposer en parties réelles et
imaginaires :
- 138 -

Chapitre 2 : Matériels et Méthodes
G (ω )=G' (ω )+i G' ' (ω ) Equation 2-27
Le module de conservation G’ correspond alors à la partie réelle de G , et le module de perte
G’’ à la partie imaginaire.
L’étude de l’évolution de G’ et de G’’ selon la fréquence permet ainsi d’explorer le comportement
rhéologique du polymère étudié à différentes échelles de temps d’observation et, dans certains cas,
d’accéder à des temps de relaxation caractéristiques du système. En particulier, cette technique offre
l’avantage de pouvoir caractériser l’échantillon dans son état d’équilibre, dès lors que les mesures
sont effectuées dans le domaine linéaire. La figure 2-12 A indique l’évolution de G’ et de G’’ en
fonction de la fréquence à différentes températures pour une solution à 2 g/L de XDan1 dans NaCl
0,01 M.
Figures 2-12 A : Evolution des modules G’ (symboles pleins) et G’’ (symboles ouverts) en fonction de la
fréquence à différentes températures pour une solution de XDan1 à 1 g/L dans NaCl 0,01 M. B : Courbe
maîtresse fréquence-température de XDan1 à 1g/L dans NaCl 0,01 M (Tref = 20°C).
- 139 -
w*aT (rad/s)
10-2 10-1 100 101
G'*b
T, G
"*b
T (Pa)
10-1
100
G' 5°CG' 10°CG' 20°CG' 30°CG' 40°CG' 50°CG' 60°C
w (rad/s)
10-1 100 101
G',
G" (P
a)
10-1
100
G' 20°CG' 30°CG' 40°CG' 50°CG' 60°C
A B
aT
bT

Chapitre 2 : Matériels et Méthodes
A partir de ces données, il est possible d’appliquer le principe d’équivalence temps-température
afin de construire une courbe maîtresse fréquence-température. Ce principe permet de savoir si les
processus de relaxation d’une solution de polymère sont identiques d’une température à une autre.
Une telle construction consiste, dans un premier temps, à fixer une température de référence, Tref
(dans notre cas, Tref = 20°C). Les valeurs de fréquences et de modules obtenues aux autres
températures sont alors multipliées respectivement par un facteur aT (décalage horizontal) et bT
(décalage vertical), de manière à superposer les courbes associées à celles de référence. La courbe
maîtresse ainsi construite (cf. figure 2-12 B) permet de modéliser le comportement du polymère
étudié sur une gamme de fréquences plus étendue que celle accessible expérimentalement. a T et bT
représentent respectivement la variation du temps de relaxation et celle de l’amplitude des modules
avec la température. Ce type de construction a été réalisé, dès que possible, sur toutes les solutions
de xanthane analysées en rhéologie en solution.
2.5.4. Rhéologie interfaciale
2.5.4.1. Définition d’une interface
Par définition, une interface est la limite entre deux phases immiscibles (liquide/liquide dans le
cas de notre étude), comme illustré sur la figure 2-13 :
Figure 2-13 : Schéma d’une interface entre deux phases immiscibles (d’après Shaw39).
- 140 -

Chapitre 2 : Matériels et Méthodes
A l’interface, les molécules ont un environnement chimique différent de celui entourant les
molécules situées au cœur des phases (dites en solution). Par conséquent, l’interface présente des
propriétés physiques très distinctes de celles observées en solution, comme, par exemple,
l’apparition d’une force supplémentaire et caractéristique, la tension interfaciale. De plus, l’interface
est le siège de l’absorption des molécules amphiphiles (tensioactifs, polymères, protéines) :
Figure 2-14 : Absorption de molécules amphiphiles à l’interface liquide/liquide (d’après Shaw39).
Les molécules absorbées forment une monocouche à l’interface, qui diminue la tension
interfaciale du système. Ce processus est dynamique : les molécules situées à l’interface et celles en
solution ne cessent de s’échanger au cours du temps. Par ailleurs, la formation de la couche
interfaciale est un phénomène contrôlé par la diffusion des molécules amphiphiles jusqu’à l’interface.
Elle dépend donc notamment de la concentration en molécules dans le milieu, mais également de la
viscosité de ce dernier.
2.5.4.2. Principes de la rhéologie interfaciale de cisaillement
De nos jours, il existe de nombreux produits (alimentaires, cosmétiques, pharmaceutiques, etc.)
sous forme d’émulsion ou de mousse, et dont les propriétés spécifiques s’expliquent par l’existence
de ces interfaces /liquide ou liquide/gaz (qui comportent généralement une monocouche absorbée).
Le contrôle de ces propriétés, lors des phases de fabrication du produit, nécessite de caractériser le
- 141 -

Chapitre 2 : Matériels et Méthodes
comportement de ces interfaces. Cette caractérisation peut être statique, via la détermination de la
tension interfaciale, mais également dynamique.
La rhéologie interfaciale est la science qui s’intéresse à la déformation et l’écoulement d’une
interface sous l’effet d’une contrainte40. La technique d’analyse correspondante, la rhéométrie
interfaciale, permet d’accéder à un certain nombre de données concernant la structuration de
l’interface. Dans le cas d’une interface présentant une monocouche absorbée de polymère, l’étude de
ses propriétés rhéologiques peut donner des informations sur la conformation des chaînes à
l’interface, la nature des interactions entre macromolécules ou encore les cinétiques de migration
des chaînes polymériques à l’interface40, 41. L’ensemble de ces paramètres est important pour mieux
comprendre ces systèmes, et notamment prévoir leur stabilité au cours du temps.
Expérimentalement, l’étude des propriétés rhéologiques d’une interface s’effectue en déformant
cette dernière, soit par dilatométrie (« dilatational interfacial rheometry »), soit par cisaillement
(« shear interfacial rheometry » ou rhéométrie interfaciale de cisaillement)41, 42. C’est cette deuxième
méthode que nous utilisons dans le cadre de ce travail.
La rhéométrie interfaciale de cisaillement consiste à soumettre l’interface à une contrainte et à
mesurer la réponse du système. Cette technique permet donc d’accéder directement au
comportement rhéologique de l’interface. Ce comportement est notamment caractérisé par une
viscosité dite interfaciale, ηs, et des modules interfaciaux Gs’ et Gs’’41, 43, 44.
- 142 -

Chapitre 2 : Matériels et Méthodes
Soit une interface soumise à une contrainte σs dans le plan xy :
Figure 2-15 : Déformation d’une interface soumise à une contrainte.
La déformation ɣs et la vitesse de cisaillement ɣ s (s-1) sont données par :
ɣ s=dxyet ɣ s=
∂ ɣ s∂t
Equation 2-28
La viscosité interfaciale ηs (en N.s/m) est alors définie par :
ηs=σ sɣ s
Equation 2-29
Tout comme en solution, il est possible d’effectuer des analyses en oscillation dans le domaine
linéaire afin de déterminer le module complexe interfacial de cisaillement Gs .
Gs (ω )=Gs' (ω )+iGs
' ' (ω ) Equation 2-30
Avec Gs’ le module interfacial de conservation et Gs’’ le module interfacial de perte (tous deux en
N/m ou Pa.m). La détermination de Gs’ et de Gs’’ en fonction de la fréquence permet ainsi de
caractériser les propriétés rhéologiques de l’interface étudiée à différentes échelles de temps
d’observation.
- 143 -

Chapitre 2 : Matériels et Méthodes
2.5.5. Rhéomètre utilisé et préparation des échantillons
2.5.5.1. Préparation des solutions
Les mesures de viscoélasticité citées ont été réalisées sur des solutions aqueuses de xanthane
(modifié ou non) à 2 g/L. Ces dernières sont obtenues par dissolution, pendant 24 h sous agitation à
température ambiante, de la masse requise de polymère dans de l’eau ultrapure. Le pH et la
conductivité du milieu sont ensuite ajustés respectivement à 4,3 (par HCl et/ou NaOH à 0,1 M) et 1
mS/cm (par NaCl(s)). Les solutions ont enfin été centrifugées à une force centrifuge relative de 362 g
pendant 3 minutes afin d’enlever toute bulle d’air.
2.5.5.2. Analyses en solution
Le rhéomètre utilisé, à contrainte imposée, est un DHR2 (TA Instrument) avec un système de
régulation de température à effet Peltier. La caractérisation des propriétés rhéologiques des solutions
de polymère est réalisée à l’aide d’une géométrie à cylindre concentrique, aussi appelée géométrie
Couette (diamètre du rotor de 27,98 mm, longueur de 42,11 mm, entrefer de 1,2 mm). La plupart des
analyses sont effectuées à haute température. Pour limiter les phénomènes d’évaporation, de l’huile
de silicone de faible viscosité (Wacker) est ajoutée sur l’échantillon avant la mesure. Le
comportement viscoélastique des solutions de xanthane est étudié en sollicitations harmoniques à
différentes fréquences (de 100 à 0,1 rad/s) dans le domaine linéaire de déformation pour une gamme
de températures allant de 5 à 80°C. Pour chaque température, la solution est maintenue à la
température requise pendant 10 minutes avant mesure.
- 144 -

Chapitre 2 : Matériels et Méthodes
2.5.5.3. Analyses à l’interface eau/huile
Le rhéomètre DHR2 présenté précédemment est également employé pour les analyses de
rhéologie interfaciale de cisaillement.
La rhéométrie interfaciale par cisaillement permet d’effectuer les mêmes analyses que celles
réalisées en solution. Elle nécessite cependant un appareillage adapté40, 44. En effet, les viscosités et
modules interfaciaux sont très souvent bien inférieurs à ceux en solution. La détermination des
propriétés viscoélastiques interfaciales requiert l’utilisation d’un rhéomètre avec une forte sensibilité
en termes de couple et une faible inertie. De la même manière, la géométrie employée doit
également ne pas imposer une inertie supplémentaire au système, au risque de ne pas pouvoir
observer les phénomènes voulus. Dans ce but, plusieurs types de géométrie sont actuellement
disponibles sur le marché pour ces analyses. Dans notre cas, nous utilisons l’anneau à double paroi45
pour réaliser nos mesures (cf. figures 2-16).
Figures 2-16 A : Coupe de profil du dispositif de l’anneau à double paroi (d’après Vandebril et al.45). Rayon de
l’anneau interne R1 = 34,5 mm ; rayon de l’anneau externe R2 = 35,5 mm ; rayon du godet interne R3 = 31 mm ;
rayon du godet externe R4 = 39,5 mm ; hauteur H = 10 mm. B : Coupe de profil de la section de l’anneau en
contact avec l’interface eau/huile.
La géométrie se compose d’un anneau de section carrée en platine/iridium, sujet à rotation lors
de l’analyse, placé à l’interface entre les deux liquides immiscibles, et d’un godet fixe en téflon. Cette
géométrie est qualifiée de double paroi, car elle présente deux entrefers dans lesquels l’interface est
cisaillée : le premier entre la paroi externe du godet et la section de l’anneau en contact avec
- 145 -

Chapitre 2 : Matériels et Méthodes
l’interface, et le second entre la paroi interne du godet et la section de l’anneau. Les dimensions de
l’anneau et du godet sont conçues de manière à ce que la vitesse de cisaillement soit identique au
niveau des deux entrefers. Les dimensions de la géométrie utilisée dans le cadre de ces travaux sont
données sur la figure 2-16 A.
La largeur du godet varie suivant la hauteur : à partir d’une certaine hauteur H, le récipient se
rétrécit. Ce rétrécissement est localisé au niveau de l’interface étudiée, et s’accompagne d’une
marche verticale le long des parois du godet. Cette conception particulière du godet permet d’éviter
la formation d’un ménisque à ce niveau, réduisant ainsi les effets de capillarité le long des parois. En
outre, l’anneau présente également une géométrie spécifique. En effet, sa section au niveau de
l’interface a la forme d’un carré (cf. figure 2-16 B), avec un bord anguleux au contact de l’interface, ce
qui permet une meilleure « accroche » de l’interface à l’anneau. Du fait de ces caractéristiques
structurelles, l’anneau à double paroi présente l’avantage de générer une très faible inertie. Cette
géométrie est donc particulièrement adaptée pour la caractérisation des propriétés rhéologiques des
interfaces très fragiles. Actuellement, elle peut être utilisée pour l’étude d’interfaces de viscosité
supérieure ou égale à 10-5 Pa.s.m45.
Le godet étant en téflon, il conduit très peu la chaleur. C’est pourquoi la majorité des mesures
viscoélastiques de la littérature sont réalisées à température ambiante. Dans notre étude, nous nous
intéressons cependant à l’évolution de Gs’ et Gs’’ en fonction de la fréquence et de la température.
Pour pouvoir faire ce type d’analyse, nous avons, au préalable, établi une courbe d’étalonnage entre
la température consigne et celle mesurée dans le godet. De l’eau a été introduite dans le godet, et ce
dernier a été chauffé par étapes successives de 20 à 95°C. L’équilibrage thermique entre les
différentes étapes varie entre 45 minutes et 1 heure. Au bout de cette durée, la température au sein
de la solution est stabilisée. La figure 2-17 représente l’évolution de la température mesurée au sein
- 146 -

Chapitre 2 : Matériels et Méthodes
de la solution aqueuse contenue dans le godet en fonction de la température consigne après
stabilisation thermique :
Température consigne (°C)30 40 50 60 70 80 90
Tem
péra
ture
mesu
rée
(°C
)
30
40
50
60
70
Figure 2-17 : Courbe d’étalonnage température mesurée dans le godet en fonction de la température consigne.
Cette courbe sert d’étalonnage, et est utilisée systématiquement pour toutes les analyses
viscoélastiques interfaciales en température. Dans la suite du manuscrit, les températures
mentionnées en rhéologie interfaciale correspondent aux températures mesurées.
Les propriétés interfaciales sont très sensibles à la présence de contaminants. L’utilisation de
l’anneau nécessite que ce dernier soit parfaitement propre. Dans ce but, l’anneau est préalablement
flambé. Le chargement du godet avant la mesure s’effectue en trois phases. La solution aqueuse
contenant le polymère à étudier est en premier introduite dans le godet. L’anneau est ensuite déposé
à l’interface eau/air. Afin que l’analyse soit efficace, l’anneau doit être complètement mouillé. Le
positionnement de la géométrie à l’interface est donc une étape délicate. Elle s’effectue
manuellement, avec contrôle visuel. Une fois l’anneau installé, la deuxième phase liquide (dans notre
cas la même huile de silicone que celle employée en analyse rhéologique en solution) est déposée au
- 147 -

Chapitre 2 : Matériels et Méthodes
dessus de la phase aqueuse, jusqu’à recouvrir complètement l’anneau. Un couple est alors imposé à
cette interface via le mouvement de la géométrie. Le système mesure le cisaillement résultant et
transcrit cette donnée dans les grandeurs désirées (modules, viscosité, etc.).
Les analyses dynamiques sont réalisées à l’interface eau/huile de silicone de solutions à 2 g/L de
xanthane dans le domaine linéaire de couple. Elles consistent en des sollicitations harmoniques à
différentes fréquences (de 0, 01 à 1 rad/s) sur une gamme de températures allant de 22,2 à 73,5°C
(température déterminée dans le godet selon la courbe d’étalonnage présentée dans la figure 2-17).
Plus précisément, nous avons effectué ces mesures en deux étapes : la phase dite « aller », de 22,2 à
73,5°C, puis la phase dite « retour » de de 73,5 à 22,2 °C. L’ensemble de ces deux phases constitue un
« cycle thermique ». L’échantillon est qualifié de « natif » lors de la première phase de mesure, et est
dit « renaturé » pendant la phase de refroidissement. Pour chaque température, la solution est
maintenue à la température requise entre 45 minutes et 1 heure avant mesure.
- 148 -

Chapitre 2 : Matériels et Méthodes
Références bibliographiques
1. Bradford, M. M., A rapid and sensitive method for the quantitation of microgram quantities ofprotein utilizing the principle of protein-dye binding. Analytical Biochemistry 1976, 72, 248-254.
2. Aguni, Y. Contribution à la compréhension des mécanismes d'interactionxanthane/galactomannane. thèse, Université du Havre, Le Havre, 2009.
3. Akoka, S., Principes de base. In Une introduction à la Résonance Magnétique Nucléaire,CEISAM: Université de Nantes, 2009; pp 1-11.
4. Maes, E. La Résonance Magnétique Nucléaire; Université Lille 1: Lille, 2009; pp 1-11.
5. Akoka, S., La RMN impulsionnelle. In Introduction à la Résonance Magnétique Nucléaire,CEISAM: Université de Nantes, 2009; pp 1-27.
6. Wu, S.-J.; Wu, J.-H.; Xia, L.-Z.; Chu, C.; Liu, D.; Gong, M., Preparation of xanthan-derivedoligosaccharides and their hydroxyl radical scavenging activity. Carbohydrate Polymers 2013, 92,1612-1614.
7. Rinaudo, M.; Milas, M.; Lambert, F.; Vincendon, M., 1H and 13C NMR investigation of xanthangum. Macromolecules 1983, 16, 816-819.
8. Hamcerencu, M.; Desbrieres, J.; Popa, M.; Khoukh, A.; Riess, G., New unsaturated derivativesof Xanthan gum: Synthesis and characterization. Polymer 2007, 48, 1921-1929.
9. Esquenet, C. Propriétés structurales et dynamiques des solutions de polyelectrolytes rigideset semi-rigides et de polysaccharides associatifs. thèse, Université Joseph Fourier, Grenoble, 2003.
10. Pass, G.; Sutcliffe, H., Polarimetry. In Practical Inorganic Chemistry, Springer Netherlands:1974; pp 221-225.
11. Boeyens, J. C. A., Chapter 5 - Quantum Theory. In The Theories of Chemistry, Elsevier ScienceB.V.: Amsterdam, 2003; pp 177-259.
12. Robert, E. G.; Jeffrey, A., Chapter 2 Analytical methods. In Tetrahedron Organic ChemistrySeries, Elsevier: 1996; Vol. Volume 14, pp 45-74.
13. Holzwarth, G., Conformation of the extracellular polysaccharide of Xanthomonas campestris.Biochemistry 1976, 15, 4333-4339.
14. Milas, M.; Rinaudo, M., Conformational investigation on the bacterial polysaccharidexanthan. Carbohydrate Research 1979, 76, 189-196.
15. Norton, I. T.; Goodall, D. M.; Frangou, S. A.; Morris, E. R.; Rees, D. A., Mechanism anddynamics of conformational ordering in xanthan polysaccharide. Journal of Molecular Biology 1984,175, 371-394.
16. Morris, E. R.; Rees, D. A.; Young, G.; Walkinshaw, M. D.; Darke, A., Order-disorder transitionfor a bacterial polysaccharide in solution. A role for polysaccharide conformation in recognitionbetween Xanthomonas pathogen and its plant host. Journal of Molecular Biology 1977, 110, 1-16.
17. Liu, W.; Sato, T.; Norisuye, T.; Fujita, H., Thermally induced conformational change of xanthanin 0.01M aqueous sodium chloride. Carbohydrate Research 1987, 160, 267-281.
- 149 -

Chapitre 2 : Matériels et Méthodes
18. Renou, F.; Petibon, O.; Malhiac, C.; Grisel, M., Effect of xanthan structure on its interactionwith locust bean gum: Toward prediction of rheological properties. Food Hydrocolloids 2013, 32, 331-340.
19. Azzi, A.; Sidney Fleischer, L. P., The use of fluorescent probes for the study of membranes. InMethods in Enzymology, Academic Press: 1974; Vol. Volume 32, pp 234-246.
20. Gabellieri, E.; Strambini, G. B., ANS fluorescence detects widespread perturbations of proteintertiary structure in ice. Biophysical Journal 2006, 90, 3239-3245.
21. Gasymov, O. K.; Glasgow, B. J., ANS fluorescence: Potential to augment the identification ofthe external binding sites of proteins. Biochimica et Biophysica Acta 2007, 1774, 403-411.
22. Abuin, E. B.; Lissi, E. A.; Aspée, A.; Gonzales, F. D.; Varas, J. M., Fluorescence of 8-anilinonaphthalene-1-sulfonate and properties of sodium dodecyl sulfate micelles in water–ureamixtures. Journal of Colloid and Interface Science 1997, 186, 332-338.
23. Birdi, K. S.; Krag, T.; Klausen, J., Notes: determination of critical micelle concentration ofanionic micellar systems by anilinonaphthalenesulfonate (ANS) in aqueous solutions. Journal ofColloid and Interface Science 1977, 62, 562-563.
24. Mast, R. C.; Haynes, L. V., The use of the fluorescent probes perylene and magnesium 8-anilinonaphthalene-l-sulfonate to determine the critical micelle concentration of surfactants inaqueous solution. Journal of Colloid and Interface Science 1975, 53, 35-41.
25. Amiji, M. M., Pyrene fluorescence study of chitosan self-association in aqueous solution.Carbohydrate Polymers 1995, 26, 211-213.
26. Creuzet, C.; Kadi, S.; Rinaudo, M.; Auzély-Velty, R., New associative systems based onalkylated hyaluronic acid. Synthesis and aqueous solution properties. Polymer 2006, 47, 2706-2713.
27. Fischer, A.; Houzelle, M. C.; Hubert, P.; Axelos, M. A. V.; Geoffroy-Chapotot, C.; Carre, M. C.;Viriot, M. L.; Dellacherie, E., Detection of intramolecular associations in hydrophobically modifiedpectin derivatives using fluorescent pobes. Langmuir 1998, 14, 4482-4488.
28. Vieira, N. A. B.; Moscardini, M. S.; Tiera, V. A. d. O.; Tiera, M. J., Aggregation behavior ofhydrophobically modified dextran in aqueous solution: A fluorescence probe study. CarbohydratePolymers 2003, 53, 137-143.
29. Martin, G.; Ross, J.; Minteer, S. D.; Jameson, D. M.; Cooney, M. J., Fluorescencecharacterization of chemical microenvironments in hydrophobically modified chitosan. CarbohydratePolymers 2009, 77, 695-702.
30. Valeur, B., Caractéristiques de l'émission de fluorescence. In Invitation à la fluorescencemoléculaire, De Boeck: Bruxelles, 2004; pp 21-44.
31. Valeur, B., Les sondes fluorescentes. In Invitation à la fluorescence moléculaire, De Boeck:Bruxelles, 2004; pp 133-174.
32. Vasilescu, M.; Caragheorgheopol, A.; Caldararu, H.; Bandula, R.; Lemmetyinen, H.; Joela, H.,Micropolarity and order in the reverse micelles of L62 and L64 pluronic copolymers, as studied bymolecular probe techniques. The Journal of Physical Chemistry B 1998, 102, 7740-7751.
33. Griffiths, P. C.; Roe, J. A.; Bales, B. L.; Pitt, A. R.; Howe, A. M., Fluorescence probe studies ofgelatin sodium dodecyl sulfate interactions. Langmuir 2000, 16, 8248-8254.
34. Parul, D. A.; Bokut, S. B.; Milyutin, A. A.; Petrov, E. P.; Nemkovich, N. A.; Sobchuk, A. N.;Dzhagarov, B. M., Time-resolved fluorescence reveals two binding sites of 1,8-ANS in intact humanoxyhemoglobin. Journal of Photochemistry and Photobiology B: Biology 2000, 58, 156-162.
- 150 -

Chapitre 2 : Matériels et Méthodes
35. Barnes, H. A.; Hutton, J. F.; Walters, K., Introduction. In An Introduction to Rheology, Elsevier:Amsterdam, 1989; Vol. 3, pp 1-10.
36. Couarraze, G.; Grossiord, J.-L.; Huang, N., Viscoélasticité linéaire. In Initiation à la Rhéologie:bases théoriques et applications expérimentales, 4e éditon; Lavoisier: Paris, 2014; pp 17-50.
37. Couarraze, G.; Grossiord, J.-L.; Huang, N., Généralités. In Initiation à la Rhéologie: basesthéoriques et applications expérimentales, 4 ed.; Lavoisier: Paris, 2014; pp 3-15.
38. Barnes, H. A.; Hutton, J. F.; Walters, K., Linear viscoelasticity. In Rheology Series, Elsevier:1989; Vol. Volume 3, pp 37-54.
39. Shaw, D. J., Liquid-gas and liquid-liquid interfaces. In Introduction to Colloid and SurfaceChemistry (Fourth Edition), Butterworth-Heinemann: Oxford, 1992; pp 64-114.
40. Krägel, J.; Derkatch, S. R., Interfacial shear rheology. Current Opinion in Colloid & InterfaceScience 2010, 15, 246-255.
41. Karbaschi, M.; Lotfi, M.; Krägel, J.; Javadi, A.; Bastani, D.; Miller, R., Rheology of interfaciallayers. Current Opinion in Colloid & Interface Science 2014, 19, 514-519.
42. Miller, R.; Fainerman, V. B.; Petsev, D. N., Interfacial rheology of adsorbed layers. In InterfaceScience and Technology, Elsevier: 2004; Vol. Volume 4, pp 61-90.
43. Langevin, D., Surface shear rheology of monolayers at the surface of water. Advances inColloid and Interface Science 2014, 207, 121-130.
44. Couarraze, G.; Grossiord, J.-L.; Huang, N., Principe et description des principaux rhéomètres.In Initiation à la Rhéologie: bases théoriques et applications expérimentales, 4 ed.; Lavoisieir: Paris,2014; pp 79-172.
45. Vandebril, S.; Franck, A.; Fuller, G.; Moldenaers, P.; Vermant, J., A double wall-ring geometryfor interfacial shear rheometry. Rheologica Acta 2010, 49, 131-144.
- 151 -

Chapitre 2 : Matériels et Méthodes
- 152 -

Chapitre 3 : Modification chimique du xanthane
Chapitre 3 : Modification chimique du xanthane
3.1. Introduction 155
3.2. Couplage peptidique par des carbodiimides 156
3.3. Modification chimique du xanthane sous forme ordonnée 1663.3.1. Processus de modification à température ambiante 166
3.3.1.1. Description du procédé 1663.3.1.2. Mise en évidence de la conformation ordonnée du xanthane lors de sa modification 1683.3.1.3. Analyse RMN 1H des échantillons et quantification de la densité de greffage 170
3.3.1.3.1. Description des spectres RMN et détermination des taux d’acétate et de pyruvate 1723.3.1.3.2. Densité de greffage des xanthanes modifiés suivant le protocole à température ambiante 175
3.3.2. Processus de modification à basse température (première étape) 179
3.3.2.1. Influence de la température sur l’efficacité du couplage peptidique 1793.3.2.2. Densité de greffage des xanthanes modifiés suivant le protocole à basse températurelors de la première étape 181
3.4. Modification chimique du xanthane sous forme désordonnée 1873.4.1. Choix du solvant pour le protocole de modification 187
3.4.2. Mise en évidence de la conformation désordonnée du xanthane dans le DMSO 188
3.4.3. Protocole de modification du xanthane dans le DMSO 190
3.4.3.1. Choix des conditions expérimentales lors de la modification chimique 1903.4.3.2. Choix des étapes de purification 1923.4.3.3. Interprétation des spectres RMN 1H pour les xanthanes modifiés dans le DMSO 1923.4.3.4. Densité de greffage estimée des xanthanes modifiés suivant le protocole dans le DMSO
1963.4.3.5. Comparaison de la densité de greffage entre les protocoles de modification sous formeordonnée et sous forme désordonnée 198
3.5. Conclusion 200
Références bibliographiques 202
- 153 -

Chapitre 3 : Modification chimique du xanthane
- 154 -

Chapitre 3 : Modification chimique du xanthane
3.1. Introduction
Ce chapitre décrit les différents protocoles de modification chimique du xanthane mis au point au
cours de ce travail afin de conférer des propriétés amphiphiles à ce polysaccharide.
Pour rappel, le xanthane existe sous deux conformations différentes en solution, suivant les
conditions opératoires : une forme ordonnée hélicoïdale, très rigide, à température ambiante et force
ionique élevée, et une conformation désordonnée flexible à haute température et/ou force ionique
très faible. Ces deux conformations ont des propriétés rhéologiques bien distinctes en solution. Deux
procédés de modification chimique distincts, chacun adapté à une conformation donnée, ont donc
été développés dans le but d’étudier l’influence de ce paramètre sur le comportement du polymère
greffé. Ainsi, la modification du xanthane sous forme ordonnée est réalisée dans l’eau, celle sous
forme désordonnée dans le diméthylsulfoxyde (DMSO). Dans les deux cas, la modification chimique
consiste à greffer des chaînes alkyle linéaires sur le xanthane via une réaction de couplage peptidique
entre une amine et les fonctions acide carboxylique situées sur les chaînes latérales du
polysaccharide. Comme présenté dans la partie bibliographique, la plupart des articles traitant de la
modification chimique du xanthane ciblent comme site de greffage les fonctions hydroxyle de ce
polymère. Ces dernières, très nombreuses, rendent de ce fait délicat le contrôle précis de la
régiosélectivité des réactions mises en jeu et donc des propriétés apportées. L’utilisation d’une
réaction de couplage peptidique, spécifique des fonctions acide carboxylique localisées uniquement
sur les chaînes latérales, permet de s’affranchir de ces problèmes et offre un meilleur contrôle de la
réaction.
Il est également possible de greffer des chaînons hydrophobes sur les fonctions acide
carboxylique du xanthane par une réaction d'estérification. Néanmoins, le xanthane possédant à la
fois des groupements acide et alcool, l’estérification pourrait conduire à une réticulation indésirable
des chaînes de polymère entre elles. L’usage d’une amine porteuse des chaînons hydrophobes lors de
la réaction de greffage élimine cet inconvénient. De plus, la fonction amide résultant du couplage est
- 155 -

Chapitre 3 : Modification chimique du xanthane
capable, tout comme la fonction carboxylique initiale du polysaccharide, de former des liaisons
hydrogène, liaisons qui interviennent dans le maintien de la conformation hélicoïdale du xanthane.
Ainsi, le remplacement des fonctions acide carboxylique par des amides ne modifie pas le nombre de
liaisons hydrogène potentielles, quelle que soit la densité de greffage.
3.2. Couplage peptidique par des carbodiimides
Avant de présenter les procédés de modification chimique du xanthane, quelques rappels sur le
couplage peptidique par l’utilisation des carbodiimides sont effectués dans cette partie.
Le couplage peptidique est une réaction chimique entre une amine et un acide carboxylique
préalablement activé, pour former une liaison amide. Cette liaison est également appelée liaison
peptidique, car elle est majoritairement présente dans les protéines, biopolymères d’acides aminés
très courants dans les systèmes biologiques. Le couplage peptidique tient d’ailleurs son nom de ces
composés. En effet, historiquement, ce type de réaction a été développé pour synthétiser
chimiquement des peptides en laboratoire. De nos jours, le couplage peptidique est utilisé également
dans d’autres domaines que la synthèse peptidique, comme par exemple dans la modification
chimique de polysaccharides1-5 ou encore la fonctionnalisation de nanoparticules6, 7.
Il faut savoir qu’un acide carboxylique est naturellement peu réactif, car la délocalisation des
électrons rend le carbone du carbonyle peu électrophile. De plus, le groupement OH est un mauvais
groupe partant. Ces paramètres rendent difficiles les réactions directes entre une amine et un acide.
C’est pourquoi le couplage peptidique comprend tout d’abord une étape d’activation de la fonction
acide carboxylique. Cette étape consiste à remplacer la fonction OH par un groupement X à la fois
davantage électro-attracteur et bon groupe partant, afin de rendre le carbone plus électrophile.
L’amine s’additionne ensuite sur le carbonyle, puis le groupement X est éliminé :
- 156 -

Chapitre 3 : Modification chimique du xanthane
Figure 3-1: Mécanisme de réaction d’un couplage peptidique.
Le groupe partant X provient d’une ou plusieurs espèces chimiques qualifiées d’agents de
couplage, comme des carbodiimides, des acylazoles, des sels de phosphonium ou d’uronium 8. Dans le
cadre de notre étude, nous avons utilisé la chimie des carbodiimides pour activer les fonctions acide.
En effet, les carbodiimides sont l’une des catégories d’agents de couplage les plus étudiées et
utilisées en synthèse peptidique. Leur succès s’explique par leur grande réactivité (notamment à base
température), leur disponibilité commerciale (grande variété de substrats, ce qui implique
notamment un large choix de solvants pour les réactions) et leur prix peu élevé par rapport à d’autres
réactifs appartenant à la même catégorie. Les différentes voies d’accès aux fonctions amide via un
couplage peptidique avec un carbodiimide sont indiquées dans la figure suivante8 :
- 157 -

Chapitre 3 : Modification chimique du xanthane
Figure 3-2 : Voies d’accès aux fonctions amides via un couplage peptidique avec un carbodiimide (d’après El-
Faham et al.8).
L’addition de l’acide carboxylique sur le carbodiimide entraîne la formation d’une O-acylisourée.
Cette espèce, très réactive, peut réagir directement avec l’amine pour donner l’amide désiré (voie A).
D’autre voies d’accès, indirectes, sont également possibles. Ainsi, en présence d’un excès d’acide, l’O-
acylisourée forme un intermédiaire anhydride (voie B). Ce dernier est ensuite attaqué par l’amine
pour aboutir à l’amide voulu. L’O-acylisourée peut également se cycliser, et former une oxalozone, qui
réagit par la suite avec l’amine (voie C). Ce réarrangement n'est toutefois possible qu'au sein d'une
molécule présentant deux fonctions amide liées consécutivement. Dans tous les cas, ces réactions
entraînent la libération de dérivés d’urée. Certains de ces dérivés peuvent s’avérer très difficiles à
éliminer. Par exemple, la N,N’-dicyclohexylurée, obtenue par réaction du dicyclohexylcarbodiimide
- 158 -
+ CN N R2R3 NHR3 C
O
N R2
O
R1
Carbodiimide O-acylisourée O
R1OX
HOX
N
O
R1 R
O
Oxazolone
R1O
O
OR1
Anhydride
AB C
C NHR1O
R4
Amide
E
D
NHR3 C
O
N R2
O R1
R1 COOH
R1COOH
R4 NH2 R4 NH2
R4 NH2
R4 NH2
Dérivé d'urée
Dérivé d'urée
Dérivé d'urée
HOX
N-acylurée
NH NH
O
R3 R2
NH NH
O
R3 R2
NH NH
O
R3 R2

