
Mott insulatorsMott insulators
• Mott-Hubbard type vs charge-transfer type • Cluster-model description • Chemical trend• Band theory• Self-energy correction• Electron-phonon interaction

Mott insulatorsMott insulators
• Mott-Hubbard type vs charge-transfer type

Correlated electron systems/Correlated electron systems/Complex materials Complex materials
La2-xSrxCuO4
Ga1-xMnxAs

Lattice models for transitionLattice models for transition--metal compounds metal compounds
p-d modelHubbard model
Transition metal ion (with d orbitals)
Non-metal anion (with p orbitals)

Lattice models for transitionLattice models for transition--metal compounds metal compounds
p-d model(degenerate) Hubbard model
t-J model
no double occopancy
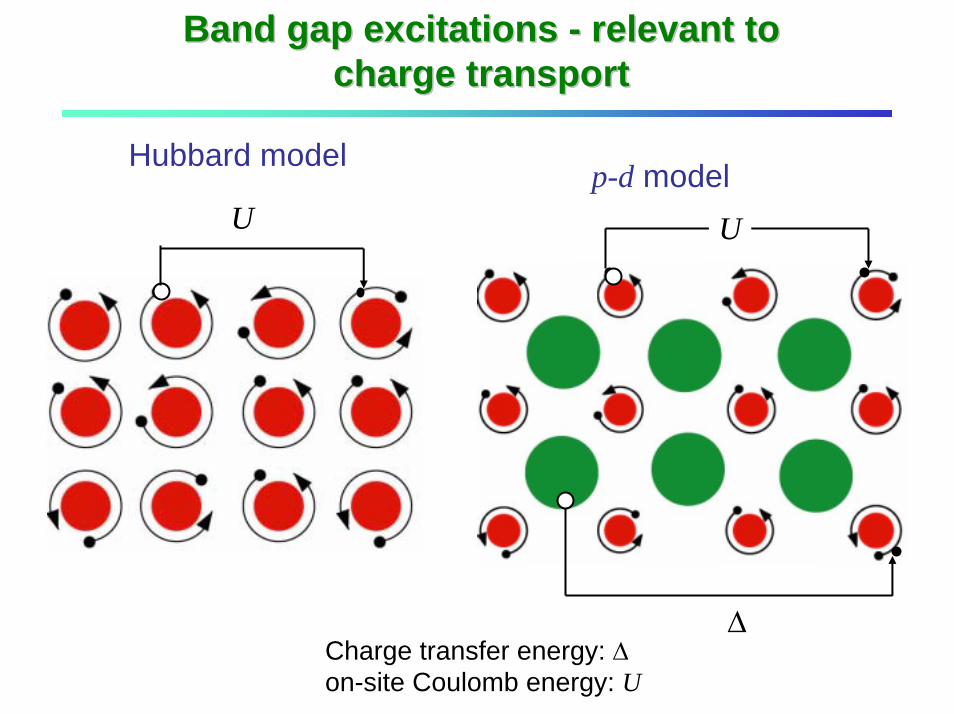
Band gap excitations Band gap excitations -- relevant to relevant to charge transportcharge transport
Hubbard modelp-d model
U U
∆Charge transfer energy: ∆on-site Coulomb energy: U
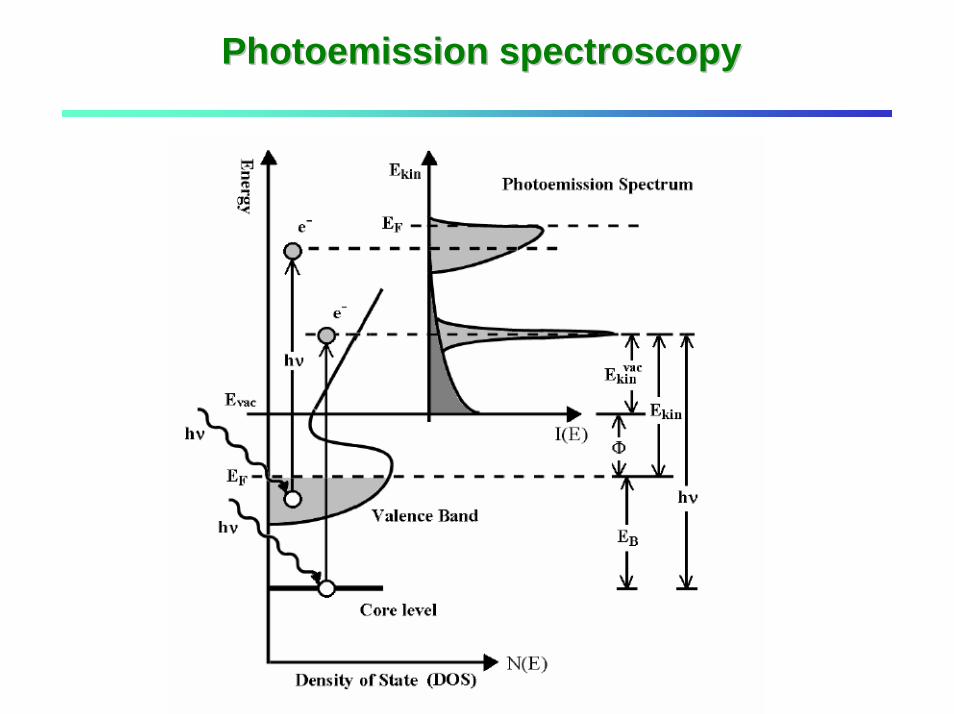
