
Protein Modelin
g

The Outline• Introduction•Comparative Modeling•Conclusion•References

Introduction

3D Structure of ProteinsStudying the 3D
Structure of Proteins
Understanding of Protein
Function
Drug Design Process
•The number of solved 3D structures increases slowly compared to the rate of sequencing DNA.
•Predictive methods have gained much interest.

3D Structure of ProteinsNatural Protein
Structure is guided by:
Laws of PhysicsChemical bonds,
H-bonds, electonic
interactions
Theory of Evolution
Families of similar proteins share
similar sequences,
structure, and often function
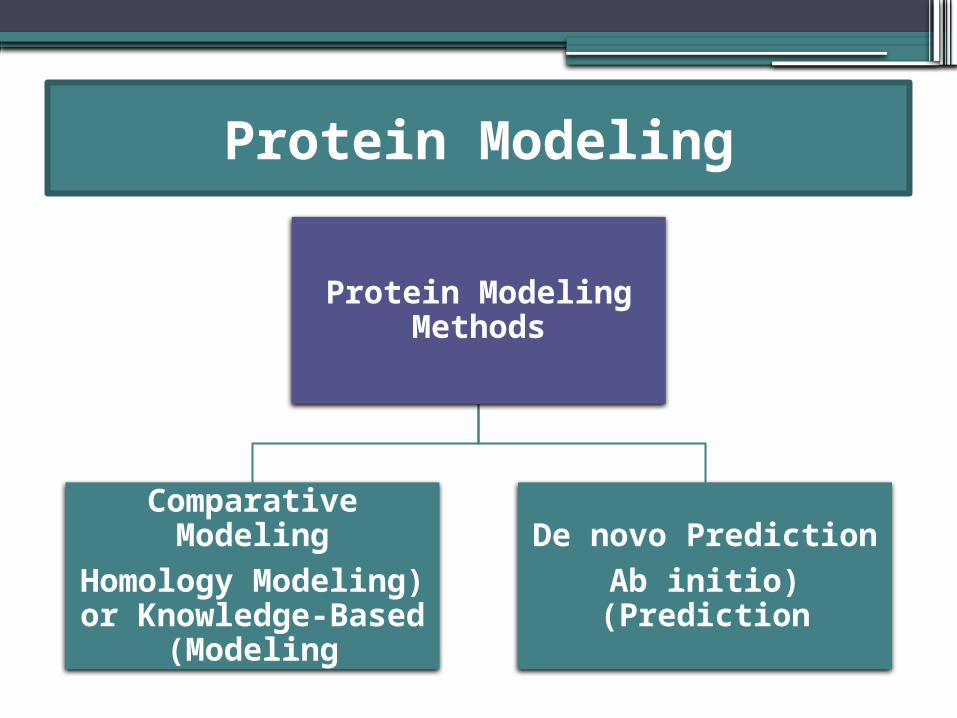
Protein Modeling
Protein Modeling Methods
Comparative Modeling
(Homology Modeling or Knowledge-Based
Modeling)
De novo Prediction(Ab initio
Prediction)

Comparative Modeling
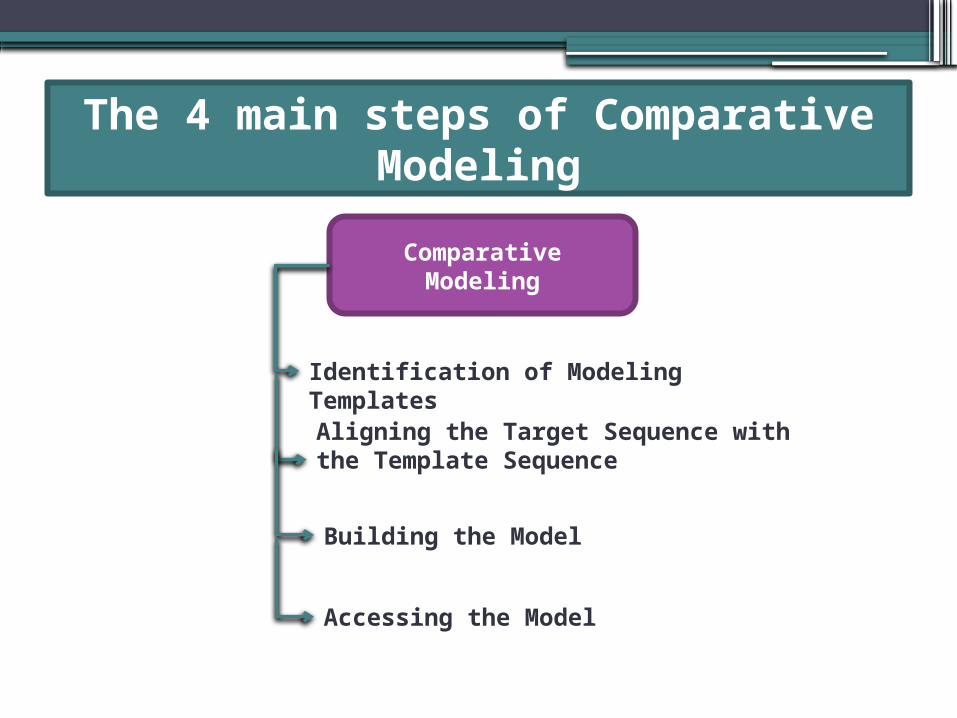
The 4 main steps of Comparative Modeling
Comparative Modeling
Identification of Modeling TemplatesAligning the Target Sequence with the Template Sequence
Building the Model
Accessing the Model