Chapitre 3 : Modification chimique du xanthane
(DCC, R2 = R3 = cyclohexyle) sur un acide carboxylique, est partiellement soluble dans tous les
solvants, sauf l’acide trifluoroacétique, ce qui complique fortement les étapes de purification 8. Pour
pallier à cette difficulté, plusieurs agents de couplage dérivés du DCC, mais solubles dans une large
gamme de solvants (ainsi que leurs dérivés urées), ont été mis au point. Parmi ces réactifs, le 1-éthyl-
3-(3-diméthylaminopropyl)carbodiimide (EDAC) est très majoritairement employé en synthèse
peptidique, car ce composé et son urée sont solubles dans l’eau, et, par conséquent, faciles à éliminer
en fin de couplage.
Comme indiqué précédemment, les O-acylisourées sont des espèces chimiques très réactives et
instables. En particulier, elles s’hydrolysent très rapidement dans l’eau pour donner un acide
carboxylique : la constante de vitesse d’hydrolyse9, à pH = 4,75, est comprise entre 2 et 3 s -1. Ce
phénomène est beaucoup plus rapide que l’addition de l’amine sur le site actif. L’acide obtenu peut
néanmoins réagir de nouveau avec un carbodiimide pour recréer une O-acylisourée. Par conséquent,
ces cycles d’activation/désactivation de l’acide ralentissent la cinétique d’amidation (et consomment
le carbodiimide). De plus, l’O-acylisourée est le siège d’un réarrangement, comme indiqué sur la
figure 3-2 (voie D). Cette réaction parasite est moins rapide que l’hydrolyse et que la réaction
d’amidation9. Elle est cependant irréversible, et présente aussi l’inconvénient de former une N-
acylurée10, qui est inerte chimiquement. La réaction entre l’amine et l’acide devient alors impossible.
Ces deux processus diminuent fortement l’efficacité du couplage peptidique.
Une méthode pour augmenter l’efficacité du couplage est de transformer l’O-acylisourée en un
ester activé (voie E). Ce type de composé présente l’avantage d’être plus stable que l’O-acylisourée.
En effet, il se caractérise par une cinétique d’hydrolyse beaucoup plus lente11, 12 (de l’ordre de l’heure
et non plus de la seconde). La stabilité accrue des sites actifs favorise donc la réaction avec l’amine.
De plus, cette fonctionnalisation offre aussi l’avantage d’empêcher le réarrangement indésirable de
l’O-acylisourée en N-acylurée. C’est pourquoi les réactions de couplage peptidique avec les esters
- 159 -
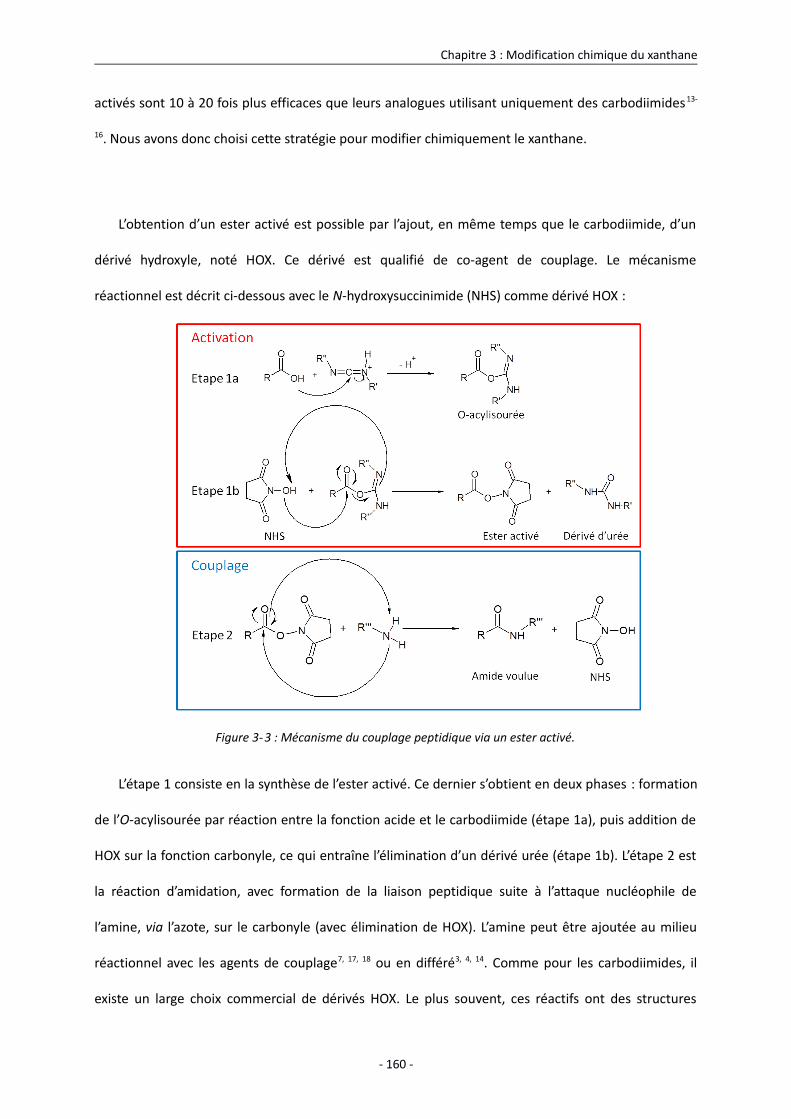
Chapitre 3 : Modification chimique du xanthane
activés sont 10 à 20 fois plus efficaces que leurs analogues utilisant uniquement des carbodiimides13-
16. Nous avons donc choisi cette stratégie pour modifier chimiquement le xanthane.
L’obtention d’un ester activé est possible par l’ajout, en même temps que le carbodiimide, d’un
dérivé hydroxyle, noté HOX. Ce dérivé est qualifié de co-agent de couplage. Le mécanisme
réactionnel est décrit ci-dessous avec le N-hydroxysuccinimide (NHS) comme dérivé HOX :
Figure 3-3 : Mécanisme du couplage peptidique via un ester activé.
L’étape 1 consiste en la synthèse de l’ester activé. Ce dernier s’obtient en deux phases : formation
de l’O-acylisourée par réaction entre la fonction acide et le carbodiimide (étape 1a), puis addition de
HOX sur la fonction carbonyle, ce qui entraîne l’élimination d’un dérivé urée (étape 1b). L’étape 2 est
la réaction d’amidation, avec formation de la liaison peptidique suite à l’attaque nucléophile de
l’amine, via l’azote, sur le carbonyle (avec élimination de HOX). L’amine peut être ajoutée au milieu
réactionnel avec les agents de couplage7, 17, 18 ou en différé3, 4, 14. Comme pour les carbodiimides, il
existe un large choix commercial de dérivés HOX. Le plus souvent, ces réactifs ont des structures
- 160 -

Chapitre 3 : Modification chimique du xanthane
similaires à celle du phénol ou de l’hydroxylamine. Par ailleurs, certains de ces co-agents sont
solubles dans l’eau, comme le N-hydroxysuccinimide.
Comme mentionné précédemment, le couplage peptidique par les carbodiimides, avec ou sans
ester activé, est couramment employé. Néanmoins, même si le mécanisme réactionnel est bien
compris, le choix des conditions opératoires optimales pour ce type de réaction reste sujet à
controverse. En effet, la littérature comprend de nombreux exemples de couplages avec les
carbodiimides, qui se caractérisent par une très grande variété de critères expérimentaux (choix des
substrats, du solvant, des réactifs, des concentrations, de la température, etc.)1, 2, 4, 7, 12, 14-16, 19-23. Par
conséquent, il est difficile d’identifier avec précision tous les paramètres clefs qui régissent l’efficacité
du couplage. A ce jour, les paramètres identifiés sont le solvant24, 25, la température14, 16, 20, la
concentration des réactifs1, 7, 13, 14, 20, 26, 27 et, dans le cas d’une réaction dans l’eau, le pH14, 16, 20, 26.
Le choix du solvant conditionne celui des agents de couplage à utiliser. Par exemple, en milieu
aqueux, l’EDAC et le NHS sont très majoritairement employés, alors que le DCC, non soluble dans
l’eau, et l’hydroxybenzotriazole (HOBt) comme co-agent sont très courants pour des couplages en
milieu organique. De plus, la nature du solvant impacte également le mécanisme réactionnel, et par
conséquent l’efficacité du couplage. En effet, la formation de la N-acylurée, inerte chimiquement, est
favorisée en milieu polaire24, 25. L’ajout de co-agents de couplage permet de limiter, voire d’éliminer, ce
phénomène.
L’efficacité du couplage peptidique avec les carbodiimides varie avec la température. La réaction
d’amidation est plus rapide à haute qu’à basse température11, 19. Cependant, en présence d’eau, une
température élevée favorise également la cinétique d’hydrolyse de l’acide activé11. En règle générale,
si le milieu n’est pas anhydre, il est observé que le rendement de la réaction diminue lorsque la
- 161 -

Chapitre 3 : Modification chimique du xanthane
température augmente14, 16, 20, 26. C’est pourquoi, le plus souvent, le couplage en milieu aqueux est
effectué à 4°C ou à température ambiante. Certains auteurs4, 18, 21 parviennent cependant à réaliser
cette réaction dans l’eau à des températures supérieures. Bejenariu et al.4, en particulier, remarquent
que la réaction de réticulation du xanthane avec l’hexanedihydrazide est plus efficace à 90°C qu’à
25°C. Ils attribuent ce phénomène à une diminution de la viscosité du milieu pour T > Tm, du fait de la
transition conformationnelle, ce qui facilite l’accès des agents de couplage et de l’hexanedihydrazide
aux fonctions carboxyliques. Dans tous ces exemples néanmoins, l’amine est introduite en même
temps que les agents de couplage dans le milieu réactionnel, ce qui favorise son addition sur l’acide
activé avant hydrolyse de ce dernier.
De nombreuses études mettent en avant l’impact de la concentration des réactifs sur l’efficacité
du couplage. Cependant, ces mêmes études ont été réalisées sur divers substrats et avec des
concentrations en agents de couplage et/ou amines très variables, ce qui rend la comparaison des
données délicate. Par exemple, le ratio ncarbodiimidenCOOH
(avec ncarbodiimide nombre de moles de
carbodiimide et nCOOH celui d’acide carboxylique) peut varier de 0,220 à plus de 1013, 22, 26, et le ratio
nco−agentncarbodiimide
de 0,05 à 2013, 14, 26. On peut néanmoins remarquer certaines tendances générales. En
premier lieu, l’évolution du rendement selon la concentration des réactifs n’est pas toujours linéaire,
et varie d’un système à l’autre. Ainsi, il a été observé que l’efficacité de la réaction diminue lorsque le
carbodiimide est introduit en large excès par rapport aux fonctions acide carboxylique7, 20, 26 :
l’activation d’un trop grand nombre de fonctions acide favorise la formation de la N-acylurée, inerte
chimiquement24, 28. Comme vu auparavant, pour limiter ce problème, un co-agent de couplage est
ajouté au milieu afin de former un ester activé. Le plus souvent, l’efficacité du couplage augmente
avec la concentration en co-agent13, 14. Certains auteurs14, 21, 22 obtiennent d’ailleurs un rendement
- 162 -

Chapitre 3 : Modification chimique du xanthane
optimal lorsque le ratio nco−agentncarbodiimide
est supérieur ou égal à 1, soit dans des conditions pour
lesquelles l’O-acylisourée est censée être totalement convertie en ester activé. Des résultats inverses
existent cependant dans la littérature, sans qu’il soit possible d’expliquer cette divergence. Sehgal et
al.26, par exemple remarquent que le rendement de la réaction est optimal pour
nco−agentncarbodiimide
=0,07 , puis diminue avec la hausse du ratio. Dulong et al.1 parviennent au même
constat, pour un ratio optimal nco−agentncarbodiimide
=0,04 .
Une autre façon de limiter la formation de la N-acylurée est d’augmenter la quantité d’amine
dans le milieu. Cette méthode implique toutefois que l’amine soit ajoutée en même temps que les
agents de couplage. Dulong et al.27, Sehgal et al.26 et Madison et al.14 constatent ainsi que la densité
de greffage ou la conversion évoluent linéairement avec le ratio namine
ncarbodiimide, mais jusqu’à une
valeur critique, qui diffère fortement d’un auteur à l’autre. Au-delà de cette valeur critique, la densité
de greffage/conversion reste stable, voire diminue. Au final, malgré les quelques tendances globales
notées, il ressort de la comparaison des données bibliographiques que le choix des conditions
optimales de concentration des réactifs pour le couplage dépend fortement du système acide/amine
étudié, et doit être traité au cas par cas.
Enfin, en milieu aqueux, le couplage dépend également du pH. En effet, la réaction entre le
carbodiimide et l’acide carboxylique est optimale lorsque le pH est compris entre 4,5 et 4,75 17, 27, 29. Le
pH acide entraîne la protonation de l’un des deux azotes du carbodiimide, comme indiqué à l’étape
1a de la figure 3-3. Ce phénomène rend le carbone situé entre les deux azotes plus électrophile et
- 163 -

Chapitre 3 : Modification chimique du xanthane
favorise l’attaque de la fonction hydroxyle située sur l’acide. Globalement, l’étape 1 est donc
avantagée à pH acide. Néanmoins, de telles conditions de pH peuvent être défavorables pour l’étape
2, en particulier quand les amines sont constituées de chaînes carbonées sans groupement électro-
attracteur. En effet, ces amines possèdent des valeurs de pKa qui varient entre 10 et 11. Par
conséquent, elles se trouvent sous forme d’ammonium à pH acide, et réagissent très peu avec l’ester
activé. Les deux étapes du couplage présentent donc des pH optimum qui s’avèrent difficilement
compatibles, ce qui est particulièrement problématique lorsque tous les réactifs sont introduits en
même temps dans le milieu.
Pour contourner cette difficulté, certains auteurs4, 14, 27 utilisent des dérivés aminés de faible pKa,
de façon à exécuter l’ensemble du couplage à pH acide. Dulong et al.27 et Bejenariu et al.4 emploient
ainsi l’hexanedihydrazide (pKa compris entre 4 et 5) pour réticuler respectivement le pullulan à pH =
4,6 et le xanthane à pH = 3. A l’inverse, Bartczak et al.7 effectuent le couplage d’un peptide sur des
nanoparticules d’or porteuses de groupements acide à un pH égal à 9, soit proche du pKa de l’amine à
greffer. Néanmoins, plusieurs études14, 20, 26 soulignent que l’efficacité de la réaction diminue lorsque le
pH du milieu réactionnel est trop basique : le pH trop élevé augmente la cinétique d’hydrolyse de l’O-
acylisourée30 et de l’ester activé11. C’est pourquoi la majorité des couplages réalisés en une seule
étape s’effectuent à un pH intermédiaire entre le pKa du carbodiimide et celui de l’amine 7, 18, 21, 23, 26, 31,
32. En règle générale, la gamme de pH retenue varie entre 5,518, 23, 32 et 821, 31.
Une autre méthode consiste à faire varier le pH entre les deux étapes. Les agents de couplage
sont d’abord introduits dans le milieu acide, puis le pH est ajusté pour devenir basique et l’amine est
ajoutée (la situation inverse est aussi envisageable). Lewis et al.16 ont testé cette technique pour le
couplage peptidique, via un ester activé, d’un anticorps humain, cT84.66, sur l’acide 1,4,7,10-
tétraazacyclododécane-1,4,7,10-tétraacétique (ou DOTA), un agent chélatant. Ils observent ainsi que
la densité de greffage maximale est obtenue quand le pH de la première étape est de 5,5, et celui de
la deuxième étape de 7. Madison et al.14 constatent également que le couplage peptidique de l’EDAN
- 164 -

Chapitre 3 : Modification chimique du xanthane
(dichlorhydrate de N-(1-naphthyl) éthylènediamine), une sonde de fluorescence, sur la
carboxyméthylcellulose, est le plus efficace quand le pH est augmenté de 4,75 à 7 entre les deux
étapes.
Pour résumer :
Le couplage peptidique via des esters activés en présence de carbodiimides permet de former
des liaisons amide par un procédé bien connu de la littérature. Ce dernier fait appel à des réactifs
commerciaux bon marché. De plus, il est applicable à des systèmes très divers (de la modification du
polymère à la fonctionnalisation de nanoparticules) et compatible avec une large gamme de solvants
(dont l’eau). Les nombreux exemples bibliographiques indiquent cependant que le choix et
l’optimisation des paramètres expérimentaux dépendent fortement du substrat acide et/ou de
l’amine à greffer. Dans le cadre de ce travail, nous nous sommes appuyés sur cette méthodologie
pour greffer des amines hydrophobes sur le xanthane, sous forme ordonnée ou désordonnée. Dans le
premier cas, la modification chimique est effectuée dans l’eau, dans le second, dans le DMSO, ce qui
implique, à chaque fois, de déterminer les meilleures conditions opératoires pour le trio
polymère/amine/solvant.
- 165 -

Chapitre 3 : Modification chimique du xanthane
3.3. Modification chimique du xanthane sous forme ordonnée
Cette section décrit le processus de modification du xanthane dans l’eau, milieu dans lequel, dans
nos conditions, le polysaccharide est sous forme ordonnée. Plus spécifiquement, deux protocoles ont
été développés : le procédé de greffage à température ambiante et celui à plus basse température.
3.3.1. Processus de modification à température ambiante
3.3.1.1. Description du procédé
Comme mentionné en début de chapitre, le couplage peptidique peut être effectué en une seule
étape, à un pH défini et constant, même lorsque la différence de pKa entre le carbodiimide et l’amine
est importante. Des expériences préliminaires de couplage entre le xanthane et l’octylamine en
appliquant ce type de protocole ont été réalisées. Nous avons testé la réaction à pH = 4,5 (meilleures
conditions pour les agents de couplage), à pH = 7 (intermédiaire entre le pH optimal pour l’EDAC et le
pKa de l’octylamine, égal à 10,6) et à pH = 10 (pH favorable pour l’amine).
A pH = 4,5, le produit obtenu est surtout greffé par des dérivés de l’EDAC, ce qui indique que ces
conditions de pH sont effectivement propices à l’activation de l’acide. En revanche, la chaîne alkyle
provenant de l’amine n’y est présente qu’à l’état de traces. Le pH acide bloque le couplage peptidique
à l’étape 1. Comme nous l’avons vu, à pH = 4,5, l’amine est très majoritairement sous forme
ammonium, perdant ainsi son caractère nucléophile. Elle ne peut donc pas s’additionner sur le
carbone électrophile de l’acide activé. A pH = 7, le polymère modifié comporte un mélange
d’octylamine greffée et de dérivés EDAC sur ses chaînes (densité de greffage d’environ 15 %, avec la
présence de nombreuses impuretés greffées). A pH = 10, le xanthane comporte également des
signaux correspondant à des chaînes octyle greffées et des dérivés d’EDAC. Ces derniers sont en
proportions plus faibles qu’à pH = 7. Néanmoins, la densité de greffage de l’échantillon est
relativement faible (inférieure à 5 %).
- 166 -

Chapitre 3 : Modification chimique du xanthane
Suite à ces résultats, il a été décidé de réaliser le couplage en deux étapes, avec deux pH
différents, comme présenté sur la figure 3-4. L’étape 1 est donc réalisée à pH = 4,5, pour favoriser la
formation de l’ester activé. L’étape 2, quant à elle, a lieu à pH = 10, proche du pKa de l’amine, afin de
faciliter son addition sur l’ester activé.
Figure 3-4 : Procédé de modification chimique du xanthane sous forme ordonnée.
Avant modification chimique, une solution aqueuse de xanthane à 5 g/L est dialysée contre eau
acide à pH = 3 pendant 24 h. Cette étape préliminaire permet de protoner les fonctions acide
carboxylique du xanthane (pKa compris entre 2,6 et 433, 34) et aussi d’éliminer les éventuelles
impuretés, notamment ioniques, qui pourraient être présentes (cations mono et divalents par
exemple). Le milieu réactionnel est obtenu par dilution, à 1 g/L dans de l’eau ultrapure, de la solution
dialysée.
La première étape du procédé consiste en l’activation des fonctions acide carboxylique du
xanthane via la formation d’esters activés. Dans ce but, l’EDAC et le NHS, tous deux solubles dans
l’eau, sont ajoutés dans le milieu à température ambiante. Le pH est ensuite ajusté à 4,5 pour
protoner les atomes d’azote du carbodiimide et ainsi faciliter l’attaque de l’acide (comme décrit
précédemment). Le milieu est alors laissé sous agitation pendant 2 heures.
La deuxième étape correspond à l’addition de l’octylamine sur l’ester activé afin de former la
liaison amide désirée. L’amine est ajoutée dans le milieu, puis le pH est immédiatement ajusté à 10
- 167 -

Chapitre 3 : Modification chimique du xanthane
afin de maintenir son caractère nucléophile et faciliter son addition sur l’ester. La réaction est laissée
sous agitation à température ambiante pendant la nuit.
La purification du milieu réactionnel est réalisée par dialyses à température ambiante. La
première dialyse contre eau acidifiée permet de protoner la chaîne de xanthane, et ainsi d’éliminer
une partie des impuretés ioniques. Par cette méthode, l’octylamine n’ayant pas réagi est également
protonée pour former l’ion ammonium, davantage soluble dans l’eau. Néanmoins, ce dernier, du fait
de sa charge, peut être en interaction avec les chaînes latérales du xanthane. C’est pourquoi cette
étape est suivie d’une dialyse contre NaCl 0,1 M, afin d’effectuer un échange de contre-ions au niveau
du polysaccharide, et plus spécifiquement de remplacer l’ammonium par Na+. En effet, il a été
observé que, en l’absence de cette étape, l’octylamine non greffée restait en interaction de façon
conséquente avec le polymère, même après dialyse intensive contre eau. Le milieu est ensuite mis à
dialyser contre eau distillée 7 jours, puis ultrapure pendant 1 jour, afin d’éliminer les dernières traces
d’octylamine et de diminuer la salinité du milieu, et le produit final récupéré par lyophilisation.
Le terme « précurseur » désigne un xanthane non modifié qui a subi toutes les étapes du
processus de greffage et de purification, mais sans l’ajout des agents de couplage lors de la première
étape du procédé. Il s’agit donc de notre référence pour un xanthane non greffé.
3.3.1.2. Mise en évidence de la conformation ordonnée du xanthane lors de sa
modification
Afin de déterminer la conformation du xanthane au cours du processus de modification, une
fraction du milieu réactionnel a été prélevée à chacune des étapes et analysée par polarimétrie :
- 168 -

Chapitre 3 : Modification chimique du xanthane
Figure 3-5 : Evolution du pouvoir rotatoire normalisé en fonction de la température du milieu réactionnel
(étapes 1 et 2).
L’évolution du pouvoir rotatoire avec la température est semblable pour les deux étapes. De 20 à
50°C environ, le pouvoir rotatoire reste relativement stable. Pour T > 50°C, on observe une hausse
marquée du pouvoir rotatoire avec la température, suivie d’une stabilisation aux alentours des 70-
75°C. Ce signal sigmoïdal est caractéristique de la transition ordre-désordre du xanthane, de sa forme
ordonnée hélicoïdale rigide à basse température, à sa forme désordonnée flexible à haute
température35-40. Dans nos conditions opératoires, le greffage est effectué à température ambiante :
le xanthane est donc modifié sous forme ordonnée. On constate également que la Tm est légèrement
plus basse lorsque le milieu réactionnel est basique (2ème étape). En effet, à pH élevé, les fonctions
acide carboxylique sont sous forme carboxylate, donc chargées. Les répulsions électrostatiques
générées par ces sites défavorisent la conformation hélicoïdale, d’où la diminution de Tm observée,
en accord avec les données de la littérature37, 41, 42.
Au chapitre 1, il a été mentionné que le xanthane sous forme ordonnée adopte une conformation
de simple ou double hélice, constituée de un ou deux brins, suivant les conditions opératoires. Dans
- 169 -
T (°C)
20 30 40 50 60 70 80
norm
alis
é
0.0
0.2
0.4
0.6
0.8
1.0 étape 1étape 2

Chapitre 3 : Modification chimique du xanthane
le cadre de ces travaux, la structure ordonnée exacte du xanthane, modifié ou non, n’a pas été
étudiée. Dans la suite du manuscrit, nous employons le terme « hélice » ou « hélicoïdal(e) » pour
désigner le xanthane en conformation ordonnée, sans présumer de la structure simple ou double de
l’hélice, ni du nombre de brins la composant.
3.3.1.3. Analyse RMN 1H des échantillons et quantification de la densité de greffage
La densité de greffage des xanthanes modifiés a été quantifiée par RMN 1H à 80°C en présence de
références internes, le 2.2.3.3-d(4)-3-(Triméthylsilyl)propanoate de sodium (TSP, pic à 0), et l’acétate
de sodium (pic à 1,91 ppm), pour les polymères décrits dans cette section du manuscrit.
Comme mentionné dans le chapitre 2, il a été observé que la viscosité très importante des
solutions de xanthanes modifiés ralentit fortement la relaxation des protons du polymère : les
spectres obtenus ne permettent pas de quantifier la densité de greffage. Pour s’affranchir de ce
problème, la viscosité des solutions est diminuée par dépolymérisation chimique du polymère via
l’ajout de H2O2 à 80°C en milieu basique pendant une heure. Les échantillons sont ensuite lyophilisés,
puis dissous de nouveau dans une solution de D2O contenant le TSP et l’acétate de sodium. Ce
processus permet d’échanger les protons labiles des groupements hydroxyle par des atomes de
deutérium, et également de limiter le signal de l’eau.
Dans le cadre de ce travail, le xanthane commercial utilisé a des DSA et DSP respectivement égaux
à 87 et 49 % : ces groupements ne sont pas présents sur toutes les unités de répétition du
polysaccharide. De plus, il est possible que les conditions de modification affectent ces degrés de
substitution. Par conséquent, on ne peut pas utiliser directement les pics des fonctions acétate et/ou
pyruvate comme références intrinsèques pour l’intégration, et donc pour la quantification de la
densité de greffage.
- 170 -

Chapitre 3 : Modification chimique du xanthane
Afin de pallier à cet inconvénient, l’utilisation d’une référence interne est une méthode courante
en RMN pour la quantification des spectres. Pour le xanthane, l’acétate de sodium est souvent
employé43-45. Dans notre cas cependant, le protocole de dépolymérisation mis au point avant analyse
RMN entraîne parfois une désacétylation partielle du polymère. En effet, la fonction acétate peut
facilement être ôtée du xanthane par un traitement en milieu basique, et ce, même à température
ambiante37, 46, 47. Des acétates libres provenant du polymère peuvent donc être présents dans le
milieu. Leur signal, en RMN, se superpose à celui de l’acétate de sodium et, par conséquent, fausse
l’intégration de la référence interne et le calcul du degré de substitution en acétate. C’est pourquoi le
TSP a été utilisé comme référence interne pour la quantification des degrés de substitution et de la
densité de greffage des xanthanes modifiés.
Il faut savoir que l’identification de ce problème de référence interne n’a pas été immédiate. De
ce fait, un certain nombre d’échantillons ont été analysés en RMN en présence des deux références
internes. De plus, nous n’avons pas disposé de suffisamment de produit pour analyser de nouveau
ces polymères en l’absence d’acétate de sodium. En outre, il a été observé que le degré de
substitution en acétate pour un xanthane donné conservé sous forme de poudre (après
lyophilisation) diminuait au cours du temps. Par conséquent, il n’a pas été possible de quantifier de
manière précise le DSA des polymères modifiés lorsque l’analyse RMN a été réalisée en présence
d’acétate de sodium. En ce qui concerne la quantification de la densité de greffage, la méthode
présentée dans le chapitre 2 fait intervenir la masse molaire de l’unité de répétition, qui nécessite au
préalable la détermination de DSA. Toutefois, l’erreur sur ce paramètre affecte peu le résultat final. En
effet, faire varier la valeur de DSA entre 39 et 87 % entraîne une variation inférieure à un point sur la
valeur de la densité de greffage, ce qui est acceptable au regard de l’erreur liée à la méthode de
quantification par RMN.
- 171 -

Chapitre 3 : Modification chimique du xanthane
3.3.1.3.1. Description des spectres RMN et détermination des taux d’acétate et de
pyruvate
Les figures 3-6 présentent des exemples de spectres RMN de xanthanes modifiés et de
précurseur synthétisés par ce protocole :
Figures 3-6 : Spectres RMN 1H A : du précurseur. B : d’un xanthane modifié sous forme ordonnée avec une
densité de greffage de 15 %.
- 172 -

Chapitre 3 : Modification chimique du xanthane
Le spectre du précurseur comporte les pics caractéristiques du xanthane, avec un pic à 1,47 ppm
correspondant aux protons du groupement pyruvate, un pic à 2,16 ppm pour ceux de la fonction
acétate et un massif entre 3,39 et 5,25 ppm attribué aux protons des unités osidiques. On constate
que ce composé ne présente aucun signal pouvant être attribué à l’octylamine. Sachant que le
précurseur a été mis en présence de l’amine lors de la deuxième étape du protocole de modification,
ce résultat indique que les différentes phases de dialyse sont très efficaces pour retirer toute trace
d’octylamine non greffée du milieu. Pour le précurseur, le degré de substitution en acétate, DSA, est
estimé à 67 %. La différence de valeur par rapport au DSA du xanthane commercial (de 87 %) fera
l’objet d’une discussion plus loin dans le manuscrit. Le degré de substitution en pyruvate, DSP, est
quant à lui évalué à 45 %. Aux erreurs de quantification RMN près, cette valeur est quasi-identique à
celle du xanthane commercial (DSP égal à 49 %). Les conditions de modification et de dialyse subies
par le précurseur n’affectent donc pas (ou peu) le degré de substitution en pyruvate du xanthane.
En ce qui concerne l’échantillon greffé à hauteur de 15 %, des pics supplémentaires à 0,88 et 1,31
ppm sont observables. Ces derniers correspondent à des protons de l’octylamine greffée : les 10
protons CH2 de la chaîne alkyle à 1,31 ppm (en vert sur la partie B des figures 3-6) et les trois protons
du groupement méthyle terminal à 0,88 ppm (en orange sur la partie B des figures 3-6). Le rapport
des intégrations de ce dernier signal et de celui de la référence TSP nous permet d’accéder à la
densité de greffage du xanthane modifié, suivant la méthode décrite dans le chapitre 2. On peut aussi
noter l’apparition d’un signal mal défini, vers 1,57 ppm, ainsi que d’un pic à 1,66 ppm. Ces derniers
peuvent correspondre aux deux protons du CH2 situé en position β de la fonction amide (en violet sur
les figures 3-6). Ces deux protons ne sont pas équivalents, du fait de leur proximité avec le carbone
asymétrique lié au groupement carbonyle. Ils peuvent donc générer deux signaux différents en RMN :
« l’épaulement » vers 1,57 ppm et le pic à 1,66 ppm. Toutefois, il faut souligner que ce dernier pic
n’est pas systématiquement présent dans les spectres RMN 1H de tous les échantillons. Par
conséquent, il est également possible que le pic à 1,66 ppm corresponde au signal émis par des
- 173 -

Chapitre 3 : Modification chimique du xanthane
impuretés. Les deux protons localisés en position α du groupement amide (en gris sur la partie B des
figures 3-6), par contre, ne sont pas observables sur le spectre. Il est probable que les pics associés à
ces protons se superposent à ceux du massif, entre 3,39 et 5,25 ppm, correspondant aux protons des
unités osidiques.
On remarque également un dédoublement du pic du pyruvate (avec apparition d’un pic
supplémentaire à 1,51 ppm) non présent sur le spectre du précurseur. La fonction pyruvate comporte
des acides carboxyliques, qui forment des amides lors de la réaction avec l’octylamine.
L’environnement chimique des protons du méthyle du pyruvate est différent suivant que le méthyle
est à proximité d’une fonction amide ou acide, ce qui expliquerait ce dédoublement. Ce phénomène,
couplé au recouvrement partiel des signaux des protons du CH2 en position β de la fonction amide
avec ceux du pyruvate, rend la quantification du DSp plus difficile à mettre en œuvre que dans le cas
du précurseur. Dans le cas de cet échantillon, le degré de substitution en pyruvate est estimé à 53 % :
aux erreurs de quantification RMN près, il est identique à celui du xanthane commercial (DS p égal à
49 %). Par ailleurs, en intégrant le pic à 1,51 ppm, on peut grossièrement estimer la proportion de
pyruvates modifiés. Les fonctions acide carboxylique du xanthane sont aussi situées sur l’acide
glucuronique de la chaîne latérale. On connaît la densité de greffage globale du polymère, via le
signal à 0,88 ppm. Il est donc possible d’accéder à la quantité d’acides glucuroniques greffés. Les
résultats indiquent qu’environ la moitié des chaînes alkyle greffées sont localisées sur le groupement
pyruvate. Etant donné que les fonctions pyruvate sont deux fois moins nombreuses que les acides
glucuroniques (DSP = 49 %), on en déduit qu’elles sont deux fois plus réactives. Ce résultat
s’expliquerait par leur plus grande accessibilité dans la conformation hélicoïdale : les pyruvates sont
localisés à l’extrémité de la chaîne latérale, contrairement aux acides glucuroniques. Ce résultat se
retrouve pour tous les xanthanes greffés. En ce qui concerne le degré de substitution en acétate de ce
polymère, il est estimé à 79 %. Cette différence de valeur avec le xanthane commercial sera expliquée
ultérieurement.
- 174 -

Chapitre 3 : Modification chimique du xanthane
3.3.1.3.2. Densité de greffage des xanthanes modifiés suivant le protocole à
température ambiante
Le rapport des intégrales entre le pic des protons du méthyle terminal de la chaîne alkyle greffée
(à 0,88 ppm) et celui de la référence TSP permet de quantifier la densité de greffage de chacun des
xanthanes modifiés suivant le protocole décrit dans le chapitre 2. La densité de greffage est comprise
entre 0 et 22 %, suivant les conditions opératoires utilisées lors du processus de modification. Le
tableau 3-1 résume les degrés de substitution en acétate et pyruvate suivant les conditions
opératoires (avec les réserves émises précédemment sur les valeurs de ce paramètre) et les densités
de greffage obtenues :
Entrée nEDACnCOOH
noctylaminenCOOH
DSA (%) DSP (%) %octyle (%)
1 0 3 60 45 0 (précurseur)
2 1 1 61 40 0
3 2 2 63 39 ≈ 6
4 2,2 2,2 60 41 7
5 2,5 2,5 44 42 13
6 2,7 2,7 67/79 48/53 13/15
7 3,5 3,5 57/44/54 41/40/39 15/18,5/22
8 5 5 19 42 20
Tableau 3-1 : DSA, DSP et densité de greffage des xanthanes modifiés via le processus à température ambiante.
Les / représentent plusieurs essais de modification réalisés dans les mêmes conditions.
Les données relatives au précurseur (entrée 1) correspondent à un échantillon issu du mélange
de deux précurseurs de DSA respectivement égaux à 54 et 67 % et de DSP égaux à 46 et 44,5 %.
Globalement, on peut noter que la densité de greffage augmente avec la quantité de réactifs
(autres que le xanthane) introduits. Néanmoins, on constate également que le procédé de
modification n’est pas toujours répétable. Par exemple, 3 essais de réaction avec un rapport
nEDACnCOOH
égal à 3,5 (entrée 7 du tableau 3-1) donnent trois résultats différents en termes de densité
- 175 -

Chapitre 3 : Modification chimique du xanthane
de greffage, avec un écart maximal entre les densités de 7 points. Ce phénomène sera discuté plus en
détail dans la suite du manuscrit.
On remarque aussi que les valeurs de DSA, comprises entre 19 et 79 %, varient fortement d’un
polymère à l’autre. Comme indiqué précédemment, la fonction acétate peut facilement être
hydrolysée en milieu basique à température ambiante37, 46, 47. Dans le cadre de notre protocole de
modification, la deuxième étape se déroulant pendant une nuit à pH = 10, une fraction des acétates
du xanthane peut être éliminée lors de cette étape. Toutefois, la méthode de dépolymérisation
affecte aussi le DSA, et rend difficile la quantification de ce paramètre. Par conséquent, il est difficile
d’attribuer les variations de DSA observées entre échantillons à la méthode de préparation pour
l’analyse RMN et/ou à la dépolymérisation. Le degré de substitution en pyruvate, quant à lui, est
compris entre 39 et 53 % suivant les échantillons, et ne varie pas au cours du temps. Au regard de la
difficulté, évoquée précédemment, pour estimer ce paramètre et de la précision de la quantification
par RMN, on peut conclure que les conditions de modification et de dialyse n’ont que peu d’impact
sur le DSp. En effet, le groupement pyruvate est beaucoup plus difficile à éliminer que la fonction
acétate. La dépyruvatation du xanthane exige de travailler à pH acide à haute température pendant
plusieurs heures37, 47, 48, conditions non employées dans notre protocole.
Les quantités des réactifs (EDAC, NHS et octylamine) sont calculées par rapport au nombre de
moles de fonctions acide carboxylique du xanthane. En règle générale, il a été observé, dans la
littérature, que le couplage peptidique est plus efficace lorsque le rapport nco−agentncarbodiimide
est
supérieur ou égal à 1. En ce qui concerne l’amine, l’efficacité de la réaction tend à augmenter avec le
rapport namine
ncarbodiimide, cependant la grande variabilité des données bibliographiques sur ce point
- 176 -

