
M A J O R A R T I C L E
Protecting Against Post-influenza BacterialPneumonia by Increasing PhagocyteRecruitment and ROS Production
Renuka Subramaniam,1 Peter F. Barnes,1 Kalyn Fletcher,1 Vijay Boggaram,2 Zachary Hillberry,1 Pierre Neuenschwander,2
and Homayoun Shams1
1Center for Pulmonary and Infectious Diseases Control (CPIDC), and 2Biomedical research, The University of Texas Health Science Center at Tyler,Tyler, Texas
Seasonal and especially pandemic influenza predispose patients to secondary bacterial pneumonias, which area major cause of deaths and morbidity. Staphylococcus aureus is a particularly common and deadly form ofpost-influenza pneumonia, and increasing staphylococcal drug resistance makes the development of new thera-pies urgent. We explored an innate immune-mediated model of the lung to define novel mechanisms by whichthe host can be protected against secondary staphylococcal pneumonia after sub-lethal influenza infection. Wefound that stimulating the innate immunity in the lung by overexpression of GM-CSF will result in resistanceto S. aureus pneumonia after sublethal influenza infection. Resistance was mediated by alveolar macrophagesand neutrophils, and was associated with increased production of reactive oxygen species (ROS) by alveolarmacrophages. Resistance was abrogated by treatment with agents that scavenged ROS. We conclude that stimu-lating innate immunity in the lung markedly reduces susceptibility to post-influenza staphylococcal pneumo-nia and that this may represent a novel immunomodulatory strategy for prevention and treatment of secondarybacterial pneumonia after influenza.
Keywords. Staphylococcus aureus; influenza; alveolar macrophages; ROS.
Influenza increases susceptibility to bacterial pneumo-nia from Staphylococcus aureus, Streptococcus pneumoniaeandHaemophilus influenzae, suggesting a post-influenzalocal immune defect [1]. Much of the morbidity andmortality from influenza results from bacterial superin-fection, particularly with S. aureus [1, 2], and bacterialcoinfection was confirmed in nearly all of 8398 autop-sies during the 1918 influenza pandemic [3, 4]. Duringthe 2009 influenza A pandemic, bacterial coinfectionoccurred in 18%–34% of cases that were admitted tothe intensive care unit [2, 5–8] and in up to 55% of fatalcases [9–11].
Studies in animal models have confirmed that influ-enza infection greatly lowers resistance to subsequent
bacterial pneumonia [12–14], due to several factors, in-cluding influenza-induced glucocorticoid and IL-10production, reduced neutrophil function, and increasedbacterial adherence [15–19]. In addition, IFN-γ pro-duced by lung T cells after influenza infection inhibitsthe capacity of alveolar macrophages (AMs) to clearmicrobes [12, 15–19]. Bacterial coinfection commonlyoccurs within the first 6 days of influenza, suggestingthat susceptibility results from defects in innate ratherthan adaptive immunity [12, 16, 18].
Granulocyte/macrophage colony-stimulating factor(GM-CSF) contributes to maturation of AMs [20],which are an important component of innate defensesagainst bacterial pneumonia [12, 21]. Administration ofGM-CSF to septic patients reversed monocyte immu-nosuppression and yielded clinical benefits [22]. Fur-thermore, mice that overexpress GM-CSF in the lungsarehighly resistant to influenza [23].Therefore,wedeter-mined if GM-CSF also protected against post-influenzasecondary bacterial pneumonia. We found that miceoverexpressing GM-CSF in the lungs are highly resis-tant to S. aureus pneumonia after sublethal influenza
Received 11 November 2013; accepted 12 December 2013; electronically pub-lished 23 December 2013.
Correspondence: Homayoun Shams ([email protected]).
The Journal of Infectious Diseases 2014;209:1827–36© The Author 2013. Published by Oxford University Press on behalf of the InfectiousDiseases Society of America. All rights reserved. For Permissions, please e-mail:[email protected]: 10.1093/infdis/jit830
Secondary Bacterial Pneumonia and ROS • JID 2014:209 (1 June) • 1827
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from

infection. This resistance is mediated through activating reac-tive oxygen species (ROS) in AMs and by enhancing ROS-independent mechanisms in neutrophils.
MATERIAL ANDMETHODS
MiceSix-eight week-old wild type C57BL/6 (WT) mice and trans-genic SPC-GM mice that lack systemic GM-CSF but constitu-tively overexpress GM-CSF only in the lung [24] were bred inthe vivarium at the University of Texas Health Science Centerat Tyler, as described [23]. SPC-GM mice were backcrossed toC57Bl/6 mice for >10 generations, and DNA typing confirmedthat they had the C57Bl/6 genetic markers. All experimentalprotocols were approved by the Institutional Animal Care andUse Committee.
Influenza AVirus (IAV)Mouse-adapted IAV/Puerto Rico/8/34 (PR8) H1N1 strain wasused in all experiments.
BacteriaStaphylococcus aureus Newman strain (ATCC 25904) was cul-tured overnight at 37°C in Brain heart infusion (BHI) brothsupplemented with 2% NaCl. Fresh bacterial cultures were pre-pared from overnight cultures and resuspended in PBS.
Cell DepletionClodronate- liposome and PBS-liposome were used to depletealveolar phagocytes. To deplete neutrophils, anti-Ly-6G (Gr-1,RB6-8C5, e-Biosciences), or IA 8 antibodies (BD Pharmingen)were used. T-cells were depleted by intraperitoneal administra-tion of anti-Thy1.2.
Quantitation of BacteriaMice were euthanized, bronchoalveolar lavage (BAL) fluid andlungs were collected aseptically and bacterial burden was deter-mined by counting colony forming units (CFU). Briefly, lungswere mechanically homogenized in 1 mL of 1X PBS on ice, andten-fold serial dilutions from lung homogenates and BALs wereplated on BHI agar plates. After overnight incubation, CFUwere counted and the number of bacteria in the originalsamples were calculated.
Total and Differential Cell CountBAL fluid was collected on day 0, and 6 days post- IAV infec-tion (DPV), and 1 and 2 days post-bacterial infection (DPB).Total cell counts were performed with an automated cellcounter. Cytospin preparations were stained with Wright-Giemsa and cells were differentiated morphologically by asingle laboratory technician who had no information about thesamples.
Quantitation of Albumin, Amphiregulin and CytokinesBAL fluid and lung homogenate were collected at different timepoints and stored at −80°C. Inteleukin-1β and tumor necrosisfactor –α levels were measured by enzyme-linked immunoassay(eBiosciences, CA). Albumin levels in BAL fluid and amphire-gulin levels in lung homogenates were measured by enzyme-linked immunoassays (Genway Biotech, CA and R&D systems,MN, respectively).
Quantitation of ROSBAL cells were incubated with 2′7′-dichlorofluorescein diace-tate (DCFDA. Abcam, MA) 20 µm of DCFDA for 30 minutesat 37°C, followed by staining with anti-CD11c and anti-Ly-6G(eBiosciences, CA) to identify AMs and neutrophils, respective-ly. Cells were subjected to flow cytometry (FACSCalibur, BD,CA) and analyzed using FlowJo Software (TreeStar, Inc).
