American Journal of Medical Genetics 49:333-336 (1994)
Brief Clinical Report
Stratton-Parker Syndrome: Confirmation of a New Entity
~~
Orazio Gabrielli, Ines Carloni, Carlo Catassi, Giuliana Natalini, Giovanni V. Coppa, and Pierluigi Giorgi Department of Pediatrics, University of Ancona, Ancona, Italy
Recently, Stratton and Parker [Am J Med Genet 32:169-173, 19891 reported on a child with a previously undescribed combination of growth hormone deficiency, wormian bones, dextrocardia, brachycamptodactyly, and other midline defects. We report on another patient with similar clinical signs. 0 1994 Wiley-Liss, Inc.
KEY WORDS: wormian bones, growth hor- mone deficiency, midline de- fects
INTRODUCTION We report on a 10-year-old boy with short stature due
to growth hormone deficiency, facial anomalies, body and facial asymmetry, congenital heart defect, genital hypoplasia, intestinal atresia, hemimegalencephaly, and wormian bones. This case may confirm the existence of a new syndrome first described by Stratton and Par- ker [19891.
CLINICAL REPORT A.M. is the third child of healthy, nonconsanguineous
parents (Italian father and English mother). He was born at term after a normal pregnancy and spontaneous delivery. Birthweight was 4,090 g, length 46 cm, head circumference (OFC) 36.5 cm. At birth facial anomalies were noted, including hypotelorism, downslanting pal- pebral fissures, depressed nasal bridge, small mouth, apparently low-set and posteriorly angulated ears, small penis, undescended testes, and generalized lymphedema. Due to retention of meconium and abdom- inal distention, he underwent surgery at 24 hours of life and was found to have stenosis of the ascending colon
Received for publication February 15, 1993; revision received September 13, 1993.
Address reprint requests to Orazio Gabrielli, M.D., Department of Pediatrics, University of Ancona, Via Corridoni 11 60123 An- cona, Italy.
0 1994 Wiley-Liss, Inc.
and atresia of the descending colon, with normal bowel in between. Postoperatively he developed Staphylococ- cus pyogenes sepsis with pneumonia and generalized seizures.
Renal ultrasound scan findings were normal, while an echocardiogram showed the presence of a small ven- tricular septa1 defect (VSD j. The chromosomes were normal male (46,XYj by G-banding, both in peripheral lymphocytes and skin fibroblasts. The baby was dis- charged one month after birth in satisfactory physical condition. At 6 months he had another episode of gener- alized seizures and was treated with anticonvulsant drugs. At this time the baby could sit unaided at a normal level of psychomotor development. Several elec- troencephalograms (EEGs) showed diffuse slow-wave abnormalities. A brain computed tomographic (CT) scan showed an asymmetrical dilation of the right lateral ventricle, especially the posterior horn, and hypoplasia of the left cerebral hemisphere. Clinical examination of the child at 12 months documented abundant adipose tissue, a length below the third centile (68 cm), and an evident decrease in growth velocity. Scrotum and penis were relatively hypoplastic. The characteristic facial traits had become more pronounced than at birth. Al- though the child could not walk freely, Denver develop- mental screening demonstrated that his development was minimally retarded in the fine motor skills but more markedly so in his gross motor performance. The psychological development was acceptable for a child of his age. A 316 systolic murmur could be heard at the lower sternal edge. Bone age was reported to correspond to the chronological age. At age 30 months an endocrine investigation was carried out due to his small stature. Arginine and insulin tests showed low levels of growth hormone. Bone age was around 24 months. A skull film showed a small pituitary fossa, wormian bones, and no abnormal intracranial calcification. The neurocranium was proportionately bigger than the splanchnocranium.
The child first came to our attention at the age of 29/12
years. He was below the third centile for weight and height (10.5 kg and 79 cm, respectively), and between the third and tenth centile for OFC (47.5 cm). His skin was thick and his muscles appeared normal. The head was slightly dolichocephalic with an anterior fontanelle
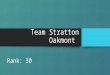
334 Gabrielli et al.
Fig. 1. The patient at 29/12 years (a$).
