
Surface Science 602 (2008) 2955–2962
Contents lists available at ScienceDirect
Surface Science
journal homepage: www.elsevier .com/locate /susc
Surface Science Prospectives
Surface science of complex environmental interfaces: Oxide and carbonatesurfaces in dynamic equilibrium with water vapor
Vicki H. Grassian *
Departments of Chemistry and Chemical and Biochemical Engineering, University of Iowa, Iowa City, IA 52246, United States
a r t i c l e i n f o a b s t r a c t
Article history:Received 20 June 2008Accepted for publication 28 July 2008Available online 12 August 2008
Keywords:Adsorbed waterOxidesCarbonatesEnvironmental interfacesEnvironmental surface science
0039-6028/$ - see front matter � 2008 Elsevier B.V. Adoi:10.1016/j.susc.2008.07.039
* Tel.: +1 319 335 1392; fax: +1 319 353 1115.E-mail address: [email protected]
Surface scientists are dealing more and more with complex systems that are challenging to investigatefrom both experimental and theoretical perspectives. The surface science of complex interfaces, suchas environmental interfaces under ambient conditions of temperature and relative humidity, requiresboth advances in experimental and theoretical methods in order for conceptual insights to emerge. In thisprospective, several aspects of environmental interfaces and the field of environmental surface scienceare discussed. These include: (i) adsorbed water on oxide and carbonate interfaces; (ii) surface chemistryof oxide and carbonate interfaces in the presence of co-adsorbed water; (iii) solvation of ions by co-adsorbed water on environmental interfaces; and (iv) research needs and challenges in environmentalsurface science.
� 2008 Elsevier B.V. All rights reserved.
1. Introduction
Much of the groundbreaking work on surfaces began with Ir-ving Langmuir who received the Nobel Prize in Chemistry in1932 ‘‘for his discoveries and investigations in surface chemistry”.As the field continued, there was a push toward more controlledexperiments in ‘‘pristine” environments done under ultra-highvacuum conditions on ideal, model systems such as single crystalsurfaces. These surface science studies from 1965 until now con-tinue and have provided a wealth of information on the nature ofsurfaces in ultra-high vacuum; and detailed information on surfacesites, including defect sites that are present on nearly all surfaces,even ideal surfaces, and surface structure, surface atom rearrange-ment from surface relaxation and surface reconstruction. The useof scanning tunneling microscopy to investigate the adsorptionand chemistry of molecules on specific sites has led to a great dealof insight into the behavior of molecules on well-defined surfaces.Thus the field continued on and The Nobel Prize in Chemistry wasawarded in surface chemistry an unprecedented second time in2007 when Gerhard Ertl was awarded the 2007 Nobel Prize inChemistry ‘‘for his studies of chemical processes on solid surfaces”.On December 8, 2007, Gerhard Ertl gave his Nobel lecture ‘‘Reac-tions at solid surfaces: From atoms to complexity” at StockholmUniversity.
The surface science of increasing complex interfaces is a chal-lenging area that is expected to continue for years to come. Com-plex interfaces include: nanoparticles surfaces with edge and
ll rights reserved.
corner sites contributing to more than 25% of the atoms presenton the surface; catalyst surfaces under operando conditions; envi-ronmental interfaces under conditions of ambient temperatureand relative humidity; liquid interfaces and biological interfacesincluding biofilms.
As a co-organizer of a recent symposium on the physical chem-istry of environmental interfaces and co-editor of a journal issue onthe symposium topic, I was struck by the number of high qualitysurface science studies of these complex interfaces. One prominenttheme that emerged in the symposium is the importance of waterinteractions with metal oxides surfaces. Metal oxide surfaces areimportant environmental interfaces for a number of reasons [1–3]. Metal oxides are widely used as catalysts and catalyst supportsin environmental remediation. Metal oxide minerals found in nat-ure can adsorb contaminants and control the transport of pollu-tants in ground water systems. Metal oxides are a component ofmineral dust aerosol in the Earth’s atmosphere [4]. Furthermore,semiconductor metal oxides are important photocatalysts in bothengineered and natural systems and water plays an important rolein these photocatalytic reactions.
Thus, there is increasing interest in understanding metal oxidesurfaces under ambient conditions of temperature and relativehumidity and the role of co-adsorbed water on the chemistry ofinterfaces under ambient conditions. These studies can provide in-sight into heterogeneous catalysis, environmental processes andcorrosion. Specific topics of discussion at this recent symposiumon environmental interfaces included both experimental and theo-retical analyses of: (i) oxides (e.g. TiO2) [5] in photocatalysis andthe role of co-adsorbed water; (ii) oxide minerals in aqueous andhumid environments (e.g. a-Fe2O3 and a-Al2O3); (iii) oxide miner-

2956 V.H. Grassian / Surface Science 602 (2008) 2955–2962
als with adsorbed organic layers and the influence of these layerson water adsorption; and (iv) the influence of co-adsorbed wateron the reactivity of oxide surfaces.
Although there is a rich literature on the surface science andsurface chemistry of oxides, many of these studies are done in ul-tra-high vacuum. Complementing these UHV surface science stud-ies is a large body of literature on oxide surfaces in aqueousenvironments. The surface science of oxides at gas–solid interfaceunder ambient conditions as a function of temperature and relativehumidity is not as well understood.
Fig. 1 shows these different regimes of water activity, a(H2O),defined as
aðH2OÞ ¼ PðH2OÞ=P0ðH2OÞ ¼ RH; ð1Þ
where RH is the relative humidity, and P(H2O) and P0(H2O) are thewater vapor pressure and the water saturated vapor pressure,respectively. These different regimes of water activity span manyorders of magnitude. The nature of the oxide surface being exploredand the conceptual framework in which to understand the proper-ties of oxide surfaces change within these different regimes of wateractivity. It is the middle regime, i.e. the humid conditions with ad-sorbed water, which is of interest here. In particular, this surfacescience prospective focuses on the structure and reactivity of oxidesurfaces, and other environmental interfaces, in the presence of a dy-namic adsorbed water layer, i.e. in equilibrium with water vapor, asa function of relative humidity (RH). Because of the very nature ofsurface science prospectives, this article will discuss and presentonly a few examples of some recent studies that strive to gain fun-damental information about the molecular nature of oxide surfacesunder ambient conditions of temperature and relative humidity,how this dynamic adsorbed water layer influences the reactionchemistry of environmental interfaces and solvates adsorbed ions.Finally, a brief discussion of future research needs and directionsis presented.