Inhibiting ROS and NOS in vivoSPC-GM mice were given 500 µg/g of N-acetylcysteine solu-tion, USP 20% (APP Pharmaceuticals, Schaumburg, IL) intra-nasally, 3 and 14 hours before S. aureus infection, and byintraperitoneal injection, 1 and 2 days after S. aureus infection.Dimethythiourea (100 µg/g body weight) was given by intra-peritoneal injection one hour before S. aureus infection. L-NGmonomethyl L-arginine (L-NMMA, 80 µg/g body weight) wasgiven intraperitoneally, starting 2 hours before and daily forfour days after S. aureus infection. All animals were monitoredfor weight loss and mortality for at least four weeks after bacte-rial infection.
RESULTS
Murine Model of Primary and Secondary S. aureus PneumoniaWe established the published murine model of S. aureus pneu-monia [25] in 8–10-week-old WT mice, which received intra-nasal PBS or S. aureus Newman strain (ATCC), under mildgeneral anesthesia (ketamine and xylazine), as described [23].S. aureus caused dose-dependent mortality, beginning with10% mortality in mice infected with 108 organisms (Figure 1A).WT mice that received only 0.1 LD50 of IAV PR8 survived, butsecondary infection with low doses of S. aureus (2.5 × 108 bac-teria) killed all WT mice (Figure 1B).
Overexpression of GM-CSF in the Lungs Protects AgainstS. aureus PneumoniaWe used SPC-GM transgenic mice that constitutively overex-press GM-CSF only in the lung [24]. After infection withS. aureus (5 × 108 bacteria/mouse) intranasally, 40% of WTmice died, but all SPC-GM mice survived (Figure 1B). SPC-GM mice also survived infection with 109 bacteria/mouse (datanot shown). Next, we evaluated the effect of pulmonary GM-CSF on secondary S. aureus pneumonia. SPC-GM and WTmice were infected with a sublethal dose (0.1 LD50) of IAV
1828 • JID 2014:209 (1 June) • Subramaniam et al
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
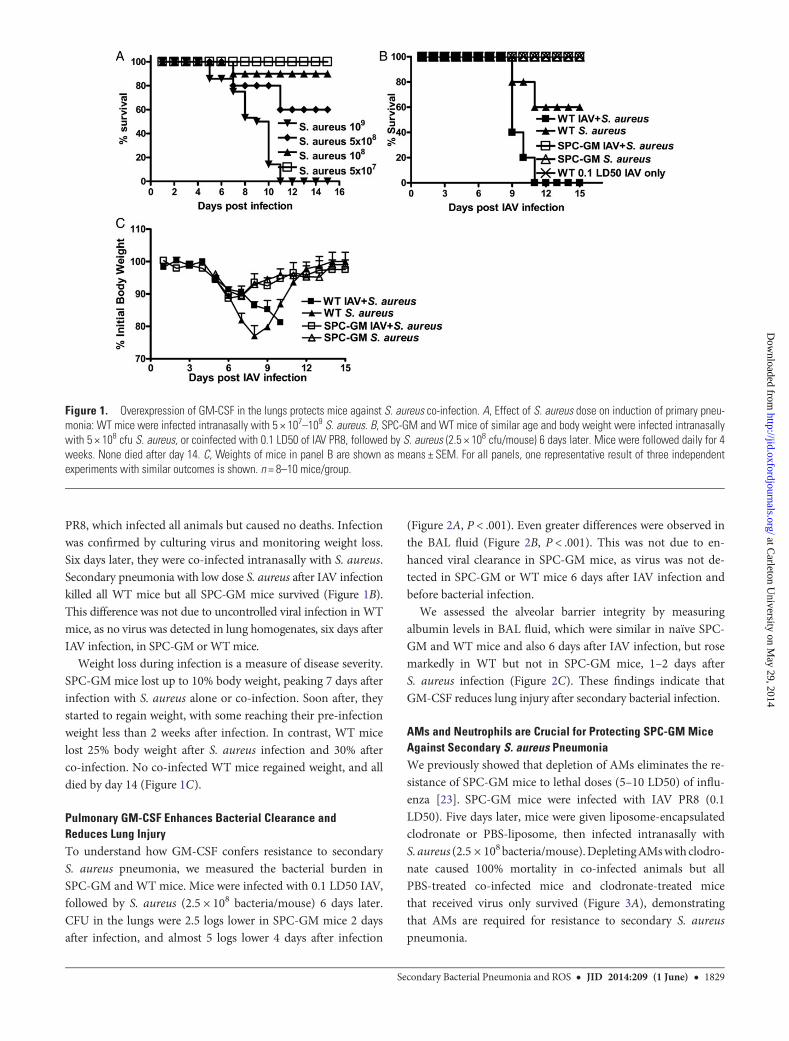
PR8, which infected all animals but caused no deaths. Infectionwas confirmed by culturing virus and monitoring weight loss.Six days later, they were co-infected intranasally with S. aureus.Secondary pneumonia with low dose S. aureus after IAV infectionkilled all WT mice but all SPC-GM mice survived (Figure 1B).This difference was not due to uncontrolled viral infection in WTmice, as no virus was detected in lung homogenates, six days afterIAV infection, in SPC-GM orWTmice.
Weight loss during infection is a measure of disease severity.SPC-GM mice lost up to 10% body weight, peaking 7 days afterinfection with S. aureus alone or co-infection. Soon after, theystarted to regain weight, with some reaching their pre-infectionweight less than 2 weeks after infection. In contrast, WT micelost 25% body weight after S. aureus infection and 30% afterco-infection. No co-infected WT mice regained weight, and alldied by day 14 (Figure 1C).
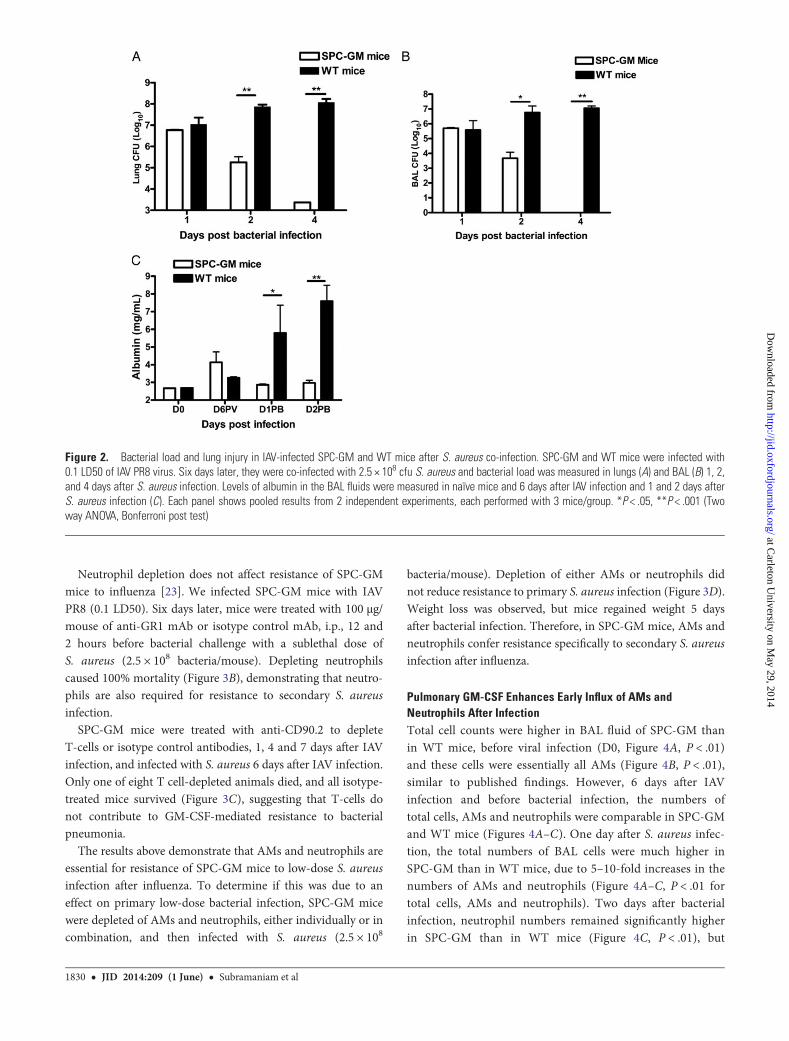
Pulmonary GM-CSF Enhances Bacterial Clearance andReduces Lung InjuryTo understand how GM-CSF confers resistance to secondaryS. aureus pneumonia, we measured the bacterial burden inSPC-GM and WT mice. Mice were infected with 0.1 LD50 IAV,followed by S. aureus (2.5 × 108 bacteria/mouse) 6 days later.CFU in the lungs were 2.5 logs lower in SPC-GM mice 2 daysafter infection, and almost 5 logs lower 4 days after infection
(Figure 2A, P < .001). Even greater differences were observed inthe BAL fluid (Figure 2B, P < .001). This was not due to en-hanced viral clearance in SPC-GM mice, as virus was not de-tected in SPC-GM or WT mice 6 days after IAV infection andbefore bacterial infection.