Chapitre 3 : Modification chimique du xanthane
rend difficile le choix précis d’une valeur de ce rapport. Dans notre cas, afin de limiter le nombre de
paramètres expérimentaux, nous avons travaillé avec les rapports nNHSnEDAC
= noctylaminenEDAC
= 1.
La figure 3-7 représente l’évolution de la densité de greffage (moyennée suivant le nombre
d’échantillons dans le cas des entrées 6 et 7 du tableau 3-1) par rapport à la quantité des différents
réactifs ajoutés :
nEDAC/nCOOH
0 1 2 3 4 5
%o
ctyl
e
0
5
10
15
20
Figure 3-7 : Evolution de la densité de greffage avec la quantité de réactifs introduits.
Pour nEDACnCOOH
compris entre 0 et 1, la quantité de chaînes alkyle présentes dans les échantillons
est trop faible pour pouvoir être quantifiée par RMN 1H. On observe ensuite une augmentation
marquée, et quasi-linéaire, de la densité de greffage avec les proportions d’agents de couplage et
d’amine jusqu’à un rapport nEDACnCOOH
égal à 3,5 environ. Au-delà, la densité de greffage semble se
stabiliser aux alentours de 20 %.
- 177 -

Chapitre 3 : Modification chimique du xanthane
En premier lieu, le greffage de chaînons alkyle nécessite au minium un rapport nEDACnCOOH
compris entre 1 et 2. Une fraction non négligeable des réactifs ajoutés est donc consommée sans
donner l’amide voulue. Plusieurs explications sont possibles pour ce phénomène. Comme décrit
auparavant, les acides et esters activés s’hydrolysent facilement : une partie des agents est
probablement consommée via cette réaction parasite. Par ailleurs, la modification intervient sur un
polysaccharide de structure complexe : il est probable que les acides soient plus difficiles d’accès que
sur une petite molécule organique ou que sur un polymère linéaire. Le couplage peptidique selon
notre protocole nécessite de travailler en excès non négligeable d’agents de couplage et d’octylamine
afin d’obtenir des densités de greffage mesurables.
Entre 1 ≤ nEDACnCOOH
≤ 3,5, la hausse presque linéaire de la densité de greffage avec la quantité de
réactifs indique qu’il est possible de contrôler à façon le taux de greffons hydrophobes sur le
xanthane suivant la concentration des réactifs. Au-delà de 3,5 équivalents, la densité de greffage
paraît se stabiliser alors que la quantité des différents réactifs (autres que le polymère) augmente.
L’efficacité du couplage peptidique semble moindre lorsque la proportion des réactifs devient trop
importante par rapport aux fonctions acide. Ce résultat est en accord avec les données de la
littérature mentionnées en début de chapitre1, 7, 14, 20, 26, 27, qui attribuent cet état de fait à l’existence
de réactions parasites (formation de N-acylurées, hydrolyse des esters activés, etc.) davantage
favorisées quand les acides sont en large défaut.
- 178 -

Chapitre 3 : Modification chimique du xanthane
Pour résumer :
Le protocole de modification du xanthane à température ambiante décrit dans ce manuscrit
permet de greffer, à façon, des chaînons hydrophobes octyle sur les acides carboxyliques du
polysaccharide sous forme ordonnée. Dans certains cas cependant, il a été remarqué que le procédé
n’est pas complètement répétable. L’identification des paramètres, autres que la concentration en
réactifs, influençant la densité de greffage a donné lieu au développement d’un nouveau protocole
de modification : le processus à basse température lors de la première étape.
3.3.2. Processus de modification à basse température (première étape)
3.3.2.1. Influence de la température sur l’efficacité du couplage peptidique
Comme mentionné auparavant, le protocole de modification du xanthane sous forme ordonnée
présente quelques problèmes de répétabilité. Les entrées 1 et 2 du tableau 3-2 indiquent les densités
de greffage obtenues dans le cas de 3 réactions de couplage peptidique réalisées avec le même
rapport nEDACnCOOH
, égal à 2,7. Les deux premiers échantillons obtenus ont des densités très proches.
La divergence observée dans les valeurs peut s’expliquer par la sensibilité de la quantification par
RMN. Le troisième polysaccharide par contre, présente une densité de greffage inférieure à 5 %.
L’étude des conditions opératoires mises en œuvre lors de ces trois couplages révèle que la première
étape donnant un xanthane modifié à 5 % a été effectuée à 27°C, soit une température plus élevée
que les deux autres synthèses (20°C). Augmenter la température nuirait à l’efficacité du couplage
peptidique14, 16, 20, 26. Afin de confirmer ou non cette hypothèse, plusieurs réactions, effectuées selon le
protocole décrit plus haut, mais avec des températures variables à chaque étape, ont été réalisées :
- 179 -

Chapitre 3 : Modification chimique du xanthane
Entrée nEDACnCOOH
Température (°C)
Etape 1
Température (°C)
Etape 2
%octyle
1 2,7 20 20 13/15
2 2,7 27 24 5
3 2,7 3 25 38
4 3 59 57 4
5 3,3 87 87 0
Tableau3-2 : Evolution de la densité de greffage suivant la température des étapes 1 et 2.
L’entrée 3 correspond à un couplage effectué à basse température lors de la première étape du
protocole. La régulation de ce paramètre est faite via l’utilisation d’un bain de glace, dans lequel le
milieu est plongé au minimum trente minutes pour équilibrage thermique avant ajout des agents de
couplage. Au bout de deux heures, le bain de glace est ôté, puis l’amine ajoutée, le pH ajusté à 10 et
le milieu laissé sous agitation à température ambiante.
Pour les réactions des entrées 1 et 2, le milieu a été laissé sous agitation à température ambiante
non régulée pendant les deux étapes. En ce qui concerne les entrées 4 et 5, il s’agit de couplages
effectués à température constante et égale respectivement à environ 60°C et 87°C pendant les deux
étapes du protocole.
Les températures indiquées dans le tableau 3-2 pour les étapes 2 correspondent à celles relevées
dans le milieu réactionnel après une nuit.
La meilleure densité de greffage est obtenue lorsque la première étape est réalisée à basse
température (entrée 3). Au fur et à mesure que la température augmente, la densité de greffage
diminue. Ainsi, les plus faibles densités de greffage sont observées pour les réactions effectuées à
température élevée (entrée 4 et 5). Les résultats corroborent donc notre hypothèse : accroître la
température impacte négativement l’efficacité du couplage peptidique. Comme mentionné en début
de chapitre, les acides/esters activés peuvent facilement s’hydrolyser9, 11, 12 : en particulier, la cinétique
de cette réaction parasite est d’autant plus importante que la température est élevée 11. Il est, de ce
- 180 -

Chapitre 3 : Modification chimique du xanthane
fait, fort probable que la baisse de rendement du couplage à température élevée s’explique par
l’instabilité thermique des acides/esters activés.
Ces expériences prouvent que le contrôle de la température est primordial pour l’efficacité du
couplage. De plus, elles mettent en évidence que le rendement de la réaction peut être fortement
amélioré en travaillant à basse température au moins lors de la première étape du protocole
(comparaison des %octyle entre les entrées 1 et 3). Dans ce cas précis, la densité de greffage est
multipliée par plus de deux lorsque la température de la première étape est abaissée de 20 à 3°C, ce
qui s’explique par le ralentissement de la cinétique d’hydrolyse des acides activés à basse
température en faveur de l’activation des acides carboxyliques.
3.3.2.2. Densité de greffage des xanthanes modifiés suivant le protocole à basse
température lors de la première étape
Suite à cette étude, le protocole de modification du xanthane décrit précédemment a été
remanié pour prendre en compte l’influence de la température, ce qui a donné lieu au
développement du processus de modification à basse température pendant la première étape (cf.
chapitre 2). Le tableau 3-3 présente les densités de greffage et les degrés de substitution en acétate
et pyruvate des différents xanthanes modifiés obtenus par ce nouveau protocole :
- 181 -

Chapitre 3 : Modification chimique du xanthane
EntréenEDACnCOOH
DSA (%) DSP (%) %octyle
1 0 66 46 0
2 0,3 97 56 4
3 0,5 78 43 8
4 0,7 81 53 8
5 1 90 54 10
6 1,1 66/68 42/48 26/27
7 1,3 69/71/72 50/42/41 24/29/30
8 1,5 79 48 35
9 2 77/70 47/51 27/34
10 2,758/75/77/7
749/49/64/5
135/38/38/39
Tableau 3-3 : DSA, DSP et densité de greffage des xanthanes modifiés via le processus à basse température lors
de la première étape.
Certaines réactions ont été testées plusieurs fois, afin de vérifier la répétabilité du protocole.
L’entrée 7, par exemple, résume les degrés de substitution en acétate (DSA) et pyruvate (DSP) et les
densités de greffage (%octyle) obtenus dans le cas de trois réactions pour lesquelles le rapport
nEDACnCOOH
= 1,3. Ces trois répétitions ont donné trois xanthanes modifiés avec des DSA/%octyle
respectivement de 69/24, 71/29 et 72/30. Les degrés de substitution en acétate/pyruvate et les
densités de greffage ont été quantifiés par RMN 1H suivant la méthode présentée dans la partie
précédente. Toutefois, pour les échantillons de cette section, seul le TSP a été introduit dans les
solutions RMN comme référence interne (sauf l’échantillon de DSA/DSP/%octyle = 77/64/38 de l’entrée
10). Par conséquent, il a été possible d’observer le pic, à 1,91 ppm, correspondant aux acétates
libérés par le xanthane suite à la dépolymérisation à chaud en milieu basique. Le degré de
substitution indiqué dans ce tableau a, de ce fait, été calculé en prenant en compte à la fois
l’intégration du pic des acétates liés au polymère (à 2,16 ppm) et celui des acétates libres.
- 182 -

Chapitre 3 : Modification chimique du xanthane
En premier lieu, le degré de substitution en pyruvate de la majorité des xanthanes modifiés est
compris entre 41 et 56 %, aux erreurs de quantification près. Par ailleurs, comme pour les xanthanes
modifiés avec le protocole à température ambiante, le greffage s’effectue majoritairement sur les
fonctions acide carboxylique du groupement pyruvate. Le degré de substitution en acétate, quant à
lui, varie essentiellement entre 66 et 90 %. Néanmoins, le polymère de l’entrée 3 présente un DSA de
97 %, soit très supérieur à celui du polysaccharide commercial (DSA = 87 %). De même, l’échantillon
de DSA/DSP/%octyle = 77/64/38 de l’entrée 10 se caractérise par un degré de substitution en pyruvate
très élevé. Les méthodes de modification et de dépolymérisation utilisées ne créent pas de nouvelles
fonctions pyruvate et ou acétate. La surestimation des DSP et DSA observée est probablement due à la
présence d’impuretés au sein du polymère, qui génèrent un signal se superposant à celui des protons
du pyruvate et/ou de l’acétate. En règle générale, la plupart des xanthanes greffés ont des DS A et DSP
relativement proches, et voisins de celui du xanthane commercial. C’est pourquoi nous considérons,
par la suite, que ces variations de degré de substitution entre échantillons sont acceptables.
Globalement, à quelques exceptions près, le protocole de modification du xanthane à basse
température influence peu le degré de substitution en acétate, et n’a que peu d’effet sur les
groupements pyruvate.
Comme indiqué précédemment, certaines réactions ont été testées plusieurs fois afin de vérifier
la répétabilité du protocole. Généralement, les différents xanthanes modifiés obtenus pour une
réaction donnée ont des densités de greffage équivalentes (à 2 ou 3 points près). Quelques
exceptions sont toutefois observables. Ainsi, le polymère de DSA/DSP/ %octyle = 69/50/24 (entrée 7), a
une densité de greffage inférieure d’au minimum 5 points à celles des deux autres polysaccharides. Le
xanthane modifié de DSA/DSP/ %octyle = 58/49/35 se caractérise par une densité de greffage
légèrement inférieure à celle des trois échantillons, bien que la différence soit moins marquée que
dans le cas du polymère de l’entrée 7. En règle générale, le protocole de modification du xanthane à
basse température est davantage reproductible que celui à température ambiante.
- 183 -

Chapitre 3 : Modification chimique du xanthane
La figure 3-8 compare l’évolution de la densité de greffage avec la quantité de réactifs introduits,
pour le protocole de modification à température ambiante et celui à basse température :
Figure 3-8 : Evolution de la densité de greffage avec la quantité de réactifs pour le protocole à température
ambiante (symboles fermés) et celui à basse température (symboles ouverts).
Dans le cas des entrées 6 et 9, la densité de greffage indiquée sur le graphique correspond à la
moyenne du %octyle des expériences de répétition. Pour les entrées 7 et 10, les résultats % octyle = 24 %
et %octyle = 35 % n’ont pas été pris en compte dans le calcul.
La courbe associée au protocole à basse température se caractérise par une augmentation
globalement linéaire de la densité de greffage avec la quantité de réactifs. La comparaison des deux
courbes montre que le nouveau protocole de modification permet d’atteindre des densités de
greffage plus élevées que celui à température ambiante. Par exemple, pour nEDACnCOOH
= 2,7, le
protocole à température ambiante permettait d’obtenir un xanthane modifié avec une densité de
greffage comprise entre 13 et 15 %. Dans les mêmes conditions de concentration, le contrôle de la
température lors de la première étape permet de synthétiser un xanthane greffé avec un % octyle
- 184 -
nEDAC/nCOOH
0 1 2 3 4 5
%oc
tyle
0
5
10
15
20
25
30
35
40
Température AmbianteBasse Température

Chapitre 3 : Modification chimique du xanthane
compris entre 35 et 39 %, soit plus du double. De plus, la densité de greffage maximale passe de 22 à
39 % suite au contrôle thermique lors de la première étape. On constate également que le nouveau
protocole est plus économique, car il nécessite moins de réactifs. De plus, il devient possible de
greffer quantitativement des chaînons octyle sur le xanthane pour des rapports nEDACnCOOH
inférieurs
à 1. Enfin, cette caractéristique signifie aussi que le milieu réactionnel est plus facile à purifier, car il
contient moins de réactifs et moins de sous-produits issus des réactions parasites.
- 185 -

Chapitre 3 : Modification chimique du xanthane
Pour résumer :
L’abaissement de la température lors de la première étape du procédé de modification permet de
ralentir les réactions d’hydrolyse parasites des acides/esters activés. Par conséquent, le nouveau
protocole développé permet de greffer de manière plus efficace (que ce soit en termes de
rendement, de purification et/ou de concentrations en réactifs) l’octylamine sur les fonctions acide
carboxylique du xanthane. De plus, le processus n’a pas d’influence non plus sur le degré de
substitution en pyruvate (tout comme celui à température ambiante), et les variations de DS A
observées entre les échantillons restent acceptables. Il serait néanmoins intéressant de mettre au
point un protocole qui permet d’obtenir des xanthanes modifiés avec le même DSA, afin de
s’affranchir complètement de ce paramètre. L’option envisagée est de désacétyler au préalable le
polysaccharide par un traitement en milieu basique avant de le modifier. Globalement, le contrôle de
la température lors de la première étape du couplage permet d’obtenir un processus de modification
plus répétable que le protocole à température ambiante.
Pour finir, il est important de souligner que les xanthanes modifiés sous forme ordonnée par le
procédé à température contrôlée ou non restent solubles dans l’eau, et ce, même à des densités de
greffage élevées. La caractérisation des propriétés physico-chimiques, et en particulier rhéologiques,
de ces composés fera l’objet du chapitre 4.
- 186 -

Chapitre 3 : Modification chimique du xanthane
3.4. Modification chimique du xanthane sous forme désordonnée
Un des objectifs de ces travaux est d’étudier l’impact de la conformation du xanthane greffé sur
ses propriétés en solution, ce qui nécessite de développer également un protocole de modification
de ce polysaccharide sous sa forme désordonnée. Pour rappel, cette conformation est accessible en
solution aqueuse lorsque la température du milieu est supérieure à la température de transition
conformationnelle et/ou la concentration en sel très faible (inférieure à 10-3 M).
3.4.1. Choix du solvant pour le protocole de modification
Greffer des chaînons octyle sur le xanthane sous forme désordonnée en milieu aqueux requiert
de travailler à force ionique nulle et/ou haute température. Or, d’après nos résultats, le xanthane est
sous forme ordonnée dans l’eau à température ambiante lors des deux étapes des protocoles (cf.
figure 3-5). L’ajout des réactifs suffit pour atteindre une force ionique critique qui permet la
stabilisation de la forme hélicoïdale. Il n’est donc pas possible de moduler ce critère. L’augmentation
de la température est également inenvisageable, car elle diminue l’efficacité du couplage
peptidique14, 16, 20, 26. De plus, dans nos conditions de concentration (1 g/L, soit en régime semi-dilué),
il est probable que la dénaturation de l’hélice ne soit que partielle. Il ressort donc, au vu de ces
contraintes, que la modification chimique du xanthane sous forme pelote ne peut pas être réalisée
dans l’eau.
Ce constat impose de trouver un autre solvant de réaction, dans lequel le xanthane est soluble et
sous conformation désordonnée. Certains auteurs49-51 s’accordent à dire que le xanthane existe sous
forme pelote dans le cadoxen, un solvant constitué d’hydroxyde de sodium, de cadmium et
d’éthylènediamine. Toutefois, ce solvant n’est clairement pas adapté pour notre réaction :
l’éthylènediamine entrerait en compétition avec l’octylamine lors du couplage peptidique, et la
présence de NaOH favoriserait l’hydrolyse des fonctions acétate.
- 187 -

Chapitre 3 : Modification chimique du xanthane
Une étude, menée par Dumitriu et al.52, décrit une réaction d’estérification du xanthane réalisée
dans le diméthylsulfoxyde (DMSO) à température ambiante, en présence de DCC. Les auteurs
précisent que, dans leurs conditions, le polysaccharide est partiellement gonflé dans le DMSO. Dans
un premier temps, nous avons donc testé la solubilité de notre xanthane commercial dans ce solvant.
Contrairement à Dumitriu et al.52, nous n’avons pas observé de gonflement de notre polymère qui
s’est avéré non soluble dans le DMSO, que ce soit à température ambiante ou à 80°C.
Il faut savoir que la solubilisation d’espèces chargées dans le DMSO, solvant aprotique, est plus
difficile que dans l’eau, solvant protique. Or, notre xanthane commercial est sous forme de sel de
sodium. L’existence de charges négatives sur le squelette du polysaccharide peut expliquer son
absence de solubilisation dans le DMSO. Ecranter les charges négatives du xanthane, via notamment
une protonation des acides carboxyliques, pourrait faciliter sa solubilisation dans le DMSO. Pour
vérifier cette hypothèse, un xanthane préalablement acidifié par dialyse contre eau acide à pH = 3 a
été mis à solubiliser à 80°C dans le DMSO. Il a été observé que, dans ces conditions, le polysaccharide
est effectivement soluble dans le DMSO. De plus, la solution obtenue reste homogène après retour à
température ambiante. L’acidification du xanthane est, par conséquent, essentielle pour sa
solubilisation dans ce solvant.
3.4.2. Mise en évidence de la conformation désordonnée du xanthane dans le DMSO
Des analyses de polarimétrie et de dichroïsme circulaire ont été réalisées sur les échantillons de
xanthane dissous dans le DMSO, afin de déterminer la conformation adoptée par les chaînes dans ce
solvant. Les résultats sont représentés dans les figures 3-9 :
- 188 -

Chapitre 3 : Modification chimique du xanthane
Figures 3-9 A : Evolution du pouvoir rotatoire de XDan1 1g/L dans l’eau ([NaCl] = 0,01 M, symboles fermés) ou
le DMSO (symboles ouverts). B : Evolution de l’ellipticité, en fonction de la longueur d’onde λ pour les mêmes
solutions à température ambiante, chemin optique de 0,2 cm (même légende que la figure A).
La figure 3-9 A représente le pouvoir rotatoire d’une solution de xanthane, dans une solution
aqueuse de NaCl à 0,01 M et dans le DMSO, en fonction de la longueur d’onde. Dans le cas du
xanthane solubilisé dans l’eau, l’évolution du pouvoir rotatoire de la solution de xanthane selon la
température suit une allure sigmoïdale, caractéristique de la transition ordre-désordre 35-40. A
température ambiante, le polysaccharide est sous forme ordonnée, et subit sa transition
conformationnelle entre 60°C et 70°C dans ces conditions. En ce qui concerne le xanthane solubilisé
dans le DMSO, le pouvoir rotatoire se caractérise par une évolution avec la température très
différente de celle du même polymère dans l’eau. En effet, reste relativement constant sur
l’ensemble de la gamme de températures. Par conséquent, le polymère est soit sous forme ordonnée,
soit sous forme désordonnée dans le DMSO sur toute la gamme de températures.
Afin de répondre à cette question, les mêmes solutions ont été analysées en dichroïsme
circulaire. La figure 3-9 B montre l’évolution de l’ellipticité, , en fonction de la longueur d’onde λ
- 189 -
T (°C)
20 30 40 50 60 70 80 90
Pouvo
ir ro
tato
ire (°)
-0.03
-0.02
-0.01
0.01
EauDMSO
l (nm)
200 210 220 230 240 250 260 270q
(mdeg)
-4
-2
0
2
4EauDMSO
A B

Chapitre 3 : Modification chimique du xanthane
pour les mêmes solutions de xanthane. Dans le cas du polymère dans l’eau, on observe une bande
positive de forte intensité à 205 nm, correspondant aux fonctions acide carboxylique, suivie d’une
bande négative moins intense vers 220 nm, attribuée aux groupements acétate. Cette allure du signal
confirme que les chaînes adoptent en solution aqueuse une conformation ordonnée hélicoïdale 35, 39, 46,
53. L’allure de la bande à 205 nm, en particulier, nous renseigne sur le degré d’organisation des
chaînes. Capron et al.53 et Morris et al.35 ont ainsi étudié l’évolution de cette bande avec la
température. Dans les deux cas, son intensité diminue quand la température augmente, ce qui
traduit l’adoption, par les chaînes, d’une structure de moins en moins ordonnée. Dans le cas du
xanthane solubilisé dans le DMSO, on remarque l’absence totale de cette bande, ainsi que celle à 220
nm : le signal reste négatif et constant entre 196 et 240 nm. Ce résultat montre donc que les chaînes
sont sous forme désordonnée dans le DMSO.
3.4.3. Protocole de modification du xanthane dans le DMSO
3.4.3.1. Choix des conditions expérimentales lors de la modification chimique
Le DMSO est un solvant usuel pour réaliser un couplage peptidique. Généralement, le
dicyclohexylcarbodiimide (DCC), en combinaison ou non avec l’hydroxybenzotriazole (HOBt) et/ou la
4-diméthylaminopyridine (DMAP) sont employés. Dumitriu et al.52 utilisent d’ailleurs ce carbodiimide
pour estérifier le xanthane dans le DMSO. Dans notre cas cependant, il a été observé que le couplage
peptidique est très peu efficace en présence de DCC, couplé ou non avec un co-agent. En effet, la
meilleure densité de greffage obtenue ne dépasse pas 8 % avec ce carbodiimide, pour des quantités
de réactifs pouvant aller jusqu’à 12 équivalents par rapport aux fonctions acide. De plus, lors de la
purification, on note l’apparition d’une poudre blanche très fine, impossible à éliminer du milieu.
Cette dernière correspond probablement au dérivé urée du DCC, formé en présence d’eau et
partiellement soluble dans tous les solvants. De même, l’efficacité de la réaction est relativement
faible lorsque l’EDAC, seul ou avec le NHS, est utilisé : l’analyse des spectres RMN révèle des densités
- 190 -

Chapitre 3 : Modification chimique du xanthane
de greffage qui ne dépassent pas les 5 %, et la présence de nombreuses impuretés fixées sur le
polymère.
La mise au point du protocole de modification du xanthane dans le DMSO a, de ce fait, nécessité
d’identifier le système d’activation le plus efficace dans ce solvant. Dans notre cas, il s’est avéré que le
couple EDAC/HOBt était le plus efficace pour notre couplage dans le DMSO. De plus, comme
expliquée dans la section précédente, la solubilisation du xanthane dans ce solvant requiert que le
polysaccharide soit préalablement acidifié. Cette opération est effectuée par dialyse contre eau acide,
à pH = 3, pendant 24 h, d’une solution aqueuse de xanthane, ensuite lyophilisée. Le solide est dissous
à 80°C dans du DMSO jusqu’à solubilisation complète du polysaccharide (entre 2 et 4 h, contrôle
visuel). Le milieu est alors laissé au repos à température ambiante jusqu’à ce que la température soit
comprise entre 20 et 25°C. En effet, tout comme le procédé de modification du xanthane sous forme
ordonnée, il a été remarqué que l’efficacité du couplage diminue d’autant plus que la température
réactionnelle est élevée. Néanmoins, le DMSO cristallise à partir de 19°C, ce qui nous empêche de
travailler à plus basse température, contrairement au processus de modification dans l’eau.
Le couplage s’effectue à température ambiante, selon les deux étapes décrites auparavant :
activation des acides carboxyliques (via la formation d’esters activés) par ajout du 1-éthyl-3-(3-
diméthylaminopropyl)carbodiimide (EDAC) et de l’hydrozybenzotriazole (HOBt) pendant 2 h, puis
addition de l’octylamine dans le milieu, laissé ensuite sous agitation à température ambiante pendant
la nuit. Les différents réactifs sont tous ajoutés dans les mêmes proportions par rapport au nombre
de moles de fonctions acide carboxylique, soit des rapports nEDACnCOOH
= nHOBtnCOOH
= noctylaminenCOOH
(pour plus de détails, se référer au chapitre 2).
- 191 -

Chapitre 3 : Modification chimique du xanthane
3.4.3.2. Choix des étapes de purification
Les étapes de purification sont plus nombreuses et complexes que celles employées pour les
procédés sous forme ordonnée. En effet, une première dialyse contre DMSO, pendant 24 h, est
réalisée, afin d’éliminer une partie de l’octylamine non greffée. Elle est suivie d’une dialyse contre un
mélange eau/isopropanol (70/30 en volume) à pH = 3, pour protoner le xanthane et l’amine en
ammonium. Puis une dialyse eau salée (NaCl 0,1 M)/isopropanol (70/30 en volume) est réalisée afin
d’effectuer des échanges de contre-ions au niveau des chaînes, et notamment remplacer
l’ammonium par Na+. Une deuxième dialyse contre DMSO est alors effectuée pour ôter l’ammonium.
Le milieu est par la suite purifié par dialyses successives contre eau distillée puis ultrapure. Le choix
de telles étapes de purification s’explique par le fait que nous envisageons ultérieurement de pouvoir
greffer des chaînes alkyle plus longues sur le xanthane. En effet, le processus de modification dans le
DMSO offre l’avantage de permettre de solubiliser plus facilement de longues chaînes alkyle aminées
que celui dans l’eau. C’est pourquoi nous avons cherché à développer des étapes de dialyse qui
permettraient d’éliminer ce type de composés très peu, voire pas solubles dans l’eau. Des
expériences préliminaires de greffage de dodécylamine sur le xanthane dans le DMSO ont montré
que les étapes de purification sont efficaces pour éliminer en majorité cette amine non greffée du
milieu.
3.4.3.3. Interprétation des spectres RMN 1H pour les xanthanes modifiés dans le
DMSO
Les échantillons ont été caractérisés par RMN du proton à 80°C, en présence de TSP comme
référence interne pour la quantification de la densité de greffage, selon le protocole décrit dans le
chapitre 2. Les figures 3-10 représentent le spectre RMN du précurseur (partie A) et celle d’un
xanthane greffé à hauteur de 19 % dans le DMSO (partie B) :
- 192 -

Chapitre 3 : Modification chimique du xanthane
Figures 3-10 : Spectres RMN 1H A : du précurseur. B : d’un xanthane modifié dans le DMSO avec une densité de
greffage de 19 %.
Pour le précurseur, on observe les pics caractéristiques du xanthane : celui des protons du
méthyle du pyruvate à 1,46 ppm, et ceux du groupement acétate (libre suite à la dépolymérisation
avant analyse à 1,91 ppm et lié au polymère à 2,16 ppm), ainsi que les protons des unités osidiques
entre 3,39 et 5,23 ppm. Globalement, le spectre est très similaire à celui du précurseur obtenu par le
protocole dans l’eau. Les degrés de substitution en acétate et pyruvate (respectivement de 87 et 54
- 193 -

Chapitre 3 : Modification chimique du xanthane
%) sont identiques à ceux du xanthane commercial, aux erreurs de quantification RMN près : le
protocole de modification dans le DMSO ainsi que les étapes de dialyse n’ont pas d’impact sur ces
groupements. Par ailleurs, l’absence de tout pic correspondant à l’octylamine confirme que les étapes
de purification utilisées permettent d’éliminer efficacement l’amine non greffée du milieu.
En ce qui concerne le xanthane greffé à hauteur de 19 %, des pics caractéristiques de l’octylamine
greffée sont observables (à 0,88 ppm, 1,29 et l’épaulement vers 1,57 ppm, qui correspondent
respectivement aux protons du méthyle terminal et aux protons des CH2). On constate aussi que les
signaux associés aux protons du groupement pyruvate (entre 1,46 et 1,67 ppm) sont plus complexes
que dans le cas des xanthanes modifiés dans l’eau. En effet, on observe les deux pics de forte
intensité, à 1,46 et 1,51 ppm, pour les pyruvates non greffés et ceux greffés par la chaîne octyle, mais
également deux signaux mal définis et de plus faible intensité à 1,55 et 1,57 ppm, au niveau de
l’épaulement. Ces deux signaux ne sont observés ni dans le spectre de l’octylamine, ni dans celui des
agents de couplage ou du précurseur, ce qui laisse penser qu’ils sont spécifiques des échantillons
modifiés. En outre, le degré de substitution en pyruvate est de 48 %, soit identique à celui du
polysaccharide commercial, si l’ensemble des pics entre 1,46 et 1,67 ppm est pris en compte. De ce
fait, il semble que ces signaux supplémentaires correspondent aux protons du méthyle du pyruvate
en présence d’un environnement chimique autre que l’acide ou la chaîne octyle. Cependant,
l’intensité de ces deux signaux est relativement faible. Par ailleurs, on remarque de nouveau un pic à
1,67 ppm, qui pourrait correspondre soit à un des protons non équivalents du CH2 en position béta de
la fonction amide, soit à une impureté. Le degré de substitution en acétate, quant à lui, est de 72 %,
soit inférieur à celui de l’échantillon commercial (DSA = 87 %). Ici également, il n’est pas possible de
dire si cette différence est due au protocole de modification et/ou à l’étape de dépolymérisation.
D’autres pics supplémentaires sont également observables : deux triplets (respectivement à 1,09
et 1,17 ppm) et deux pics partiellement dédoublés à 2,91 et 3,18 ppm. Ces pics indiquent la présence
- 194 -

Chapitre 3 : Modification chimique du xanthane
d’impuretés en proportion non négligeables sur le xanthane (notées Imp. sur la partie B des figures 3-
10). De plus, ces impuretés sont présentes dans l’échantillon même après plusieurs cycles de dialyses
contre DMSO, et contre des mélange 70/30 en volume d’eau acide/isopropanol ou d’eau
salée/isopropanol, ce qui suggère qu’elles sont greffées sur le polymère.
La comparaison de ce spectre avec celui des agents de couplage (EDAC et HOBt), le précurseur et
un xanthane ayant subi uniquement la première étape du protocole de modification dans le DMSO
semble indiquer que ces impuretés sont des dérivés de l’EDAC (un anhydride de l’acide activé et/ou
une N-acylurée). Ce constat montre, en premier lieu, que l’HOBt ne réagit pas complètement avec
l’acide activé, alors qu’il est ajouté en quantités stœchiométriques par rapport au carbodiimide. En
effet, si la réaction entre l’acide activé et l’HOBt était totale, l’espèce parasite observée serait un
dérivé de l’HOBt. Il est possible qu’une partie du co-agent soit perdue lors de réactions parasites, d’où
le résultat obtenu.
De plus, il se pose également la question de la nature des dérivés d’EDAC greffés. En effet, la
situation est moins problématique si ces composés sont un anhydride que la N-acylurée
correspondante. Dans le premier cas, cette espèce est toujours réactive, et peut être éliminée en
favorisant la deuxième étape (via l’augmentation de la quantité d’amine introduite par exemple).
Dans le dernier cas par contre, la N-acylurée nuit à l’efficacité du couplage peptidique : étant inerte
chimiquement, elle empêche l’addition de l’amine sur l’acide. L’obtention de la N-acylurée peut être
provoquée soit par un défaut en HOBt (suite à des réactions parasites), soit par le fait que la
formation de cette espèce est plus rapide que l’addition du HOBt sur l’acide activé. Pour rappel,
certaines des études24, 25 présentées en début de chapitre ont montré que la cinétique d’obtention de
la N-acylurée est accélérée avec la polarité du milieu. Si ce cas de figure s’avère exact, il peut être
alors nécessaire de revoir entièrement le processus de modification, afin de limiter autant que
possible la formation de ce composé. Les pistes envisagées peuvent être la modification des
- 195 -

Chapitre 3 : Modification chimique du xanthane
quantités des différents réactifs, les réactifs eux-mêmes, la durée de chacune des étapes, voire leur
nombre.
Il faut savoir que ces impuretés greffées se retrouvent sur tous les spectres des xanthanes
modifiés dans le DMSO. De plus, leur proportion n’est pas négligeable. Par conséquent, il est difficile
de savoir si le comportement en solution des xanthanes modifiés dans le DMSO (caractérisé dans le
chapitre suivant) a purement pour origine les chaînons alkyle, ou bien si ces impuretés y participent
également (et à quelle hauteur). Il apparaît donc nécessaire, en perspectives, de parvenir à identifier
la nature de ces composés et d’optimiser le procédé de modification afin de limiter leur formation,
voire la prévenir totalement. Par ailleurs, ces impuretés rendent l’interprétation du spectre RMN 1H,
ainsi que les quantifications des degrés de substitution (en particulier des pyruvates) et de la densité
de greffage plus délicates à mettre en œuvre.
3.4.3.4. Densité de greffage estimée des xanthanes modifiés suivant le protocole dans
le DMSO
Le tableau 3-4 indique les degrés de substitution en acétate et pyruvate et les densités de
greffage obtenues pour différents xanthanes modifiés dans le DMSO :
EntréenEDACnCOOH
DSA DSP %octyle
1 1 87 54 0
2 0,3 83 51 11
3 0,4 86 42 4
4 0,5 72/104 48/63 19/23
5 0,6 78 47 27
6 0,7 100 56 35
7 1 70/71 44/44 40/44
Tableau 3-4 : DSA, DSP et densité de greffage estimée des xanthanes modifiés selon le processus dans le DMSO.
- 196 -