2. Adsorbed water on environmental interfaces
Recent experimental and theoretical studies of water adsorp-tion on surfaces have provided new insights into our understand-ing of environmental interfaces under ambient conditions oftemperature and relative humidity. Some of these advances havecome about through of the development of new experimental toolsincluding synchrotron-based X-ray methods [6].
One such advance is the ambient pressure photoelectron spec-troscopy (APPES). The details of the instrument, its mode of oper-ation and a number of environmental applications using APPEShave been recently reviewed by Salmeron and Schlög [7]. Quanti-fying water adsorption on oxide surfaces under ambient conditions
Fig. 1. The diagram shows that as the environment goes from dry to humid to wet, i.e. asurfaces under these different environmental conditions changes. The conceptual theoresurfaces in the presence of increasing water activity changes and for the intermediate regand other environmental interfaces.
of temperature and relative humidity is one area that APPES hasbeen successfully applied [7,8]. Water adsorption on TiO2(110) isshown here as an example of the use of APPES. The spectra shownin Fig. 2 are of the O1s region at low and high relative humidity.Multiple peaks observed in the spectra represent photoelectronsof different binding energies for lattice oxygen atoms, surface O–H groups and two forms of water: molecular adsorbed and gas-phase. From these data, it can be seen that the binding energyfor the adsorbed water peak decreases at higher coverage. Thechange in binding energy is a result of water molecules initiallypreferentially binding to surface hydroxyl groups at low coveragesand once these sites are filled water molecules bind to other siteson the surface. The low %RH XPS data, where %RH = RH � 100, arecurve fit as shown in Fig. 2a. Furthermore once the spectra aredeconvoluted into different spectral components, the intensitiescan be analyzed and surface coverages of the individual adsorbedcomponents, O–H and H2O, can be obtained, as shown in Fig. 2band c.
It can be seen that the coverage of molecular water increases asa function of increasing RH where as the hydroxyl groups do not. Inaddition, it is seen that the formation of hydroxyl groups occurimmediately upon exposing TiO2(110) to water at a very low RHof �5 � 10�4. As shown by the data plotted in Fig. 2c, the onsetof molecular water adsorption occurs after the formation of surfacehydroxyl groups. Based on these results as well as additionallymeasured isotherms and isobars, a model was proposed by Ketteleret al. whereby at very low RH water first dissociates on the surfacein oxygen vacancies [8]. The dissociation results in O–H occupyingbridge sites. Water molecules then hydrogen bond to these O–Hgroups, thus surface hydroxyls are proposed to be nucleation sitesfor water adsorption. The initial O–H:H2O complexes that form arecharacterized by high enthalpies of adsorption (�72 kJ/mol). Afterthese sites are saturated, only then does water adsorption occupyother sites and configurations on the surface. For additional wateradsorption above 0.5 ml, the enthalpy of adsorption approachesthe bulk enthalpy of condensation of �45 kJ/mol. These data andquantitative insights on the behavior of water adsorbed on TiO2