We assessed the alveolar barrier integrity by measuringalbumin levels in BAL fluid, which were similar in naïve SPC-GM and WT mice and also 6 days after IAV infection, but rosemarkedly in WT but not in SPC-GM mice, 1–2 days afterS. aureus infection (Figure 2C). These findings indicate thatGM-CSF reduces lung injury after secondary bacterial infection.
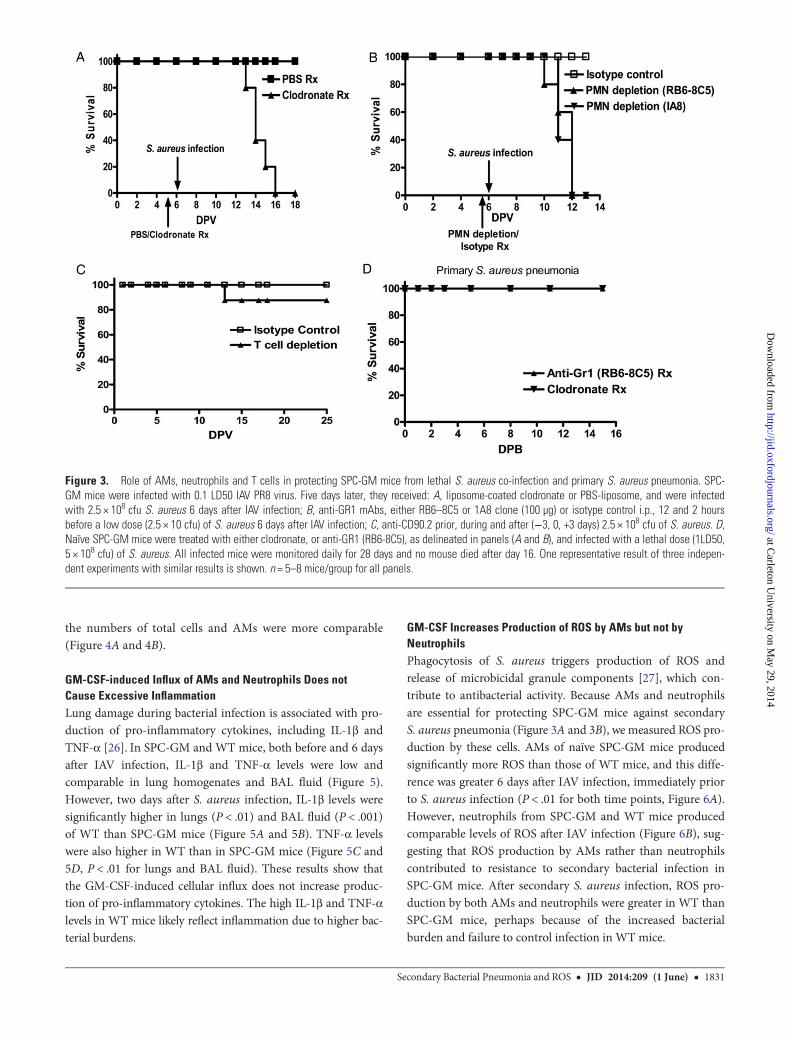
AMs and Neutrophils are Crucial for Protecting SPC-GM MiceAgainst Secondary S. aureus PneumoniaWe previously showed that depletion of AMs eliminates the re-sistance of SPC-GM mice to lethal doses (5–10 LD50) of influ-enza [23]. SPC-GM mice were infected with IAV PR8 (0.1LD50). Five days later, mice were given liposome-encapsulatedclodronate or PBS-liposome, then infected intranasally withS. aureus (2.5 × 108bacteria/mouse).DepletingAMswith clodro-nate caused 100% mortality in co-infected animals but allPBS-treated co-infected mice and clodronate-treated micethat received virus only survived (Figure 3A), demonstratingthat AMs are required for resistance to secondary S. aureuspneumonia.
Figure 1. Overexpression of GM-CSF in the lungs protects mice against S. aureus co-infection. A, Effect of S. aureus dose on induction of primary pneu-monia: WT mice were infected intranasally with 5 × 107–109 S. aureus. B, SPC-GM and WT mice of similar age and body weight were infected intranasallywith 5 × 108 cfu S. aureus, or coinfected with 0.1 LD50 of IAV PR8, followed by S. aureus (2.5 × 108 cfu/mouse) 6 days later. Mice were followed daily for 4weeks. None died after day 14. C, Weights of mice in panel B are shown as means ± SEM. For all panels, one representative result of three independentexperiments with similar outcomes is shown. n = 8–10 mice/group.
Secondary Bacterial Pneumonia and ROS • JID 2014:209 (1 June) • 1829
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
Neutrophil depletion does not affect resistance of SPC-GMmice to influenza [23]. We infected SPC-GM mice with IAVPR8 (0.1 LD50). Six days later, mice were treated with 100 µg/mouse of anti-GR1 mAb or isotype control mAb, i.p., 12 and2 hours before bacterial challenge with a sublethal dose ofS. aureus (2.5 × 108 bacteria/mouse). Depleting neutrophilscaused 100% mortality (Figure 3B), demonstrating that neutro-phils are also required for resistance to secondary S. aureusinfection.
SPC-GM mice were treated with anti-CD90.2 to depleteT-cells or isotype control antibodies, 1, 4 and 7 days after IAVinfection, and infected with S. aureus 6 days after IAV infection.Only one of eight T cell-depleted animals died, and all isotype-treated mice survived (Figure 3C), suggesting that T-cells donot contribute to GM-CSF-mediated resistance to bacterialpneumonia.
The results above demonstrate that AMs and neutrophils areessential for resistance of SPC-GM mice to low-dose S. aureusinfection after influenza. To determine if this was due to aneffect on primary low-dose bacterial infection, SPC-GM micewere depleted of AMs and neutrophils, either individually or incombination, and then infected with S. aureus (2.5 × 108
bacteria/mouse). Depletion of either AMs or neutrophils didnot reduce resistance to primary S. aureus infection (Figure 3D).Weight loss was observed, but mice regained weight 5 daysafter bacterial infection. Therefore, in SPC-GM mice, AMs andneutrophils confer resistance specifically to secondary S. aureusinfection after influenza.