Identification of modeling templates
• It requires at least one sequence of known 3D structure with significant similarity to the target sequence.
•BLAST, PSI-BLAST, or HHSearch Programs are used.

Identification of modeling templates
The Reference=
The template with the highest sequence similarity
Maximizing the number of C∞ pairs in the common core while minimizing
their relative mean square deviation

Aligning the target sequence with the template sequence
•Using the best-scoring diagonals obtained by SIM.
•Residues which should not be used for model building will be ignored during the modeling process.

Building the model
•Framework Construction Averaging the position
of each atom in the
target sequence, based on
the location of the
corresponding atoms in the template
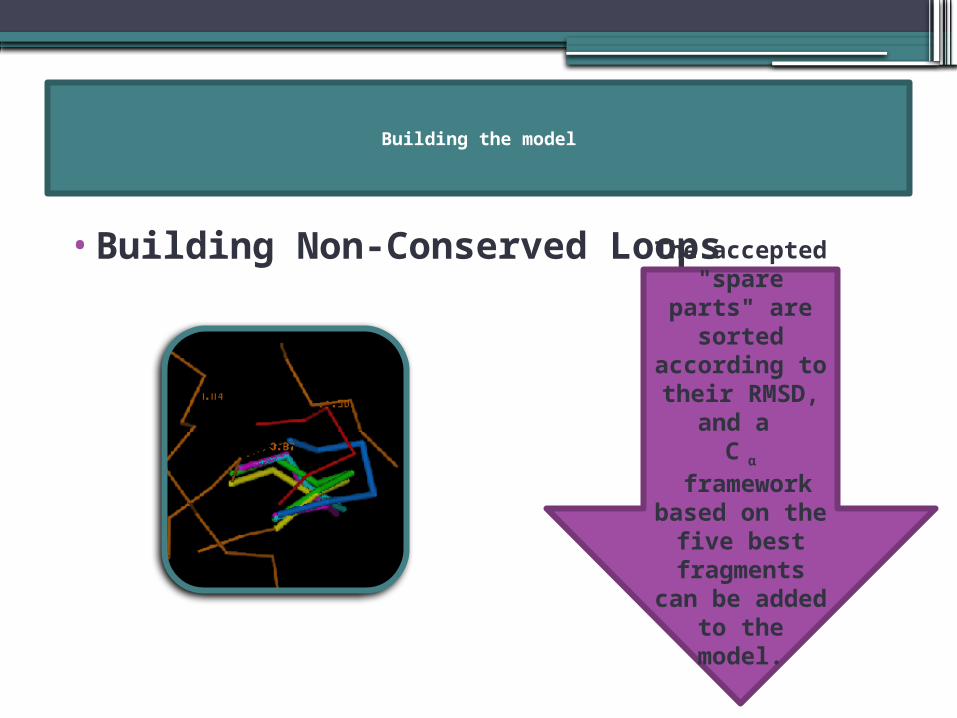
Building the model
•Building Non-Conserved Loops
WHY?
The accepted
"spare parts" are
sorted according to their RMSD,
and a C α
framework based on
the five best fragments
can be added to the
model.
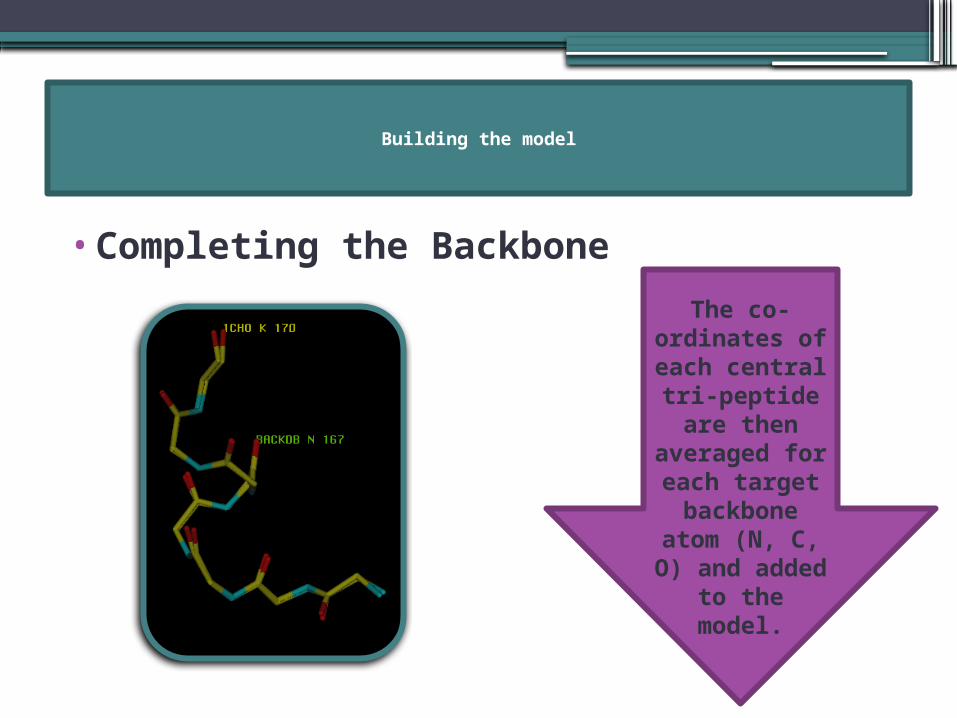
Building the model
•Completing the Backbone
The co-ordinates of each central tri-peptide are then