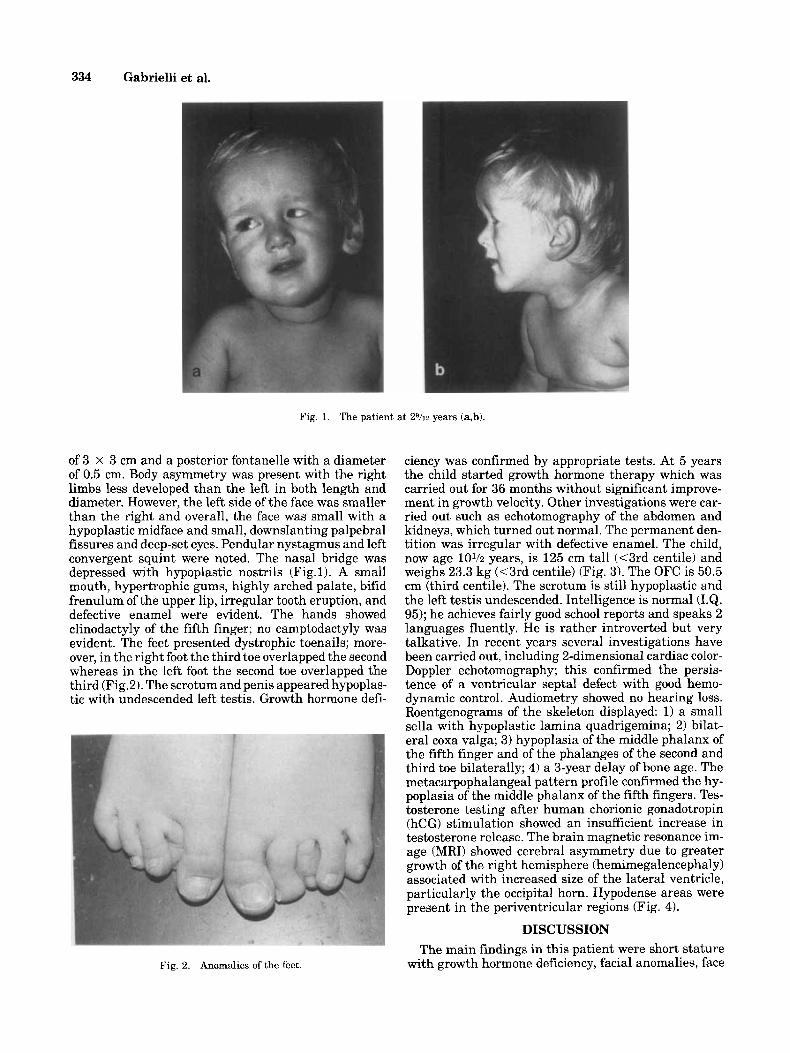
of 3 x 3 cm and a posterior fontanelle with a diameter of 0.5 cm. Body asymmetry was present with the right limbs less developed than the left in both length and diameter. However, the left side of the face was smaller than the right and overall, the face was small with a hypoplastic midface and small, downslanting palpebral fissures and deep-set eyes. Pendular nystagmus and left convergent squint were noted. The nasal bridge was depressed with hypoplastic nostrils (Fig.1). A small mouth, hypertrophic gums, highly arched palate, bifid frenulum of the upper lip, irregular tooth eruption, and defective enamel were evident. The hands showed clinodactyly of the fifth finger; no camptodactyly was evident. The feet presented dystrophic toenails; more- over, in the right foot the third toe overlapped the second whereas in the left foot the second toe overlapped the third (Fig.2). The scrotum and penis appeared hypoplas- tic with undescended left testis. Growth hormone defi-
Fig. 2. Anomalies of the feet.