would have been difficult without the type of quantitative dataprovided by APPES.
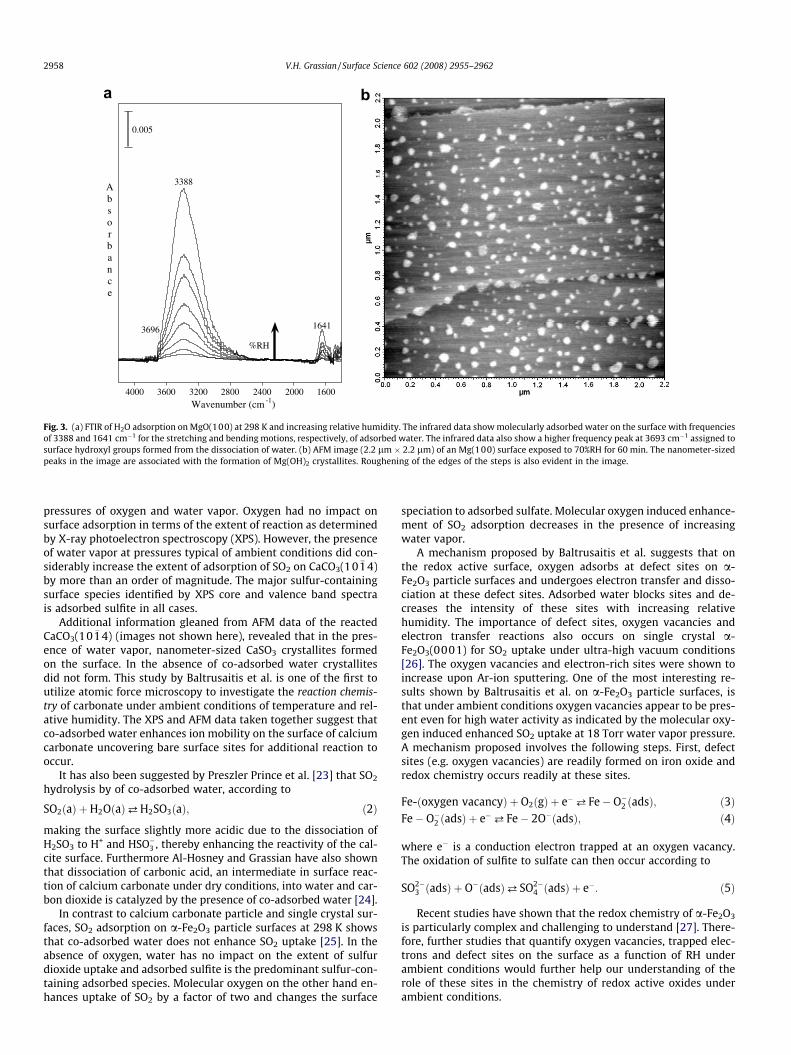
Other techniques to study water adsorption on surfaces includeinfrared spectroscopy. Fig. 2 shows water adsorption on MgO(100)as a function of increasing RH [9]. Foster and co-workers have pre-viously measured water uptake on MgO(100) at ambient temper-atures [10]. Based on their analysis of the vibrational bands due tomolecular adsorbed water at 3388 and 1641 cm�1, they suggestedthat the adsorbed water layer grows in via three-dimensional is-land formation. The presence of a new higher wavenumber bandat 3696 cm�1, whose intensity increases with repeated water
s the water activity increases over several orders of magnitude, the nature of oxidetical framework in which to understand the atomic and molecular nature of oxide
ime of humid environments there is clearly a lack of understanding of oxide surfaces
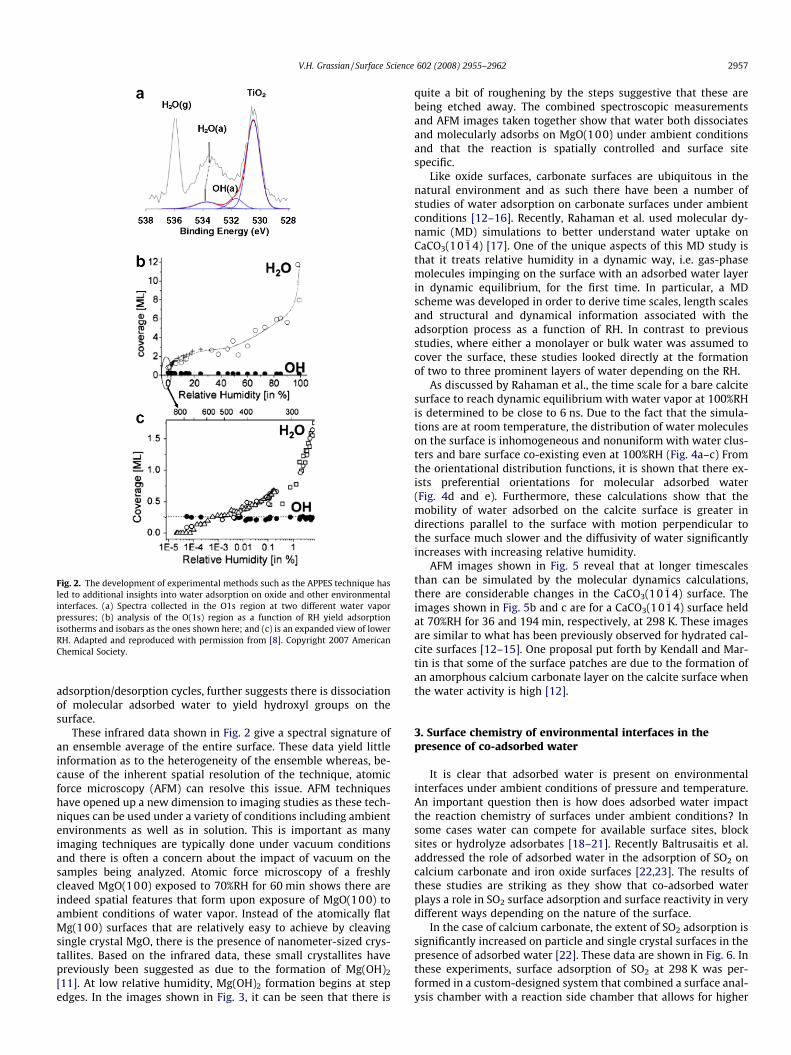
Fig. 2. The development of experimental methods such as the APPES technique hasled to additional insights into water adsorption on oxide and other environmentalinterfaces. (a) Spectra collected in the O1s region at two different water vaporpressures; (b) analysis of the O(1s) region as a function of RH yield adsorptionisotherms and isobars as the ones shown here; and (c) is an expanded view of lowerRH. Adapted and reproduced with permission from [8]. Copyright 2007 AmericanChemical Society.
V.H. Grassian / Surface Science 602 (2008) 2955–2962 2957
adsorption/desorption cycles, further suggests there is dissociationof molecular adsorbed water to yield hydroxyl groups on thesurface.
These infrared data shown in Fig. 2 give a spectral signature ofan ensemble average of the entire surface. These data yield littleinformation as to the heterogeneity of the ensemble whereas, be-cause of the inherent spatial resolution of the technique, atomicforce microscopy (AFM) can resolve this issue. AFM techniqueshave opened up a new dimension to imaging studies as these tech-niques can be used under a variety of conditions including ambientenvironments as well as in solution. This is important as manyimaging techniques are typically done under vacuum conditionsand there is often a concern about the impact of vacuum on thesamples being analyzed. Atomic force microscopy of a freshlycleaved MgO(100) exposed to 70%RH for 60 min shows there areindeed spatial features that form upon exposure of MgO(100) toambient conditions of water vapor. Instead of the atomically flatMg(100) surfaces that are relatively easy to achieve by cleavingsingle crystal MgO, there is the presence of nanometer-sized crys-tallites. Based on the infrared data, these small crystallites havepreviously been suggested as due to the formation of Mg(OH)2
[11]. At low relative humidity, Mg(OH)2 formation begins at stepedges. In the images shown in Fig. 3, it can be seen that there is
quite a bit of roughening by the steps suggestive that these arebeing etched away. The combined spectroscopic measurementsand AFM images taken together show that water both dissociatesand molecularly adsorbs on MgO(100) under ambient conditionsand that the reaction is spatially controlled and surface sitespecific.