Chapitre 3 : Modification chimique du xanthane
Globalement, le degré de substitution en acétate des xanthanes greffés est compris entre 70 et
87 %, soit relativement proche, voire identique, à celui du polymère commercial. Le procédé de
modification et de purification n’entraîne donc pas, ou peu, de désacétylation. On remarque
cependant que deux échantillons ont des valeurs de DSA supérieures ou égales à 100 % (entrées 4 et
6). Les conditions de modification et de dépolymérisation ne créant pas de nouveaux groupements
acétate, cette surestimation du DSA implique la présence d’impuretés dans le milieu. On constate
d’ailleurs que les deux échantillons concernés présentent des degrés de substitution en pyruvate de
56 et 63 %, ce qui confirme l’existence de nombreux sous-produits dans ces polymères. En ce qui
concerne le DSP, ce dernier est compris entre 42 et 54 % (sans prendre en compte les deux polymères
précédents). Au regard de la difficulté à estimer ce paramètre et aux erreurs de quantification RMN
près, nous pouvons conclure que les groupements pyruvate ne sont pas, ou peu affectés, par le
procédé de modification du xanthane dans le DMSO
On constate que la densité de greffage augmente avec le rapport nEDACnCOOH
, tout comme pour
les protocoles de greffage dans l’eau. Par ailleurs, les quelques essais de répétition menés sur
certaines expériences (entrées 4 et 8) semblent indiquer que le protocole est relativement répétable.
- 197 -

Chapitre 3 : Modification chimique du xanthane
3.4.3.5. Comparaison de la densité de greffage entre les protocoles de modification
sous forme ordonnée et sous forme désordonnée
A partir de ces données, il est possible de tracer l’évolution de la densité de greffage en fonction
de la quantité de réactifs introduits, comme illustré sur la figure 3-11 :
Figure 3-11 : Evolution de la densité de greffage suivant la quantité de réactifs pour le protocole de modification
dans le DMSO (symboles pleins) et celui dans l’eau à basse température (symboles ouverts).
Dans le cas du processus de modification dans le DMSO, la densité de greffage augmente très
rapidement, et presque linéairement, avec la quantité d’agents de couplage et d’amine. On obtient
ainsi des densités de greffage relativement élevées, aux alentours des 40 %, ce qui correspond à la
fonctionnalisation d’environ 1/4 des acides carboxyliques du polymère. Pour rappel, le xanthane
comporte 1,5 fonction carboxylique par unité de répétition, ce qui équivaut à une densité de greffage
maximale de 150 %.
La comparaison des courbes entre le protocole dans l’eau et celui dans le DMSO montre que le
procédé de modification dans le DMSO est beaucoup plus efficace que celui dans l’eau, même à
température contrôlée. En effet, les densités de greffage des xanthanes modifiés dans le DMSO sont
- 198 -
nEDAC/nCOOH0,0 0,5 1,0 1,5 2,0 2,5
%oc
tyle
0
10
20
30
40
DMSOBasse Température

Chapitre 3 : Modification chimique du xanthane
supérieures en tout point, et pour des équivalents en réactifs bien moindres, que ceux obtenus dans
l’eau. Ce phénomène peut s’expliquer par le fait que les chaînes du polysaccharide, dans le DMSO,
sont sous forme désordonnée. Par conséquent, les acides carboxyliques sont davantage accessibles
pour réagir. De plus, il est aussi possible que les réactifs, notamment l’amine, soient davantage
réactifs dans le DMSO que dans l’eau. Dans tous les cas, le protocole de modification dans le DMSO
requiert moins de réactifs que celui dans l’eau.
Pour résumer :
Le protocole développé dans le DMSO permet donc de greffer des chaînes octylamine sur les
fonctions acide carboxylique du xanthane sous sa conformation désordonnée. A notre connaissance,
il s’agit du premier exemple de modification chimique du xanthane sous cette forme. De plus, le
procédé s’avère beaucoup plus efficace, en termes de quantité de réactifs à ajouter et de densité de
greffage finale, que les processus dans l’eau. Toutefois, le processus de modification dans le DMSO
présente des inconvénients. En premier lieu, l’analyse des spectres RMN des composés obtenus
révèlent la présence d’impuretés greffées qui semblent correspondre à des dérivés de carbodiimide,
et ce en proportions non négligeables. Actuellement, la nature exacte de ces impuretés (N-acylurée
ou anhydride) n’a pas été identifiée. De plus, nous ignorons si ces espèces influencent, ou non, le
comportement des polymères greffés dans l’eau. Il apparaît donc que l’optimisation du protocole
développé soit nécessaire pour empêcher la formation de ces produits secondaires sur le xanthane.
De plus, il a été observé que tous les xanthanes modifiés obtenus par le protocole dans le DMSO,
même à faible densité de greffage, ne soient que partiellement solubles dans l’eau. Ceci complique la
caractérisation de leurs propriétés physico-chimiques et rhéologiques. En particulier, certains
échantillons très greffés (de l’ordre de 40 %) présentent des phénomènes de séparation de phase
lorsqu’ils sont mis à solubiliser en milieu aqueux. Ces résultats seront discutés plus en détail dans le
chapitre suivant.
- 199 -

Chapitre 3 : Modification chimique du xanthane
3.5. Conclusion
Les résultats présentés dans ce chapitre montrent qu’il est possible de greffer chimiquement et à
façon des chaînons hydrophobes de longueur C8 sur les fonctions acide carboxylique du xanthane soit
sous forme ordonnée, soit sous forme désordonnée. A notre connaissance, la modification du
xanthane sous cette dernière conformation n’a, à ce jour, pas été décrite dans la littérature. Le choix
du solvant, l’eau ou le DMSO, et des agents de couplage adaptés, permet de modifier le xanthane
sous l’une ou l’autre de ses formes. Dans les deux cas, la réaction employée est un couplage
peptidique qui fait appel aux carbodiimides, des agents de couplage très courants en chimie
peptidique et en modification chimique de polymères naturels ou synthétiques, ainsi que des co-
agents usuels, le NHS (dans l’eau) ou le HOBt (dans le DMSO).
En ce qui concerne le protocole de modification sous forme ordonnée, certains problèmes de
répétabilité rencontrés avec le procédé à température ambiante ont permis de mettre en évidence
l’influence majeure de la température sur l’efficacité du couplage. Ce constat a donné lieu au
développement du processus de modification à basse température, qui permet d’obtenir plus
efficacement des xanthanes amphiphiles avec des densités de greffage pouvant atteindre 40 %. De
plus, de tels échantillons restent solubles dans l’eau, même avec des densités de greffage aussi
élevées. Ce nouveau procédé offre notamment l’avantage, par rapport au précédent, d’utiliser moins
de réactifs et d’être davantage répétable.
Le processus de modification du xanthane sous forme désordonnée permet, quant à lui, de
synthétiser des polysaccharides amphiphiles greffés à hauteur de 40 % à des concentrations en
réactifs moindres par rapport au protocole de modification dans l’eau. Néanmoins, les polymères
obtenus comportent a priori des impuretés (probablement des dérivées de l’EDAC) greffées sur leurs
chaînes, qui peuvent interférer lors de la caractérisation du comportement en solution de ces
xanthanes. Une optimisation de la méthode de modification dans le DMSO est donc nécessaire pour
limiter, voire prévenir, la formation de ces produits secondaires. Par ailleurs, tous les polysaccharides
- 200 -

Chapitre 3 : Modification chimique du xanthane
amphiphiles synthétisés via ce protocole ne sont que partiellement solubles dans l’eau. Ce point sera
décrit en détail dans le chapitre suivant, consacré à l’étude des propriétés physico-chimiques des
xanthanes amphiphiles en milieu aqueux.
- 201 -

Chapitre 3 : Modification chimique du xanthane
Références bibliographiques
1. Dulong, V.; Mocanu, G.; Picton, L.; Le Cerf, D., Amphiphilic and thermosensitive copolymersbased on pullulan and Jeffamine®: Synthesis, characterization and physicochemical properties.Carbohydrate Polymers 2012, 87, 1522-1531.
2. Yang, L.; Zhang, B.; Wen, L.; Liang, Q.; Zhang, L.-M., Amphiphilic cholesteryl grafted sodiumalginate derivative: synthesis and self-assembly in aqueous solution. Carbohydrate Polymers 2007,68, 218-225.
3. Laurienzo, P.; Malinconico, M.; Motta, A.; Vicinanza, A., Synthesis and characterization of anovel alginate-poly(ethylene glycol) graft copolymer. Carbohydrate Polymers 2005, 62, 274-282.
4. Bejenariu; Popa, M.; Le Cerf, D.; Picton, L., Stiffness xanthan hydrogels: synthesis, swellingcharacteristics and controlled release propertis. Polymer Bulletin 2008, 61, 631-641.
5. Colinet, I.; Dulong, V.; Hamaide, T.; Le Cerf, D.; Picton, L., New amphiphilic modifiedpolysaccharides with original solution behaviour in salt media. Carbohydrate Polymers 2009, 75, 454-462.
6. Barras, A.; Szunerits, S.; Marcon, L.; Monfilliette-Dupont, N.; Boukherroub, R.,Functionalization of diamond nanoparticles using click chemistry. Langmuir 2010, 26, 13168-13172.
7. Bartczak, D.; Kanaras, A., Preparation of peptide-functionalized gold nanoparticules usingone-pot EDC/sulfo-NHS coupling. Langmuir 2011, 27, 10119-10123.
8. El-Faham, A.; Albericio, F., Peptide coupling reagents, more than a letter soup. ChemicalReview 2011, 111, 6557-6602.
9. Hoare, D. G.; Koshland, D. E., A method for the quantitative modification and estimation ofcarboxylic acid groups in proteins. Journal of Biological Chemistry 1967, 242, 2447-2453.
10. Timkovich, R., Detection of the stable addition of carbodiimide to proteins. AnalyticalBiochemistry 1977, 79, 135-143.
11. Anjaneyulu, P. S. R.; Staros, J. V., Reactions of N-hydroxysulfosuccinimide active esters.International Journal of Peptide and Protein Research 1987, 30, 117-124.
12. Grabarek, Z.; Gergely, J., Zero-length crosslinking procedure with the use of active esters.Analytical Biochemistry 1990, 185, 131-135.
13. Staros, J. V.; Wright, R. W.; Swingle, D. M., Enhancement by N-hydroxysulfosuccinimide ofwater-soluble carbodiimide-mediated coupling reactions. Analytical Biochemistry 1986, 156, 220-222.
14. Madison, S. A.; Carnali, J. O., pH optimization of amidation via carbodiimides. Industrial &Engineering Chemistry Research 2013, 52, 13547-13555.
15. Lewis, M. R.; Raubitschek, A.; Shively, J. E., A facile, water-soluble method for modification ofproteins with DOTA. Use of elevated temperature and optimized pH to achieve high specific activityand high chelate stability in radiolabeled immunoconjugates. Bioconjugate Chemistry 1994, 5, 565-576.
16. Lewis, M. R.; Kao, J. Y.; Anderson, A.-L. J.; Shively, J. E.; Raubitschek, A., An improved methodfor conjugating monoclonal antibodies with N-hydroxysulfosuccinimidyl DOTA. BioconjugateChemistry 2001, 12, 320-324.
- 202 -

Chapitre 3 : Modification chimique du xanthane
17. Souguir, Z.; Roudesli, S.; About-Jaudet, E.; Le Cerf, D.; Picton, L., Synthesis and physico-chemical characterization of a novel ampholytic pullulan derivative with amphiphilic behavior inalkaline media. Journal of Colloid & Interface Science 2007, 313, 108-116.
18. Tiong, W. H. C.; Damodaran, G.; Naik, H.; Kelly, J. L.; Pandit, A., Enhancing amine terminals inan amine-deprived collagen matrix. Langmuir 2008, 24, 11752-11761.
19. Tsubokawa, N.; Hosoya, M.; Kurumada, J., Grafting reaction of surface carboxyl groups oncarbon black with polymers having terminal hydroxyl or amino groups using N,N'-dicyclohexylcarbodiimide as a condensing agent. Reactive and Functional Polymers 1995, 27, 75-81.
20. Wang, Y.; Hsieh, Y.-L., Enzyme immobilization to ultra-fine cellulose fibers via amphiphilicpolyethylene glycol spacers. Journal of Polymer Science Part A: Polymer Chemistry 2004, 42, 4289-4299.
21. Follain, N. g.; Montanari, S.; Jeacomine, I.; Gambarelli, S.; Vignon, M. R., Coupling of amineswith polyglucuronic acid: Evidence for amide bond formation. Carbohydrate Polymers 2008, 74, 333-343.
22. Decostaire, I. P.; Lelièvre, D.; Zhang, H.; Delmas, A., Controlling the outcome of overacylationof N-protected aminooxyacetic acid during the synthesis of an aminooxy-peptide for chemicalligation. Tetrahedron Letters 2006, 47, 7057-7060.
23. Mendes, A. C.; Baran, E. T.; Reis, R. L.; Azevedo, H. S., Fabrication of phospholipid - xanthanmicrocapsules by combining microfluidics with self-assembly. Acta Biomaterialia 2013, 9, 6675-6685.
24. Palazon, F.; Montenegro Benavides, C.; Léonard, D.; Souteyrand, E.; Chevolot, Y.; Cloarec, J.-P.,Carbodiimide/NHS derivatization of COOH-terminated SAMs: Activation or byproduct formation?Langmuir 2014, 30, 4545-4550.
25. Singh, J.; Rich, D. H., The carbodiimide method. In The Peptides: Analysis, Synthesis, Biology,Academic Press: New York, 1979; Vol. 1, pp 241-261.
26. Sehgal, D.; Vijay, I. K., A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Analytical Biochemistry 1994, 218, 87-91.
27. Dulong, V.; Le Cerf, D.; Picton, L.; Muller, G., Carboxymethylpullulan hydrogels with an ionicand/or amphiphilic behavior: Swelling properties and entrapment of cationic and/or hydrophobicmolecules. Colloids and Surfaces A: Physicochem. Eng. Aspects 2006, 274, 163-169.
28. Sam, S.; Touahir, L.; Salvador Andresa, J.; Allongue, P.; Chazalviel, J. N.; Gouget-Laemmel, A.C.; Henry de Villeneuve, C.; Moraillon, A.; Ozanam, F.; Gabouze, N.; Djebbar, S., Semiquantitativestudy of the EDC/NHS activation of acid terminal groups at modified porous silicon surfaces.Langmuir 2009, 26, 809-814.
29. Kuo, J. W.; Swann, D. A.; Prestwich, G. D., Chemical modification of hyaluronic acid bycarbodiimides. Bioconjugate Chemistry 1991, 2, 232-241.
30. Lei, P. Q.; Lamb, D. H.; Heller, R. K.; Shannon, A. G.; Ryall, R.; Cash, P., Kinetic studies on therate of hydrolysis of N-ethyl-N'-(dimethylaminopropyl)carbodiimide in aqueous solutions using massspectrometry and capillary electrophoresis. Analytical Biochemistry 2002, 310, 122-124.
31. Bokias, G.; Mylonas, Y.; Staikos, G.; Bumbu, G. G.; Vasile, C., Synthesis and aqueous solutionproperties of novel thermoresponsive graft copolymers based on a carboxymethylcellulose backbone.Macromolecules 2001, 34, 4958-4964.
32. Mendes, A. C.; Baran, E. T.; Numes, C.; Coimbra, M.; Azevedo, H.; Reis, R. L., Palmitoylation ofxanthan polysaccharide for self-assembly microcapsule formation and encapsulation of cells inphysiological conditions. Soft Matter 2011, 7, 9647-9658.
- 203 -

Chapitre 3 : Modification chimique du xanthane
33. Holzwarth, G., Conformation of the extracellular polysaccharide of Xanthomonas campestris.Biochemistry 1976, 15, 4333-4339.
34. Milas, M.; Rinaudo, M., Investigation on conformational properties of xanthan in aqueoussolutions. In Solution Properties of Polysaccharides, American Chemical Society: 1981; Vol. 150, pp25-30.
35. Morris, E. R.; Rees, D. A.; Young, G.; Walkinshaw, M. D.; Darke, A., Order-disorder transitionfor a bacterial polysaccharide in solution. A role for polysaccharide conformation in recognitionbetween Xanthomonas pathogen and its plant host. Journal of Molecular Biology 1977, 110, 1-16.
36. Norton, I. T.; Goodall, D. M.; Frangou, S. A.; Morris, E. R.; Rees, D. A., Mechanism anddynamics of conformational ordering in xanthan polysaccharide. Journal of Molecular Biology 1984,175, 371-394.
37. Holzwarth, G.; Ogletree, J., Pyruvate-free xanthan. Carbohydrate Research 1979, 76, 277-280.
38. Foss, S., Thermal stability and chain conformational studies of xanthan at different ionicstrengths. Carbohydrate Polymers 1987, 7, 421-433.
39. Milas, M.; Rinaudo, M., Conformational investigation on the bacterial polysaccharidexanthan. Carbohydrate Research 1979, 76, 189-196.
40. Liu, W.; Sato, T.; Norisuye, T.; Fujita, H., Thermally induced conformational change of xanthanin 0.01M aqueous sodium chloride. Carbohydrate Research 1987, 160, 267-281.
41. Christensen, B.; Smidsrod, O., Hydrolysis of xanthan in dilute acid: Effects on chemicalcomposition, conformation, and intrinsic viscosity. Carbohydrate Research 1991, 214, 55-69.
42. Agoub, A. A.; Smith, A. M.; Giannouli, P.; Richardson, R. K.; Morris, E. R., "Melt-in-the-mouth"gels from mixtures of xanthan and konjac glucomannan under acidic conditions: A rheological andcalorimetric study of the mechanism of synergistic gelation. Carbohydrate Polymers 2007, 69, 713-724.
43. Renou, F.; Petibon, O.; Malhiac, C.; Grisel, M., Effect of xanthan structure on its interactionwith locust bean gum: Toward prediction of rheological properties. Food Hydrocolloids 2013, 32, 331-340.
44. Milas, M.; Rinaudo, M.; Tinland, B., Comparative depolymerization of xanthan gum byultrasonic and enzymic treatments. Rheological and structural properties. Carbohydrate Polymers1986, 6, 95-107.
45. Callet, F.; Milas, M.; Rinaudo, M., Influence of acetyl and pyruvate contents on rheologicalproperties of xanthan in dilute solution. International Journal of Biological Macromolecules 1987, 9,291-293.
46. Dentini, M.; Crescenzi, V.; Blasi, D., Conformational properties of xanthan derivatives in diluteaqueous solution. International Journal of Biological Macromolecules 1984, 6, 93.
47. Bradshaw; Nisbet; Kerr; Sutherland, Modified xanthan: its preparation and viscosity.Carbohydrate Polymers 1983, 3, 23-38.
48. Shatwell, K.; Sutherland, I. W.; Dea, I. C. M.; Ross-Murphy, S. B., The influence of acetyl andpyruvate substituents on the helix coil transition behaviour of xanthan. Carbohydrate Research 1990,206, 87-103.
49. Liu, W.; Norisuye, T., Thermally induced conformation change of xanthan: interpretation ofviscosity behavior in 0.01M aqueous sodium chloride. International Journal of BiologicalMacromolecules 1988, 10, 44-50.
- 204 -

Chapitre 3 : Modification chimique du xanthane
50. Sato, T.; Norisuye, T.; Fujita, H., Double-stranded helix of xanthan: dimensional andhydrodynamic properties in 0.1M aqueous sodium chloride. Macromolecules 1984, 17, 2696-2700.
51. Hatakenaka, K.; Liu, W.; Norisuye, T., Stability of xanthan in aqueous sodium chloride atelevated temperature. International Journal of Biological Macromolecules 1987, 9, 346-348.
52. Dumitiru, S.; Popa, M.; Dumitriu, M.; Pandele, T., Synthesis and characterization of nalidixicacid conjugate. Journal of Bioactive and Compatible Polymers 1990, 5, 310-315.
53. Capron, I.; Brigand, G.; Muller, G., About the native and renatured conformation of xanthanexopolysaccharide. Polymer 1997, 38, 5289-5295.
- 205 -

Chapitre 3 : Modification chimique du xanthane
- 206 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.1. Introduction 209
4.2. Comportement en solution des xanthanes modifiés sous forme ordonnée 2104.2.1. Conformation en solution des chaînes modifiées sous forme ordonnée 2104.2.2. Etude de l’organisation en solution des chaînes modifiées sous forme ordonnée parfluorescence 2134.2.3. Etude des propriétés rhéologiques dynamiques en solution des xanthanes modifiés sousforme ordonnée 221
4.2.3.1. Courbes maîtresses fréquence-température 2224.2.3.2. Loi d’Arrhenius 2294.2.3.3. Viscosité dynamique 231
4.2.4. Modèle d’organisation des chaînes de xanthane modifié sous forme ordonnée en solution233
4.2.5. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous formeordonnée 236
4.2.5.1. Comportement du précurseur à l’interface eau/huile 2384.2.5.2. Comportement des xanthanes modifiés sous forme ordonnée à l’interface eau/huile
2464.2.5.3. Modèles d’organisation des chaînes de xanthane modifié sous forme ordonnée àl’interface eau/huile 251
4.3. Comportement en solution des xanthanes modifiés sous forme désordonnée 2564.3.1. Etude des propriétés rhéologiques en solution des xanthanes greffés sous formedésordonnée 256
4.3.1.1. Comportement en solution du précurseur « désordonné » 2574.3.1.2. Comportement en solution des xanthanes modifiés dans le DMSO 259
4.3.2. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous formedésordonnée 267
4.3.2.1. Comportement à l’interface eau/huile du précurseur « désordonné » 2684.3.2.2. Comportement à l’interface eau/huile des xanthanes modifiés dans le DMSO 270
4.4. Conclusion 275
Références bibliographiques 277
- 207 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
- 208 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.1. Introduction
Ce chapitre présente les principaux résultats obtenus lors de la caractérisation des propriétés en
solution et à l’interface eau/huile des xanthanes modifiés soit sous forme ordonnée, soit sous forme
désordonnée. En particulier, on s’intéresse à montrer, autant que possible, le lien entre l’organisation
des macromolécules en solution et les propriétés qui en découlent.
La première partie concerne l’organisation et les propriétés viscoélastiques en solution des
xanthanes modifiés sous forme ordonnée. Dans un premier temps, des analyses de polarimétrie, de
dichroïsme circulaire, de fluorescence et de rhéologie en solution ont été réalisées sur les xanthanes
greffés suivant le protocole à température ambiante. Nous nous sommes également intéressés au
comportement rhéologique à l’interface eau/huile des xanthanes modifiés selon le procédé à basse
température. Par manque de matière (et les problèmes de reproductibilité de certains protocoles
évoqués dans le chapitre précédent), il n’a pas été possible de réaliser toutes les analyses sur les
mêmes échantillons.
La deuxième partie décrit des résultats préliminaires concernant le comportement rhéologique,
en solution et à l’interface, des xanthanes modifiés sous forme désordonnée. Il ressort des premières
mesures viscoélastiques que le comportement de ces échantillons est bien distinct de ceux obtenus
via les procédés dans l’eau. A priori, la flexibilité des chaînes modifiées, qui gouverne l’organisation de
ces dernières en solution, serait à l’origine de ces différences de propriétés. Néanmoins, le manque
de résultats sur cette partie ne nous permet pas de statuer sur ce point à ce jour.
- 209 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.2. Comportement en solution des xanthanes modifiés sous forme ordonnée
4.2.1. Conformation en solution des chaînes modifiées sous forme ordonnée
Dans le chapitre 3, il a été montré que les méthodes de greffage développées dans l’eau
permettent de modifier le xanthane sous forme ordonnée. En fin de purification, le polymère est
récupéré sous forme solide par lyophilisation, puis mis à solubiliser dans l’eau pour des analyses
physico-chimiques. Il se pose alors la question de savoir si les chaînes amphiphiles conservent ou non
une telle conformation lors du retour en solution. Pour répondre à cette interrogation, des analyses
de dichroïsme circulaire ont été réalisées sur les polymères suivants :
EntréenEDACnCOOH
DSA (%) DSP (%) %octyle (%) Notation
1 0 89 49 0 XDan1
2 0,5 78 43 8 BTX8C8FO
3 1,3 71 42 29 BTX29C8FO
Tableau 4-1 : Caractéristiques des échantillons étudiés en dichroïsme circulaire.
XDan1 correspond au polysaccharide commercial. Les échantillons modifiés présentés dans le
tableau ont été obtenus via le protocole de modification dans l’eau à basse température lors de la
première étape (cf. chapitre 3). La notation suivante est utilisée pour désigner les échantillons :
- BTXAC8FO désigne un xanthane (X) greffé à basse température (BT) par des chaînons
hydrophobes constitués de 8 atomes de carbone, sous forme ordonnée (FO), avec une
densité de greffage (déterminée par RMN 1H) de A.
- 210 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La conformation adoptée par les xanthanes greffés en solution à température ambiante a été
étudiée par dichroïsme circulaire. La figure 4-1 montre l’évolution de l’ellipticité en fonction de la
longueur d’onde λ pour XDan1, BTX8C8FO et BTX8C29FO :
Figure 4-1 : Evolution de l’ellipticité en fonction de la longueur d’onde λ pour XDan1, BTX8C8FO1 et BTX29C8FO à 1
g/L dans NaCl 0,01 M.
Pour les trois polymères, on observe une bande positive de forte intensité vers 205 nm, suivie
d’une bande négative moins intense vers 220 nm. Ces deux bandes sont attribuées respectivement
aux fonctions acide carboxylique et fonctions acétate, qui sont des groupements chromophores. La
présence de ces deux bandes est caractéristique de la forme ordonnée du xanthane1-4. A température
ambiante, les chaînes amphiphiles sont donc capables d’adopter une conformation hélicoïdale. On
remarque également que l’intensité des deux bandes augmente avec la densité de greffage, ce qui
traduit un système de plus en plus ordonné1, 3. Par conséquent, la présence de groupements
hydrophobes en proportions croissantes renforce la forme hélicoïdale rigide des chaînes en solution.
Des analyses de polarimétrie ont également été effectuées sur des échantillons de xanthane,
modifiés ou non. Les caractéristiques de ces polymères sont indiquées dans le tableau 4-2 :
- 211 -
(nm)
200 210 220 230 240 250 260 270
(m
deg)
-2
0
2
4
6
8
10Xdan1
BTX8C8FO1
BTX29
C8FO

Chapitre 4 : Propriétés en solution des xanthanes modifiés
EntréenEDACnCOOH
DSA (%) DSP (%) %octyle (%) Notation
1 0 66 46 0 (précurseur) BTX0C8FO
2 0,7 81 53 8 BTX8C8FO
3 1 90 54 10 BTX10C8FO
Tableau 4-2 : Caractéristiques des échantillons étudiés en polarimétrie.
La figure 4-2 montre l’évolution du pouvoir rotatoire en fonction de la température pour ces
xanthanes :
Figure 4-2 : Pouvoir rotatoire en fonction de la température pour des solutions à 1 g/L de précurseur,
BTX8C8FO2 et BTX10C8FO (pH = 4,3 et σ = 1mS/cm).
Dans le cas du précurseur, le pouvoir rotatoire reste relativement constant entre 20 et 50°C, puis
augmente fortement entre 50 et 70°C et se stabilise à plus haute température. Cette allure
sigmoïdale est caractéristique de la transition ordre-désordre du xanthane. En utilisant la méthode
des tangentes, il est possible de déterminer la Tm du précurseur (Tm = 62°C). La même allure est
observée en ce qui concerne les polymères modifiés. On constate néanmoins que la Tm semble se
décaler vers des températures de plus en plus importantes avec la densité de greffage. Ces résultats
indiquent que la transition conformationnelle se produit aussi au sein des chaînes amphiphiles, mais
- 212 -
T (°C)
20 30 40 50 60 70 80 90
Alp
ha (
°)
-0,035
-0,030
-0,025
-0,020
-0,015
-0,010
-0,005
BTX0C8
BTX8C8
BTX10C8

Chapitre 4 : Propriétés en solution des xanthanes modifiés
à des températures plus élevées que celles du précurseur. Ce phénomène est accentué quand la
densité de greffage augmente. Ces données confirment les observations faites en dichroïsme
circulaire. D’une part, à température ambiante, les chaînes modifiées sont bien sous forme ordonnée.
D’autre part, les greffons hydrophobes ont pour effet de renforcer et de stabiliser thermiquement la
forme hélicoïdale, un phénomène d’autant plus marqué que la densité de greffage est importante.
4.2.2. Etude de l’organisation en solution des chaînes modifiées sous forme
ordonnée par fluorescence
En règle générale, les polymères amphiphiles possèdent la capacité de s’organiser en solution
aqueuse via des interactions inter et/ou intramoléculaires entre leurs chaînons hydrophobes5-7. La
conformation adoptée par les chaînes modifiées en solution est, le plus souvent, très différente de
celle de leurs analogues non greffés. En particulier, au-delà d’une concentration critique c crit, les
interactions hydrophobes intermoléculaires résultent en la formation de microdomaines, qui sont à
l’origine des propriétés épaississantes supérieures des polymères amphiphiles8-10.
La fluorescence est une technique usuelle pour caractériser l’organisation des systèmes
amphiphiles en solution. En effet, certaines sondes de fluorescence, sensibles à la polarité et/ou
l’hydrophobicité de leur milieu, permettent de mettre en évidence la présence (ou non) des agrégats
hydrophobes. L’acide 8-anilinonaphtalène-1-sulphonique, ou ANS, est, par exemple, une des sondes
dont l’émission varie suivant ces paramètres. En effet, cette sonde est dite hypsochrome : son spectre
d’émission se caractérise par un décalage des longueurs d’onde vers le bleu lorsque la polarité du
milieu diminue. De plus, elle est aussi sensible à la présence d’eau. Ainsi, en milieu non aqueux,
l’émission de fluorescence est très importante. Au contraire, en présence d’eau, l’intensité mesurée
est très faible. L’eau est donc une molécule d’extinction de fluorescence de l’ANS. C’est pourquoi l’ANS
est également utilisé comme sonde d’hydrophobicité.
- 213 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Dans le cadre de cette étude, les échantillons caractérisés sont ceux obtenus par le processus de
modification du xanthane sous forme ordonnée à température ambiante. Les notations suivantes ont
été adoptées pour désigner les xanthanes :
- TAXAC8FO désigne un xanthane (X) greffé à température ambiante (TA) par des chaînons
hydrophobes constitués de 8 atomes de carbone, sous forme ordonnée (FO), avec une
densité de greffage (déterminée par RMN 1H) de A %.
- Précurseur (ou TAX0C8FO) désigne un xanthane non modifié qui a subi toutes les étapes du
processus de greffage et de purification, mais sans l’ajout des agents de couplage lors de la
première étape du procédé. Il s’agit donc de notre référence pour un xanthane non modifié.
Le tableau 4-3 indique les degrés de substitution en acétate (DSA), en pyruvate (DSP) et densités
de greffage (%octyle) des échantillons étudiés dans cette section :
EntréenEDACnCOOH
noctylaminenCOOH
DSA (%) DSP (%) %octyle (%) TAXAC8FO
1 0 3 60 45 0 (précurseur) TAX0C8FO
2 2,2 2,2 60 41 7 TAX7C8FO
3 2,7 2,7 67 48 13 TAX13C8FO
4 2,7 2,7 79 53 15 TAX15C8FO
5 3,5 3,5 44 40 18,5 TAX18,5C8FO
Tableau 4-3 : Caractéristiques des échantillons étudiés en fluorescence.
- 214 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La figure 4-3 représente le spectre d’émission de fluorescence de l’ANS pour TAX18,5C8FO et le
précurseur à différentes concentrations en polysaccharide :
Figure 4-3 : Spectre d’émission de fluorescence de l’ANS en présence de X29C8 (traits pleins) et le précurseur
(traits pointillés) à différentes concentrations.
Pour toutes les courbes, l’intensité de fluorescence I augmente lorsque la longueur d’onde
décroît, jusqu’à une longueur d’onde critique à partir de laquelle I diminue. On définit ainsi I max
comme étant l’intensité maximale d’émission et λmax la longueur d’onde au maximum d’émission. On
remarque également que l’intensité de fluorescence augmente avec la concentration pour les deux
xanthanes. Ce phénomène est davantage marqué pour TAX18,5C8FO que pour le précurseur. Il est
accompagné d’un décalage de la longueur d’onde d’émission vers le bleu quand la concentration
croît. Les spectres d’émission des autres échantillons, non indiqués sur la figure 4-3 pour des raisons
de clarté, présentent ces mêmes caractéristiques.
- 215 -
(nm)
400 450 500 550 600
Inte
nsité
(u.
a.)
0
10
20
30
40
50
60
70TAX
18,5C8FO 0,1g/L
TAX18,5
C8FO 1g/L
TAX18,5
C8FO 5g/L
TAX0
C8FO 0,1g/L
TAX0
C8FO 1g/L
TAX0
C8FO 5g/L

Chapitre 4 : Propriétés en solution des xanthanes modifiés
A partir de ces données, il est possible de tracer l’évolution de la longueur d’onde au maximum
d’émission en fonction de la concentration pour tous les polymères étudiés, comme illustré sur la
figure 4-4 :
Figure 4-4 : Evolution de la longueur d’onde maximale d’émission en fonction de la concentration pour les
xanthanes modifiés et le précurseur.
Toutes les courbes ont une allure similaire : λmax diminue fortement (de 520-530 nm à 460-470
nm) entre 0 et 1 g/L, puis se stabilise ensuite aux alentours des 465 nm au-delà de cette
concentration. Il faut savoir que, dans l’eau, l’ANS émet à 520 nm, alors que dans un environnement
apolaire, l’émission maximale est observée entre 460 et 480 nm11-13. La comparaison des données de
la littérature avec nos résultats indique que les molécules d’ANS sont situées dans un environnement
de plus en plus apolaire lorsque la concentration en polysaccharide augmente. De plus, ce constat est
valable pour tous les xanthanes, qu’ils soient greffés ou non : a priori, tous les échantillons forment
des domaines apolaires qui interagissent avec l’ANS. Par ailleurs, la valeur de λ max reste quasiment
constante et égale à 465 nm à partir de 1 g/L pour tous les xanthanes : à partir de cette
concentration, l’environnement autour de la sonde n’évolue plus.
- 216 -
c (g/L)
0 1 2 3 4 5 6
ma
x (u
. a.
)
460
470
480
490
500
510
520
530TAX
0C8FO
TAX7
C8FO
TAX13
C8FO
TAX15
C8FO
TAX18,5
C8FO

