
UK ISSN 0032-1 400
PLATINUM METALS REVIEW
A quarterly survey of research on the platinum metals and of developments in their application in industry
VOL. 42 OCTOBER 1998
Con tents
Platinum Metals Review and the Internet
Biphasic Homogeneous Catalysis By Paul 3. Dyson, David J. Ellis and Thomas Welton
Progress in Dye-Sensitised Photovoltaics By R. 3. Potter
Formation and Decomposition of Palladium Hydride Particles By P. D. Cobhn, B. E. Nieuwenhuys, V. V. Gorodetskii and V. N. Pamzon
Carbon Monoxide Sensing Technology By Gavin Troughwn
Platinum Labware Catalog
Aqueous-Organic Biphasic Catalysis By Paul 3. Dyson
The Build-Up of Bimetallic Transition Metal Clusters By Paul R. Raithby
Construction of Miniature Organo-Rhodium Boxes
Conferences Report Progress in Catalysis By C. F. 3. Barnard and W. Weston; K. E. Simons and A. F. Chafley
Combinatorial Chemistry Identifies Fuel Cell Catalyst
Catalysts for Butane Reforming in Zirconia Fuel Cells By K. Kendall and D. S. Williams
Geoffrey Wilkinson and Platinum Metals Chemistry By M. L. H. Green and W. P. GnfSlth
Abstracts
New Patents
Indexes to Volume 42
NO. 4
134
135
140
141
144
144
145
146
157
158
163
164
168
174
179
183
Communications should be addressed to The Edizor, Susan V. Ashton, Platinum Metals Rev&
Johnson Matthey Public Limited Company, Hatton Garden, London ECl N 8EE

PLATINUM METALS REVIEW AND THE INTERNET
Johnson Matthey is pleased to announce that a full text online version of "Platinum Metals Review" can now be accessed on the Internet. The electronic version of the journal is accessible without charge, as is the printed version. The journal is being hosted on the website of the Royal Society of Chemistry's Turpin Distribution Services Limited, TOPS, at http://www.turpin-distribution.com. It may also be accessed from the "Platinum Metals Review" button on the Johnson Matthey website, at http://www.matthey.com, which is linked to the Turpin website. To read the journal it will first be necessary to download the free Adobe@ Acrobat@ Reader software held on the Turpin site.
At present, besides the October 1998 issue, the full texts of the April 1998 and July 1998 issues of "Platinum Metals Review'' are also available on the Internet, and the website will archive all future issues of the journal as they are published. In some areas of the world the electronic version of the journal may be available before the printed version is delivered.
The electronic journal and the printed journal are identical, providing our coverage of papers, conference reports, book reviews, small items and reports, and the Abstracts and Patents on the science and technology of the platinum group metals. The 1998 Name and Subject Indexes will also be accessible for downloading. The website offers readers the facility of searching with the MUSCAT@ search engine using natural language: by subject, across titles and the full text, and by names, which OCCUT as authors of papers or as part of the text or the references. Patentees, abstract authors and journals cited can also be searched across all the issues of the electronically held journal. At present the documents retrieved are ranked in order of relevance to the words used in the search query. More facilities will become available as the Turpin website is developed; for instance, readers will shortly be able to register for E-mail alerting to advise them that a new issue of the journal is available.
In addition to access to the electronic version of "Platinum Metals Review", the Johnson Matthey website also carries a leaflet for each issue of the journal. The leaflets, which were first published on the Johnson Matthey website in April 1997, outline the contents of the journal and carry html links to the websites of the authors, to their places of work or institutions, to topics related to the papers and to background information and other relevant items. The Johnson Matthey website also carries an E-mail form to enable readers to pass on comments or suggestions about the journal to the editor. Susan V. Ashton EDITOR
Platinum Metals Rev., 1998,42, (4), 134 134

Biphasic Homogeneous Catalysis By Paul J. Dyson, David J. Ellis and Thomas Welton Department of Chemistry, Imperial College of Science, Technology and Medicine. London
Biphasic catalysis is becoming an area of environmentally responsible catalysis, but its development and use have until recently been somewhat neglerted. Here, the basic principles and the design of features going into such systems are explained, and ageneral overview is presented with the intention of encour- aging greater interest in this under utilised technique. Some well-established aqueous-organic regimes are described and there is a discussion of some possible future directions involving ionic-liquidlorganic systems.
There are many benefits to be gained by using homogeneous catalysis in place of heterogeneous catalysis in organic synthesis, the most notable being the use of less aggressive reaction condi- tions and increased selectivity.
The main disadvantages of traditional organic phase reactions employing homogeneous tran- sition metal catalysts are the difficulties asso- ciated with separating the catalyst ii-om the prod- uct and solvent. Separation techniques, such as distillation, require an extra expenditure of energy and can, in certain instances, lead to degradation of both the products and the cat- alyst used. As the catalyst requires extraction before a new reaction run can be performed, the ‘turn around time’ between runs also becomes a prime factor.
These problems coupled with the inevitable loss of the catalyst species (allowing for some imperfection in the separation techniques employed) tend to redress the balance between heterogeneous and homogeneous catalysis.
One possible solution to these problems is to heterogenise the catalyst and product into two separate and immiscible phases. Reactions may
then be performed as shown in Figure 1. Here the catalyst resides in solution in one of the two phases and the substrate resides in the other phase. During reactions, the two layers are vig- orously stirred, thus allowing suitable interac- tion of catalyst and substrate. Once the reaction has reached the appropriate stage, the stirring is stopped and the mixture of phases separates into two layers, one containing the product and the other containing the catalyst. Separation of the two is then carried out by simple decanta- tion and, in principle, the catalyst solution is available for immediate reuse. Clearly, these biphasic reactions offer a potential answer to the problems mentioned above.
This type of approach was first used com- mercially for the polymerisation of ethylene (Shell Higher Olefins Process (1)) although in this case the catalyst and substrate are initially in a single phase and the product forms the sec- ond, immiscible phase; the principal, however, is the same. Clearly, this approach is not suitable for many other processes and has thus lead to the selection of water as the preferred catalyst solvent for biphasic conditions.
Fig. 1 A schematic
Product solution
the initial reactant solution and product solution are
solution
Catalyst solution
Platinum Metals Rev., 1998, 42, (4), 135-140 135
Fig. 1 A schematic representation of a two- phase process showing how the initial reactant solution and product solution are immiscible with the catalyst solution
Reactant Solution
Catalyst SoIuticn

s09" Fig. 2 Two water-soluble phosphines: (a) triphenylphosphine mono-sulfonate and (b) triphenylphosphine
The latter phosphine has a
has been used extensively
SOjNa
~ p~ tri-sulfonate.
solubility of 1.1 kg I-' and thus
/ /
SOjNa
The selection of water is straightforward and offers many benefits. First, wide ranges of organic solvents are immiscible with water; water is cheap, easily purified, and readily obtained and disposed of. However, despite the many advantages offered by aqueouslorganic bipha- sic systems, the level of commercial exploita- tion is still relatively low. This is probably due, at least in part, to a lack of suitable water-sol- uble catalysts. However, having said this, inter- est in fundamental research has escalated rapidly in recent years. There have been a number of reviews published (2-6), for instance, an entire volume of the Journal of Molecular Catalysis, with an excellent editorial dedicated to the subject (7), and a .recent book, reviewed here on page 145, which examines aqueous phase catalysts from an industrial perspective (8).
Sulfonated Phosphines and Their Industrial Use
The main consideration when attempting to design a water-soluble complex is how to ren- der hydrophilic a typical hydrophobic organo- metallic complex. In order to do this, an appro- priate ligand (or ligands) must be placed around the metal centre (or centres); alternatively, ionic catalysts, such as Dipamp Rh(cod)+ and (binap)Ru cations, could be used. One class of ligands that are widely used in homogeneous single phase catalysis are phosphines and diphos- phines and it is therefore not surprising that the synthesis of water-soluble phosphine derivatives is attractive and has been the focus of much lig- and design. Inducing hydrophilicity into a phos- phine may be achieved by the introduction of
Platinum Metals Rev., 1998,42, (4)
polar groups onto the phosphine substituent. In this respect, one of the most widely used groups is a sulfate (SO,') group which can be attached to the phenyl rings in PPh,. These represent, at the current time, the most widely commercially exploited ligand system.
The first of these sulfonated phosphines, triph- enylphosphine mono-sulfonate (TPPMS, see Figure 2(a)) was reported as early as 1958 and was produced by the oleum sulfonation of triph- enylphosphine (9). Modification of this syn- thesis led to the production of the tri-sulfonated ligand (TPPTS, see Figure 2(b)) which is now the most common ligand in use (10, 11). The tri-sulfonated ligand has an extremely high water solubility of ca. 1.1 kg 1.' (2). Formation of the catalyst complex is then carried out by co-ordi- nation of the sulfonated phosphine ligand. Although direct sulfonation of pre-complexed triphenylphosphine ligands should be possible, the extremely acidic conditions needed to effect the change make the process unreliable.
Concentrating for the moment on the tri-sul- fonated ligands, a whole range of water-soluble catalysts based on monometallic and cluster compounds has been reported, and a review by Kalck and Monteil includes a comprehensive list of these compounds ( 3 ) .
Of particular interest here is the rhodium com- plex which is used in the RuhrchemielRhBne- Poulenc process for the biphasic hydroformy- lation of propene to n-butyraldehyde (1 2), a process which is used to produce about 330,000 tons of n-butyraldehyde per year. This process is highly selective and gives a linear aldehyde to branched aldehyde (nliso) ratio of 9515 with
136

99 per cent substrate conversion. Side reactions and loss of catalyst are both negligible. Since the use of this process commenced, catalyst development has continued and more active catalysts have been reported, although the selectivity has decreased ( 13, 14).
RhBne-Poulenc have also expanded the use of their biphasic production facilities into the man- ufacture of alcohols by hydrogenation and hydrodimerisation (5). A similar process with a rhodium/TPPTS catalyst is also used for the production of valeraldehyde from butene (1 5), which is the basis of n-valeric acid, used in the manufacture of CFC-free refrigerants.
Although the majority of work has concen- trated on sulfonated phosphines there are other polar groups that can be used to induce water solubility. These include (in no particular order) hydroxyl, ether, carboxylate and amine groups. Ligands with these water solubilising groups will not be discussed in detail, due to the large num- ber which exist, and in most cases the catalytic properties of complexes with these ligands have not been investigated in detail. The reviews men- tioned previously present a more comprehen- sive and fuller picture of these ligands (2-6).
Metal Clusters as Water-Soluble Cat a 1 y s t s
The use of metal clusters in conventional homogeneous processes has been widely stud- ied because they are considered to have prop- erties intermediate between homogeneous and heterogeneous catalysts. As a cluster consists of several metal atoms, activation of an organic substrate may take place at more than one metal atom and this can have a profound effect on the activity. The number of catalytic processes in which clusters are effective is extensive, although examples where they are used in commercial processes are rare (1 6). Also, there is often uncer- tainty as to whether the cluster is broken down into mononuclear fragments during catalytic processes or whether the metal core remains intact.
A new aspect of biphasic catalysis has been the synthesis of water-soluble clusters. There are only a few water-soluble clusters at present and
co co
60 co Fig. 3 The structures of (a) Rm(CO),,(TI'PTS) and (b) RU~(CO)~(TPPTS), ; both catalyse the hydrogenation of non-activated olefins
two main types have been reported (1 7, 18). The first of these is based on TPPTS deriva- tives, two examples of which are shown in Figure 3. In most cases the synthesis of this type of clus- ter is simple and is achieved by the replacement of one or more carbonyl ligands with the water- soluble phosphine ligand under reflux in a suitable solvent.
The use of these compounds in the water- gas shift reaction (the reduction of water to hydrogen) has also been examined (19). This is a particularly valuable reaction, as it would potentially allow water to be the source of hydro- gen for biphasic hydrogenations. AU of the clus- ters tested were found to catalyse the water-gas shift reaction, although no quantitative mea- surements were performed, and some changes in the catalysts were found to have taken place during the course of the reaction.
We have since directed our attention to the use of this range of catalysts in both hydroformy- lation and hydrogenation reactions. Early results for hydrogenations of olefins have proved pos- itive although catalytic turnovers are quite low. A similar change in the catalyst also occurs dur- ing hydrogenation and this is now thought to be independent of the catalytic reaction. However, the change in species does not seem
Platinum Metals Rev., 1998, 42, (4) 137

a I
1 *+
Fig. 4 The structure of [HBu,(q-C,H,),]a'; this cluster catalyses the hydrogenation of benzene and other arenes in water and ionic liquid biphasic reactions
to affect catalytic activity. A detailed analysis of our findings will be published in due course.
An alternative series of water-soluble clus- ters has also been reported (1 7). These are not based on phosphine ligands but on cationic tetraruthenium clusters with arene and hydride ligands. Several have been synthesised and char- acterised and one of the clusters, see Figure 4, has been-successfully used to hydrogenate ben- zene and some simple arenes -which is encour- aging from an industrial perspective (20). The clusters are reported to be quite active, especially for the hydrogenation of benzene to cyclohexane, but not unexpectedly the conversion of the arene derivatives showed slightly lower activity and was not particularly selective. Indeed, where the substrate molecules possessed unsaturated side- groups, these were preferentially hydrogenated.
Ionic Liquids - the Future of Biphasic Catalysis?
While water has proved to be the most widely used solvent for biphasic reactions there are sev- eral problems associated with it. For example, the chemical modifications to the catalyst which are needed to induce water solubility often reduce the catalytic activity, and certain active homogeneous catalysts are unsuited to any type
of conversion. Also, catalysts which are sensi- tive to moisture (as many are) cannot be used in water. The ideal situation would be an alter- native solvent, capable of dissolving a wide range of metal compounds without reacting with the metal centre and so deactivating the catalyst. Even without modification, ionic liquids may represent an alternative solvent.
Ionic liquids are fundamentally molten salts. Molten sodium chloride (m.p. S O S O C ) is an example of a single component ionic liquid. It is clear to see - from its high melting point - that sodium chloride would not be suitable as a solvent for biphasic catalysis. However, a range of dual-component ionic liquids is available which are molten at and near room tempera- ture (2 1). The physical properties of these liq- uids are quite interesting but the main proper- ties of interest here are:
their immiscibility with a wide range of organic solvents (making them ideal for bipha- sic systems);
their polar nature (making them good solvents); and
the low nucleophilicity of their component ions (preventing deactivation of the catalyst).
The ionic liquids we are interested in using are formed from two chemical components: an organic component (either 1 -ethyl-3-methyl- imidazolium chloride, see Figure 5(a), or
L J
'a-
Fig. 5(a) 1-ethyl-3-methyl-imidazolium chloride and (b) 1-butyl-3-methyl-imida- zolium chloride
Platinum Metals Rev., 1998, 42, (4) 138

1 -butyl-3-methyl-imidazolium chloride, see Figure 5(b)) and an inorganic salt (aluminium chloride or sodium tetrafluoroborate).
A number of studies in biphasic catalysis have already been performed using an ionic liquid as the catalyst solvent (22-26). Ionic liquid sys- tems have been used for hydrogenations (with rhodium, ruthenium and cobalt complexes), hydroformylations (with rhodium complexes), Heck coupling (with palladium complexes) and oligomerisations (with nickel complexes).
So far our interest has concentrated on the use of metal clusters in the ionic liquid. We have carried out a preliminary investigation into the effect of the chloroaluminate acid melt (a 2: 1 molar ratio of AlCl, and 1-butyl-3-methyl- imidazolium chloride) on a range of metal car- bonyls. The strongly Lewis acidic environment presented by the ionic liquid causes a change in the metal carbonyls over a period of time, but we have been unable to draw any conclusions at the present time.
Since our preliminary work into the effect of the acidic melt and with the experience of sev- eral low-pressure hydrogenation attempts (in which the melt has initiated rapid oligomeri- sation of the olefins used) we have shifted our attention to ionic liquids based on the tetra- fluoroborate ion. These ionic liquids, unlike those based on chloroaluminates, are air sta- ble and are thus much easier to handle. They also do not cause oligomerisation of the olefin substrates. The main disadvantage concerned with the tetrafluoroborate range of ionic liquids is that they exhibit considerably greater vis- cosity than the chloroaluminate variety and thus very aggressive agitation is required. Another
disadvantage is that uncharged species have decreased solubility in this ionic liquid whereas a wide range of neutral compounds can dissolve in the chloroaluminate melts. The tetrafluoro- borate melts are still, however, very strongly polar in character and as such will dissolve charged species easily. We are currently design- ing new catalysts based on these requirements.
Conclusions Biphasic catalysis is an under-exploited tech-
nique, but with increasingly demanding envi- ronmental legislation the opportunity for this technique to become more widespread in indus- try is quite clear. In addition to those discussed above, other biphasic regimes are also available, for example, a group of perfluorinated ethers which are chemically inert, non-toxic and gen- erally immiscible with other organic solvents, has been reported (27-29). As with aqueous biphasic catalysis, the catalytic species have to be modified to achieve solubility, but in this case, such modifications are based on the replace- ment of traditional ligands with partially fluo- rinated or perfluorinated ligands. However, the high cost of both the fluorous solvent phase and the catalyst systems required has, at the present time, made this type of biphasic system less attractive to industry even though they are envi- ronmentally friendly compared to the systems currently available.
The use of biphasic catalysis is gradually becoming increasingly acceptable and it can be expected that the number of processes involv- ing it will continue to grow as the benefits that it offers are shown to be both environmentally sound and cost effective.
References W. Keim, Chem. Ing. Tech., 1984,56,850 E. G. Kuntz, Chemtech, 1987, 17, 570 P. Kalck and F. Monteil, Adv. Organomet. Chem., 1992,34,219 W. A. Herrmann and C. W. Kohlpainter, Angew. Chem., Int. Ed. Engl., 1993,32, 1524 B. Cornils, W. A. Herrman and R. Eckl, J. Mol. Catal. A: Chem., 1997, 116, 27
6 F. JOC, and A. Katho,J Mol. C a d . A: Chem., 1997,
7 I. T. Horvath,J. MoZ. Catal. A: Chem., 1997,116, 116,3
Editorial
8 B. Cornils and W. A. Herrman, “Applied Homogeneous Catalysis by Organometallic Catalysts”, Wiley-VCH, Weinheim, 1998
9 S. Ahrland, J. Chatt, N. R. Davies and A. A. Williams,J. Chem. SOL., 1958, 276
10 E. G. Kuntz, French Patent 2,314,910; 1975 11 J. L. Sabot, European Patent 61104,967; 1982 12 C. Larpent, R. Dabard and H. Patin, lnorg. Chem.,
1987,22,2922 13 Y. Amrani, L. Lecomte, D. Sinou, J. Bakos, I. Toth
and B. Heil, Organometallics, 1989, 8, 542
Platinum Metals Rev., 1998, 42, (4) 139
1 W. Keim, Chem. Ing. Tech., 1984,56,850 2 E. G. Kuntz, Chemtech, 1987,17,570 3 P. Kalck and F. Monteil, Adv. Organomet. Chem.,
4 W. A. Herrmann and C. W. Kohlpainter, Angew.
5 B. Cornils, W. A. Herrman and R. Eckl, J. Mol.
6 F. JOC, and A. Katho,J MoZ. C u d . A: Chem., 1997,
7 I. T. Horvath,J. MoZ. Cutul. A: Chem., 1997,116,
1992,34,219
Chem., Int. Ed. Engl., 1993,32, 1524
CutuZ. A: Chem., 1997, 116, 27
116,3
Editorial

14
15
16 17
18
19
20
21
A. Avey, D. M. Schut, T. J. R. Weakley and D. R. Tyler, Inorg. Chem., 1993, 32, 233 H. Bahrmann, C. D. Frohning, P. Heymanns, H. Kalbfell, P. Lappe and D. Peters, 3. Mol. Catal. A: Chem., 1997, 116,35 L. N. Lewis, Chem. Rev., 1993,93,2693 L. Plasseraud and G. Suss-Fink, J. Organomet. Chem., 1997, 163, 539 B. Fontal, J. Orlewski, C. C. Santini and J. M. Basset, Inorg. Chem., 1986, 25, 4320 D. F. Bryce, P. J. Dyson, B. K. Nicolson and D. Parker, Polyhedron, in press G. Meister, G. Rheinwald, H. Stoeckli-Evans and G. Siiss-Fink,J. Chem. SOC., Ddwn Trans., 1994,3215 C. L. Hussey, Adv. Molten Salt Chem., 1983, 5, 185
22 Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed. Engl., 1995, 34, 2698
23 P. Suarez, J. E. L. Dullius, S. Einloft, R. F. DeSouza and J. Dupont, Polyhedron, 1996, 15, 1216
24 A. L. Montiero, F. K. Zinn, R. F. DeSouza and J. Dupont, Tetrahedron Asymm., 1997, 8, 177
25 J. E. L. Dullius, P. A. Z. Suarez, S. Einloft, R. F. DeSouza, J. Dupont, J. Fischer and A. DeCian, Organometallics, 1998, 17, 8 15
26 Y. Chauvin, Actual. Chim., 1996, 44 27 I. T. Horvath and J. Rabai, Science, 1994,266,72 28 J. R. Gladysz, Science, 1994, 266, 5 5 29 B. Cornils, Angew. Chem., Int. Ed. Engl., 1997,
36,2057
Progress in Dye-Sensitised Photovoltaics The 12th International Conference on the diffusive processes, although under some
conditions this is likely to be field-assisted. The extraordinarily slow time-constants
(typically hundreds of ms) of the cell in response to chopped illumination is almost certainly due
Conversion of Solar Energy into Photovoltaic Power and Storage, IPS-12, was held in Berlin from 9th to 14th August. This is the principal technical conference on photovoltaics and solar energy storage worldwide, and is held every two years. This year there were over 400 delegates, with most coming from academic institutions. The major surprise of the conference was the growth in activity in dye-sensitised photovoltaics (DSPVs), with over half of the presentations and posters being related to this topic.
The basic science behind dye-sensitised photovoltaic cells is well known (1). Cells are typically constructed from a glass/ITO electrode coated with a thin layer of dyed titania (TiO,). The TiO, is dyed with ruthenium-based com- pounds, such as R u o (2,2'-bipyridyl-4,4'-dicar- boxylate),(NCS),, and then impregnated with a liquid electrolyte containing the I-/I; couple as a regenerative redox shuttle between the dye and the counter electrode (platinum-coated glass/ITO). The cell power-conversion efficiency can be remarkably high, with figures of > 10 per cent under AM1.5 conditions being quoted, due to the broad absorption spectrum of the dye (extending into the infrared region for some dyes (2)) and the absence of significant charge recombination in the (n-type) semiconducting TiO,.
While the complex photophysics and chem- istry of this system are still challenging, some consensus emerged as to what makes these cells work as well as they do:
to extensive trapping of electrons in surface states on the TiO,. I Increased cation (for example Li') penetra- tion into the pores of the TiO, probably improves the efficiency of the electron transfer process and certainly assists the ionic (iodide) current in the liquid phase. The net benefit is an increase in cell current, although the type of cation also affects the cell open-circuit potential in ways that are not yet clearly understood. 0 The electron-hole recombination may be retarded by virtue of the fact that the iodide 'hole-carrier' is negatively charged. I The electron-injection kinetics (not the efficiency) are relatively insensitive to the dye type and very sensitive to the surface condi- tion of the TiO,.
Perhaps the highlight of the conference was a 'live' demonstration of DSPV technology by Dr K. P. Hanke, Institut fiir Angewandte Photo- voltaik, Gelsenkirchen, Germany, who used a prototype module to turn an electric fan, during his lecture on issues involved in cell scale-up.
In summary, the work presented at this conference has shown that dye-sensitised pho- tovoltaic cells are continuing to show promise as practical devices, and may, in the longer term, open up a new market for ruthenium and platinum-based materials.
The more advanced ruthenium-based dyes
that proper sealing of the cell is achieved to
References show evidence for provided 1 M, GrHael, platinum Metals Rev., 1994, 38, (4),
1 ct - I J I - - prevent ingress of oxygen and water (which initiate free-radical attack of the dye).
Electrons move primarily through TiO, by 7 19 R. J. POlTER
2 Md. K. Nazeeruddin, R. Humphry-Baker, M. Gratzel and B. A. Murrer, Chem. Commun., 1998,
Platinum Metals Rev., 1998, 42, (4) 140

Formation and Decomposition of Palladium Hvdride Particles
J
IMAGING PICTURES ON THE NANOMETRE SCALE
By €? D. Cobden and B. E. Nieuwenhuys Leiden Institute of Chemistry, Leiden University, The Netherlands
and Y V. Gorodetskii and Y N. Parmon Boreskov Institute of Catalysis, Novosibirsk, Russia
Nanoscale changes in surface structure that accompany the low temperature exposure of pa l lad ium to hydrogen are reported. Field Emiss ion Microscopy, a method for rapid in situ imaging of surfaceprocesses, has been used to exam- inepal ladium tips of radius - 200 nm, produced by a novel technique. Images are presented of the initial stages of the uptake of hydrogen. Subsurface hydrides were init ially formed when pal ladium t ips were exposed to hydrogen gas at low temperatures, starting a t highly open surfaces present o n the t ip . Ex truding P d H particles were also formed on top of the pal ladium t i p and their growth was observed to proceed in a ‘staccato’-like manner. Pa l lad ium crystallites remained on the surface after most of the hydrogen had been removed f r o m the pal ladium sample by heating in vacuum. O n heating the crystallites remained quite stable up to a temperature of - 700 K , but then melted back into the tip.
A detailed understanding of the process of hydrogen absorption by metallic phases (met- als, alloys and intermetallic compounds) is important for the development of new materi- als for hydrogen storage. The interaction of hydrogen with palladium in particular has been extensively studied (1,2), since the first report of the absorption of hydrogen into palladium in 1866 and since the first measurements of the palladium-hydrogen pressure-constitution-tem- perature relationship in 1895 (3). Two non-sto- ichiometric hydride phases can co-exist below the critical temperature (- 3OO0C), and on going from the a- to the P-phase there are large increases in volume, with the lattice constant increasing by - 3.3 per cent, which is a volume increase of - 11 per cent. These changes occur continuously over the phase transition.
It is surprising that comparatively few stud- ies have been focused on the structural changes occurring at the metal surface under the strain of the expanding lattice. Triangular shapes, of
- 10 to 20 pm in size, have been seen developing during the early stages of hydrogen loading of a Pd( 1 1 1) single crystal (4). In addi- tion, networks in parallel lines have been observed on large single crystals by Sugeno and Kawabe (5). These patterns became apparent on complete transformation to the P-phase and could sometimes be seen after desorption of hydrogen (5).
Field Emission Microscopy (FEM) (resolu- tion - 2 nm) images of surface processes were observed in situ during exposure of stable clean palladium tips to hydrogen, see Figure 1. The tips, produced by a novel technique (6), were held in hydrogen at a pressure of 2.6 x 10” mbar and 147 K. Figure l(a) is characteristic of a clean palladium tip with the (1 10) plane in its centre. In general, a FEM pattern represents a work function map of the various crystal faces on the end of a hemispherically shaped single crystal tip. Figure 1 (b) shows the growth of high intensity patches (that is, patches of increased
Platinum Metals Rev., 1998, 42, (4), 141-144 141
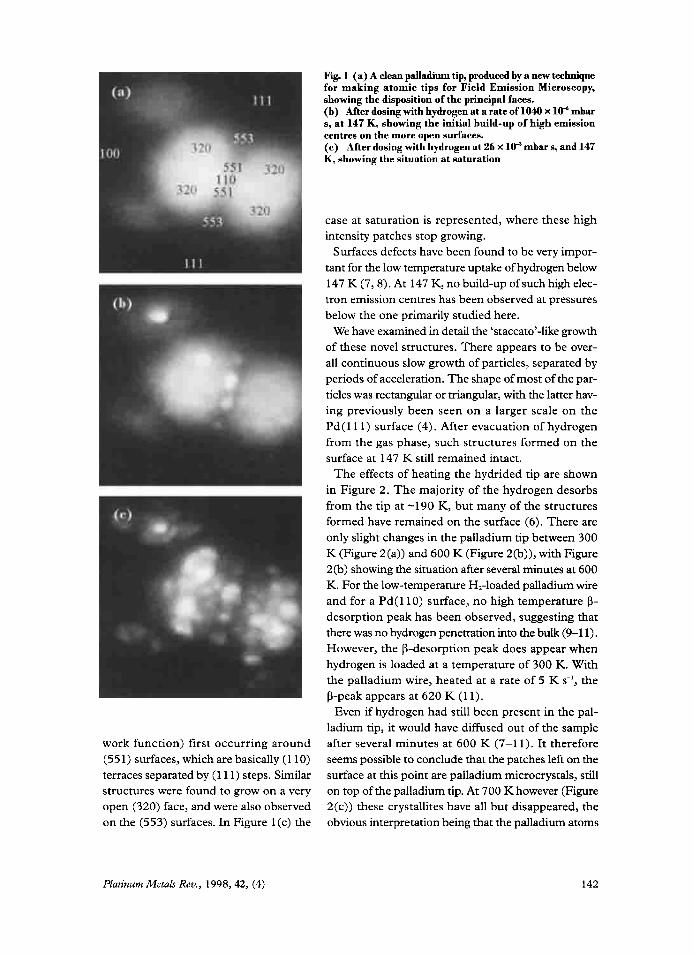
Fig. 1 (a) A clean palladium tip, produced by a new technique for making atomic tips for Field Emission Microscopy, showing the disposition of the principal faces. (b) After dosing with hydrogen at a rate of 1040 x lo" mbar s, at 147 K, showing the initial build-up of high emission centres on the more open surfaces. (c) After dosing with hydrogen at 26 x lo3 mbar s, and 147 K, showing the situation at saturation
case at saturation is represented, where these high intensity patches stop growing.
Surfaces defects have been found to be very impor- tant for the low temperature uptake of hydrogen below 147 K (7,s). At 147 K, no build-up of such high elec- tron emission centres has been observed at pressures below the one primarily studied here.
We have examined in detail the 'staccato'-like growth of these novel structures. There appears to be over- all continuous slow growth of particles, separated by periods of acceleration. The shape of most of the par- ticles was rectangular or triangular, with the latter hav- ing previously been seen on a larger scale on the Pd( 1 1 1) surface (4). After evacuation of hydrogen from the gas phase, such structures formed on the surface at 147 K still remained intact.
The effects of heating the hydrided tip are shown in Figure 2. The majority of the hydrogen desorbs from the tip at -190 K, but many of the structures formed have remained on the surface (6). There are only slight changes in the palladium tip between 300 K (Figure 2(a)) and 600 K (Figure 2(b)), with Figure 2(b) showing the situation after several minutes at 600 K. For the low-temperature H,-loaded palladium wire and for a Pd( 1 10) surface, no high temperature 0- desorption peak has been observed, suggesting that there was no hydrogen penemtion into the bulk (9-1 1). However, the P-desorption peak does appear when hydrogen is loaded at a temperature of 300 K. With the palladium wire, heated at a rate of 5 K s-', the P-peak appears at 620 K (1 1).
Even if hydrogen had still been present in the pal- ladium tip, it would have diffused out of the sample after several minutes at 600 K (7-1 1). It therefore seems possible to conclude that the patches left on the surface at this point are palladium microcrystals, still on top of the palladium tip. At 700 K however (Figure 2(c)) these crystallites have all but disappeared, the obvious interpretation being that the palladium atoms
work function) first occurring around (55 1) surfaces, which are basically (1 10) terraces separated by (1 11) steps. Similar structures were found to grow on a very open (320) face, and were also observed on the (553) surfaces. In Figure 1 (c) the
Platinum Metals Rev., 1998, 42, (4) 142
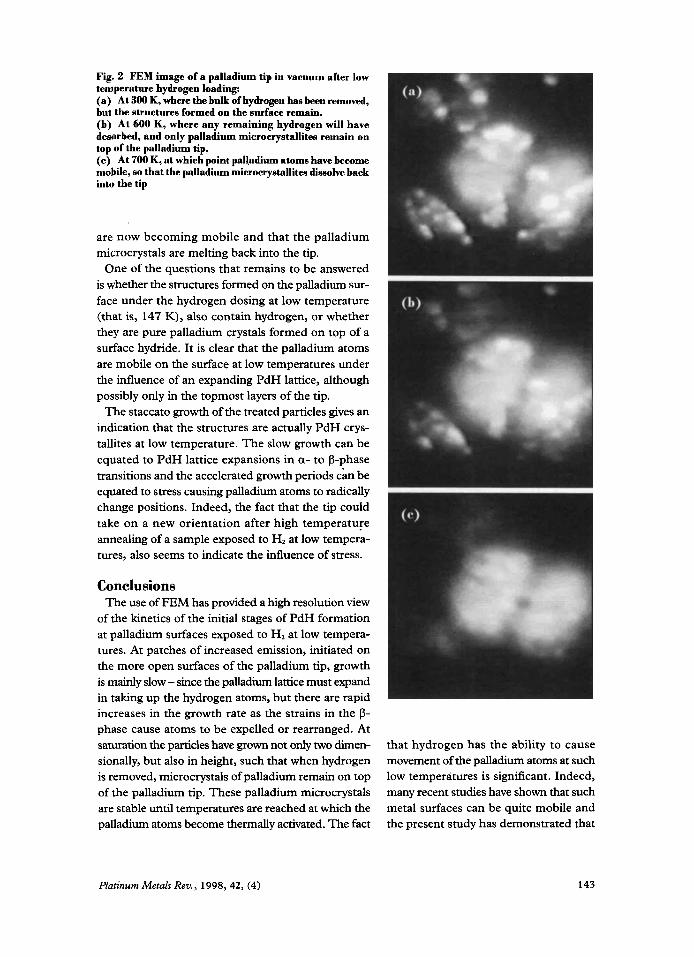
Ttemperature hydrogen loading: (a) At 300 K, where the bulk of hydrogen has been removed, but the structures formed on the surface remain. (b) At 600 K, where any remaining hydrogen will have desorbed, and only palladium microcrystallites remain on top of the palladium tip. ( c ) At 700 K, at which point pal1,adium atoms have beeome mobile, so that the palladium microerystallitea dissolve back into the tip
are now becoming mobile and that the palladium microcrystals are melting back into the tip.
One of the questions that remains to be answered is whether the structures formed on the palladium sur- face under the hydrogen dosing at low temperature (that is, 147 K), also contain hydrogen, or whether they are pure palladium crystals formed on top of a surface hydride. It is clear that the palladium atoms are mobile on the surface at low temperatures under the influence of an expanding PdH lattice, although possibly only in the topmost layers of the tip.
The staccato growth of the treated particles gives an indication that the structures are actually PdH crys- tallites at low temperature. The slow growth can be equated to PdH lattice expansions in a- to p-phase transitions and the accelerated growth periods c'an be equated to stress causing palladium atoms to radically change positions. Indeed, the fact that the tip could take on a new orientation after high temperatuFe annealing of a sample exposed to H, at low tempera- tures, also seems to indicate the influence of stress.
Conclusions The use of FEM has provided a high resolution view
of the kinetics of the initial stages of PdH formation at palladium surfaces exposed to H2 at low tempera- tures. At patches of increased emission, initiated on the more open surfaces of the palladium tip, growth is mainly slow - since the palladium lattice must expand in taking up the hydrogen atoms, but there are rapid increases in the growth rate as the strains in the p- phase cause atoms to be expelled or rearranged. At satura~on the particles have grown not only two dimen- sionally, but also in height, such that when hydrogen is removed, microcrystals of palladium remain on top of the palladium tip. These palladium microcrystals are stable until temperatures are reached at which the palladium atoms become thermally activated. The fact
that hydrogen has the ability to cause movement of the palladium atoms at such low temperatures is significant. Indeed, many recent studies have shown that such metal surfaces can be quite mobile and the present study has demonstrated that
Platinum Metals Rev., 1998, 42, (4) 143
Fig 2 FEM image of a palladium tip in vacuum after low temperature hydrogen loading: (a) At 300 K, where the bulk of hydrogen has been removed, but the structures formed on the surface remain. (b) At 600 K, where any remaining hydrogen will have desorbed, and only palladium microcrystallites remain on top of the palladium tip. ( c ) At 700 K, at which point pal1,adium atoms have h o m e mobile, so that the palladium microerystallitea dissolve back into the tip