Like oxide surfaces, carbonate surfaces are ubiquitous in thenatural environment and as such there have been a number ofstudies of water adsorption on carbonate surfaces under ambientconditions [12–16]. Recently, Rahaman et al. used molecular dy-namic (MD) simulations to better understand water uptake onCaCO3(10 �14) [17]. One of the unique aspects of this MD study isthat it treats relative humidity in a dynamic way, i.e. gas-phasemolecules impinging on the surface with an adsorbed water layerin dynamic equilibrium, for the first time. In particular, a MDscheme was developed in order to derive time scales, length scalesand structural and dynamical information associated with theadsorption process as a function of RH. In contrast to previousstudies, where either a monolayer or bulk water was assumed tocover the surface, these studies looked directly at the formationof two to three prominent layers of water depending on the RH.
As discussed by Rahaman et al., the time scale for a bare calcitesurface to reach dynamic equilibrium with water vapor at 100%RHis determined to be close to 6 ns. Due to the fact that the simula-tions are at room temperature, the distribution of water moleculeson the surface is inhomogeneous and nonuniform with water clus-ters and bare surface co-existing even at 100%RH (Fig. 4a–c) Fromthe orientational distribution functions, it is shown that there ex-ists preferential orientations for molecular adsorbed water(Fig. 4d and e). Furthermore, these calculations show that themobility of water adsorbed on the calcite surface is greater indirections parallel to the surface with motion perpendicular tothe surface much slower and the diffusivity of water significantlyincreases with increasing relative humidity.
AFM images shown in Fig. 5 reveal that at longer timescalesthan can be simulated by the molecular dynamics calculations,there are considerable changes in the CaCO3(10 �14) surface. Theimages shown in Fig. 5b and c are for a CaCO3(10 �14) surface heldat 70%RH for 36 and 194 min, respectively, at 298 K. These imagesare similar to what has been previously observed for hydrated cal-cite surfaces [12–15]. One proposal put forth by Kendall and Mar-tin is that some of the surface patches are due to the formation ofan amorphous calcium carbonate layer on the calcite surface whenthe water activity is high [12].
3. Surface chemistry of environmental interfaces in thepresence of co-adsorbed water
It is clear that adsorbed water is present on environmentalinterfaces under ambient conditions of pressure and temperature.An important question then is how does adsorbed water impactthe reaction chemistry of surfaces under ambient conditions? Insome cases water can compete for available surface sites, blocksites or hydrolyze adsorbates [18–21]. Recently Baltrusaitis et al.addressed the role of adsorbed water in the adsorption of SO2 oncalcium carbonate and iron oxide surfaces [22,23]. The results ofthese studies are striking as they show that co-adsorbed waterplays a role in SO2 surface adsorption and surface reactivity in verydifferent ways depending on the nature of the surface.
In the case of calcium carbonate, the extent of SO2 adsorption issignificantly increased on particle and single crystal surfaces in thepresence of adsorbed water [22]. These data are shown in Fig. 6. Inthese experiments, surface adsorption of SO2 at 298 K was per-formed in a custom-designed system that combined a surface anal-ysis chamber with a reaction side chamber that allows for higher
1600200024002800320036004000Wavenumber (cm -1)
0.005
Absorbance
%RH
3388
16413696
Fig. 3. (a) FTIR of H2O adsorption on MgO(100) at 298 K and increasing relative humidity. The infrared data show molecularly adsorbed water on the surface with frequenciesof 3388 and 1641 cm�1 for the stretching and bending motions, respectively, of adsorbed water. The infrared data also show a higher frequency peak at 3693 cm�1 assigned tosurface hydroxyl groups formed from the dissociation of water. (b) AFM image (2.2 lm � 2.2 lm) of an Mg(100) surface exposed to 70%RH for 60 min. The nanometer-sizedpeaks in the image are associated with the formation of Mg(OH)2 crystallites. Roughening of the edges of the steps is also evident in the image.
2958 V.H. Grassian / Surface Science 602 (2008) 2955–2962
pressures of oxygen and water vapor. Oxygen had no impact onsurface adsorption in terms of the extent of reaction as determinedby X-ray photoelectron spectroscopy (XPS). However, the presenceof water vapor at pressures typical of ambient conditions did con-siderably increase the extent of adsorption of SO2 on CaCO3(10 �14)by more than an order of magnitude. The major sulfur-containingsurface species identified by XPS core and valence band spectrais adsorbed sulfite in all cases.
Additional information gleaned from AFM data of the reactedCaCO3(10 �14) (images not shown here), revealed that in the pres-ence of water vapor, nanometer-sized CaSO3 crystallites formedon the surface. In the absence of co-adsorbed water crystallitesdid not form. This study by Baltrusaitis et al. is one of the first toutilize atomic force microscopy to investigate the reaction chemis-try of carbonate under ambient conditions of temperature and rel-ative humidity. The XPS and AFM data taken together suggest thatco-adsorbed water enhances ion mobility on the surface of calciumcarbonate uncovering bare surface sites for additional reaction tooccur.
It has also been suggested by Preszler Prince et al. [23] that SO2
hydrolysis by of co-adsorbed water, according to
SO2ðaÞ þH2OðaÞ¢ H2SO3ðaÞ; ð2Þ
making the surface slightly more acidic due to the dissociation ofH2SO3 to H+ and HSO�3 , thereby enhancing the reactivity of the cal-cite surface. Furthermore Al-Hosney and Grassian have also shownthat dissociation of carbonic acid, an intermediate in surface reac-tion of calcium carbonate under dry conditions, into water and car-bon dioxide is catalyzed by the presence of co-adsorbed water [24].
In contrast to calcium carbonate particle and single crystal sur-faces, SO2 adsorption on a-Fe2O3 particle surfaces at 298 K showsthat co-adsorbed water does not enhance SO2 uptake [25]. In theabsence of oxygen, water has no impact on the extent of sulfurdioxide uptake and adsorbed sulfite is the predominant sulfur-con-taining adsorbed species. Molecular oxygen on the other hand en-hances uptake of SO2 by a factor of two and changes the surface
speciation to adsorbed sulfate. Molecular oxygen induced enhance-ment of SO2 adsorption decreases in the presence of increasingwater vapor.