Photoemission spectroscopyPhotoemission spectroscopy
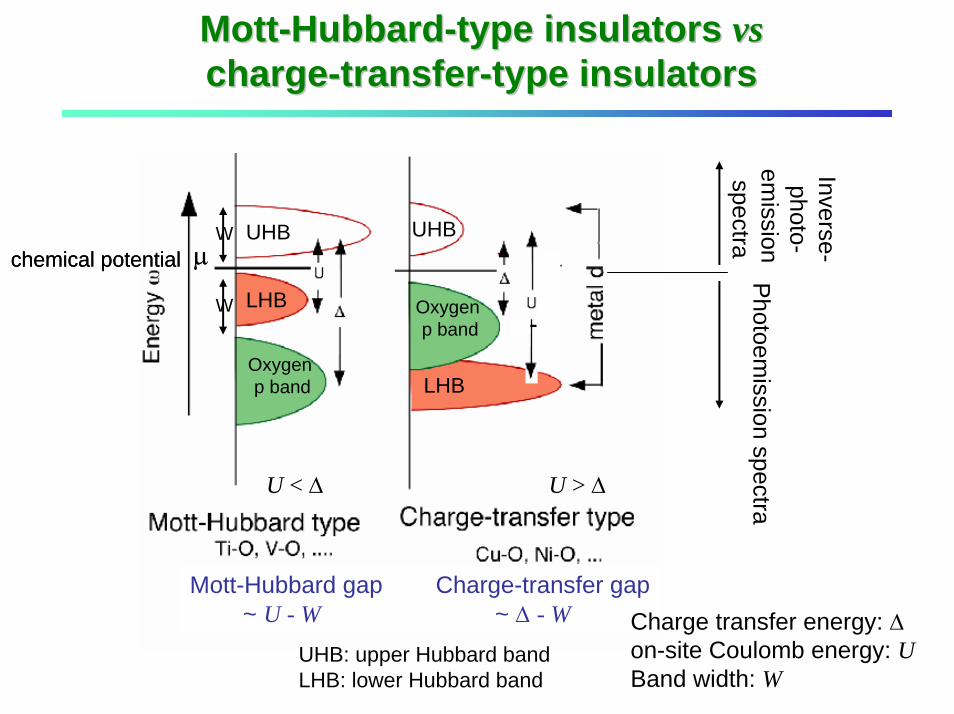
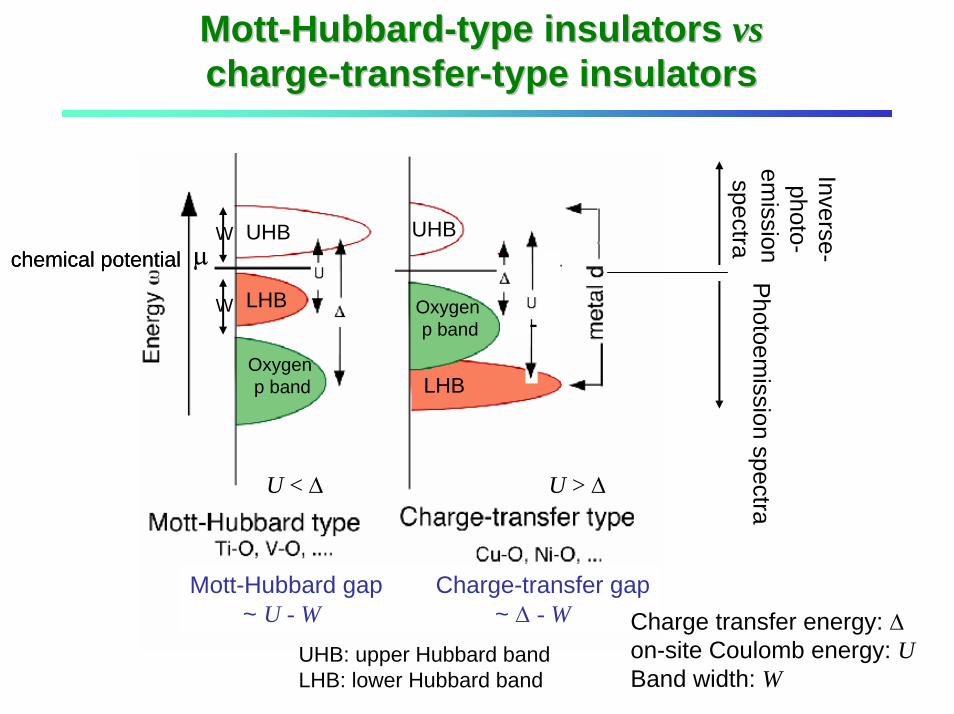
MottMott--HubbardHubbard--type insulators type insulators vsvschargecharge--transfertransfer--type insulatorstype insulators
µ
Mott-Hubbard gap Charge-transfer gap~ U - W ~ ∆ - W
chemical potential Photoem
ission spectra
Inverse-photo-
emission
spectra
U < ∆ U > ∆
Charge transfer energy: ∆on-site Coulomb energy: UBand width: W
µchemical potentialW
W
UHB
LHB
Oxygen p band
Oxygen p band
UHB
LHB
UHB: upper Hubbard bandLHB: lower Hubbard band

Mott insulatorsMott insulators
• Cluster-model description
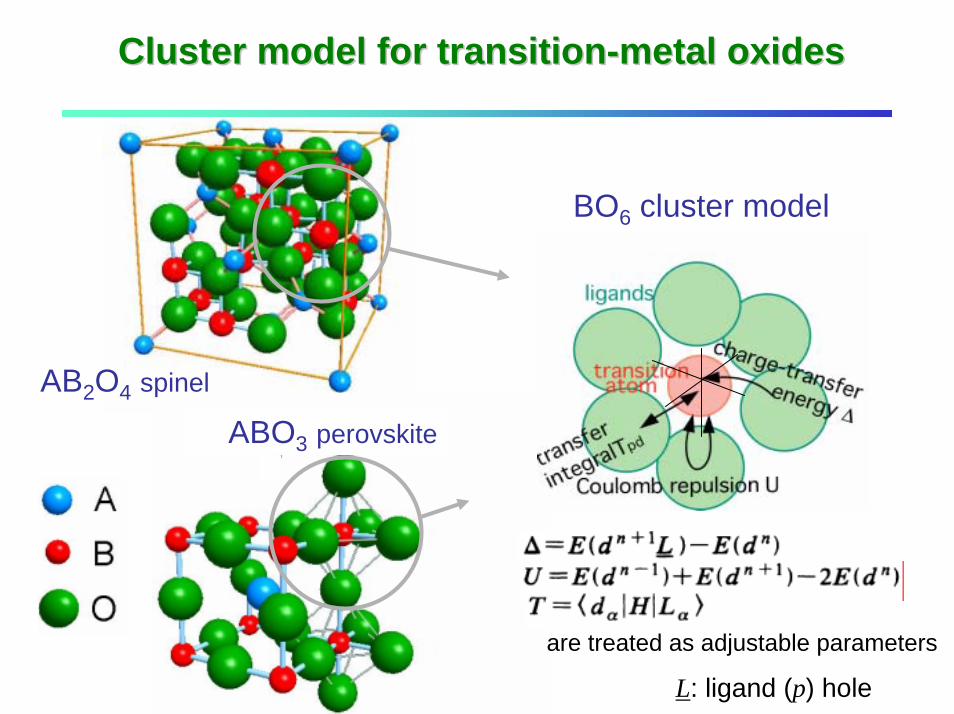
Cluster model for transitionCluster model for transition--metal oxidesmetal oxides
are treated as adjustable parameters
perovskite
AB2O4 spinel
L: ligand (p) hole
ABO3 perovskite
BO6 cluster model

ManyMany--electron energy levels electron energy levels vsvs singlesingle--particle energy level particle energy level
Photoemission
Inverse
photoemission