large reconstructions can occur when palladium interacts with hydrogen at 150 K. However, in the bulk of a metal in which a hydride is being formed, there are few places for the atoms to go
prevent material degradation, when metal- hydrogen reservoirs are being designed.
Acknowledgement
Netherlands Organisation for Scientific Research when the lattice expands. As a consequence it
appear that more thought needs to
The authors acknowledge financial Support from the
[ W O ) in the framework ofthe ‘Russia Propramme’ be
1
2 3 4
5
6
7
- given to stabilising the surface in order to and of the Priority Programme “on-Linear Systems’.
References F. A. Lewis, “The Palladium Hydrogen System”, Academic Press, 1967, LondonNew York
8 H. Okuyama, W. Siga, N. Takagi, M. Nishijma and T. Aruga, Sud Sci., 1998,401,344
F. A. Lewis, Inz. J. Hydrogen Energy, 1981,6, 319 C. Hoitsema, Z. Phys. Chem., 1895, 17, 1
9 R. J. Behm, v. Penka, M.-G. Cattaniaj K. Christmann and G. Ertl, J. Chem. Phys., 1983, 78.7486
T. J. Tiedema, B. C. de Jong and W. G. Burgers, 10 M.-G. Cattania. V. Penka. R. 1. Behm. K. Proc. Kon. Ned. Akad. Wet., 1960, 63B, 422 T. Sugeno and H. Kawabe, Mem. Inst. Scient. Ind. Res. Osaka Univ., 1957, 14, 25 P. D. Cobden, V. V. Gorodetskii and B. E. Nieuwenhuys, to be published R. Dus, E. Nowicka and Z. Wolfram, Surf Sci., 1989,216, 1
11
Christmann and*G. Ertl, Suh ScI., 1983,126, 382 0. M. Ilinitch, F. P. Cuperus, V. V. Gorodetskii, M. Yu. Smirnov, 0. P. Burmatova and I. 0. Ilinitch, Proc. 4th Workshop “Optimisation of Catalytic Membrane Reactor Systems, European Science Foundation, 1997, SINTEFF Materials Technology, Oslo, p. 89
Carbon Monoxide Sensing Technology Growing awareness of the hazard of carbon
monoxide (CO) in the home environment has aroused great interest in detector alarms in the U.K. and North America. Various sensing tech- nologies have been used to detect the gas.
The first commercial sensor, the Taguchi sen- sor, correlated the change in conductivity of a heated tin oxide pellet to the concentration of CO present. However, due to its high power require- ments, this sensor required mains wiring. The first battery powered CO detectors used an opti- cal detection technique based on colour chem- istry, the colour change being the same as in the formation of carboxyhaemoglobin in the blood.
Recently, electrochemical units, suitable for use in battery powered alarms, have become commercially available. These have significant advantages over prior technologies in their accu- racy and reliability over a wide range of gas con- centrations. Some instruments have visual dis- plays to differentiate between acute high CO concentrations and hazardous chronic low con- centrations. Carbon monoxide and oxygen dif- fuse into the sensor &om the ambient air to react:
Anode: Cathode: Overall:
The current flowing between the anode and cathode through an external circuit is pro- portional to the C O present over a wide con- centration range. The carbon dioxide (COJ that
CO + H,O 4 CO, + 2H’ + 2e %O, + 2 H + 2 e ~ + HZO CO + %02 + C 0 2
is produced diffuses out from the sensor. The electrode reactions take place under acidic
conditions to avoid a build up of CO, in the sen- sor. Under these conditions platinum is required to catalyse the electrode reactions. Platinum has the ability to form a range of chemisorbed sur- face species, thereby lowering the activation energy of intermolecular reactions. Platinum forms car- bony1 species and surface bound hydroxyl species required for the overall anode reaction.
In practice porous electrodes made from a high surface area platinum material are used. This provides a three-phase boundary between the gas, the electrolyte and the electrode where the electrode reactions can occur rapidly in the presence of CO. GAVIN TROUGHTON
Platinum Labware Catalog Alfa Aesar in North America has just pub-
lished a “Platinum Labware Catalog” which describes a range of laboratory products incor- porating platinum, platinum group metals and Zirconia Grain Stabilised (ZGS) platinum, util- ising the inertness and malleability of platinum.
The catalogue describes typical uses of the equipment and contains reference data and information on a recycling programme.
To obtain a copy of the catalog contact Alfa Aesar; in North America, tel: 800-343-0660 ext. 6404, fax: 800-322-4757; Rest of the World, tel: 978-521-6404, fax: 978-521-6350.
Platinum Metals Rev., 1998, 42, (4) 144

Aqueous-Organic Biphasic Catalysis Aqueous-Phase Organometallic Catalysis: Concepts and Applications EDITED BY BOY C0RNIL.S AND WOLFGANG A. HERRMA”, Wiley-VCH, Weinheim, 1998,615 pages, ISBN 3-527-29478-3, E140.00
This is the first book devoted entirely to the subject of aqueous-organic biphasic catalysis and is both timely and important for this envi- ronmentally clean technology. Biphasic catal- ysis involves two immiscible liquid phases, one containing the catalyst and the other the sub- strate, so that the separation of the catalysts is drastically simplified. Many eminent scientists contribute chapters, including F. Joo and E. G. Kuntz whose papers are seminal. The editors are well-known experts, one from academia and one from industry, and t h i s is reflected in the range of contributions. The book covers other catalysts besides those of the platinum group metals, but since it describes many industri- ally important catalytic processes, ruthenium, rhodium and palladium frequently feature.
The book contains eight main chapters made up from between one and twenty-five individ- ual contributions. Certain chapters, such as that on environmental and safety aspects of bipha- sic catalysis, are essential reading for a rounded picture of the subject.
The platinum metals feature most prominently in the chapter entitled “Typical Reactions” which is more than 250 pages in length, with contri- butions from many authors. Hydroformylation is discussed first, with B. Cornils and E. G. Kuntz providing a resume of the development of a com- mercial biphasic 0x0 plant employing a water- soluble rhodium catalyst. Hydroformylation of lower and higher oletins, as well as functionalised olefins, is described, and not surprisingly, rhodium catalysts feature prominently. F. Joo and A. Katho write a section on hydrogenation which is dominated by rhodium and ruthenium catalysts. After this comes a series of shorter sec- tions beginning with carbonylation and carbon- carbon coupling reactions, the emphasis being firmly on palladium-based catalysts. Allylic sub- stitution, hydrodimerisation, asymmetric syn- thesis, fine chemical syntheses, polymerisation
and olefin metathesis are all reviewed and the r81e of the platinum metals in these reactions is described. In keeping with the underlying theme of clean catalysis, the hydrogenation and hydrogenolysis of organosulfur compounds, and dehalogenations using hydrophilic catalysts, are also covered. These sections are written by notable experts including M. Beller, J. G. E. Krauter, W. A. Herrmann, C.-P. Reisinger, D. Sinou, N. Yoshimura, S. Haber, W. C. Schattenmann, R. H. Grubbs, D. M. Lynn, C. Bianchini, A. Meli, M. Bressan and A. Morvillo.
The penultimate chapter on other biphasic concepts includes non-aqueous biphasic regimes. The section on fluorous-organic systems by I. T. Horvath covers rhodium and iridium cata- lysts. Ionic liquid-organic systems are described by H. Olivier who illustrates the use of ruthe- nium, rhodium, palladium and platinum cata- lysts. P. C. J. Kamer and P. W. N. M. van Leeuwen describe an amphiphilic approach and M. Beller and J. G. E. Krauter conclude with a section on water-soluble, polymer-bound cat- alysts. These methodologies are emerging as important - but related - alternatives to the aqueous-organic protocol.
The book is full of factual data presented in tables and graphs and as such is an invaluable source of information when coupled with the extensive bibliographies at the end of each sec- tion. There are also a large number of figures and schemes which help to clarify the text.
The editors have succeeded in producing a book of interest to everyone working with plat- inum metals in homogeneous catalysis and com- pliments their earlier volume entitled “Applied Homogeneous Catalysis with Organometallic Compounds”. It sets out ways in which organometallic catalysts can be made hydrophilic and shows their wide range of uses in biphasic catalysis for small-scale synthesis and industrial- scale work. PAUL J. DYSON
Platinum Metals Rev., 1998, 42, (4), 145 145

The Build-Up of Bimetallic Transition Metal Clusters By Paul R. Raithby Department of Chemistry, University of‘ Cambridge, England
The synthesis and reaction chemistry of high nuclearity transition metal carbonyl clusters is briejly reviewed, and new synthetic strategies leading to the “rational” synthesis of bimetallic clusters containing metal cores of over 1 nm in dimension are described. The solid state structures of a number of usmiuml mercury, osmiumlgold and rutheniumlcopper bimetallic clusters are discussed with regard to the nature of their formation, und of their bonding and redox properties. Suggestions are made as to how the synthetic strategies can be adapted to prepare bimetallic clusters of industrially useful combinations of metals. Recent work showing that bimetallic nunuparticles prepared frum clusters are catalytically active when anchored inside mesoporous silica is also discussed.
Transition metal carbonyl cluster chemistry has been an important and developing topic of research in organometallic chemistry for the last three decades (1). One of the main appeals of clusters is that they lie at the interface between ‘‘conventional” organometallic chemistry and the chemistry of colloids and of the bulk metal. Figure 1 illustrates the progression in particle size from a single atom through clusters, with metal core sizes of around 1 nm; nanoparticles, with sizes up to 100 nm; leading into the col- loid regime; and then on to the bulk metal.
Indeed, at what size (number of metal atoms) does a metal cluster stop behaving like an organometallic complex, with bonding proper- ties that can be described in terms of discrete molecular orbitals, and take on metallic prop- erties, where the bonding can be described in terms of band structure? There is no immedi- ate answer to this question, but there is a clear progression towards the clusters taking on metal-
lic properties as the nuclearity increases, although different sizes of cluster exhibit dif- ferent types of metal-like properties under different conditions (2).
One of the main thrusts of cluster chemistry at Cambridge has been to prepare ever larger transition metal clusters and to investigate their physical and chemical properties. A range of clusters containing more than ten metal atoms has now been prepared and crystallographically characterised ( 3 ) and examples in which the metal atoms “condense” to form structures cor- responding to the hexagonal, cubic and body- centred cubic packings found in bulk metal have been observed, as well as other clusters, such as [Pt19(C0)22]4- (4), which exhibit five-fold sym- metry packing.
The diameters of the metal cores in the largest of these clusters, such as [Ni,,Pt,(CO)4aH,,]”~ (n = 5, 4) are of the order of 2 nm (5). Even larger clusters containing copper and selenium
* . .. . .. ;.;: - .*. 4 -
Single Cluster Nanoparticle Colloid Bulk metal metal atom Fig. 1 The progression in particle
size from a single metal atom to Particle diameter r o i - lo2i 4 0 3 i )lo% the bulk metal
Platinum Metals Rev., 1998, 42, (4), 146-157 146

have been prepared, and the largest of these to have been crystallographically characterised is [ C U ~ ~ ~ S ~ ~ ~ ( P P ~ ~ ) ~ ~ ] in which the selenium atoms exhibit “ABA” stacking and the copper atoms occupy interstitial sites (6). There are also reports of transition metal clusters, for instance those containing Auss (7) and Rhss (8) , Pt,09 (9) and Pd,,, (1 0) units, and a series of palladium clusters containing up to 2000 metal atoms ( I I), which have not yet been crystallographically characterised, but which must have dimensions of the order of 4 nm.
Several research groups have proposed that clusters can act as good building blocks in nanoscale architecture and thus will find appli- cation in the fabrication of single electron devices (1 2 ) . Small metal particles and other transition metal clusters have also been clearly shown to form densely packed monolayers on electron microscope grids when they are ligated by organic surfactant molecules (1 3).
From the viewpoint of catalysis, metal clus- ters can be considered as fragments of a metal surface surrounded by a layer of “adsorbed” lig- and molecules. Even the largest osmium clus- ter carbonyl so far characterised, [Os,,(CO),]’~, where the metal framework is approximately 0.9 x 0.9 x 0.9 nm in size, contains only surface atoms, with each osmium atom being bonded to at least one carbonyl ligand, see Figure 2, (14).
However, can clusters be regarded as good models of metal surfaces in heterogeneous cat- alytic reactions? The “cluster/surface” analogy was pointed out quite early in the development of cluster chemistry (1 5), and ever since then clusters have been used as models for catalytic systems (1 6).
Certainly, organic molecules bond to catalyt- ically active metal surfaces in the same way as to metal clusters, and analysis of cluster systems is easier because they can be subjected to the full range of solution spectroscopic techniques, such as IR and multinuclear NMR spectro- scopies, mass spectrometry and, in the solid state, single crystal X-ray crystallography, whereas analysing the bonding modes of co-ordinated molecules on a metal surface
Fig. 2 The metal core structure of [Os,(CO),]*- showing the four triangular faces of the Osm tetrahedron
under catalytic conditions is rather more challenging. However, for the majority of clus- ter models, for example [Os,,(CO),,]’- (14), only “surface” atoms are present, and since the sub-layers of the bulk metal influence the chem- istry of the surface atoms on a catalytic surface, some aspects of the model are not valid, so the “analogy” should be treated with caution.
Therefore, the simple answer to the question of whether clusters are good models for heterogeneous catalysts would be “no”.
“Rational” Synthesis of High Nuclearity Mixed-Metal Clusters
With a view to preparing precursor materials that could have applications in catalysis and nanoparticle technology, the “ c l ~ ~ t e r ” group in Cambridge has been developing strategies for synthesising high nuclearity mixed-metal clus- ters containing ten or more metal atoms in their cluster cores. Even the smallest of these clus- ters should have a core diameter in excess of 0.5 nm and have the advantage, unlike bimetallic particles prepared by other routes, that the exact ratio of the two metallic elements is known. As much of the early cluster synthesis work
Platinum Metals Rev., 1998, 42, (4) 147

Fig. 3 The formation of the “spiked”-triangular cluster [Os,H,(CO),,] from the reaction of the activated cluster [OS~(CO)~~(M~CN)~] with [OsHZ(CO)d] I
involved pyrolysis or thermolysis techniques and resulted in a range of products, all in low yields, fiom a single reaction, it has been necessary to develop synthetic strategies where one target cluster molecule can be obtained in good yield (17).
In order to achieve this, two fairly straight- forward synthetic routes are available, given that the starting materials for the production of high nuclearity clusters are usually low nuclearity carbonyl clusters.
The first route, illustrated in Figure 3 by the formation of [ O S ~ H ~ ( C O ) ~ ~ ] , involves the acti- vation of the binary carbonyl [Os,(CO),,] with Me,NO, in the presence of MeCN and results in the oxidation of carbonyl ligands to carbon dioxide and the occupation of the vacant co- ordination sites with labile MeCN ligands. The subsequent addition of the neutral mononuclear complex [OSH,(CO)~] displaces the MeCN groups and affords the “spiked”-triangular clus- ter [OS,H~(CO),~] (18). In this method the cluster nuclearity is increased by one, by the reaction of a neutral, activated, low nuclearity
cluster with a neutral monometal complex. The second route involves the ionic coupling
reaction between a carbonyl cluster anion and a monometal cationic species, again to increase the cluster nuclearity by one. In Figure 4, the tetranuclear osmium cluster [Os,H,(CO) ,,I is initially reduced with WPh,CO to form the dianion [Os,H,(CO) ,,I ‘-, and then treated immediately with the labile cation [M(q6- C6H6)(MeCN),I2+ (M = Ru, 0 s ) to form the pentanuclear, neutral cluster [Os&W,(CO),,(q6- C6H6)] (1 9). By choosing appropriate cation and anion charges and ratios, the cluster nuclear- ity can be increased by two units, as in the reaction of [Os,(CO),,]’~ with two equivalents of [Ru(q5-CsH5)(MeCN),]’ to give the pen- tanuclear cluster [Os3Ru,(CO),, (q’-C,H,),] (20). This latter ionic coupling route has been par- ticularly successful, and has been extensively exploited to prepare a wide range of higher nuclearity clusters containing carbocyclic ligand groups (21).
The method has also been used by a number of research teams for synthesising mixed-metal
Insertion M = Ru, 05 ..!. \ a NCMe (Oc)2
[H,Os,CCO),,]
(C0)3
H ,,OS,,M(CO)~I(C~H~)
Fig. 4 [ O S ~ H ~ ( C O ) ~ ~ ] ~ ~ with [M(t16-C6H6)(MeCN),]2+ (M = Ru, 0 s )
The synthesis of [OsdMH,(CO),,(~6-C6H6)] (M = Ru, 0s) from the coupling of
Platinum Metals Rev., 1998,42, (4) 148

% P
b
Fig. 5 The structure of the “raft” cluster [{Os,(CO)rlHg}s] showing the linking ofthree Os, triangles to the central Hg3 triangle
clusters containing the coinage metals by the reaction of carbonyl cluster anions with cationic copper, silver and gold complexes (22).
Osmium-Mercury Clusters So far, our most extensive series of studies into
the formation of mixed-metal clusters contain- ing ten or more metal atoms have also involved the reaction of a range of ruthenium and osmium cluster carbonyl anions with late transition metal cations, such as [ H a ] ’ (X = C1, CF,), [AuPR,]’ (R = alkyl, aryl) and [CU(NCM~)~]’. The first indication that cluster build-up could occur to produce higher nuclearity clusters came from the metathesis reaction of [OS,H(CO)~~]~ with mercury(I1) salts. The product of the reaction was the extremely photolabile, dodecanuclear ‘‘rafl” cluster [ { Os,(CO) IHg} ,] shown in Figure 5 (23). It is significant that the three mercury atoms form a central triangle with Hg-Hg distances in the range 3.08-3.12 A. The “OS,(CO)~~” fragments bridge this central tri- angle, with each “Os(CO),” group forming bonds to two mercury atoms (0s-Hg in the range 2.71-2.76 A); the co-ordinated “Os(CO),” groups each form one bond to a mercury atom (0s-Hg in the range 2.98-3.05 A). This clus- ter can be viewed as a model for a bimetallic surface, and while being only one layer thick
has a core diameter in the surface plane of approximately 1.2 nm (12 A).
This coupling process, more correctly called redox condensation, resulting in cluster build- up, can also be achieved between the carbido- stabilised, decanuclear cluster anion [OsloC(CO)z4]z~ and [Hg(O,SCF,),] to afford the anion [ { OsloC(CO)z4}zHg]2 , Figure 6. Here the mercury atom links the two decanuclear clusters by bridging an edge of each Oslo unit, forming a cluster containing 2 1 metal atoms (24). In this reaction, the appropriate choice of the mercury(I1) salt, [Hg(O,SCF,),], is the key to the formation of the higher nuclearity mixed- metal cluster. This is partly because the deca- osmium dianion precursor has low nucleo- philicity (the negative charge being delocalised over the ten metal centres) and because there is an increased propensity for the reaction to follow alternative pathways involving partial degradation or rearrangement of the metal
L b
Fig. 6 The molecular structure of the mercury linked cluster dianion [{Os,,C(CO),,},Hg]*~
Platinum Metals Rev., 1998,42, (4) 149
0 0 0 4 P L P 4
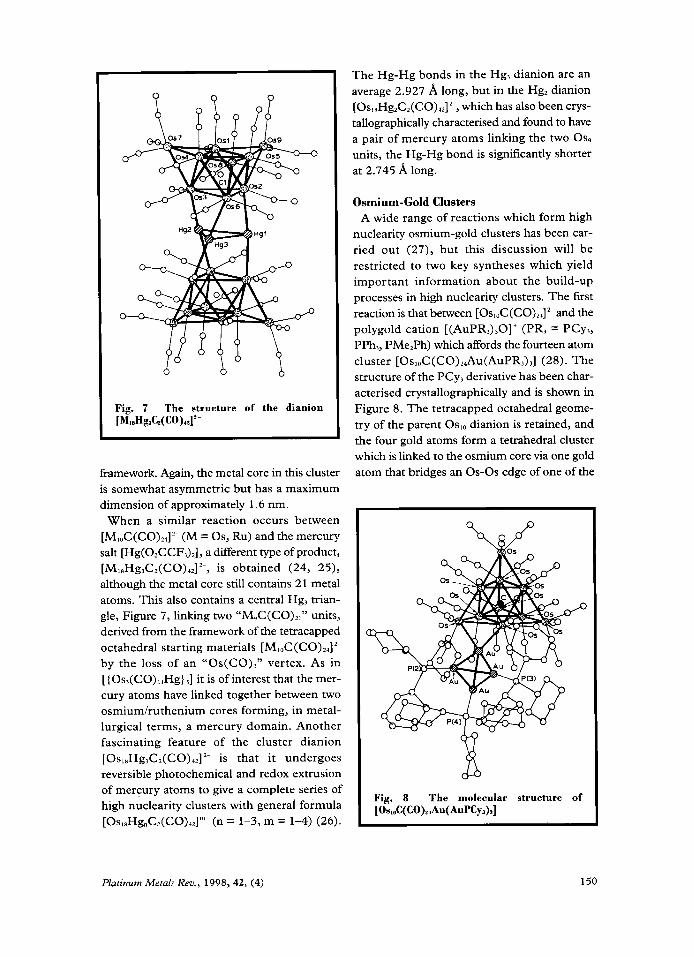
Fig. 7 The structure of the dianion [MisHgX~(Co)u]’-
framework. Again, the metal core in this cluster is somewhat asymmetric but has a maximum dimension of approximately 1.6 nm.
When a similar reaction occurs between [M,,C(CO),,]’~ (M = Os, Ru) and the mercury salt [Hg(O,CCF,),], a different type of product, [M18Hg3CZ(CO)IZ]2~, is obtained (24, 25), although the metal core still contains 21 metal atoms. This also contains a central Hg, trian- gle, Figure 7, linking two “M9C(CO)ZI” units, derived from the framework of the tetracapped octahedral starting materials [MI& ( CO)zr] 2-
by the loss of an “Os(CO),” vertex. As in [{OS,(CO),~H~} ,I it is of interest that the mer- cury atoms have linked together between two osmiumhthenium cores forming, in metal- lurgical terms, a mercury domain. Another fascinating feature of the cluster dianion [ O S , , H ~ , C ~ ( C O ) ~ ~ ] ~ - is that it undergoes reversible photochemical and redox extrusion of mercury atoms to give a complete series of high nuclearity clusters with general formula [Os,sHg,Cz(CO),2]”~ (n = 1-3, m = 1-4) (26).
The Hg-Hg bonds in the Hg, dianion are an average 2.927 A long, but in the HgZ dianion [Os,,Hg2C,(CO)42]2~, which has also been crys- tallographically characterised and found to have a pair of mercury atoms linking the two Os, units, the Hg-Hg bond is significantly shorter at 2.745 A long.
Osmium-Gold Clusters A wide range of reactions which form high
nuclearity osmium-gold clusters has been car- ried out (27), but this discussion will be restricted to two key syntheses which yield important information about the build-up processes in high nuclearity clusters. The first reaction is that between [OS,~C(CO),,]~~ and the polygold cation [(AuPR,),O]+ (PR, = PCy,, PPh,, PMeJ’h) which affords the fourteen atom cluster [OsloC (CO) ,,Au(AuPR,) ,] (28). The structure of the PCy, derivative has been char- acterised crystallographically and is shown in Figure 8. The tetracapped octahedral geome- try of the parent Os,, dianion is retained, and the four gold atoms form a tetrahedral cluster which is linked to the osmium core via one gold atom that bridges an 0 s - 0 s edge of one of the
Q I)
\8P
Fig. 8 The molecular structure of [ O S ~ ~ C ( C O ) , ~ U ( A ~ P C ~ ~ ) : ~ ]
Platinum Metals Rev., 1998, 42, (4) 150
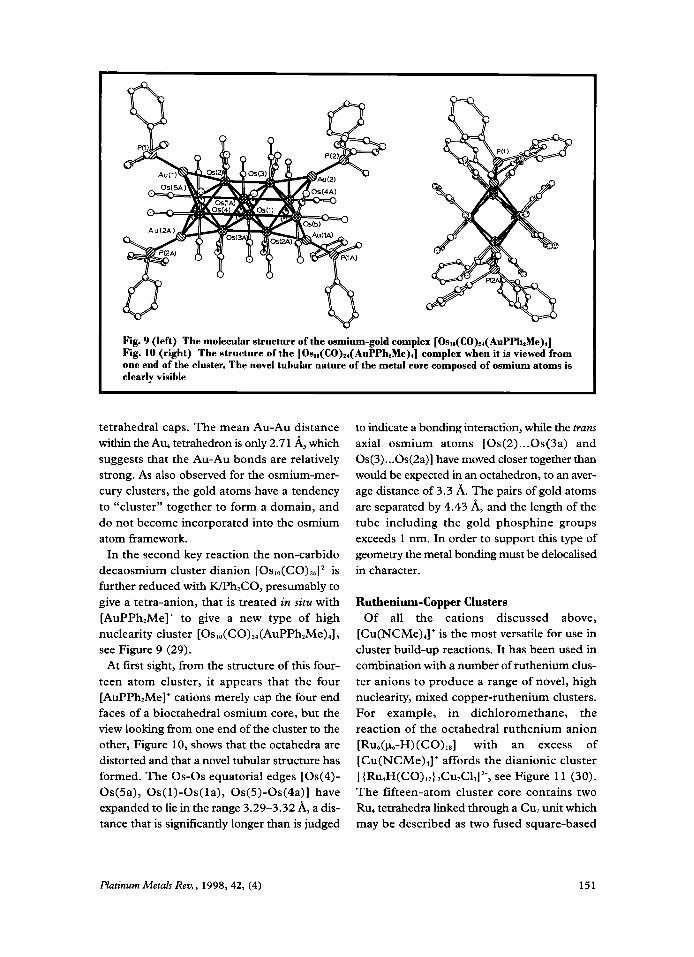
Fig. 9 (left) The molecular structure of the osmium-gold complex [Osla(CO),,(AuPPhzMe),] Fig. 10 (right) The structure of the [OS,~(CO)~,(AUPP~~M~),] complex when it is viewed from one end of the cluster. The novel tubular nature of the metal cnre composed of osmium atoms is clearly visible
tetrahedral caps. The mean Au-Au distance within the Au, tetrahedron is only 2.7 1 A, which suggests that the Au-Au bonds are relatively strong. As also observed for the osmium-mer- cury clusters, the gold atoms have a tendency to “cluster” together to form a domain, and do not become incorporated into the osmium atom framework.
In the second key reaction the non-carbido decaosmium cluster dianion [0~10(c0)26]’~ is further reduced with KPh,CO, presumably to give a tetra-anion, that is treated in situ with [AuPPh,Me]+ to give a new type of high nuclearity cluster [Oslo(CO),,(AuPPh2Me),], see Figure 9 (29).
At first sight, from the structure of this four- teen atom cluster, it appears that the four [AuPPh,Me]+ cations merely cap the four end faces of a bioctahedral osmium core, but the view looking from one end of the cluster to the other, Figure 10, shows that the octahedra are distorted and that a novel tubular structure has formed. The 0s-0s equatorial edges [Os(4)- Os(5a), Os(1)-Os(la), Os(5)-0s(4a)] have expanded to lie in the range 3.29-3.32 A, a dis- tance that is significantly longer than is judged
Platinum Metals Rev., 1998, 42, (4)
to indicate a bonding interaction, while the truns axial osmium atoms [Os(2) ... Os(3a) and Os(3) ... Os(2a)l have moved closer together than would be expected in an octahedron, to an aver- age distance of 3.3 A. The pairs of gold atoms are separated by 4.43 A, and the length of the tube including the gold phosphine groups exceeds 1 nm. In order to support this type of geometry the metal bonding must be delocalised in character.
Ruthenium-Copper Clusters Of all the cations discussed above,
[Cu(NCMe),]’ is the most versatile for use in cluster build-up reactions. It has been used in combination with a number of ruthenium clus- ter anions to produce a range of novel, high nuclearity, mixed copper-ruthenium clusters. For example, in dichloromethane, the reaction of the octahedral ruthenium anion [ R U ~ ( ~ ~ - H ) ( C O ) , ~ ] ~ with an excess of [Cu(NCMe),]’ affords the dianionic cluster [{Ru,H(C0)12)2Cu7C1,]2~, see Figure 11 (30). The fifteen-atom cluster core contains two Ru, tetrahedra linked through a Cu7 unit which may be described as two fused square-based
151