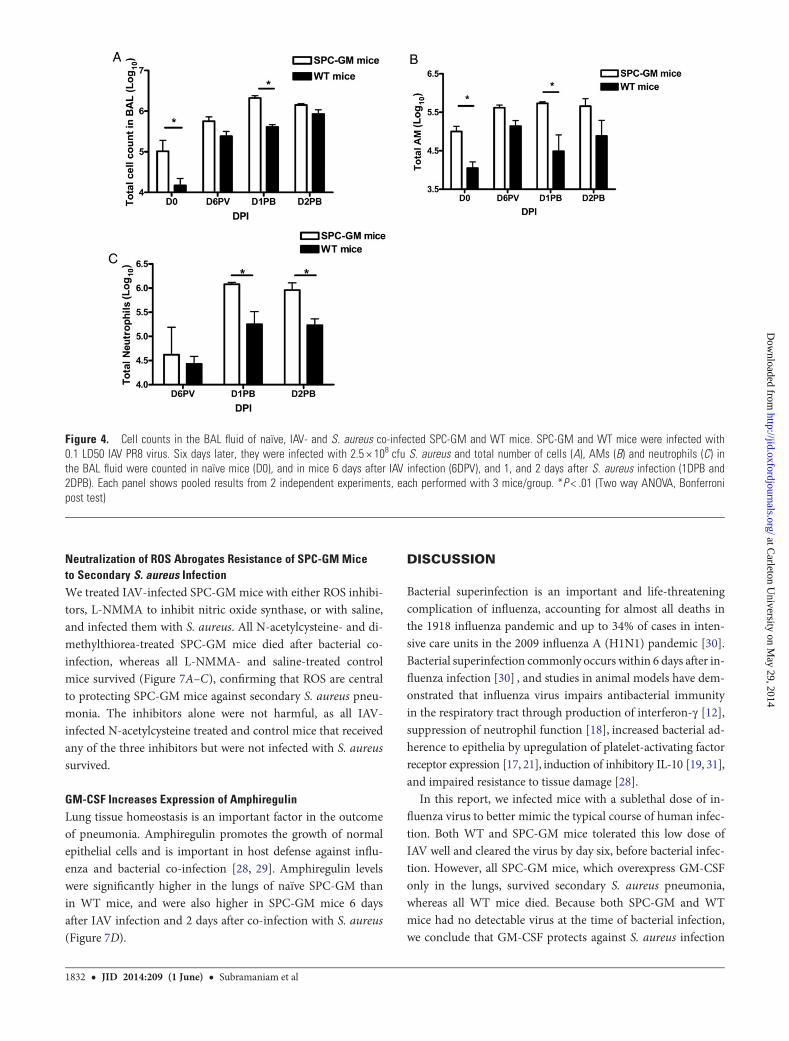
Pulmonary GM-CSF Enhances Early Influx of AMs andNeutrophils After InfectionTotal cell counts were higher in BAL fluid of SPC-GM thanin WT mice, before viral infection (D0, Figure 4A, P < .01)and these cells were essentially all AMs (Figure 4B, P < .01),similar to published findings. However, 6 days after IAVinfection and before bacterial infection, the numbers oftotal cells, AMs and neutrophils were comparable in SPC-GMand WT mice (Figures 4A–C). One day after S. aureus infec-tion, the total numbers of BAL cells were much higher inSPC-GM than in WT mice, due to 5–10-fold increases in thenumbers of AMs and neutrophils (Figure 4A–C, P < .01 fortotal cells, AMs and neutrophils). Two days after bacterialinfection, neutrophil numbers remained significantly higherin SPC-GM than in WT mice (Figure 4C, P < .01), but
Figure 2. Bacterial load and lung injury in IAV-infected SPC-GM and WT mice after S. aureus co-infection. SPC-GM and WT mice were infected with0.1 LD50 of IAV PR8 virus. Six days later, they were co-infected with 2.5 × 108 cfu S. aureus and bacterial load was measured in lungs (A) and BAL (B) 1, 2,and 4 days after S. aureus infection. Levels of albumin in the BAL fluids were measured in naïve mice and 6 days after IAV infection and 1 and 2 days afterS. aureus infection (C). Each panel shows pooled results from 2 independent experiments, each performed with 3 mice/group. *P < .05, **P < .001 (Twoway ANOVA, Bonferroni post test)
1830 • JID 2014:209 (1 June) • Subramaniam et al
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
the numbers of total cells and AMs were more comparable(Figure 4A and 4B).
GM-CSF-induced Influx of AMs and Neutrophils Does notCause Excessive InflammationLung damage during bacterial infection is associated with pro-duction of pro-inflammatory cytokines, including IL-1β andTNF-α [26]. In SPC-GM and WT mice, both before and 6 daysafter IAV infection, IL-1β and TNF-α levels were low andcomparable in lung homogenates and BAL fluid (Figure 5).However, two days after S. aureus infection, IL-1β levels weresignificantly higher in lungs (P < .01) and BAL fluid (P < .001)of WT than SPC-GM mice (Figure 5A and 5B). TNF-α levelswere also higher in WT than in SPC-GM mice (Figure 5C and5D, P < .01 for lungs and BAL fluid). These results show thatthe GM-CSF-induced cellular influx does not increase produc-tion of pro-inflammatory cytokines. The high IL-1β and TNF-αlevels in WT mice likely reflect inflammation due to higher bac-terial burdens.
GM-CSF Increases Production of ROS by AMs but not byNeutrophilsPhagocytosis of S. aureus triggers production of ROS andrelease of microbicidal granule components [27], which con-tribute to antibacterial activity. Because AMs and neutrophilsare essential for protecting SPC-GM mice against secondaryS. aureus pneumonia (Figure 3A and 3B), we measured ROS pro-duction by these cells. AMs of naïve SPC-GM mice producedsignificantly more ROS than those of WT mice, and this diffe-rence was greater 6 days after IAV infection, immediately priorto S. aureus infection (P < .01 for both time points, Figure 6A).However, neutrophils from SPC-GM and WT mice producedcomparable levels of ROS after IAV infection (Figure 6B), sug-gesting that ROS production by AMs rather than neutrophilscontributed to resistance to secondary bacterial infection inSPC-GM mice. After secondary S. aureus infection, ROS pro-duction by both AMs and neutrophils were greater in WT thanSPC-GM mice, perhaps because of the increased bacterialburden and failure to control infection in WT mice.
Figure 3. Role of AMs, neutrophils and T cells in protecting SPC-GM mice from lethal S. aureus co-infection and primary S. aureus pneumonia. SPC-GM mice were infected with 0.1 LD50 IAV PR8 virus. Five days later, they received: A, liposome-coated clodronate or PBS-liposome, and were infectedwith 2.5 × 108 cfu S. aureus 6 days after IAV infection; B, anti-GR1 mAbs, either RB6–8C5 or 1A8 clone (100 µg) or isotype control i.p., 12 and 2 hoursbefore a low dose (2.5 × 10 cfu) of S. aureus 6 days after IAV infection; C, anti-CD90.2 prior, during and after (−3, 0, +3 days) 2.5 × 108 cfu of S. aureus. D,Naïve SPC-GM mice were treated with either clodronate, or anti-GR1 (RB6-8C5), as delineated in panels (A and B), and infected with a lethal dose (1LD50,5 × 108 cfu) of S. aureus. All infected mice were monitored daily for 28 days and no mouse died after day 16. One representative result of three indepen-dent experiments with similar results is shown. n = 5–8 mice/group for all panels.