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La figure 4-5 A montre l’évolution de l’intensité maximale de fluorescence en fonction de la
concentration pour tous les xanthanes étudiés :
Figures 4-5 A : Intensité maximale de fluorescence de l’ANS en fonction de la concentration en polymère
pour les xanthanes modifiés et le précurseur. B : Evolution de l’intensité maximale de fluorescence en fonction
de la concentration normalisée pour les xanthanes modifiés et le précurseur. Les symboles sont identiques à
ceux utilisés dans les figures précédentes.
Toutes les courbes sont semblables. Aux faibles concentrations en polymère, l’intensité de
fluorescence est quasiment nulle et constante : l’ANS est majoritairement en contact avec des
molécules d’eau, qui quenchent la fluorescence de cette sonde. A partir d’une concentration critique,
notée ccrit, l’intensité de fluorescence augmente de manière significative. L’ANS est de moins en moins
en contact avec le solvant, ce qui indique la formation de régions hydrophobes et apolaires au sein du
milieu. La concentration critique est déterminée graphiquement comme l’intersection des deux
tangentes, comme indiqué sur la figure 4-5 A. On remarque également que c crit diminue d’autant plus
que la densité de greffage est élevée. Ainsi, le précurseur est caractérisé par une concentration
critique d’approximativement 0,8 g/L, alors qu’elle est d’environ 0,5 g/L pour TAX18,5C8FO. De plus,
l’évolution de ccrit avec la densité de greffage montre que la création des régions apolaires est facilitée
- 217 -
c (g/L)0.1 1 10
Imax
(u.
a.)
10
20
30
40
50
60TAX
0C8FO
TAX7C8FO
TAX13
C8FO
TAX15
C8FO
TAX18,5
C8FO
c/ccrit
0.1 1 10
Imax
(u.
a.)
10
20
30
40
50
60TAX
0C8FO
TAX7C8FO
TAX13
C8FO
TAX15
C8FO
TAX18,5
C8FO
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
quand le %octyle augmente. On peut néanmoins noter que les valeurs de ccrit restent relativement
proches pour tous les échantillons. Ce point sera discuté plus en détail par la suite. Par ailleurs, pour c
> ccrit, l’intensité de fluorescence ne cesse de croître avec la concentration, contrairement à ce qui est
observé pour λmax. Ce phénomène s’explique par la formation de régions hydrophobes de plus en plus
denses (et donc davantage « imperméables » à l’eau) avec la concentration.
Au premier abord, ces résultats semblent surprenants pour le précurseur. En effet, ils indiquent
que ce dernier est capable de former des zones apolaires et hydrophobes à partir d’une
concentration critique d’environ 0,8 g/L. Or le spectre RMN 1H de ce polymère (cf. chapitre 3) montre
l’absence de toute chaîne octyle, greffée ou non, au sein de cet échantillon. Deux hypothèses
peuvent être avancées pour expliquer ces données.
La viscosité du milieu étudié peut avoir une influence sur la fluorescence des sondes14, 15.
Cependant, l’ANS n’étant pas un rotor moléculaire, il n’est pas conçu pour être une sonde de
microviscosité. Par conséquent, l’effet de viscosité n’est probablement pas le phénomène
majoritairement responsable des résultats observés.
Une autre possibilité est que le précurseur est bien capable de former des régions apolaires. Le
xanthane est un polysaccharide hydrophile. Cependant, des expériences de rétention d’arôme ont
montré que le squelette cellulosique de ce polymère est capable de retenir des composés
hydrophobes16-18. Il est, par conséquent, fort probable que les molécules d’ANS interagissent de
manière similaire avec les chaînes du précurseur. De plus, la concentration critique déterminée pour
le précurseur, de 0,8 g/L, est très proche de la concentration de recouvrement, c** du xanthane en
solution aqueuse (de l’ordre de 1 g/L)19-21. c** correspond à la concentration à partir de laquelle les
chaînes de polymère forment un réseau tridimensionnel viscoélastique au sein de la solution. Dans le
cas du xanthane, ce réseau serait créé via des interactions hydrogène intermoléculaires entre les
chaînes, les interactions intramoléculaires intervenant dans la stabilisation de la conformation
- 218 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
hélicoïdale. L’existence de ce réseau au-delà de c** pourrait favoriser l’expulsion des molécules d’eau
du voisinage des sondes d’ANS, d’où l’augmentation de l’intensité de la fluorescence pour c > ccrit.
En ce qui concerne les xanthanes modifiés, les données pourraient s’expliquer par le fait que ces
polysaccharides adoptent, au-delà de ccrit, une conformation en solution semblable à celle des
polymères amphiphiles flexibles. En effet, comme décrit dans le premier chapitre, ces derniers
forment en solution des microdomaines hydrophobes bien définis, suite aux interactions
intermoléculaires entre les greffons. Ce phénomène modifie fortement l’organisation des chaînes.
Ainsi, les polymères amphiphiles flexibles se caractérisent, le plus souvent, par des conformations
bien distinctes de celles observées pour leurs analogues non modifiés22-25.
Néanmoins, dans notre cas, cette hypothèse paraît peu vraisemblable. En effet, l’étude de nos
xanthanes modifiés par polarimétrie montre que ces derniers présentent une température de
transition conformationnelle, Tm, égale ou supérieure à celle du précurseur (cf. figure 4-2). En
d’autres termes, tous les xanthanes greffés sont capables, à température ambiante et/ou une force
ionique suffisamment élevée, d’adopter une conformation ordonnée hélicoïdale très rigide (longueur
de persistance entre 100 et 150 nm26-29), malgré la modification subie. Or, dans nos conditions
d’analyse de fluorescence, la force ionique du milieu tamponné est suffisamment importante pour
permettre au xanthane d’être sous forme ordonnée. Au regard de la forte rigidité des hélices, il paraît
très difficile de concevoir que les chaînes de xanthane puissent se replier sur elles-mêmes pour créer
des microdomaines hydrophobes bien définis semblables à ceux des polymères amphiphiles flexibles.
De plus, comme mentionné précédemment, la courbe du précurseur (figure 4-5 A) présente de
fortes similitudes avec celles des xanthanes greffés, alors que ce polymère n’est pas modifié. La seule
différence réside en la concentration critique ccrit, qui décroît quand le %octyle augmente. En
normalisant les courbes de l’intensité d’émission en fonction de la concentration de la figure 4-5 A
- 219 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
par c/ccrit, il est possible d’obtenir une seule et unique courbe, identique à celle du précurseur (figure
4-5 B). Ce résultat indique que le comportement en émission de l’ANS est globalement le même, que
la sonde soit en présence du précurseur ou d’un xanthane greffé : la sonde ressent un environnement
essentiellement apolaire pour c > ccrit. La présence des chaînons hydrophobes en proportions
croissantes a uniquement pour effet de diminuer la valeur de ccrit, soit, en d’autres termes, d’autoriser
le passage d’un milieu polaire à apolaire à des concentrations de plus en plus faibles. Par conséquent,
nous en déduisons que l’organisation des chaînes modifiées en solution est très similaire à celle du
précurseur, et que la concentration critique correspond à c**. Cette dernière diminue quand la
densité de greffage augmente, car les interactions intermoléculaires attractives entre les chaînons
hydrophobes favorisent la formation du réseau tridimensionnel. Ce phénomène a été observé pour
d’autres polymères amphiphiles25, 30-33. Pour autant, dans le cas du xanthane, les valeurs de c** pour
tous les échantillons sont relativement proches (c** compris entre 0,8 et 0,5 g/L), ce qui montre que
l’influence de la densité de greffage sur la formation du réseau est faible. Pour les échantillons
amphiphiles, les liaisons hydrogène semblent être de nouveau les interactions majoritaires dans la
création du réseau tridimensionnel : la présence des greffons affecte peu le caractère associatif du
xanthane.
- 220 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Pour résumer :
Les analyses de fluorescence du précurseur et des xanthanes modifiés prouvent que la présence
de chaînons hydrophobes n’a pas, ou peu, d’impact sur l’organisation des chaînes du polysaccharide
en solution. Au-delà d’une concentration critique, correspondant probablement à la concentration
d’enchevêtrement c**, tous les échantillons forment un réseau tridimensionnel viscoélastique. Dans
le cas du précurseur, ce réseau est stabilisé par des liaisons hydrogène intermoléculaires. Pour les
polysaccharides amphiphiles, à ces interactions majoritaires s’ajoute la contribution modérée des
greffons hydrophobes. En effet, les interactions intermoléculaires entre ces greffons favorisent la
création du réseau à de plus faibles concentrations que les valeurs usuelles de c** pour ce polymère.
Néanmoins, ces interactions ne modifient pas l’organisation globale des chaînes, contrairement à ce
qui est observé pour les polymères amphiphiles flexibles. Ce phénomène s’explique par la forte
rigidité des hélices de xanthane, qui empêche ces dernières de se replier pour former des
microdomaines hydrophobes. L’organisation des chaînes en solution est donc contrôlée, dans notre
cas, par la rigidité du polysaccharide et non pas par les interactions attractives entre les greffons
hydrophobes. Une conséquence de ce phénomène est que le caractère associatif des échantillons
greffés est peu différent de celui du précurseur. Notre xanthane modifié est donc un polysaccharide
amphiphile, mais non associatif.
4.2.3. Etude des propriétés rhéologiques dynamiques en solution des xanthanes
modifiés sous forme ordonnée
Les polymères amphiphiles flexibles se caractérisent par des propriétés rhéologiques supérieures
à celles de leurs homologues non modifiés au-delà d’une concentration critique9, 10, 34, 35. Dans la
majorité des cas, cette différence dans les propriétés est liée à l’organisation bien spécifique des
chaînes amphiphiles en solution, suite aux interactions hydrophobes attractives entre les chaînons.
Or, l’étude de nos xanthanes greffés par fluorescence a montré que les chaînons n’ont pas (ou peu)
- 221 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
d’influence sur la conformation des chaînes en solution. Il se pose alors la question de savoir si la
modification chimique a un impact sur le comportement rhéologique du xanthane. Cette
interrogation est l’objet de cette partie, qui s’intéresse à la caractérisation des propriétés
viscoélastiques du xanthane (précurseur et modifié) en solution, notamment suivant la température.
Les échantillons étudiés dans cette section ont été obtenus par le protocole de modification à
température ambiante. Leurs caractéristiques sont indiquées dans le tableau 4-4 :
EntréenEDACnCOOH
noctylaminenCOOH
DSA (%) DSP (%) %octyle (%) TAXAC8FO
1 0 3 60 45 0 (précurseur) TAX0C8FO
2 2,2 2,2 60 41 7 TAX7C8FO
3 2,7 2,7 67 48 13 TAX13C8FO
5 2,7 2,7 79 53 15 TAX15C8FO
6 3,5 3,5 44 40 18,5 TAX18,5C8FO
Tableau 4-4 : Caractéristiques des échantillons étudiés en rhéologie en solution.
4.2.3.1. Courbes maîtresses fréquence-température
Avant d’étudier les propriétés viscoélastiques en solution des xanthanes modifiés, nous avons
caractérisé celles du précurseur via des sollicitations harmoniques à différentes fréquences (de 0,1 à
100 rad/s) dans le domaine linéaire de déformation pour une gamme de températures allant de 5 à
80°C. La figure 4-6 A représente la dépendance en fréquence de G’, module élastique, et G’’, module
visqueux d’une solution de précurseur à 2 g/L (soit en régime enchevêtré) à 5, 30, 40 et 60°C. Pour
des raisons de clarté, les autres températures n’ont pas été indiquées.
- 222 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Figures 4-6 A : Evolution des modules G’ (symboles pleins) et G’’ (symboles ouverts) en fonction de la
fréquence à différentes températures pour le précurseur à 2 g/L. B : Courbe maîtresse fréquence-température
du précurseur à 2 g/L (Tref = 20°C).
Le précurseur présente les caractéristiques typiques d’une solution de xanthane à cette
concentration19, 36-38. En effet, à 5°C, G’ est supérieur à G’’ pour ω ≥ 0,37 rad/s. De plus, la dépendance
en fréquence des deux modules est faible, ce qui traduit un comportement de type gel faible, dû aux
liaisons hydrogène intermoléculaires entre les chaînes. Par ailleurs, les deux modules se croisent à la
fréquence ωref = 0,37 rad/s, qui correspond à la fréquence de relaxation. Cette fréquence se décale
vers des valeurs de plus en plus hautes lorsque la température augmente, et n’est plus accessible
expérimentalement à partir de 60°C. A cette température, G’’ est supérieur à G’ sur toute la gamme
de fréquences étudiée, ce qui est typique d’un liquide viscoélastique.
A partir de ces données, il est possible de construire une courbe maîtresse fréquence-
température selon le protocole décrit dans le chapitre 2. La figure 4-6 B montre la courbe maîtresse
obtenue dans le cas du précurseur. Les données se superposent très bien les unes aux autres dans les
gammes de températures et de fréquences étudiées. De plus, il est possible de construire cette
- 223 -
aT (rad/s)
10-3 10-2 10-1 100 101G
'*b
T, G
"*b
T (P
a)
10-4
10-3
10-2
10-1
100
G' 5°CG' 10°CG' 20°CG' 30°CG' 40°CG' 50°CG' 60°CG' 70°C
B
(rad/s)
10-1 100 101
G',
G" (P
a)
10-2
10-1
100
G' 5°CG' 20°CG' 40°CG' 60°C
A

Chapitre 4 : Propriétés en solution des xanthanes modifiés
unique courbe maîtresse jusqu’à une température de 70°C, soit supérieure à la Tm du précurseur. Ce
résultat est en accord avec ceux de Pelletier et al.39, de Choppe et al.36 et de Renou et al.37, qui ont
montré que la transition conformationnelle ordre-désordre du xanthane n’a que peu d’impact sur les
mécanismes de relaxation des chaînes en régime enchevêtré.
A hautes fréquences et basses températures, le précurseur a le comportement d’un gel faible,
avec G’ supérieur à G’’ et une faible dépendance en fréquence des deux modules : G’ ~ ω0,45 et G’’ ~
ω0,3. Au contraire, à basses fréquences et hautes températures, le système se comporte comme un
liquide viscoélastique, avec G’’ supérieur à G’. De plus, la dépendance en fréquence des deux
modules est davantage marquée : G’ ~ ω1,8 et G’’ ~ ω0,94, ce qui est proche des dépendances en
fréquence observées pour des liquides maxwelliens. Ces observations sont en accord avec celles
décrites dans la littérature36-38.
Ce type de construction a également été réalisé sur les xanthanes modifiés. La figure 4-7
représente ainsi les courbes maîtresses fréquence-température pour le précurseur TAX7C8FO et TAX18,5C8FO
(les autres courbes ne sont pas tracées pour des raisons de clarté) :
Figure 4-7 : Courbes maîtresses fréquence-température de G’ (symboles pleins) et G’’ (symboles ouverts) pour le
précurseur, TAX7C8, et TAX18,5C8 à 2 g/L (Tref = 20°C).
- 224 -
*aT (rad/s)
10-5 10-4 10-3 10-2 10-1 100 101 102
G'*b
T, G
"*b
T (
Pa)
10-4
10-3
10-2
10-1
100
G' TAX0C8FO
G' TAX7C8FO
G' TAX18,5C8FO

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La superposition est également satisfaisante pour tous les xanthanes greffés. A haute fréquence,
tous les échantillons ont un comportement de gel faible similaire à celui du précurseur. En outre, le
module élastique augmente légèrement avec la densité de greffage, alors que le module visqueux
reste constant. A basse fréquence cependant, le comportement des dérivés diffère fortement de celui
du précurseur : la dépendance en fréquence des deux modules est de moins en moins marquée au
fur et à mesure que la densité de greffage augmente. Ce phénomène s’accompagne d’un fort
décalage de la fréquence de relaxation vers des valeurs de plus en plus faibles, ce qui traduit un
ralentissement prononcé de la dynamique de relaxation du système. La dynamique de relaxation est
notamment caractérisée par le temps de relaxation τ = 1/ω ref, dont l’évolution suivant la densité de
greffage est indiquée dans la figure 4-8 :
Figure 4-8 : Evolution du temps de relaxation en fonction de la densité de greffage pour le précurseur et les
xanthanes modifiés.
Le temps de relaxation évolue avec la densité de greffage suivant la relation suivante :
τ=1,63exp (0,23∗ octyle ) . Il est, de ce fait, possible de contrôler le comportement des xanthanes
modifiés selon la densité de greffage. En outre, la dynamique de relaxation des chaînes modifiées
dépend fortement de la densité de greffage. Plus spécifiquement, cette dynamique est
- 225 -
%octyle
0 5 10 15
(s
)
100
101
102
103
104
%octyle t (s)
0 0,77
7 16,4
13 190
15 1 400
18,5 21 000
X ~ 30 000

Chapitre 4 : Propriétés en solution des xanthanes modifiés
considérablement ralentie par la présence de greffons hydrophobes, un phénomène d’autant plus
accentué que la densité de greffage est élevée. Par exemple, TAX18,5C8FO se caractérise ainsi par un
temps de relaxation 30 000 fois plus important que le précurseur.
Il faut savoir que la dynamique de relaxation des polymères amphiphiles peut être décrite par le
modèle de la reptation collante40, 41, présenté dans le premier chapitre de ce manuscrit. Pour rappel,
cette théorie prédit que la relaxation des polymères amphiphiles associatifs présente deux plateaux
caoutchoutiques avant la zone terminale d’écoulement, auxquels sont respectivement associés deux
temps de relaxation distincts : la durée des interactions entre les greffons hydrophobes, τ sticker, et le
temps de reptation τrep, avec τsticker < τrep. Au premier plateau, à haute fréquence, pour t < τ sticker, les
chaînons hydrophobes n’ont pas le temps de se libérer des microdomaines. Par conséquent, ces
derniers agissent comme des nœuds élastiquement actifs, et leur contribution au module élastique
s’ajoute à celle des enchevêtrements topologiques, eux aussi nœuds actifs dans cette gamme de
fréquences. Pour τsticker < t < τrep, la contrainte est relaxée uniquement au sein des microdomaines, les
enchevêtrements étant toujours élastiquement actifs. On atteint alors le deuxième plateau
caoutchoutique, de module identique à celui observé pour un polymère analogue non modifié. Ce
plateau se caractérise également par un temps de reptation beaucoup plus important que celui de
son analogue non greffé. En effet, tout chaînon hydrophobe, bien que « libéré » de son
microdomaine, est capable d’interagir avec un autre chaînon si ce dernier est suffisamment proche.
Or, ce type de situation est fréquent au cours du mouvement de reptation de la chaîne modifiée hors
de son tube. Par conséquent, les greffons hydrophobes agissent comme des points collants qui
ralentissent le processus de relaxation de la chaîne, ce qui se traduit par un temps de reptation τ rep
plus élevé.
- 226 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Au regard des résultats expérimentaux obtenus pour les xanthane amphiphiles, il apparaît que ce
modèle paraît approprié pour décrire nos systèmes. En effet, le comportement viscoélastique étudié
semble correspondre à une partie du deuxième plateau caoutchoutique et à la zone terminale
d’écoulement. A haute fréquence, la valeur du module élastique des xanthanes modifiés est proche
de celle du précurseur. De plus, on observe bien le ralentissement de la dynamique des chaînes
greffées, phénomène d’autant plus marqué que la densité de greffage est importante.
Nos données expérimentales présentent néanmoins quelques divergences par rapport au modèle
de la reptation collante. En premier lieu, un seul plateau caoutchoutique est obtenu, et non pas deux.
Toutefois, ce résultat s’explique par l’absence de formation de microdomaines hydrophobes, ainsi que
par le temps très court d’association entre les stickers. De ce fait, il très difficile, expérimentalement,
d’observer le premier plateau caoutchoutique prédit par le modèle de la reptation collante. Par
ailleurs, selon la théorie, le module élastique du polymère amphiphile au deuxième plateau est censé
être identique à celui de l’analogue non modifié. Or, dans notre cas, le G’ du polymère modifié est
légèrement supérieur à celui du précurseur. Des résultats similaires ont été obtenus par Caputo et
al.42 pour des polyacrylamides amphiphiles. Une hausse de G’ signifie que le nombre de nœuds
élastiquement actifs est plus important. Or, ces nœuds ne peuvent pas provenir des microdomaines
hydrophobes, puisque ces derniers ont complètement relaxé dans cette gamme de fréquences. Pour
Caputo et al.42, la valeur plus grande de G’ s’explique par l’existence d’enchevêtrements topologiques
supplémentaires pour le polyacrylamide amphiphile. En effet, les interactions hydrophobes
attractives entre les greffons favorisent une plus grande proximité entre les chaînes, ce qui résulte en
un nombre d’enchevêtrements élastiques plus élevé. Dans notre cas, il est possible qu’un mécanisme
similaire soit à l’œuvre, d’où la légère augmentation du module élastique à haute fréquence avec la
densité de greffage.
- 227 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Pour autant, on peut noter que la valeur du module élastique des xanthanes modifiés est peu
différente de celle du précurseur. A haute fréquence le G’ de TAX18,5C8FO est, par exemple, à peine deux
fois plus élevé que le G’ du précurseur. En comparaison, Caputo et al.42 obtiennent, pour un
polyacrylamide greffé à hauteur seulement de 11 %, un module élastique sept fois plus grand que
celui de l’analogue non modifié. Ce phénomène peut s’expliquer par le fait que les chaînes de
xanthane modifié ont la même organisation en solution que celle du précurseur, comme l’ont montré
les analyses de fluorescence présentées auparavant. Pour rappel, en milieu concentré, l’organisation
des chaînes consiste en un réseau transitoire, constitué des hélices semi-rigides de xanthane
arrangées parallèlement les unes aux autres et en interactions entre elles via des liaisons hydrogène19,
36, 43, 44. Ces liaisons sont considérées comme des nœuds de jonction, ou élastiquement actifs, de
temps de vie fini. De plus, l’alignement parallèle des hélices de xanthane a pour conséquence de
créer un nombre fini de liaisons hydrogène. La présence de chaînons hydrophobes facilite sans doute
un rapprochement plus important des hélices, et par conséquent la création ponctuelle de nouveaux
nœuds, mais ne peut toutefois pas contrer l’organisation globale du système au regard de sa forte
rigidité et des contraintes stériques qu’elle engendre. De plus, selon la littérature 19, 36, 43, 44, la
dynamique de relaxation des chaînes de xanthane modifié en régime concentré est contrôlée par les
liaisons hydrogène, soit des interactions spécifiques, et non pas par de simples enchevêtrements
topologiques. C’est pourquoi nous préférons utiliser le terme de « relaxation collante » pour décrire
la dynamique de nos polymères modifiés au lieu du modèle de la reptation collante, développé à
l’origine pour des polymères associatifs flexibles.
- 228 -
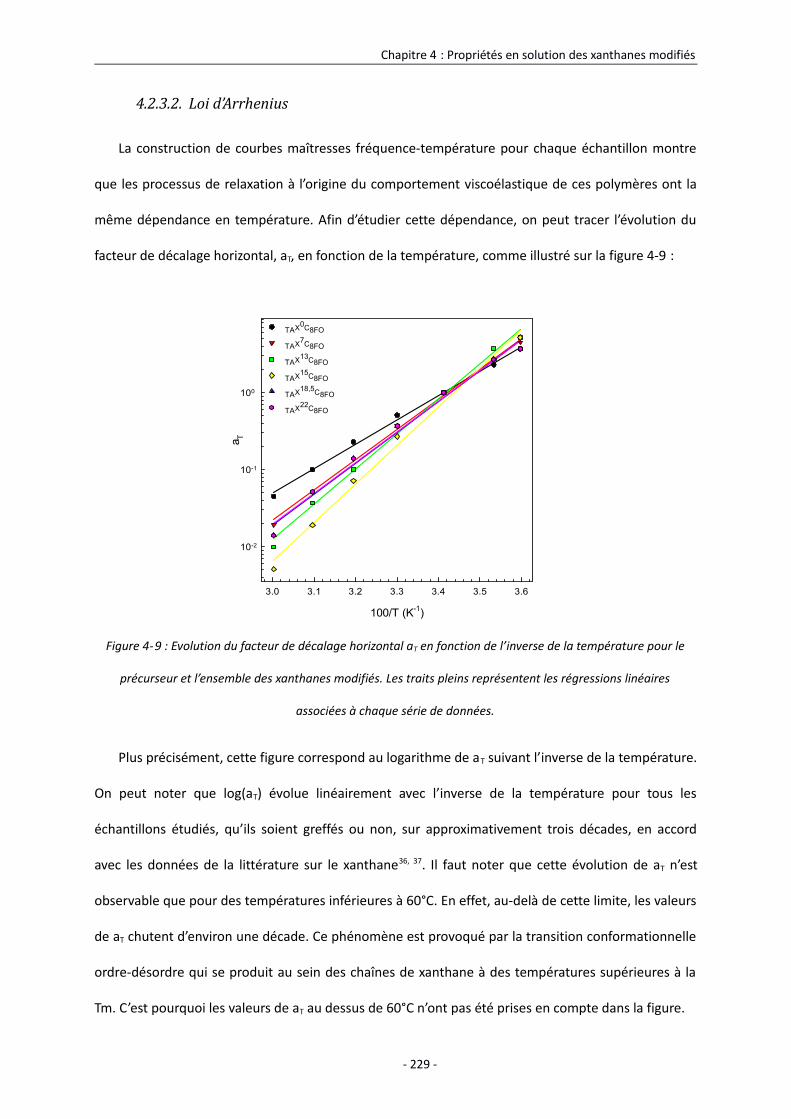
Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.2.3.2. Loi d’Arrhenius
La construction de courbes maîtresses fréquence-température pour chaque échantillon montre
que les processus de relaxation à l’origine du comportement viscoélastique de ces polymères ont la
même dépendance en température. Afin d’étudier cette dépendance, on peut tracer l’évolution du
facteur de décalage horizontal, aT, en fonction de la température, comme illustré sur la figure 4-9 :
Figure 4-9 : Evolution du facteur de décalage horizontal aT en fonction de l’inverse de la température pour le
précurseur et l’ensemble des xanthanes modifiés. Les traits pleins représentent les régressions linéaires
associées à chaque série de données.
Plus précisément, cette figure correspond au logarithme de aT suivant l’inverse de la température.
On peut noter que log(aT) évolue linéairement avec l’inverse de la température pour tous les
échantillons étudiés, qu’ils soient greffés ou non, sur approximativement trois décades, en accord
avec les données de la littérature sur le xanthane36, 37. Il faut noter que cette évolution de aT n’est
observable que pour des températures inférieures à 60°C. En effet, au-delà de cette limite, les valeurs
de aT chutent d’environ une décade. Ce phénomène est provoqué par la transition conformationnelle
ordre-désordre qui se produit au sein des chaînes de xanthane à des températures supérieures à la
Tm. C’est pourquoi les valeurs de aT au dessus de 60°C n’ont pas été prises en compte dans la figure.
- 229 -
100/T (K-1)
3.0 3.1 3.2 3.3 3.4 3.5 3.6
a T
10-2
10-1
100
TAX0C8FO
TAX7C8FO
TAX13C8FO
TAX15C8FO
TAX18,5C8FO
TAX22C8FO

Chapitre 4 : Propriétés en solution des xanthanes modifiés
L’obtention de ces droites indique que les propriétés viscoélastiques de nos xanthanes, greffés ou
non, ont une dépendance en température qui peut être décrite par une loi d’Arrhenius. Cette loi
permet de définir, pour chaque échantillon, une unique énergie d’activation, Ea (en kJ/mol). Les
valeurs de Ea pour chaque polymère sont données dans le tableau 4-5 :
Echantillon Ea (kJ/mol)
TAX0C8FO 60
TAX7C8FO 78
TAX13C8FO 99
TAX15C8FO 100
TAX18,5C8FO 83
Tableau 4-5 : Valeurs de Ea (en kJ/mol) pour le précurseur et les xanthanes modifiés.
L’énergie d’activation des xanthanes greffés est supérieure, dans tous les cas, à celle du
précurseur. Les processus de relaxation au sein des polymères modifiés nécessitent davantage
d’énergie pour être mis en œuvre, ce qui s’explique par les interactions intermoléculaires
supplémentaires générées par les greffons. Des résultats similaires sont décrits dans la littérature par
Caputo et al.42 et Durand et al.45, respectivement pour des polyacrylamides et des dextranes modifiés
hydrophobiquement. On constate cependant que la différence d’énergie entre nos échantillons est
modeste, et qu’aucune corrélation n’est possible entre les valeurs de Ea et la densité de greffage. Les
valeurs obtenues sont comprises entre 60 et 100 kJ/mol, ce qui correspond à l’ordre de grandeur de
l’énergie d’activation du xanthane non modifié dans la littérature36, 37. Il ressort de ces données que
l’impact des chaînons hydrophobes sur l’énergie d’activation est plutôt modéré. Il faut savoir que,
dans le cas du précurseur, la dépendance en température des propriétés rhéologiques de cet
échantillon est due aux liaisons hydrogène. Les valeurs similaires de Ea entre les xanthanes modifiés
et le précurseur signifient que ces mêmes liaisons contribuent majoritairement à la dépendance en
température des propriétés viscoélastiques des polymères modifiés, et que la contribution des
interactions hydrophobes est limitée. Par conséquent, la dépendance en température des propriétés
du xanthane apparaît identique, quelle que soit la densité de greffage. Ces résultats montrent donc
- 230 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
que les processus de relaxation au sein des chaînes des polymères greffés sont identiques à ceux du
précurseur, ce qui confirme une fois de plus que les échantillons modifiés adoptent, en régime
concentré, la même organisation en solution que le xanthane non greffé.
4.2.3.3. Viscosité dynamique
A partir des courbes maîtresses fréquence-température de la viscoélasticité de chaque
échantillon, il est possible d’obtenir des courbes maîtresses fréquence-température de la viscosité
dynamique, comme illustré sur la figure 4-10 :
Figure 4-10 : Courbes maîtresses fréquence-température de la viscosité dynamique du précurseur et des
xanthanes modifiés à 2g/L (Tref = 20°C).
Ces courbes permettent de prédire le comportement en écoulement des solutions de nos
polymères modifiés. En effet, plusieurs auteurs ont montré que la loi de Cox-Merz est applicable pour
le xanthane dans le régime concentré21, 36, 38, 39, 46. Pour rappel, cette loi empirique énonce que les
courbes, viscosité de cisaillement η en fonction de la vitesse de cisaillement γ , et viscosité
complexe η* en fonction de la fréquence ω, se superposent lorsque le polymère n’est soumis qu’à des
- 231 -
*aT
10-5 10-4 10-3 10-2 10-1 100 101 102
' (
Pa.
s)
10-1
100
101
102
103
TAX0C8FO
TAX7C8FO
TAX13
C8FO
TAX15
C8FO
TAX18,5
C8FO
TAX22
C8FO

Chapitre 4 : Propriétés en solution des xanthanes modifiés
enchevêtrements. Au contraire, si l’organisation des chaînes en solution résulte d’interactions
particulières, la superposition des courbes n’est alors plus possible. Dans le cas du xanthane, cette loi
a été démontrée pour le plateau newtonien et jusqu’à quatre décades de vitesses de cisaillement au-
delà de γ , bien que le xanthane en solution n’obéisse pas au modèle des simples
enchevêtrements topologiques.
A haute vitesse de cisaillement, tous les polysaccharides présentent un comportement fortement
rhéofluidifiant, ce qui est caractéristique de ce polysaccharide en solution à cette concentration. On
remarque d’ailleurs que l’ensemble des courbes se confondent dans cette gamme de fréquences : la
présence de chaînons hydrophobes ne semble donc pas influencer la rhéofluidification du xanthane.
A l’inverse, à basse vitesse de cisaillement, le précurseur exhibe un plateau newtonien, avec η0 = 2
Pa.s pour ω < 0,01 rad/s, alors que les xanthanes modifiés se comportent toujours comme des fluides
rhéofluidifiants. Plus précisément, le comportement rhéofluidifiant s’étend sur une gamme de
fréquences de plus en plus étendue avec la densité de greffage. Par exemple, à 10 -5 rad/s, TAX18,5C8FO
montre une viscosité au moins 250 fois supérieure à celle du précurseur. La présence de chaînons
alkyle impacte fortement le comportement du xanthane dans cette gamme de fréquences : les
interactions hydrophobes qui se développent entre les groupements greffés renforcent de manière
considérable le pouvoir suspensif du xanthane au repos (ou sous très faibles sollicitations). De plus,
cet effet est d’autant plus accentué que la densité de greffage est élevée. Ce type de comportement
est typiquement observé dans le cas des polymères amphiphiles associatifs30, 31, 47, 48.
- 232 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Pour résumer :
Les interactions hydrophobes entre les groupements greffés ne modifient pas les processus de
relaxation au sein des chaînes de xanthane. Ainsi, les polymères amphiphiles présentent une
dépendance en température de leurs propriétés rhéologiques très proche de celle du précurseur, et
ce, quelle que soit la densité de greffage. Cette dépendance est notamment caractérisée par une
énergie d’activation Ea, qui est du même ordre de grandeur pour tous les polysaccharides. Il apparaît
donc que l’organisation des chaînes modifiées en solution, en régime concentré, est peu différente de
celle du précurseur. Néanmoins, les greffons hydrophobes ralentissent considérablement la
dynamique de relaxation des chaînes amphiphiles, selon un processus qualifié de « relaxation
collante ». Ce phénomène est d’autant plus marqué que la densité de greffage est élevée. Les
interactions entre les chaînons hydrophobes sont également à l’origine du pouvoir suspensif accru du
xanthane au repos, contrôlable suivant la densité de greffage, et sans pour autant modifier son
comportement rhéofluidifiant à haute vitesse de cisaillement. Le xanthane modifié reste donc aussi
facile à cisailler, à haute vitesse, que le précurseur. Cette caractéristique du xanthane amphiphile lui
confère des possibilités d’applications intéressantes dans le domaine de la formulation comme agent
stabilisant et épaississant.
4.2.4. Modèle d’organisation des chaînes de xanthane modifié sous forme ordonnée
en solution
En solution, les chaînes modifiées adoptent une conformation ordonnée de type hélicoïdale,
similaire à celui du précurseur. De plus, il apparaît que l’organisation des chaînes greffées en solution
est très similaire à celle du xanthane. Au-delà de ccrit,, tous les échantillons forment un réseau
tridimensionnel viscoélastique stabilisé majoritairement par des liaisons hydrogène, et par des
interactions hydrophobes dans le cas des dérivés amphiphiles. Leur contribution, toutefois, est
modeste au regard des liaisons hydrogène. Par ailleurs, ces interactions ne modifient pas
- 233 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
l’organisation globale des chaînes, contrairement à ce qui est observé pour les polymères
amphiphiles flexibles. Ce phénomène s’explique par la forte rigidité des hélices de xanthane, qui
empêche ces dernières de se replier pour former des microdomaines hydrophobes. L’organisation des
chaînes en solution est donc contrôlée, dans notre cas, par la rigidité du polysaccharide et non pas
par les interactions attractives entre les greffons.
La caractérisation des propriétés viscoélastiques des xanthanes greffés semble confirmer cette
hypothèse. En premier lieu, tous les échantillons ont le même comportement de gel faible que le
précurseur à haute fréquence. En particulier, les xanthanes modifiés se caractérisent par un module
élastique à peine plus élevé que celui du précurseur. Par conséquent, le nombre de nœuds
élastiquement actifs est quasiment identique entre les polymères, quelle que soit la densité de
greffage. Ceci implique que la présence des greffons hydrophobes modifie peu l’organisation des
chaînes en solution en régime enchevêtré. De plus, la dépendance en température des propriétés
viscoélastiques est universelle pour tous les xanthanes. Elle est notamment caractérisée par une
énergie d’activation Ea, dont la valeur, pour chaque dérivé, est proche de celle d’un xanthane non
modifié. Le mécanisme de relaxation des polymères greffés est, de ce fait, identique à celui du
précurseur, ce qui corrobore notre supposition.
La présence des chaînons hydrophobes n’a donc que peu d’impact sur l’organisation des
macromolécules en solution en régime enchevêtré. Néanmoins, elle ralentit considérablement la
dynamique de relaxation des chaînes. De plus, ce phénomène est d’autant plus accentué que la
densité de greffage est élevée. Afin d’expliquer ces données, nous proposons le modèle
d’organisation suivant en solution :
- 234 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Figure 4-11 : Schéma d’organisation des chaînes de xanthane modifié sous forme ordonnée en solution, en
régime concentré.
L’organisation des chaînes consiste en un réseau transitoire, constitué des hélices semi-rigides de
xanthane arrangées parallèlement les unes aux autres et en interaction entre elles via des liaisons
hydrogène19, 36, 43, 44. Ces liaisons sont considérées comme des nœuds de jonction, ou élastiquement
actifs, de temps de vie fini36. De plus, l’alignement parallèle des hélices de xanthane a pour
conséquence de créer un nombre bien défini de liaisons hydrogène. Ce modèle correspond à la partie
gauche du schéma.
Lors du protocole de modification, le xanthane est sous forme ordonnée. De plus, les analyses de
dichroïsme circulaire et de polarimétrie montrent que, dans nos conditions opératoires, les chaînes
modifiées adoptent également une conformation hélicoïdale, plus ordonnée que celle du précurseur
du fait de la présence des greffons hydrophobes. Au regard de nos données, les dérivés amphiphiles
forment très probablement un réseau transitoire analogue à celui du précurseur en solution (cf.
partie droite de la figure 4-11). L’organisation de ce réseau est majoritairement contrôlée par les
liaisons hydrogène. La présence des chaînons hydrophobes facilite sans doute un rapprochement plus
important des hélices, et par conséquent la création ponctuelle de nouveaux nœuds. Elle ne peut pas
toutefois contrer l’organisation globale du système au regard de sa forte rigidité. Une conséquence de
ce phénomène est que le caractère associatif des échantillons greffés est peu différent de celui du
- 235 -
Précurseur Xanthane amphiphile
Modification chimique