Fig. 11 The structure of the [{RulH(CO)lr}rCu,Cld]2~ dianion showing the central Cu: unit
- v CIO)
pyramids sharing a common triangular face. Three chloro ligands each symmetrically bridge pairs of copper atoms, while the remaining cop- per atom, Cu(5), forms no bonds with ligands but has eight metal contacts. Thus, the imme- diate environment around Cu(5) is similar to that in metallic copper and, overall, again it is seen that the element with the formal d'O elec- tron configuration has formed the central domain, and the transition metal units are fused to its periphery.
The product of this reaction is very sensitive to the nature of the solvent used. In the presence of [(Ph,P),N]Cl in MeCN, when [Ru,H(CO),J is treated with a large excess of [Cu(NCMe),]+, a different product, [{RU,H(CO),,),CU~C~,]~, is obtained in good yield, see Figure 12 (30). In this case degrada-
tion of the original Ru, octahedron has not occurred, and the two octahedra are linked through two Cu, tetrahedra which share a com- mon edge, generating two butterfly arrange- ments which bond to the ruthenium units. Overall, the eighteen-atom cluster can be viewed as a linear condensation of four octahedra. Two edges of the Cu, unit are symmetrically bridged by chloride ions.
For both reactions, the presence of chloride ions is apparently necessary, even if, in the first case, they are abstracted from the solvent. However, by simply altering the solvent, from dichloromethane to [(Ph,P),N] C1 in MeCN, good yields of high nuclearity clusters with ruthe- nium:copper ratios of 8:7 (approximately 1: 1) and 2: 1, respectively, are obtained from a room temperature reaction.
Fig. 12 The structure of the O(21 I [ {Ru,H(CO)17}2C~CI,]2~ dianion
Platinum Metals Rev., 1998, 42, (4) 152
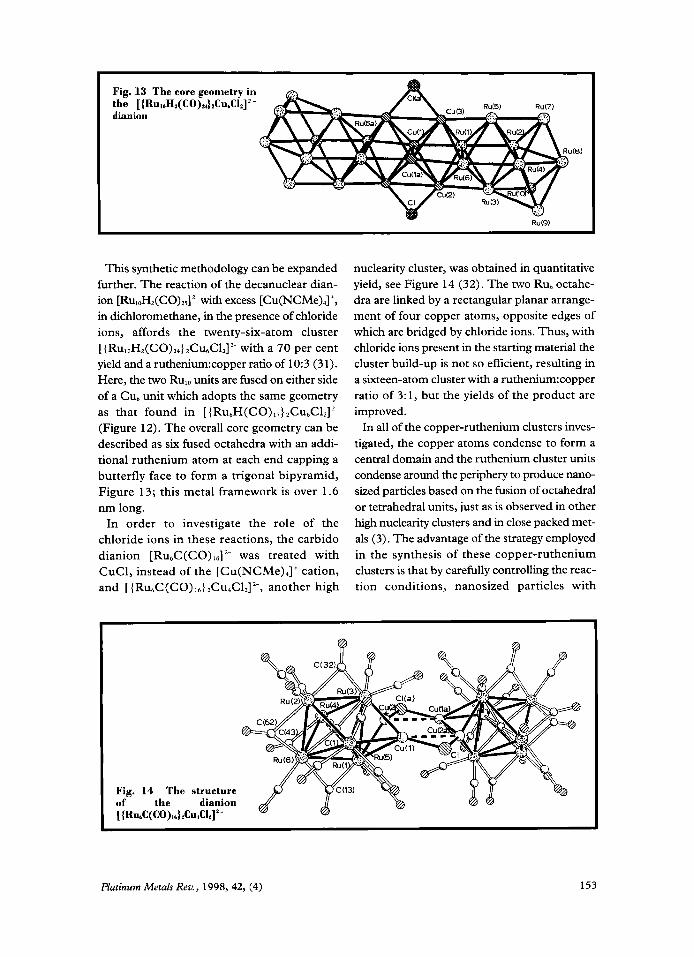
Fig. 13 The core geometry in
dianion the [ {R~~OHZ(CO)~~}~CU,CI~]~ -
This synthetic methodology can be expanded further. The reaction of the decanuclear dian- ion [RU,,H,(CO)~~]~~ with excess [CU(NCM~)~]+, in dichloromethane, in the presence of chloride ions, affords the twenty-six-atom cluster [ {RuloH,(CO),,}2C~~ClZ]~~ with a 70 per cent yield and a ruthenium:copper ratio of 10:3 (31). Here, the two Rule units are fused on either side of a Cu, unit which adopts the same geometry as that found in [ { Ru,H(CO) !,} ,CU,CI,]~- (Figure 12). The overall core geometry can be described as six fused octahedra with an addi- tional ruthenium atom at each end capping a butterfly face to form a trigonal bipyramid, Figure 13; this metal framework is over 1.6 nm long.
In order to investigate the role of the chloride ions in these reactions, the carbido dianion [RU~C(CO)~~]*- was treated with CuCl, instead of the [Cu(NCMe)J+ cation, and [ {Ru,C(CO),,)2Cu,Cl,]2~, another high
nuclearity cluster, was obtained in quantitative yield, see Figure 14 (32). The two Ru, octahe- dra are linked by a rectangular planar arrange- ment of four copper atoms, opposite edges of which are bridged by chloride ions. Thus, with chloride ions present in the starting material the cluster build-up is not so efficient, resulting in a sixteen-atom cluster with a ruthenium:copper ratio of 3: 1, but the yields of the product are improved.
In all of the copper-ruthenium clusters inves- tigated, the copper atoms condense to form a central domain and the ruthenium cluster units condense around the periphery to produce nano- sized particles based on the fusion of octahedral or tetrahedral units, just as is observed in other high nuclearity clusters and in close packed met- als (3). The advantage of the strategy employed in the synthesis of these copper-ruthenium clusters is that by carefully controlling the reac- tion conditions, nanosized particles with
Fig. 14 The structure of the dianion [ {hC( C0)w) iC~,Clz] *
Platinum Metals Rev., 1998, 42, (4) 153

Fig. 15 The network of reversible redox transformations of [Os,,Hg,,C,(CO),,]”’- (11 = 1-3, m = M), Fr = ferroeene
particular ruthenium:copper ratios can be obtained in high yields. This control has not been evident in previous studies (3).
Some Electrochemical Considerations
If higher nuclearity bimetallic clusters become “metallic” in character they would be expected to undergo extensive redox chemistry, with the cluster cores behaving as “electron sinks”. For all of the copper-ruthenium clusters described above it is possible to assign a formal oxidation state of + 1 to each of the copper atoms in the structures, despite the view that the bonding must be delocalised over the metal core. Similarly, for the tubular osmium-gold clus- ter [Osin(CO)?,(AuPPh,Me),] (Figure 9) each gold atom can be assigned an oxidation state of + 1 (29). Cyclic voltammetry studies show that this cluster undergoes two reversible one- electron reductions, indicating that there is no major change in core geometry with the uptake of two electrons (27). This is consistent with the cluster being able to act as an “elec- tron sink” since it is able to reversibly take up and release a pair of electrons repeatedly, with- out it degrading, or without any major struc- tural change that would cause the redox process to become irreversible.
The situation for the osmium-mercury sys- tems is more complicated. As noted earlier, all
the species [OS,,H~,C~(CO)~,]”’ (n = 1-3, m = 1-4) (26) have been characterised, and it is not possible to assign realistic oxidation states for the individual mercury atoms in these species. For example, in [OS~,H~,C~(CO),,]~-, in order to balance the formal -4 charge from each “OS,C(CO)~,” fragment, each mercury atom would have to be assigned an unrealistic oxi- dation state of +3. The network of reversible redox transformations shown in Figure 15 con- firms that this series of clusters can act as “elec- tron sinks” (26) with a description of the bond- ing within the metal core best described in terms of delocalised molecular orbitals.
Clusters as Nanoparticle Precursors For the chemistry outlined above it is clear
that it is now possible to prepare a wide vari- ety of bimetallic, nanosized clusters in good yield with specific, known ratios of the two metals. In principle, this strategy can be applied with- out difficulty to other combinations of transi- tion metal atoms, such as palladium-rhodium and platinum-rhodium, and high nuclearity clusters could be prepared. However, would these systems have real uses with industrial appli- cations?
It has long been established that metal clus- ters can be anchored to oxide surfaces such as silica and alumina, and even encapsulated in zeolite cages; after heating the residual metal particles have high catalytic activity (33). It is also known that bimetallic catalysts often exhibit superior operating stability to monometal sys- tems, and experiments have shown that rhenium- platinum clusters, supported on alumina, are effective catalysts for naphtha reforming (34). However in these experiments the exact nature of the cluster precursors and the catalytically active materials were not known.
Recently, Shapley and co-workers have pre- pared a set of supported bimetallic catalysts from two structural isomers of [RejIrC(C0)2,]’ by deposition onto high surface area alumina and activation in dihydrogen at 773 K (35). The spe- cific activities of the catalysts depend on both the metal framework structure and the coun- terion present in the precursor (either [NEt,]’
Platinum Metals Rev., 1998, 42, (4) 154

or [N(PPh,)2]+). Interpretation of EXAFS data has enabled specific models to be developed for the catalyst particle nanostructures which cor- relate with their catalytic activities. The more active catalysts are modelled by a hemisphere of close packed metal atoms, with an average diameter of 1 nm, with iridium at the core. In a series of related studies, Shapley has also shown that [PtRu,C(CO),,] can be used as a neutral cluster precursor for the formation of carbon- supported platinum-ruthenium nanoparticles with exceptionally narrow size and composition distributions (36). The bimetallic particles are obtained by reduction of the carbido cluster with hydrogen. A detailed structural model of the nanoparticles was deduced on the basis of in situ EXAFS, scanning transmission electron microscopy, microprobe energy-dispersive X- ray analysis and electron microdifiaction stud- ies. These experiments show that the nanopar- ticles have a Ru:Pt ratio of 5:1, an average diameter of approximately 1.5 nm and adopt a face centred cubic close packed structure. This is in contrast to the stable phase of the bulk alloy which is hexagonal close packed. The EXAFS studies also show that there is a non-statistical distribution of different metal atoms in the nanoparticles: the platinum atoms exhibit pref- erential migration to the surface of the particles under an atmosphere of dihydrogen.
Of particular interest is a recent report by Johnson, Thomas and colleagues that a clus- ter anion [ A ~ , R u , ~ C ~ ( C O ) , , C ~ ] ~ - (Figure 16) with a structure in which a central Ag, triangle links two Ru, square based pyramids, closely related to the copper-ruthenium systems
described above, had been successfully anchored inside mesoporous silica (37). Activation and anchoring of the adsorbed cluster on the MCM- 41 silica support was achieved by heating the sample under dynamic vacuum. EXAFS spec- troscopy confirmed the presence of a bimetal- lic particle anchored to the silica oxygen atoms through the silver atoms of the cluster. High- resolution electron microscopy of the heat- treated material shows a uniform distribution of the bimetallic nanoparticles aligned along the zeolite channels. The catalytic performance of the activated, supported bimetallic particles was tested for hydrogenation of hex-1-ene to hexane. Initial experiments showed a high selec- tivity (in excess of 99 per cent) and a turnover frequency of at least 6300 mol hexane per mol [Ag,RuIo] per hour.
The success of these studies led to the incorporation of the copper-ruthenium cluster anion, [ {RU,C(CO),,}~CU~CI.']~~, described pre- viously (Figure 14) (32), into the mesoporous channels of silica (38). Gentle thermolysis of the anchored clusters gives the bimetallic nanoparticles, characterised by X-ray absorp- tion and FT-IR spectroscopies, and high-reso- lution scanning transmission electron micro- scopy. The copper and ruthenium K-edge X-ray absorption spectra show that these catalytically active particles have diameters of approximately 1.5 nm and display a rosette-shaped structure with 12 exposed ruthenium atoms that are con- nected to a square base composed of relatively concealed copper atoms. In turn, these are anchored by four oxygen bridges to four silicon atoms of the mesopore. The nanoparticles are
Fig. 16 The structure of the metal core of dianion: [ A ~ J ~ & ( C O ) U C ~ ] * -
Platinum Metals Rev., 1998, 42, (4) 155

active catalysts for the hydrogenation of hex- 1-ene, diphenylacetylene, phenylacetylene, stil- bene, cis-cyclooctene and D-limonene, with turnover frequencies of 22400, 17610,70, 150 and 360, respectively, at 373 K and 65 bar of dihydrogen. The catalysts showed no tendency to sinter, aggregate of fragment into their component metals during these experiments.
Conclusions In this review it has been shown that synthetic
strategies to prepare high nuclearity, bimetallic clusters in good yields have been developed. The metal cores of these clusters have dimen- sions in excess of 1 nm. By careful control of reaction conditions it is possible to obtain spe- cific target molecules with known ratios of the two metallic components, and the methodol- ogy may be extended further to encompass the majority of the late transition elements. In the “condensed” clusters obtained, for the major-
ity of the osmiudmercury, rutheniudmercury, osmiumigold and copperiruthenium systems investigated, the mercury, gold or copper atoms form a central domain and the osmium or ruthe- nium cluster units are fused onto the periphery of these central units. In no case did the two metallic components become dispersed through- out the metal core. Lastly, evidence is begin- ning to emerge that nanoparticles derived from these and related clusters may prove to be active catalysts when anchored on silica or alumina supports.
Acknowledgements My grateful thanks go to Professor the Lord Lewis
and Professor Brian F. G. Johnson for their support and encouragement over the years, and for initiating the research described in this review. I am also indebted to the many research workers in the Department of Chemistry, at Cambridge, who have carried out the synthetic and structural work described, and to Johnson Matthey for the generous loan of the heavy transition metal salts.
References “Transition Metal Clusters”, ed. B. F. G. Johnson, Wiley, New York, 1980; “Metal Clusters”, ed. M. Moskovits, Wiley, New York, 1986; ‘‘The 9 G. Schmid, B. Morun and J.-0. Malm, Angew. Chemistry of Metal Cluster Complexes”, eds. D. F. Shriver, H. D. Kaesz and R. D. Adams, VCH publishers, Weinheim, 1990; D, M. p, ~i~~~~ 10 N. M. Vargaftik, I. I. Moiseev, D. I. Kochubey and D, J. wales, Introduction cluster and K. I. Zamaraev, Faraday Discuss. Cheni. SOC., Chemistry”, Prentice-Hall, New York, 1990; 1991,92, 13; M. N. Vargaftik, V. P. Zagorodnikov, -Clusters and Colloids. F~~~ Theory I. P. Stolyarov, I. I. Moiseev, V. A. Likholobov, D. Application”, ed. G. Schmid, VCH Publishers, I. Kochubey, A. L. Chuvilin, V. I. Zaikovsky, K. weinhei,-,-,, 1994; c, E. ~ ~ ~ ~ ~ ~ ~ ~ f t , “Metal-Metal I. Zamaraev and G. I. Timofeeva, j? Chem. Soc., Bonded Carbonyl Dimers and Clusters”, Oxford Chenz. Conzmun.y 1985, 937 University Press, Oxford, 1996
1 1 G. Schmid, in “Clusters and Colloids. From G. Schmid, J. Chem. SOC., Dalton Trans., 1998, Theory to Application”, ed. G. Schmid, VCH 1077 Publishers, Weinheim, 1994, p. 178
8 G. Schmid, Chenz. Rev., 1992, 92, 1709
Chem., Int. Ed. Engl., 1989, 28, 778
D. M. P. Mingos and A. S. May, in “The Chemistry of Metal Cluster Complexes”, eds. D. F. Shriver, H. D. Kaesz and R. D. Adams, VCH Publishers, Weinheim, 1990, pp. 11-1 19
D. M. Washecheck, E. J. Wucherer, L. F. Dahl, A. Ceriotti, G. Longoni, M. Manassero, M. Sansoni and P. ChiniJ Ant. Chem. Soc., 1979, 101,6110
A. Ceriotti, F. Demartin, G. Longoni, M. Manassero, M. Marchionna, G. Piva and M. Sansoni, Angew. Chem., Int. Ed. Engl., 1985, 24, 697
H. Krautscheid, D. Fenske, G. Baum and M. Semmelmann, Angew. Chem., Int. Ed. Engl., 1993, 32, 1303
G. Schmid, U. Giebel, W. Huster and A. Schwenk, Inorg. Chim. Acta, 1984, 85, 97
12 J. de Jongh, in “Physics and Chemistry of Metal Cluster Compounds. Model Systems for Small Metal Particles. Series on Physics and chemistry of Materials with Low-Dimensional Structures”, ed. J. de Jongh, Reidel, Dordrecht, 1994
13 M. T. Reetz, W. Winter and B. Tesche, Chenz. Conzmun., 1997, 147; T. Sato, D. Brown and B. F. G. Johnson, Chem. Conznzun., 1997, 1007; B. Dusemund, A. Hoffmann, T. Salzmann, U. Kreibig and G. Schmid, Z. Phys. D , 1991,20,305
14 L. H. Gade, B. F. G. Johnson, J. Lewis, M. McPartlin, H. R. Powell, P. R. Raithby and W.- T. Wong,J. Chem. SOC., Dalton Trans., 1994, 521
15 E. L. Muetterties, T. N. Rhodin, E. Band, C. F. Brucker and W. R. Pretzer, Chenz. Rev., 1979,19, 91
Platinuni Metals Rev., 1998,42, (4) 156
1 “Transition Metal Clusters”, ed. B. F. G. Johnson, Wiley, New York, 1980; “Metal Clusters”, ed. M. Moskovits, Wiley, New York, 1986; “The Chemistry of Metal Cluster Complexes”, eds. D. F. Shriver, H. D. Kaesz and R. D. Adams, VCH Publishers, Weinheim, !990; D. M. P. Mingos and D. J. Wales, “An Introduction to Cluster Chemistry”, Prentice-Hall, New York, 1990; “Clusters and Colloids. From Theory to Application”, ed. G. Schmid, VCH Publishers, Weinheim, 1994; C. E. Housecroft, “Metal-Metal Bonded Carbonyl Dimers and Clusters”, Oxford University Press, Oxford, 1996
2 G. Schmid, J. Chem. SOC., Dalton Trans., 1998, 1077
3 D. M. P. Mingos and A. S. May, in “The Chemistry of Metal Cluster Complexes”, eds. D. F. Shriver, H. D. Kaesz and R. D. Adams, VCH Publishers, Weinheim, 1990, pp. 11-1 19
4 D. M. Washecheck, E. J. Wucherer, L. F. Dahl, A. Ceriotti, G. Longoni, M. Manassero, M. Sansoni and P. ChiniJ Ant. Chem. SOC., 1979, 101,6110
5 A. Ceriotti, F. Demartin, G. Longoni, M. Manassero, M. Marchionna, G. Piva and M. Sansoni, Angew. Chem., Int. Ed. Engl., 1985, 24, 697
6 H. Krautscheid, D. Fenske, G. Baum and M. Semmelmann, Angew. Chem., Inr. Ed. Engl., 1993, 32, 1303
7 G. Schmid, U. Giebel, W. Huster and A. Schwenk, Inorg. Chim. Acta, 1984,85, 97

16
17
18
19
B. F. G. Johnson, J. Lewis, C. E. Housecroft, M. A. Gallop, M. Martinelli, D. Braga and F. Grepioni,J. Mol. Catal., 1992, 74, 61; B. F. G. Johnson, M. A. Gallop and Y. V. Roberts,J. Mol. Catal., 1994, 86, 51
B. F. G. Johnson and J. Lewis, Adv. Inorg. Chem. Radiochem., 1981,24, 225
E. J. Ditzel, B. F. G. Johnson, J. Lewis, P. R. Raithby and M. J. Taylor, J. Chem. SOC., Dalton Trans., 1985, 555 J. Lewis, C.-K. Li, M. C . Ramirez de Arellano, P. R. Raithby and W.-T. Wong, J. Chem. Soc., Dalton Trans., 1993, 1359
20 R. Buntem, J. Lewis, C. A. Morewood, P. R. Raithby, M. C. Ramirez de Arellano and G. P. Shields, J. Chem. SOC., Dalton Trans., 1998, 109 1
21 D. Braga, P. J. Dyson, F. Grepioni and B. F. G. Johnson, Chem. Re%, 1994,94, 1585; P. R Raithby and G. P. Shields, Polyhedron, 1998, in press
22 I. D. Salter, Adv. Organomet. Chem., 1989, 29, 249
23 M. Fajardo, H. D. Holden, B. F. G. Johnson, J. Lewis and P. R. Raithby, J. Chem. SOC., Chem. Commun., 1984,24
24 L. H. Gade, B. F. G. Johnson, J. Lewis, M. McPartlin and H. R. Powel1,J Chem. Sac., Chem. Commun., 1990, 110
25 P. J. Bailey, B. F. G. Johnson, J. Lewis, M. McPartlin and H. R. Powel1,J. Chem. SOC., Chem. Commun., 1989, 1513; P. J. Bailey, M. J. Duer, B. F. G. Johnson, J. Lewis, G. Conole, M. McPartlin, H. R. Powell and C. E. Anson, J. Organomet. Chem., 1990,383,441
26 E. Charalambous, L. H. Gade, B. F. G. Johnson, T. Kotch, A. J. Lees, J. Lewis and M. McPartlin, Angew. Chem., Znt. Ed. Engl., 1990,29, 1137; L. H. Gade, B. F. G. Johnson, J. Lewis, M. McPartlin, T. Kotch and A. J. Lees,J. Am. Chem. SOC., 1991, 113,8698
Construction of Miniature Many types of molecular cages exist in which
ions, atoms or molecules can be trapped. These cages are usually held in suspension and are typ- ically constructed from bifunctional ligands, with square planar or tetrahedral metal centres at the vertices. Until now there have been no cubic shaped organometallic cages. However, if octa- hedral transition metal building blocks could be constructed, then the assembly of cubic- shaped structures should be possible.
Now, researchers fi-om the University of Illinois have succeeded in constructing a molecular box from a cubic array of cyano-linked rhodium and cobalt octahedra (K. K. Klausmeyer, T. B. Rauchfuss and S. R. Wilson, Angew. Chem.
Tricyanometalates Et4N[Cp’Rh(CN),] and K[CpCo(CN),] (where Cp‘ = C5Me5, Cp =
Znt. Ed., 1998, 37, (12), 1694-1696).
I 1
27 Z. Akhter, Ph. D. Thesis, University of Cambridge, 1995
28 V. Dearing, S. R. Drake, B. F. G. Johnson, J. Lewis, M. McPartlin and H. R. Powell, J. Chem. SOC., Chem. Commun., 1988, 1331
29 Z. Akhter, S. L. Ingham, J. Lewis and P. R. Raithby, Angew. Chem., In?. Ed. Engl., 1996, 35, 992
30 M. A. Beswick, J. Lewis, P. R. Raithby and M. C. Ramirez de Arellano, Angm. Chem., Int. Ed. Engl., 1997,36,291
3 1 M. A. Beswick, J. Lewis, P. R. Raithby and M. C. Ramirez de Arellano, Angm. Chem., Int. Ed. Engl., 1997,36,2227
32 M. A. Beswick, J. Lewis, P. R. Raithby and M. C. Ramirez de Arellano, J. Chem. SOC., Dalton Trans., 1996,4033
33 S. Kawi and B. C. Gates, in “Clusters and Colloids. From Theory to Applications”, ed. G. Schmid, VCH Publishers, Weinheim, 1994, p. 299
34 J. H. Sinfelt, “Bimetallic Catalysts: Discoveries, Concepts and Applications”, Exxon Monograph, Wiley, New York, 1983
35 M. S. Nashner, D. M. Somerville, P. D. Lane, D. L. Adler, J. R. Shapley and R. G. Nuzzo, J. A m . Chem. SOC., 1996, 118, 12964
36 M. S. Nashner, A. I. Frenkel, D. L. Adler, J. R. Shapley and R. G. Nuzzo, J. A m . Chem. SOC., 1997,119,7760
37 D. S. Shephard, T. Maschmeyer, B. F. G. Johnson, J. M. Thomas, G. Sankar, D. Ozkaya, W. Zhou, R. D. Oldroyd and R. G. Bell, Angew. Chem., Inz. Ed. Engl., 1997, 36, 2242
38 D. S. Shephard, T. Maschmeyer, G. Sankar, J. M. Thomas, D. Ozkaya, B. F. G. Johnson, R. Raja, R D. Oldroyd and R. G. Bell, Chem. Eur. 3,1998, 4, 1214
Organo-Rhodium Boxes C5H5) were used to prepare a series of molec- ular “squares”, by reaction with [Cp’RhCl,] or [(cymene)RuCl,] (cymene = 4-isopropyl- toluene). To assemble the box from the “squares” the chloride ligands were removed by AgPF,. The “molecular boxes” of most inter- est have the structure [(C5R5)8M8(p-CN),z] (M = Rh or Co) and are a subunit of hexa- cyanometalates, of which Prussian blue is one example.
The most interesting box has alternate rhodium and cobalt atoms at the vertices, linked by CN groups. Each metal atom can adopt its preferred octahedral position. The box has edges 5.1 A long with a volume of - 132 A’, giving enough space inside to encapsulate a caesium atom. The box is also soluble so it could therefore be used for trapping molecules in solution.
Platinum Metals Rev., 1998, 42, (4) 157

Conferences Report Progress in Catalysis l lTH INTERNATIONAL SYMPOSIUM ON HOMOGENEOUS CATALYSIS
This series of meetings, which began in 1978, has regularly drawn around 400 researchers from around the world to discuss new developments in homogeneous catalysis. For this eleventh meeting, held from July 12th to 17th, the del- egates gathered at St. Andrews University, Scotland, and enjoyed traditional Scottish hos- pitality and a very full programme of both oral and poster presentations.
Professor Peter M. Maitlis On this occasion, the opening day of the sym-
posium was a celebration of the work of Professor Peter M. Maitlis, marking his 65th birthday and 45 years of chemical research. The organising committee had reflected the global reach of his influence by inviting speakers from Canada, Mexico, Japan, Russia and Israel, as well as Europe, including his own group at the University of Sheffield, U.K. The topics dis- cussed covered a wide range of applications of homogeneous catalysis in organic synthesis (for example carbonylation, cycloaddition, addition to alkenes, oxidation chemistry and C-C cou- pling). The association of Professor Maitlis’s research with current industrial developments was emphasised in the presentation by M. J. Howard (BP Chemicals, U.K.) on the iridium- catalysed Ca&aTM process for the carbonylation of methanol to give acetic acid. This has replaced the rhodium (Monsanto) process in plants in the U.S.A. (from 1995) and more recently in Korea, increasing capacity there from 200 kt to 350 kt per annum. Retrofitting of the CativaTM process in BP plants in the U.K. is underway and on completion the process will account for almost 20 per cent of worldwide acetic acid production. The first new plant using this technology is planned for operation in Malaysia in 2000.
The day concluded with a retrospective lec- ture by Professor Maitlis, reviewing some of his early collaborations. However, he did not deny himself the opportunity to describe some new results on the chemistry of complexes contain-
ing functionalised cyclopentadienyl ligands, Me,CpCH,X. The ruthenium complex [RuCl(Me,CpCH,Cl)(CO),] (I) is sufficiently
stable to allow the CH,Cl functional group to undergo a wide range of organic transforma- tions while leaving the parent complex intact. However, the carbonyl and chloride ligands are sufficiently reactive to undergo the expected reactions for such organometallic compounds, for example, substitution reactions. The complexes are active catalysts for the cyclo- propanation of styrene with ethyldiazoacetate.
Palladium-Catalysed Coupling Reactions
The considerable progress made recently in the area of palladium-catalysed coupling reac- tions was emphasised by several talks and many posters. J. F. Hartwig (Yale University, U.S.A.) discussed the use of 1,l’-bis(phosphin0) ferrocene ligands for C-C and C-X (X = N or 0) coupling. Mechanistic considerations sug- gested that the large bite angle of this ligand was effective in stabilising the reaction intermediate and that increasing the basicity of the phosphine would lead to rate enhancements. This was borne out by the comparison of 1,l’-bis- (dipheny1phosphino)ferrocene and 1, 1’-bis(di- t-buty1phosphino)ferrocene (11) ligands. The
latter proved to be an effective ligand for the coupling of aryl chlorides which were otherwise unreactive. 1,2-Disubstituted ferrocenes are also suitable ligands for this reaction and provide a convenient route for varying the substituents on
Platinum Metals Rev., 1998, 42, (4), 158-163 158

each of the phosphorus atoms. A mixed di-t- butyl/diphenyl (111) was used for the selective
mono-arylation of primary amines. J. M. Brown (University of Oxford, U.K.)
described mechanistic studies employing low temperature heteronuclear NMR to characterise intermediate species.in the catalytic cycle for the Heck reacdon involving oxidative addition, alkene insertion and reductive elimination steps. Factors influencing the insertion of palladium and rhodium into C-X (X = C, H, 0 or halogen) bonds were discussed by D. Milstein weizmann Institute of Science, Israel) for complexes con- taining 1,3-disubstituted aryl ligands (IV)
('pincer' ligands). The palladium complexes are highly stable catalysts for Heck reactions.
The application of MeO-Biphep ligands (V)
M=o@wh2
for enantioselective Heck reactions was described by P. S. Pregosin (ETH Zurich, Switzerland), while, by contrast, J. G. de Vries (DSM Research, The Netherlands) described the development of phosphine-free Heck chemistry for industrial applications. Through the use of a decarbonylative reaction with benzoic anhy- dride, the arylation of olefins is possible in good yield with easy reprocessing of the reaction by- products (CO and benzoic acid). This elimi- nates the environmental problems caused by the waste streams associated with an equivalent
Friedel-Crafts reaction. Evidence supporting both Pd(O)/Pd(II) and Pd(II)/Pd(IV) cycles as possible mechanisms for Heck reactions was presented during these many presentations and this topic provided some lively discussion.
Polymerisation Catalysts In the area of polymerisation catalysts, R. H.
Grubbs (California Institute of Technology, U.S.A.) and others described the development and application of ruthenium-alkylidene com- plexes, such as [RuCl,(CHPh) {P(c-C,H,,),},] (VI), for olefin metathesis. Improvements in the
preparation of these catalysts have been made by several groups so that they can now be pre- pared in high yield, one-pot processes and are available commercially. The initial industrial application of these catalysts will be for the poly- merisation of dicyclopentadiene, but due to the ability of the complexes to tolerate water and the presence of a wide variety of functional groups further applications will soon follow. The use of this type of catalyst for ring closing metathesis in synthetic organic chemistry was described by A. Fiirstner (Max-Planck-Institut fiir Kohlenforschung, Germany). Using an dlyli- dene complex (also described by P. H. Dixneuf, Universite de Rennes, France, along with other metallacumulenes LRu(C=C,=CRJ]) the syn- thesis of a number of large ring molecules from a,o-dienes was achieved. Functional groups in the diene play an important role in the com- plexing of the olefin to the metal allowing ring closure to predominate over oligomerisation of the diene.
Polyketone Synthesis The use of palladium catalysts for polyketone
synthesis by alternating copolymerisation of CO and olefins is being developed by Shell International. Modelling of the reaction inter- mediates was described by K. Vrieze (Universiteit van Amsterdam, The Netherlands).
Platinum Metals Rev., 1998, 42, (4) 159