Secondary Bacterial Pneumonia and ROS • JID 2014:209 (1 June) • 1831
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
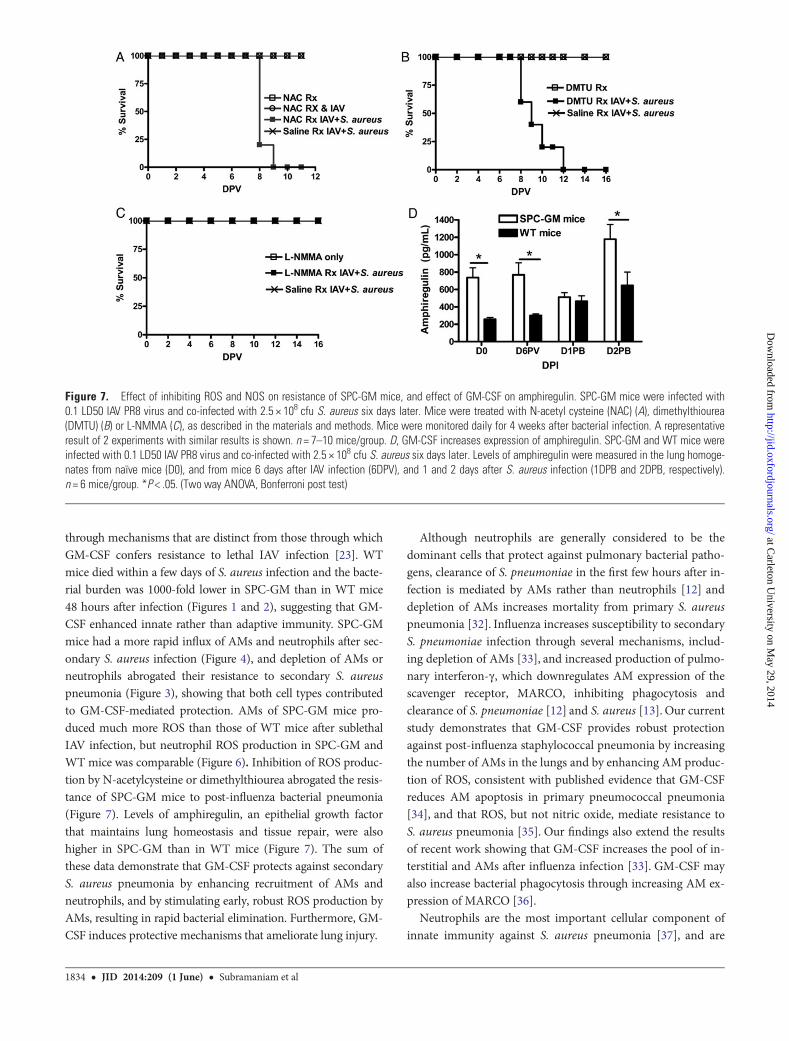
Neutralization of ROS Abrogates Resistance of SPC-GM Miceto Secondary S. aureus InfectionWe treated IAV-infected SPC-GM mice with either ROS inhibi-tors, L-NMMA to inhibit nitric oxide synthase, or with saline,and infected them with S. aureus. All N-acetylcysteine- and di-methylthiorea-treated SPC-GM mice died after bacterial co-infection, whereas all L-NMMA- and saline-treated controlmice survived (Figure 7A–C), confirming that ROS are centralto protecting SPC-GM mice against secondary S. aureus pneu-monia. The inhibitors alone were not harmful, as all IAV-infected N-acetylcysteine treated and control mice that receivedany of the three inhibitors but were not infected with S. aureussurvived.
GM-CSF Increases Expression of AmphiregulinLung tissue homeostasis is an important factor in the outcomeof pneumonia. Amphiregulin promotes the growth of normalepithelial cells and is important in host defense against influ-enza and bacterial co-infection [28, 29]. Amphiregulin levelswere significantly higher in the lungs of naïve SPC-GM thanin WT mice, and were also higher in SPC-GM mice 6 daysafter IAV infection and 2 days after co-infection with S. aureus(Figure 7D).
DISCUSSION
Bacterial superinfection is an important and life-threateningcomplication of influenza, accounting for almost all deaths inthe 1918 influenza pandemic and up to 34% of cases in inten-sive care units in the 2009 influenza A (H1N1) pandemic [30].Bacterial superinfection commonly occurs within 6 days after in-fluenza infection [30] , and studies in animal models have dem-onstrated that influenza virus impairs antibacterial immunityin the respiratory tract through production of interferon-γ [12],suppression of neutrophil function [18], increased bacterial ad-herence to epithelia by upregulation of platelet-activating factorreceptor expression [17, 21], induction of inhibitory IL-10 [19, 31],and impaired resistance to tissue damage [28].
In this report, we infected mice with a sublethal dose of in-fluenza virus to better mimic the typical course of human infec-tion. Both WT and SPC-GM mice tolerated this low dose ofIAV well and cleared the virus by day six, before bacterial infec-tion. However, all SPC-GM mice, which overexpress GM-CSFonly in the lungs, survived secondary S. aureus pneumonia,whereas all WT mice died. Because both SPC-GM and WTmice had no detectable virus at the time of bacterial infection,we conclude that GM-CSF protects against S. aureus infection
Figure 4. Cell counts in the BAL fluid of naïve, IAV- and S. aureus co-infected SPC-GM and WT mice. SPC-GM and WT mice were infected with0.1 LD50 IAV PR8 virus. Six days later, they were infected with 2.5 × 108 cfu S. aureus and total number of cells (A), AMs (B) and neutrophils (C) inthe BAL fluid were counted in naïve mice (D0), and in mice 6 days after IAV infection (6DPV), and 1, and 2 days after S. aureus infection (1DPB and2DPB). Each panel shows pooled results from 2 independent experiments, each performed with 3 mice/group. *P < .01 (Two way ANOVA, Bonferronipost test)
1832 • JID 2014:209 (1 June) • Subramaniam et al
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
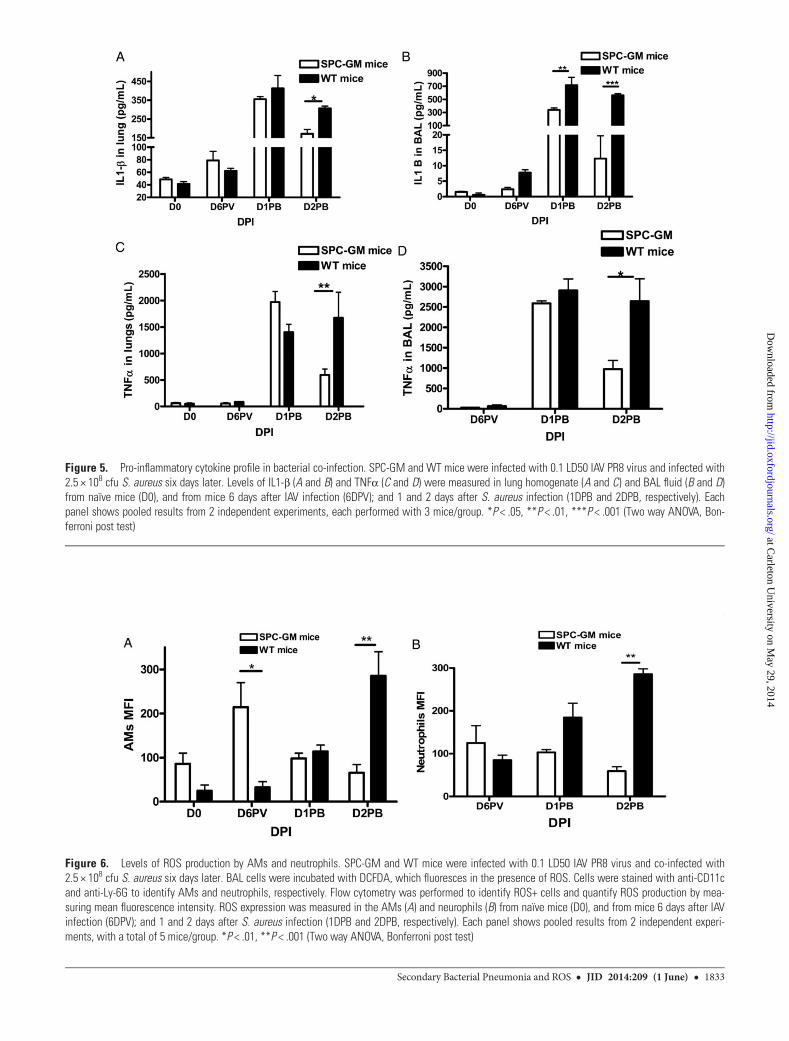
Figure 6. Levels of ROS production by AMs and neutrophils. SPC-GM and WT mice were infected with 0.1 LD50 IAV PR8 virus and co-infected with2.5 × 108 cfu S. aureus six days later. BAL cells were incubated with DCFDA, which fluoresces in the presence of ROS. Cells were stained with anti-CD11cand anti-Ly-6G to identify AMs and neutrophils, respectively. Flow cytometry was performed to identify ROS+ cells and quantify ROS production by mea-suring mean fluorescence intensity. ROS expression was measured in the AMs (A) and neurophils (B) from naïve mice (D0), and from mice 6 days after IAVinfection (6DPV); and 1 and 2 days after S. aureus infection (1DPB and 2DPB, respectively). Each panel shows pooled results from 2 independent experi-ments, with a total of 5 mice/group. *P < .01, **P < .001 (Two way ANOVA, Bonferroni post test)
Figure 5. Pro-inflammatory cytokine profile in bacterial co-infection. SPC-GM and WT mice were infected with 0.1 LD50 IAV PR8 virus and infected with2.5 × 108 cfu S. aureus six days later. Levels of IL1-β (A and B) and TNFα (C and D) were measured in lung homogenate (A and C) and BAL fluid (B and D)from naïve mice (D0), and from mice 6 days after IAV infection (6DPV); and 1 and 2 days after S. aureus infection (1DPB and 2DPB, respectively). Eachpanel shows pooled results from 2 independent experiments, each performed with 3 mice/group. *P < .05, **P < .01, ***P < .001 (Two way ANOVA, Bon-ferroni post test)
Secondary Bacterial Pneumonia and ROS • JID 2014:209 (1 June) • 1833
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
