
S. Vajda, 2005
Computational mapping of proteins for fragment based drug design
Sandor Vajda, Spencer Thiel, Michael Silberstein, Melissa Landon, and David Lancia
Boston University, Boston, MA & SolMap Pharmaceuticals, Cambridge MA

S. Vajda, 2005
Dennis, S., Kortvelyesi T., and Vajda. S. Computational mapping identifies the binding sites of organic solvents on proteins. Proc. Natl. Acad. Sci. USA., 99: 4290-4295, 2002.
Silberstein, M., Dennis, S., Brown III, L., Kortvelyesi, T., Clodfelter, K., and Vajda, S. Identification of substrate binding sites in enzymes by computational solvent mapping, J. Molec. Biol. 332, 1095-1113, 2003.
Mattos C, Ringe D: Locating and characterizing binding sites on proteins. Nat. Biotechnol. (1996) 14(5):595-599.
Hajduk PJ, Huth JR, Fesik SW: Druggability indices for protein targets derived from NMR-based screening data. J Med Chem (2005) 48(7):2518-2525.
Small molecule binding druggability of the
binding site

S. Vajda, 2005
Computational Mapping Step 1: Placing the probes

S. Vajda, 2005
Step 2: Move the probes around to find binding positions

S. Vajda, 2005
Step 3: Remove high energy clusters of the ligand
S. Vajda, 2005
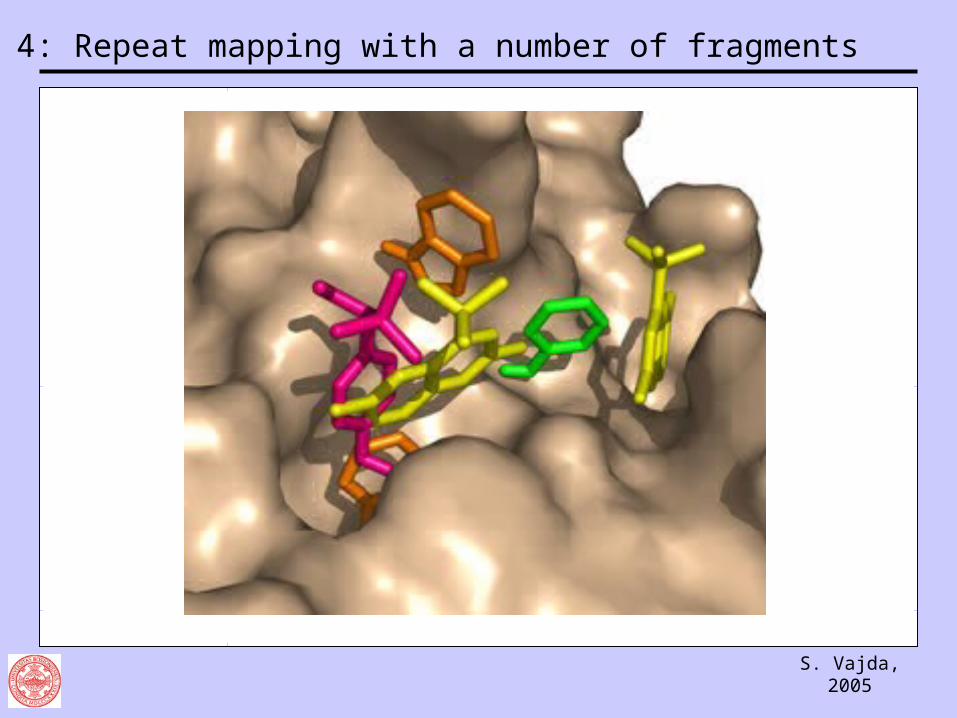
Step 4: Repeat mapping with a number of fragments
S. Vajda, 2005
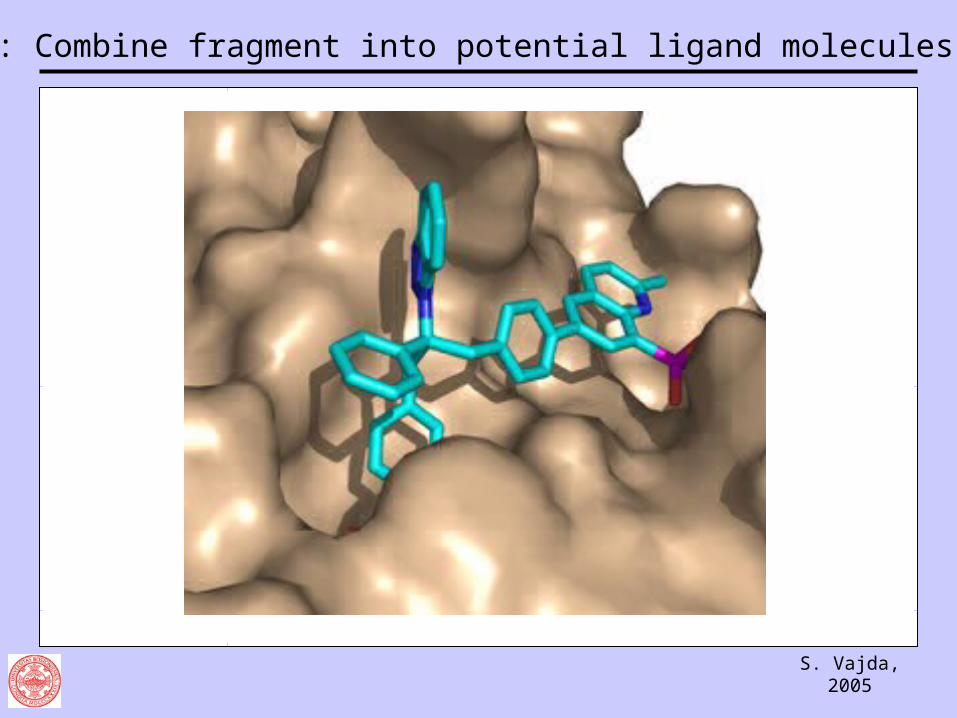
Step 5: Combine fragment into potential ligand molecules