The use of rigid, potentially terdentate nitro- gen donor ligands (VII) gave very high rates for
C O insertion. The strain created by the rigid backbone leads to one nitrogen donor being eas- ily displaced to create a site for C O or olefin binding. Development of a catalytic system for COktyrene copolymerisation and COiethanei styrene terpolymerisation was described by B. Milani (University of Trieste, Italy). With [Pd(bipy),] [PF,], as catalyst, the use of triflu- oroethanol instead of methanol as solvent resulted in significantly greater stability for the catalytic intermediate and hence high produc- tivity from the catalyst, and high molecular weight (-75,000) for the polymer.
Unusual Solvents Presentations on catalysis in unusual media as
solvents reflected the growing interest in this area. K. R. Seddon (Queen's University of Belfast, U.K.) discussed the potential of ionic liquids. These are good solvents for many organic, inorganic and polymeric compounds. They remain liquid over a much wider tem- perature range than conventional solvents and by selection of the constituents they may be
moisture sensitive or tolerant. The wide choice of quaternary ammonium and phosphonium salts as cations, combined with different anions, such as AICI, , BF,- and organic carboxylates, allows the properties of the solvent to be tuned to suit the needs of a specific reaction, for instance to give easy product separation.
The potential of supercritical carbon dioxide, scCO,, as a medium for catalysis was discussed in a number of posters and by a presentation by T. Sakakura (National Institute of Materials and Chemical Research, Japan). Arylphosphine com- plexes have inadequate solubility in scCOz for satisfactory catalysis, so modification of the phos- phine ligands with perfluoroalkyl substituents has been used to improve their solubility. These ligands are also applicable to catalysis in per- fluorohydrocarbon solvents, as described by A. M. Stuart (University of Leicester, U.K.). Examples of rhodium-catalysed hydroformyla- tion for higher olefins in each of these media were given in poster presentations, the main ben- efit being the easier separation of the product from the catalyst.
Many other excellent presentations were made on homogeneous catalysis using platinum group metals (pgms) and non-pgm transition metal catalysts. This high standard and the sustained interest of the many delegates will no doubt lead to continued success for these symposia. The 12th meeting is scheduled for Stockholm, from August 27th to September lst, 2000.
C . F. J. BARNARD AND W. WESTON
9TH INTERNATIONAL SYMPOSIUM ON RELATIONS BETWEEN HOMOGENEOUS AND HETEROGENEOUS CATALYSIS
The 9th meeting in this series was held in Southampton from 20th to 24th July 1998 and attracted over 200 participants, the vast major- ity coming from overseas. As is often the case, the experience and knowledge gained from studying one type of system can often be applied to others, so this Symposium on homogeneous and heterogeneous catalysis, was aimed at help- ing the flow of ideas between these two very sim- ilar areas of catalysis.
The opening plenary talk, by Jean-Mane Basset
(University of Lyon- 1, France) examined the field of immobilised homogeneous catalysis, a theme that was well represented in the remain- der of the conference. Basset's remarks that syn- ergy - as opposed to relations - between homo- geneous and heterogeneous catalysis, should be what is considered, were thus rather prophetic.
A highlight of the first day was undoubtedly the presentation by Richard Lambert (University of Cambridge, U.K.) who has developed STM (scanning tunnelling microscopy) techniques to
Platinum Metals Rev., 1998, 42, (4) 160

the point where he is able to apply surface science techniques to fine chemical synthesis. Lambert described the palladium-catalysed trimerisation of acetylene to benzene. For the Pd(0) surface, under low coverage the ben- zene molecules lie flat, whereas at high cover- age they are tilted to the palladium surface. Several isotopic studies have been conducted using C2D, to gain insight into the possible mechanism for this reaction. In conjunction with several surface techniques, the statistical distri- bution of products has indicated that the mech- anism proceeds via a C,H, metallocycle inter- mediate. Lambert found that the desorption temperature for the flat benzene coverage is higher than for the tilted configuration.
Using pseudo-real time STM he was able to identify the likely active site for alkyne coupling reactions on various Au/Pd surfaces. Extensive mixing of the two metals occurred around 500°C and palladium was found to deposit onto the gold particles in an orderly fashion. The activ- ity of these particles towards alkyne coupling increased as the palladium coverage approached a monolayer. At a coverage above one mono- layer the activity began to fall, as the palladium surface became rough (with steps and terraces, and so on) and was no longer the regular Pd( 1 1 1) surface.
Enantioselective Hydrogenation The second day was dominated by enantio-
selective hydrogenation. A. Baiker (ETH Ziirich, Switzerland) gave a broad review of the work performed on the asymmetric hydrogenation of a-ketoesters over chirally modified supported platinum metals catalysts during the last decade. His more recent work on the application of the technology to new reactants, including ketopan- tolactones, (the hydrogenated product of which is useful in the manufacture of vitamins) was also presented.
John Bradley (Max-Planck-Institut fur Kohlenforschung, Miilheim, Germany) and Robin Whyman (University of Liverpool, U.K.) reported their respective studies into the use of colloids as model catalysts for the study of enantioselective hydrogenation. As always, the
use of model catalysts to try to explain catalytic behaviour under real reaction conditions gen- erated controversy. However, Whyman’s obser- vation that minute quantities ofwater were essen- tial to attain a rate enhancement, although not yet understood, must be relevant to the rate enhancements seen under normal operating conditions.
Peter Wells (University of Hull, U.K.) pre- sented molecular modelling which supported the generally accepted mechanism for the asym- metric reaction. The same ideas were also used to explain the mechanism for the functioning of oxycodone as a modifier, the major differ- ence being that a step-site on the metal sur- face is required to enable the enantioselective site to form.
Martin Wills (University of Warwick, U.K.) spoke on two aspects of the asymmetric reduc- tion of ketones to secondary alcohols. Firstly using chiral phosphinamide catalysts in the pres- ence of a borane Lewis acid. The catalysts are air- and moisture-stable, with the reactions pro- ceeding in good yield. However, the reactions require 10 mol per cent of catalyst and high temperature conditions (1 10°C).
The second part of his talk involved ruthe- nium(I1) catalysed hydrogenation reactions. These systems have the advantage of running at room temperature which is advantageous for less stable ketones. The active species is derived from [RuCl, (p-cymene)] and a hydroxylamine ligand, such as 1 -amino-2-indanol. The rigid- ity of the indanol ligand structure is necessary for achieving the desired high stereoselective con- trol, see Figure 1. Enantiomeric excesses (e.e.) of up to 98 per cent have been obtained using just 1 mol per cent of ligand and 0.5 mol per
Fig. 1 Ruthenium catalyst giving optimal results fur ketone hydrogenation
Platinum Metals Rev., 1998, 42, (4) 161

cent of ruthenium. Both yield and e.e. were shown to decrease with different, less rigid hydroxylamine ligands.
Polymer Synthesis A series of ruthenium catalysts for ring open-
ing metathesis polymerisation (ROMP) and ring closure metathesis reactions (RCM) have been developed by R. H. Grubbs (California Institute of Technology, U.S.A.), who talked at length about his work. Some of the ruthenium carbene catalysts (VI) have been commercialised and are now being sold in kg quantities.
He stated that the activity of the catalyst depends on the phosphine ligand. Bulkier groups tend to give higher activity catalysts such that P(Cy), >P'Pr, > PPh,.
Heterogeneous analogues to these systems have been attempted by attaching the ruthe- nium to a phosphine polymer support. Unfor- tunately the supported phosphine is typically labile and undergoes ligand exchange reactions. This leads to the ruthenium becoming detached from the support and consequently leaching from the catalyst. Grubbs has also incorporated functionalised phosphine ligands into the cat- alysts to obtain water soluble metathesis cata- lysts, see Figure 2. Such catalysts require the presence of HCl to give acceptable rates and yields but do offer a great benefit for carrying out aqueous phase reactions.
L. Delaude (University of Liege, Belgium) con- tinued with the subject of ROMP catalysts in a paper on the polymerisation of 2,3-dicar- boalkoxy-norbornadienes. The active catalytic species was similar to Grubbs' Ru(I1) carbene, but without co-ordinated phosphine ligands. The catalyst was derived from [RuC12(p-cymene)],
CIh, . , ! Ph R"-/
PR 3 tl' I
Fig. 2 Water soluble ruthenium catalyst with functionalised quaternary arnine
I Fig. 3 Trimethylsilyldiazomethane (TMSD)
and was activated by the carbene precursor, trimethylsilyldiazomethane see Figure 3. The polymers produced from this reaction have high stereo-regularity reaching 99 per cent zruns con- figuration. Even in the absence of stabilising phosphines the catalysts are still quite stable. The activities of these catalysts were not signif- icantly different to those produced by Grubbs. Interestingly, the addition of a phosphine (tri- cyclohexylphosphine) to the reaction actually leads to a decrease in both yield and selectivity.
Clusters in Catalysis G. Schmid (Universitat G H Essen, Germany)
spoke of recent developments in the field of metallic cluster catalysis. Cluster molecules can be thought of as being somewhere between dis- crete molecules and bulk metal. Schmid explained how the absorbance relaxation of gold clusters changed with size. Au,, clusters exhib- ited molecular optical behaviour whereas Auss clusters acted like a bulk metal.
The transition from molecular to bulk prop- erties occurs somewhere between the two clus- ter sizes. The different clusters should exhibit properties relating to homogeneous and het- erogeneous catalysis, respectively. Recently Schmid has put clusters into nanoporous alu- mina membranes. These membranes are formed by the anodisation of aluminium and contain small channels running perpendicular to the surface. The pore walls can be functionalised with alkoxysilanes and used to trap catalytically active (cluster) species. These systems are still under development and show good potential for gas phase catalysts.
Ionic Liquids H. Olivier (Institut FranCais du Pktrole, France)
gave a lecture on the use of ionic liquids and how they can be used in reactions that involve
Platinum Metals Rev., 1998, 42, (4) 162

complexes or ligands that are either poorly sol- uble in water or unstable in water. Ionic liquids have been explored as solvents for transition metal catalysts for over 20 years, and used in hydrogenation and hydroformylation reactions (DuPont, Texaco, Unilever). A new range of room temperature non-aqueous ionic liquids (NAILS) were presented by Olivier based on organic cations (such as quaternary phospho- nium or ammonium ions) and inorganic anions (such as AlCl, , Al,Cl;, BF,, PF;).The transi- tion metal catalysts remain in the ionic liquid, giving the benefits of reduced catalyst consump- tion and disposal. By changing the combina- tions of anions and cations the ionic liquids can be tailored to industrial reactions, thus provid- ing new solvents for hard or soft metal catalysts.
Other novel ideas were presented in the two poster sessions and some of these will undoubt- edly mature in time for the next conference in this series, which is to be held in Lyon, France in 2001.
One poster, which won a poster prize was by R. L. Augustine (Seton Hall University, U.S.A.), who described the immobilisation of homoge- neous catalysts on high surface area supports such as carbon, silica and alumina. It will be interesting to see whether his work will justify the opening remarks on synergy made by Professor Basset.
The proceedings of this conference will be published in a special issue of the Journal of Molecular Catalysis.
K. E. SIMONS AND A. F. CHIFFEY
Combinatorial Chemistry Identifies Fuel Cell Catalvst The combinatorial chemical screening of large
numbers of samples has received much atten- tion in recent years, particularly due to its use in drug discovery for the identification of new leads, although the approach has been used for about twenty years to find new inorganic materials. The technique is now being adapted and applied to finding new materials (1).
Direct methanol fuel cells (DMFCs) have an advantage over other fuel cells in converting methanol directly at the anode to electricity, but poor performance has limited their commer- cialisation, the major limitations being anode and membrane performances (2). Combinatorial screening can be used to find more active elec- trochemical catalysts, but presently-used cur- rent-voltage methods are time-consuming and cumbersome for such large numbers of sam- ples. In other combinatorial screenings, a flu- orescent indicator has been used to detect the presence of ions (such as H+), the intensity of the fluorescence being an indication of activity.
Now, fluorescent detection and combinator- ial chemistry have been used by scientists at Pennsylvania State University, Illinois Institute of Technology and ICET in Massachusetts to identify a combination of platinum group met- als with improved properties, which they say may be used as the anode in a DMFC (3).
Platinum, ruthenium, osmium, iridium and rhodium were combined in a 645-electrode array, deposited onto carbon paper and screened in a methanol/fluorescent indicator medium. The most active components were selected; bulk
J
samples were characterised by various tech- niques and tested as anode catalysts in DMFCs. The best combination was Pt(44)/Ru(41)/ Os(lO)/Ir(5), which had a current density 40 per cent higher than presently used Pt-Ru at 400 mV and more than double the value under short circuit conditions.
With this method a wide range of composi- tions was searched rapidly and thoroughly, allow- ing areas of apparent inactivity to be investi- gated. In fact, the activity of this particular combination does not lie in the expected active regions. Such optical screening may be useful to identify other electrochemical materials.
References 1 Chem. Week,Aug. 12, 1998, 501, p. 18 2 M. P. Hogarth and G. A. Hards, Platinum Metals
Reo., 1996, 40, (4), 150 3 E. Reddington, A. Sapienza, B. Gurau, R.
Viswanathan, S. Sarangapani, E. S. Smotkin and T. E. Mallouk, Science, 1998, 280, (5370), 1735
The Development of the Platino-Calixarenes In the January 1998 issue of Platinum Metals
Review, on page 15, right hand column, the sev- enth line should read “obtained by reacting com- plex 3 with 4,4’-bipyridine, see Scheme v.” In the legend of Scheme V L* must be replaced by 3.
Polymers of Platinum Metals Complexes Immobilised on Electrodes
In the April 1998 issue of PIaFinum Metals Rmkw, on page 6 1, right hand column, the thirteenth line should read “[Rh(bpy)(PPhzEt)z(CI)(H)]”’.
Platinum Metals Rev., 1998, 42, (4) 163

Catalysts for Butane Reforming in Zirconia Fuel Cells By K. Kendall and D. S. Williams Birchall Centre for Inorganic Chemistry and Materials Science, Keele University, England
The ability of f ue l cells to use hydrocarbon fuels efficiently is important i f they are to compete with battery power. Solid oxide fue l cells, particularly zirconia fuel cell devices, are generally well suited to utilise a variety of fuels. They are commercially attractive, especially in remote locations where battery supply and muintenunce costs are prohibitive but where f w l , particularly butane, is readily available. Butane can be safely stored at high energy density and is thus a useful fwl for zirconia fuel cells in remote areas. Partial oxidation would be the preferred route to reform butane, but this requires a suitable catalyst. Ruthenium i s a n excellent partial oxidation catalyst, giving nearly total reformation of butane and producing high levels of hydrogen. However, prob- lems such as carbon deposition and catalyst optimisation need to be addressed. Here, work with a zirconia fuel cell successfully fuelled by butane and using a ruthenium catalyst under controlled reaction conditions is discussed.
Zirconia fuel cell devices can be small-scale and portable. They can generally use a variety of fuels, including hydrocarbons, since they are not susceptible to carbon monoxide poisoning. They do not require external reformation of the fuel.
Butane is a particularly attractive fuel because it is cheap, easily stored and is available at remote sites, where battery power is expensive. The main problem with butane, when fed directly into zirconia devices, is its tendency to deposit carbon on the fuel cell anodes. A possible solu- tion to eliminate this is by reforming the butane through the addition of steam (l) , carbon diox- ide (2), or air to bring about partial oxidation (3). For the fuel cell device described here par- tial oxidation of butane by air has been used, as this provides the simplest device construction. The system in Figure 1 shows the position of the partial oxidation catalyst upstream of the fuel cell electrodes on the zirconia tubes (4).
Catalytic Reactions Partial catalytic oxidation is widely used for
the reformation of butane (5) and other paraf- fins (6,7) to higher value chemical feedstocks,
such as formaldehyde or butadiene (8,9). Such products are not suitable as feeds for fuel cells. Therefore, new catalysts must be found to con- vert the butane to synthesis gas - the preferred form of butane reaction product for fuel cell use (10):
C,H,, + 2 0 , + 4CO + 5H, (1)
while preventing the total oxidation of the butane to water and carbon dioxide:
C,H,, + 6.502 + 4C02 + 5Hz0 (ii)
This reaction requires more than simple restric- tion of the availability of oxygen (1 l), as carbon tends to be deposited on the anode during the reaction:
C4H10 + xOz + aCO + bCO, + CC + dHz + eH,O ( i )
where the numbers x, a, b, c, d and e vary according to the conditions.
A number of catalysts are known to convert methane to synthesis gas (1 2-1 6) on a variety of catalyst supports (1 7-2 l), but methane reacts in a much simpler way than butane since it has no C-C bonds to give the intermediate hydro- carbon products formed by butane. Even so,
Platinum Metak Rev., 1998, 42, (4), 164-167 164
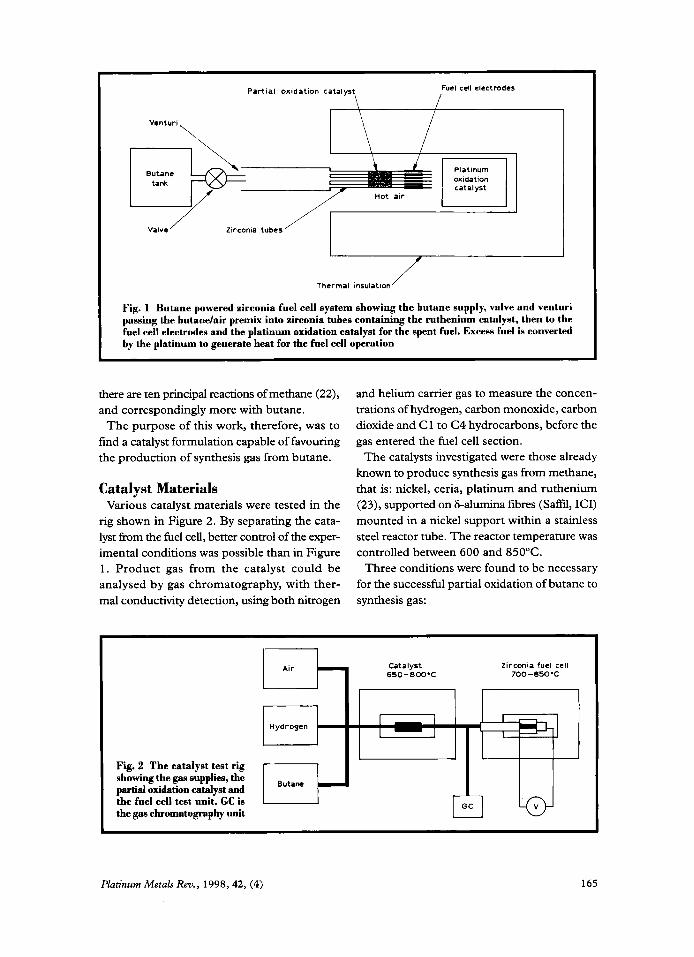
Partial Oxidation catalyst Fuel cell electrodes I
Ztrconia tubes
Thermal insulation / Fig. 1 Butane powered zirconia fuel cell system showing the butane supply, valve and veuturi passing the butanelair premix into zirconia tubes containing the ruthenium catalyst, then to the fuel cell electrodes and the platinum oxidation catalyst for the spent fuel. Excess fuel is converted by the platinum to generate heat for the fuel cell operation
there are ten principal reactions of methane (22), and correspondingly more with butane.
The purpose of this work, therefore, was to find a catalyst formulation capable of favouring the production of synthesis gas from butane.
Catalyst Materials Various catalyst materials were tested in the
rig shown in Figure 2. By separating the cata- lyst from the fuel cell, better control of the exper- imental conditions was possible than in Figure 1. Product gas from the catalyst could be analysed by gas chromatography, with ther- mal conductivity detection, using both nitrogen
and helium carrier gas to measure the concen- trations of hydrogen, carbon monoxide, carbon dioxide and C1 to C4 hydrocarbons, before the gas entered the fuel cell section.
The catalysts investigated were those already known to produce synthesis gas from methane, that is: nickel, ceria, platinum and ruthenium (23), supported on &alumina fibres (Saffil, ICI) mounted in a nickel support within a stainless steel reactor tube. The reactor temperature was controlled between 600 and 850°C.
Three conditions were found to be necessary for the successful partial oxidation of butane to synthesis gas:
Hydrogen
Fig. 2 The catalyst test rig r-I sh&iog the. gas supplies, thi d a l oxidation catalyst and I Butam
;he fuel cell test uni;. GC is the gas chromatography unit
Catalyst 650-800%
Zirconia fuel cell 700-850.C
I . I
Platinum Metals Rev., 1998,42, (4) 165
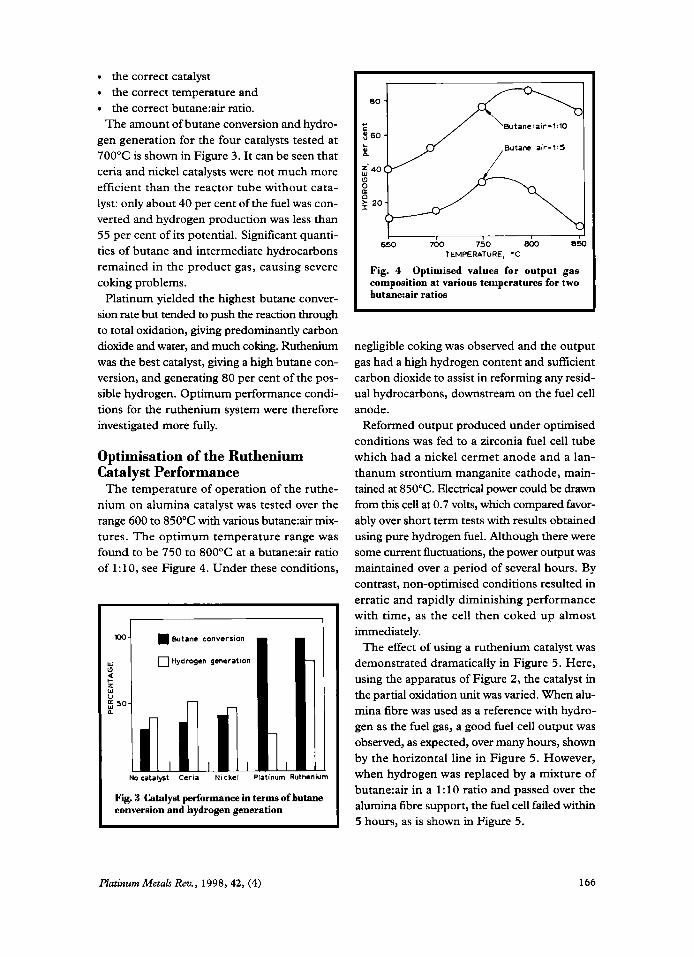
the correct catalyst the correct temperature and the correct butane:air ratio.
The amount of butane conversion and hydro- gen generation for the four catalysts tested at 700°C is shown in Figure 3. It can be seen that ceria and nickel catalysts were not much more efficient than the reactor tube without cata- lyst: only about 40 per cent of the fuel was con- verted and hydrogen production was less than 55 per cent of its potential. Significant quanti- ties of butane and intermediate hydrocarbons remained in the product gas, causing severe coking problems.
Platinum yielded the highest butane conver- sion rate but tended to push the reaction through to total oxidation, giving predominantly carbon dioxide and water, and much coking. Ruthenium was the best catalyst, giving a high butane con- version, and generating 80 per cent of the pos- sible hydrogen. Optimum performance condi- tions for the ruthenium system were therefore investigated more fully.
Optimisation of the Ruthenium Catalyst Performance
The temperature of operation of the ruthe- nium on alumina catalyst was tested over the range 600 to 850°C with various butane:air mix- tures. The optimum temperature range was found to be 750 to 800°C at a butane:air ratio of 1 : 10, see Figure 4. Under these conditions,
[7 Hydrcgen generation
NO catalyst Ceria Nickel Platinum Ruthenium
Fig. 3 Catalyst performance in terms of butane conversion and hydrogen generation
650 760 750 8k 850 TEMPERATURE, .C
Fig. 4 Optimised values for output gas composition at various temperatures for two butane:air ratios
negligible coking was observed and the output gas had a high hydrogen content and sufficient carbon dioxide to assist in reforming any resid- ual hydrocarbons, downstream on the fuel cell anode.
Reformed output produced under optimised conditions was fed to a zirconia fuel cell tube which had a nickel cermet anode and a lan- thanum strontium manganite cathode, main- tained at 850°C. Electrical power could be drawn from this cell at 0.7 volts, which compared favor- ably over short term tests with results obtained using pure hydrogen fuel. Although there were some current fluctuations, the power output was maintained over a period of several hours. By contrast, non-optimised conditions resulted in erratic and rapidly diminishing performance with time, as the cell then coked up almost immediately.
The effect of using a ruthenium catalyst was demonstrated dramatically in Figure 5. Here, using the apparatus of Figure 2, the catalyst in the partial oxidation unit was varied. When alu- mina fibre was used as a reference with hydro- gen as the fuel gas, a good fuel cell output was observed, as expected, over many hours, shown by the horizontal line in Figure 5. However, when hydrogen was replaced by a mixture of butane:air in a 1 : l O ratio and passed over the alumina fibre support, the fuel cell failed within 5 hours, as is shown in Figure 5 .
Platinurn Metals Rev., 1998, 42, (4) 166
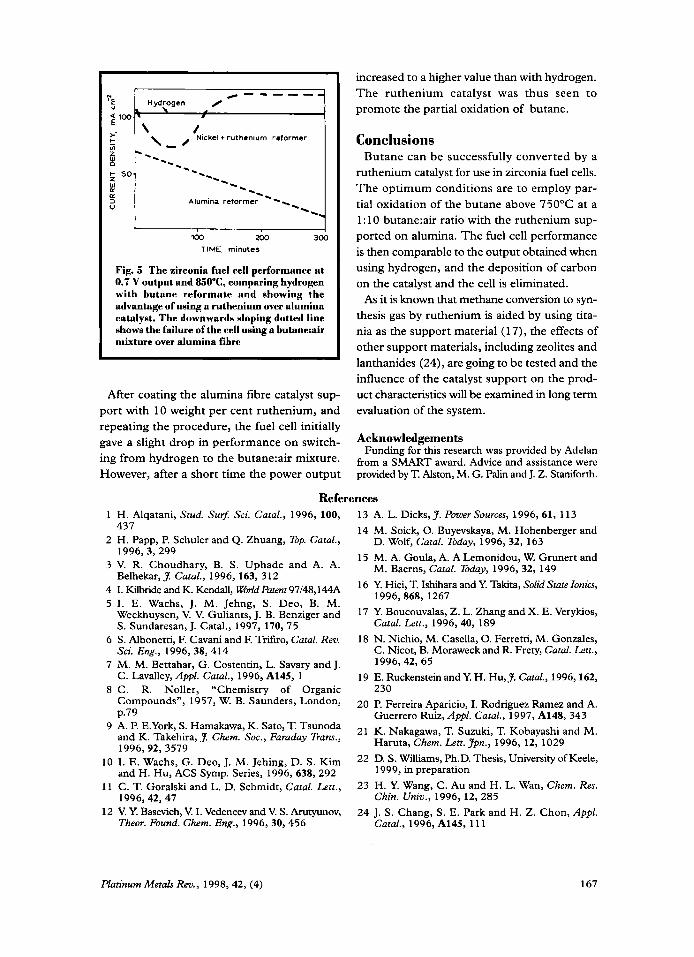
7: 4 roo.+ >- € \ E
6 I- so. w a U
After coating the alumina fibre catalyst sup- port with 10 weight per cent ruthenium, and repeating the procedure, the fuel cell initially gave a slight drop in performance o n switch- ing from hydrogen to the butane:air mixture. However, after a short time the power output
/ - - - - - -
Hydrogen / \ ,
/ Nickel + ruthenium reformer \ - / --.- -I.
-* *-I -- -- .
Alumina reformer -- --\.
increased to a higher value than with hydrogen. T h e ruthenium catalyst was thus seen t o promote the partial oxidation of butane.
Conclusions Butane can be successfully converted by a
ruthenium catalyst for use in zirconia fuel cells. T h e opt imum conditions are to employ par- tial oxidation of the butane above 750°C at a 1 : 10 butane:air ratio with the ruthenium sup- ported on alumina. The fuel cell performance is then comparable to the output obtained when using hydrogen, and the deposition of carbon on the catalyst and the cell is eliminated.
As it is known that methane conversion to syn- thesis gas by ruthenium is aided by using tita- nia as the support material (1 7), the effects of other support materials, including zeolites and lanthanides (24), are going to be tested and the influence of the catalyst support on the prod- uct characteristics will be examined in long term evaluation of the system.
Acknowledgements Funding for this research was provided by Adelan
from a SMART award. Advice and assistance were provided by T. Alston, M. G. Palin and J. Z. Staniforth.
References 1 H. Alqatani, Scud. S u ~ Sci. Catal., 1996, 100,
437 2 H. Papp, P. Schuler and Q. Zhuang, T q . Catal.,
1996,3,299 3 V. R. Choudhary, B. S. Uphade and A. A.
4 I. Kilbride and K. Kendall, W P a t a r 97/48,144A 5 I. E. Wachs, J. M. Jehng, S. Deo, B. M.
Weckhuysen, V. V. Guliants, J. B. Benziger and S. Sundaresan, J. Catal., 1997, 170, 75
6 S. Albonetti, F. Cavani and F. Trifiro, Cacal. Rev. Sci. Eng., 1996, 38, 414
7 M. M. Bettahar, G. Costentin, L. Savary and J. C. Lavalley, Appl. Catal., 1996, A145, 1
8 C. R. Noller, “Chemistry of Organic Compounds”, 1957, W. B. Saunders, London,
9 A. P. E.York, S. Hamakawa, K. Sato, T. Tsunoda and K. Takehira, J. Chem. Soc., Faraday Trans., 1996,92, 3579
10 I. E. Wachs, G. Deo, J. M. Jehing, D. S. Kim and H. Hu, ACS Symp. Series, 1996, 638, 292
11 C. T. Goralski and L. D. Schmidt, Catal. Lett., 1996,42,47
12 V. Y. Basevich, V. I. Vedeneev and V. S. Arutyunov, Theor. Found. Chem. Eng., 1996, 30, 456
Belhekar,J. Cacal., 1996, 163, 312
p.79
13 A. L. Dicks, 3. Power Sources, 1996, 61, 113 14 M. Soick, 0. Buyevskaya, M. Hohenberger and
D. Wolf, Catal. Today, 1996, 32, 163 15 M. A. Goula, A. A Lemonidou, W. Grunert and
M. Baerns, Catal. Today, 1996,32, 149 16 Y. Hiei, T. Ishihara and Y. Takita, Sold State lonics,
1996,868,1267 17 Y. Boucouvalas, 2. L. Zhang and X. E. Verykios,
Catal. Lect., 1996, 40, 189 18 N. Nichio, M. Casella, 0. Ferretti, M. Gonzales,
C. Nicot, B. Moraweck and R. Frety, Catal. Lett., 1996,42,65
19 E. Ruckenstein and Y. H. Hu,3. C a d . , 1996,162, 230
20 P. Ferreira Aparicio, I. Rodriguez Ramez and A. Guerrero Ruiz, Appl. Catal., 1997, A148, 343
21 K. Nakagawa, T. Suzuki, T. Kobayashi and M. Haruta, Chem. Letc.Tpn., 1996, 12, 1029
22 D. S. Williams, Ph.D. Thesis, University of Keele, 1999, in preparation
23 H. Y. Wang, C. Au and H. L. Wan, Chem. Res. Chin. Univ., 1996, 12, 285
24 J. S. Chang, S. E. Park and H. 2. Chon, Appl. Catal., 1996, A145, 11 1
Platinum Metals Rev., 1998,42, (4) 167
Fig. 5 The zirconia fuel cell performance at 0.7 V output and 850C, comparing hydrogen with butane reformate and showing the advantage of using a ruthenium over alumina catalyst. The downwards sloping dotted line shows the failure of the cell using a butaneair mixture over alumina fibre

Geoffrey Wilkinson and Platinum Metals Chemistry By M. L. H. Green University of Oxford, England
and W. P. Griffith Imperial College of Science, Technology and Mrtlic.inr. London
At this time, the second anniversar?,of Geoffrey WZkinson’s death on 26th September 1996, his work and influence on the development ojinorganic chemistry and the chemistry of the platinum group metals are recalled by two of his former students and colleagues. Geoffrey Wilkinson’.s early life and career, important areas of his platinurn metals research and work leading to the award in 1973 of the Nobel Prize are surveyed. He is remembered by his relationship with Johnson Matthex his work at Imperial College and by affectionute anecdotes,from the laboratory
Professor Sir Geoffrey Wilkinson, F.R.S. (or Geoff as he was always called by his students and colleagues) was one of the greatest inter- national post-war inorganic chemists; he made remarkably original contributions to many areas of transition metal chemistry, especially homo- geneous catalysis, organometallic and co-ordi- nation chemistry. His research career, spanning some 54 years, involved well over half of the ele- ments of the Periodic Table - he worked with almost every d-block transition metal, most of the lanthanides and some of the actinides and main group elements. Much of his work con- cerned the six platinum group metals, and indeed many of his most important discoveries involved them. Ofhis 557 publications (l), well over one third are concerned with the platinum metals.
The Johnson Matthey Connection The platinum metals Geoff used were invari-
ably supplied by Johnson Matthey through the loan scheme (inaugurated in 1955 by his great friend, the late Frank Lever). This scheme has done much to foster university research in plat- inum metals chemistry in the U.K. and over- seas. Over the years Geoff developed a close connection with Johnson Matthey: not only were there the patents (involving for the most part his rhodium catalysts) and consultancy, but also he had many kiends in the company - and some
of his ex-students came to work for it. On his twice-yearly visits to the Technology Centre in Sonning Common he would always take a plas- tic shopping bag filled with platinum metals residues, garnered from his research group and from his colleagues - the used materials from the loan scheme.
On his retirement in 1988 to become Professor Emeritus, Johnson Matthey expressed their appreciation of this mutually productive rela- tionship by providing Geoff with the spacious Johnson Matthey laboratory at Imperial College. Here he continued very productive work with a small, creative team to the day before his death.
In 1964 Geoffrey Wilkinson wrote an article for Platinum Metals Review on platinum group organometallic n-aromatic complexes (2). This article, however, preceded his important dis- coveries in the catalytic chemistry of these ele- ments. As a fuller account of Geoff’s chemistry has already been published (l), this report will focus on his life and work with the platinum metals in the order of their interest to him, list- ing items published in Platinum Metals Review.
Early Life and Education Geoffrey’s grandfather (also Geoffrey
Wilkinson) came to Todmorden, a small ‘cot- ton town’ in the West Riding of Yorkshire, close to Lancashire, from the Yorkshire town of
Platinum Metals Rev., 1998, 42, (4), 168-173 168

Professor Sir Geoffrey Wilkinson
A Yorkshireman by birth, Geoffrey Wilkinson started his working life, during World War 11, on the atomic bomb project in Canada and then the United States. He returned to England in 1956 to the Sir Edward Frankland Chair of Inorganic Chemistry, at Imperial College in London. He was awarded the Nobel Prize for Chemistry in 1973
1921-1996
Boroughbridge. Geoffrey’s father, Harry, married Ruth Crowther, a weaver, and Geoffrey was born on 14th July, 1921, the first of three children, in the village of Springside on the outskirts of Todmorden.
In 1926 the family moved into Todmorden, which lies at the junction of three deep valleys in the heart of the Pennines, where the sur- rounding moors rise 1000 feet above the town. The present population of 13,000 is about half that before the decline of the cotton industry, but there is still a strong local pride and sense of community which Geoff shared all his life. His eyes would light up when he spoke of “Tod” or of the superb countryside close by. He often returned to the town to see friends and family, and enjoyed walking and climbing in the area, in the Lake District and the Yorkshire Dales.
Geoff‘s interest in chemistry began early. At the age of six he was fascinated to see his father - a house painter and decorator - mixing his materials. His uncle managed a factory making Epsom and Glauber’s salts in Todmorden, and Geoff would recall how he loved to go on Saturday mornings to tinker in the small labo- ratory at t he factory. Indeed, the family hoped that he would eventually become its manager. His parents, like most people at that time, had left full-time education by the age of 12 and they were determined that their children should be
better taught. Geoff won a County Scholarship in 193 1 to Todmorden Secondary School (later Todmorden High School). A remarkable num- ber of its pupils later became famous, including Sir John Cockcroft, who worked with Rutherford at Cambridge and was to become, in 195 1, the first of the school’s two Nobel Laureates. Geoff made exceptional progress and in 1939 won a Royal Scholarship to the Imperial College of Science and Technology, London University.
At Imperial his main subject was chemistry but he also studied geology as an ancillary subject, indeed in those early days he almost gave up chemistry in favour of geology. He graduated in 1941 with a first class honours B.Sc. degree, the top student of his year, and went on to do a Ph.D. under H. V. A. Briscoe (at that time the only Professor of Inorganic Chemistry in the country) on “Some Physico- chemical Observations on Hydrolysis in the Homogeneous Vapour Phase”. This rather Delphic title conceals the fact that the main sub- strate studied was phosgene (Geoff later remarked that Briscoe “directed his Ph.D. research from a safe distance”).
In 1942 he was selected by the Joint Recruiting Board as a scientific officer at the Atomic Energy project in Canada, and sailed to Halifax, Nova Scotia in January 1943. In Canada he worked at the University of Montreal and then at Chalk
Platinum Metals Rev., 1998, 42, (4) 169