through mechanisms that are distinct from those through whichGM-CSF confers resistance to lethal IAV infection [23]. WTmice died within a few days of S. aureus infection and the bacte-rial burden was 1000-fold lower in SPC-GM than in WT mice48 hours after infection (Figures 1 and 2), suggesting that GM-CSF enhanced innate rather than adaptive immunity. SPC-GMmice had a more rapid influx of AMs and neutrophils after sec-ondary S. aureus infection (Figure 4), and depletion of AMs orneutrophils abrogated their resistance to secondary S. aureuspneumonia (Figure 3), showing that both cell types contributedto GM-CSF-mediated protection. AMs of SPC-GM mice pro-duced much more ROS than those of WT mice after sublethalIAV infection, but neutrophil ROS production in SPC-GM andWT mice was comparable (Figure 6). Inhibition of ROS produc-tion by N-acetylcysteine or dimethylthiourea abrogated the resis-tance of SPC-GM mice to post-influenza bacterial pneumonia(Figure 7). Levels of amphiregulin, an epithelial growth factorthat maintains lung homeostasis and tissue repair, were alsohigher in SPC-GM than in WT mice (Figure 7). The sum ofthese data demonstrate that GM-CSF protects against secondaryS. aureus pneumonia by enhancing recruitment of AMs andneutrophils, and by stimulating early, robust ROS production byAMs, resulting in rapid bacterial elimination. Furthermore, GM-CSF induces protective mechanisms that ameliorate lung injury.
Although neutrophils are generally considered to be thedominant cells that protect against pulmonary bacterial patho-gens, clearance of S. pneumoniae in the first few hours after in-fection is mediated by AMs rather than neutrophils [12] anddepletion of AMs increases mortality from primary S. aureuspneumonia [32]. Influenza increases susceptibility to secondaryS. pneumoniae infection through several mechanisms, includ-ing depletion of AMs [33], and increased production of pulmo-nary interferon-γ, which downregulates AM expression of thescavenger receptor, MARCO, inhibiting phagocytosis andclearance of S. pneumoniae [12] and S. aureus [13]. Our currentstudy demonstrates that GM-CSF provides robust protectionagainst post-influenza staphylococcal pneumonia by increasingthe number of AMs in the lungs and by enhancing AM produc-tion of ROS, consistent with published evidence that GM-CSFreduces AM apoptosis in primary pneumococcal pneumonia[34], and that ROS, but not nitric oxide, mediate resistance toS. aureus pneumonia [35]. Our findings also extend the resultsof recent work showing that GM-CSF increases the pool of in-terstitial and AMs after influenza infection [33]. GM-CSF mayalso increase bacterial phagocytosis through increasing AM ex-pression of MARCO [36].
Neutrophils are the most important cellular component ofinnate immunity against S. aureus pneumonia [37], and are
Figure 7. Effect of inhibiting ROS and NOS on resistance of SPC-GM mice, and effect of GM-CSF on amphiregulin. SPC-GM mice were infected with0.1 LD50 IAV PR8 virus and co-infected with 2.5 × 108 cfu S. aureus six days later. Mice were treated with N-acetyl cysteine (NAC) (A), dimethylthiourea(DMTU) (B) or L-NMMA (C), as described in the materials and methods. Mice were monitored daily for 4 weeks after bacterial infection. A representativeresult of 2 experiments with similar results is shown. n = 7–10 mice/group. D, GM-CSF increases expression of amphiregulin. SPC-GM and WT mice wereinfected with 0.1 LD50 IAV PR8 virus and co-infected with 2.5 × 108 cfu S. aureus six days later. Levels of amphiregulin were measured in the lung homoge-nates from naïve mice (D0), and from mice 6 days after IAV infection (6DPV), and 1 and 2 days after S. aureus infection (1DPB and 2DPB, respectively).n = 6 mice/group. *P < .05. (Two way ANOVA, Bonferroni post test)
1834 • JID 2014:209 (1 June) • Subramaniam et al
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from

recruited to the lung rapidly, where they eliminate bacteriathrough production of ROS and through non-oxidative mecha-nisms, including antimicrobial peptides, cathelicidins, cathep-sins and proteases [37]. We found that neutrophils wereessential for resistance of SPC-GM mice to post-influenzastaphylococcal pneumonia (Figure 3B). However, neutrophilROS expression was not increased in SPC-GMmice (Figure 6B),indicating that GM-CSF elicited neutrophil-dependent protec-tion through other mechanisms. GM-CSF substantially in-creased the number of neutrophils in the lungs, 24–48 hoursafter secondary S. aureus infection (Figure 4C), probablythrough a combination of increased recruitment and delayedapoptosis [38, 39], which enhance antibacterial activity. GM-CSF may also boost granule-dependent bactericidal mecha-nisms, as influenza virus inhibits fusion of neutrophil azurophilgranules with phagosomes containing S. aureus, reducing bac-terial killing [40], and GM-CSF increases neutrophil expressionof cathepsin C [41], which enhances killing of S. aureus [42].