A mechanism proposed by Baltrusaitis et al. suggests that onthe redox active surface, oxygen adsorbs at defect sites on a-Fe2O3 particle surfaces and undergoes electron transfer and disso-ciation at these defect sites. Adsorbed water blocks sites and de-creases the intensity of these sites with increasing relativehumidity. The importance of defect sites, oxygen vacancies andelectron transfer reactions also occurs on single crystal a-Fe2O3(0001) for SO2 uptake under ultra-high vacuum conditions[26]. The oxygen vacancies and electron-rich sites were shown toincrease upon Ar-ion sputtering. One of the most interesting re-sults shown by Baltrusaitis et al. on a-Fe2O3 particle surfaces, isthat under ambient conditions oxygen vacancies appear to be pres-ent even for high water activity as indicated by the molecular oxy-gen induced enhanced SO2 uptake at 18 Torr water vapor pressure.A mechanism proposed involves the following steps. First, defectsites (e.g. oxygen vacancies) are readily formed on iron oxide andredox chemistry occurs readily at these sites.
Fe-ðoxygen vacancyÞ þ O2ðgÞ þ e�¢ Fe� O�2 ðadsÞ; ð3ÞFe� O�2 ðadsÞ þ e�¢ Fe� 2O�ðadsÞ; ð4Þ
where e� is a conduction electron trapped at an oxygen vacancy.The oxidation of sulfite to sulfate can then occur according to
SO2�3 ðadsÞ þ O�ðadsÞ¢ SO2�
4 ðadsÞ þ e�: ð5Þ
Recent studies have shown that the redox chemistry of a-Fe2O3
is particularly complex and challenging to understand [27]. There-fore, further studies that quantify oxygen vacancies, trapped elec-trons and defect sites on the surface as a function of RH underambient conditions would further help our understanding of therole of these sites in the chemistry of redox active oxides underambient conditions.
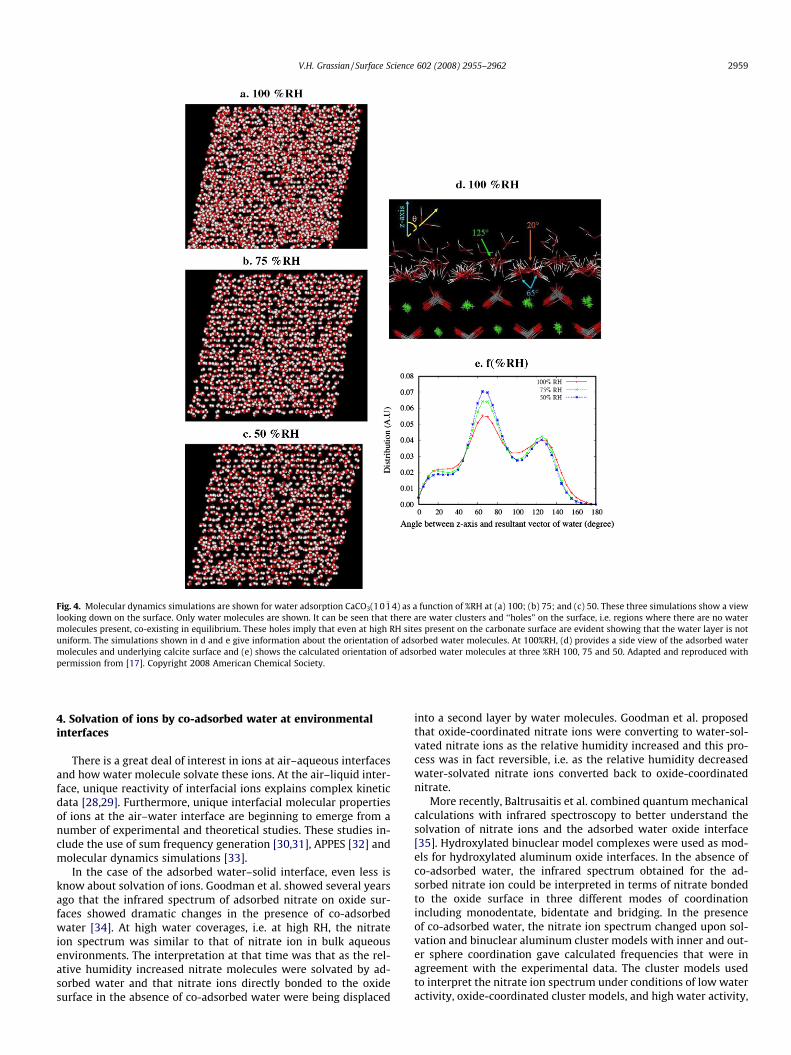
Fig. 4. Molecular dynamics simulations are shown for water adsorption CaCO3(10 �14) as a function of %RH at (a) 100; (b) 75; and (c) 50. These three simulations show a viewlooking down on the surface. Only water molecules are shown. It can be seen that there are water clusters and ‘‘holes” on the surface, i.e. regions where there are no watermolecules present, co-existing in equilibrium. These holes imply that even at high RH sites present on the carbonate surface are evident showing that the water layer is notuniform. The simulations shown in d and e give information about the orientation of adsorbed water molecules. At 100%RH, (d) provides a side view of the adsorbed watermolecules and underlying calcite surface and (e) shows the calculated orientation of adsorbed water molecules at three %RH 100, 75 and 50. Adapted and reproduced withpermission from [17]. Copyright 2008 American Chemical Society.
V.H. Grassian / Surface Science 602 (2008) 2955–2962 2959
4. Solvation of ions by co-adsorbed water at environmentalinterfaces
There is a great deal of interest in ions at air–aqueous interfacesand how water molecule solvate these ions. At the air–liquid inter-face, unique reactivity of interfacial ions explains complex kineticdata [28,29]. Furthermore, unique interfacial molecular propertiesof ions at the air–water interface are beginning to emerge from anumber of experimental and theoretical studies. These studies in-clude the use of sum frequency generation [30,31], APPES [32] andmolecular dynamics simulations [33].