River, Ontario, on nuclear fission with many celebrated scientists -John Cockcroft (from his old school), Bertrand Goldschmidt, Charles Coryell, Alfred Maddocks (later to go to Cambridge), Jules Gueron and Pierre Auger being amongst them, and two scientists later convicted of being spies for the Soviet Union, Alan Nunn May and Bruno Pontecorvo.
After the war Geoff returned briefly to Britain and then went to the Lawrence Livermore Laboratory at the University of California, Berkeley, to work with Glenn T. Seaborg on the production of neutron-deficient isotopes of the transition elements and the lanthanides. It was said by Seaborg (and Geom that he made more artificial isotopes - eighty nine - than anyone has ever made. From this time he started to amass his vast knowledge of descriptive inor- ganic chemistry, since in those days it was essen- tial for nuclear chemists to have a profound knowledge of the chemistry of the transition metals, the lanthanides and the actinides in order to devise appropriate means of separating and identifying the products of nuclear fission reac- tions. One of his nuclear transmutations was that of platinum into gold, which caught the public imagination after a report in 1948 in the ‘San Francisco Chronicle’ (“Scientist discovers gold mine in the cyclotron”).
In 1950 he went to M.I.T. and turned to co- ordination chemistry research. His first paper on this concerned the isolation of the unusual zerovalent complex, [Ni(PCl,),] (3). In 1951 he was appointed Assistant Professor of Chemistry at Harvard, and it was here that he did the research on ferrocene and other cyclopentadi- enyl compounds which was to lay the corner- stone of his Nobel Prize. In 1976, after this award in 1973, he received his knighthood.
The Structure of Ferrocene and Early Platinum Metals Work
In 1951, the joint recognition by Wilkinson and R. B. Woodward of the unique “sandwich” structure of ferrocene (bis(cyclopentadienyl)iron, Cp,Fe) was perhaps the most crucial point in his career; it launched the new wave of ‘organo- transition metal chemistry’ which remains to
this day. Twenty years later Geoff wrote a vivid personal account of the discovery (4). From 1952 to 1953 he made a number of other bis(cyclopentadieny1) complexes, including those of ruthenium, rhodium and iridium. During this period he used the fledgling technique of nuclear magnetic resonance (NMR) to show that cova- lent metal hydrides (in this case Cp2ReH) gave high-field ‘H NMR shifts. This was crucial to his later work on rhodium hydrido complexes and their catalytic properties.
Return to Imperial College In 1955 Geoff was appointed to Briscoe’s old
chair at Imperial College ~ still the only estab- lished chair of inorganic chemistry in Britain - and arrived there in January 1956. At 34 he was one of the youngest professors that the College has ever had, and here he did most of his plat- inum metals work. It is tempting to trace this profound interest in platinum metals chemistry to his wartime and early peacetime radiochem- ical work, when he had made new radioisotopes of rhodium and ruthenium. However, it is much more likely that his fascination with these met- als derived from his early experience and knowl- edge of their general chemistry, and in partic- ular with the remarkable versatility of oxidation state changes exhibited by the metals, later harnessed for his catalytic work.
Rhodium Chemistry Geoff once said that much of his chemistry
concerned the ‘three Rs’ - rhodium, ruthenium and rhenium. At Berkeley, as part of his radio- chemical work, he isolated the short-lived ‘%h, one of the many fission products of ‘W, and in 1953 he made salts of the [CpzRh]’ cation. Then in 1961, in work that he himself carried out, he reacted cis- and tr~ns-[RhCl~(en)~]’ with sodium borohydride in aqueous solution to give [RhHCl(en)’]’ (detected by the high-field shift of the hydridic proton by ‘H NMR) (5). It was on this occasion that he rushed into the labo- ratory, demanded a Bunsen burner and a test tube, and returned later with the tube full of a foaming brown liquid which he brandished about, calling “Who wants a Ph.D?” This early
Platinum Metals Rev., 1998, 42, (4) 170

work led to a paper on the isolation of [RhH2(en),] (BPh,), and the establishment of the reduction of quinone to quinol by hydride transfer from [RhH(trien)Cl]+. In collaboration with that wizard of platinum metals chemistry, A. R. Powell of Johnson Matthey, salts of [~UIH(NH,)~]’’ and [RhH(H20)(NH,)4]” were isolated; these materials were used to prepare hydrido complexes of ethylenediamine and propylenediamine (6).
Hydrogenation and Hydroformylation with Rhodium Complexes
Geoff’s work on this topic has revolutionised our view of homogeneous catalysis effected by transition metal complexes and constitutes some of his most celebrated work. It is well reviewed in articles by his graduate student, Fred Jardine, who was the first to isolate the compound RhCI(PPh,),, universally known as Wilkinson’s catalyst (7).
In 1965 Geoff reported that catalytically small amounts of reducing agents (such as hypophos- phorous acid, zinc amalgam and dihydrogen itself) would catalyse the otherwise slow sub- stitution reactions of rhodium(II1) complexes. He had earlier shown that RhCl,.nH,O would absorb dihydrogen and convert hex-1 -ene to hexane, and in 1965 he found that fac- RhCI,(PPh,), would convert hex-1-ene to n- heptaldehyde, with dihydrogen and carbon monoxide under pressure at 55°C. However RhCI,(PPh,), is difficult to make, and it was dur- ing an attempt to make some that RhCI(PPh,), was produced. This compound was a much more effective catalyst for the hydrogenation of alkenes and alkynes and also hydroformylated hex-l- yne to n-heptaldehyde and 2-methylhexalde- hyde. RhCl(PPh,), was made from the surpris- ingly simple reaction between RhCl,.nH,O in ethanol with excess triphenylphosphine. It is described in a classic paper of 1966 (8).
Although RhCI(PPh,), is a hydrogenation cat- alyst (and subsequently chiral analogues were developed by others for asymmetric synthesis, for example for L-Dopa) Geoff later showed that it was not a hydroformylation catalyst, and that the compound responsible for the latter process
was RhH(CO)(PPh,),. Nowadays most of the butyraldehyde used for synthesis of bis(2-ethyl- hexyl)phthalate, a plasticiser for PVC, uses RhH(CO)(PPh,), as the catalyst.
The early hydrogenation and hydroformyla- tion work was documented in a short article in this journal exactly thirty years ago, with fuller papers in 1975 and 1988 (9). There is no doubt that this catalysis work, together with his work in so many other areas, contributed to his Nobel prize of 1973, though the citation was for “sand- wich” compounds.
Other Work with Rhodium One of Geoff‘s major fascinations (foreshad-
owed in his only paper in this journal (2)) was with homoleptic alkyl and aryl complexes, mainly of the early transition metals. In 1968 he made [Rh(CZHj)(NH,)5]” and, much later, in 1988 and 1990, isolated salts of the remarkable methyl complex [Rh(CH,),]’ and of the dimeric oxo- bridged neopentyl complex R ~ & I - O ) ~ [ C H ~ C - (CH,),],, respectively. Another achievement was the synthesis in 1991 of Rh(mesityl),, which has a pyramidal structure in the solid state.
Ruthenium Chemistry Ruthenium he called “an element for the con-
noisseur’’. Again his first approach to this metal was via its radiochemistry in his early work in Canada and the United States, followed by the preparation of ruthenocene Cp,Ru and the ruthenicinium [Cp,Ru]+ cation as a logical fol- low-up to his classic ferrocene paper.
In the 1960s he isolated a number of ruthe- nium(I1) and (111) complexes of phosphines, arsines and stibines, including RuX,(LPh,), (X = C1, Br; L = P, Sb) and RUX,(LR,)~(CH,OH) (L = P, As). These were precursors for many other complexes with a wide variety of ligands, such as dithiocarbamates, amines, nitriles and carboxylates, and a number of them had use- ful catalytic activities. Another highlight was the isolation of the first paramagnetic second-row transition metal complexes, Ru2(OCOR),C1 (R = Me, Et, n-Pr). A variety of carboxylato com- plexes of the form RuH(OCOCH,)(FPh,), were found to be efficient hydrogenation catalysts for
Platinum Merals Rev., 1998, 42, (4) 171

The Nobel Prize for chemistry awarded to Sir Geoffrey Wilkinson in 1973 for his work on “sandwich” compounds
alk-1-enes. A review of this and other work on ruthenium carboxylates has been described by Steve Robinson, one of his former students ( 10).
His later work with ruthenium was also very productive. In 1986 the novel aluminohydride complexes L,HRuAIH(p-H),AlH(p-H)2RuHLj (L = PMe,, PEtPh2, PPh,) were obtained from R u C l L and lithium aluminium hydride. In the mid-1 980s he isolated the alkyl complexes RU~(IV)(~-O)~% (R = neopentyl, CH,SiMe,), the tetrahedral homoleptic RuR., (R = o- CH,C,H,, mesityl), salts of [Ru(CH,),]’-, and some unusual ruthenium(n3 and (V) imido com- plexes. Alkyl complexes had become of increas- ing interest to him; in fact, his Nobel Prize award speech in 1973 was entitled “The long search for transition metal alkyls” (1 1) and in 1993 he wrote a review paper on the homoleptic alkyls and aryls of the platinum group metals ( 12).
Osmium Chemistry Although he made ruthenocene and the
ruthenicinium cation in 1952, soon after his fer- rocene work, it was E. 0. Fischer (with whom he shared the Nobel Prize) who first made osmocene, Cp20s, in 1958. Geoff‘s first paper at Imperial College concerned K[OsO,N], but apart from this he came to osmium chemistry relatively late in his research career.
In the 1980s he made the acetato complexes: [Os(OCOCH3),(PMe~)ll Cl, OS~OCOCH,),C~@Y), and sky-blue K[OsO2(0COCH,),] .2CH,COOH, the X-ray crystal structure of which was obtained
in 1982. His most celebrated work with osmium however lay with the aryl and imido com- plexes -he liked to refer to such highly unusual species as “text- book cases”. He made the alkyl complexes Os(VI)O(CH2SiMe,), and also the dimeric neo-pentyl
In 1984 the tetrakis phenyl com- plex Os(IV)Ph, was isolated and in 1988, the tetrahedral complexes Os(IV) (2-CH3C,H ,), and [Os(V)(2-CH,C6H,),]+ were prepared. At that time, tetrahedral co-ordination was unprece- dented for the tetra- or pentavalent oxidation states of second or third-row transition elements.
For imido chemists, the ‘Holy Grail’ was the isolation of a homoleptic complex containing the =NR ligands, and in 1991 Geoff achieved this with the isolation and structural character- isation, by electron diffraction, of the tetrahe- dral tertbutylimido complex Os(1V) (NBu),.
OS*(vI) (O~CCHI)~(CH~S~M~J)+
Iridium Chemistry Geoff did some work in the late 1960s on “irid-
ium iodate” (1 3), but he then seems to have neglected the metal until 1989 when he isolated salts of [Ir(CH,),]’-.
In 1991 he made the tetrahedral complexes Ir(IV)R.,, where R is a sterically hindered aryl, such as 2-tolyl, 2,5-xylyl; and in 1992 he made Ir(mesityl),. In the same year he made Ir(mesityl), which, like its rhodium analogue, has a pyramidal structure in the solid state. Also in 1992 he isolated Ir(mesityl)2(SEt2)2, a very rare example of a planar iridium(I1) complex, by reaction of mer-IrC1,(SEt2), with the Grignard reagent Mg(rnesityl),(SEtJ2.
Palladium and Platinum Chemistry Of the six platinum group metals, palladium
and platinum received far less attention from Geoff than did the other four, perhaps because
Platinum Metals Rev., 1998, 42, (4) 172

these elements are less versatile in their oxida- tion states and also, perhaps, because other well- known chemists in the country were doing much palladium and platinum work. But, between 1966 and 1970, he did show that the zerovalent complex Pt(PPh,), reacts with CS, to give Pt(PPh3)z(CSz) , while the reaction of Pt(PPh,)2(0,) with CO, CO, and CS2 yielded Pt(PPh,)2(C0,), Pt(PPh,),(C04) (a peroxocar- bonate) and Pt(PPh,),(OZCS2), respectively.
Wilkinson the Man For both of the authors of this article, Geoff
was an academic supervisor in the late 1950s. We wrote, in our joint obituary of him for ‘The Independent’: “The spirit in his research group was more like that of an urgent gold rush in the West than the scholarly and disciplined calm expected in academia.” (1 4). If anything this understates the truth: he expected his students to work as hard as he did - seven days a week or at least six, from early morning to late evening. He was, however, not a slave driver and was gen- erally tolerant of eccentric behaviour. When thwarted or stirred he made creative and inge- nious use of expletives - quite unsuitable for quotation here - and he always had a ready sense of fun. His enthusiasm was always infectious, and he was an excellent raconteur and anec- dotalist, with a remarkable memory. In those days (the late 1950s) Geoff would often emerge from his office in the late afternoon and wander up to each student in turn and say “Well, what’s new?”: his whole ethos being the search for some new aspect of chemistry.
He was a severe critic of derivative chemistry
which he would dismiss as ‘stamp collecting’. Sometimes when a reaction seemed not to be working he would offer the suggestion “Why don’t you goose it up” meaning, raise the tem- perature. Geoff was not sympathetic to theo- retical chemistry and would often cite the story of how the brilliant young Harvard theoretician Bill Mofit t had advised him that bis-benzene chromium would be unlikely to be stable. E. 0. Fischer shortly afterwards reported this famous compound.
Geoff was always in a great hurry to publish new results and, from time to time, this led to errors; one example was the reaction product between thiophene and iron pentacarbonyl which was published as thiopheneirontricar- bonyl. Gordon Stone later showed that there was no sulfur present and the product was the unexpected butadieneiron tricarbonyl. Geoff was not given to sulking over such matters but looked forward to the next new compound.
Geoff was a doughty fighter for chemistry in the U.K., writing in blunt style to Prime Ministers, ministers of education, vice chan- cellors and others charged with the care of fun- damental scientific research. He would refer to such powerful people in administration dismissively as “the apparatchiks”.
Geoffrey Wilkinson was a remarkable scien- tist and an unforgettable person. His belief that innovative and creative synthesis is a powerful tool for new chemistry is borne out by his vast range of scientific achievements. His legacy to his former students and his enthusiastic influ- ence on chemistry have given us all many long- term, far reaching benefits.
References 1 M. A. Bennett, A. A. Danopoulos, W. P. Griffith
and M. L. H. Green,J. Chem. SOC. Dalton Trans., 1997,3049
2 G. Willdnson, Platinum Metals Rev., 1964,8, (l), 16 3 J. W. Irvine and G. Wilkinson, Science, 195 1,113,
4 G. Wilkinson, J. Organomet. Chem., 1975,100,
5 G. Wilkinson, Proc. Chem. Soc., 1961, 72 6 K. Thomas, J. A. Osborn, A. R. Powell and G.
Wilkinson,J. Chem. Soc. (A), 1968, 1801; A. R. Powell, Platinurn Metals Rev., 1967, 11, (2), 58
742
273
7 F. Jardine, Rhodium Express, 1997,16,4; F. Jardine, Prog. Inorg. Chem., 1981, 28, 63
8 J. A. Osborn, F. H. Jardine, J. F. Young and G. Wil!&son,J. Chem. Soc. (A), 1966, 171 1
9 Platinum Metals Rev., 1968, 12, (4), 135; F. J. Smith, op. cit., 1975,19, (3), 93; M. J. H. Russell, op. cit., 1988, 32, (4), 179
10 A. Dobson and S. D. Robinson, Platinum Metals Rev., 1976, 20, (2), 56
1 1 G. Wilkinson, Nobel Foundation, 1974 12 G. Wilkinson, Sci. Progress, 199314, 71, 15 13 Platinum Metals Rev., 1969, 13, (4), 152 14 “The Independent”, October 1, 1996
Platinum Metals Rev., 1998, 42, (4) 173

ABSTRACTS of current literature on the platinum metals and their alloys
PROPERTIES The Pd I Spectrum, Term System, Isotope Shift and Hyperfine Structure - Revised and Extended Analysis Based on FTS Emission Spectroscopy R. ENGLEMAN, U. LITZEN, H. LUNDBERGandJ.-F. W A R T , Phys. Scr., 1998, 57, (3), 345-364 The spectrum of a neutral Pd atom emitted from hol- low cathode discharges was studied by Fourier trans- form spectroscopy in the 1750-55000 t f region. 684 lines were identified as transitions between 67 even and 76 odd levels. Isotope shift and hyperfine struc- ture were seen and interpreted in 24 lines. Forbidden lines caused by Stark-effect mixing were observed. Most Pd I lines below 64000 cm-' are now known.
Formation of Thin Single-Wall Carbon Nanotubes by Laser Vaporization of RhlPd- Graphite Composite Rod
Y. MANIWA, T. HANYU and Y. ACHIBA,Jpn. j? Appl. Phys.,
Single-wall C nanotubes (1) were prepared in high yield by laser vaporisation of a Rh/Pd-graphite com- posite rod at 1200°C. Lattice constants of the bundle were found to be 1 .&1.5 nm. Nine Raman peaks orig- inating from the breathing modes were observed, and these frequencies and lattice constants indicate the presence of the (1) indexed from (53) to (8,s) which are thinner than (1) obtained with a NiiCo catalyst.
H. KATAURA, A. KIMURA, Y. OHTSUKA, S. SUZUKI,
1998,37, (5B), L616-L618
CHEMICAL COMPOUNDS Facile Synthesis of Isomerically Pure cis- Dichlorodiammineplatinum(II), Cisplatin V. YU. KUKUSHKIN, A. OSKARSSON, L. I . ELDING and N. F A R R E L Inorg. Synth., 1998, 32, 141-144 The rapid and facile one-step synthesis of isomeri- cally pure cis-[PtCl,(NH,),] (cisplatin), an important anticancer agent, is described. This involves heating a mixture of K,[PtCl,], NH,CO,CH, and KCl in H,O under reflux.
Pd(I1) and Pt(I1) Complexes with Chalcogenide Derivatized Phosphathia Ligands J. CONNOLLY, A. R. J. GENGE, s. J. A. POPE and G. REID, Polyhedron, 1998, 17, (13-14), 2331-2336 The mixed phosphine sulfide/thioether and phosphine selenide/thioether ligands L'-L' react with PdC1, or PtC1, in the presence of TIFF, in MeNO, solution to give the distorted square planar complexes [Pd(L)](PF,), or [Pt(L)](PF&. The complexes are characterised by IR spectroscopy and "'Pt NMR stud- ies and the X-ray crystal structure of L' is reported.
Face-Coordinated C, Complexes with Carbido Peutarutheuium Cluster Cores Including a Bimetallic Platinum-Pentaruthenium Complex K. LEE and J. R. SHAPLEY, Organometallics, 1998, 17,
The interaction of C,,, with RU,C(CO),~ or PtRusC- (CO),,(COD) in hot chlorobenzene, followed by treat- ment with solubilising phosphines, gave compounds with a hexahapto co-ordination of C,,) to a Ru, face of the square pyramidal Ru,C or octahedral PtRu,C cluster framework. The C,,-cluster bond is robust.
Convergent and Divergent Noncovalent Synthesis of Metallodendrimers
NIBBERING, F. c. J. M. VAN VEGGEL and D. N. REINHOUDT, J. Am. Chenz. SOL., 1998, 120, (25), 6240-6246 A new building block, with one pyridine and two kinet- ically inert complexed Pd(I1) ions, is reported for con- trolling the assembly of metallodendrimers by a con- vergent or a divergent route. A double pincer ligand was cyclopalladated with Pd[CHICN],(BF,)2 and con- verted to a neutral bis-I'd chloride complex (1). The pyridine moiety of ( l ) , covalently attached to the spacer bridging the two pincer complexes, co-ordi- nates to activated Pd centres. Via pyridine- and cyano- based building blocks, dendrons up to the third gen- eration were assembled and characterised, by divergent and convergent routes, respectively.
Preparation and Structure of [Ru(CO),- ( PPh3)(q-CsMe5)] [Fe3( ps-C2Bu')(CO)9] M. I . BRUCE, N. N. ZAITSEVA, B. w. SKELTON and A. H. WHITE,AUSt. 3 Chew., 1998, 51, (5), 433435 The reaction between RuCI(C=CHBu')(PPh,)(q- C,Me,) and Fe2(C0)9 produced the Ru salt [Ru(CO),(PPh,)(q-CSMe,)][Fe,(p,-CZBu')(CO).]. X- ray structure studies showed a piano-stool structure for the cation while the anion contained a CCBu' ligand sitting on a triangular Fe, cluster.
A Simple and Convenient Synthesis of cis/trans-RuH2(Ph2PCH2PPh,), and of trans-
G. s. HILL, D. G. HOLAH, A. N. HUGHES and E. M. PROKOPCHUK, Inorg. Chim. Acta, 1998, 278, (2), 22 6-2 2 8 Treatment of a suspension of cis-RuC1,(Ph2PCH,Ph,), (cis-1) in EtOH or 2-propanol with a large excess of KOH, gave cisitrans-RuH,(Ph,PCH,PPh,)2 (2) in 5 90% yields, with the reaction taking < 2 min in EtOH. A similar reaction with a small excess of KOH gives trans-RuHCI(Ph,PCH,PPh2)2 in 64% yield, which can be converted into (2) in moderate yields. Under these conditions, (trans-1) is unreactive.
(14), 3020-3026
W. T. S . HUCK, 1.. 1. PRINS, R. H. FOKKENS, N. M. M.
RuHCI(Ph2PCHZPPh2)z
Platinum Metals Rev., 1998, 42, (4), 174-178 174

Ruthenium Nitrosyl Complexes with N- Heterocyclic Ligands
J. Z-SCHPECTOR, s. c. SILVA and D. w. FRANCO, Znorg. Chem., 1998,37, (1 l), 2670-2677 The synthesis of a series of Ru nitrosyl complexes of formula rrans-[Ru(NH,),L(NO)] (BF,),, where L = imidazole, L-histidine, pyridine or nicotinamide, is described. The compounds have relatively high v(N0) stretching frequencies showing that a high degree of positive charge resides on the co-ordinated nitrosyl group. The nitrosyl complexes react with OH-. The crystal structure of trans-[Ru(NH,)mi~NO]~(SiF~), confirms the presence of a Ru"-NO' moiety.
Reaction of (Ru(PPh,),Cp},(p-C,) with Tetracyanoethene: Macrocycle Formation by Intermolecular CN Coordination M. I. BRUCE, r. 1. LOW, B. w. SKELTON and A. H. WHITE, N e w 3 Chem., 1998,22, (5), 419-422 Reactions between (Ru(PPh,),Cp},(p-C4) and C,(CN), add the cyanocarbon to one of the C-C bonds forming the allylic complex Ru{q3-
(1). In solution, (1) is in equilibrium with its dimer (2). Structural studies of (2) show the presence of a ten-membered macrocyclic ring formed by displace- ment of the co-ordinated double bond in one mole- cule of (1) by a CN group from a second molecule.
S. DA S. S. BORGES, C. U. DAVANZO, E. E. CASTELLANO,
C(CN)zC [CEC { Ru(PP~&CP}] C=C(CN),} (PPh3)Cp
A Catalytic Hydrogen Wave of the Osmium- Cysteine System M. KAWASAKI, T. KAKIZAKI, W. H U , K. TOYOTA and K. HASEBE, Electroanalysis, 1998, 10, (4), 276-278 Os(VIII)O, in the presence of cysteine in HC1, exhibits a well-defined maximum wave at -0.85 V (vs. SCE). The effects of the 0 s valence states and the coexist- ing anions on the reduction wave have been studied polarographically in acidic solution. The high, as opposed to low, axidation states (Os(VII1) and (VI)), greatly influence the height of the H, wave.
PHOTOCONVERSION Photocatalytic Degradation of Trichloro- benzene using Immobilized Ti0, Films Containing Poly(tetrafluoroethy1ene) and Platinum Metal Catalyst H. UCHIDA, S. KATOH and M. WATANABE, Electrochim. Acta, 1998, 43, (1P15), 2111-21 16 A new photocatalyst film is described which has Pt- loaded Ti0, and poly(tetrafluoroethy1ene) particles immobilised on an In-Sn oxide glass substrate. This gives rapid and complete degradation of trichloroben- zene in dilute aqueous solutions under illumination, with higher catalytic activity than Pt-TiO, or TiOJNi- PTFE films. The decomposition rate was enhanced by the efficient consumption of photogenerated elec- trons and holes in the reduction of 0, and oxidative
Enhancement of ~ ~ l ~ ~ ~ l ~ ~ ~ ~ ~ d ~ ~ ~ i ~ degradation of trichlorobenzene, respectively.
Hyperpolarizabilities in Ruthenium(I1) 4,4'- Large Enhancement in Photocurrent BipYridinim Complexes by N-PhenYlation Efficiency Caused by UV Illumination B. J . COE, J. A. HARRIS, L. J. HARRINGTON, J. c. JEFFERY, of the Dye-Sensitized Heterojunction L. H. REES, s. HOUBRECHTS and A. PERSOONS, Inorg. Chem., 1998,37, (13), 3391-3399 Dipolar Ru(I1) tetra- or pentaammine complexes of N-substituted 4,4'-bipyridinium ligands show static first hyperpolarisabilities, Po, among the largest reported for transition metal complexes. The d6, 18- electron Ru(I1) centres function as powerful rr-elec- tron donors, and the nonlinear optical properties are readily tuned by ligand changes. N-phenylation of 4,4'-bipyridinium ligands is shown as an effective means to increase Po in MLCT-based chromophores.
ELECTROCHEMISTRY Electrocatalytic Dehalogenation of chloroaromatics on Palladium-Loaded Carbon Felt Cathode in Aqueous Medium A. I. TSYGANOK, I. YAMANAKA and K. OTSUKA, Chem. Lett. Jpn., 1998, (4), 303-304 The selective dechlorination of highly toxic chloroaro- matic herbicides based on phenoxyacetic acid is reported. This was achieved under mild conditions in H 2 0 by electrocatalytic reduction using a Teflon membrane-separated flow-through cell with a 5 wt.% P d C felt cathode and a Pt foil anode. After 4 hours, all the compounds underwent > 90% conversion giv- ing 80-93% yield of C1-free phenoxyacetic acid. This method is also applicable to other chloroaromatics.
Ti02/RuLL%CS/CuSCN: Initiation and Potential Mechanisms B. O'REGAN and D. T. SCHWARTZ, Chem. Muter., 1998,
When subjected to low-power UV illumination for 10-30 min, the wide band-gap, dye-sensitised het- erojunctions, n-Ti0,lRu-dyelp-CuSCN (Ru-dye = Ru polypyridyl dyes) undergo a dramatic increase in efficiency. The W illurnination increases the incident photon-to-current efficiency for light absorbed by the dye by a factor of 5-1 0 and increases the open circuit voltage by 1OC-300 mV. This effect is stable for months after the W illumination has ceased.
Photocatalytic Activity of RuSz/SiO, for Water Decomposition K. HARA, K. SAYAMA and H. ARAKAWA, Chem. Lett. B n . ,
HI and 0, were produced from the photocatalytic decomposition of H 2 0 using a RuS, powder catalyst (1) in the presence of sacrificial agents. The activity towards H, production was greatly improved by sup- porting (1) on SiO,, with a 1 wt.% RuS,/SiO, catalyst giving 213 pmol of Hz after 46 hours. No 0, was formed over a non-supported (l), but 0, was pro- duced over 1 wt.% RuS2/Si0, (121 pnol 0, after 25 hours) and 0.2 wt.% Pt/l wt.% RuS2/Si0, with the yields increasing with increasing irradiation time.
10, (9, 1501-1509
1998, (9, 387-388
Platinum Metals Rev., 1998, 42, (4) 175

Fine-Tuning the Electronic Properties of Binuclear Bis(terpyridyl)ruthenium(II) Complexes
R. ZIESSEL,Angezo. Chem. Int. Ed., 1998, 37, (12),
Two Ru(terpy)-based binuclear chromophores with vastly improved photophysical properties are described in which the butadiynylene bridge is interspersed with either a 1,4-phenylene or a 5,5’-(2,2‘-bipyridylene) spacer. Further improvement in the photoproper- ties was achieved by complexation of cations, such as Zn‘+, Cd” or BaZ+, to vacant co-ordination sites of the aromatic nucleus in the central unit, due to a better blending of the respective LUMO levels.
M. HISSLER, A. EL-GHAYOURY, A. HARRIMAN and
1717-1720
APPARATUS AND TECHNIQUE Sulphur Dioxide Gas Detection by Reversible q’-SO,-Pt Bond Formation as a Novel Application for Periphery Functionalised Metallo-Dendrimers M. ALBRECHT, R. A. GOSSAGE, A. L. SPEK and G. VAN KOTEN, Chem. Commun., 1998, (9), 1003-1004 Multimetallic dendrimers, functionalised at their periphery with square planar Pt(I1) metal centres, reversibly absorb SO, to yield macromolecules with significantly enhanced solubility. Drastic colour changes from colourless to bright orange occur in the presence of traces of SO2 as low as 10 mg dm-’, giv- ing highly active sensors for toxic SO, gas detection.
Pd-Doped SnO, Thin Films Deposited by Assisted Ultrasonic Spraying CVD for Gas Sensing: Selectivity and Effect of Annealing D. BRIAND, M. IABEAU, J. F. CURRIE and G. DELABOUGLISE, Sens. Actuators B: Chem., 1998, 48, (1-3), 395-402 Polycrystalline Pd-doped SnOl thin films (1) (0.25-1.75 pm) have been deposited on Si nitride by spray pyrolysis at 460-540°C. The gas sensitivity of (1) was tested in air for CO (300 ppm), EtOH (100 ppm) and CH, (1000 ppm). Those synthesised at 46&500”C are most sensitive to CO and, in the steady state, sensitivities 5 4500 were obtained for the thinnest films at 100°C. Cross-sensitivity to EtOH and CH, was observed. Annealing under air at 500°C for 12 hours stabilises the microstructure and gives a 2-10 fold increase in CO sensitivity.
Effect of Plasticizer Viscosity on the Sensitivity of an [Ru(bpy),2’(Ph4B-),] -Based Optical Oxygen Sensor A. MILLS and M. D. THOMAS, Analyst, 1998, 123, (5), 11 35-1 140 The quenching of the electronically-excited, lumophoric state of [Ru(bpy),Z’(Ph,B-),] by 0, was studied in various neat plasticisers. The compatibil- ity of the polymer-plasticiser combination is the dom- inant factor in determining the O2 sensitivity. For highly compatible combinations, such as TPP-PMMA, the plasticiser with the lowest viscosity, TPP, produces films of the highest O2 sensitivity.
Electrochemiluminescence Oxalic Acid Sensor Having a Platinum Electrode Coated with Chitosan Modified with a Ruthenium (11) Complex C:Z. ZHAO, N. EGASHIRA, Y. KURAUCHI and K. OHGA, Electrochim. Acta, 1998, 43, (1&15), 2167-2173 An electrochemiluminescence (ECL) sensor con- taining a Pt electrode coated with a tris(2,2’-bipyri- dine)Ru(II)-modified chitosan responded to oxalic acid (1) more strongly than to other substrates, includ- ing malkylamines. The ECL response to (1) was repro- ducible within 5% over 10 runs and the calibration curve gave a straight line in the concentration range 0.1-10 mM with a detection limit of 3 x 10 M.
HETEROGENEOUS CATALYSIS Mechanistic Considerations for the Reduction of NO, over PtlA1203 and AI2O3 Catalysts under Lean-Burn Conditions R. BURCH, J. A. SULLIVAN and T. c . WATLING, Catal.
The reduction of NOx under lean-burn conditions is compared over a series of catalysts and the reaction mechanisms are divided into 2 classes. In (l), NOx reduction occurs on the Pt surface (such as C,H,-NO- 0, over Pt/Al,O,), and is active at the lowest tem- peratures and resistant to S poisoning. In (2), DeNOx reactions OCCUT on ALO, with a weakly adsorbed reduc- tant (such as C,H,-NO-02 over Pt/Al,O, and A120,, and C,H,-N02-0, over Al,O,). They are strongly poi- soned by S and occur via formation of a surface nitrate species on the ALO, which activates the reductant.
Pt/MCM-41 Catalyst for Selective Catalytic Reduction of Nitric Oxide with Hydrocarbons in the Presence of Excess Oxygen R. LONG and R. T . YANG, CUtal. Lett., 1998, 52, (1, 2), 91-96 0.5-5 wt.% PtlMCM-41 (1) catalysts were used for the selective catalytic reduction of NO with CW, C& C,H, and C,H, in the presence of excess 02. High activity was seen with C2H, or C,H, as the reductant, with a maximum NO reduction rate of 4.3 mmol g-’ h-’ achieved with 1000 ppm NO, 1000 ppm C,H,, 2% O2 and He as the balance. Little or no activity was observed with CH, or C,H,. (1) showed good stabil- ity and H,O and SO2 did not cause deactivation.
Activity and Stability of Two Polymer- Supported Rhodium-Based Catalysts for the Vapour Phase Carbonylation of Methanol
Today, 1998,42, (l-z), 13-23
N. DE BLASIO, E. TEMPESTI, A. KADDOURI, C. MAZZOCCHIA and D. J. COLE-HAMILTON, 3. Card., 1998, 176, (I), 2 5 3-2 5 9 Rh catalysts supported on a diphenylphosphinated copolymer of styrene and divinylbenzene (SDT) or polyvinylpyrrolidone (PVP) were tested for the car- bonylation of MeOH at 80 bar and 180-190°C. WPVP catalysts showed excellent activity and selec- tivity, as well as very high stability, with no Rh leach- ing during 50 h testing, unlike W S D T catalysts.
Platinum Metals Rev., 1998, 42, (4) 176