µ Photoem
ission spectra
Eg
Inverse-photoem
ission spectra
Ground state
EN
+1
EN
-1 µ : chemical potentialEg : band gap
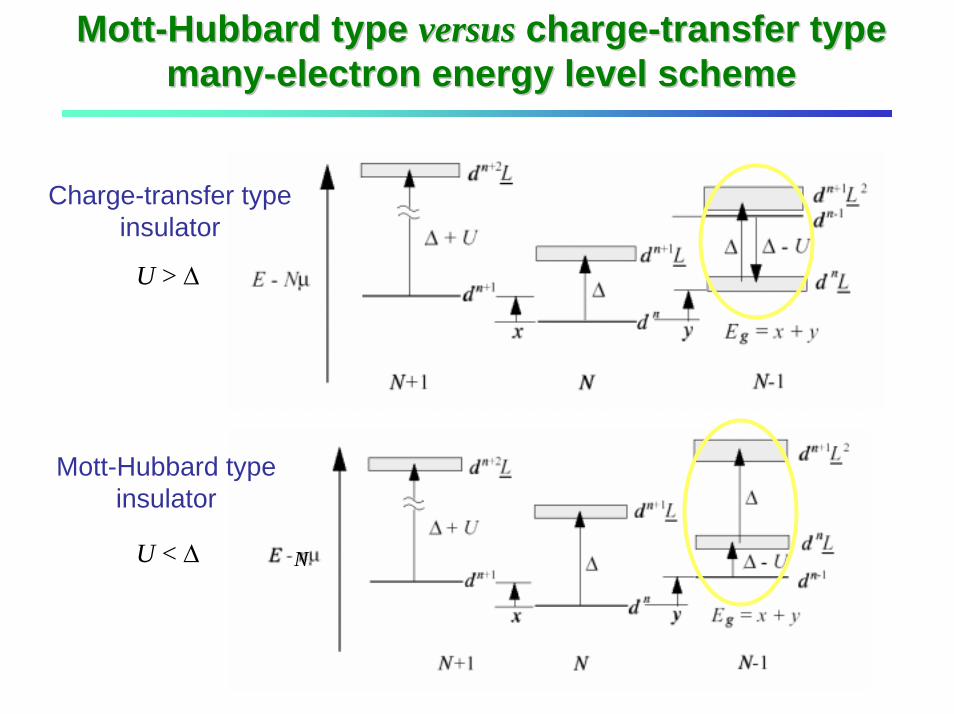
MottMott--Hubbard type Hubbard type versusversus chargecharge--transfer type transfer type manymany--electron energy level scheme electron energy level scheme
Mott-Hubbard typeinsulator
NU < ∆
Charge-transfer typeinsulator
U > ∆
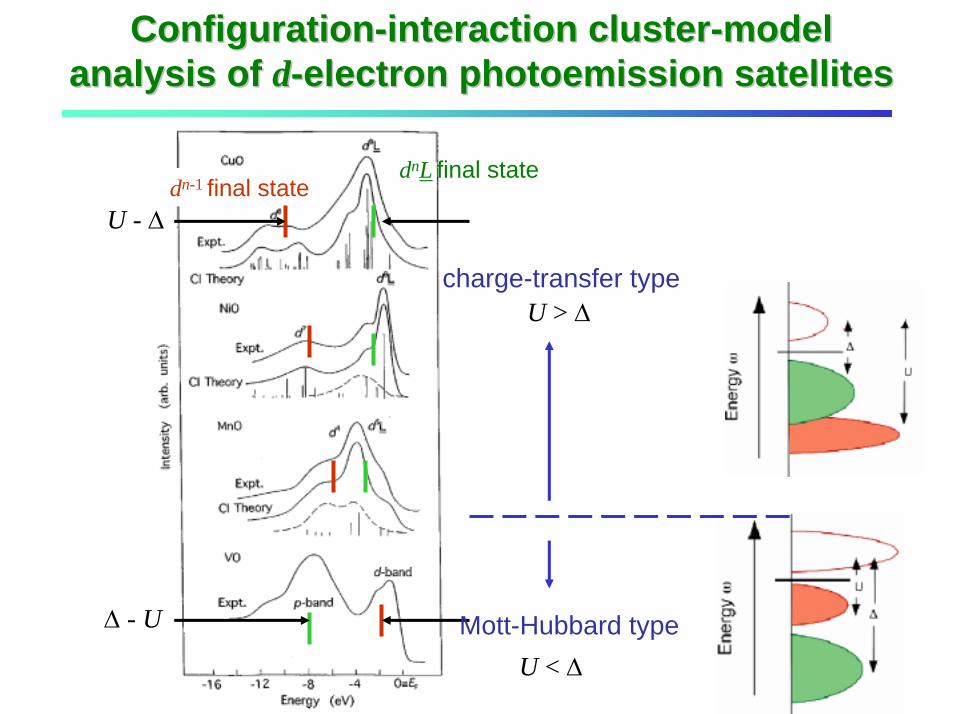
ConfigurationConfiguration--interaction clusterinteraction cluster--model model analysis of analysis of dd--electron photoemission satelliteselectron photoemission satellites
dn-1 final statednL final state
U - ∆
∆ - U
U > ∆
U < ∆
charge-transfer type
Mott-Hubbard type
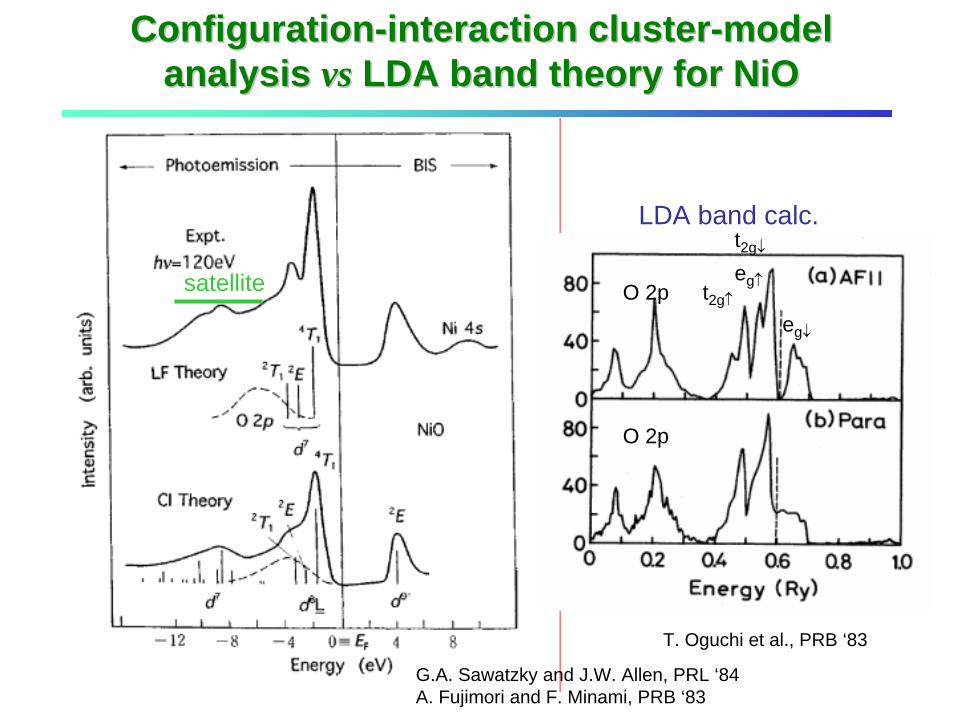
ConfigurationConfiguration--interaction clusterinteraction cluster--model model analysis analysis vsvs LDA band theory for LDA band theory for NiONiO
G.A. Sawatzky and J.W. Allen, PRL ‘84A. Fujimori and F. Minami, PRB ‘83
T. Oguchi et al., PRB ‘83
satellite
LDA band calc.