In the case of the adsorbed water–solid interface, even less isknow about solvation of ions. Goodman et al. showed several yearsago that the infrared spectrum of adsorbed nitrate on oxide sur-faces showed dramatic changes in the presence of co-adsorbedwater [34]. At high water coverages, i.e. at high RH, the nitrateion spectrum was similar to that of nitrate ion in bulk aqueousenvironments. The interpretation at that time was that as the rel-ative humidity increased nitrate molecules were solvated by ad-sorbed water and that nitrate ions directly bonded to the oxidesurface in the absence of co-adsorbed water were being displaced
into a second layer by water molecules. Goodman et al. proposedthat oxide-coordinated nitrate ions were converting to water-sol-vated nitrate ions as the relative humidity increased and this pro-cess was in fact reversible, i.e. as the relative humidity decreasedwater-solvated nitrate ions converted back to oxide-coordinatednitrate.
More recently, Baltrusaitis et al. combined quantum mechanicalcalculations with infrared spectroscopy to better understand thesolvation of nitrate ions and the adsorbed water oxide interface[35]. Hydroxylated binuclear model complexes were used as mod-els for hydroxylated aluminum oxide interfaces. In the absence ofco-adsorbed water, the infrared spectrum obtained for the ad-sorbed nitrate ion could be interpreted in terms of nitrate bondedto the oxide surface in three different modes of coordinationincluding monodentate, bidentate and bridging. In the presenceof co-adsorbed water, the nitrate ion spectrum changed upon sol-vation and binuclear aluminum cluster models with inner and out-er sphere coordination gave calculated frequencies that were inagreement with the experimental data. The cluster models usedto interpret the nitrate ion spectrum under conditions of low wateractivity, oxide-coordinated cluster models, and high water activity,
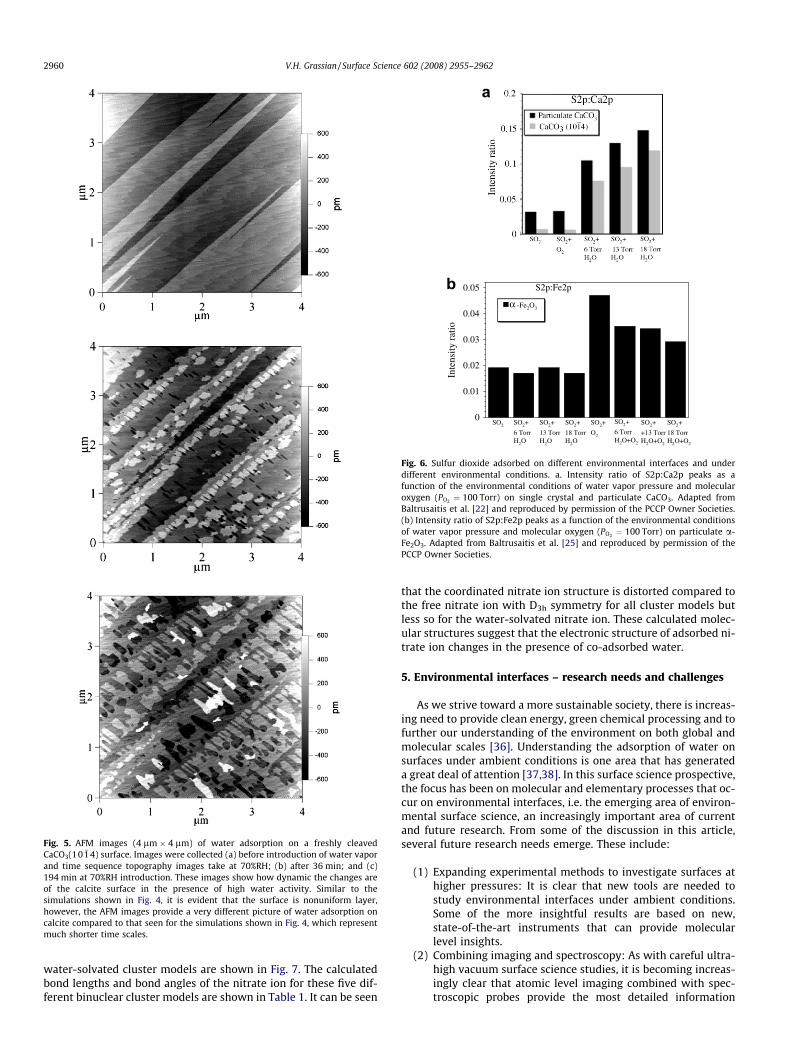
Fig. 5. AFM images (4 lm � 4 lm) of water adsorption on a freshly cleavedCaCO3(10 �14) surface. Images were collected (a) before introduction of water vaporand time sequence topography images take at 70%RH; (b) after 36 min; and (c)194 min at 70%RH introduction. These images show how dynamic the changes areof the calcite surface in the presence of high water activity. Similar to thesimulations shown in Fig. 4, it is evident that the surface is nonuniform layer,however, the AFM images provide a very different picture of water adsorption oncalcite compared to that seen for the simulations shown in Fig. 4, which representmuch shorter time scales.
0
0.01
0.02
0.03
0.04
0.05
-Fe2O3
-FeOOH
Inte
nsity
rat
io
S2p:Fe2p
SO2 SO2+
O2
SO2+
6 TorrH2O
SO2+
13 TorrH2O
SO2+
18 TorrH2O
SO2+
+13 TorrH2O+O2
SO2+
6 TorrH2O+O2
SO2+
18 TorrH2O+O2
α
Fig. 6. Sulfur dioxide adsorbed on different environmental interfaces and underdifferent environmental conditions. a. Intensity ratio of S2p:Ca2p peaks as afunction of the environmental conditions of water vapor pressure and molecularoxygen (PO2 ¼ 100 Torr) on single crystal and particulate CaCO3. Adapted fromBaltrusaitis et al. [22] and reproduced by permission of the PCCP Owner Societies.(b) Intensity ratio of S2p:Fe2p peaks as a function of the environmental conditionsof water vapor pressure and molecular oxygen (PO2 ¼ 100 Torr) on particulate a-Fe2O3. Adapted from Baltrusaitis et al. [25] and reproduced by permission of thePCCP Owner Societies.