Intrapulmonary delivery of GM-CSF reduced bacterialburdens during primary infection with less invasive strains ofS. pneumoniae [34]. GM-CSF also ameliorated AM depletionduring influenza infection, and reduced the incidence of sec-ondary pneumococcal pneumonia [33]. Our current findingsprovide the first evidence that GM-CSF markedly reduces mor-bidity and mortality from secondary staphylococcal pneumoniapost-influenza. Clinical and laboratory studies suggest that dif-ferent mechanisms mediate susceptibility to S. aureus andS. pneumoniae after influenza infection. The pneumococcus isthe most common bacterial cause of community-acquired pneu-monia, whereas S. aureus causes only 2%–5% of cases [43]. Incontrast, after influenza infection, S. aureus is much morecommon, and was isolated from 58% of cases of bacterial pneu-monias in critically ill children with 2009 pandemic influenzaA H1NI, compared to 12% for S. pneumoniae [44]. This sug-gests that influenza increases susceptibility to S. aureus to agreater extent than to S. pneumoniae, and studies in murinemodels suggest different mechanisms for susceptibility to thesebacteria [12, 19, 31, 45]. Given its pleiotropic effects, GM-CSFmay act through distinct mechanisms to protect against differ-ent bacteria. GM-CSF increased macrophage but not neutro-phil recruitment to the lung, and increased expression of nitricoxide in response to S. pneumoniae infection [34]. In contrast,we found that GM-CSF increased both macrophage and neu-trophil numbers after secondary S. aureus infection, and pro-tection was mediated by ROS rather than nitric oxide. Ourresults extend previous findings and delineate additional mech-anisms by which GM-CSF protects against microbial infection.
The severity of lung disease from bacterial infection dependsnot only on the microbial burden but on the severity of inflam-mation. The S. aureus α-hemolysin increases lung necrosis byactivating the nucleotide-binding domain and leucine-richrepeat containing gene family, pyrin domain containing 3
inflammasome without increasing the bacterial load [46]. Fur-thermore, S. aureus strains that cause more severe disease post-influenza elicit more inflammation and necrosis without higherbacterial numbers, whereas the virulence of S. pneumoniaestrains correlates more closely with bacterial burden [14]. Post-influenza, lung innate lymphoid cells produce amphiregulin,which is essential for restoring epithelial integrity and tissuerepair [29], and for preventing lung damage from secondary bac-terial infection with Legionella pneumophila [28, 29]. We presentthe first evidence that GM-CSF in the lung increases expressionof amphiregulin, providing an additional potential mechanismto reduce the severity of secondary bacterial pneumonia. Thismay be particularly important because GM-CSF also favors re-cruitment of AMs and neutrophils, and stimulates production ofROS, which can cause inflammation and cellular toxicity.
Because GM-CSF acts by stimulating innate immunity, itshould act against multiple microbial pathogens. If our resultsin mice also hold in humans, GM-CSF could be used to treat orprevent post-influenza bacterial pneumonia. GM-CSF is ap-proved by the FDA and aerosolized GM-CSF effectively treatsalveolar proteinosis in humans [47–49]. Also, aerosolized drugsare used to treat pneumonia, indicating the feasibility of usingaerosolized GM-CSF to treat pulmonary infections.
Notes
Acknowledgments. We thank Dr Amy R. Tvinnereim for her assistancein Flow Cytometry and animal studies.Financial support. This work was in part supported by grants from the
Flight Attendant Medical Research Institute (092015-Clinical InnovatorAward), to H. Shams, and the Cain Foundation for Infectious DiseaseResearch.Potential conflicts of interest. H. S. is the inventor of two pending
patents filed by the Board Of Regents, The University of Texas System, foruse of GMCSF to prevent influenza and its secondary bacterial pneumonia.All authors have submitted the ICMJE Form for Disclosure of Potential
Conflicts of Interest. Conflicts that the editors consider relevant to thecontent of the manuscript have been disclosed.
References
1. Brundage JF. Interactions between influenza and bacterial respiratorypathogens: implications for pandemic preparedness. Lancet Infect Dis2006; 6:303–12.
2. Kumar A, Zarychanski R, Pinto R, et al. Critically ill patients with 2009influenza A(H1N1) infection in Canada. JAMA 2009; 302:1872–9.
3. Johnson NP, Mueller J. Updating the accounts: global mortality of the1918–1920 “Spanish” influenza pandemic. Bull Hist Med 2002; 76:105–15.
4. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterialpneumonia as a cause of death in pandemic influenza: implications forpandemic influenza preparedness. J Infect Dis 2008; 198:962–70.
5. Estenssoro E, Rios FG, Apezteguia C, et al. Pandemic 2009 influenza Ain Argentina: a study of 337 patients on mechanical ventilation. Am JRespir Crit Care Med 2010; 182:41–8.
6. Farias JA, Fernandez A, Monteverde E, et al. Critically ill infants andchildren with influenza A (H1N1) in pediatric intensive care units inArgentina. Intensive Care Med 2010; 36:1015–22.
7. Rice TW, Rubinson L, Uyeki TM, et al. Critical illness from 2009 pan-demic influenza A virus and bacterial coinfection in the United States.Crit Care Med 2012; 40:1487–98.
Secondary Bacterial Pneumonia and ROS • JID 2014:209 (1 June) • 1835
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from

8. Webb SA, Pettila V, Seppelt I, et al. Critical care services and 2009 H1N1influenza in Australia and New Zealand. N Engl J Med 2009; 361:1925–34.
9. Bacterial coinfections in lung tissue specimens from fatal cases of 2009pandemic influenza A (H1N1) - United States, May-August 2009.MMWRMorb Mortal Wkly Rep 2009; 58:1071–4.
10. Gill JR, Sheng ZM, Ely SF, et al. Pulmonary pathologic findings of fatal2009 pandemic influenza A/H1N1 viral infections. Arch Pathol LabMed 2010; 134:235–43.
11. Mauad T, Hajjar LA, Callegari GD, et al. Lung pathology in fatal novelhuman influenza A (H1N1) infection. Am J Respir Crit Care Med 2010;181:72–9.
12. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense byinterferon-gamma during recovery from influenza infection. Nat Med2008; 14:558–64.
13. Hang do TT, Choi EJ, Song JY, Kim SE, Kwak J, Shin YK. Differentialeffect of prior influenza infection on alveolar macrophage phagocytosisof Staphylococcus aureus and Escherichia coli: involvement of interfer-on-gamma production. Microbiol Immunol 2011; 55:751–9.
14. Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA.Influenza virus primes mice for pneumonia from Staphylococcusaureus. J Infect Dis 2011; 203:880–8.
15. Hall MW, Geyer SM, Guo CY, et al. Innate immune function and mor-tality in critically ill children with influenza: a multicenter study. CritCare Med 2013; 41:224–36.
16. Jamieson AM, Yu S, Annicelli CH, Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a sec-ondary bacterial infection. Cell Host Microbe 2010; 7:103–14.