Hydroformylation of I-Octene under Atmospheric Pressure Catalyzed by Rhodium Carbonyl Thiolate Complexes Tethered to Silica H. GAO and R. J. ANGELICI, Organometallics, 1998, 17,
The SO,-tethered Rh thiolate complex catalysts Rh-SISiO, and Rh-S-P/SiO, were prepared by the con- densation of SiO, with Rh,[pS(CH2),Si(OCH,),],- (CO), or R~,[C~-S(CH,),S~(OCH,),] ,[Ph2P(CH,),- Si(OC,H,),],(CO),. These catalysts were highly active for the hydroformylation of 1-octene in the presence of phosphine donor ligands at 60°C and 1 atm. The high activity resulted from the stabilisation of Rh(SR)(CO),(PR3) species on the catalyst surfaces.
Effect of Support on the Conversion of Methane to Synthesis Gas over Supported Iridium Catalysts
T. SUZUKI, Y. TENG, T. KOBAYASHI and M. HARUTA, Cad.
The production of synthesis gas from CH, (1) via par- tial oxidation was studied using Ir catalysts supported on various metal oxides. The reaction proceeded via a two-step process consisting of combustion of (1) to give H,O and CO, followed by the reforming of (1) from CO, and steam. The combustion and reform- ing of (1) from steam was not dependent on the cat- alyst support, but reforming of (1) from CO, with Ir was highly dependent on the support in the order: TiO,> ZrO, 2 Y,O, > LazO, > MgO 2 AlzO, 5 SiO,.
(14), 3063-3069
K. NAKAGAWA, K. ANZAI, N. MATSUI, N. IKENAGA,
Lett., 1998, 51, (3, 4), 163-167
HOMOGENEOUS CATALYSIS Asymmetric Direct a,p-Functionalization of Allenes v ia Asymmetric Carbopalladation K. HIRoI, F. KATO and A. YAMAGATA, Chem. Lett. Jpn.,
The asymmemc direct a$-functionalisation of allenes with chiral phosphine ligands in the presence of Pd(dba), in various solvenrs is described. The reac- tion of racemic allenes with iodobenzene and a nucle- ophile (malonate carbanion) using chiral phosphines occurred with extremely high enantioselectivity. However, a similar reaction of the chiral allene using an achiral phosphme ligand proceeded with complete enantiospecificity,
Heterocycles via Pd Catalysed Molecular Queuing Processes. Relay Switches and the Maximisation of Molecular Complexity R. GRIGG and v. SRIDHARAN, Pure Appl. Chem., 1998,
Pd(0) catalysts facilitate the orderly assembly of com- plex heterocycles and carbocycles containing 3-7 membered rings from diverse building blocks (allenes, CO, alkenes, etc.) by polymolecular queuing. Certain compounds are identified as relay switches because they extend the relay phase of the cyclisation-anion capture cascade while the Pd catalysed cascades switch between inter- and intra-molecular processes.
1998, (3, 397-398
70, (5), 1047-1057
Palladium-Catalyzed Cross-Coupling Reactions in Supercritical Carbon Dioxide D. K. MORITA, D. R. PESIRI, s. A. DAVID, w . H. GLAZE and W.TUMAS, Chem. Commun., 1998, (13), 1397-1398 The C-C bond coupling Heck and Stille reactions are reported in supercritical C 0 2 (scC0,) with various phosphines giving rates and selectivities comparable to those in toluene. Fluorinated phosphines, in par- ticular tris [3,5-bis(trifluoromethy)phenyl] phosphine (l), give high conversions (> 99%) as they enhance the solubility of metal complexes in scC0,.
A Highly Active Palladium Catalyst System for the Arylation of Anilines J. P. SADIGHI, M. c. HARRIS and s. L. BUCHWALD, Tetrahedron Lett., 1998,39, (30), 5327-5330 A 0.5 mol% Pd(OAc),/DPEphos system, where DPEphos is bis [2-(diphenylphosphmo)phenyl] ether, was a highly active catalyst for the arylation of pri- mary anilines by aryl bromides giving products in 5 99% yield. This system is effective for electron-poor anilines and electron-rich aryl bromides.
Palladium-Catalyzed Carbon-Nitrogen Bond Formation: A Novel, Catalytic Approach Towards N- Arylated Sulfoximines C. BOLM and J. P. HILDEBRAND, Tetrahedron Lett., 1998,
Pd(OAc),, in the presence of chelating bisphosphines, catalyses the coupling of sulfoximines with aryl bro- mides (l), giving N-arylated products. High yields ( I 96%) were obtained with (1) containing electron- withdrawing groups in the ortho- or para-positions.
Rhodium Cationic Complexes Using Dithioethers as Chiral Ligands. Application in Styrene Hydroformylation
39, (32), 573 1-5734
A. OREJON, A. M. MASDEU-BULTO, R. ECHARRI, M. D&GUU, J. FO&S-ChER, C. CIAVER and C. J. CARDIN, J. Organomet. Chem., 1998,559, (1-2), 23-29 The dithioethers (-)-DIOSR, (R = Me, 'Pr) (2,3-0- isopropylidene-1,4-dimethyl (and diisopropyl) thioether-L-threitol) react with [Rh(COD),] Clop (COD = 1,5-cyclooctadiene) in CH,Cl, to give [Rh(COD)(DIOSR2)]C10,. These were active cata- lyst precursors for styrene hydroformylation, giving conversions of 2 99% at 30 atm and 65"C, with regio- selectivity in 2-phenylpropanal as high as 74%.
Transition Metal Catalysis in Fluorous Media: Application of a New Immobilization Principle to Rhodium-Catalyzed Hydrogenation of Alkenes
I. T. HORVATH and J. A. GLADYSZ, Catal. Today, 1998,
Biphase systems comprise toluene solutions of vari- ous alkenes and CF,C,F,, solutions (1) of the pre-cat- alyst, CIRh[P(CH2CH2(CF,)rCF,)3]3 (1.1-0.8 mol%) (1). After 8-26 hours under 1 atm of H, at 45"C, hydrogenation products were extracted in 98-87% yields from (l), which could be reused.
D. RUTHERFORD, 1. I . J. JULIETTE, C. ROCABOY,
42, (4), 381-388
Platinum Metals Rev., 1998, 42, (4) 177

New Catalysts and Methods for Highly Enantioselective Metal Carbene Reactions M. P. DOYLE, Pure Appl. Chem., 1998, 70, (5), 1123-1 128 Chiral diRh(I1) carboxamidate catalysts, with bridg- ing chiral pyrrolidone, oxazolidinone, azedinone or imidazolidinone ligands, are effective for highly enan- tio-, diastereo- and regioselective syntheses of lac- tones and lactams by cyclopropanation, cycloprope- nation, C-H insertion, and ylide derived reactions of diazoacetates and diazoacetamides. Reactions occur with high turnover numbers and give products in high yield with enantiomeric excesses 2 90%.
Rhodium Complex-Catalysed Allylic Alkylation of Allylic Acetates R. TAKEUCHI and N. KITAMURA, New J. Chem., 1998,
[Rh(COD)Cl],-P(OPh), (P:Rh = 2-3) is an efficient catalyst for the allylic alkylation of allylic acetates giving products in < 90% yield. Alkylation at the more substituted allylic terminus is predominant.
22, (7), 659-660
Selective Aerobic Oxidation of Primary Alcohols Catalyzed by a Rh(PPh3)3C12/ Hydroquinone System A. HANYU, E. TAKEZAWA, s. SAKAGUCHI and Y. ISHII, Tetrahedron Lett., 1998, 39, (31), 5557-5560 The selective aerobic oxidation of primary alcohols to aldehydes, even in the presence of secondary alco- hols, was catalysed by a Ru(PPh,) ,Cl,/hydroquinone system under atmospheric 0, at 60°C. Aliphatic and cyclic primary alcohols gave aldehydes in good yields, while allylic alcohols gave unsaturated aldehydes in high yields without intramolecular H transfer.
Homogeneous Catalysis. Use of a Rnthenium(I1) Complex for Catalysing the ene Reaction w. w . ELLIS, w. ODENKIRK and B. BOSNICH, Chem. Commun., 1998, (12), 1311-1312 The complex rrans-[Ru(salen)(NO)(H,O)]' (1) catal- yses the ene reaction between activated enophiles and olefins to give homoallylic alcohols by a stepwise process. Using 1 mol% of (l), (+)-citronella1 was con- verted 10 I-isopulegol after 6 h in 80% yield. It may be possible to use chiral analogues of this catalyst for asymmetric catalytic intramolecular ene reactions.
and Mechanism Of Catal~sed Oxidation of Formaldehyde by Cerium(1V) in Aqueous Sulfuric Acid Media . .
D. KAR, s. K. MONDAL, M. DAS and A. K. DAS, 3. Chem. Res. (S), 1998, (7), 394-395
FUEL CELLS Studies of the kinetics and mechanism of the Ir(II1) (-1 0-6 mol dm-') catalysed oxidation of formaldehyde to formic acid by Ce(IV) were performed in aque- ous H,SO,. An intermediate, involving an association of the catalyst, substrate and oxidant, was formed prior to the electronic transfer step and the Ir(III)/Ir(IV) catalytic cycle.
Cationic Ruthenium Allenylidene Complexes as a New Class of Performing Catalysts for Ring Closing Metathesis A. FURSTNER, M. PICQUET, c . BRUNEAU and P. H. DIX~TEUF, Chem. Commun., 1998, (12), 1315-1316 The cationic 18-electron allenylidene Ru complexes [Ru=C=C=CR,Q(CI)(arene)]PF, (L = PCy,, PPt,), were found to be excellent catalyst precursors for ring closing olefm metathesis. Particularly important are the smooth cyclisations ofthe conformationally flex- ible dienes to 16- and 18-membered cycloalkenes.
Catalysis in Aqueous Solution: Hydrogenation of Benzene Derivatives Catalysed by (@- CJh)2R~2Clr E. G. FIDALGO, L. PLASSERAUD and G. SUSS- FINK,^ Mol. Catal. A: Chem, 1998, 132, (l), 5-12 The catalyst precursor (q"-C,H,),Ru2Cl, was used for the hydrogenation of benzene and various alkyl-ben- zene derivatives. Under biphasic conditions, cyclo- hexane derivatives were obtained with turnover rates of 20-2000 cycles per hour. The less active species, [(qo-CbH,),Ru,H,]" and [(rl~'-C,H,),Ru,H,]'+, were found in the reaction mixture after the catalytic runs. A more active intermediate, [Ru,(~~'-C,H~)~(CI~-CI)~~,- O)(pm-H),]' was also detected.
ELECTRICAL AND ELECTRONIC ENGINEERING
Carbon Supported and Unsupported Pt-Ru Anodes for Liquid Feed Direct Methanol Fuel Cells L. LIU, c . PU, R. VISWANAI'HAN, Q. FAN, R. LIU and E. s. SMUI'KIN, Elecmchinz. Acta, 1998,43, (24), 3657-3663 The performance of supported (1) (< 0.8 mg cm-l, Pt-Ru (l : l) /C) and unsupported (2) (Pt-Ru (1:l)) catalysts was compared in DMFCs having a reversible H reference electrode. The measured specific activi- ties of (1) were 3 times higher than (2) but mem- brane electrode assemblies made with (1) showed no improvement with loadings > 0.5 mg c d . Fuel cells with 0.46 mg cm-' supported electrodes performed as well as unsupported electrodes with 2 mg cm-'.
Electrical and Structural Properties and Phase Diagram of a Molecular Superconductor P- [(CH&N] [Pd(dmit),I2 A. KOBAYASHI, A. MIYAMOTO, R. KATO, A. SATO and H. KOBAYASHI, Bull. Chem. SOC. Jpn., 1998, 71, (5), 997-1006 P-[(CH,),N] [Pd(dmit),], (1) is isomorphic to [(CHJ,N] mi(dmit)2], which is the first pure n accep- tor molecular conductor exhibiting a superconduct- ing transition. The phase diagram of (1) resembles that of typical organic superconductors. The super- conducting phase appeared at 6 9 kbar. (1) has a char- acteristic "pre-superconducting region" at - 5.5 kbar, where resistivity decreases very rapidly with lowering temperature. The highest Tc was 6.5 K.
Platinum Metals Rev., 1998, 42, (4) 178

NEW PATENTS ELECTROCHEMISTRY Insoluble Anode TOBATA SEISAKUSHO K.K. Japanese Appl. 10172,690 An insoluble anode has an electrode substrate with at least one of Ti, Ta, Nb, Zr, and an electrode active layer of Pt group metal, such as Ru, Rh, Pd, Os, Ir or Pt, supported on the electrode substrate through a diffusion layer. The anode is used in electrolytic sur- face treatment, including electroplating. The anode displays superior durability at high current densities and during electrolysis.
ELECTRODEPOSITION AND SURFACE COATINGS Activation Bath METAL ARTS CO. INC. US. Patent 5,753,304 An activation bath comprises 0.1-2 g of a Pd salt, 20-250 g of an alkali metal fluoride or hydrofluoric acid, 0.05-0.5 1 of carboxylic acid as a complexing agent, 1-3 g of an alkali metal salt of gluconic acid, 1-5 g of an Fe salt, 10-30 g of a Ni salt and suffi- cient deionised HzO to make 1 gallon. The bath is used for the electroless plating of Ni onto Al-containing substrates, such as automobile wheels and computer discs. The process is efficient and requires fewer steps.
Oxidation Resistant Coatings GENERAL ELECTRIC CO. US. Patent 5,759,380 A protective CrRuAl-based coating is formed on a shaped substrate by electrodeposition of Ru and Cr, followed by aluminising by heating in a powder pack to form the final coating. The Cr is 55-70 vol.% of the Cr and Ru layers. The coating may be formed on internal surfaces, especially on Nb-based substrates in jet engine components.
Glossy Palladium Plating Bath OKUNO PHARM. m. K.K. Japanese Appl. 9/235,69 1 A plating bath contains 1-40 g 1 ' of Pd, 0.04-6 mol 1 ' of an NH, compound and aromatic sulfonamide or sulfobenzoic acid imides. The pH of the HZO- soluble Pd salt mixture is > 10. The plating is per- formed at 20-50°C with a current density of 0.1-10 A dm'. The plating has superior corrosion resistance, antiwear and electrical properties. It is used for electric contact points and connector circuit substrates.
Thin Platinum Films TONG YANG CEMENT c o w . Japanese Appl. 10184,086 A thin Pt film is formed on a substrate used in elec- tronic components by depositing Pt on an insulated oxide layer on a substrate under an oxidising amos- phere, followed by heating to form an 02-free Pt layer with a specified orientation, preferably (200). This method forms pure Pt thin films positively oriented in the polarisation direction. The electronic compo- nent has greatly enhanced performance and an improved fatigue effect.
APPARATUS AND TECHNIQUE Electric Connection for Oxygen Detection
A high melting point electrically conductive connec- tion for I.C.E. lambda probes, used for detecting the O2 content in exhaust gas, connects a contact point with a contact member. These are separated by a dif- fusion active layer of a Pd-Ni alloy, 2-20 pm thick. The contact member is heated to a welding temper- ature at the contact point. The contacts have high temperature and mechanical stability with relatively large surface area electric contact.
Electrode for Detection of Nitric Oxide UNN. DUKE World Appl. 98114,639A An electrode for detecting NO, especially in biolog- ical samples, has a surface, preferably comprising Ru and/or Ru oxide, which forms a complex, preferably a nitrosyl complex, with NO. A specific electrode is formed by conditioning such an electrode in saline at +675 mV for 2 hours. The electrode has a sensitiv- ity for NO in the nM range, a response time of a few seconds, and is stable in biological fluids and tissue.
Exhaust Gas Sensor GENERAL MOTORS c o w . US. Patent 5,733,504 An exhaust gas sensor for I.C.E. comprises inner and outer electrodes separated by a solid, porous elec- trolyte. A porous protective coating which covers the outer electrode is coated with a microporous com- posite layer made of 80-99.998 wt.% ceramic and the remaining 0.002-20 wt.% is a catalyst material selected from Pt, Pd, Rh or other transition metals. This layer is 10-500 pm thick to reduce H, induced lean shift.
Nitrogen Dioxide Sensor SHIMADZU COW. Japanese Appl. 10190,22 1 A controlled potential electrolysis type NO, sensor has a metal layer and a Pt layer formed on a gas per- meable diaphragm, which is in the contact surface of an electrolyte and a tested gas. A counter electrode and a reference pole are formed on a detection pole on the diaphragm. The concentration of NO2 detected is based on the current between the counter electrode and the detection pole.
ROBERT BOSCH G.m.b.H. European Appl. 831,565A
HETEROGENEOUS CATALYSIS Palladium Composite Catalyst AKIN AW CO. LTD. European Appl. 826,419A A composite ZnO-Pd catalyst is prepared by dispersing and fixing Pd on the surface of a ZnO (1) substrate, by adsorption of Pd" ions from an acid solution, followed by reduction of the adsorbed Pd ions to metal- lic Pd. Active C fibres and/or TiO, are additionally integrated in, and deposited on, the surface of (1). The catalyst is used in the elimination of hazardous components, such as CO and NOx, from automobile exhaust gases.
Platinum Metals Rev., 1998, 42, (4), 179-182 179

Solid Bed Catalyst BASF A.G. European Appl. 841,090A Solid bed catalysts containing either Pd and Se and/or Te on a SiO, carrier have a BET surface area of 80-380 m* g-’, a pore volume of 0.6-0.95 cm’ g-’ and a pore diameter of 3 nm-300 pm. They are used as iso- merisation catalysts, especially for 3-buten-1-01 com- pounds. The process gives fewer hydrogenation reac- tion by-products or low boiling point compounds.
Diesel Engine Catalyst CATALER IND. CO. LTD. European Appl. 842,692A A catalyst for punfylng diesel engine exhaust gas com- prises SiO, and Al,O, supports in a mixing weight ratio of 98:2-72:28, and 0.01-0.55 g Pd per litre of support. The catalyst can oxidise SO, in the exhaust gas at higher temperatures, so it can effectively inhibit the formation of sulfates, as well as simultaneously control the emission of particulates.
Purification of Exhaust Gases INST. FRANCAIS DU PETROLE
European Appl. 842,693A A process for the low temperature purification of exhaust gases from I.C.E. comprises the incorpora- tion of a Pd-based catalyst or an absorbent up-stream of the conventional three-way catalyst to eliminate alkynes, particularly acetylene, at 220-250°C. The system eliminates hydrocarbon pollution from cold start either by hydrogenation or absorption of the acetylene using the Pd-based compound. The process should meet the envisaged EC legislation for levels of CO, HCs and NOx in the years 2000 and 2005.
Nitrogen Oxide Trap for I.C.E. FORD GLOBAL TECHNOLOGIES INC.
European Appl. 845,289A A NOx trap for I.C.E. exhaust gases comprises a porous support loaded with (in wt.%): 6-15 Sr oxide; 0.5-5 Pt, Pd and/or Rh; 3.5-15 Zr and 15-30 sulfate. It traps NOx during lean burn operation and releases absorbed NOx when the O2 concentration falls; the desorbed NOx is converted to N, and 02.
Noble Metal Support ASAHI KASEI KOGYO K.K. World Appl. 98126,867A A noble metal support comprises a Pd-free layer inside the support, and a layer where Pd is supported in a region > 100 pm deep from the outer surface of the support. It has high activity and resistance to wear and is useful as a catalyst for oxidation, reduction and hydrogenation, esterification of acrolein and/or metha- crolein, or as a catalytic converter for car exhausts.
Preparation of High Octane Paraffin TEXACO INC. US. Patent 5,744,667 A high octane paraffin for use as a blending compo- nent in gasoline is prepared by reacting a 5-10 C acceptor olefin and a 3-1 0 C donor parafin (l), with different backbone structures, in the presence of a Pt catalyst supported on a Li neutralised large pore B p- zeolite. The catalyst contains 0.1-2 wt.% P t and 0.05-2 wt.% B, to dehydrogenate a portion of (1).
Catalytic Reforming of Hydrocarbons UOP US. Patent 5,755,956 Reforming a gasoline-range hydrocarbon feedstock to an aromatics-rich effluent stream, involves con- tacting it with a catalyst comprising a multigradient noble metal of Pt and surface-layer Ru, a non-acidic large-pore molecular sieve, and an inorganic oxide binder. The process has increased selectivity for the conversion of paraffins to aromatics and improved catalyst stability, particularly in the presence of S .
Hydrogenation Catalyst
A hydrogenation catalyst comprises a C support and Ru, Sn and optionally another Group VIII metal, uni- formly distributed inside the support. The catalyst is used for the catalytic hydrogenation of carboxylic acids giving high yields of 1,4-butanediol andlor tetrahydrofuran, from maleic anhydride, maleic acid, etc., under relatively mild conditions.
Diesel Engine Catalyst HINO MOTORS LTD. Japanese Appl. 10176,159 A purification catalyst has a fine particle-like carrier comprising Rh, Pt, Ir, Pd, Au, Ag or Ru, with a grain size of 0.1-50 pm; a metallic oxide fine particle, such as A1203, Ti02 or SiO,; and an inorganic binder, such as sols of SiO, or A1,0,, etc. The rate of NOx reduc- tion of exhaust gas from a diesel engine is improved.
Diesel Exhaust Purification Catalyst NISSAN MOTOR CO. LTD. Japanese Appls. 10176,162-3 A catalyst for exhaust gas purification for diesel and lean burn engines, has a catalyst layer chosen from Rh, Pd and Pt, and a ceramic component selected from Si carbide, Si nitride and B nitride along with at least one rare earth element selected from lan- thanum, neodymium, etc. The catalyst shows high temperature durability and excellent purification capacity for hydrocarbons, CO and NOx.
Exhaust Gas Purification from I.C.E.
MITSUBISHI CHEM. COW. Japanese Appl. 10171,332
TOYOTA CHUO KENKYUSHO K.K. Japanese Appl. 10/85,600
A catalyst, for the oxidation and purification of hydro- carbons contained in exhaust gas from I.C.E. com- prises Zr oxide particles and a catalytic noble metal, with 2 50% of the latter being in a high oxidation state. Pt carrying hydroxide, obtained by adding Pt to Zr hydroxide, is also included. The purification capac- ity of SOF in a low temperature region is good and the oxidation of SO2 and the formation of sulfates is suppressed.
Removal of NOx KYOCERA C O W . Japanese Appl. 10185,602 An oxide catalyst material for removal of NOx from exhaust gas contains 0.5-20 wt.% Pd oxide and oxides of Ga and Ni. The NOx contained in the exhaust gas is directly decomposed into N, and O2 without the use of a reducer. The catalyst is used for the removal of NOx from exhaust gas from factories, power stations and motor vehicles.
Platinum Metals Rev., 1998, 42, (4) 180

Purification of Exhaust Gas DENKI KAGAKU KOGYO K.K.Japanese Appl. 10185,604 A catalyst, for purifymg exhaust gas from I.C.E., com- prises a catalyst component containing Si nitride andlor B nitride, Si carbide, Rh and Pt and/or Pd. The Si nitride andor B nitride are in the Pt- andor Pd-containing layer while the Si carbide is in the Rh-containing layer. The catalyst improves exhaust gas purification. Catalytic activity and endurance under a stoichiometric environment are also improved.
Hydrogenation of Aromatic Amines BAYER A.G. German Appl. 1196141,688 A catalyst for the hydrogenation of aromatic amines to cycloaliphatic amines comprises an alkalised sup- port impregnated with 0.05-10 wt.% Ru and Pd in a weight ratio of Ru:Pd of (1:30)-(30:1), and con- tains no halogen. Aromatic amines can be completely converted even at high catalyst loadings, with a high selectivity for primary cycloaliphatic amines, without addition of NH, and with no hydrogenolysis or methanation.
Hydrogen Peroxide Production BASF A.G. German Appl. 1196142,770 The production of H,02 solutions containing 2 2.5 wt.% H20, involves continuously reacting H2 and 0, on catalysts containing Pd as an active component. The reaction takes place in H 2 0 or 1-3 C alkanol on moulded catalyst bodies. The catalysts used have a long service life.
HOMOGENEOUS CATALYSIS Aromatic Haloamino Compounds NOVARTIS A.G. European Appl. 842,920A A catalyst is comprised of a Rh, Ru, Ir, Pt or Pd cat- alyst modified with an inorganic or organic P com- pound with an oxidation state e 5, and a V compound. The catalyst is used in the preparation of aromatic haloamino compounds by hydrogenation of the cor- responding halonitro compounds. Haloaminos are intermediates in the production of dyestuffs and pes- ticides. The reaction gives very high selectivity with few side products, high yields and short reaction times, at low pressures (5 bar) and temperatures ( 1 00%).
Supported Phase Chiral Catalyst CALIFORNIA INST. OF TECHNOLOGY
U S . Patent 5,736,480 A supported phase catalyst, with a metal selected from Rh, Ru, Ir, Pd, Pt, V, Pb, Sn and Ni, comprises an organometallic compound of chiral2,2'-bis(diphenyl phosphino)-1,1 '-binaphthyl (BINAP) solubilised in a solvent having two alcohol groups. Each phenyl group is at least monosulfonated. Also claimed is the use of the above catalyst in the asymmetric hydro- genation of 2-arylacrylic acids, especially dehydrona- proxen. The catalyst system is soluble in highly polar solvents but not in non-polar solvents so the catalyst may be solvated on a solid catalyst support, facilitat- ing easy separation from the product after synthesis.
Hydrosilation of Unsaturated Compounds DOW CORNING CORP. US. Patent 5,756,795 The hydrosilation of unsaturated organic and Si com- pounds, such as Si hydrides, in the presence of a Pt compound or Pt complex catalyst, and a specific accel- erator, such as 1,7-octadiyne or maleic anhydride, is described. The accelerators improve yields in the pres- ence or absence of 0, and are very effective in the hydrosilation of internal unsaturated bonds.
Platinum Complex Catalysts RHONE-POULENC CHIM. French Appl. 2,750,349 Pt complexes with various olefin ligands are claimed for use as homogeneous and thermo-activatable cat- alysts in hydrosilylation reactions between silanes or siloxanes, and compounds with reactive unsaturated aliphatic andor polar functional groups. The catalyst is stable at 3040°C for long periods. The Si oil prod- uct is used for anti-adhesive coatings on fibres, tak- ing dental impressions, adhesives, etc.
Selective Chlorine Component Removal GES BESEITIGUNG VON UMWELTSCHAEDEN
German Appl. 2/97122,33 1 A catalyst for the selective hydrogenative removal of fluorochlorohydrocarbons and halones from gases comprises a carrier with a fixed active component of Os.Ru&Y, (where X = Group VIII metal; Y = Group 111 or IV metal or rare earth metal; and a, b, c and d = 0-100, with a + b # 0). The catalyst is used for the removal of ozone-damaging C1-containing com- ponents, such as FCHCs from gases evolved in FHC production. The catalyst has higher activity and longer life than the PdAlF, previously reported.
FUEL CELLS Platinum Electrocatalyst for Fuel Cells NE CHEMCAT CORP. European Appl. 827,225A An electrocatalyst (1) comprises a skeleton alloy of Pt with Ga, V, Cr, Mn, Fe, Co, Ni or Cu. Also claimed is an electrode and its production comprising the elec- trocatalyst and a H,O repellent binder, bound to a conductive and gas permeable support substrate. (1) is a cathode for proton exchange membrane or phos- phoric acid type fuel cells and may also be a gas but- ton cell diffusion electrode, etc. It has high activity and long term stability for 0, reduction compared to conventional electrocatalysts.
Catalyst for Use in Fuel Cells JOHNSON MATTHEY PLC European Appl. 838,872A A catalyst, for use in gas diffusion electrodes for fuel cells, particularly PEMFCs, comprises a Pt-M alloy in intimate contact with Y, where M is one or more transition metal, Group IIIA or Group IVA metal, and Y is a bronze-forming element or oxide (M is not Ru i fY is WO,; M is optionally two or more metals where one metal is Ru). A catalyst comprising Pt- Ru alloy alloyed with W is also claimed. The cata- lyst is tolerant of poisons and can be used as the electrocatalyst on the anode and the cathode.
Platinum Metals Rev., 1998, 42, (3) 181

Electrocatalyst Particles UNIV. MASSACHUSETTS U S . Patent 5,702,836 Particles for a fuel cell electrocatalyst for oxidising alcohols comprise an Fe oxide core with an outer Pt oxide shell. The electrocatalyst is colloidal, lightweight and cheaper to manufacture due to the non-Pt core.
Anode Catalyst for Fuel Battery TOSHIBA K.K. Japanese Appl. 10174,523 A catalyst for a fuel battery has Pt or fine Pt alloy par- ticles for H, storage distributed on the surface of a conductive C powder carrier. A highly active anode is obtained and battery durability is improved.
Hydrogen Fuel Cell Accumulator R. WOLLHERR German Appl. 1196144,864 A hydrogen fuel cell accumulator, for use in electric vehicles, comprises a polymer electrolyte membrane cell with a Ru catalyst, an air filter, a metal hydride storage unit, a small compressor and a thermoelec- tric heat exchanger. The H, from the hydride storage unit is converted electrochemically with O2 from the air. The air is cleansed by passing through a filter. The accumulator is economical to produce.
ELECTRICAL AND ELECTRONIC ENGINEERING Glass Circuit Substrate CANON K.K. European Appl. 838,980A A glass circuit substrate comprises a layer of Pd nuclei carrying a Pd-P plating layer. Also claimed is the pro- duction of a semiconductor device, by fabricating the above substrate, patterning the plating layers to define wiring and electrodes, and bonding IC chips to the electrodes. A wiring pattern can be formed by wet plating, without degrading the smoothness of the glass surface or the adhesion against external thermal effects. Adhesion to the substrate is enhanced, thus resistance to peeling is improved.
Magnetic Recording SHOWA DENKO K.K.
Medium U S . Patent 5,731,070
A magnetic recording medium comprises a Si layer on a non-magnetic substrate and a layer comprising at least one Pt group metal or alloy, or C, with the Pt being partly silicified and the C being partly amor- phous, both due to Si diffusion. An undercoat is formed over the Pt/C layer, followed by a magnetic layer and a protective overcoat. The medium, used for magnetic recording disks, has a high coercive force and squareness ratio which increases the magnetic recording density.
Electrically Conducting Body FURUKAWA ELECTRIC CO. LTD.
Japanese Appl. 10184,065 The body comprises an electrically conductive base carrying a Ni or Ni alloy foundation layer on its sur- face. Upon this is an interface layer of Pd or Pd alloy, 0.005-0.1 pm thick, carrying a monoatomic layer of Au or Au alloy. The member has excellent soldering properties and oxidation of the interface layer is pre- vented. It is used in diodes, transistors, etc.
TEMPERATURE MEASUREMENT Temperature Sensor HONEYWELL M C . U S . Patent 5,726,624 A tubular temperature sensor for use in ovens at - lOOO"F, has conductive strips of Pt-Ag alloy deposited on a rigid Al oxide substrate, with insulating glass lay- ers over and between the conductive strips. A resis- tive temperature detector (RTD) is placed on one end region of the substrate and attached electrically to the conductive strips. Fibreglass sleeved wires are not needed and electrical connections can be made to the RTD in parts where the sensor is at high temperatures.
MEDICAL USES Crosslinkable Silicone Dental Compound ZHERMACK S.P.A. European Appl. 822,233A A crosslinkable Si compound for use in dentistry com- prises a crosslinkable Si polymer, a crosslinking agent, a Pt catalyst and a Na-A1 zeolite. The composition remains stable under storage conditions, even at tem- peratures higher than those normally recommended. The Pt catalyst is protected by the zeolite against the action of potential contaminants.
Precious Metal Dental Alloy BEG0 BREMER GOLDSCHLAEGEREI
German Appl. 1197119,677 A precious metal dental alloy contains Au, Ag, Pt, Pd and Mn, with the Ag, and preferably the Mn, content higher than the Pt content. The alloy is free from Cu, Sn, In, Mg and Ca. The use of the above Cu-free pre- cious metal alloy is claimed as material for false teeth, bridges, crowns, etc., which may be covered with ceramic and supported by implants, etc. The alloy has high hardness (250 HV5) and strength, resists corrosion and tarnishing, and is free from toxic Cu.
Dental Palladium-Based Alloy SUPERMETALL RES. PRODN. COMPLEX
Russian Patent 2,092,603
palladium-containing hi^ ~ ~ ~ d i ~ ~ wire Japanese Appl. 10183,71
An alloy for use in dentistry contains in wt.%: 45-70 Pd, 10-25 Au, 10-15 Cu and 10-15 Sn, at a Cu:Sn ratio of 1 : 1. The allov is non-toxic. strong. has good STEEL
_I .2
A thin Au alloy wire for bonding the electrode of a semiconductor device and an external lead includes 0.005-1 wt.% Pd and 0.005-0.3 wt.% Mn, the remainder being impure Au. The wire improves cor- rosion resistance, ensures long-term reliability, improves junction properties and allows high density mounting.
flowability for casting complex dental prostheses, and good adhesion to a wide range of ceramic coatings.
The New Patents abstracts have been prepared from material published by Derwent Information Limited.
Platinum Metals Rev., 1998, 42, (4) 182