2960 V.H. Grassian / Surface Science 602 (2008) 2955–2962
water-solvated cluster models are shown in Fig. 7. The calculatedbond lengths and bond angles of the nitrate ion for these five dif-ferent binuclear cluster models are shown in Table 1. It can be seen
that the coordinated nitrate ion structure is distorted compared tothe free nitrate ion with D3h symmetry for all cluster models butless so for the water-solvated nitrate ion. These calculated molec-ular structures suggest that the electronic structure of adsorbed ni-trate ion changes in the presence of co-adsorbed water.
5. Environmental interfaces – research needs and challenges
As we strive toward a more sustainable society, there is increas-ing need to provide clean energy, green chemical processing and tofurther our understanding of the environment on both global andmolecular scales [36]. Understanding the adsorption of water onsurfaces under ambient conditions is one area that has generateda great deal of attention [37,38]. In this surface science prospective,the focus has been on molecular and elementary processes that oc-cur on environmental interfaces, i.e. the emerging area of environ-mental surface science, an increasingly important area of currentand future research. From some of the discussion in this article,several future research needs emerge. These include:
(1) Expanding experimental methods to investigate surfaces athigher pressures: It is clear that new tools are needed tostudy environmental interfaces under ambient conditions.Some of the more insightful results are based on new,state-of-the-art instruments that can provide molecularlevel insights.
(2) Combining imaging and spectroscopy: As with careful ultra-high vacuum surface science studies, it is becoming increas-ingly clear that atomic level imaging combined with spec-troscopic probes provide the most detailed information

Fig. 7. Binuclear Al cluster models provide insights into the bonding of nitrate ion on aluminum oxide surfaces as a function of water activity. (a) Oxide-coordinated nitrateion cluster models are used to interpret the infrared spectrum of adsorbed nitrate under dry conditions, i.e. at low water activity in the absence of co-adsorbed water and (b)water-solvated nitrate ion cluster models are used to interpret the infrared spectrum of adsorbed nitrate under humid conditions, i.e. at increasing water activity in thepresence of co-adsorbed water. Adapted from Baltrusaitis et al. [35] and reproduced by permission of the PCCP Owner Societies.
Table 1Structural parameters and vibrational modes frequencies (cm�1) for free nitrate ion and coordinated nitrate ion in binuclear cluster models, calculated at B3LYP/6-31+G(d) levelof theorya
Optimized structural parameters
Bond length (Å) Angle (�)
O1–N1 O2–N1 O3–N1 O1–N1–O2 O2–N1–O3 O1–N1–O3
Free nitrate ion1.27 1.27 1.27 120 120 120
Coordinated nitrate ionOxide-coordinated nitrate cluster models
(I) Monodentate 1.33 1.24 1.23 117.9 125.0 117.1(II) Bidentate 1.28 1.30 1.22 113.4 122.6 124.1(III) (O–O)-bridged 1.29 1.29 1.20 118.7 120.7 120.5
Water-solvated nitrate cluster models(V) Inner sphere coordinated nitrate ion 1.28 1.25 1.24 117.3 121.8 120.9(VI) Outer sphere coordinated nitrate ion 1.27 1.24 1.27 121.3 120.8 117.9
a Adapted from Baltrusaitis et al. [35] and reproduced by permission of the PCCP Owner Societies.
V.H. Grassian / Surface Science 602 (2008) 2955–2962 2961
needed for additional insights of environmental interfaces asthese surfaces are nonuniform and spatially heterogeneous.Spatial heterogeneity plays an import role in the surfacereactivity of environmental interfaces.
(3) Water adsorption on modified surfaces: Oxide surfaces andother environmental interfaces often coated with thin filmsor monolayers of organic layers, the detailed nature of howthese coatings impact water uptake is an area that needsadditional attention.
(4) Nanoparticles: Single crystal surfaces, with well-definedindex planes and atomic spatial arrangements, are extre-mely important from fundamental surface science studies.Nanoparticles are found in many natural and engineeredsystems and as such are important environmental interfaces.With many of the atoms located at corner and edge sites,there is the need to further our efforts in obtaining quantita-tive information on the structure of these more complex yetubiquitous interfaces.
(5) Merging theory and experiment: From the examples shownhere and many others, it is clear that theory and experimentneed to converge to similar time scales and spatial complex-ity. This is not a problem inherent just to environment inter-faces but to other complex systems.
Acknowledgements
I would like to thank my co-organizers, Professors Franz Geiger,Howard Fairbrother and John Hemminger, of the symposium‘‘Physical Chemistry of Environmental Interfaces” in New Orleansat the American Chemical Society National Meeting in April 2008for putting together such an exciting program. I thank the partici-pants as well for their excellent contributions to the symposium. Iam grateful for the contributions of my students, postdocs and col-laborators to the research I have reviewed here in this surface sci-ence prospective. I am especially grateful to Professor Claudio

2962 V.H. Grassian / Surface Science 602 (2008) 2955–2962
Margulis, Dr. Asif Rahaman, Professor Hind Al-Abadleh and Dr.Jonas Baltrusaitis for their theoretical and experimental contribu-tions to environmental surface science research at the Universityof Iowa. This material is based upon work supported by the Na-tional Science Foundation under Grant No. CHE-0503854 (VHG).Any opinions, findings and conclusions or recommendations ex-pressed in this material are those of the author and do not neces-sarily reflect the views of the National Science Foundation. Theauthor also gratefully acknowledges the donors of the PetroleumResearch Fund, administered by the American Chemical Society,for partial support of this research through Grant No. 42820-AC5.