Chapitre 4 : Propriétés en solution des xanthanes modifiés
précurseur. Ces greffons, par contre, influencent fortement le temps de relaxation du réseau. En
particulier, nous avons prouvé que la dynamique de ces systèmes peut être décrite par un mécanisme
de « relaxation collante ». Les processus de relaxation au sein des chaînes modifiées sont les mêmes
que ceux présents dans le précurseur, mais très fortement ralentis par les interactions entre les
chaînons hydrophobes.
Il faut savoir que Mendes et al.49 ont greffé des groupements hydrophobes de palmitoyle sur le
xanthane en conformation ordonnée. Ils observent que le dérivé obtenu amphiphile forme, en
solution, une structure lamellaire à plusieurs couches. Afin d’expliquer ces résultats, ils proposent un
modèle d’organisation en solution dans lequel les greffons hydrophobes sont supposés être situés sur
les chaînes latérales du xanthane et interagissent entre eux via des interactions hydrophobes pour
créer une double couche d’hélices. D’après leur modèle, la présence des greffons hydrophobes
n’affecte pas l’organisation globale des chaînes en solution, et est donc en accord avec nos données.
4.2.5. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous
forme ordonnée
L’analyse du comportement en écoulement des xanthanes modifiés hydrophobiquement a mis en
évidence le pouvoir suspensif supérieur de ces composés, d’où un potentiel intéressant en tant
qu’agent stabilisant dans le domaine de la formulation. Par ailleurs, du fait de leur caractère
amphiphile, il serait intéressant de déterminer si les xanthanes modifiés ont des propriétés
interfaciales, tout comme les tensioactifs. Ils pourraient ainsi contribuer à la stabilisation d’émulsion.
Afin de mettre ou non en évidence de telles propriétés pour nos échantillons, nous avons réalisé des
analyses de rhéologie interfaciale. Pour rappel, cette technique permet d’étudier le comportement
viscoélastique d’une interface liquide/gaz ou liquide/liquide (dans notre cas, eau/huile). Pour une
interface présentant une monocouche absorbée de polymère, l’étude de ses propriétés rhéologiques
- 236 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
peut donner des informations sur la conformation des chaînes à l’interface, leur cinétique de
migration ou encore la nature des interactions entre macromolécules.
Les échantillons analysés dans cette section ont été obtenus par le protocole de modification
dans l’eau à basse température. La notation suivante est employée pour désigner ces polymères :
- BTXAC8FO, avec X pour xanthane, BT pour Basse Température, A la densité de greffage et FO
pour Forme Ordonnée.
Le tableau 4-6 donne les caractéristiques des polysaccharides étudiés par rhéologie interfaciale :
EntréenEDACnCOOH
DSA (%) DSP (%) %octyle BTXAC8FO
1 0 66 46 0 BTX0C8FO
(précurseur)
3 0,7 81 53 8 BTX8C8FO
4 1 90 54 10 BTX10C8FO
5 1,3 69 50 24 BTX24C8FO
7 2,7 77 51 39 BTX39C8FO
Tableau 4-6 : Caractéristiques des échantillons étudiés en rhéologie interfaciale.
Les mesures dynamiques ont été effectuées à l’interface eau/huile de solutions à 2 g/L de
xanthane dans le domaine linéaire de déformation, sur une gamme de températures allant de 22,2 à
73,5°C (aller et retour, cf. chapitre 2). L’aller et retour constituent un « cycle thermique ». L’échantillon
est qualifié de « natif » lors de la première phase de mesure, et est dit « renaturé » pendant la phase
de refroidissement.
- 237 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.2.5.1. Comportement du précurseur à l’interface eau/huile
Les figures 4-12 représentent l’évolution du module de conservation Gs’ et de perte Gs’’ en
fonction de la fréquence de l’interface eau/huile de silicone, pour le précurseur à 2 g/L, à différentes
températures, en phase « aller » (partie A) et en phase « retour » (partie B).
Figures 4-12 A : Evolution de Gs’ (symboles fermés) et Gs
’’ (symboles ouverts) en fonction de la fréquence
pour le précurseur à 2 g/L (pH = 4,3 et σ = 1 mS/cm) de 22,2°C à 73,5°C, phase « aller ». B : Evolution de Gs’ et
Gs’’ en fonction de la fréquence pour le précurseur de 73,5 à 22,2°C, phase « retour ». Pour des raisons de clarté,
toutes les températures ne sont pas reportées sur les figures.
En phase « aller » (figure 4-12 A), les propriétés viscoélastiques de l’interface eau/huile sont
similaires à celles décrites pour les solutions de précurseur en solution. En effet, à 22,2°C, le module
de conservation est supérieur à celui de perte pour ω > 0,25 rad/s : l’interface est majoritairement
élastique. Les deux modules se croisent à 0,25 rad/s, ce qui correspond à la fréquence de relaxation.
Cette dernière se décale vers des valeurs de plus en plus grandes quand la température augmente, et
s’accompagne d’une baisse des modules. Ainsi, à partir de 42,7°C, Gs’’ est supérieur à Gs
’ sur
quasiment toute la gamme de fréquences étudiée : le comportement observé est celui d’une
interface liquide viscoélastique. Pour rappel, le précurseur a le même comportement en solution.
- 238 -
(rad/s)
10-2 10-1 100
Gs'
, G
s" (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' 22,2°C aller
Gs' 42,7°C aller
Gs' 63,2°C aller
Gs' 73,5°C
(rad/s)
10-2 10-1 100
Gs'
, G
s'' (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' 73,5°C
Gs' 63,2°C retour
Gs' 42,7°C retour
Gs' 22,2°C retour
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
D’après ces résultats, on peut conclure que le précurseur est bien présent et forme un réseau adsorbé
à l’interface eau/huile. On peut noter que pour des fréquences supérieures à 0,1 rad/s, les modules à
73,5°C sont plus élevés que ceux à 63,2°C, phénomène non observé en solution.
En phase retour, la diminution de température entraîne une augmentation des modules, et un
décalage de la fréquence de relaxation vers des valeurs de plus en plus faibles. Le comportement de
l’interface évolue de liquide viscoélastique à celui de solide élastique en refroidissant. Ici également,
ces données indiquent que le précurseur est toujours présent à l’interface eau/huile, même après le
cycle thermique. On constate néanmoins que les propriétés de l’interface « retour » sont différentes
de celles de la phase « aller ». En effet, lors du refroidissement, le comportement élastique est
observé dès 63,2°C pour ω > 0,16 rad/s. En comparaison, à la même température, l’interface « aller »
se comporte comme un liquide viscoélastique. De même, à 22,2°C, G s’ est supérieur à Gs
’’ sur toute la
gamme de fréquences lors de la phase « retour », contrairement à la phase « aller ». Par ailleurs, la
dépendance en fréquence des modules à 22,2°C lors de la phase « retour » est beaucoup moins
marquée qu’en phase « aller ». En outre, les valeurs des modules, dans le cas de l’interface « retour »,
sont supérieures à toutes les températures à celles de l’interface « aller » (d’au minimum une
décade). Il apparaît donc que le cycle thermique subi par le précurseur a pour effet de renforcer le
caractère élastique de l’interface à température ambiante. Ce phénomène semble montrer que
l’organisation des chaînes de xanthane n’est plus la même à l’interface eau/huile suite au traitement
thermique.
- 239 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
A partir de ces données, il est possible de tracer l’évolution de G s’ et Gs
’’ en fonction de la
température pour une fréquence fixée (ici, ω = 0,1 rad/s), comme indiqué sur la figure 4-13 A :
Figures 4-13 A : Evolution de Gs’ (symboles fermés) et Gs’’ (symboles ouverts) en fonction de la température
à une fréquence fixe de 0,1 rad/s, lors des phases « aller » et « retour », pour une solution à 2 g/L de précurseur.
B : Evolution de G’ (symboles fermés), de G’’ (symboles ouverts) en fonction de la température à une fréquence
fixe de 1 rad/s pour le même échantillon en analyse rhéologique en solution (symboles bleus : évolution du
pouvoir rotatoire normalisé, normalisé, selon la température pour le précurseur).
En premier lieu, à ω = 0,1 rad/s, l’évolution avec la température des modules G s’ et Gs’’ du
précurseur à l’interface en phase « aller » (figure 4-13 A) est très semblable à celle de G’ et G’’ de la
solution de précurseur (figure 4-13 B), pour des températures comprises entre 20 et 70°C. Dans les
deux cas, on constate une diminution monotone des valeurs des modules jusqu’à 55-60°C, suivie
d’une chute brutale pour des températures supérieures. Cette évolution particulière des modules se
produit, en solution et à l’interface, dans la gamme de températures où intervient la transition ordre-
désordre du xanthane (courbe bleue de la figure 4-13 B). Par conséquent, il apparaît que la
viscoélasticité du précurseur à l’interface en phase « aller » est régie par les mêmes processus que
ceux en œuvre lorsque ce polymère est en solution, jusqu’à 70°C. Au-delà de cette température, les
- 240 -
T (°C)
20 30 40 50 60 70
Gs',
Gs" (N
/m)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' 1er aller
Gs' 1er retour
T (°C)
10 20 30 40 50 60 70 80
G',
G" (P
a.s
)
10-1
100
norm
alis
é
0.0
0.2
0.4
0.6
0.8
1.0G' allerG' retournormalisé
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
valeurs des modules interfaciaux remontent fortement, alors que ceux en solution semblent se
stabiliser. Sous l’effet de la dénaturation, les chaînes de xanthane, davantage flexibles, s’adsorbent
sans doute plus facilement à l’interface que sous forme ordonnée très rigide. Par ailleurs, en
conformation désordonnée, le squelette cellulosique apolaire est probablement plus accessible et
sujet à s’adsorber à l’interface eau/huile, ce qui expliquerait la hausse des modules dans cet intervalle
de température. Le traitement thermique lors de la phase « aller » modifie donc l’organisation des
chaînes à l’interface eau/huile.
La phase « retour » se caractérise par une augmentation graduelle des modules lorsque la
température diminue de 70 à 22,2°C : les modules évoluent sur une décade et demi dans cette
gamme de températures. Le refroidissement entraîne un renforcement progressif de l’élasticité de
l’interface. Entre 73,5 et 70°C, on note une hausse prononcée de G s’. Ce phénomène se produit aux
alentours de la Tm du précurseur, ce qui semble indiquer que les chaînes adoptent de nouveau une
conformation hélicoïdale lors du refroidissement, fait qui expliquerait l’augmentation de la
viscoélasticité interfaciale. Pour des températures inférieures à 70°C, les modules croissent de
manière monotone, ce qui suggère que l’organisation des chaînes du précurseur à l’interface ne subit
plus de modifications majeures pour T < Tm.
La forme « renaturée » diffère de la forme « native ». En effet, les modules plus élevés observés
en phase « retour » par rapport à la phase « aller » montrent que la conformation « renaturée » est
davantage adsorbée à l’interface que celle « native ». En premier lieu, il a été noté, lors de la phase
« aller », que le précurseur s’adsorbait davantage à l’interface à 73,5°C, soit sous forme désordonnée.
De plus, dans le cadre de notre étude, nous travaillons en régime enchevêtré. Or, plusieurs auteurs50-
52 ont prouvé que, dans ce régime de concentration, le processus de dénaturation du xanthane est
incomplet. Les formes dénaturées et renaturées sont des hélices plus ou moins dissociées28, 50, 53, 54,
constituées de zones désordonnées de type pelote maintenues ensembles par des « nœuds » de
- 241 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
double hélice28, 51. De ce fait, il est probable que la forme ordonnée « renaturée » soit une hélice
imparfaite, qui comporte davantage de zones désordonnées que la conformation « native ». Cette
proportion plus importante de zones désordonnées serait à l’origine de la meilleure adsorption de la
forme « renaturée » à l’interface eau/huile.
Globalement, le traitement thermique effectué sur le précurseur améliore les propriétés
viscoélastiques de ce dernier à l’interface eau/huile suite à la modification de la conformation des
chaînes. Capron et al.51 et Lee et al.52 ont également observé un renforcement du comportement
rhéologique du xanthane en solution en régime concentré. Ces auteurs attribuent ce fait à la
renaturation des hélices partiellement dissociées, qui aboutit à la formation d’un réseau comportant
davantage de nœuds de jonction qu’à l’état initial. Dans notre cas, il est possible qu’un tel processus
se produise à l’interface eau/huile entre les chaînes de précurseur lors du refroidissement.
Nous avons, dans un second temps, cherché à déterminer si l’organisation des chaînes à
l’interface eau/huile après ce premier traitement est modifiée ou non lorsque le polymère subit de
nouveau un processus thermique de dénaturation et de renaturation. Dans ce but, nous avons
soumis le précurseur à deux cycles thermiques consécutifs (soit deux phases « allers » et deux phases
« retour »). Entre les deux cycles, l’échantillon est maintenu à 22,2°C pendant une nuit. L’évolution de
Gs’ et de Gs’’ en fonction de la température à une fréquence donnée pour les deux cycles est
représentée dans les figures 4-14 :
- 242 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Figures 4-14 : Evolution de Gs’ (symboles fermés) et Gs’’ (symboles ouverts) en fonction de la température
de 0,1 rad/s pour le précurseur à 2 g/L à l’interface eau/huile après 2 traitements thermiques consécutifs A : à
0,1 rad/s. B : à 0,01 rad/s.
Sur la figure 4-14 A, Gs’ est supérieur à Gs’’ sur toute la gamme de températures étudiée lors des
deux phases du 2ème cycle thermique : le comportement de l’interface est celle d’un solide
viscoélastique. Par comparaison, l’interface présente les propriétés d’un liquide pendant la 1 ère phase
« aller » sur toute la gamme de températures étudiée, et d’un solide lors de la 1 ère phase « retour »
pour T < 30°C. De plus, les valeurs des modules mesurées lors du 2ème cycle sont supérieures, sur
toute la gamme de températures, à celles obtenues lors du 1er cycle. On peut noter que Gs’ diminue
pendant la 2ème phase « aller » (ou augmente pendant la 2ème phase « retour ») de manière monotone
sur toute la gamme de températures lorsque la température croît (ou décroît). G s’’, quant à lui, reste
constant sur tout l’intervalle de températures pendant l’ensemble du cycle. Les dépendances en
température des modules sont, par ailleurs, semblables entre les deux phases du 2 ème cycle, et
diffèrent de celles du 1er cycle. On remarque également que les modules sont quasiment identiques
entre les deux phases du 2ème cycle, contrairement à ce qui a été observé pendant le 1er cycle
thermique.
- 243 -
T (°C)20 30 40 50 60 70
Gs',
Gs" (N
/m)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' 1er aller
Gs' 1er retour
Gs' 2ème aller
Gs' 2ème retour
T (°C)20 30 40 50 60 70
Gs',
Gs" (N
/m)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' 1er aller
Gs' 1er retour
Gs' 2ème aller
Gs' 2ème retour
0,1 rad/s 0,01 rad/sA B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La figure 4-14 B représente l’évolution des modules lors des deux cycles thermiques pour ω =
0,01 rad/s. L’allure de Gs’ et Gs’’ avec la température lors du 1er cycle thermique est la même que celle
décrite à 0,1 rad/s. Lors de la 2ème phase « aller », le comportement de l’interface est celle d’un solide
viscoélastique jusqu’à 56°C environ. De plus, Gs’ diminue très progressivement avec la température,
avec une dépendance en température très différente de celle de la 1ère phase « aller », alors que Gs’’
reste constant. Ces caractéristiques sont identiques à celles relevées à 0,1 rad/s. Au-delà de 56°C par
contre, le comportement de l’interface à 0,01 rad/s présente des divergences par rapport à celui
décrit à 0,1 rad/s. En effet, l’interface se comporte comme un liquide viscoélastique (Gs’’ > Gs’). Gs’
décroît également plus brutalement aux alentours de la Tm. Néanmoins, l’amplitude de variation de
Gs’, qui couvre moins d’une décade, est plus faible que celle observée lors de la 1 ère phase « aller »
(2,5 décades). Pendant la 2ème phase « retour », Gs’ augmente fortement entre 73,5 et 70°C, et le
comportement passe de liquide à solide viscoélastique. Pour T < 70°C, Gs’ croît de manière monotone
lorsque la température augmente, ce qui est similaire au comportement observé à 0,1 rad/s. Les
valeurs de Gs’ de la phase « retour » sont supérieures à celles de la phase « aller ». Pour autant, la
différence de valeurs entre les modules est peu marquée entre les deux phases, notamment lorsque
la température est inférieure à 50°C.
Il ressort de la comparaison des données que l’influence de la transition sur le comportement du
xanthane à l’interface est beaucoup moins prononcée lors du 2ème cycle thermique par rapport au 1er.
En outre, cette influence est minime, voire négligeable, à haute fréquence. Une explication possible
est que les chaînes adsorbées à l’interface eau/huile se dénaturent moins lors de la 2 ème phase
« aller ». En premier lieu, la formation d’un réseau avec un plus grand nombre de nœuds de jonction
lors du 1er cycle thermique limite probablement les mouvements des chaînes lorsqu’elles se
dénaturent. De plus, il est probable que les chaînes adsorbées à l’interface après le 1 er cycle forment
des hélices imparfaites, avec davantage de zones désordonnées que la forme « native ». De ce fait, le
processus de dénaturation agit sur une quantité de chaînes bien moindre que lors de la 1ère phase
« aller », au point de ne pas être observable à haute fréquence. Cette hypothèse explique également
- 244 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
l’évolution de Gs’ lors de la 2ème phase « retour : la dénaturation des chaînes à l’interface étant plus
limitée suite au premier traitement thermique, le processus de renaturation affecte par conséquent
moins de chaînes que lors du 1er cycle. En outre, les valeurs des modules de la phase « retour » sont
similaires à celles de la phase « aller », soit sur toute la gamme de températures (pour ω = 0,1 rad/s),
soit pour T < 50°C (pour ω = 0,01 rad/s). Ce résultat semble confirmer que l’organisation globale des
chaînes à l’interface eau/huile est peu modifiée lors du 2ème traitement thermique, ce qui confirmerait
notre supposition concernant la dénaturation très limitée des chaînes après un 1er cycle thermique.
- 245 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.2.5.2. Comportement des xanthanes modifiés sous forme ordonnée à l’interface
eau/huile
Les mêmes analyses rhéologiques interfaciales ont été réalisées sur les xanthanes modifiés sous
forme ordonnée. Les résultats obtenus sont résumés dans la figure 4-15 A pour les xanthanes greffés
jusqu’à 10 %, et la figure 4-15 B pour les échantillons modifiés entre 24 et 39 %. Pour des raisons de
clarté, les modules de perte n’ont pas été représentés.
Figures 4-15 : Evolution de Gs’ en fonction de la température à une fréquence fixe de 0,1 rad/s, lors des
phases « aller » (symboles fermés) et « retour » (symboles ouverts) pour A : le précurseur, BTX8C8FO et BTX10C8FO à 2
g/L. B : le précurseur, BTX24C8FO et BTX39C8FO à 2 g/L.
Pour BTX8C8FO, en phase « aller », Gs’ diminue de manière monotone lorsque la température
augmente, puis, à partir de 70°C, remonte brutalement. Les valeurs du module de cet échantillon
sont, jusqu’à 56°C, légèrement supérieures à celles du précurseur. A 73,5°C, le module est supérieur à
celui de tous les autres points de la phase « aller ». En phase « retour », le module augmente
fortement entre 73,5 et 70°C, soit dans la gamme de températures ou survient la transition
conformationnelle de ce polymère (cf. figure 4-2). Pour T < 70°C, G s’ continue de croître, mais avec
une dépendance en température plus faible, et qui est différente de celle exhibée par le xanthane
- 246 -
(rad/s)
20 30 40 50 60 70
Gs' (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' BTX0C8FO aller
Gs' BTX24C8FO aller
Gs' BTX29C8FO aller
0.1rad/s
(rad/s)20 30 40 50 60 70
Gs' (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' BTX0C8FO aller
Gs' BTX8C8FO aller
Gs' BTX10C8FO aller
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
non greffé. En outre, les valeurs des modules en phase « retour » sont supérieures en tout point à
celles de la phase « aller ».
Pour BTX10C8FO, le module reste relativement constant sur toute la gamme de températures en
phase « aller ». Lors de la phase « retour », Gs’ croît progressivement lorsque la température diminue,
avec une dépendance en température proche de celle de BTX8C8FO. Globalement, les modules en phase
« retour » sont plus élevés que ceux de la phase « aller ». Néanmoins, on n’observe pas de variation
brutale de Gs’ à haute température, contrairement à BTX8C8FO.
BTX24C8FO et BTX39C8FO, quant à eux, ont des allures similaires : le module reste relativement constant
en phase « aller ». En phase « retour », le module n’évolue quasiment pas et sa valeur est proche de
celle de la phase « aller » entre 73,5 et 50°C environ. Pour T < 50°C, Gs’ augmente progressivement
quand la température décroît, et devient supérieur à celui de la phase « aller ».
On remarque également qu’à 22,2°C en phase « aller », le module Gs’ augmente
exponentiellement avec la densité de greffage ( G'=0.0247e0.033 octyle ), comme illustré dans la
figure 4-16 :
%octyle
0 5 10 15 20 25 30 35 40
Gs'
(N
/m)
10-4
10-3
10-2
Gs' aller
Gs' retour
Figure 4-16 : Evolution de Gs’ en fonction de la densité de greffage à 22,2°C en phase « aller » (symboles fermés)
et « retour » (symboles ouverts).
- 247 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Plus le xanthane est greffé, plus le caractère élastique de l’interface est prononcé à 22,2°C avant
le traitement thermique : la présence de groupements hydrophobes en proportions croissantes
favorise l’adsorption des chaînes modifiées à l’interface eau/huile, même sous forme ordonnée. En
phase « retour », la valeur du module présente un maximum pour %C8 = 8 %, diminue brutalement
entre 8 et 10 % et augmente progressivement ensuite. Il n’y a pas de corrélation possible entre la
valeur du module et la densité de greffage. L’origine de ces évolutions sera écrite plus loin dans le
manuscrit.
L’organisation à l’interface eau/huile des chaînes modifiées évolue au cours du traitement
thermique, selon un processus plus complexe que celui en œuvre pour le précurseur. Les analyses de
polarimétrie et de dichroïsme circulaire effectuées sur les xanthanes amphiphiles mettent en
évidence que ces macromolécules adoptent en solution une conformation ordonnée d’autant plus
rigide et stable thermiquement que la densité de greffage augmente. Les propriétés rhéologiques
interfaciales de ces systèmes sont alors contrôlées par deux mécanismes antagonistes : la perte de
rigidité des chaînes sous l’effet de la température, qui tend à diminuer le caractère élastique de
l’interface, et l’adsorption des zones désordonnées et/ou des zones amphiphiles à l’interface, qui
renforce l’élasticité de cette dernière. Ces deux mécanismes s’exercent conjointement au sein des
chaînes lors du chauffage. De plus, ils dépendent directement de la transition ordre-désordre du
xanthane. De ce fait, plus le polysaccharide est greffé, plus sa dénaturation est partielle, et la quantité
de zones désordonnée formées est faible. Ainsi, pour les échantillons les plus modifiés, les
contributions de la rigidité et de l’adsorption des zones désordonnées et/ou amphiphiles sont, après
traitement thermique, sensiblement identiques à celles de la forme «native », ce qui explique le
faible gain d’élasticité observé après le cycle thermique (cf. figure 4-16). Au contraire, pour les
polymères les moins greffés, l’augmentation de Gs’ en phase « aller » à haute température indique
que l’adsorption des chaînes l’emporte sur la perte de rigidité au passage de la transition
conformationnelle, ce qui est à l’origine de la forte augmentation de l’élasticité interfaciale après le
- 248 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
cycle thermique. Par conséquent, la viscoélasticité interfaciale après traitement thermique est très
fortement dépendante du passage de la forme ordonnée à la forme désordonnée lors du chauffage,
passage qui est lui-même assujetti à la densité de greffage du polysaccharide.
Les xanthanes modifiés ont été soumis à deux cycles thermiques (soit deux phases « aller » et
deux phases « retour »), selon les mêmes conditions que celles employées pour le précurseur. Les
résultats pour le précurseur, BTX8C8FO et BTX39C8FO sont représentés dans les figures 4-17 :
Figures 4-17 : Evolution de Gs’ en fonction de la température lors de deux cycles thermiques pour des
solutions à 2 g/L de A : précurseur et BTX8C8FO. B : précurseur et BTX39C8FO (phases « aller » : symboles fermés,
phases « retour » : symboles ouverts).
Les autres polymères n’ont pas été indiqués pour des raisons de clarté. Globalement, leur
comportement est identique à celui du précurseur, de BTX8C8FO et BTX39C8FO : le module diminue très
progressivement lors de la 2ème phase « aller », puis augmente doucement pendant la 2ème phase
« retour ». La conformation adoptée par les chaînes modifiées après le 1er cycle thermique n’est pas
affectée par ce 2ème cycle. De plus, les valeurs de Gs’ sont presque identiques entre la 1ère phase
- 249 -
(rad/s)
20 30 40 50 60 70
Gs' (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' BTX0C8FO 1er aller
Gs' BTX0C8FO 2ème aller
Gs' BTX8C8FO 1er aller
Gs' BTX8C8FO 2ème aller
(rad/s)
20 30 40 50 60 70
Gs' (
N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' BTX0C8FO 1er aller
Gs' BTX0C8FO 2ème aller
Gs' BTX39C8FO 1er aller
Gs' BTX39C8FO 2ème aller
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
« retour »et la 2ème phase « aller », ce qui signifie que l’organisation des macromolécules est la même
entre le 1er et le 2ème traitement thermique.
Au regard de la similitude de comportement avec le précurseur, il semble que l’hypothèse émise
pour le xanthane soit valable pour les échantillons greffés. Pour rappel, nous avons supposé que les
chaînes adsorbées à l’interface eau/huile se dénaturent moins lors de la 2ème phase « aller ». Une des
conséquences est que le processus de dénaturation lors de la 2ème phase « aller » affecte moins de
chaînes. Nos résultats obtenus après un cycle thermique semblent donc être à l’équilibre
thermodynamique.
Pour résumer :
Nos analyses de rhéologie interfaciales effectuées sur le précurseur en régime concentré à
différentes températures montrent que la conformation adoptée par le xanthane influence fortement
ses propriétés à l’interface eau/huile. En particulier, nous avons observé que le traitement thermique
réalisé sur le polymère (cycle de chauffage et de refroidissement) permet d’améliorer fortement le
caractère élastique de l’interface. Ce résultat s’explique par un processus de dénaturation partielle
des chaînes lors de la phase « aller », qui entraîne la formation de zones désordonnées s’adsorbant à
l’interface eau/huile pour T > Tm. Lors de la phase « retour », la renaturation d’une partie de ces
zones entraîne une amélioration de l’élasticité.
Les xanthanes modifiés, quant à eux, possèdent des propriétés rhéologiques interfaciales accrues
par rapport au précurseur à température ambiante, avant tout traitement thermique. Ce résultat est
dû à la présence des greffons hydrophobes, qui favorisent l’adsorption des chaînes amphiphiles à
l’interface eau/huile. Comme pour le précurseur, l’organisation des macromolécules amphiphiles à
l’interface eau/huile évolue sous l’effet de la température. Ces dernières adoptent en solution une
conformation ordonnée d’autant plus rigide que la densité de greffage est élevée. Le comportement
- 250 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
interfacial de ces polymères est régi par deux mécanismes antagonistes : la perte de rigidité des
chaînes sous l’effet de la température, et l’adsorption des zones désordonnées et/ou amphiphiles,
favorisée à haute température. Ces deux mécanismes, qui s’exercent conjointement, dépendent de la
densité de greffage du polymère. Ainsi, plus le xanthane est greffé, plus la dénaturation partielle est
difficile et la conformation des chaînes dénaturées est similaire à celle des macromolécules
« natives ». Pour les échantillons les moins modifiés en revanche, l’adsorption des zones
désordonnées et/ou amphiphiles est majoritaire par rapport au gain de flexibilité. Pendant la phase
« retour », la contribution des zones greffées adsorbées en phase « aller », et la renaturation des
chaînes améliorent le caractère élastique de l’interface. Cependant, l’amélioration des propriétés
élastiques suite au traitement thermique est d’autant moins marquée que la densité de greffage est
élevée.
4.2.5.3. Modèles d’organisation des chaînes de xanthane modifié sous forme
ordonnée à l’interface eau/huile
Nous avons étudié le comportement rhéologique de nos xanthanes greffés à l’interface eau/huile.
Les résultats indiquent que le traitement thermique imposé à l’échantillon, amphiphile ou non,
modifie son organisation à l’interface. Cette modification se produit essentiellement lors de la phase
de chauffage. Le refroidissement a pour effet de renforcer le caractère élastique de l’interface. Par
ailleurs, la réorganisation des polymères modifiés dépend de la densité de greffage de ces derniers.
Tout comme les analyses réalisées en solution, les mesures à l’interface eau/huile sont effectuées
sur des solutions de xanthane en régime concentré. Par conséquent, il est probable que les chaînes
s’adsorbent à l’interface eau/huile sous forme d’hélice. Par ailleurs, l’évolution particulière des
propriétés rhéologiques avec la température suggère que la transition conformationnelle du
polymère, plus ou moins complète, est à l’origine du changement d’organisation constatée à
- 251 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
l’interface suite au traitement thermique. A partir de ces hypothèses, nous proposons le modèle
suivant pour expliquer nos résultats :
Figure 4-18 : Organisation des chaînes de précurseur (cas A), xanthane modifié à faible densité de greffage (cas
B) et xanthane modifié à forte densité de greffage (cas C) à l’interface eau/huile après dénaturation et
renaturation.
Pour le précurseur, la dénaturation partielle entraîne la formation de zones désordonnées au sein
des chaînes. Ces zones, plus flexibles, s’adsorbent à l’interface eau/huile à haute température (partie
2 A) pour T > Tm, d’où l’augmentation du module au-delà de cette température. Lors de la
renaturation (partie 3 A), certaines de ces zones restent sous forme désordonnée et contribuent à
l’élasticité, d’autres adoptent de nouveau une conformation hélicoïdale. Ces phénomènes expliquent
la hausse de Gs’ en phase « retour ». Par ailleurs, la renaturation est également un processus partiel.
- 252 -
Thuile
huilehuile
A« Natif »
1 2
T
3
B
huile huileT
huile
C
T T
huile
C
huile
Dénaturation Renaturation
Dénaturation Renaturationhuile T

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Par conséquent, le système renaturé comporte davantage de chaînes à l’état désordonné que le
système « natif ».
Pour les xanthanes amphiphiles à faible densité de greffage, l’augmentation de la température
entraîne une diminution de la rigidité des chaînes adsorbées, ce qui explique la baisse du module lors
de la phase « aller ». De plus, les chaînes se dénaturent partiellement à haute température, et les
zones désordonnées ainsi créées peuvent s’adsorber à l’interface eau/huile (partie 2 B). C’est
pourquoi, pour T > 70°C, le caractère élastique de l’interface est renforcé. Au cours de la phase
« retour », une fraction des zones désordonnées adsorbées se renature (partie 2 C), et contribue à
l’élasticité de l’interface. L’organisation globale des chaînes est donc modifiée suite au traitement
thermique, ce qui a un impact direct sur le comportement interfacial mesuré.
En revanche, le processus de dénaturation est très partiel, voire nul, pour les polymères à forte
densité de greffage, car la Tm est supérieure à la température expérimentale appliquée lors des
analyses. Par conséquent, il n’y a pas de modifications majeures dans la conformation et
l’organisation des chaînes modifiées lors du cycle thermique (partie C), d’où la faible évolution des
propriétés interfaciales observée entre les formes « natives » et « renaturées ».
En ce qui concerne les xanthanes modifiés de faible à moyenne densité de greffage (entre 10 et
24 %), l’évolution des propriétés rhéologiques avec la température dépend de la compétition entre la
perte de rigidité des chaînes, liée à la dénaturation partielle du système, et l’adsorption à l’interface
des zones dénaturées et/ou greffées, qui est favorisée par le gain de flexibilité.
- 253 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La figure 4-19 compare l’évolution du module interfacial Gs’ (à température ambiante en phase
« aller ») et du module élastique G’ (à 20°C également) en fonction de la densité de greffage pour les
xanthanes modifiés sous forme ordonnée :
Figure 4-19 : Comparaison entre l’évolution de Gs’ (symboles fermés) et G’ (symboles ouverts) en fonction de la
densité de greffage.
Le module élastique en solution, G’, reste quasiment constant lorsque la densité de greffage croît.
L’augmentation de la quantité de greffons hydrophobes ne permet pas de créer (ou peu) de nouveaux
nœuds de jonction, du fait de la rigidité très importante des chaînes en conformation ordonnée. Ce
résultat corrobore notre modèle d’organisation des xanthanes amphiphiles en solution, à savoir un
réseau transitoire d’hélices qui interagissent majoritairement entre elles via des liaisons hydrogène. A
l’interface eau/huile en revanche, le module interfacial Gs’ augmente exponentiellement avec la
densité de greffage. La présence de chaînons hydrophobes en proportions croissantes favorise
fortement l’adsorption des chaînes à l’interface, renforçant ainsi le réseau formé. Ainsi, bien que les
greffons hydrophobes ne contribuent pas à l’élasticité en solution, ils permettent au xanthane
modifié de s’adsorber davantage à l’interface, stabilisant cette dernière. Les comportements en
solution et à l’interface sont donc découplés.
- 254 -
%octyle
0 5 10 15 20 25 30 35 40
Gs'
(N
/m)
10-4
10-3
10-2
G' (
Pa)
100
101
102