Abarra, E. N. Abe., K. Abid, Z. A c h i i Y. Ahlafi, H. Ahmed, G. Aissi, F. Aizel, G. Ajjoy A. N.
Alam,M.R Albrecht, F. Albrecht, M. Al-4 E. I. Allen, F. M. A W , H. Anderson, N. L. Angelici, R J. Angell,C.H. 27, Antonid, M.
ALashiM.
Antonmi P. L. Antonmi v. AnzaiK.
Page 123 83 34 174 38 127 37 I27 127 125 39 57 176 38 126. 127 55 177
38 34 34 177
46, i16
Aonuma, S. 78 M. 128
Arakawa, H. I75 -K. 125 AKlR P. 38 Aricd, A. S. 34 Artemenko,Yu.A. 99 Asahw,K. 38 Ashton, S. V. 24,98,
134, 163 Asomom, R 80 Attard, G. S. 36 AuguStine, R L. 163
Bm, D.4. Baker, A. Bajusz, 1.4. Barlow, S. Barnard, C. F. J. Barth, J. V. Bar t le P. N. Beshilov, V. V.
Baur, J. E. B e , D. Becker, R Bea,H.B. 27, Beldarain, T. R BeUer, M. -A.K Benniston, A. C.
Basset, J.-M.
37 161 80 81
17, 158 24 36 18 160 124 127 56
46, 116 34
39, 145 38
100
Page Berzias, A. R I24 Bianchi, D. 38 Bhchini,C. 35, 145 B h q M. C. 34 Bockholt, A. 37 Bogdanovic, s. 39 Bolm, c. 177 Bon, M. 81 Bond, A. M. 36 B6m- H. 78 Borges, S. D. S. S. I75 Bosnich, B. 178 Braddock-Wilhng, J. 128 Bradley, J. S. Briise, S. Bnssan, M. Breuer, K. Briand, D. Briscoe, H. V. A. Britz, P. Bronger, W. Brook, T. E. Brown, D. A. Brown, J. M. Brown, S. N. Bmce, M. I. Brick, R Bnmeay C. B m e r , H. Buchwald, S. L. Bulgakov, R G. Bmh, R Burkov, A. Buschinger, B.
Cai, M.-2. Caiazza, A. Calvo, R L. Cameron, D. C. cardin, C. J.
Casaw, J. A. castau* Y. castellano, E. E. than, H. S. 0.
Carpentier, J.-F.
cyhan, M. C.-W. chang, J.-R chao, s. che, C.-M. Chen, c.-w. Chen, D. Chm,K.Y.
Chen, M.-Q. chen, L.
161 126 145 39 176 169 78 78 37 78 159 34
174, 175 56 178 I24 I77 35 I76 78 78
126 82 127 83 177 127 35 127 175 124 123 81 125
123, 124 125 128 128 124 125
Platinum Metals Rev., 1998, 42, (4), 183-186
Page then, W. 82 Chen, Y.-w. 126 Chen, z. 36
Cheung,K.-K. 123, 124 Cheung, T.4. 124 Chiba, A. 68 Chickos, J. S. 128 chiffey, A. F. 25, 160 (30, H.-J. 83, 124
Chmg, C.-H. 81
Ch04 S.-H. 37 Chong, E. K. 55 christofides,C. 36, 125 C!h.auow&i,W. 128 Chuang, K T. 81 Chuman, T. 10 chun& c. w. 80 churchill, D. G. 127 Churchill, M. R 127 claver, c. 177
cobdQ1, P. D. 141 Cockcroft, J. 169 Coe, B. J. 175 Col+Hamilton, D. J. 176 Coleman,N.RB. 36 Connolly, J. 174 Cook, J. D. 125 comilp, B. 145 Conon. J. B. 27,46, 116 Curie, J. F. 176 W W A. 69
Clegg, w. 100
D& L.-X. 82 Das,A.K. 178 Dap, M. 178 Davanzo, C. U. 175 David, S. A. 177 Davidson, L. 90 Daviea, H. M. L. 127 Daviea, H. 0. 78 Davies, R J. H. 79 Davydov, A. A. 81 De Blasio, N. 176 DeGiovaui,W.F. 79 de Meijere, A. 126 De Souza, J. P. I. 79 DeVries J.G. 127, 159 Degi- L. 78
Delabouglise,G. 176 Delaude, L. 162 Demes, J. N. 80
DeGraff, B. A. 80
Denisov, N. N. 79
Page
Deronzier,A. 60, 163 Maz, V. 38 Maz&da,M.E. 80
Denisovich, L. I. 35
DiCguez, M. Dixneuf, P. H. Donnerstag, A. Doyle, M. P. Driver, R -is, D. L. I)lmn, B. Dyer, S. E. Dyson, P. J.
%itmi, K Echani, R Eckert, M. Egashira, N. Egaw4 T. Eicher, S. El G M i , A. Elding, L. I. El-Glmyoury, A. Elliott, J. M. Ellis, D. J. Ellis, W. W. Emori, M. Enders. D. EnglematZR Eremenlro, I. L. Eremenko, L. T. Ertz G. Estcruelas, M. A.
Faltermeier, G. Fan, J. C. Fan, Q. Famll, N. Farrugin, L. J. Fenga, P. G. Fidalgo, E. G. Fischer, A. Fodor, K Foklrens, R H. Fords, J. FomiW%mer, J. Fkter, S. France, D. W. Fremy, G. Fry, B. E. Fy W. Fq Y. Fujie, Y. Fujii, H.
177 159, 178
56 178 55 79 83 125
135, 145
38,82 177 39 176 128 34 37 174 176 36 135 178 128 39 174
83, 123 83 24 82
56 128 178 174 100 79 178 128 108 174 78 I77 38 175 127 123 73 81 82 98
183
NAME INDEX TO VOLUME 42

Page
Fukuda, H. 36
FulNshima, N. 83 Fukuyama, T. 126 Fiastner,A. 159, 178 Fumya, N. 83
Fujisaki, Y. 39
Fukui, M. 37
Gamble, S. 98 Gao, H. 177 Geibl, c . 78
Genge, A. R J. I74 Geissler, H. 39
Gengembre, L. 37 Gielen, E. 39 Gladysz, J. A. 39, 177 Goldshleger, N. F. 79 Goltsov, V. A. 99 Goltsoq M. V. 99 Goluboichaya, M. A. 83 Golunski, S. E. 2 &eZ, P. 59 G6rnez-Sas0, M. A. 78 Goodwin, J. G. 80 -V.V. 141 Goaaage, R A. 176 Gottberl3, I. 56 Gozzi, c. 127
Graze, W. H. 177 Green, M. L. H. 168 Gregory, 0. J. 125 Greiner, A. 38 Glifti& w . P. 168
R 177
Grove, D. E. 16 Grubbs, R H. 34, 145, 159 Gu, J.-H. 124
Guo, A. 123 Guraq B. 163 Gu& w. 78
Grkel, M. 35
Grimblot, J. 37
Guelton, M. 37
HabR, s.
Han, K-s. -wK. HanasaLi, N. M e , K. P. Hsnkin, D. M. Hauyu, A. Hanylz T. Hara,K. Hara, s. Harding, I. s.
Haberman, J. X. 145 128 37 35 78
140 105 178 174 175 78 37
Ih l imaq A. Haniugton, L. J. Harris, J. A. Harris, M. C. Hartwig, J. F. Hmuta, M. Hasebe, K. Hascgawa, Y. Hashiguchi, S. Haasan, J. Hayashi, H. Hayfield, P. C. S.
Heck, R M. Hegedlls, L. Heinrich, A. Heitz, W. Herman, R. G. Herrero, J. Hemnann, W. A. Hess, J. S. Hibbert, D. B. Hildebraud, J. P. Kiu, A. F. Hill, G. S. Hiratani, M. mi, K. Hissler, M. Hockaday, R Holah, D. G. Hor, T. S. A. Horii, H. Homes, J. HorvAth, I. T.
HOU, X.-L. Houbrechtp, S. Howard, M. J. Hu, W. Hu, Z. Huan, Z. Huang, X. Hmk, W. T. S. Hughes, A. N. Hwang, C. S.
Idoffsson, T. Ikenaga, N. Imam= s. JJlabaI Y. Inoue, H. Inow, s.4. Iorio, L. E. Isaac, I. Ishiguro, K. Wii, Y.
Platinum Metals Rev., 1998, 42, (4)
Page
I76 175 175 177 I58 177 175 39 39
127 I26 27,
46, 116 126 108 78 38 34 82
145 34 36
I77 39
174 39
I77 176 115 174 I24 83 78
39, 145, I71 82
I75 I58 I75 126 124 126 174 I74
83, 124
56 177 126 82 35 39
123 127 82
178
Ishikawa, H. Islam, M. S. Ito, H. Ito, s.4 Ito, Y. Itoh, N. Ivanov, A. V. Ivey, D. G. Iwakura, c. Iwasaki, s. Iwasawa, Y. Iwuoha, E. I. Jzawa, Y. w M.
JwW L. Jeffery, J. C. Jezkova, J. J i q M. Jimbo, T. Jhdra, J. Job, F.
Juliette, J. J. J.
Kaddouri, A. Kaji, H. Kakaki, T. Kalli, K. h e d a , N. h e r , P. KandaSamy, K. K a m h K . m g , c. s. Kqlunov, M. G. Kar, D. Kaphimura, Y. Kataura, H. Kath6, A. Kato, F. Kato, R.
Katsnel'son, A. A. Kawasaki, M. Keiderw, T. A. Kelling, S. Kelly, J. M. Kendall, K. Khaselev, 0. Kikuchi, H. Kim, H. J. Kim, W. D. Kim, w.-Y. Kimura, A. Kimura, H. M. Kimura, M.
JOO, S.-K.
Kato4 s.
Page
I28 83 10 38 82 78 81 34 35 10 38 37 39 83
79 I75 37
Page King, D. A. 8 Kirsch-De Mesmaeker,
A. 79
Kishida, M. 126 Kitamura, N. 178 Klausmeyer, K. K. 1 57
KlingeMfer, S. 38 Kobayashi, A. 178 Kobayashi, H. I78 Kobayaahi, S. 82 Kobayashi, T. 177
Kolesnichenko, N. V. 38
Kisenyi, M. 57
Klier, K. 34
Kohle, 0. 35
Koncki, R 37 Kmdo, T. 35 Konovalova N. P. 83
81 -is, S. I28 Krauter, J. G. E. 128 K ~ k d k i n , 145 V. Yu.
39, 171 Kumar,A. 34 Kulilrov,A.V.
Kung, H. H. 176 KunimOri,K. 35 Kuntz,E.G.
175 Kuramhi,Y. 36, 125 Kurosaka,T. 39, 127 Kurt,R
145 Kwtov, L. M. 99 Kuwano,R
38, 82 Kuzin,A.P. 83 Kuzina,I.A. 19 Kw&D.J.
178 78 Labeau,M.
174 Lahav,M. 145 Lahoz,F. J. 177 Lai,S.-W.
78, 178 -,RE. 175 Lam,M.H.W. 99
175 128
8 79
164 124 82
124 83
126 174 78 35
Lambert,RM.
Latef, A. Lattes, A. Laurell, M. LaVenOG L. Lluzaron, R. Leclercq, G.
Lan, M.-H.
Leclercq, L. Lee, B.-I. Lee, B. T. Lee, C.-H. Lee, c. P. Lee, C.-T.
36 145
106, 174
79 39
125 38
145 176 125 78 81 82 99 99 80
176 81 82
123 126 124 160 36 37 81 56
127 82 37 37 34 83
126 128 36
184

Lee, J.-H. Lee, J.-K. Lee, J. M. Lee, K. Lee, K. H. Lee, M. Y. Lee, s. I. Lee, s.-K. Lemaire, M. Lester, T. P. Lev- w. Lewis. F. A. Li, A.-W.
Li, M. Li, T. Li, x Li, X. G. Li, z. Liebl, J. Lima, E. C. Lin, G. Lin, I. Lin, T.B. Likh , U. Liu, L. Liu, Q. Liu, R Liu, W.
Li, C.-J.
Long, R W-kla,
M. A. Low, P. J. Lu, z. L-, H. LuIldM@ I. Lymao, C. E. Lynn, D. M.
Ma, Y. H. Mackie, P. R Maitlis, P. M. Mallouk, T. E. Maugiaracina,A.
Marder, S. R Mali- Y.
Mardilovicb P. P. Martin,A. Martin, M. Martinez, F.
Page 125 37
124 I74 83 83 83 36
I27 34
124 99
124 128 124 36
125 68 81 57 79
128 124 81
174 178 124 178 73
176 34
175 124 174 I25 126 145
80 100
25, 158 163 39
I74 81 80 78 82 78
Matsui, N.
Matsumura, Y. Matsushita,K. Matt, D. Maw, w. Mazzocchia, C. McGrath, R B. McNally, P. J. McNamara, K. P. Meli, A. Menj6n, B. Meyer, A. F. Meyer, T. B.
Miki, H. Milani, B. Miller, J. M. Mills, A.
Mitchell, S. Mitose, K. Miyamoto, A. Mizugaki, T. MocaL, J. Modica, E. Mondal, S. K. M d e r , E. Montanaro, L. Mo~imoto, Yu. Monta, D. K. Morhmx, A. M d o , A. Momer, M. Moulijn, J. A. M o w J. C. Mi~ller, P. Muralcam, Y. Murase, K. Murray, R W.
Matsui Y.
Miedaaer, A.
Milstein, D.
Page 177 39
I26 80
1 1 , 163 56
176 68 83
125 145 78 35 35 79 39
160 83
176 81, 159
36 83
178 38 36 34
178 127 58
123 I77 127 145 127 69
60, 163 78 35 83 34
Nacltochenk0,V.A. 79 Nagata, H. 126 Naksgawa, K. 177 Nakamura,K. 128 Nakamura, T. 35 Narayanaswamy,R 37 Nart, F. C. 79 Navarrete, J. 78 Nawdali, M. 38
Mas&& M. 37 N e c h , D . C . 123 Masdeu-Bult6,A.M. 177 Nefedov, S. E. 83, 123
Masumoto, T. 78 N e p , A . 58 M&M, T. 108 Nestetenlr0,D.A. 83 Matmhdi, H. 125 Ng,S. C. 124
Manu& H. 34 Nephi ,N. 10
Platinum Metals Rev., 1998, 42, (4)
Page Nibbering, N. M. M. 174 Nieuwenhuys, B. E. 141 Nishio, T. 126 Nishiyama, Y. 128
Nolan, K. B. 78 Nix, R M. 37
Noll, B. C. 79 Nomum, K. 39 Noto, V. D. 79 Novikov, Y. N. 35 Noyori, R 39 Nyokong, T. 79
odenkirk, W. 178 oestreich, s. 38 Ogasawara, K. 10 Oh, H.-S. 34
O b k a m h i , H. 35
Ohtsuka, Y. 174
Ohga, K. 176
Ohtsuka, K. 36
Okano, M. 82 O h - K . 38 OkUmura,Y. I26 O M , E. 83 0- I. 36
oklIyam& s. 80 Olivier, H. S. 145, I62 Olvera, M. D. L. L. 80 mte, E. 82 Ono, T. 68 O'Regaq B. 175 W b A. 177 Om, L. A. 82 oskarsson, A. 174
Othonos, A. 36, 125 OtsUkrsK. 83, 175
okuyama, K. 80
-R 35
Otto, E. Owen, J. R
Parmon, V. N. Parsons, s. Passalacqua, E. Pastor, E. PattiSon, D. I.
Pekala,RW.
Peregudova, S. M. Perez, E. Periana, R A. Persoons, A. Peruk, R N. P-, M.
Paulose, K. v.
Penalva, v.
57 36
141 100 34 79 35 55 83
127 35 81 98
175 35 35
Ped, D. R Peters, A. F'falzgd, B.
Picquet, M. wrmz,K.
Pi- s. Pitachke, w. Plassaaud, L. Pletcher, D. Ponina, M. 0. Pope, S. J. A. Popov, B. N.
Potgieter, J. H. Potter, R J. Regosin, P. s. Price, J. M. Pringle, P. Prins, L. J. Pmkopchuk, E. M. pu, c. pu, Y. m W R J. puech, L.
PwVik-Biro, R
Quinn, Y.
Raithby, P. R Rani V. Rao, z. ~~, T. B. Reddington, E. Rees, L. H.
Reid, G. Rehhoudt, D. N.
Rei, M.-H.
ReiSinger. C.P. Reshetnikov, A. v. Rh-, H.-K. Ric- J. T. Rico-Laaes, I. Rivadulla, J. F. Rims, J.
Rodriguea, I. D. A. Romero, J. R Romero, T.
Rothe, J. Ruck, M. Rucnpf, T. Rusov, V. D. Rutherford, D. Ryumstana, T. A.
RocaboY, c.
Rosenzweig, z.
sadahiro, Y.
Page 177 57 56
124 178 37 78
178 124 123 174 123 81
123 I40 159 80 26
174 174 178 128 I23 81
90
146 125 55
157 163 175 80
174 174 145 123 37 69 81 34 34
177 79 79 37
125 78 34
123 99
177 99
82
185

Page Sadigbi, J. P. 177 Sakaguchi, s. 178 Sakahaa, T. 82, 160 Sako, T. Sanger, A. R Santhanam, K S. V. Sapienza, A. Sarmgapani, s. Sato, A. Satoh, T. Sawabe, A. Sayana, K scha- w. c. Scheers, P. V. T. Schmid, G. Fichumaan, J. Schwartz, D. T. Schwaq P. F. Seaborg, G. T. Seddon,K.R Sekota, M. Serizawa, T. Settambolo, R Sbaharulzamwn, M. Shamsuzmha, M. Shapley, J. R She, Y. Shibata, M. shimifll, s. shim- Y. Shirai, H. Shirai, M.
82 81
125 163 163 178 98 36
I75 I45 123 162 78
175 79
I70 160 79
125 82 28 39
174 80 83 83 35 35
128 Sidorov, A. A. Sigan, A. L. Silva, R A. G. D. Silva, s. c. Simons, K. E. S h u , D. Skelton, B. W. Skryabiha, N. E. Sliviapky, E. V. smirnov, L. I. Smith, A. M. Smotkin, E. S. Smyth, M. R Sokolov, V. I. Sone, T. song, c.4. Song, H. G. Spaine, T. W. Spek, A. L. Spek, A. Spivak, L. V. s r i u v. stasilr, I. Steele, D. F.
83, 123 35
128 175 160 145
174, 175 99 38 99
124 163, 178
37 18 82
126 80
124 176 125 99
177 I27 90
Steger, J. J. Steglich, F. Stephan, M. S. Stevens, M. G. Skoh, N. Stuart, A. M. Stull, A. D. Suga, M. suk, c.4. Sullivan, J. A. SiiSs-Fink, G. S W K . Suzuki, M. Suzuki, s. Suzuki, T.
Tabata, M. Tajima, H. Takahashi, M. Takahashi, S.
Page 126 78
127 55
I24 I60 125 39
124 I76 I78 36 35
174 123, 177
82 78 82
68.82 Takapy Y. Takeuchi, R Takeznwa, E. Tam, C. N.
Tanaka, S. Tanaka, T. Tamai, T. Taube, D. J. Taube, H. Teat, S. J. Teleahev, A. T. Tempesti, E. Ten& Y.
Teny, M.
Thayumanavan,s.
Tan, K-L.
TerekhOVa, G. V.
Teunissen, A. J. J. M.
Thomas, J. Thomas, M. D. Thompson, D. T. Tishin, B. A. Tokuyama, H. Ttillner, K. Tomalia, D. A. Toyota, K. Tran, T. D. Troughton, G. Trzmiak, A. M.
Tsai, C. M. Tsyganok, A. I. Turnas, w. Tunglcr, A. Turner, J. A. Turro, N. J.
Tsai, C.-D.
Platinum Metals Rev., 1998, 42, (4)
35 I78 I78 128 I24 83 37
I08 98 98
I00 38
I76 I77 38 83
127 81 78
176 71 35
126 81 79
175 83
144 127 36
128 175 177 108 I24 79
Page Twigg, M. V. 56
Uccello-Barretta, G. 82 Uchida, H. 128. 175 Umeno, M. 128 Uno, M. 82 Usami, H. 80 Usatov, A. V. 35 utani, K. 126
Valencia-Godez, XO
Van Koten, G. 176 VanLeeuwen, 25. 145
Van Veggel, I74
Velasco-Garcia, N. 80
M. J.
P. W. N. M.
F. C. J. M.
Vergara, M. C. Venijl, G. K. M. vescoli, v. Viswanathan, R Vogel, W. Volkova, L. M. VollmUer, F. Vrieze, K. Vytras, K.
Wadowhi, A. Waegell, B. Wakabayashi, K Walsh, M. wan, c. z. Wmg, D. Wang, E. Wang, J. H. Wang, R
Wang, X. F. Watanabe, M. Watling, T. C. Watson,R Wells, P. B. Welton, T. Wendt, H. weston, w. wetzig, K. White, A. H.
Wan& s. Y.
34 I27 78
163, 178 78 83 39
I59 37
127 126 126 56
126 128 36 36
38,81 I28 126
128, 175 176 56
161 135 128 I58 78
174, I75 White, P. 10,55, 157 White, R E. 123 Whyman,R 161 Wieckowski, A. 128 WieSer-Jeune~pe. C. 1 1,163 Wilhelm, T. E. 34 Wmams, D. S. 164 Wills, M. 161
Page Wilkinson, G. 168 Wilson, S. R 157 Wilton-Ely, J. D. E. T. 39 Winner& J. Woodward, R. B. WoH, I. M. Wnesien, K. wu, L.-z. W y a J.-F.
xiao, z. Xie, Q.C. Xiong, G.-X. Xiong, H. Xu, G . 4 xu, w. xu, w.-c. xu, Y.
Yamada, H. Kamada, Y.
Kamaoaka, I. Kamashiica, T. Yang, R T.
Yamagata,A.
Yang, w. Yang, Y. Yanowky, A. I. YarimiZy T. Yeager, E. B. Yokoshima, S. Yokota, K. Yon& T. Yoshida, Y. Yoshjkawa, T. Yoshimura, N. Yu, R Yuzaki, K.
Zaitseva, N. N. Zambelli, T. Zelentsova, T. N. Zeng, H. C. m, M. m, R zhan& x. zhang, x.4. zhao, c.-z. Zhao, H.B. Zheng, G.
Zhu, B. Zhu, G. Zieasel, R Zi6lkowski, J. J. Z-Schpector, J.
zhitomlrq, I.
24 I70 I23 78
124 174
124 82
124 73
124 80 78 83
80 123 177 175 I26 176 82 73 78 38
123 126
31.82 39, 127
35 I0
145 I24 38
174 24 99
126 81 34 83 35
176 I24 I23 80
38,8l 83
176 127 175
186

SUBJECT INDEX TO VOLUME 42
a = abstract Acid Chlorides, phenylation, a Acoustic Waves, for catalytic rate enhancement,
N-Acyl Amino Acids, synthesis, a Alcohols, allyl, hydrogenation, over Pt colloids, a
on Pt surfaces
ethyl, from ethyl acetate, a from aldehydes, over Rh clusters, a from methyl ketones, Rh catalysed, a homoallylic, isomerisation, a
methyl, carbonylation, a synthesis, a
from syngas, a on-board Hi production, for fuel cell vehicles oxidation, in fuel cells, a
primary allylic, oxidation, a primary, oxidation, a 1 -propanol, 2-propano1, electrooxidation, a propargyl, reaction with norbomene, a cis-4-substituted cyclohexane- 1 -methanoh, a synthesis, over Rh-Mo-WAI,O,, a
Aldehydes, hydrogenation, over Rh clusters, a synthesis, a a$-unsaturated, from alcohols, a
Alfa Aesar, “Platinum Labware Catalog” Alkanes, dehydrogenation, by (PCP)lrH, Allmres, hydrogenation, biphasic, a Akylation, allylic, allylic acetates, a Allmes, a$-functionalisation, asymmetric, a Allylic Acetatea, allylic alkylation, a Amidea, reduction, via hydrosilylation, a Amidowrbonylation, Kacyl amino acid synthesis, a Am@on, Pd,(dba), catalysed, a Ammes, tertiary, from amides, a
triatyl, synthesis, a Anilines, arylation, a h a , reactions with I-bromoadamantane. a Aryl Halides, homocoupling reactions, a Aryl Iodi&s,.phenylation, a Arylation, anilines, a
over Pdy-Al,O,, a
Benzene, hydrogenation, biphasic, a
Benzylic Compounds, electrooxidation, a Book Reviews, “Aqueous-Phase Organometallic
Catalysis: Concepts and Applications” lnorganica Chimica Acta: Special Volumes on
Platinum Chemistry “Structured Catalysts and Reactors”
oxidation, over Pt-F/AI,O,, a
Idromonrlnmantaue, reaction with styrenes,
Butanal, from propane, a Butane, reforming, zirconia fuel cells
arenes, a
Page
126
8 39
I25 38 38 39
128 I78 I76 I26
2 128
38,81 82
178 79 82 82 81 38
178 82
144 71
177 I78 I77 178 82 39 81 82 81
I77 I26 127 126 177
I78 81 79
145
17 69
126 82
164
Cdhrenea, complexes with Pt metals 11, 163 Cancer, drugs, a 78,83, 174
supercapacitors, RuOJC, a 83 thin film, Pt/(Ba, Sr)TiO,/Pt, a 83
(SrRuOJBa,Sr, ,TiO,/SrRuO,), a 83
Carbocycles, Pd-catalysed synthesis, a 177 carbon oxides, CO,, reduction, electrochemical, a 79
sensors, I#*-, WO,-based, a 125 supercritical, as catalysis medium I58
Heck, Stille reactions, a 177 38
electrooxidation, on Pt-RdC, a 34 hydrogenation, over PdZrO,, a 126
capacitors, a 35,39
carbenes, (PCy,),Cl,Ru=CHR, a 34
0, chemisorption, on RdALO,, a
Page
oxidation, motorcycle emissions, a 126
123 8 2
sensors, a 80, 176
176 propane to butanal, a 82
biphasic, a 177, 178 book review 145 homogeneous 135
heterogeneous, a 37-38,8041, 125-126, 176-177
conference reports 158 72 56
formaldehyde oxidation, a 178 177
Carbon oxides, CO, (contd.)
over Pd-WSiO,, a 38
on Pt, sonochemical rate enhancement
battery powered 144
Catalpis, acoustic enhancement 8
on Pt, Pt-Sn, Pt-Ru electrodes, a
removal, from fuel cell vehicles
carbonylation. MeOH, over RhlSDT, W V P , a
homogeneous, a 39,81-83, 126-128,177-178
“Catalysis Technical Guide‘‘, Johnson Matthey Catalp, automotive, at the SAE congress Catalysts, Iridium, lr(IIl)/Ce(lV),
metal oxide-supported, synthesis gas production, a Pt(44)/Ru(41)/Os( IO)/Ir(5), for DMFCs
asymmetric transfer, of aromatic ketones, a [I~(TFB)(PJP~),]BF,, olefin hydrogenation, a (PCP)lrHI, alkane dehydrogenation
Catalysts, Iridium Complexes, hydrogenation,
on polymer electrodes, hydrogenation Catalysts, Osmium, Pt(44)/Ru(41)/0s( IO)/Ir(5),
Catalysts, Osmium Complexes, (DMS0)20s”Pc,
Catalysts, Palladium, colloids, Heck reactions, a
for DMFCs
cysteine oxidation, a
on gas diffusion electrodes, in fuel cells, a motorcycle emissions control, a PdG-AI,O,, isoprene hydrogenation, a Pd/y-Al,O,, MeOH oxidation, a P d C felt cathode, dechlorination, a PdC, Heck reactions, a Pd(O)/SiO,, phenylation reactions, a Pd-Rh/Si02, CO oxidation, a PdZrO,, CO hydrogenation, a Pd/Z#,, Pd/SOdZrO,, a in supercritical CO,, Heck, Stille reactions, a
Catalysts, pall ad@^ Complexes, C,Pd(PPh,),, hydrogenation of tnple bonds
halides, for amidocarbonylation, a on polymer electrodes, hydrogenation Pd(O), heterocycle assembly, a [Pd(Bu,PetpE)Br](BF,),, CO, reduction, a PdCI,, Heck reactions, a
163
39 82 71
60, 163
163
79 38 83
126 81
38,81 I75 I26 126 38
I26 81
177
18 39
60. 163 177 79
127 81
I26 norbomene polymerisation, a
PdCli(PPh,),, ketone synthesis, a Pd(dba)2, allenes, asymmetric carbopalladation, a Pd2(dba),, triarylamine synthesis, a Pd(OAc),, Karylated sulfoximines, a Pd(OAc),iDPEphos, aniline arylation, a Pd(OAc),/nBaNBr, homocoupling, aryl halides, a PdJs,phen,(OAc),,, alcohol oxidation, a Pd phosphine complexes, C 0 2 electroreduction, a selectivity, towards hydrogenation
Catalysts, Platinum, motorcycle emissions control, a in PEMFCs, a for photovoltaic devices, a Pt(44)/Ru(41)/0s( 10)/1r(5), for DMFCs Pt( 110) crystal, CO oxidation, sonochemistry Pt colloids, allyl alcohol hydrogenation, a Pt, Pt-lO%Rh, HCN synthesis, a Pt/AIIO,, NOx reduction, a Pt/y-Al,O,, NO reduction, a Pt-F/AI,O, benzene oxidation, a Pt-K /SiO,, methanation, a
I i 7 81
177 177 127 82 79
I08 126 I28 124 I63
R I25 37
176 125 81 80
Platinum Metals Rev., 1998, 42, (4), 187-192 187