S. Vajda, 2005
Why does CS-Map give better results than earlier methods ?
Properties:
• Improved sampling of the regions of interest• A scoring potential that accounts for desolvation• Clusters are ranked, not individual conformations• Consensus site: The binding of different solvents
reducesthe probability of finding false positives
Comparison to:
• Geometric: Flood-fill, PASS • Energetic: QsiteFinder, PocketFinder• Mapping/Docking: GRID, MCSS
S. Vajda, 2005
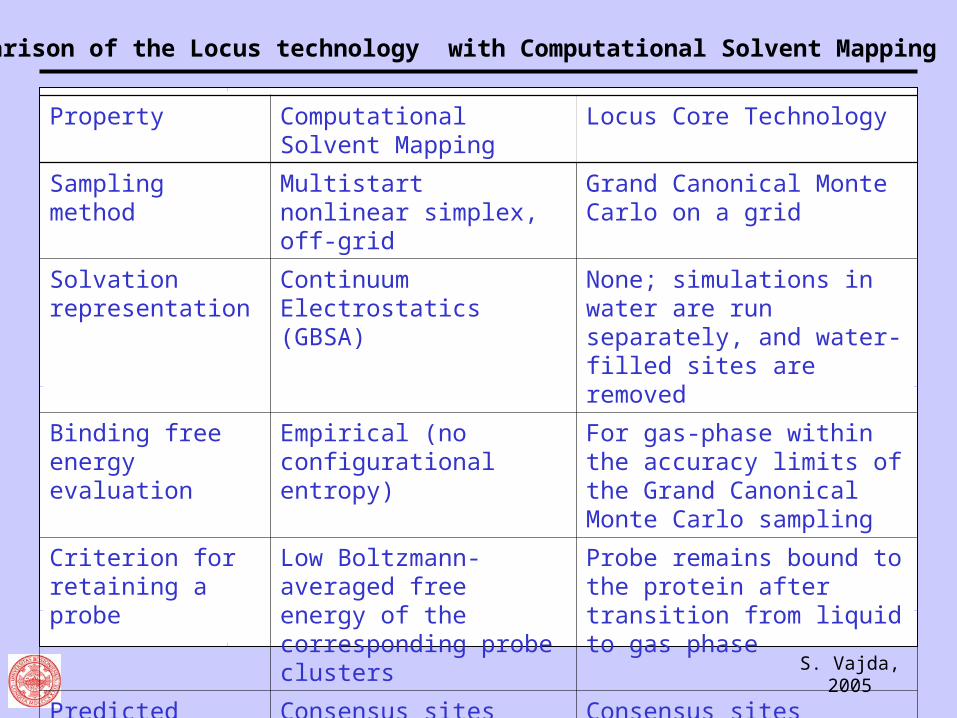
Comparison of the Locus technology with Computational Solvent Mapping
Property Computational Solvent Mapping
Locus Core Technology
Sampling method Multistart nonlinear simplex, off-grid
Grand Canonical Monte Carlo on a grid
Solvation representation
Continuum Electrostatics (GBSA)
None; simulations in water are run separately, and water-filled sites are removed
Binding free energy evaluation
Empirical (no configurational entropy)
For gas-phase within the accuracy limits of the Grand Canonical Monte Carlo sampling
Criterion for retaining a probe
Low Boltzmann-averaged free energy of the corresponding probe clusters
Probe remains bound to the protein after transition from liquid to gas phase
Predicted druggable binding sites
Consensus sites Consensus sites
CPU time About 1 hour About 7 days
S. Vajda, 2005
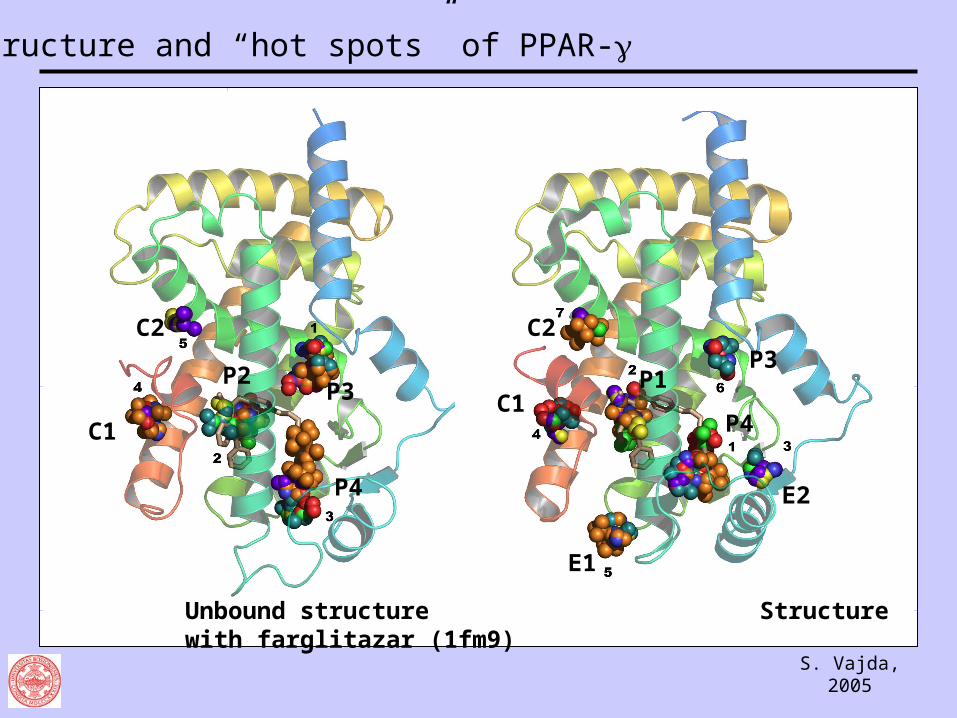
Unbound structure Structure with farglitazar (1fm9)
C2
C1
P2P3
P4
C2
C1
E1
P1P3
P4
E2
Structure and “hot spots” of PPAR-