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Pour résumer :
La modification chimique du xanthane sous forme ordonnée n’affecte pas l’organisation en
solution des chaînes en milieu concentré. Ces dernières forment toujours un réseau transitoire
d’hélices stabilisé très majoritairement par des liaisons hydrogène. Les chaînons hydrophobes
ralentissent cependant considérablement la dynamique de relaxation des chaînes, qui peut être
décrit par un processus de « relaxation collante ». Ces caractéristiques du xanthane « ordonné »
s’expliquent par la très forte rigidité de la conformation hélicoïdale, qui empêche les chaînes de se
replier pour créer des microdomaines hydrophobes. La rigidité l’emporte donc sur les interactions
hydrophobes attractives, et définit la structuration du système. De ce fait, les xanthanes greffés
présentent un comportement associatif identique à celui du précurseur, mais avec une dynamique
fortement ralentie.
La présence des groupements hydrophobes influence le comportement des xanthanes modifiés à
l’interface eau/huile. En effet, les greffons facilitent l’adsorption des fragments hydrophobes à
l’interface, ce qui explique l’augmentation du caractère élastique de cette dernière avec la densité de
greffage. Par ailleurs, les xanthanes amphiphiles adoptent en solution une conformation ordonnée
dont la rigidité et la stabilité thermique augmentent avec la densité de greffage. La hausse de la
température entraîne une dénaturation et renaturation partielle des hélices, ce qui entraîne une
restructuration des chaînes adsorbées à l’interface. Cette restructuration dépend de la densité de
greffage du polymère : plus ce paramètre est élevé, plus la forme dénaturée des macromolécules est
similaire à celle de la forme « native ». La viscoélasticité interfaciale après traitement thermique est
donc très fortement dépendante du passage de la forme ordonnée à la forme désordonnée lors du
chauffage, passage qui est lui-même fonction de la densité de greffage du polysaccharide.
- 255 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.3. Comportement en solution des xanthanes modifiés sous forme désordonnée
Cette partie présente des résultats préliminaires concernant les propriétés rhéologiques, en
solution et à l’interface eau/huile, des xanthanes modifiés sous forme désordonnée. Pour rappel, ces
échantillons sont obtenus par le procédé de modification du xanthane dans le DMSO, décrit dans les
chapitres deux et trois. Les analyses RMN 1H de ces composés ont notamment montré que de
nombreuses impuretés, correspondant à des dérivés de l’EDAC, sont greffées sur les chaînes. A ce
jour, nous ignorons si elles ont un impact sur le comportement viscoélastique de ces polymères en
solution.
Dans la suite du manuscrit, nous qualifions « d’ordonné » un xanthane synthétisé par le protocole
de modification dans l’eau, et de « désordonné » un polymère obtenu par le processus dans le DMSO.
Cette terminologie se réfère à l’état du xanthane lors des étapes de modification et ne présume en
rien la conformation adoptée par les macromolécules une fois remise en solution aqueuse.
4.3.1. Etude des propriétés rhéologiques en solution des xanthanes greffés sous
forme désordonnée
La notation suivante est employée pour désigner les xanthanes modifiés dans le DMSO :
- XAC8FD, avec X pour xanthane, A la densité de greffage et FD pour Forme Désordonnée.
- 256 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Le tableau 4-7 donne les caractéristiques des polysaccharides étudiés dans cette section :
EntréenEDACnCOOH
DSA DSP %octyle XAC8FD
1 1 80 58 0 X0C8FD (précurseur)
2 0.5 104 63 23 X23C8FD
3 0,7 100 56 35 X35C8FD
4 1 71 44 44 X44C8FD
Tableau 4-7 : Caractéristiques des xanthanes modifiés dans le DMSO étudiés en rhéologie en solution.
4.3.1.1. Comportement en solution du précurseur « désordonné »
Avant d’examiner les propriétés des xanthanes modifiés sous forme désordonnée, nous nous
sommes intéressés à celles du précurseur synthétisé par le protocole dans le DMSO. Cette étude a
été réalisée afin d’avoir une référence non modifiée pour l’interprétation des données des polymères
greffés dans les mêmes conditions. Elle nous permet aussi de déterminer si les étapes de
solubilisation utilisées dans le processus de modification sous forme désordonnée a un impact ou
non sur le comportement en solution aqueuse du xanthane initial non greffé (dépolymérisation,
perte d’acétate et/ou de pyruvate, etc.). Dans ce but, le précurseur dit « désordonné » a été solubilisé
dans l’eau dans les mêmes conditions de concentration, pH et salinité que le précurseur « ordonné »,
et soumis aux mêmes analyses pour comparaison.
Le comportement viscoélastique en solution du précurseur « désordonné » a été caractérisé via
des sollicitations harmoniques à différentes fréquences (de 0,1 à 100 rad/s) dans le domaine linéaire
de déformation pour une gamme de températures allant de 5 à 80°C. La figure 4-20 A montre la
dépendance en fréquence de G’ et G’’ de la solution aqueuse à 2 g/L de ce polymère à 5, 20, 40 et
60°C. Pour des raisons de clarté, les autres températures n’ont pas été représentées.
- 257 -
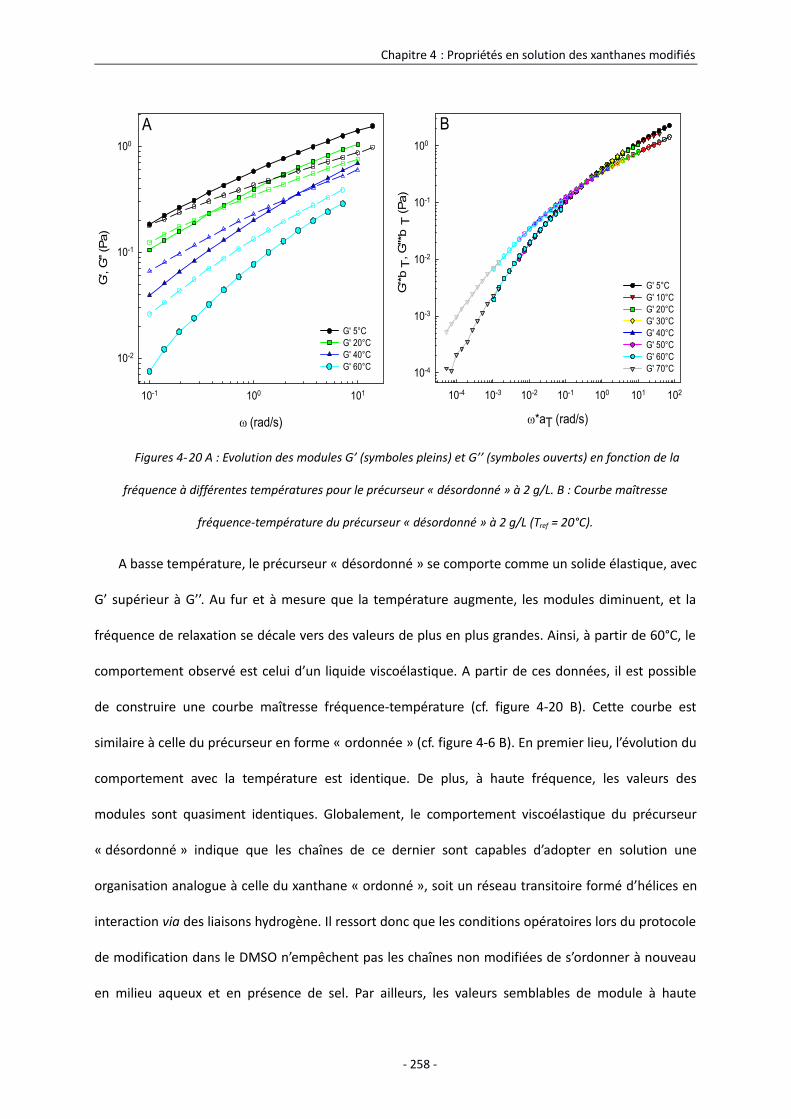
Chapitre 4 : Propriétés en solution des xanthanes modifiés
Figures 4-20 A : Evolution des modules G’ (symboles pleins) et G’’ (symboles ouverts) en fonction de la
fréquence à différentes températures pour le précurseur « désordonné » à 2 g/L. B : Courbe maîtresse
fréquence-température du précurseur « désordonné » à 2 g/L (Tref = 20°C).
A basse température, le précurseur « désordonné » se comporte comme un solide élastique, avec
G’ supérieur à G’’. Au fur et à mesure que la température augmente, les modules diminuent, et la
fréquence de relaxation se décale vers des valeurs de plus en plus grandes. Ainsi, à partir de 60°C, le
comportement observé est celui d’un liquide viscoélastique. A partir de ces données, il est possible
de construire une courbe maîtresse fréquence-température (cf. figure 4-20 B). Cette courbe est
similaire à celle du précurseur en forme « ordonnée » (cf. figure 4-6 B). En premier lieu, l’évolution du
comportement avec la température est identique. De plus, à haute fréquence, les valeurs des
modules sont quasiment identiques. Globalement, le comportement viscoélastique du précurseur
« désordonné » indique que les chaînes de ce dernier sont capables d’adopter en solution une
organisation analogue à celle du xanthane « ordonné », soit un réseau transitoire formé d’hélices en
interaction via des liaisons hydrogène. Il ressort donc que les conditions opératoires lors du protocole
de modification dans le DMSO n’empêchent pas les chaînes non modifiées de s’ordonner à nouveau
en milieu aqueux et en présence de sel. Par ailleurs, les valeurs semblables de module à haute
- 258 -
*aT (rad/s)
10-4 10-3 10-2 10-1 100 101 102G
'*b
T, G
"*b
T (P
a)
10-4
10-3
10-2
10-1
100
G' 5°CG' 10°CG' 20°C G' 30°CG' 40°CG' 50°CG' 60°CG' 70°C
(rad/s)
10-1 100 101
G',
G" (P
a)
10-2
10-1
100
G' 5°CG' 20°CG' 40°CG' 60°C
A B

Chapitre 4 : Propriétés en solution des xanthanes modifiés
fréquence entre les deux précurseurs indiquent que le processus de modification dans le DMSO soit
ne dégrade pas les chaînes, soit que cette dégradation est suffisamment limitée pour ne pas avoir
d’influence notable sur le comportement viscoélastique du xanthane à la concentration étudiée.
4.3.1.2. Comportement en solution des xanthanes modifiés dans le DMSO
Les mêmes analyses rhéologiques en solution ont été effectuées sur les échantillons modifiés
dans le DMSO. La figure 4-21 illustre l’évolution de G’ et de G’’ en fonction de la fréquence de X 23C8FD
à 5, 20, 40, 70 et 80°C :
Figure 4-21 : Evolution de G’ (symboles pleins) et G’’ (symboles ouverts) en fonction de la fréquence à
différentes températures pour X23C8FD à 2 g/L dans les mêmes conditions de pH et de salinités que les
précurseurs.
A basse température, on constate que les valeurs des modules sont trop faibles pour pouvoir être
mesurées par le rhéomètre. Les résultats présentés à 5 et 20°C ne sont pas représentatifs de
l’échantillon, mais correspondent à l’inertie de la géométrie. Le module élastique associé aux
propriétés rhéologiques de X23C8FD devient quantifiable à partir de 40°C, pour des fréquences
- 259 -
(rad/s)10-2 10-1 100 101
G',
G"
(Pa)
10-4
10-3
10-2
10-1
G' 5°CG' 20°CG' 40°CG' 70°CG' 80°C

Chapitre 4 : Propriétés en solution des xanthanes modifiés
inférieures à 1 rad/s. La valeur du module visqueux, par contre, est inférieure à la limite de sensibilité
de l’appareil. A partir de 40°C, l’augmentation de la température entraîne une hausse de la valeur des
deux modules, avec G’ supérieur à G’’ sur toute la gamme de fréquences. De plus, la dépendance en
fréquence des deux modules est très faible.
Le comportement viscoélastique de X23C8FD est, par conséquent, très différent de celui de son
précurseur et des xanthanes greffés sous forme ordonnée. En premier lieu, la valeur des modules, à
basse température, est beaucoup plus faible pour le dérivé modifié dans le DMSO que pour les autres
polymères. Les modules deviennent du même ordre de grandeur que ceux du précurseur (soit
compris entre 10-1 et 10-2 Pa) à partir de 70°C. Par ailleurs, l’évolution des propriétés viscoélastiques
avec la température, dans le cas de X23C8FD, est à l’opposé de celle décrite pour les précurseurs et les
polysaccharides modifiés sous forme hélice. En effet, pour ces derniers, il a été noté que les modules
diminuent avec la température et que la dépendance en fréquence augmente. De plus, le
comportement passe de solide à liquide viscoélastique. En ce qui concerne X23C8FD, les valeurs des
modules augmentent avec la température, et le comportement observé correspond à celui d’un gel
fort (faible dépendance en fréquence des modules) dont le caractère élastique est renforcé sous
l’effet de la température. Enfin, il n’est pas possible, pour X23C8FD, de construire de courbe maîtresse
fréquence-température.
Ces données nous permettent d’affirmer que l’organisation des chaînes de X23C8FD en solution
diverge fortement de celle du précurseur et des dérivés « ordonnés ». De plus, l’évolution très
particulière des modules avec la température suggère que la structuration des chaînes greffées dans
la solution fluctue lors du traitement thermique. Par ailleurs, il est probable que chaque organisation
se caractérise par un processus de relaxation spécifique, avec une dépendance en température qui lui
est propre, ce qui expliquerait l’impossibilité de construire une courbe maîtresse fréquence-
température.
- 260 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Afin d’étudier plus en détail l’impact de la température sur les propriétés en solution des
xanthanes modifiés sous forme désordonnée, des rampes en température, de 5 à 85°C (rampe
« aller ») puis de 85 à 5°C (rampe « retour »), ont été effectuées sur les différents échantillons. Entre
les deux rampes, un balayage en fréquence de 100 à 0,1 rad/s a été réalisé à 85°C après 15 minutes
d’équilibrage thermique : la solution a été maintenue à haute température pendant environ une
heure avant la rampe « retour ». Les résultats de cette mesure sont indiqués dans les figures 4-22
pour X23C8FD :
T (°C)10 20 30 40 50 60 70 80
G',
G" (
Pa)
10-4
10-3
10-2
10-1
100
101
G' allerG' retour
w (rad/s)10-2 10-1 100 101 102
G',
G" (
Pa)
10-4
10-3
10-2
10-1
100
G' 20°C allerG' 85°C G' 20°C retour
A B
1
Figures 4-22 A : Evolution de G’ (symboles fermés) et G’’ (symboles ouverts) en fonction de la température à ω =
1 rad/s pour X23C8FD à 2 g/L. B : Evolution de G’ et G’’ (mêmes symboles que la figure A) en fonction de la
fréquence à 20°C (rampes « aller » et « retour ») et 85°C.
Entre 5 et 60°C, les valeurs des modules élastique et visqueux pendant la phase de chauffage sont
faibles. De plus, G’’ est du même ordre de grandeur que celui de l’eau. A partir de 60°C par contre, les
modules augmentent fortement. On constate également que G’ devient supérieur à G’’ vers 65°C
environ. Ces données sont en accord avec les résultats de la figure 4-21, et confirment que la
modification de l’organisation des chaînes greffées se produit entre 60 et 85°C.
- 261 -
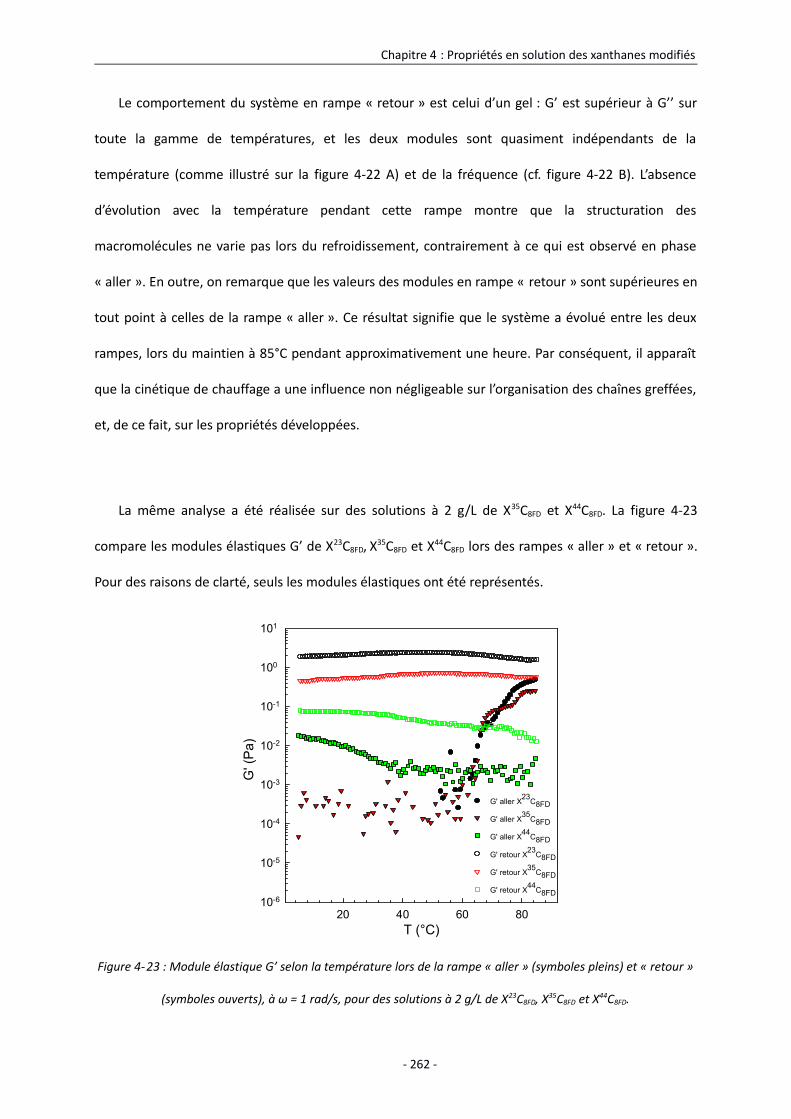
Chapitre 4 : Propriétés en solution des xanthanes modifiés
Le comportement du système en rampe « retour » est celui d’un gel : G’ est supérieur à G’’ sur
toute la gamme de températures, et les deux modules sont quasiment indépendants de la
température (comme illustré sur la figure 4-22 A) et de la fréquence (cf. figure 4-22 B). L’absence
d’évolution avec la température pendant cette rampe montre que la structuration des
macromolécules ne varie pas lors du refroidissement, contrairement à ce qui est observé en phase
« aller ». En outre, on remarque que les valeurs des modules en rampe « retour » sont supérieures en
tout point à celles de la rampe « aller ». Ce résultat signifie que le système a évolué entre les deux
rampes, lors du maintien à 85°C pendant approximativement une heure. Par conséquent, il apparaît
que la cinétique de chauffage a une influence non négligeable sur l’organisation des chaînes greffées,
et, de ce fait, sur les propriétés développées.
La même analyse a été réalisée sur des solutions à 2 g/L de X35C8FD et X44C8FD. La figure 4-23
compare les modules élastiques G’ de X23C8FD, X35C8FD et X44C8FD lors des rampes « aller » et « retour ».
Pour des raisons de clarté, seuls les modules élastiques ont été représentés.
T (°C)20 40 60 80
G' (
Pa)
10-6
10-5
10-4
10-3
10-2
10-1
100
101
G' aller X23
C8FD
G' aller X35
C8FD
G' aller X44
C8FD
G' retour X23
C8FD
G' retour X35
C8FD
G' retour X44
C8FD
Figure 4-23 : Module élastique G’ selon la température lors de la rampe « aller » (symboles pleins) et « retour »
(symboles ouverts), à ω = 1 rad/s, pour des solutions à 2 g/L de X23C8FD, X35C8FD et X44C8FD.
- 262 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Le comportement de X35C8FD est identique à celui de X23C8FD : les modules augmentent fortement à
partir d’une température critique, aux alentours des 65°C. X44C8FD par contre ne présente pas
d’augmentation de module avec la température lors de la rampe « aller ». Le module de la rampe
« retour » est néanmoins supérieur à celui de la phase « aller », tout comme les deux autres
échantillons. Il ressort donc que l’organisation des chaînes de X44C8FD est également modifiée par le
traitement thermique de l’aller, mais dans une proportion moindre que les deux autres polymères.
Nous supposons que, dans le cas de X44C8FD, le phénomène de restructuration survient à une
température critique plus importante que dans les deux autres cas. Il est probable que les chaînes de
X44C8FD adoptent en solution un degré d’organisation moindre que celui des deux autres échantillons,
ce qui expliquerait la diminution des valeurs de G’ avec la densité de greffage en rampe « retour ». Ce
phénomène peut être mis en parallèle avec les résultats de dichroïsme circulaire et de polarimétrie
des xanthanes amphiphiles sous forme ordonnée. Pour rappel, les chaînes greffées adoptent une
conformation ordonnée hélicoïdale en solution, et d’autant plus rigide que la densité de greffage est
importante. Ce fait se traduit notamment par une température de transition conformationnelle Tm
qui augmente avec la densité de greffage. On peut donc supposer que les groupements hydrophobes
présents sur les chaînes modifiées dans le DMSO stabilisent également la conformation adoptée par
ces macromolécules en solution. La température critique des xanthanes « désordonnés » peut alors
être considérée comme « l’équivalent » de la Tm pour les xanthanes « ordonnés », et évolue de la
même manière que cette dernière avec la densité de greffage.
Il faut savoir que la solubilisation des échantillons modifiés sous forme désordonnée dans l’eau,
avant tout traitement thermique, est incomplète. Des « filaments » en suspension dans la solution
sont observables pour chaque polysaccharide, quelle que soit la densité de greffage. On obtient
même une séparation de phase macroscopique dans le cas de X44C8FD. Par ailleurs, des tests de
solubilisation à 80°C pendant 2 h de X44C8FD dans de l’eau ont mis en évidence qu’un tel traitement
- 263 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
thermique permet d’augmenter la solubilité des composés. Ce phénomène est illustré sur la figure 4-
24 :
AprèsAvant
Traitement thermique
A B
Figure 4-24 : Influence du traitement thermique sur la solubilité de X44C8FD en solution A : Avant traitement
thermique. B : Après 2h à 80°C puis retour à température ambiante.
Cette amélioration de la solubilité sous l’effet de la température est probablement à l’origine du
renforcement des propriétés viscoélastiques de nos dérivés lors de la rampe « aller ». Il faut toutefois
souligner que les solutions obtenues, même après traitement thermique, ne sont pas toujours
complètement homogènes. Par conséquent, les propriétés mesurées ne correspondent pas à celles
de l’ensemble de l’échantillon, impliquant de considérer ces résultats avec prudence.
Nous avons précédemment montré que, dans les conditions de salinité et de concentration
utilisées dans ce travail, les précurseurs et les xanthanes modifiés sous forme ordonnée forment un
réseau transitoire d’hélices en interaction via des liaisons hydrogène. Cette organisation s’explique
par la forte rigidité des hélices, qui régit la structuration des chaînes en solution au détriment des
interactions hydrophobes. Les résultats des échantillons issus du protocole dans le DMSO mettent
clairement en évidence que ces derniers n’ont pas cette organisation en solution. Il se pose alors la
question de la conformation des chaînes modifiées dans le DMSO lorsqu’elles sont dissous dans l’eau.
- 264 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
En premier lieu, nous avons constaté que les xanthanes modifiés sous forme désordonnée ont
tendance à fortement s’agréger en milieu aqueux avant traitement thermique. Cette caractéristique
est observée pour toutes les densités de greffage, et elle est d’autant plus marquée que ce paramètre
est élevé. En comparaison, dans les mêmes conditions opératoires, les précurseurs et xanthanes
greffés sous forme ordonnée sont complètement solubles dans l’eau. Les analyses de polarimétrie et
de dichroïsme circulaire ont mis en évidence que, dans le DMSO, les chaînes sont modifiées sous
forme désordonnée (cf. chapitre 3). Une hypothèse envisageable est que ces systèmes adoptent une
organisation similaire dans l’eau, ou du moins plus flexible que celle des polymères « ordonnés ». Plus
précisément, la configuration des chaînes serait suffisamment flexible pour que les interactions
attractives entre les groupements hydrophobes dominent l’organisation globale des macromolécules
en solution. En particulier, le nombre d’interactions entre greffons est suffisamment important pour
entraîner la création d’agrégats d’une ou plusieurs chaînes en suspension dans l’eau. C’est pourquoi le
comportement rhéologique relevé est celui du solvant, l’eau, d’où les très faibles valeurs de modules.
De plus, plusieurs auteurs48, 55, 56 ont mis en évidence que les polymères amphiphiles flexibles avec des
densités de greffage trop importantes sont insolubles dans l’eau, ce qui correspond au comportement
exhibé par nos polymères, en particulier X44C8FD. Le greffage de chaînons hydrophobes sur le xanthane
sous forme désordonnée semble donc gêner la renaturation des chaînes lorsqu’elles sont mises à
solubiliser dans l’eau, à température ambiante, dans des conditions normalement favorables à la
forme ordonnée. Malheureusement, suite aux difficultés de solubilisation des échantillons, il n’a pas
été possible de déterminer la Tm de ces derniers par polarimétrie afin de confirmer ou non cette
hypothèse.
Lors de la rampe « aller », l’énergie thermique apportée permet de rompre une partie de ces
interactions hydrophobes, en accord avec les résultats de la littérature9, 35, 42, 57, 58. Ce phénomène
provoque un déploiement des chaînes, qui deviennent également davantage solubles. Elles peuvent
alors former un réseau physique, qui comporte des enchevêtrements topologiques, mais également
des nœuds de jonction constitués de groupements hydrophobes en interaction les uns avec les
- 265 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
autres. Ceci se traduit par la hausse des modules observée à haute température et un comportement
davantage élastique. Lors du chauffage, le nombre d’interactions hydrophobes à rompre pour pouvoir
solubiliser les agrégats est sans doute proportionnel à la densité de greffage. Plus cette dernière est
élevée, plus la durée de chauffage et/ou la température de rupture des interactions sera importante.
Ainsi, dans le cas de X44C8FD, il est probable que le traitement thermique appliqué ne permette pas de
rompre toutes les liaisons hydrophobes, d’où les propriétés exhibées.
L’absence d’évolution des modules lors de la rampe « retour » indique que le réseau formé sous
l’effet du chauffage se maintient lors du refroidissement. De même, les propriétés du réseau
n’évoluent pas selon la fréquence. A priori, le réseau formé ne relaxe pas dans la gamme ni de
températures, ni de fréquences étudiée, ce qui est similaire au comportement d’un gel chimique ou
physique caractérisé par un temps de relaxation extrêmement long. A ce jour, nous n’avons pas
d’explication concernant ces résultats.
Pour résumer :
Les xanthanes modifiés sous forme désordonnée se caractérisent par des propriétés rhéologiques
très différentes de celles de leurs analogues non greffés. En premier lieu, à l’état « natif », le
comportement viscoélastique de ces polymères est analogue à celui du solvant, l’eau. Cette
caractéristique s’explique par le fait que les solutions de ces polysaccharides ne sont pas homogènes,
mais constituées d’agrégats plus ou moins isolés en suspension dans l’eau. Par comparaison, dans les
mêmes conditions opératoires, les xanthanes modifiés dans l’eau forment un réseau d’hélices
stabilisé par des liaisons hydrogène. En outre, l’évolution des propriétés rhéologiques des xanthanes
« désordonnés » sous l’effet de la température diverge fortement de celle décrite pour les
échantillons « ordonnés » : à partir d’une température critique, les modules augmentent avec la
température pour les xanthanes modifiés dans le DMSO, alors qu’ils diminuent pour ceux greffés
dans l’eau. Les échantillons « désordonnés » présentent alors le comportement d’un gel.
- 266 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Ces résultats montrent que l’organisation adoptée dans l'eau par les chaînes modifiées dans le
DMSO est bien différente, à l’état « natif », de celle des chaînes greffées dans l’eau. De plus, cette
organisation évolue sous l’effet du traitement thermique. L’hypothèse formulée pour expliquer ces
données est la suivante : dans l’eau, les xanthanes greffés dans le DMSO adoptent une conformation
plus flexible que leurs analogues « ordonnés ». De ce fait, les interactions entre les groupements
hydrophobes dominent l’organisation globale des chaînes, ce qui explique la très forte tendance à
l’agrégation de ces échantillons. Sous l’effet de la température, les interactions hydrophobes sont
partiellement détruites. Les chaînes peuvent alors se déployer et former un réseau physique, qui
comporte des enchevêtrements topologiques et des nœuds de jonction via les groupements
hydrophobes, d’où les propriétés de type gel obtenues. Cependant, la dépendance en fréquence de
ces systèmes reste encore inexpliquée. Il est également important de souligner que même après
traitement thermique, les solutions ne sont pas complètement homogènes. Par conséquent, les
propriétés rhéologiques mesurées ne sont pas celles de l’ensemble de l’échantillon, et doivent à ce
stade être considérées avec prudence.
4.3.2. Etude des propriétés rhéologiques interfaciales des xanthanes modifiés sous
forme désordonnée
Le tableau 4-8 donne les caractéristiques des xanthanes modifiés dans le DMSO étudiés en
rhéologie interfaciale :
EntréenEDACnCOOH
DSA DSP %octyle XAC8FD
1 1 87 54 0 X0C8FD (précurseur)
2 0,3 83 51 11 X11C8FD
3 0,5 72 48 19 X19C8FD
4 0,6 78 47 27 X27C8FD
5 1 70 44 40 X40C8FD
Tableau 4-8 : Caractéristiques des xanthanes modifiés dans le DMSO étudiés en rhéologie interfaciale.
- 267 -
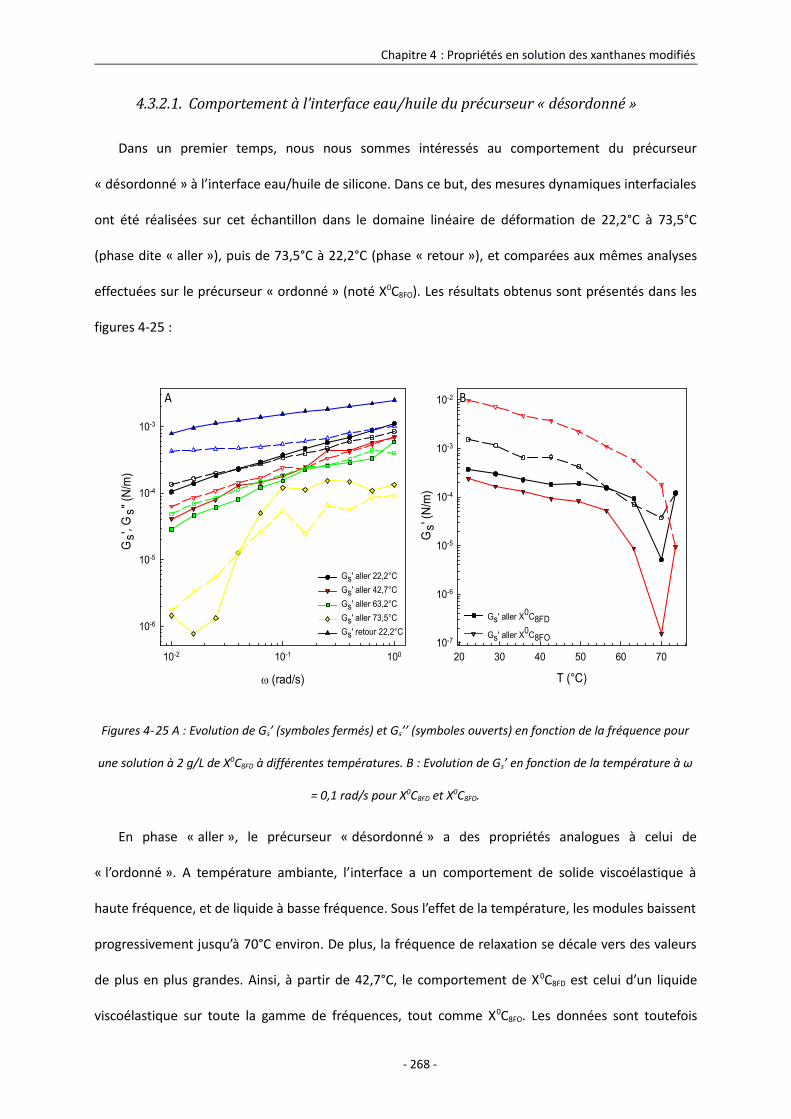
Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.3.2.1. Comportement à l’interface eau/huile du précurseur « désordonné »
Dans un premier temps, nous nous sommes intéressés au comportement du précurseur
« désordonné » à l’interface eau/huile de silicone. Dans ce but, des mesures dynamiques interfaciales
ont été réalisées sur cet échantillon dans le domaine linéaire de déformation de 22,2°C à 73,5°C
(phase dite « aller »), puis de 73,5°C à 22,2°C (phase « retour »), et comparées aux mêmes analyses
effectuées sur le précurseur « ordonné » (noté X0C8FO). Les résultats obtenus sont présentés dans les
figures 4-25 :
w (rad/s)
10-2 10-1 100
Gs'
, Gs"
(N/m
)
10-6
10-5
10-4
10-3
Gs' aller 22,2°C
Gs' aller 42,7°C
Gs' aller 63,2°C
Gs' aller 73,5°C
Gs' retour 22,2°C
T (°C)
20 30 40 50 60 70
Gs'
(N/m
)
10-7
10-6
10-5
10-4
10-3
10-2
Gs' aller X0C8FD
Gs' aller X0C8FO
A B
Figures 4-25 A : Evolution de Gs’ (symboles fermés) et Gs’’ (symboles ouverts) en fonction de la fréquence pour
une solution à 2 g/L de X0C8FD à différentes températures. B : Evolution de Gs’ en fonction de la température à ω
= 0,1 rad/s pour X0C8FD et X0C8FO.
En phase « aller », le précurseur « désordonné » a des propriétés analogues à celui de
« l’ordonné ». A température ambiante, l’interface a un comportement de solide viscoélastique à
haute fréquence, et de liquide à basse fréquence. Sous l’effet de la température, les modules baissent
progressivement jusqu’à 70°C environ. De plus, la fréquence de relaxation se décale vers des valeurs
de plus en plus grandes. Ainsi, à partir de 42,7°C, le comportement de X0C8FD est celui d’un liquide
viscoélastique sur toute la gamme de fréquences, tout comme X0C8FO. Les données sont toutefois
- 268 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
beaucoup plus bruitées, à haute température, pour le précurseur « désordonné » que pour celui
« ordonné ». En phase « retour », les modules augmentent lorsque la température décroît, et leur
dépendance en température est de moins en moins prononcée : l’interface adopte les propriétés
d’un gel interfacial faible, analogue à celui noté pour le précurseur « ordonné ».
On constate toutefois certaines divergences dans les valeurs des modules. La figure 4-25 B
montre l’évolution de Gs’ en fonction de la température lors des deux « phases » pour les deux
précurseurs. En phase « aller », l’allure est comparable pour les deux échantillons : la valeur du
module diminue progressivement avec le chauffage jusqu’à une température critique à partir de
laquelle le module chute brutalement, puis remonte ensuite. Dans les deux cas, cette température
critique est proche de la Tm des deux polymères (résultats non montrés pour X0C8FD). Pour T < Tm, les
valeurs des modules sont proches pour les deux polymères. Au-delà de la Tm par contre, on
remarque que la chute du module est beaucoup moins prononcée pour X0C8FD que pour son analogue
« ordonné ». La différence dans les modules est bien plus flagrante en phase « retour ». En effet, le
module du précurseur « désordonné » diminue entre 73,5 et 70°C (alors que celui de X0C8FO augmente
fortement). Pour T < 70°C, Gs’ croît de manière monotone avec une dépendance en température
similaire à X0C8FO, bien que les valeurs soient plus faibles.
La différence importante dans les valeurs des modules, en particulier en phase « retour », peut
avoir plusieurs origines. En premier lieu, les données des modules de X 0C8FD sont davantage bruitées
que celles du précurseur « ordonné » à haute température en phase « aller ». Il en est de même en
phase « retour ». Il faut savoir que, dans nos conditions d’analyse, X0C8FD est sous forme ordonnée.
Les analyses rhéologiques en solution ont montré que les chaînes de ce polymère adoptent une
organisation similaire à celle de X0C8FO, à savoir un réseau d’hélices stabilisé par des liaisons
hydrogène. Toutefois, la conformation hélicoïdale de X0C8FD est sans doute différente de celle de
X0C8FO. En effet, les chaînes du précurseur « désordonné » ont subi une dénaturation supplémentaire
- 269 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
par rapport à X0C8FO, vu qu’elles ont été solubilisées dans le DMSO lors du procédé de modification. La
conformation ordonnée de X0C8FD avant traitement thermique peut, par exemple, être une hélice
imparfaite, avec une proportion moindre de zones hélicoïdales que les chaînes de X 0C8FO, d’où la chute
moins prononcé du module lors de la transition conformationnelle.
Globalement, malgré les différences de modules, la similitude dans les propriétés interfaciales
des deux précurseurs suggère que les mêmes processus que ceux pour le précurseur ordonné sont à
l’œuvre pour X0C8FD.
4.3.2.2. Comportement à l’interface eau/huile des xanthanes modifiés dans le DMSO
En ce qui concerne les xanthanes modifiés dans le DMSO, leurs propriétés à l’interface eau/huile
ont été étudiées dans un premier temps via des mesures dynamiques dans le domaine linéaire de
déformation de 22,2 à 73,5°C (phase « aller »), puis à 22,2°C uniquement après 2 heures d’équilibrage
thermique (phase « retour rapide »). Les comportements du précurseur et de X27C8FD sont indiqués
sur les figures 4-26 à 22,2°C, 42,7°C, 63,2°C et 73,5°c de la phase « aller », et 22,2°C de la phase
« retour rapide ». Pour des raisons de clarté, les autres températures ne sont pas représentées.
- 270 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
w (rad/s)
10-2 10-1 100
Gs',
Gs"
(N/m
)
10-7
10-6
10-5
10-4
10-3
w (rad/s)
10-2 10-1 100
Gs',
Gs"
(N/m
)
10-5
10-4
10-3
Gs' aller 22,2°C
Gs' aller 42,7°C
Gs' aller 63,2°C
Gs' aller 73,5°C
Gs' retour rapide 22,2°C
A B
Figures 4-26 : Evolution de Gs’ (symboles fermés) et Gs’’ (symboles ouverts) en fonction de la fréquence pour une
solution à 2 g/L de A : précurseur « désordonné ». B : X27C8FD. La légende est identique pour les deux figures.
Le comportement du précurseur est identique à celui décrit précédemment. En ce qui concerne
X27C8FD, on constate qu’en phase « aller », les valeurs des modules sont très inférieures en tout point à
ceux du précurseur. L’allure bruitée des courbes montre qu’ici également, on atteint la limite de
sensibilité du rhéomètre. Aux incertitudes près, Gs’ est inférieur à Gs
’’ sur toute la gamme de
fréquences, ce qui semble traduire un comportement interfacial de type liquide. De plus, les valeurs
des modules diminuent avec la température jusqu’à 56°C environ, puis remontent légèrement
ensuite. Lors du retour rapide à 22,2°C par contre, cette hausse des modules est beaucoup plus
marquée : pour ω = 0,1 rad/s, le module Gs’ est multiplié par 100 environ entre les deux phases. Par
ailleurs, Gs’ est supérieur à Gs
’’ sur presque l’ensemble de la gamme de fréquences : le comportement
à l’interface a évolué de liquide à gel faible suite au traitement thermique. Les valeurs des modules
restent toutefois inférieures à celles du précurseur. Cette étude confirme les observations faites en
solution : la température a un très fort impact sur l’organisation des chaînes de xanthane greffés sous
forme désordonnée à l’interface eau/huile.
- 271 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
La figure 4-27 A compare l’évolution de Gs’ à 22,2°C en rampe « aller » puis en rampe « retour
rapide » pour X19C8FD, X27C8FD et X40C8FD (pour des raisons de clarté, Gs’’ n’a pas été représenté) :
w (rad/s)
10-2 10-1 100
Gs'
(N/m
)
10-6
10-5
10-4
10-3
10-2
Gs' aller X0F8FD
Gs' aller X19C8FD
Gs' aller X27C8FD
Gs' aller X40C8FD
T (°C)
20 30 40 50 60 70
Gs'
(N/m
)10-6
10-5
10-4
10-3
Gs' aller X0C8FD
Gs' aller X11C8FD
Gs' aller X19C8FD
A B
Figures 4-27 A : Evolution de Gs’ en fonction de la fréquence à 22,2°C lors de la phase « aller » (symboles fermés)
et de la phase « retour rapide » (symboles ouverts). B : Evolution de Gs’ en phase « aller » (symboles fermés) et
en phase « retour » (symboles ouverts) à 0,1 rad/s en fonction de la température pour certains échantillons.
Pour des raisons de clarté, le module Gs’’ n’est pas indiqué.
X19C8FD a des propriétés viscoélastiques similaires à celles de X27C8FD. En effet, en rampe « aller »,
les valeurs de modules sont très faibles, et à la limite de détection du rhéomètre. En rampe « retour
rapide » par contre, le module augmente très fortement suite au traitement thermique imposé. Le
comportement de X19C8FD à 22,2°C en rampe « retour rapide » est ainsi analogue à celui du
précurseur. On remarque également que les valeurs de Gs’ de cet échantillon sont légèrement
supérieures à celles de X27C8FD. Le cycle thermique a le même impact sur les propriétés de X 40C8FD.
Toutefois, ce dernier exhibe dès l’état « natif » des propriétés interfaciales quelque peu différentes.
En effet, les valeurs des modules de cet échantillon sont bien supérieures à celles des deux autres
xanthanes. De plus, le comportement de ce polymère est quasiment identique à celui du xanthane
non greffé : les modules ont des valeurs proches et diminuent avec la température. La dépendance en
- 272 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
fréquence est également similaire. En outre, le comportement évolue de solide à liquide
viscoélastique. Au « retour » néanmoins, l’accroissement des modules est bien plus marqué que celui
du précurseur. X40C8FD exhibe un comportement de gel plus prononcé (modules plus élevés,
dépendance en fréquence plus faible) que la référence non modifiée. Le traitement thermique a un
impact très important sur les propriétés interfaciales des xanthanes greffés dans le DMSO. De plus,
cet impact est beaucoup plus marqué que dans le cas du précurseur. Par exemple, pour X19C8FD, le
module à 0,1 rad/s et 22,2°C évolue de 7*10 -6 N/m à 6,5*10-4 N/m (facteur multiplicatif de 90
environ) entre les deux cycles. Pour X27C8FD et X40C8FD, le module augmente respectivement de 1,7*10 -6
à 2,5*10-4 N/m (facteur multiplicatif de 150) ; et de 2,5*10-4 à 6*10-3 N/m (facteur multiplicatif de 24),
alors que celui du précurseur croît de 3*10 -4 à 5,8*10-4 N/m (facteur multiplicatif de 2). Il n’est
cependant pas possible de corréler la valeur de Gs’ avec la densité de greffage, que ce soit en rampe
« aller ou en rampe « retour rapide », contrairement aux xanthanes amphiphiles sous forme
ordonnée. Ce constat peut s’expliquer par le fait que les solutions comportent toujours des agrégats,
même après traitement thermique. De ce fait, les propriétés mesurées ne représentent pas
l’ensemble de l’échantillon.
La figure 4-27 B représente l’évolution de Gs’ à 0,1 rad/s en fonction de la température en phase
« aller » et « retour » pour le précurseur, X11C8FD et X19C8FD. Le cas du précurseur « désordonné » a déjà
été discuté précédemment (cf. figures 4-25). L’allure du module des échantillons modifiés lors du
chauffage est bien différente de celle du précurseur. En premier lieu, les valeurs de G s’ restent
relativement faibles et constantes à basse température. A partir d’une température critique,
différente pour chaque polymère, Gs’ augmente significativement. Cette température est d’autant
plus basse que la densité de greffage est faible. Notre hypothèse, pour expliquer ces résultats, est
celle présentée précédemment lors de l’étude des propriétés en solution : en solution, les chaînes
modifiées dans le DMSO, plus flexibles que leurs analogues « ordonnés », sont majoritairement
localisées dans des agrégats plus ou moins isolés. Elles ne forment donc pas de réseau à l’interface
eau/huile. L’augmentation de la température permet de solubiliser une fraction des chaînes, qui
- 273 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
s’adsorbent alors à l’interface. Il est probable cependant qu’une partie des macromolécules restent
insolubles après le traitement thermique, ce qui explique les propriétés interfaciales inférieures de
ces polymères par rapport à celles des xanthanes « ordonnés ». En phase « retour », les valeurs du
module augmentent progressivement avec le refroidissement, comme pour le précurseur.
Pour résumer :
Les propriétés interfaciales des xanthanes greffés dans le DMSO évoluent fortement sous l’effet
du traitement thermique appliqué sur les échantillons. Ainsi, en phase « aller », les modules mesurés
sont très faibles : les chaînes ne forment pas de réseau à l’interface eau/huile, contrairement à ce qui
est observé pour les xanthanes modifiés dans l’eau dans les mêmes conditions d’analyse. Néanmoins,
au-delà d’une température critique (variable suivant la densité de greffage du polymère), les modules
augmentent fortement lors du chauffage, ce qui indique une adsorption importante de
macromolécules à l’interface. L’hypothèse formulée pour expliquer ces résultats est la même que
celle proposée en solution : à l’état « natif », les chaînes modifiées dans le DMSO sont plus flexibles
que leurs analogues dans l’eau. Par conséquent, leur organisation en solution est contrôlée par les
interactions attractives entre les groupements hydrophobes. Ce phénomène se traduit notamment
par la formation d’agrégats de chaînes greffées, qui les empêchent de s’adsorber à l’interface.
L’augmentation de la température permet de dissocier partiellement les chaînes engagées dans ces
agrégats. Elles peuvent alors s’adsorber à l’interface, d’où la hausse des modules.
En phase « retour », la diminution de la température entraîne une hausse progressive des
modules. De plus, les propriétés interfaciales évoluent de liquide à gel faible interfacial. Cette
évolution est semblable à celle décrite dans le cas des xanthanes « ordonnés » peu greffés. Dans leur
cas, ce comportement s’explique par la renaturation partielle des chaînes adsorbées à l’interface lors
du refroidissement. Il est donc possible que le même processus s’applique pour la fraction des
chaînes, modifiées dans le DMSO, qui est dissociées des agrégats.
- 274 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
4.4. Conclusion
Les xanthanes modifiés sous forme ordonnée adoptent en solution une organisation similaire à
celle du précurseur, à savoir un réseau transitoire d’hélices majoritairement en interaction via des
liaisons hydrogène. La présence des greffons hydrophobes ne modifie pas l’organisation globale des
chaînes, contrairement à ce qui est habituellement observé pour les polymères amphiphiles. Ce
phénomène s’explique par la très forte rigidité des macromolécules greffées, qui domine la
structuration globale des chaînes au détriment des interactions hydrophobes attractives. Par
conséquent, le caractère associatif de nos polysaccharides modifiés est peu différent de celui du
précurseur : le xanthane greffé est un polymère amphiphile, mais non associatif. Par ailleurs, les
groupements hydrophobes ne modifient pas les propriétés viscoélastiques du xanthane, mais
contribuent cependant à fortement ralentir la dynamique de relaxation des chaînes, selon un
processus de « relaxation collante ». Ce phénomène confère notamment au xanthane amphiphile un
pouvoir suspensif potentiel accru. A l’interface eau/huile, nous avons démontré que la viscoélasticité
interfaciale augmentait de manière significative avec la densité de greffage. De plus, les propriétés
viscoélastiques de ces xanthanes varient fortement avec la température. Ce phénomène est lié à
deux mécanismes antagonistes, qui s’exercent conjointement : la perte de rigidité des chaînes sous
l’effet de la température, et l’adsorption des zones désordonnées et/ou amphiphiles à haute
température. Ces deux mécanismes dépendent eux-mêmes de la densité de greffage : plus le
polymère est greffé, plus sa dénaturation est incomplète. Ainsi, pour les échantillons les plus
modifiés, les contributions de la rigidité et de l’adsorption des zones désordonnées et/ou amphiphiles
sont, après traitement thermique, sensiblement identiques à celles de leur forme «native ». Au
contraire, pour les polymères les moins greffés, l’adsorption des chaînes l’emporte sur la perte de
rigidité au passage de la transition conformationnelle. Par conséquent, la viscoélasticité interfaciale
après traitement thermique découle de la transition de la forme ordonnée vers la forme désordonnée
lors du chauffage. Cette transition est elle-même dépendante de la densité de greffage.
- 275 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Les xanthanes modifiés sous forme désordonné ont un comportement bien distinct de celui de
leurs analogues ordonnés. Ainsi, en solution à l’état « natif », ces polymères ne forment pas de
réseau. Au contraire, les chaînes ont tendance à s’agréger, au point d’entraîner une séparation de
phase macroscopique pour les polymères les plus greffés. Nous supposons, pour expliquer ces
données, que les chaînes modifiées dans le DMSO adoptent en solution une conformation plus
flexible que les xanthanes ordonnés, ce qui favorise les interactions hydrophobes entre les greffons et
la formation des agrégats. L’augmentation de la température permet de rompre partiellement les
agrégats, et les chaînes ainsi libérées peuvent alors former un réseau en solution, d’où
l’accroissement notable des propriétés viscoélastiques observées. Le comportement viscoélastique
interfaciale de ces polysaccharides est similaire à celui observé en solution, pour les mêmes raisons :
la formation d’un réseau interfacial nécessite de libérer les chaînes « piégées » dans les agrégats, ce
qui est partiellement possible via l’augmentation de la température.
Il ressort donc que les propriétés en solution des polymères amphiphiles dépendent fortement
de conformation de leurs chaînes et, par conséquent, de leur rigidité.
- 276 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
Références bibliographiques
1. Capron, I.; Brigand, G.; Muller, G., About the native and renatured conformation of xanthanexopolysaccharide. Polymer 1997, 38, 5289-5295.
2. Milas, M.; Rinaudo, M., Conformational investigation on the bacterial polysaccharidexanthan. Carbohydrate Research 1979, 76, 189-196.
3. Morris, E. R.; Rees, D. A.; Young, G.; Walkinshaw, M. D.; Darke, A., Order-disorder transitionfor a bacterial polysaccharide in solution. A role for polysaccharide conformation in recognitionbetween Xanthomonas pathogen and its plant host. Journal of Molecular Biology 1977, 110, 1-16.
4. Dentini, M.; Crescenzi, V.; Blasi, D., Conformational properties of xanthan derivatives in diluteaqueous solution. International Journal of Biological Macromolecules 1984, 6, 93.
5. Winnik, M. A.; Yekta, A., Associative polymers in aqueous solution. Current Opinion in Colloid& Interface Science 1997, 2, 424-436.
6. Sinquin, A.; Hubert, P.; Dellacherie, E., Amphiphilic derivatives of alginate: Evidence for intra-and intermolecular hydrophobic associations in aqueous solution. Langmuir 1993, 9, 3334-3337.
7. Taylor, K. C.; Nasr-El-Din, H. A., Water-soluble hydrophobically associating polymers forimproved oil recovery: A literature review. Journal of Petroleum Science and Engineering 1998, 19,265-280.
8. Chassenieux, C.; Nicolai, T.; Benyahia, L., Rheology of associative polymer solutions. CurrentOpinion in Colloid & Interface Science 2011, 16, 18-26.
9. Volpert, E.; Selb, J.; Candau, F., Associating behaviour of polyacrylamides hydrophobicallymodified with dihexylacrylamide. Polymer 1998, 39, 1025-1033.
10. Colinet, I.; Dulong, V.; Hamaide, T.; Le Cerf, D.; Picton, L., New amphiphilic modifiedpolysaccharides with original solution behaviour in salt media. Carbohydrate Polymers 2009, 75, 454-462.
11. Mast, R. C.; Haynes, L. V., The use of the fluorescent probes perylene and magnesium 8-anilinonaphthalene-l-sulfonate to determine the critical micelle concentration of surfactants inaqueous solution. Journal of Colloid and Interface Science 1975, 53, 35-41.
12. Vieira, N. A. B.; Moscardini, M. S.; Tiera, V. A. d. O.; Tiera, M. J., Aggregation behavior ofhydrophobically modified dextran in aqueous solution: A fluorescence probe study. CarbohydratePolymers 2003, 53, 137-143.
13. Martin, G.; Ross, J.; Minteer, S. D.; Jameson, D. M.; Cooney, M. J., Fluorescencecharacterization of chemical microenvironments in hydrophobically modified chitosan. CarbohydratePolymers 2009, 77, 695-702.
14. Valeur, B., Les sondes fluorescentes. In Invitation à la fluorescence moléculaire, De Boeck:Bruxelles, 2004; pp 133-174.
15. Thomas, T.; Mishra, A., ANS fluorescence as a tool to monitor cross-linking polymerization ofacrylamide. European Polymer Journal 2002, 38, 1805-1810.
16. Jouquand, C.; Aguni, Y.; Malhiac, C.; Grisel, M., Influence of chemical composition ofpolysaccharides on aroma retention. Food Hydrocolloids 2008, 22, 1097-1104.
- 277 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
17. Secouard, S.; Grisel, M.; Malhiac, C., Flavour release study as a way to explain xanthan–galactomannan interactions. Food Hydrocolloids 2007, 21, 1237-1244.
18. Bylaite, E.; Adler-Nissen, J.; Meyer, A. S., Effect of xanthan on flavor release from thickenedviscous food model systems. Journal of Agricultural and Food Chemistry 2005, 53, 3577-3583.
19. Cuvelier, G.; Launay, B., Concentration regimes in xanthan gum solutions deduced from flowand viscoelastic properties. Carbohydrate Polymers 1986, 6, 321-333.
20. Launay, B.; Doublier, J. L.; Cuvelier, G., Flow properties of aqueous solutions and dispersionsof polysaccharides. In Functional Properties of Food Macromolecules, 2nd edition , Hill, S. E.; Mitchell,J. R.; Ledward, D. A., Eds. Springer: 1998; pp 1-78.
21. Wyatt, N. B.; Liberatore, M. W., Rheological and viscosity scaling of the polyelectrolytexanthan gum. Journal of Applied Polymer Science 2009, 114, 4076-4084.
22. Charpentier-Valenza, D.; Merle, L.; Mocanu, G.; Picton, L.; Muller, G., Rheological propertiesof hydrophobically modified carboxymethyl celluloses varying by their hydrophili-lipophilic balance.Carbohydrate Polymers 2005, 60, 87-94.
23. Simon, S.; Dugast, J. Y.; Le Cerf, D.; Picton, L.; Muller, G., Amphiphilic polysaccharidescharacterizartion in dilute regime. Competition between intramolecular and intermolecularinterations. Polymer 2003, 44, 7917-7924.
24. Tanaka, R.; Meadows, J.; Phillips, G. O.; Williams, P. A., Viscometric and spectroscopic studieson the solution behaviour of hydrophobically modified cellulosic polymers. Carbohydrate Polymers1990, 12, 443-459.
25. McCormick, C. L.; Nonaka, T.; Johnson, C. B., Water-soluble copolymers: 27. Synthesis andaqueous solution behaviour of associative acrylamideN-alkylacrylamide copolymers. Polymer 1988,29, 731-739.
26. Sho, T.; Sato, T.; Norisuye, T., Viscosity behaviour and persistence length of sodium xanthan inaqueous sodium chloride. Biophysical Chemistry 1986, 25, (3), 307-313.
27. Sato, T.; Norisuye, T.; Fujita, H., Double-stranded helix of xanthan: Dimensional andhydrodynamic properties in 0.1M aqueous sodium chloride. Macromolecules 1984, 17, 2696-2700.
28. Liu, W.; Sato, T.; Norisuye, T.; Fujita, H., Thermally induced conformational change of xanthanin 0.01M aqueous sodium chloride. Carbohydrate Research 1987, 160, 267-281.
29. Berth, G.; Dautzenberg, H.; Christensen, B.; Harding, S.; Rother, G.; Smidsrød, O., Static lightscattering studies on xanthan in aqueous solutions. Macromolecules 1996, 29, 3491-3498.
30. Bataille, I.; Huguet, J.; Muller, G.; Mocanu, G.; Carpov, A., Associative behaviour ofhydrophobically modified carboxymethylpullulan derivatives. International Journal of BiologicalMacromolecules 1997, 20, 179-191.
31. Desbrieres, J., Autoassociative natural polymer derivatives: the alkylchitosans. Rheologicalbehaviour and temperature stability. Polymer 2004, 45, 3285-3295.
32. Colinet, I.; Dulong, V.; Mocanu, G.; Picton, L.; Le Cerf, D., New amphiphilic and pH-sensitivehydrogel for controlled release of a model poorly water-soluble drug. European Journal ofPharmaceutics and Biopharmaceutics 2009, 73, 345-350.
33. Leonard, M.; Rastello de Boisseson, M.; Hubert, P.; Dalençon, F.; Dellacherie, E.,Hydrophobically modified alginate hydrogels as protein carriers with specific controlled releaseproperties. Journal of Controlled Release 2004, 98, 395-405.
34. Regalado, E. J.; Selb, J.; Candau, F., Viscoelastic behavior of semidilute solutions ofmultisticker polymer chains. Macromolecules 1999, 32, 8580-8588.
- 278 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
35. Kujawa, P.; Audibert-Hayet, A.; Selb, J.; Candau, F., Rheological properties of multistickerassociative polyelectrolytes in semidilute aqueous solutions. Journal of Polymer Science Part B:Polymer Physics 2004, 42, 1640-1655.
36. Choppe, E.; Puaud, F.; Nicolai, T.; Benyahia, L., Rheology of xanthan as a function oftemperature, concentration and ionic strength. Carbohydrate Polymers 2010, 82, 1228-1235.
37. Renou, F.; Petibon, O.; Malhiac, C.; Grisel, M., Effect of xanthan structure on its interactionwith locust bean gum: Toward prediction of rheological properties. Food Hydrocolloids 2013, 32, 331-340.
38. Milas, M.; Rinaudo, M.; Knipper, M.; Schuppiser, J. C., Flow and viscoelasticproperties ofxanthan gum solutions. Macromolecules 1990, 23, 2507-2511.
39. Pelletier, E.; Viebke, C.; Meadows, J.; Williams, P. A., A rheological study of the order-disorderconformation transition of xanthan gum. Biopolymers 2001, 59, 339-346.
40. Rubinstein, M.; Semenov, A. N., Dynamics of entangled solutions of associating polymers.Macromolecules 2001, 34, 1058-1068.
41. Leibler, L.; Rubinstein, M.; Colby, R. H., Dynamics of reversible networks. Macromolecules1991, 24, 4701-4707.
42. Caputo, M.-R.; Selb, J.; Candau, F., Effect of temperature on the viscoelastic behaviour ofentangled solutions of multisticker associating polyacrylamides. Polymer 2004, 45, 231-240.
43. Ross-Murphy, S. B.; Morris, V. J.; Morris, E. R., Molecular viscoelasticity of xanthanpolysaccharide. Faraday Symposia of the Chemical Society 1983, 18, 115-129.
44. Rochefort, W. E.; Middleman, S., Rheology of xanthan gum: Salt, temperature, and straineffects in oscillatory and steady shear experiments. Journal of Rheology 1987, 31, 337-369.
45. Durand, A.; Dellacherie, E., Aqueous solutions of native and hydrophobically modifiedpolysaccharides: Temperature effect. Biomacromolecules 2006, 7, 958-964.
46. Carnali, J. O., A dispersed anisotropic phase as the origin of the weak-gel properties ofaqueous xanthan gum. Journal of Applied Polymer Science 1991, 43, 929-941.
47. Aubry, T.; Moan, M., Rheological behavior of a hydrophobically associating water solublepolymer. Journal of Rheology 1994, 38, 1681-1692.
48. Xie, X.; Hogen-Esch, T. E., Copolymers of N,N-dimethylacrylamide and 2-(N-ethylperfluorooctanesulfonamido)ethyl acrylate in aqueous media and in bulk. Synthesis andproperties. Macromolecules 1996, 29, 1734-1745.
49. Mendes, A. C.; Baran, E. T.; Numes, C.; Coimbra, M.; Azevedo, H.; Reis, R. L., Palmitoylation ofxanthan polysaccharide for self-assembly microcapsule formation and encapsulation of cells inphysiological conditions. Soft Matter 2011, 7, 9647-9658.
50. Matsuda, Y.; Biyajima, Y.; Sato, T., Thermal denaturation, renaturation, and aggregation of adouble-helical polysaccharide xanthan in aqueous solution. Polymer Journal 2009, 41, 526-532.
51. Capron, I.; Brigand, G.; Muller, G., Thermal denaturation and renaturation of a fermentationbroth of xanthan: Rheological consequences. International Journal of Biological Macromolecules1998, 23, 215-225.
52. Lee, H.-C.; Brant, D. A., Rheology of Concentrated Isotropic and Anisotropic XanthanSolutions: 3. Temperature Dependence. Biomacromolecules 2002, 3, 742-753.
53. Stokke, B. T.; Elgsaeter, A.; Smidsrod, O., Electron microscopic study of single-and double-stranded xanthan. International Journal of Biological Macromolecules 1986, 8, 217-225.
- 279 -