17. McCullers JA, Rehg JE. Lethal synergism between influenza virus andStreptococcus pneumoniae: characterization of a mouse model and therole of platelet-activating factor receptor. J Infect Dis 2002; 186:341–50.
18. McNamee LA, Harmsen AG. Both influenza-induced neutrophildysfunction and neutrophil-independent mechanisms contribute toincreased susceptibility to a secondary Streptococcus pneumoniaeinfection. Infect Immun 2006; 74:6707–21.
19. van der Sluijs KF, Nijhuis M, Levels JH, et al. Influenza-induced expres-sion of indoleamine 2,3-dioxygenase enhances interleukin-10 productionand bacterial outgrowth during secondary pneumococcal pneumonia.J Infect Dis 2006; 193:214–22.
20. Paine R III, Morris SB, Jin H, et al. Impaired functional activity of alve-olar macrophages from GM-CSF-deficient mice. Am J Physiol LungCell Mol Physiol 2001; 281:L1210–8.
21. van der Sluijs KF, van Elden LJ, Nijhuis M, et al. Involvement of theplatelet-activating factor receptor in host defense against Streptococcuspneumoniae during postinfluenza pneumonia. Am J Physiol Lung CellMol Physiol 2006; 290:L194–9.
22. Meisel C, Schefold JC, Pschowski R, et al. Granulocyte-macrophagecolony-stimulating factor to reverse sepsis-associated immunosuppres-sion: a double-blind, randomized, placebo-controlled multicenter trial.Am J Respir Crit Care Med 2009; 180:640–8.
23. Huang FF, Barnes PF, Feng Y, et al. GM-CSF in the lung protectsagainst lethal influenza infection. Am J Respir Crit Care Med 2011; 184:259–68.
24. Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmo-nary epithelial cell expression of GM-CSF corrects the alveolar protei-nosis in GM-CSF-deficient mice. J Clin Invest 1996; 97:649–55.
25. Bubeck WJ, Patel RJ, Schneewind O. Surface proteins and exotoxins arerequired for the pathogenesis of Staphylococcus aureus pneumonia.Infect Immun 2007; 75:1040–4.
26. Ma X, Chang W, Zhang C, Zhou X, Yu F. Staphylococcal Panton-Valentine leukocidin induces pro-inflammatory cytokine productionand nuclear factor-kappa B activation in neutrophils. PLoS ONE 2012;7:e34970.
27. Voyich JM, Braughton KR, Sturdevant DE, et al. Insights into mecha-nisms used by Staphylococcus aureus to avoid destruction by humanneutrophils. J Immunol 2005; 175:3907–19.
28. Jamieson AM, Pasman L, Yu S, et al. Role of tissue protection in lethalrespiratory viral-bacterial coinfection. Science 2013; 340:1230–4.
29. Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cellspromote lung-tissue homeostasis after infection with influenza virus.Nat Immunol 2011; 12:1045–54.
30. Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grandrounds review. JAMA 2013; 309:275–82.
31. van der Sluijs KF, van Elden LJ, Nijhuis M, et al. IL-10 is an importantmediator of the enhanced susceptibility to pneumococcal pneumoniaafter influenza infection. J Immunol 2004; 172:7603–9.
32. Martin FJ, Parker D, Harfenist BS, Soong G, Prince A. Participation ofCD11c(+) leukocytes in methicillin-resistant Staphylococcus aureusclearance from the lung. Infect Immun 2011; 79:1898–904.
33. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macro-phages during influenza infection facilitates bacterial superinfections.J Immunol 2013; 191:1250–9.
34. Steinwede K, Tempelhof O, Bolte K, et al. Local delivery of GM-CSFprotects mice from lethal pneumococcal pneumonia. J Immunol 2011;187:5346–56.
35. Kohler J, Breitbach K, Renner C, et al. NADPH-oxidase but not induc-ible nitric oxide synthase contributes to resistance in a murine Staphy-lococcus aureus Newman pneumonia model. Microbes Infect 2011;13:914–22.
36. Szeliga J, Daniel DS, Yang CH, Sever-Chroneos Z, Jagannath C,Chroneos ZC. Granulocyte-macrophage colony stimulating factor-mediated innate responses in tuberculosis. Tuberculosis (Edinb) 2008;88:7–20.
37. Rigby KM, Deleo FR. Neutrophils in innate host defense against Staph-ylococcus aureus infections. Semin Immunopathol 2012; 34:237–59.
38. Cowburn AS, Summers C, Dunmore BJ, et al. Granulocyte/macrophagecolony-stimulating factor causes a paradoxical increase in the BH3-only pro-apoptotic protein Bim in human neutrophils. Am J RespirCell Mol Biol 2011; 44:879–87.
39. Epling-Burnette PK, Zhong B, Bai F, et al. Cooperative regulation ofMcl-1 by Janus kinase/stat and phosphatidylinositol 3-kinase contrib-ute to granulocyte-macrophage colony-stimulating factor-delayed apo-ptosis in human neutrophils. J Immunol 2001; 166:7486–95.
40. Abramson JS, Lewis JC, Lyles DS, Heller KA, Mills EL, Bass DA. Inhibi-tion of neutrophil lysosome-phagosome fusion associated with influen-za virus infection in vitro. Role in depressed bactericidal activity. J ClinInvest 1982; 69:1393–7.
41. Yousefi S, Cooper PR, Mueck B, Potter SL, Jarai G. cDNA representa-tional difference analysis of human neutrophils stimulated by GM-CSF.Biochem Biophys Res Commun 2000; 277:401–9.
42. Liu W, Yan M, Liu Y, McLeish KR, Coleman WG Jr, Rodgers GP. Ol-factomedin 4 inhibits cathepsin C-mediated protease activities, therebymodulating neutrophil killing of Staphylococcus aureus and Escheri-chia coli in mice. J Immunol 2012; 189:2460–7.
43. Donowitz GR. Principles and practice of infecious diseases. 7th ed.Churchill Livingstone Elsevier, 2010.
44. Randolph AG, Vaughn F, Sullivan R, et al. Critically ill children duringthe 2009–2010 influenza pandemic in the United States. Pediatrics2011; 128:e1450–8.
45. Small CL, Shaler CR, McCormick S, et al. Influenza infection leads toincreased susceptibility to subsequent bacterial superinfection by im-pairing NK cell responses in the lung. J Immunol 2010; 184:2048–56.
46. Kebaier C, Chamberland RR, Allen IC, et al. Staphylococcus aureusalpha-hemolysin mediates virulence in a murine model of severe pneu-monia through activation of the NLRP3 inflammasome. J Infect Dis2012; 205:807–17.
47. Tazawa R, Hamano E, Arai T, et al. Granulocyte-macrophage colony-stimulating factor and lung immunity in pulmonary alveolar proteino-sis. Am J Respir Crit Care Med 2005; 171:1142–9.
48. Wylam ME, Ten R, Prakash UB, Nadrous HF, Clawson ML, AndersonPM. Aerosol granulocyte-macrophage colony-stimulating factor forpulmonary alveolar proteinosis. Eur Respir J 2006; 27:585–93.
49. Tazawa R, Inoue Y, Arai T, et al. Duration of benefit in patients withautoimmune pulmonary alveolar proteinosis after inhaled GM-CSFtherapy. Chest 2013.
1836 • JID 2014:209 (1 June) • Subramaniam et al
at Carleton U
niversity on May 29, 2014
http://jid.oxfordjournals.org/D
ownloaded from
Recommended

