S. Vajda, 2005
Structure and “hot spots” of PPAR-
Site Description
P1 Head group of agonists, interacting with helix H12
P2 Overlapping the middle of agonists
P3 Upper distal end of the binding site, reached only by the partial agonist
P4 Hydrophobic pocket close to the entrance
B Surface pocket in the back, overlapping the dimerization region
F Surface pocket of unknown role
C1 Surface pocket, possibly contributing to cofactor binding
C2 Overlapping with the binding site of the co-activator peptide SRC-1
E1 Pocket defined by the lower ends of helices H3, H7, and H10
E2 Putative ligand entrance between H2’ and the -sheet
H12
Sheu, S-H., Kaya, T., Waxman D. J., and Vajda, S. Exploring the binding site structure of the PPAR-g ligand binding domain by computational solvent mapping. Biochemistry, 44, 1193-1209, 2005.
S. Vajda, 2005
Current work: A prototype fragment library

S. Vajda, 2005
Credits
Poster: Hot spots in the binding site of renin
Vajda, S. and Guarnieri, F. Characterization of protein-ligand interaction sites using experimental and computational methods. Current Opinion in Drug Design and Development. In press (May 2006).
Dr. Sheldon DennisDr. Tamas Kortvelyesi Shu-Hsien SheuKarl Clodfelter
Dr. Dagmar Ringe (Brandeis University)
National Institute of Health National Institute of Environmental HealthNational Science FoundationSolMap Pharmaceuticals, Inc.
Recommended

