O 2p
O 2peg↓
t2g↑
t2g↓
eg↑

Mott insulatorsMott insulators
• Chemical trend
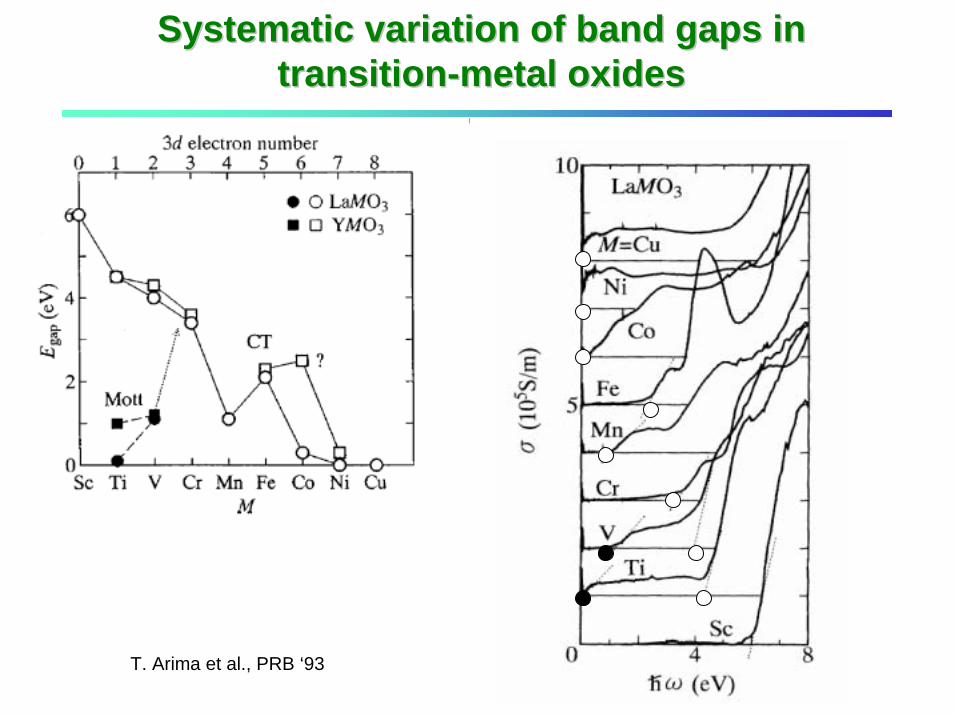
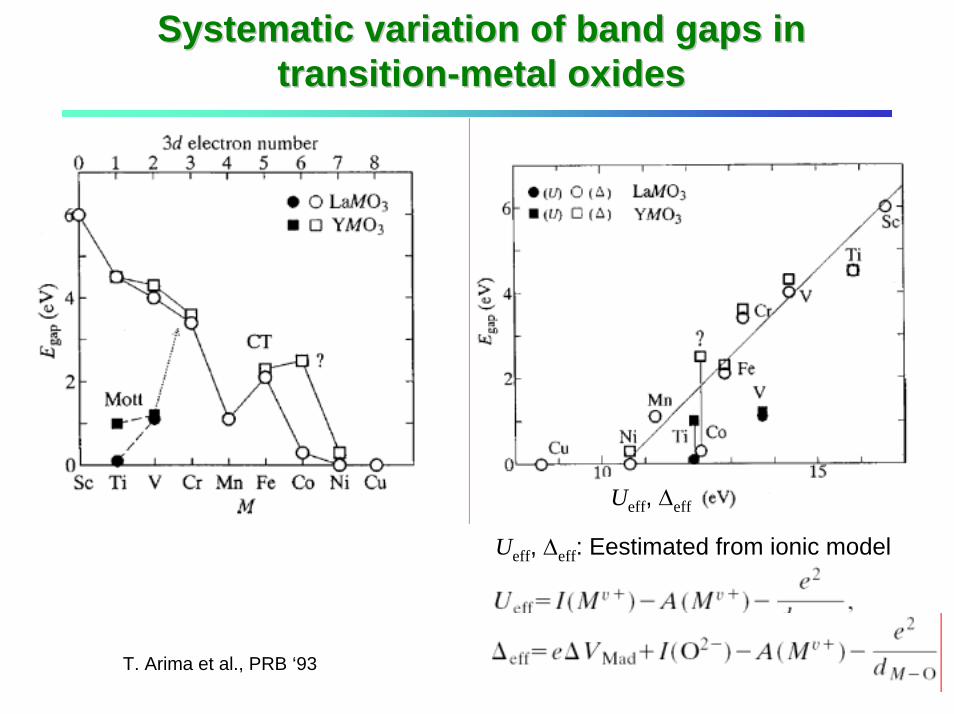
Systematic variation of band gaps in Systematic variation of band gaps in transitiontransition--metal oxidesmetal oxides
Ueff, ∆eff
T. Arima et al., PRB ‘93
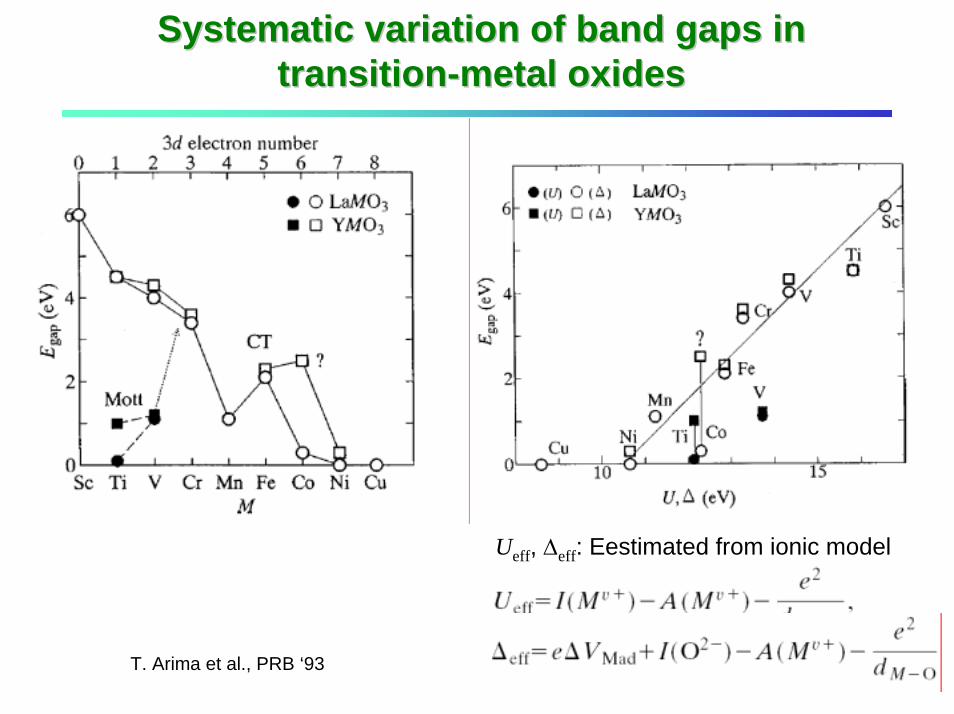
Systematic variation of band gaps in Systematic variation of band gaps in transitiontransition--metal oxidesmetal oxides
Ueff, ∆eff: Eestimated from ionic model
T. Arima et al., PRB ‘93
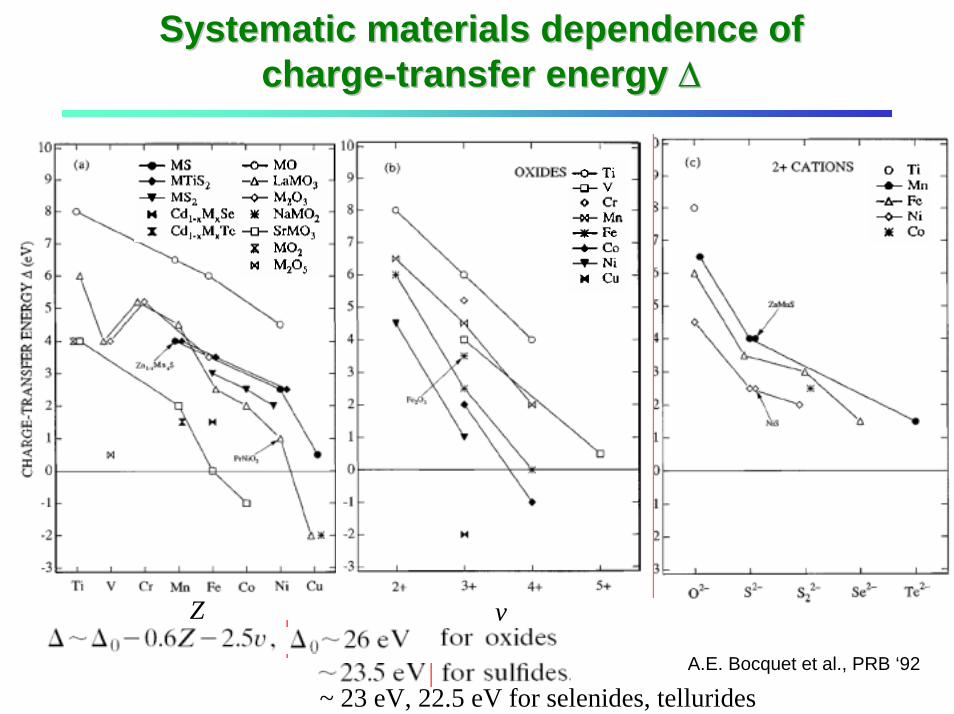
Systematic materials dependence of Systematic materials dependence of chargecharge--transfer energy transfer energy ∆∆
Z v
A.E. Bocquet et al., PRB ‘92
~ 23 eV, 22.5 eV for selenides, tellurides
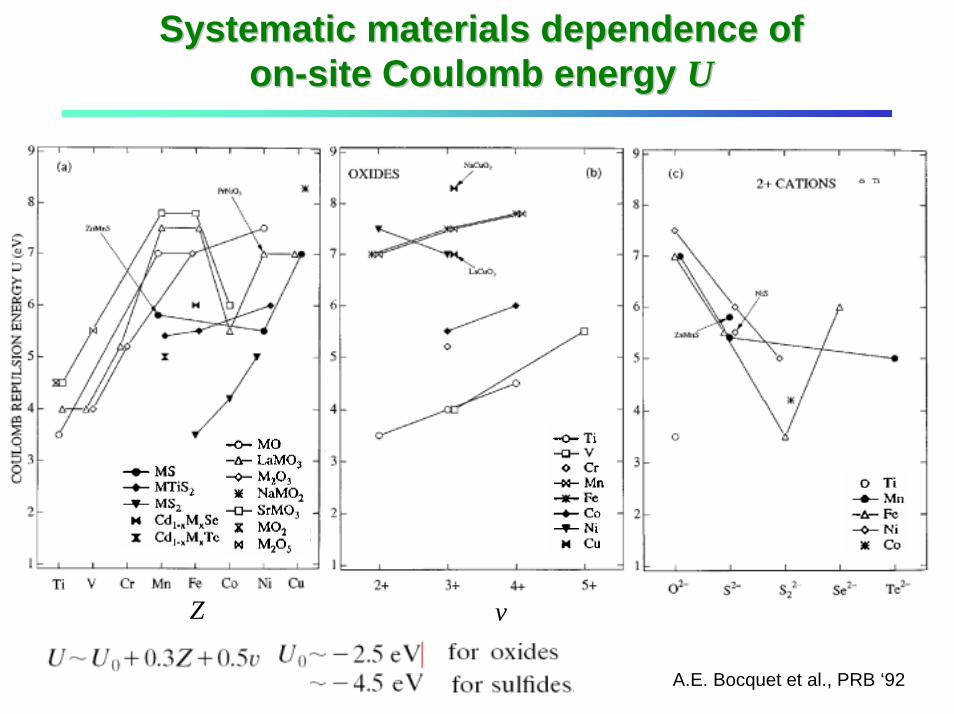
Systematic materials dependence of Systematic materials dependence of onon--site Coulomb energy site Coulomb energy UU
Z v
A.E. Bocquet et al., PRB ‘92
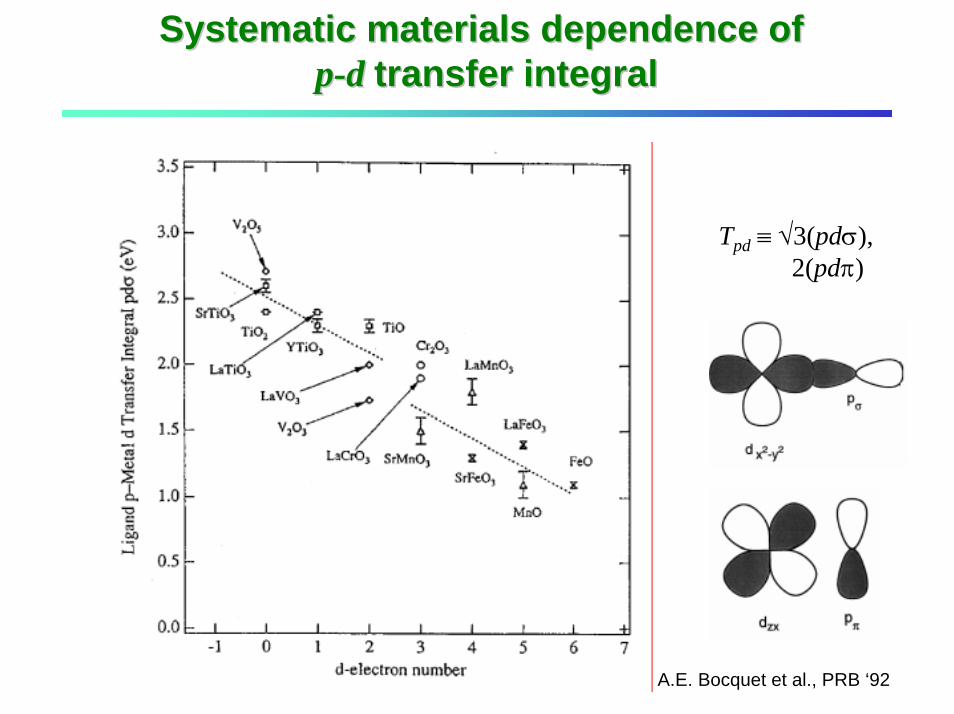
Systematic materials dependence of Systematic materials dependence of pp--d d transfer integral transfer integral