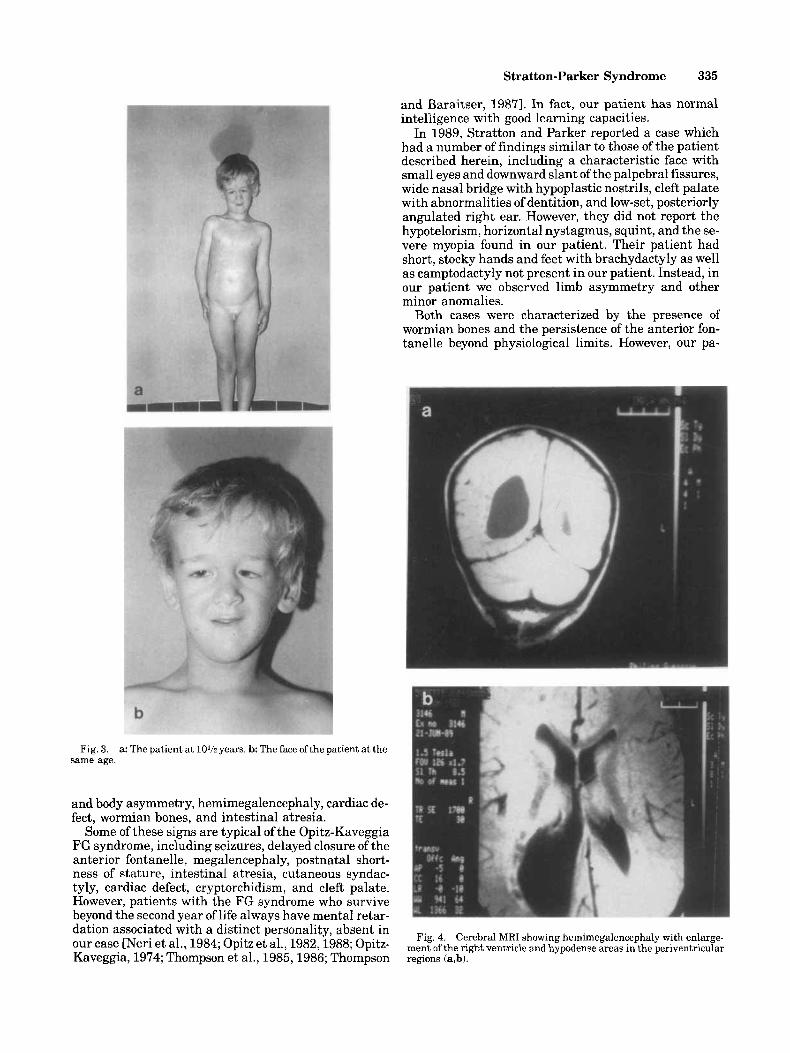
ciency was confirmed by appropriate tests. At 5 years the child started growth hormone therapy which was carried out for 36 months without significant improve- ment in growth velocity. Other investigations were car- ried out such as echotomography of the abdomen and kidneys, which turned out normal. The permanent den- tition was irregular with defective enamel. The child, now age 101h years, is 125 cm tall (<3rd centile) and weighs 23.3 kg (<3rd centile) (Fig. 3). The OFC is 50.5 cm (third centile). The scrotum is still hypoplastic and the left testis undescended. Intelligence is normal (I.&. 95); he achieves fairly good school reports and speaks 2 languages fluently. He is rather introverted but very talkative. In recent years several investigations have been carried out, including 2-dimensional cardiac color- Doppler echotomography; this confirmed the persis- tence of a ventricular septa1 defect with good hemo- dynamic control. Audiometry showed no hearing loss. Roentgenograms of the skeleton displayed: 1) a small sella with hypoplastic lamina quadrigemina; 2) bilat- eral coxa valga; 3) hypoplasia of the middle phalanx of the fifth finger and of the phalanges of the second and third toe bilaterally; 4) a 3-year delay of bone age. The metacarpophalangeal pattern profile confirmed the hy- poplasia of the middle phalanx of the fifth fingers. Tes- tosterone testing after human chorionic gonadotropin (hCG) stimulation showed an insufficient increase in testosterone release. The brain magnetic resonance im- age (MRI) showed cerebral asymmetry due to greater growth of the right hemisphere (hemimegalencephaly) associated with increased size of the lateral ventricle, particularly the occipital horn. Hypodense areas were present in the periventricular regions (Fig. 4).
DISCUSSION The main findings in this patient were short stature
with growth hormone deficiency, facial anomalies, face
Stratton-Parker Syndrome 335
and Baraitser, 19871. In fact, our patient has normal intelligence with good learning capacities.