References
[1] H.A. Al-Abadleh, V.H. Grassian, Surface Science Reports 52 (2003) 63.[2] G.E. Brown, V.E. Henrich, W.H. Casey, D.L. Clark, C. Eggleston, A. Felmy, D.W.
Goodman, M. Gratzel, G. Maciel, M.I. McCarthy, K.H. Nealson, D.A. Sverjensky,M.F. Toney, J.M. Zachara, Chemical Reviews 99 (1999) 77.
[3] D.L. Sparks, in: V.H. Grassian (Ed.), Environmental Catalysis, CRC Press, BocaRaton, FL, 2005, p. 3.
[4] C.R. Usher, A.E. Michel, V.H. Grassian, Chemical Reviews 103 (2003) 4883.[5] U. Diebold, Surface Science Reports 48 (2003) 53.[6] P.J. Eng, T.P. Trainor, G.E. Brown, G.A. Waychunas, M. Newville, S.R. Sutton, M.L.
Rivers, Science 288 (2000) 1029.[7] M. Salmeron, R. Schlögl, Surface Science Reports 63 (2008) 169.[8] G. Ketteler, S. Yamamoto, H. Bluhm, K. Andersson, D.E. Starr, D.F. Ogletree, H.
Ogasawara, A. Nilsson, M. Salmeron, Journal of Physical Chemistry C 111(2007) 8278.
[9] H.A. Al-Abadleh, Ph.D. Dissertation, University of Iowa, 2004.[10] M. Foster, M. Furse, D. Passno, Surface Science 502 (2002) 102.[11] B.J. Krueger, J. Ross, V.H. Grassian, Langmuir 21 (2005) 8793.[12] T.A. Kendall, S.T. Martin, Journal of Physical Chemistry A 111 (2007) 505.[13] D.B. Hausner, R.J. Reeder, D.R. Strongin, Journal of Colloid and Interface Science
305 (2007) 101.[14] P. Geissbuhler, P. Fenter, E. DiMasi, G. Srajer, L.B. Sorensen, N.C. Sturchio,
Surface Science 573 (2004) 191.[15] S.L.S. Stipp, Geochimical et Cosmochimica Acta 63 (1999) 3121.[16] W. Neagle, C.H. Rochester, Journal of the Chemical Society – Faraday
Transactions 86 (1990) 181.
[17] A. Rahaman, V.H. Grassian, C.J. Margulis, Journal of Physical Chemistry C 112(2008) 2109.
[18] M.A. Henderson, Langmuir 21 (2005) 3443.[19] B.J. Finlayson-Pitts, L.M. Wingen, A.L. Sumner, D. Syomin, K.A. Ramazan,
Physical Chemistry Chemical Physics 5 (2003) 223.[20] D.M. Cwiertny, M.A. Young, V.H. Grassian, Annual Reviews of Physical
Chemistry 59 (2008) 27.[21] J. Szanyi, J.H. Kwak, R.J. Chimentao, C.H.F. Peden CHF, Journal of Physical
Chemistry C 111 (2007) 2661.[22] J. Baltrusaitis, C.R. Usher, V.H. Grassian, Physical Chemistry Chemical Physics 9
(2007) 3011.[23] A. Preszler Prince, P.D. Kleiber, V.H. Grassian, M.A. Young, Physical Chemistry
Chemical Physics 9 (2007) 3432.[24] H.A. Al-Hosney, V.H. Grassian, Journal of American Chemical Society 126
(2004) 8068.[25] J. Baltrusaitis, D.M. Cwiertny, V.H. Grassian, Physical Chemistry Chemical
Physics 9 (2007) 5542.[26] D.S. Toledano, V.E. Henrich, Journal of Physical Chemistry B 105 (2001)
3872.[27] S.V. Yanina, K.E. Rosso, Science 320 (2008) 218.[28] E.M. Knipping, M.J. Lakin, K.L. Foster, P. Jungwirth, D.J. Tobias, R.B. Gerber, D.
Dabdub, B.J. Finlayson-Pitts, Science 288 (2000) 301.[29] S. Gopalakrishnan, D. Liu, H.C. Allen, M. Kuo, M.J. Shultz, Chemical Reviews 106
(2006) 115.[30] E.A. Raymond, G.L. Richmond, Journal of Physical Chemistry B 108 (2004)
5051.[31] D.E. Otten, P.B. Peterson, R.J. Saykally, Chemical Physics Letters 449 (2007)
261.[32] S. Ghosal, J.C. Hemminger, H. Bluhm, B.S. Mun, E.L.D. Hebenstreit, G. Ketteler,
D.F. Ogletree, F.G. Requejo, M. Salmeron, Science 307 (2005) 563.[33] P. Jungwirth, D.J. Tobias, Chemical Reviews 106 (2006) 1259.[34] A.L. Goodman, E.B. Bernard, V.H. Grassian, Journal of Physical Chemistry A 105
(2001) 6443.[35] J. Baltrusaitis, J. Schuttlefield, J.H. Jensen, V.H. Grassian, Physical Chemistry
Chemical Physics 9 (2007) 4970.[36] V.H. Grassian, G. Meyer, H. Abruna, L.E. Achenie, T. Allison, B. Brunschwig, G.W.
Coates, G.J. Ferry, M. Garcia-Garibay, J. Gardea-Torresdey, C.P. Grey, J.Hutchison, C.J. Li, C. Liotta, A. Ragauskas, S. Minteer, K. Mueller, J. Roberts, O.Sadik, R. Schmehl, W. Schneider, A. Selloni, P. Stair, J. Stewart, D. Thorn, J.Tyson, B. Voelker, J.M. White, F. Wood-Black, Environmental Science andTechnology 41 (2007) 4840.
[37] G.E. Ewing, Chemical Reviews 106 (2006) 1511.[38] M.A. Henderson, Surface Science Reports 46 (2002) 5.
Recommended

