Tpd ≡ √3(pdσ), 2(pdπ)
A.E. Bocquet et al., PRB ‘92

ZaanenZaanen--SawatzkySawatzky--Allen diagramAllen diagram
Mott-Hubbard regime
Mott-Hubbard regime
charge-transferregime
charge-transfer regime
nega
tive-
∆re
gim
e
Eg ~ ∆ − W
Eg ~ U - Wp-metal
d-metal
U = W
∆ = W
4+
3+
3+
2+
3+
3+
3+ 3+
3+
3+3+3+
2+
2+
2+
2+4+4+
4+
5+
J. Zaanen, G.A. Sawatzky, J.W. Allen, PRL ‘85 A.E. Bocquet et al., PRB ‘96
MottMott--HubbardHubbard--type insulators type insulators vsvschargecharge--transfertransfer--type insulatorstype insulators
µ
Mott-Hubbard gap Charge-transfer gap~ U - W ~ ∆ - W
chemical potential Photoem
ission spectra
Inverse-photo-
emission
spectra
U < ∆ U > ∆
Charge transfer energy: ∆on-site Coulomb energy: UBand width: W
µchemical potentialW
W
UHB
LHB
Oxygen p band
Oxygen p band
UHB
LHB
UHB: upper Hubbard bandLHB: lower Hubbard band
Systematic variation of band gaps in Systematic variation of band gaps in transitiontransition--metal oxidesmetal oxides
Ueff, ∆eff
Ueff, ∆eff: Eestimated from ionic model
T. Arima et al., PRB ‘93

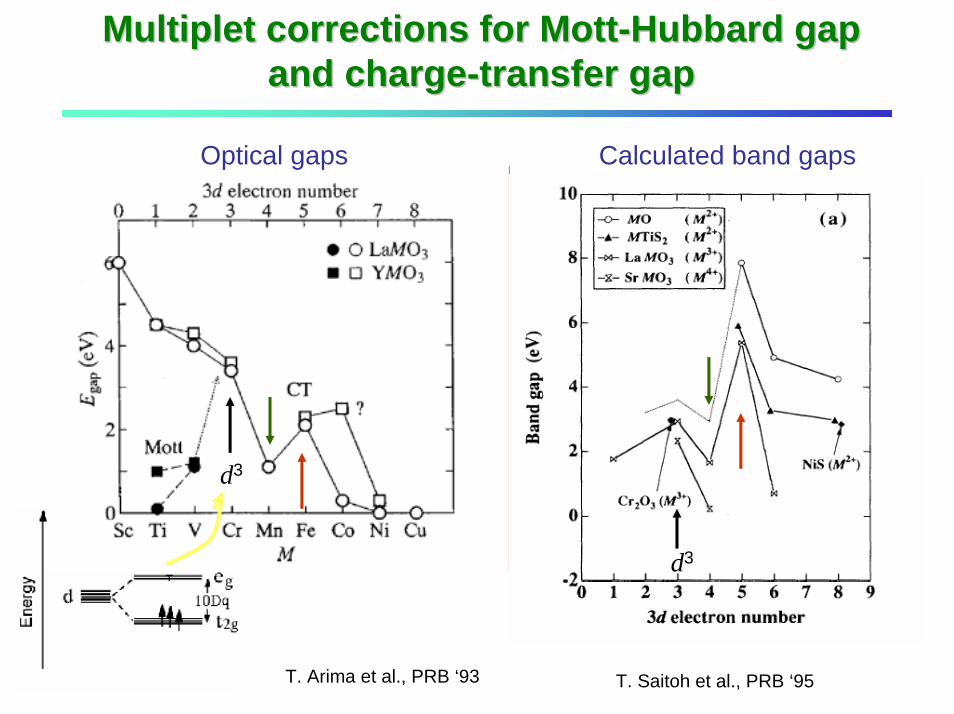