In 1989, Stratton and Parker reported a case which had a number of findings similar to those of the patient described herein, including a characteristic face with small eyes and downward slant of the palpebral fissures, wide nasal bridge with hypoplastic nostrils, cleft palate with abnormalities of dentition, and low-set, posteriorly angulated right ear. However, they did not report the hypotelorism, horizontal nystagmus, squint, and the se- vere myopia found in our patient. Their patient had short, stocky hands and feet with brachydactyly as well as camptodactyly not present in our patient. Instead, in our patient we observed limb asymmetry and other minor anomalies.
Both cases were characterized by the presence of wormian bones and the persistence of the anterior fon- tanelle beyond physiological limits. However, our pa-
Fig. 3. same age.
a: The patient at 10% years. b: The face ofthe patient at the
and body asymmetry, hemimegalencephaly, cardiac de- fect, wormian bones, and intestinal atresia.
Some of these signs are typical of the Opitz-Kaveggia FG syndrome, including seizures, delayed closure of the anterior fontanelle, megalencephaly, postnatal short- ness of stature, intestinal atresia, cutaneous syndac- tyly, cardiac defect, cryptorchidism, and cleft palate. However, patients with the FG syndrome who survive beyond the second vear of life alwavs have mental retar- dation associated with a distinct personality> absent in
Fig, 4, Cerebral MRI showing hemimegalencephaly with enlarge- Our case " x i et ale, 1984; OPitZ et & 198231988; OPitZ- Kaveggia, 1974; Thompson et al., 1985,1986; Thompson
ment of the right ventricle and hypodense areas in the periventricular regions (a,b).

336 Gabrielli et al.
tient also had hemimegalencephaly with hypodense periventricular areas, probably vascular in origin, and a shallow sella with hypoplasia of the quadrigeminal lamina.
Both cases had a defect of the gastrointestinal tract but a t different levels: in the anus, in the case reported by Stratton and Parker 119891, in the colon in our case.
Both patients had a cardiac anomaly: VSD in our patient and dextrocardia in theirs. Both patients had cryptorchidism and short stature with proven growth hormone deficiency. In our case, treatment with growth hormone gave poor results. The patient of Stratton and Parker also had left renal hypoplasia and hypospadias (hypoplastic genitalia in our case). In their report Strat- ton and Parker [1989] did not mention seizures, which our patient had suffered in the first year of life.
Slight motor delay was present in both cases; in our case this has normalized as the patient has grown.
In conclusion we think that the case presented here shows significant similarities with the case described by Stratton and Parker [1989], especially the facial appear- ance, wormian bones, short stature with growth hormone deficiency, intestinal anomalies, and cryptorchidism.
ACKNOWLEDGMENTS We thank Mrs. Phyllis Ashburn for translating the
manuscript.
REFERENCES Neri G, Blumberg B, Miles PV, Opitz JM (1984): Sensorineural deaf-
ness in the FG syndrome: Report of four new cases. Am J Med Genet 19:369-377.
Opitz JM, Kaveggia EG (1974): Studies of malformation syndromes of man XXXIII: the FG syndrome: an X-linked recessive syndrome of multiple congenital anomalies and mental reteroletion. Z Kinder- heilk 117:l-18.
Opitz JM, Kaveggia EG, Adkins WN Jr, Gilbert EF, Vioeskul C, Pet- tersen J C , Blumberg B (1982): Studies of malformation syndromes of humans. XXXIII C: The FG syndrome-further studies on three affected individuals from the FG family. Am J Med Genet 12147- 154.
Opitz JM, Richieri-da Costa A, Aase JM, Benke PJ (1988): FG syn- drome update 1988: Note of 5 new patients and bibliography. Am J Med Genet 30:309-328.
Stratton RF, Parker MW (1989): Growth hormone deficiency, Wormian bones, dextrocardia, hrachycamptodactyly, and other midline de- fects. Am J Med Genet 32:169-173.
Thompson EM, Baraitser E (1987): Syndrome of the month: FG syn- drome. J Med Genet 24:139-143.
Thompson EM, Baraitser M, Lindenbaum RH (1985): The FG syn- drome: 7 new cases. Clin Genet 27:582-594.
Thompson EM, Harding BN, Lake BD, Smith SC (1986): Necropsy findings in a child with FG syndrome. J Med Genet 23:372-373.
Recommended