Chapitre 4 : Propriétés en solution des xanthanes modifiés
54. Paoletti, S.; Cesaro, A.; Delben, F., Thermally induced conformational transition of xanthanpolyelectrolyte. Carbohydrate Research 1983, 123, 173-178.
55. Durand, A., Aqueous solutions of amphiphilic polysaccharides: Concentration andtemperature effect on viscosity. European Polymer Journal 2007, 43, 1744-1753.
56. Petit, F.; Iliopoulos, I.; Audebert, R.; Szönyi, S., Associating polyelectrolytes with perfluoroalkylside chains: Aggregation in aqueous solution, association with surfactants, and comparison withhydrogenated analogues. Langmuir 1997, 13, 4229-4233.
57. Biggs, S.; Selb, J.; Candau, F., Copolymers of acrylamideN-alkylacrylamide in aqueous solution:The effects of hydrolysis on hydrophobic interactions. Polymer 1993, 34, 580-591.
58. Hourdet, D.; Gadgil, J.; Podhajecka, K.; Badiger, M. V.; Brûlet, A.; Wadgaonkar, P. P.,Thermoreversible behavior of associating polymer solutions: Thermothinning versusthermothickening. Macromolecules 2005, 38, 8512-8521.
- 280 -

Conclusion Générale
- 281 -

Conclusion Générale
- 282 -

Conclusion Générale
L’objectif de ces travaux de thèse était de déterminer l’influence de la conformation du squelette
hydrophile sur les propriétés en solution d’un polysaccharide amphiphile. Notre choix s’est porté sur
le xanthane, car ce polysaccharide possède la caractéristique d’exister sous deux conformations
différentes en solution : une forme ordonnée hélicoïdale très rigide à température ambiante et force
ionique élevée, ou une forme désordonnée de type pelote flexible, à haute température et/ou faible
force ionique. Dans ce but, nous avons développé deux protocoles de modification nous permettant
de greffer des chaînes alkyle C8 hydrophobes sur le xanthane, sous ses deux conformations. Par la
suite, le comportement, notamment rhéologique, en solution des xanthanes amphiphiles a été
caractérisé, afin de comprendre la relation entre la conformation adoptée par les chaînes modifiées
et les propriétés observées.
Nous avons mis au point deux protocoles de modification chimique du xanthane qui permettent
de greffer des groupements hydrophobes sur ce polysaccharide, soit sous forme ordonnée, soit sous
forme désordonnée. A notre connaissance, la modification du xanthane sous cette dernière
conformation n’a pas été décrite dans la littérature. Les méthodes développées au cours de ce travail
autorisent un greffage à façon des chaînons alkyle sur le polysaccharide.
En solution, les xanthanes amphiphiles obtenus par le protocole de modification dans l’eau
adoptent une conformation hélicoïdale plus rigide que celle du précurseur. Cette rigidité gouverne
l’organisation globale des chaînes au détriment des interactions hydrophobes, contrairement à ce qui
est habituellement observé pour les polymères amphiphiles. De ce fait, l’organisation des
macromolécules modifiées en solution est similaire à celle de leur précurseur, et peut être modélisée
par un réseau transitoire d’hélices semi-rigides arrangées parallèlement les unes aux autres, et
majoritairement en interaction les unes avec les autres via des liaisons hydrogène. Les greffons
hydrophobes ne modifient pas les mécanismes de relaxation des chaînes amphiphiles, mais
ralentissent considérablement la dynamique de ces dernières. Ce phénomène est d’autant plus
- 283 -

Conclusion Générale
prononcé que la densité de greffage est élevée. Une autre conséquence est que les xanthanes
amphiphiles ont un caractère associatif peu différent de celui du précurseur.
A l’interface eau/huile en revanche, la présence des chaînons hydrophobes favorise l’adsorption
des chaînes modifiées à l’interface, renforçant ainsi son élasticité. Par ailleurs, les propriétés
interfaciales évoluent fortement avec la température, ce qui indique une restructuration des chaînes
adsorbées. Ce phénomène peut s’expliquer par le processus de dénaturation et renaturation partielle
des hélices amphiphiles, qui dépend notamment de la densité de greffage des échantillons. En effet,
plus le polymère est greffé, plus la dénaturation est difficile et donc incomplète, et la conformation
des chaînes dénaturées est similaire à celle des macromolécules « natives ». Ainsi, pour les xanthanes
les moins greffés, la dénaturation partielle des hélices entraîne la formation de zones désordonnées
qui s’adsorbent à l’interface eau/huile à haute température, et renforcent le caractère élastique de
cette dernière. Au contraire, pour les xanthanes les plus greffés, le processus de dénaturation est très
incomplet, voire nul. Les propriétés interfaciales avant et après cycle thermique sont identiques, car
le traitement thermique appliqué ne modifie pas l’organisation globale des chaînes modifiées. Pour
les xanthanes modifiés à densité de greffage intermédiaire, l’évolution des propriétés rhéologiques
avec la température dépend de la compétition entre la perte de rigidité des chaînes, liée à la
dénaturation partielle du système, et l’adsorption à l’interface des zones greffées, qui est favorisée
par le gain de flexibilité. Il ressort de cette étude que les xanthanes amphiphiles sous forme
ordonnée, du fait de leurs propriétés interfaciales spécifiques, peuvent avoir des applications
potentielles dans la stabilisation d’interface de type émulsion.
Les xanthanes greffés dans le DMSO présentent des comportements viscoélastiques en solution
et à l’interface très différents de ceux du précurseur et des xanthanes amphiphiles sous forme
ordonnée. En premier lieu, les solutions de ces polysaccharides ne sont pas homogènes, mais
comportent de nombreux agrégats plus ou moins isolés. De plus, l’évolution du comportement
- 284 -

Conclusion Générale
viscoélastique, en solution ou à l’interface eau/huile, de ces polymères sous l’effet de la température
se distingue également de celui des xanthanes « ordonnés » : les propriétés viscoélastiques
augmentent fortement au-delà d’une température critique, qui croît avec la densité de greffage. Il
ressort donc que l’organisation des chaînes modifiées dans le DMSO, en solution ou dans l’eau, est
très différente de celle des xanthanes « ordonnés ». Notre hypothèse, pour expliquer ces résultats,
est que ces macromolécules adoptent en solution une conformation plus flexible que celle des
chaînes ordonnées, ce qui favorise les interactions hydrophobes et la formation d’agrégats. La hausse
de température permet de rompre partiellement ces agrégats, et les chaînes ainsi libérées peuvent
alors former un réseau en solution ou à l’interface eau/huile.
En conclusion, les propriétés rhéologiques, en solution ou à l’interface eau/huile, des xanthanes
amphiphiles diffèrent fortement suivant le protocole de modification du polymère. Cette différence
est liée à la conformation des chaînes en solution : les chaînes amphiphiles « ordonnées » adoptent
une conformation hélicoïdale rigide, et les macromolécules « désordonnées » une forme beaucoup
plus flexible. Dans le premier cas, la rigidité des chaînes, gouvernée par les interactions hydrogène,
impose l’organisation globale du système. Dans le second cas, l’organisation des macromolécules est
régie par les interactions attractives entre les groupements hydrophobes. La conformation du
squelette hydrophile influence donc fortement les propriétés en solution du xanthane amphiphile.
Il faut toutefois rappeler que les résultats concernant les polysaccharides modifiés dans le DMSO
sont, à ce jour, préliminaires. Des analyses supplémentaires sont nécessaires pour confirmer les
observations faites. En particulier, la présence d’agrégats, même après traitement thermique, n’a pas
permis d’accéder au comportement « réel » de ces échantillons. Il serait donc intéressant de
déterminer les conditions opératoires optimales pour éliminer ces agrégats. Les paramètres pouvant
être étudiés sont la température, la cinétique de la rampe de chauffage, la densité de greffage ou
d’autres voies de solubilisation. Par ailleurs, les polymères modifiés dans le DMSO comportent de
- 285 -

Conclusion Générale
nombreux sous-produits greffés sur les chaînes. A ce jour, nous ignorons l’impact de ces impuretés
sur le comportement rhéologiques de ces systèmes. Une optimisation du procédé de modification
dans le DMSO peut être envisagée pour éclaircir la contribution de ces dérivés sur les propriétés des
xanthanes « désordonnés ». En outre, nos travaux montrent que le comportement rhéologique des
xanthanes « ordonnés » greffés par des chaînons alkyle C8 est essentiellement contrôlé par la rigidité
du squelette hydrophile. Il serait intéressant de déterminer si cette situation est toujours valable pour
les échantillons les plus greffés, ou s’il n’existe pas une densité de greffage critique à partir de
laquelle les interactions hydrophobes contrôlent l’organisation globale des chaînes. Par un
raisonnement analogue, on peut également s’interroger sur l’influence de la longueur des
groupements hydrophobes greffés sur le comportement du xanthane amphiphile. Dans ce but, on
pourrait envisager d’adapter le protocole de modification développé dans l’eau afin d’allonger la
longueur des greffons, et notamment de travailler en cosolvant, puis d’étudier les propriétés
rhéologiques des polymères ainsi obtenus.
- 286 -


Résumé :
Les polysaccharides amphiphilespossèdent des propriétés rhéologiques etinterfaciales singulières dues aux interactionsattractives entres les groupementshydrophobes greffés le long du squelettehydrophile. Leurs propriétés sont modulablessuivant certains paramètres bien identifiés(nature des groupements hydrophobes,densité de greffage, etc.). Néanmoins, peud’études s’intéressent à l’impact de laconformation du squelette sur lecomportement de ces systèmes. L’objectif dece mémoire est de déterminer l’influence de larigidité du squelette sur les propriétés ensolution d’un polysaccharide amphiphile àbase de xanthane. En solution, ce polymèrepeut adopter deux conformations distinctessuivant les conditions opératoires : une formeordonnée hélicoïdale rigide ou une formedésordonnée de type pelote flexible. Deuxprotocoles de greffage ont été développés afinde modifier le xanthane sous ces deux formes.Les xanthanes greffés sous forme ordonnéeconservent une conformation hélicoïdalerigide, qui gouverne l’organisation globale desmacromolécules. Au contraire, pour lespolymères greffés sous forme désordonnée,l’organisation des chaînes est principalementcontrôlée par les interactions attractives entreles groupements hydrophobes.
Mots-clefs : xanthane, polysaccharideamphiphile, conformation, modificationchimique, rhéologie.
Abstract:
Hydrophobically modified polysaccharidesshow unusual rheological and interfacialproperties in solution due to the self-association of hydrophobic entities graftedonto their hydrophilic backbone. Theirproperties are tunable according to some well-known parameters, such as the length of thehydrophobic moieties or the grafting density.However, very few studies deal with theinfluence of the backbone conformation onthe properties of such systems in solution.Therefore, the objective of this work is todetermine the impact of the backbone rigidityon the behavior of an amphiphilicpolysaccharide based on xanthan. Indeed, insolution, this polymer can adopt two differentconformations depending on the experimentalconditions: an ordered, rigid helix or adisordered, flexible coil. Hydrophobic moietieshave been grafted on xanthan either under itsordered and disordered conformation. Chainsmodified under the ordered conformationremain ordered and rigid after grafting and theoverall properties are controlled by the highrigidity of the polymer backbone. On thecontrary, macromolecules grafted under thedisordered form show a more flexibleconformation in solution. As a result, theorganization of these derivatives is mainlycontrolled by the attractive interactionsbetween the grafted moieties.
Key words: xanthan, amphiphilicpolysaccharide, conformation, chemicalmodification, rheology.
Recommended

