averaged for each target backbone
atom (N, C, O) and
added to the model.
Building the model
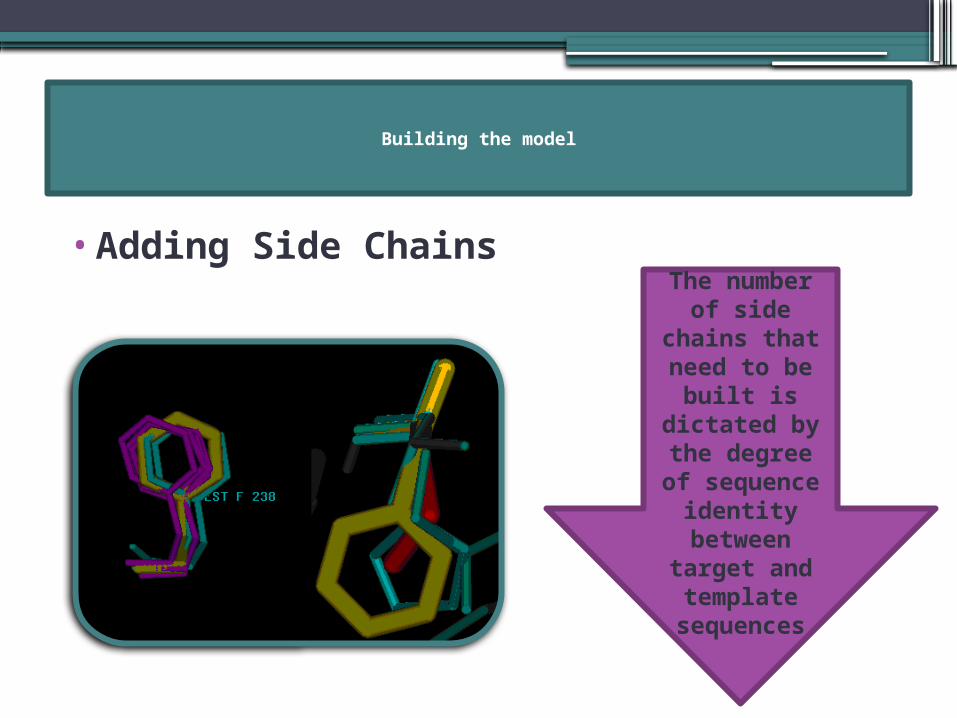
•Adding Side Chains
The number of side
chains that need to be
built is dictated by the degree of sequence
identity between
target and template
sequences

Building the model
•Model Refinement
Idealization of bond geometry and removal of
unfavorable non-bonded contacts
can be performed by
energy minimization
with force fields such as
CHARMM, AMBER or GROMOS.
Accessing the Model
•Removing Errors
PS: Error increases rapidly below 30% sequence similarity.

CONCLUSION

We got to know that
Comparative is more accurate…but requires other
structures to be known.

References• Baker, D.; Sali, A. Protein Structure Prediction and
Structural Genomics. Science 2001, 294, 93-96.
• Fiser, A.; Feig, M.; Brooks, C. L.; Sali, A. Evolution and Physics in Comparative Protein Structure Modeling. Acc. Chem. Res. 2002, 35, 413-421.
• Peitsch ;Guex.,N. Comparative protein modeling. GlaxoWellcome Experimental Research S.A. 16, chemin des Aulx 1228 Plan-les-Ouates / Switzerland.

QUESTIONS AND FEEDBACK

Thanks for
attending and
Listening
Recommended

