Page
Catalysts, Platinum, (conrd.) Pt-Mo-NdSiO, NOx removal, a 37 Pt-Mo/SiO, hydrocarbon reactions with H,, a 37 Pt-Rh, 3-way HC/CO/NO conversion, a 126 Pt-SOJZrO,, dehydrogenative CH, coupling, a 125 Pt/H-beta, PVH-MOR, n-hexane isomerisation, a 37 Pt/MCM-41, NO reduction, a 176 Pt/Ru, in fuel cells, a 128 Pfli02/€'TFE, trichlorobenzene photodegradation, a 175
CHI oxidation 98 hydrodesulfurisation 25 Pt(ll)bis(benzoylacetonate), Pt(I1) bis-
(benzoyltrifluoroacetonate), photocuring. a 123 Catalysts, Rhodium, biphasic 135
motorcycle emissions control, a 126 Rh-Mo-WAliOl, alcohol synthesis, a 81 Rh-Pt, 3-way HCICOINO conversion, a 126
Rl-Pd/SiO,, CO oxidation, a 38 Rl-SISiO,, Rh-S-PISiO,, 1 -octene
hydroformylation, a 177 Wone-atomic layer GeO,/SiOi, ethyl acetate
hydrogenation, a 38 W V P , WSDT, MeOH carhonylation, a I76 RhAJSY, Rh/AI,O,, N,O decomposition, a 38
Catalysts, Rhodium Complex?, acacRh(CO)i, 38
178
hydrogenation, a 177
Catalysts, Platinum Complexes, [(bpym)PtCL],
(trifluoroacetylacetonate), Pt(I1) bis-
Rl-Pt, HCN synthesis, a 37
hex- 1 -ene hydroformylation, a chiral diRh(I1) carboxamidate, carbene reactions, a CIRh[P(CH,CH,(CF,),CF,),],, alkene
(DMSO)(CI)~"'Pc, [(CN),Rh"'Pc] , cysteine
hydroformylation 25 on polymer electrodes, hydrogenation 60, 163 Rh,(OOct),, 2,3-dihydrofuran synthesis, a 127 %(CO),,, vinylpyrrole hydroformylation, a 82 Rh,(CO),, clusters, polymer bound, aldehyde
hydrogenation, a 38 WPA(Na')/DPPEA polymer, olefin
hydroformylation, a 127 RhCI(CO)(PMe,),, propane carbonylation, a 82 [RhCI (P[CH,CH,(CF,),CF,],},], hydroboration, a 39 [RhCI(PPhl),([9]aneS,)]PF,, ligand substitution, a 39 [Rh(COD)CI],-P(OPh),, allylic alkylation, a 178 [Rh(COD)CI],IPPh,, hydroboration, a 82 [Rh(COD)(DlOSR,)]ClO,, styrene
hydroformylation. a 177 [RhH,( Ph,N,)(PPh,),], phenylacetylene
hydrogenation, a 39, 127
[Rh(Hdmg)l(PPh,)]2, [Rh(Hdmg)(CIZndmg)(PPh,)],, I-hexenc hydroformylation, hydrogenation, a 127
[Rh(he~adiene)CI]~i4,4'-diheptadecyl-2,2'- bipyridine LB films, a 81
RhH(PPhJ),, itol oxidation, a I27 [Rh(norbornadiene)CI],, phenylacetylene
polymerisation, a 82 Rh(triazolinylidene), hydrosilylation, asymmetric, a 39 Rh trisulfonated triphenylphosphine, acrylic ester
hydroformylation, a 127 Catalysts, Ruthenium, biphasic 135
Pt(44)/Ru(41)/0s( 10)/1r(5), for DMFCs 163 Ru/AI,O,, H,O/NiO decomposition, a 126
hydrogenation isotherms, a 38 Ru/AI,OI, butane reforming, zirconia fuel cells 164 Ru/Mn/Ce, waste water oxidation, a 126 Ru/Pt, in fuel cells, a I28 RuS,/SiO,, HiO decomposition, a 175
benzene hydrogenation. a 178 (rl'-cyclopentadienyl)tris( acetonitrile)Ru,
norbornene cyclopropanation, a 82 [(DMSO),Ru"Pc].2DMSO, cysteine oxidation, a 79
electrooxidation, a 79
RhH(CO)(PPh,),, amide reduction, a 82
Catalysts, R ~ t h e n i ~ ~ Complexes, (T~'-C~H,)~RUZCL.
Page
Catalysts, Ruthenium Complexes, (mrd . ) [R~(4,4'-Me,bpy),(PPh,)(H,O)](C10,)~, benzylic
[Ru=C=C=CR,(L)(Cl)(arene)]PF,, ring closing olefin metathesis, a 178
Ru[BINAP], mono-ethyl fumarate, maleate, reduction, a 128
83 RuCl>(PPh,),, homoallylic alcohol isomerisation, a 128 Ru04, electrochemical oxidation of organic waste 90 Ru(PPh,),Cl,/hydroquinone, alcohol oxidation, a 178 frans-[Ru(salen)(NO)(H20)]~, ene reactions, a 178
"Catalytic Reaction Guide", from Johnson Matthey 16
Cathodic Protection, by Pt/Ti electrodes 27,116 Chemiluminescence, during oxidations,
see also Luminescence Chlor-Alkali, electrodes, development 27,46 Chlorine, sensor, using Ru tris bipyridyl, a 37 Chlmmmatics, herbicides, dechlorination, a 175 Cisplatin, a 174 Clusters, Bi,RhBr,, structure, a 34
metallic, catalysis 160 Os, OsIRu, OsIHg. OsIAu, RdCu, RuMg 146 Pd,,,phen,,(OAc),,,. alcohol oxidation, a 82 Rh,(CO),,, aldehyde hydrogenation, a 38 Ru,(CO),,(TPPTS), Ru,(CO),(TPPTS),, biphasic
hydroformylation, hydrogenation I35 RUC(CO),~, PtRu,C(CO),,(COD), with C,", a 174
27,46 Pt, from electroplating bath, a 124 see also Electrodeposltion and Deposition
10 2
Colloids, Pd, for Heck reactions, a 38 Pt, ally1 alcohol hydrogenation, a 125
PtRu[N(Oct),CI], alloys, a 78 I63
oxidations, a 79
[RuCI2(C,H,)li, hydrosilylation of ketones, a
[2]-catenanes, 0 s complexes 100
by Ru(bpy),'-, a 35
Coatings, electrodes, for chlor-alkali cells
Cold Cathodes, for flat panel display Cold Start, in fuel cell powered vehicles
formation, light catalysed, a 34
Combinatorid Chemistry, for fucl cell catalysts Conferences, 9th International Symposium on
Relations between Homogeneous and Heterogeneous Catalysis, Southampton,
England. 20-24 July 1998 160 I Ith International Symposium on Homogeneous
Catalysis, St Andrews, Scotland,
12th International Conference on the Conversion of Solar Energy into Photovoltaic Power and
12-17 July, 1998 158
Storage, Berlin, Germany, 9-14 August, 1998 140 SAE, Detroit, U.S.A., February, 1998 56 Second Anglo-Dutch Symposium, Amsterdam,
26 September, I997 25 Second International Conference on the Hydrogen
Treatment of Materials, Donetsk, Ukraine, 2-5 Junc, 1998 99
Corrosion Protection, by Pt/Ti electrodes 27, 116 reinforced concrete 116
Coupling Reactions, aryl halides, a 127 Pd catalysed 158
Cyclohe-, 4-substituted 1 methylidene- cyclohexanes, hydroboration, a 82
from benzenes, a I78 cis-Cyclooctene, hydrogenation 156
CycloproPanaton, for mctal carbene reactions, a 178 norbornene, a 82
Cyclophaues, Ru, 0 s complexes 100
Cysteine, electrooxidation. a 79
DechloWon, chloroaromatic herbicides, a 175 Decomposition, H,/N,O, over Ru/A120,, a 126
N?O, over Rh, a 38 Dehydrogenation, alkanes, by (PCP)IrH, 71 Dehydrogenative Coupling, CH,, over Pt-SOJZrO,, a 125 Dendrimm, Pd, synthesis, a I74
Platinum Metals Rev., 1998, 42, (4) 188

Page Page
80
80
Films, (confd.) [Ru(bpy),]” in organogel, 0, sensor, a Ru(I1)-polymer, photoproperties, a 35 RuO,, Ru0,-TiO,, on Pt, a see also Langmuir-Blodgett see also Thin Films
Flat Panel Displays 10 Formaldehyde, oxidation, a 178 Fuel Cells, a 83, 128, 178
DMFCs, catalysts 163 Pt-Ru anodes, a 178
HotSpotTM reactor, for on-board H, generation 2 NO reduction, Pd catalysts, a 83 PEMFCs, a 128 portable systems 115 Pt/Ru catalysts, MeOH oxidation, a 128 zirconia, butane reforming 164
Fullerenes, Ir complexes, a 79 Pt metal complexes 18 Pt, Ru complexes, a 174 Rh, Ir complexes, electrochemical oxidation,
reduction, a 35 Furans, 2,3-dihydrofurans, synthesis, a I27
Dendrimers, (contd.) photoproperties, with ‘Ru(4,7-(S0,C,H5),-phen),c, a Pt(II), SO, sensor, a
Deposition, chemical spray, Pt:SnO, films. a Pt, on C nanotubes, a PZT films, on Pt-coated Si, a see also Coatings and Electdeposition
Detectols, see Sensors Diesel Engines, pollution control, at SAE conference Diodes, A1-AI,OI-Pd, H, sensor, a Diphenylacetylene, hydrogenation Dissociation, CH,, at Pd single crystals, a DNA, photoaddition to Ru(tap)2(bpy)’+. a
Electrical Contacts, ohmic contacts, PdPtlAdPd, a
Electrochemistry, a 34-35, 79, 123, oxidation of toxic organic waste Rh, Ir fullerenes, oxidation, reduction, a
Ele&dpsition, Ir oxide films, a RuOi, Ru0,-TiO,, on Pt, a see also Castings and Deposition
Eleclnxh, anodes, 0, evolution Pt-foil, dechlorination, a Pt-Ru, for DMFCs, a
P d C felt. dechlorination, a
PdSn, Pd/Ge, thermal stability, a
cathodes, cold, PtlSiOJSilAI, flat panel display
79 176 80
124 39
56 80
156 34 79
34 83
175 90 35
124 80
116 175 178 10
175 PEMFCs, a 128
83 gas diffusion, in fuel cells, with Pd catalysts, a ir oxiddglassy C, a 124 noble metal/oxide coated Ti, development 27,46, 116 Osipolymer, in C paste, glucose sensor, a 31 Pd filmipoly(4-~inyl)pyridine, hydrazine detection, a 36 Pd-coated LaNi,,sAlo,l, a 123
35 79
60, 163 39
CO sensors 144 inorganic ion detection, a 36 pbotovoltaic devices 140
with Ru(I1)-modified chitosan, oxalic acid sensor, a 176 79
123 36 34 79
RuO, nanooarticlesiC ae rods . for sunercaoacitors. a 83
Pd, palladinised, for hydrogenations, a phthalocyanine modified, cysteine oxidation, a polypyrrole film, with Rh, Ir, Pd Pt, bottom, O2 diffusion, a
in capacitors, a 83
on polymers, medical implants 55
Pt, Pta,Rua ,,, electrooxidation, a Pt, Pt-Sn, Pt-Ru, CO oxidation, a Pt, Rh, Ir, for Au reduction and stripping, a Pt-RdC, electrooxidation, of CO, a Ru containing, benzylic oxidations, a
. . . . . . RUO;, PH sensors, a Ru0,-VO,, for electrochemical capacitors, a RuOJTi, improved, a
Electrogalvanising, Zn onto steel strip Electroless Plating, for Pd composite membranes, a
Pd membranes, a Pt, onto polymers, for medical implants
Electrolytes, K,PdCI&Fe(CN)dPEG 600, a Electron Trausfer, photoinduced, Ru, 0 s complexes Electroplat+g Baths, Pt coatings, a Electro- Emission Control, at SAE congress
motorcycle catalysts, a Esters, acrylic, hydroformylation, a Etclung, Pt, thin films, a Ethyl Acetate, hydrogenation, Rh catalysed, a 4Ethyltoluene. from 4-methylstyrene, a
Films, hydrous Ir oxide, a
37 35 35
116 124 80 55 79
100 I24 116 56
126 127 80 38 35
174 Pd, in H, sensor, a polymer, for fi-, Ir-, Pd-substituted, electrodes Pt. bottom electrodes. in caoacitors. a
36 60, 163
39 1 L
mesoporous, on Au, a 36 WTiOJF’TFE, trichlorobenzene degradation, a 175
Glucose, biosensor, Os/polymer based electrode, a Gold, reduction, stripping, at Pd, Rh, Ir electrodes, a
37 36
Heck Reactions, Pd catalysed 126, 127, 158
in supercritical COi, a 177 Heterocycles, Pd-catalysed synthesis, a 177 Heterojunctions, TiO,/RuLL’NCS/CuCN,
photoproperties, a 175 37 38
hydrogenation 156 hydrogenation, hydroformylation, a 127
coated Ti electrode 27,46, 116 Geoffre Wilkinson 168
fuel cell vehicles 2 36
60, 163
Pd colloid catalysed, a 38
*Hexme, isomerisation, Pt catalysed, a 1-Hexene, hydroformylation, by acacRh(CO)*, a
History, development of noble metalioxide
Hotspot 9 Reactor, H, generation, on-board
Hydrazine, detection, at Pd film electrode, a Hydridea, Rh, Ir complexes, electrocatalysis
Hydrocarbons, oxidation, motorcycle emissions, a
on, crude oil, using Pt catalysts
Hydrohtion, Rh catalysed, a 39 4-substituted 1 -methylidenecyclohexanes, a 82
reactions with HI. over Pt-Mo/SiO,, a 126 37 25
128 Hydrodimerisation, biphasic 135 Hydroformylation, acrylic esters, a 127
biphasic 135 by Rh complexes 25 1-hexene, a 38, 127 I-octene, a I77 olefins, a 127 styrene, a 177
Hydrogen, absorption, by Pd 141 2
78 128
photoproduction, a 124, 175 reactions with hydrocarbons,
over Pt-MoiSiO,, a 37 sensors, a 36,80, 125 storage, using (PCP)lrH, 71 treatment of materials, at HTM-98 conf. 99 wave, catalytic, from OsOJcysteine, a 175
37 38
alkenes, biphasic, a 177
HY*- thiophene, a
vinylpyrroles, a 82
generation, on-board a fuel cell vehicle permeability, in Pd,, &, a permeation, through Pd membranes, a
Hydrogen Cyanide, synthesis, over Pt, Pt-lO%Rh, a Hydrogenation, aldehydes, over h ( C O ) , s clusters, a
Platinum Metals Rev., 1998, 42, (4) 189

Hydrogenation, (contd.) ally1 alcohol, over Pt colloids, a aromatic ketones, asymmetric, Ir catalysed, a benzenes, biphasic, a b i p h a s i c C=O bonds, by Rh LB films, a CO, over Pd/ZrO,, a cis-cyclooctene enantioselective ethyl acetate, Rh catalysed, a 1 -hexene, a I isoprene, over Pd/S-AI,O,, a isotherms, of C species, on RdALO,, a
Page
127.
125 39
178 135 81
126 156 160 38
156 81 38
o-limonene 156 4-methylstyrene, using palladinised Pd electrode, a 35 olefins, a 82 organics, at Rh, Ir, Pd electrodes 60, 163 Pd catalysed, liquid-phase, selectivity 108 phenylacetylene, diphenylacetylene, a 39. 127, 156 stilbene 156 triple bonds, using C,Pd(PPh,), 18
Hydrosilylation, amides, giving amines, a 82 asymmetric, ketones, a 83
methyl ketones, a 39
Inorganic Ions, detection, at Pt electrodes, a Internet, Platinum Metals Review Ionic Liquids, biphasic catalysis
in catalysis 158, Iridium, Au reduction and stripping, a
films, hydrous oxide, a lr(001). with diamond thin films, a IIQ, COi sensors, a
1r.S *, thin films, a Iridium Alloys, IrAl, Ir, ,Ni,AI, structural properties
Iridium Complexes, fullerenes, electrochemical oxidation, reduction, a
H,lr(PPh,),, interaction with C,, a photoreactions
homoallylic alcohols, a Isomerisation, n-hexane, Pt catalysed, a
Isoprene, hydrogenation, over Pd/G-AI,O,, a Itols, unprotected, oxidation, a
Johason Matthcy, “Catalysis Technical Guide” “Catalytic Reaction Guide” HotSpotTM reactor Internet new autocatalyst manufacturing plant in Argentina “Platinum 1998”
Ketones, aromatic, asymmetric hydrogenation. a asymmetric hydrosilylation, a methyl, hydrosilylation, a synthesis, using PdCli(PPh,),, a
Lactams, synthesis, a Lactones, synthesis, a Langmuir-Blodgett Films, Rh complex,
see also Films Lean Bum, engines, at SAE conference
D-Limonene, hydrogenation LuminwcenCe, ECL, in oxalic acid sensor, a
Rdpolymer complexes, pH sensors, a [Ru(bpy),]’-, Oi sensor, a
see also Chemilumhmcence
Magnetic Fmpertiea, Pd/Co thin film multilayers, a Magnetism, in PtCo thin films, a Medical, cisplatin, anti-cancer, a
C=O hydrogenation, a
NOx reduction, a
implants, Pt-coated trans-[Pt(NC5H,C(O)NHC2H,oNo,)2C121.
36 I34 135 160 36
124 36
125 68 78
35 79 73 37
128 81
127
72 16 2
134 59
105
39 83 39
I26
178 178
81
57 I76 156 176 80 80
34 123 174 55
Page
antitumour properties, a 83 razoxane, anti-cancer drug, a 78
37 124
defect free, a 80 H: permeation, a I28
at HTM-98 conf. 99 in small fuel cells 115
128 Mercury, demercuration, of bis-(alkynyl)mercurials, a 39 Methanation, on Pt-K’/SiO,, a 80 Methane, conversion to synthesis gas, a I77
dehydrogenative coupling, a 125 34 37
oxidation, by [(bpym)PtCL] 98
Molecular Cages, [(C-R,)~M~(P-CN),~] 157
Medical, (conrd.)
Medical Uses, a 83 Membranes, composite, Ru-doped TiO?, a
Pd, by electroless plating, a
Pt-PEMs, for fuel cells, a
dissociation, at Pd single crystals, a HCN synthesis, over Pt, Pt- 1 O%Rh, a
CMethyMyene, hydrogenation, a 35
Nau@cles, Pt, optical properties, a [(RuC(CO),,)~CU~CI~]~ /SO,, hydrogenation catalyst R u O K electrodes, in supercapacitors, a
Nanotechnology, C nanotubes, from RhiPd-graphite, a Pt on C nanotubes, a
Nitrogen, from NO. in fuel cells, a Nitrogen Oxides, N,O, decomposition, over Rh, a
NO, reduction, by propene, over Pt/y-Al,O,, a
NOx, reduction, at the SAE conference
over Ru/A1201. a
over Pt/MCM-41, a to N,, in fuel cells, a
lean bum. over Pt/AI,O,, a removal, over Pt catalysts, a
Norbornme, cyclopropanation. a polymerisation. PdCI, catalysed, a
34 I46 83
I74 124 83 38
I26 I25 I76 83 56
176 37 82 81
1-Octene, hydroformylation, a 177 Ohmic Contacts, see Electrical Contacts Olefins, hydroformylation, a 127
hydrogenation. a 82 ring closing metathesis, a 178
Ru(l1) 4,4‘-bipyridinium complexes, a I75 RuSi, a 78
electrochemical 90
Optical properties, Pt nanoparticles 34
Organic Industrial Waste, destruction,
Osmium Complexes, bis-bipyridyl, polymer modified, glucose biosensor, a 37
[Os(bipy),(L)]”’, molecular assemblies I00 Os(PP,)H,. photochemical properties, a 35
clusters, 0s. Os/Ru, OsiHg, OsiAu 146
Os(VllI)OJcysteine, H, wave, a I75 Oxalic Acid, sensor, a I76 Oxidation, benzene, over Pt-F/AI,O,, a 81
CO, over Pd-RhiSi02, a 38 on Pt surfaces, sonochemistry 8
CO, HC, motorcycle emissions, a 126 electro. benzylic compounds, a 79
123 on Pt-RuiC, a 34
cysteine, by phthalocyanines, a 79 1 -propanol, 2-propanol, a 79
90 electrochemical, of Ir, Rh fullerene complexes, a 35 formaldehyde, a 178 MeOH, in fuel cells, a 128
over Pd/y-Al,O,, a 38,81
synthesis gas production, a 177 35
partial, butane reforming 164
CO, on Pt, Pt-Sn. Pt-Ru electrodes. a
toxic organic waste, by RuO,
methane, by [(bpym)PtCI,] 98
Na anthracenide, pyrenide. by Ru(bpy),”, a
Platinum Metals Rev., 1998, 42, (4) 190

Page
primary alcohols, a 178 primary allylic alcohols, a 82 unprotected itols, a 127 waste water, domestic, over RdMdCe, a 126
39 dissociation, at Pt surfaces, mechanism 24 evolution, lr0,/Taz05 anode coatings 116 photoproduction, from HiO, over RuS,/SiO?, a 175
Oxidation, (contd.)
Oxygm, diffusion, in Pt bottom electrodes, a
. . sensor, a
fibre-optic, Ru-based, a oDtical. a
80 125 176
photoluminescent, with Pt porphyrin, a 36
Palladium, P-[(CHI),N][Pd(dmit)J2, superconductor, a electrodes, Pd-coated LaNi,,Al,,,, a film electrode, in hydrazine detector, a film, in H, sensor, a H, treatment, at HTM-98 conf. membranes, composite, by electroless plating, a
defect free, a for fuel cells H, permeation, a
neutral atoms, Fourier transform spectrum, a palladinised Pd electrode, for hydrogenations, a Pd-AI,O,-AI, H, sensor, a Pd/Co multilayer thin films, magnetic properties, a PdGaN Schottky diode, a Pd/Ge/Pd interlayers, between n-GaAs and Si, a Pd/Pt/AdPd ohmic contacts, a Pd/Rh-graphite, C nanotube production, a Pd/Sn, Pd/Ge, ohmic contacts, a Pd/SnO, thin film, CO sensor, a PdH particles, imaging single crystals, CH, dissociation, a thin films, H2 sensors, a
Palladium Alloys, H, treatment, at HTM-98 conf. Pd,.,,Si,, H2 permeability, a PdCr, strain gauges, a Ti-Pd-Ni, shape memory properties, a
superconductor, a Palladium Complexes, P'-Et,Me,P[Pd(drnit),],,
178 123 36 36 99
124 80
115 128 I74 35 80 34
128 128 34
174 83
176 141 34
I25 99 78
I25 83
78 calixaienes 1 1 , 163 dendrimers, from Pd[CH,CN],(BF,),, a 174 fullerenes 18
[Pd(L)](PF&, synthesis, a I74 K,PdCh/K,Fe(CN)dPEG 600 electrolyte, a 79
photoreactions 73 Patents 4 W , 84-88, 129-132, 179-182 pH, sensors, a 37,80 wenylacetylene, homologs, hydrogenation, a 127
hydrogenation 39, 156 polymerisation, a 82
Phenylation, acid chlorides, aryl iodides, a 126 phoaphines, sulfonated, H,O-soluble catalysts 135 Photccatalysk,,photocuring,
of ceramic precursors, a 123 F'hotoumvemion, a 35.79, 123-124, 175 Phot&t&or, a Photonics, LEDs, a P h ~ b p p t i e s , 'R~(4,7-(SO,C~~),-phen),~,
with dendrimers, a in combinatorial chemistry PfliOJPTFE, trichlorobenzene degradation, [ (Pt(CN)(C,,H,,N!)I,l, a Pt(1I) complexes, in probes for SDS micelles Ru dye molecules, on TiO, surfaces, a Ru(PP3)H2, Os(PP,)H2, a RuS,/SiO,, H20 decomposition, a bis(terpyridyl)Ru(lI), a TiOJRuLL'NCSICuSCN heterojunction, a
Photoreadons, in cyclophanes and catenanes H,Ir(PPh,), with Cm, a Pt, Pd, Ru, Ir, Rh complexes Ru(tap)2(bpy)'', addition to DNA, a
36 81
79 163
a I75 123
, a 124 124 35
175 176 175 100 79 73 79
Photosynthesis, artificial models, Ru, 0 s complex Photovoltaic Cells, with c~s-Ru"(LH,),(NCS)~, a Photovoltaic Devices, at solar energy conference
H2 production from H,O, a "platinum 1998" Platirmm, Au reduction and stripping, a
deposition, on C nanotubes, a onto polymers, medical implants
electrodes, CO sensors inorganic ion detection, a oxalic acid sensor, a
films, bottom electrodes. 0, diffusion, a mesoporous, on Au, a
nanoparticles, optical properties, a Pd/Pt/AdPd ohmic contacts, a polymer-coated Pt plates, in transistors, a polymers, containing Pt, a Pt( 1 10) crystal, sonochemistry Pt( I 1 I ) surface, mechanism of Oi dissociation Pt:Sn02 thin films, as CO sensors, a Pt. Pb,,Ru, ,,, electrooxidation, a Pt, Pt-Sn, Pt-Ru electrodes, CO oxidation, a Pt-coated Si, PZT film growth, a Pt-PEMs, for fuel cells, a Pt-Ru anodes, for DMFCs, a Pt-RdC, electrooxidation, of CO, a PtlSiOJWAl, cold cathodes with RuOi, RuO,-TiO,, films, a thin films, by etching, a
on SiO& by MOCVD, a Ti/Pt/Au, Schottky contact with InGaP,
photodetectors, a Platinum Alloys, Pt, ,Al,.,, phase transitions, a
PtCo thin films, magnetic properties, a PtRu[N(0~t),Cl]~ colloids, a
Platinum complexes, calixarenes fullerenes [NBb][fran~-Pt"(C,F~)~Br(C0)], structure, a (OC)Pt[p-N,N'-N(NPh)CJL]r
ReCI[NH(NPh)C,H,], a
Page ;es 100
35 140 124 105 36
124 55
144 36
176 39 36 34 34
125 123
8 24 80 79
123 39
128 178 34 10 80 80
124
36 78
123 78
1 1 , 163 18 78
123 photoreactions 73 [PtlL'2(p-dppm)l~, [PtJ-'2(p-dppm)l*-,
photoproperties, a 124 Pt porphyrin, for 0, sensing, a 36 cis-[PtCI2(NH,),], cisplatin, a 174 cis-, frans-[PtCI,(PhCH,CN)i],
[Pt( EtCN),] [ S0,C FA]? 106 cis-PtCl,(razoxane), structure, a 78 [ (Pt(CN)(CloH,INl))s], photoproperties, a 123 Pt(ll) dendrimers, SO2 sensor, a 176 [Pt(L)](PFs),, synthesis, a 174 trans-[Pt(NCIH,C(0)NHC,H,ONOi)lCli],
antitumour properties, a 83 Pt(N0,)(H20)'*, in electroplating baths, a 124 PtRu5C(CO),.(COD), with C,, a I74
144 25
at the SAE conference 56 toxic waste, electrochemical destruction 90
2 Polyamtylene, from phenylacetylene. a 82 Polyketones, synthesis 158 PolymerisatiOn, norbornene, PdCI, catalysed, a 81
phenylacetylene, a 82 Ru catalysed 158
Polymm, containing Pt, a 123
films, with Pt, in 0, sensors, a 36
with Pd colloids, Heck reactions, a 38 polypyrrole film electrodes, with Rh, Ir, Pd 60, 163
55 Rh/PPA(Na+)/DPPEA, olefin hydroformylation, a 127 siloxane ring, in Ru pH sensors, a 80 synthesis, Pd catalysed 160
"platinum Labware Catalog", Alfa Aesar Pollution Control, at 2nd Anglo-Dutch Symposium
using fuel cell powered vehicles
electrolytes, a 79
with Ru(ll), photoproperties, a 35
Pt coated, for medical implants
Platinum Metals Rev., 1998, 42, (4) 191

Page Ropane, carbonylation. a 82 Protoas, reduction, by Rh. Ir. Pd complexes 60, I63
Razoxane, anti-cancer drug, a 78 Reduction, electro. CO,, Pd catalysed, a 79
electrocatalytic, of protons 60. 163 electrochemical. of Ir. Rh fullcrcnc complexes, a 35 mono-ethyl fumarate, maleate, a 128 NO, by propene. over Pt/y-Al,O,. a 125 tertiary amides, by hydrosilylation, a 82
Reformin& butane, zirconia fuel cells I64 Relay Switches, in heterocycle.
carbocycle assembly, a 177 Rhodium, Au reduction and stripping, a 36
I74
molecular cages 157 calixarenes 1 1 . 163 fullerenes, electrochemical oxidation. reduction. a 35
34 34 37
I78 I23
Rh/Pd-graphite, C nanotube production, a Rhodium Co@exe~, [(C,Rs),Mx(p-CN),,].
photoreactions 73 Rhodium Compounds, BiXhBr,, structure, a Ruthenium, Pt-RdC, electrooxidation, of CO. a
Ru-doped TiOl composite membranes. a Ru-Pt anodes, for DMFCs, a Ru-Pt electrodes, CO oxidation, a RuO:-VO,, dip-coated electrodes,
RuOJTi electrodes, from RuOl-La20,/Ti, a TiOdRuLL'NCSiCuSCN heterojunction,
Ruthenium Alloys, PtRu[N(Oct),CI], colloids, a
electrochemical capacitors. a 35 3s
photoproperties, a 175 78
Ru, ,,Ph,, electrooxidation, a 79 Ru modified Fe40Cr. Fe-35Cr-5Al. a 123 RuSi, optical properties, a 7x
with dendrimers, photoproperties, a 79 Ruthenium Complexes, 'R~(4,7-(SO,C,H,)~-phen),i . tris(2,2'-bipyridine)Ru( I I)-modified chitosan.
tris(5-acrylamid0,l ,I0 phenanthroline) Ru chloride,
tris-bipyridyl, CI2 sensor. a 37 [(bpy),Ru(phendione)I(PF,)I,
oxalic acid sensor, a 176
O2 sensor, a I25
[(bpy),Ru(phendioxime)](PF,)~, photoproperties, a I24
calixarenes 1 1 . 163 clusters, RdOs, RdCu RdHg I46
RuC(CO),,, PtRu,C(CO),,(COD), with C,. a 1 74
[Ru(bpy),]- , 0, sensor, a 80 Ru(bpy)l'*, oxidation of Na anthracenide, pyrenide, a 35 [Ru(bpy)l'(PhB )?I, optical 0, sensor, a I76 [Ru(bpy)N(bpy(C02MePEG-3S0),),.,](C10,)2.
molten salts, a 34 [Ru(CO),(PPh,)(rl-C,Mer)l[Fe~(~~-C2Bu')(CO)"], a 174 cis/trans-RuH,(Ph,PCH,PPhi)i,
~ncRuHCI(Ph,PCH.PPh,),. a I74
photoreactions 73
[R~(bipy)~(L)]". molecular assemblies 100
Page
RU(I~'-C(CN)~C[C=C { Ru(PPh,)Fp] ]C=C(CN).)-
Ru(H)(H,)CI(PCy,),, reactivity, a Ru(II)(2 2'-bipyridyl-4.4'dicarboxylate),(NCS)..
(PPhXp, a
p h o t o v o l t h Ru(I1) 4,4'-bipyridinium. optical properties, a Ru(lI), in polymer film, photoproperties. a cis-Ru"(LH,),(NCS),, sensitiser for
photovoltaic cells, a trans-[Ru(NH,),L(NO)](BF,),, a [R~(phen)~[phen(OH)~]]'-, [Ru(Ph2phen)?-
[phen(OH),]]", pH sensors, a Ru(PP,)Hi, photochemical properties, a Ru(tap),(bpy)'+, addition to DNA, a bis(terpyridyl)Ru(ll), photoproperties, a
Ruthenium Compounds, Ru02, pH sensors, a RuO?, Ru02-Ti02, films, on Pt. a RuO, nanoparticlesiC electrodes, in supercapacitors.
175 34
140 175 35
35 175
80 35 79
176 37 80
a 83
Ruthenium Compounds, ( conrd. )
Schottky Contacts, InGaP. with TiiPtlAu. photodetectors, a
Schottky Diode, PdiGaN, a Selectivity, in Pd catalysed hydrogenations Sensors, CI2. using Ru tris bipyridyl. a
CO,, Ir02-. W0,-based. a CO, battery powered
Pd!Sn02 thin films. a Sn0,:Pt films. a
glucose, with Osipolymer complcx. a H:, AI-AI?O,-Pd diode. a
(SrRuO,/Ba,Sr, ,TiOdSrRuO?). thin film capacitors.
by Pd film, a thermal, with Pd films, a
inorganic ions, at Pt elcctrodes, a O?, by [Ru(bpy),]' , in organogel. a
fibre-optic. Ru-based. a optical. a photoluminescent. a
oxalic acid, a Pd(l), properties, a pH, at RuO? electrodes, a
photo, performance. a SO,. by Pt(lI) dendrimers. a strain, a
Shape Memory, in Ti-Pd-Ni. a Sodium Antbwnide, oxidation, by Ru(bpy),", a Sodium Pyrenide, oxidation, by Ru(bpy)?'*, a Smochemislry, for catalytic rate enhancement.
Spectra, Pd(l). properties, a Sputtering, of PdKo thin film multilayers. a Stilbene, hydrogenation Stille Reactions, in supercritical CO,. a Strain Gauges, PdCr thin films, a Styrene, from phenylacetylene. Rh catalysed. a
hydroformylation, a reaction with I-bromoadamantane, a
Sulfoximines, N-arylated, synthesis, a Sulfur Dioxide, sensor, Pt( 11) dendrimers. a Supercapacitors, Ru0: nanoparticlesiC aerogels, a Superconductor, P'-Et,Me2P[Pd(drnit),],. a
Supramolecules, Ru, 0s complexes. electron transfer Surface Science, H. uptake on Pd
Synthesis Gas, from methane. a
luminescent Ru complexes. a
on PI surfaces
I.1-[(CH,),N][Pd(dmit)~]~, a
Pd(0) surfaces
giving MeOH, a
ThinFilms, capacitors. Pt/(Ba, Sr)TiO,/Pt. a (SrRuOJBaSr, ,TiOdSrRuO,), a
diamond, epitaxial. on Ir(001). a l r S , ". a Pd, H2 sensors. a PdKo multilayers, magnetic properties. a PdiSnO,, CO sensor, a PdCr, strain gauges, a Pt:SnO,, as CO sensors, a Pt. etched, a
on SiOJSi, by MOCVD, a PtCo, magnetic properties. a see also Films
Transistors, polycarbazole conducting polymer. a Trichlorobemme, photodegradation, a
Vinylpymles, hydroformylation. a
Waste, industrial, destruction Water, domestic, oxidation, a
photodecomposition, over RuS,/Si02, a waste, oxidation, over RdMniCe, a
Willrinson, Geoffrey. Prof. Sir
a 83
36 128 108 37
I25 I44 176 80 37 80
125 36 36 80
125 I76 - 36 I76 174 37 xo 36
176 125 81 .. 3s 35
8 174 34
IS6 177 12s 39
I77 126 I77 176 83 78
178 100 141 160 I77 126
83 83 36 78
12s 34
176 125 80 80
124 123
125 I75
82
90 126 I75 I26 168
Platinum Metals Rev., 1998, 42, (4) 1 9 2
Recommended

