Multiplet corrections for MottMultiplet corrections for Mott--Hubbard gap Hubbard gap and chargeand charge--transfer gaptransfer gap
Correction for charge-transfer energy: ∆ → ∆eff
Correction for on-site Coulomb energy: U → Ueff
d5
d4
M-H and CT gap is enhanced
CT gap is reduced
Multiplet corrections for ∆ and U
T. Saitoh et al., PRB ‘95
Multiplet corrections for MottMultiplet corrections for Mott--Hubbard gap Hubbard gap and chargeand charge--transfer gaptransfer gap
Optical gaps
d3
d3
Calculated band gaps
T. Arima et al., PRB ‘93 T. Saitoh et al., PRB ‘95
Mott insulatorsMott insulators
• Band theory
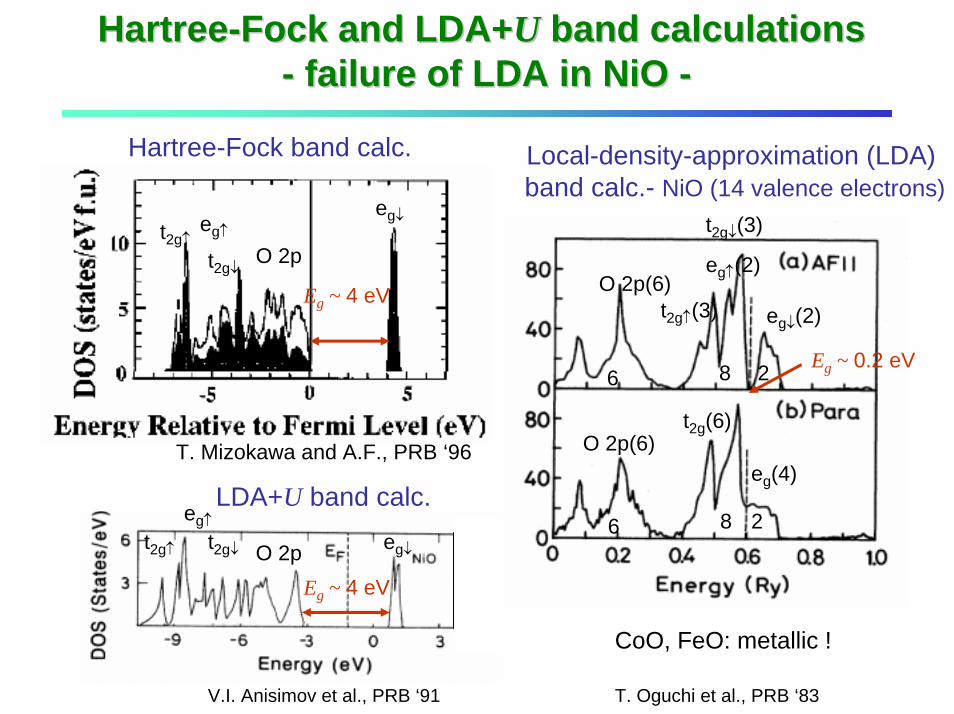
HartreeHartree--Fock and LDA+Fock and LDA+UU band calculationsband calculations-- failure of LDA in failure of LDA in NiONiO --
Hartree-Fock band calc.
O 2p(6)
O 2p(6)eg↓(2)t2g↑(3)
t2g↓(3)
eg↑(2)
eg↓
eg↓O 2p
O 2pt2g↑
t2g↓
eg↑
t2g↑ t2g↓
eg↑LDA+U band calc.
V.I. Anisimov et al., PRB ‘91
T. Mizokawa and A.F., PRB ‘96
Eg ~ 4 eV
Eg ~ 4 eV
Eg ~ 0.2 eV
eg(4)
t2g(6)
6 8 2
6 8 2
Local-density-approximation (LDA) band calc.- NiO (14 valence electrons)
CoO, FeO: metallic !
T. Oguchi et al., PRB ‘83

Failure of LDA in Mott insulatorsFailure of LDA in Mott insulators
Hartree-Fock potential energy (also for LDA+U)
: occupation number of orbital i
→ orbital-dependent self-consistent potential→ positive feedback toward orbital polarization
Local-density approximation (LDA) potential energy
: total occupation number (local density)
→ “spherically” averaged potential, unphysical self-interaction→ orbital polarization suppressed
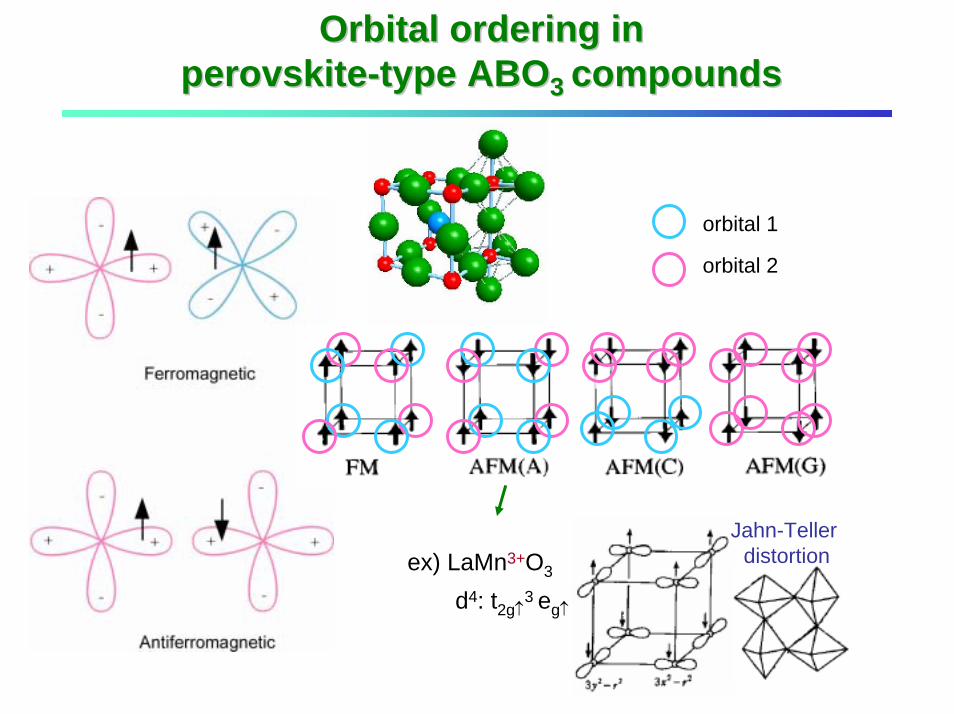
Orbital ordering in Orbital ordering in perovskiteperovskite--type ABOtype ABO3 3 compoundscompounds
Jahn-Teller distortion
orbital 1
orbital 2
ex) LaMn3+O3
d4: t2g↑3 eg↑

Mott insulatorsMott insulators
• Self-energy correction
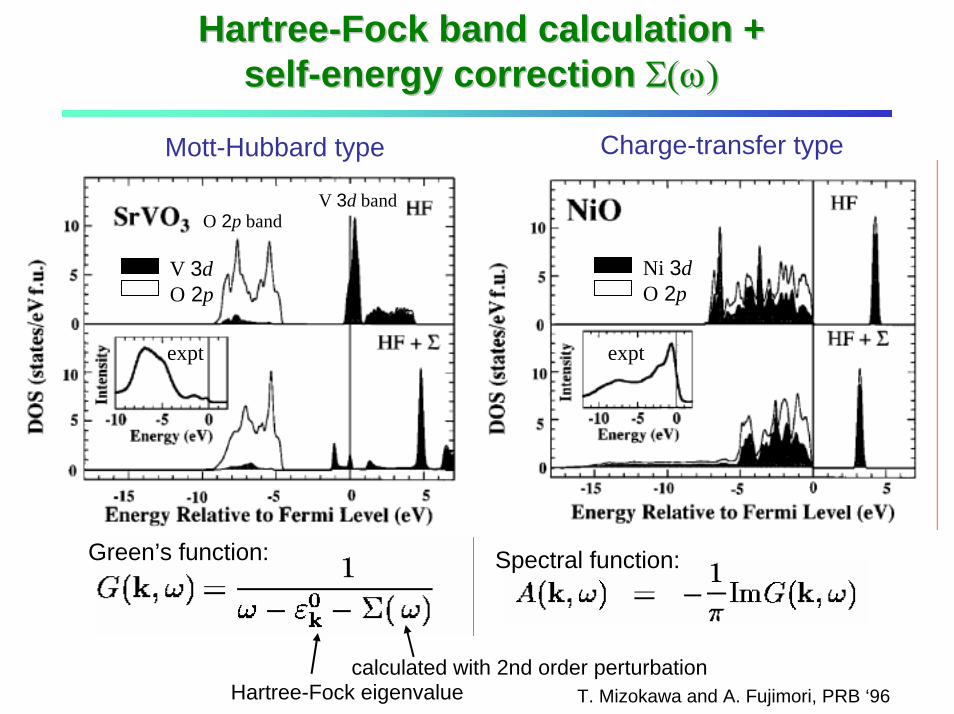
HartreeHartree--Fock band calculation + Fock band calculation + selfself--energy correction energy correction Σ(ω)Σ(ω)
T. Mizokawa and A. Fujimori, PRB ‘96calculated with 2nd order perturbation
Hartree-Fock eigenvalue
expt
Spectral function: Green’s function:
V 3dO 2p
expt
Ni 3dO 2p
Mott-Hubbard type Charge-transfer type
O 2p band V 3d band
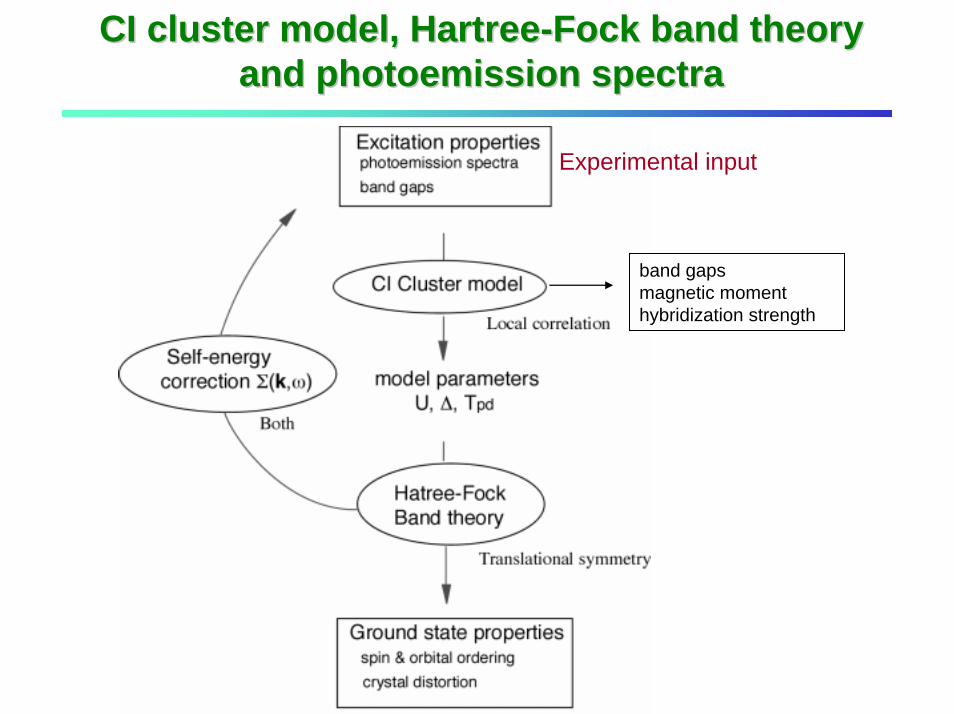
CI cluster model, HartreeCI cluster model, Hartree--Fock band theory Fock band theory and photoemission spectraand photoemission spectra
Experimental input
band gapsmagnetic momenthybridization strength

Mott insulatorsMott insulators
• Electron-phonon interaction
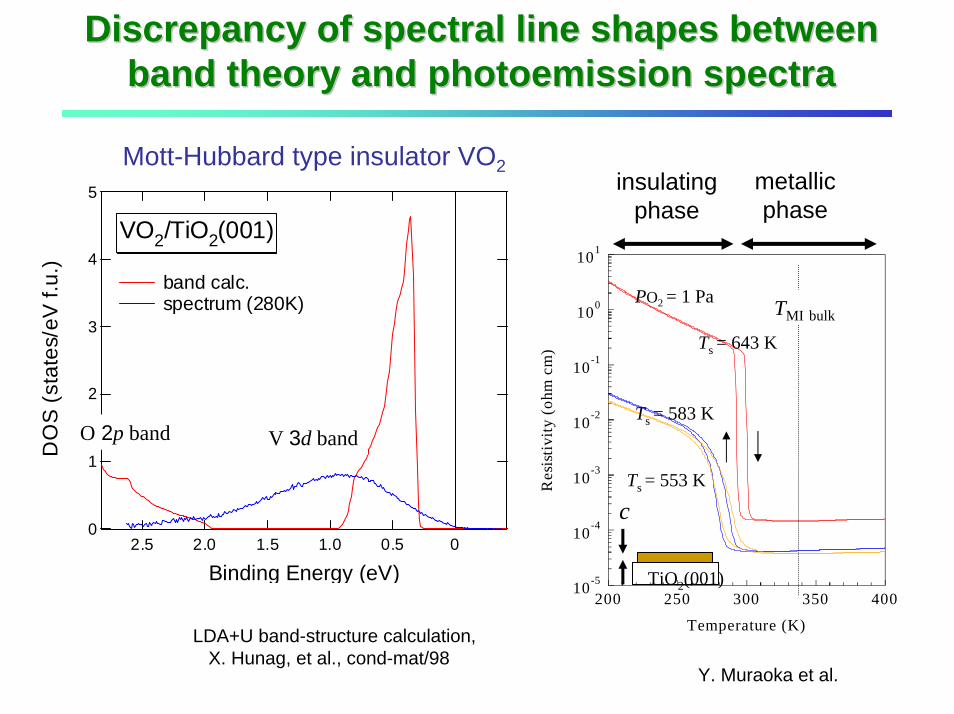
Discrepancy of spectral line shapes between Discrepancy of spectral line shapes between band theory and photoemission spectraband theory and photoemission spectra
Mott-Hubbard type insulator VO25
4
3
2
1
0
DO
S (s
tate
s/eV
f.u.
)
2.5 2.0 1.5 1.0 0.5 0
Binding Energy (eV)
band calc. spectrum (280K)
VO2/TiO2(001)
O 2p band V 3d band
10-5
10-4
10-3
10-2
10-1
100
101
Res
istiv
ity (
ohm
cm
)
400350300250200
Temperature (K)
Ts = 553 K
Ts = 583 K
Ts = 643 K
TiO2(001)
c
PO2 = 1 Pa TMI bulk
metallic phase
insulating phase
LDA+U band-structure calculation, X. Hunag, et al., cond-mat/98
Y. Muraoka et al.
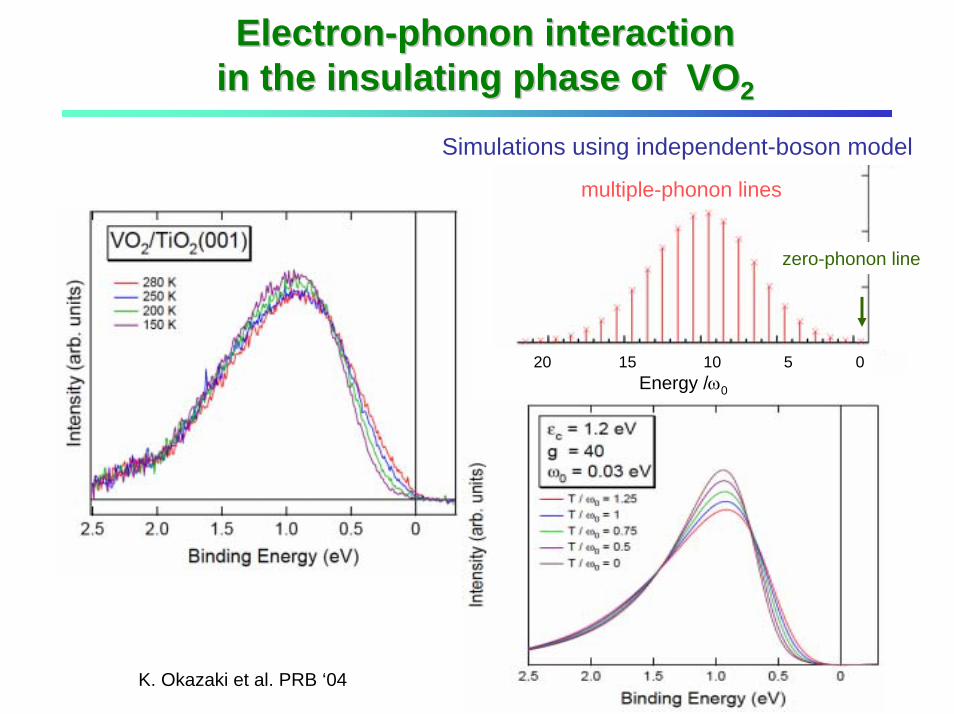
20 15 10 5 0Energy /ω0
zero-phonon line
multiple-phonon lines
ElectronElectron--phonon interaction phonon interaction in the insulating phase of VOin the insulating phase of VO22
Simulations using independent-boson model
K. Okazaki et al. PRB ‘04
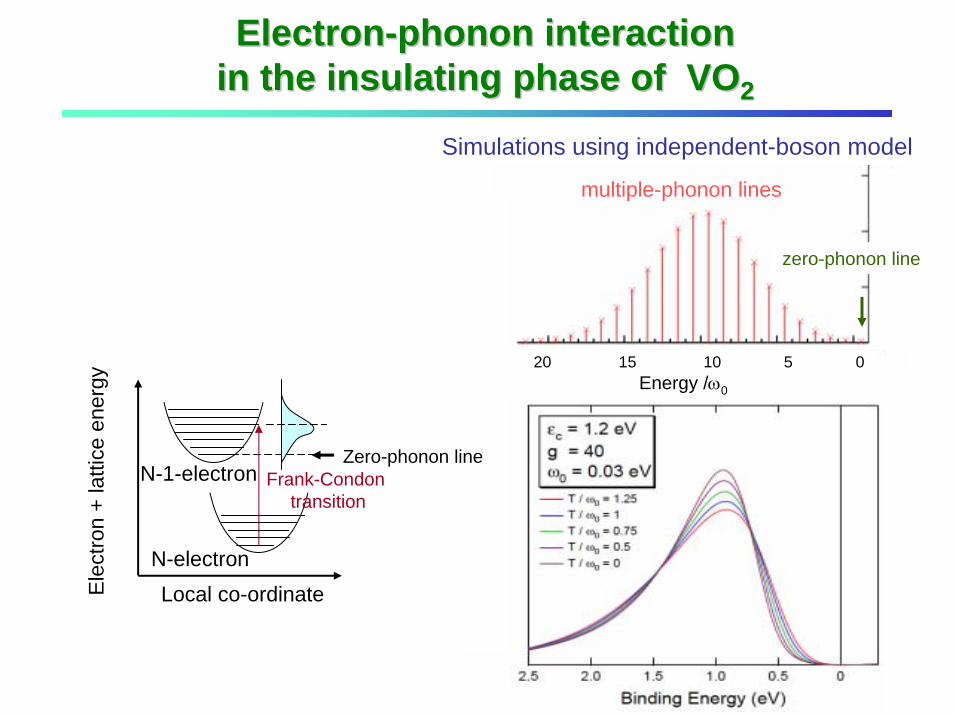
20 15 10 5 0Energy /ω0
zero-phonon line
multiple-phonon lines
Local co-ordinateEle
ctro
n +
latti
ce e
nerg
y
Zero-phonon line
N-electron
N-1-electron Frank-Condontransition
ElectronElectron--phonon interaction phonon interaction in the insulating phase of VOin the insulating phase of VO22
Simulations using independent-boson model
Recommended

















